

Significance
Influenza A viruses cause significant morbidity and mortality in humans and swine, yet we have limited understanding of how influenza A viruses evolve within pigs in Southeast Asia. Here, we analyzed influenza A virus genomic data collected from pigs in Cambodia, combined with a global dataset, and show the prolonged cryptic circulation of diverse swIAV lineages in Southeast Asia, including some that resulted from reverse zoonotic transmission to pigs that occurred up to 20 B.P. We uncover the breadth of H1 and H3 diversity present in Cambodian swine, identifying the complex genomic reassortment processes and spatial movement of viruses across geographical borders that drive the emergence of new reassortant viruses, posing an undetermined risk to both humans and pigs.
Keywords: pandemic, zoonotic, European avian-like virus, evolution
Abstract
Swine are a primary source for the emergence of pandemic influenza A viruses. The intensification of swine production, along with global trade, has amplified the transmission and zoonotic risk of swine influenza A virus (swIAV). Effective surveillance is essential to uncover emerging virus strains; however gaps remain in our understanding of the swIAV genomic landscape in Southeast Asia. More than 4,000 nasal swabs were collected from pigs in Cambodia, yielding 72 IAV-positive samples by RT-qPCR and 45 genomic sequences. We unmasked the cocirculation of multiple lineages of genetically diverse swIAV of pandemic concern. Genomic analyses revealed a novel European avian-like H1N2 swIAV reassortant variant with North American triple reassortant internal genes, that emerged approximately seven years before its first detection in pigs in 2021. Using phylogeographic reconstruction, we identified south central China as the dominant source of swine viruses disseminated to other regions in China and Southeast Asia. We also identified nine distinct swIAV lineages in Cambodia, which diverged from their closest ancestors between two and 15 B.P., indicating significant undetected diversity in the region, including reverse zoonoses of human H1N1/2009 pandemic and H3N2 viruses. A similar period of cryptic circulation of swIAVs occurred in the decades before the H1N1/2009 pandemic. The hidden diversity of swIAV observed here further emphasizes the complex underlying evolutionary processes present in this region, reinforcing the importance of genomic surveillance at the human–swine interface for early warning of disease emergence to avoid future pandemics.
Global swine populations play an integral role in the emergence of zoonotic influenza and pandemic viruses by providing a suitable environment for reassortment and human adaptation of avian and mammalian influenza viruses (1–3). As pork production has dramatically increased over the past 50 y (SI Appendix, Fig. S1), international trade and movement have further amplified the zoonotic risks at the human–pig interface (4–6). In 2020, Southeast Asian countries had estimated annual swine production of 1.9 million head in Cambodia, 4.3 million in Laos, 7.5 million in Thailand, and 22 million in Vietnam (6, 7). The impact of intensified commercial swine production for the emergence and spread of swine influenza A virus (swIAV) is still unknown, particularly in Southeast Asia where epidemiological and genetic data on swIAV are sparse.
Establishing the exact origin of the human H1N1/2009 pandemic (H1N1/pdm09) virus was prevented by the lack of available sequence data from swine (1, 2, 8, 9). Dated phylogenies showed that the swIAV ancestors of the H1N1/pdm09 virus were unsampled for at least 10 y (8). It was not until 2016 that reassortants of European avian-like (EA) and North American swine viruses found in Mexican swine were identified as H1N1/pdm09 precursors (10). Analysis of swIAV viruses collected in Hong Kong from 1998 to 2010 showed that virus population dynamics were dominated by repeated intercontinental movement and reassortment of swIAVs that produced diverse virus populations (3). Population-based simulation of virus movements conducted in 2015 suggested that swIAV diversity from large pig populations in China and the United States, which are the source of most swIAV genomes, did not represent global diversity and that surveillance efforts should be extended to other regions (11). These studies demonstrate the central role of global swine movements in facilitating the mixing of independently evolving swIAV populations that leads to reassortment and the generation of novel swIAV strains.
The H1N1/pdm09 has become a critical contributor of genetic segments for generating novel swIAV variant strains in recent years. Large-scale swIAV surveillance across 17 European countries by Henritzi et al. (12) uncovered at least 31 new genotypes generated through substantial gene reassortment with H1N1/pdm09, and showed that European avian-like (EA) H1N1 reassortant viruses were prevalent in swine populations since 2015. The same study showed that human sera expressed varying levels of cross-reactivity toward H1N1/pdm09 but reacted poorly with EA H1N1 swine viruses, suggesting a lack of preexisting immunity against EA H1N1 viruses in humans. More recent extensive surveillance by Sun et al. (13) found an IAV prevalence of 0.45% (136/29,918) in pig slaughterhouses in China between 2011 and 2018, with the majority (122/136) belonging to EA H1N1 virus. Their study revealed the circulation of 6 novel EA H1N1 reassortant viruses in pigs, designated as genotype G1–G6 viruses, which carry segments from the H1N1/pdm09 and triple-reassortant internal gene (TRIG) viruses. Since 2016, the G4 EA H1N1 virus has been found in many provinces of China and has become predominant in swine populations. Experimentally, the G4 EA H1N1 virus replicates efficiently in human airway epithelial cells and is highly infective and transmissible among ferrets, and demonstrates low antigenic cross-reactivity with circulating human H1N1/pdm09 viruses, highlighting their pandemic threat (13).
In addition to distinct swIAVs lineages discovered in Europe and China, multiple lineages of swIAV circulate in pigs across Southeast Asia (14–20). In Thailand, the H1N1/pdm09 virus was the prevailing subtype detected in swine from 2011 to 2014 (16), cocirculating with classical swine (CS) H1, EA H1N1, and H3N2 lineages (15). Multiple H1 and H3 genotypes have been generated in swine from Thailand through frequent reassortment between these lineages (14, 15). Surveillance studies from Vietnam between 2010 and 2013 detected H1N1, H1N2, and H3N2 subtype IAV in swine (17–19). While in Myanmar, H1N1/pdm09, H1N2, and multiple H3 lineages have been detected from 2017 to 2019 (20).
Influenza A viruses exhibit broad host tropism outside of the natural reservoir of wild waterfowl and can infect a wide range of species, including humans, swine, and domesticated fowl such as chicken, that have frequent human contact in live-bird markets throughout Asia (21). There have been multiple documented cases of zoonotic (8, 22–25) and reverse-zoonotic (26–30) transmission and establishment of IAV in multiple hosts. Cambodian smallholders often keep swine alongside chicken and ducks (31), creating an environment favorable to cross-species transmission (32, 33). However, there are very few swIAV sequences available from Cambodia and the rest of Southeast Asia, and the lack of systematic surveillance infrastructure has created gaps in our ability to identify emerging pathogens in pigs and their potential for zoonotic spillover (7).
In this study, we collected over 4,000 nasal swabs from pigs in Cambodia to reveal the prolonged undetected circulation and establishment of diverse swIAV lineages in Cambodia, including human H1N1/pdm09 and H3N2 viruses introduced to swine through reverse-zoonosis. We uncovered an EA H1N2 reassortant genotype generated through the intercontinental movement of different viral lineages, and phylogeographic reconstruction demonstrated that China is the leading source contributing to dissemination of EA swine viruses in Asia. Our results unmask an increasingly complex genomic landscape of swIAV in Southeast Asia that is shaped by repeated introduction and reassortment of virus lineages, a process that is known to heighten pandemic risk.
Results
Detection and Characterization of Swine Influenza A Viruses in Slaughterhouse Pigs.
From March 2020 to July 2022, swine influenza surveillance was conducted in 18 pig slaughterhouses in Cambodia. We collected a total of 4,089 nasal swabs from individual pigs from different districts in four neighboring provinces (Kampong Speu, Kandal, Phnom Penh, and Takeo) (Fig. 1). Of these, 72 sampled pigs (1.8%) were positive for influenza A virus by RT-qPCR (Table 1), in range with similar studies that sampled from slaughterhouses that had IAV detection rates of 1.3 to 1.5% in pigs (13, 34–36). Kandal province exhibited a higher positivity rate of IAV by RT-qPCR (4.5%) compared to the three other provinces (0.2 to 1.8%).
Fig. 1.

Geographical area of swine influenza surveillance study conducted in Cambodia, 2020 to 2022. Striped lines indicate the location by district (D1 to D9) of sampled pig slaughterhouses in Kampong Speu, Kandal, Phnom Penh, and Takeo provinces.
Table 1.
Quantitative reverse transcription PCR (RT-qPCR) positivity rate of influenza A viruses from pigs in Cambodia, 2020 to 2022
| RT-qPCR | ||
|---|---|---|
| Sampling locations | No. of nasal swab tested | No. of IAV-positive nasal swab (%) |
| Takeo Province | ||
| District 1 | 769 | 17 (2.2) |
| District 2 | 242 | 0 (0) |
| subtotal | 1,011 | 17 (1.7) |
| Kampong Speu Province | ||
| District 3 | 870 | 3 (0.3) |
| District 4 | 382 | 0 (0) |
| subtotal | 1,252 | 3 (0.2) |
| Phnom Penh Province | ||
| District 5 | 539 | 2 (0.4) |
| District 6 | 157 | 9 (5.7) |
| District 7 | 391 | 8 (2.1) |
| subtotal | 1,087 | 19 (1.8) |
| Kandal Province | ||
| District 8 | 258 | 19 (7.4) |
| District 9 | 481 | 14 (2.9) |
| subtotal | 739 | 33 (4.5) |
| Total | 4,089 | 72 (1.78) |
We obtained and analyzed complete or partial swine influenza genomes from 45 nasal swab samples (SI Appendix, Table S1). Both H1 and H3 HA subtypes as well as N1 and N2 NA subtypes were identified during the sampling period, in combination with 10 HA and 9 NA individual coinfections (Fig. 2A). H1N1 subtype was predominant, being present in 37 (82.2%) of 45 sequenced pig samples (Fig. 2A). To trace the evolutionary origin of individual gene segments of swIAV from Cambodia, maximum likelihood phylogenies were reconstructed for each gene segment: H1-HA (n = 1,009), N1-NA (n = 986), H3-HA (n = 766), N2-NA (n = 773), PB2 (n = 924), PB1 (n = 923), PA (n = 915), NP (n = 927), MP (n = 927) and NS (n = 927).
Fig. 2.
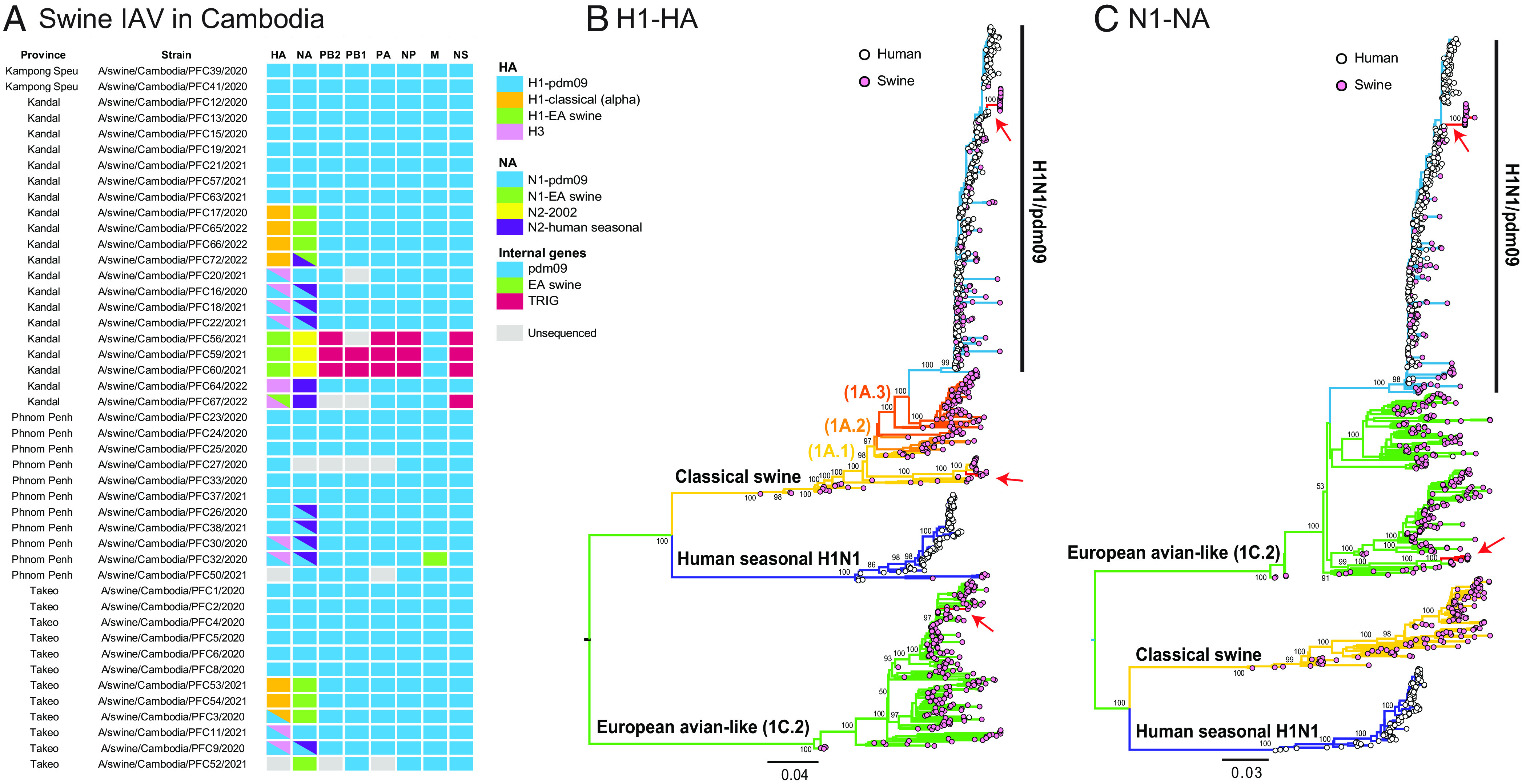
Diversity of swIAV strains. (A) Genomic constellations for each gene segment of swIAV strains from sampled pigs in Cambodia. Colored boxes represent the major swIAV lineages detected in this study. (B and C) Evolutionary relationships of the H1-HA (B) and N1-NA (C) genes of human and swine influenza viruses inferred by maximum likelihood method in RAxML. Human and swine influenza viruses are denoted by pink and white tip circles, respectively. Colored branches represent different IAV lineages; red branches and arrows indicate swIAV gene sequences generated from this study. Bootstrap support (≥50) are indicated at major nodes.
Phylogenetic inference indicated that diverse lineages of H1 were in circulation within Cambodia. The majority of the new H1 and N1 sequences were derivatives of the human H1N1/pdm09 lineage (Fig. 2 B and C). Genomic analysis revealed a unique reassortant European avian-like (EA) H1N2 swine influenza subtype in four samples. This H1N2 subtype possessed an EA H1-HA gene (shown by green boxes/branches in Figs. 2 A and B and 3A), while the acquisition of the N2 gene was from swine-like N2-2002 (denoted by yellow boxes in Fig. 2A) but nested within human seasonal N2. Classical swine (CS) H1N1 viruses were detected in seven swine samples, all with HA genes belonging to the CS H1 alpha lineage (orange boxes in Fig. 2A). Notably, these CS H1N1 viruses possessed the EA swine N1 gene (green boxes/branches in Fig. 2 A and C). In one sample (A/swine/Cambodia/PFC3/2020) the HA of CS H1 and H1N1/pdm09 were both detected.
Fig. 3.

Time-scaled phylogenies of H1-HA genes of human and swine influenza viruses. (A) European avian-like H1N1 swine viruses, represented by green branches. (B) Human H1N1/pdm09 viruses, represented by blue branches. (C) Classical H1N1 swine viruses, represented by orange/yellow branches. Red branches represent swIAV sequences from this study. Persistent amino acid substitutions are indicated at the nodes. Bayesian PP values (≥0.95) are shown at major nodes.
We detected H3 influenza subtypes in 10 (22.2%) of 45 sequenced pig samples from Cambodia. Eight swine samples were codetected with H1N1/pdm09 and H3-HA viruses (shown by blue–pink gradient boxes in Fig. 2A). Six of these samples contained both an N2-NA gene and the N1 gene of H1N1/pdm09 virus (Fig. 2A). Another two samples (A/swine/Cambodia/PFC26/2020 and A/swine/Cambodia/PFC38/2021) showed evidence of H1N1/pdm09 and coinfection with H3N2 NA genes, but an H3 sequence was not obtained. These H1N1/pdm09 viruses were present in slaughterhouse pigs in all four provinces and H3N2 viruses were present in three provinces, in contrast to CS H1N1 and EA H1N2 subtypes, which were mostly observed in Kandal and Takeo. As such, we found nine distinct swIAV HA and NA lineages are currently cocirculating in pig populations within Cambodia, facilitating the mixing of diverse genetic lineages through gene reassortment.
Genesis of EA H1N2 Reassortant Virus in Swine.
We next unraveled the parental origin and gene constellations of the European avian-like H1N2 reassortant viruses. Four swine samples collected from Kandal Province (A/swine/Cambodia/PFC56/2021, A/swine/Cambodia/PFC59/2021, A/swine/Cambodia/PFC60/2021, A/swine/Cambodia/PFC67/2022) were assigned to EA H1N2 subtype (Figs. 2A and 3A). The N2 of EA H1N2 was most closely related to the North American swine N2-2002 lineage. In contrast, the internal genes of EA H1N2 subtype were acquired from the North American triple-reassortant internal gene (TRIG) cassette, except the MP gene that was derived from the H1N1/pdm09 lineage (Fig. 2A and SI Appendix, Figs. S2–S7). The gene constellations are distinct from previously described EA swine viruses from Europe and China, which contain mostly H1N1/pdm09-origin internal genes, indicating that the contemporary EA H1N2 reasssortant viruses in Cambodia arose from diverse ancestral origins.
Our temporal phylogeny of the H1-HA gene showed that three out of four EA H1 swine sequences from Cambodia clustered to form a monophyletic group [posterior probability (PP) = 1.00, denoted by red branches in Fig. 3A], with a mean TMRCA estimated around June 2021 (95% HPD intervals: April 2021 to August 2021, Table 2). These three EA H1 sequences were most closely related to the EA H1N1 G4 genotype viruses found in swine from Liaoning, China between 2014 and 2016. The introduction of G4 H1-HA genes into pigs from Cambodia may have occurred between October 2014 and June 2021, up to 7 years before its first detection in Cambodia. Comparatively, one EA swine sample (A/swine/Cambodia/PFC67/2022) collected in 2022 appeared to be phylogenetically segregated and it was more related to recent viruses from Tianjin and Liaoning in 2018 to 2020, reflecting a possible independent introduction (Fig. 3A and SI Appendix, Fig. S8).
Table 2.
Estimated times to most recent common ancestor (TMRCA) of swIAV lineages from pigs in Cambodia
| Time to most recent common ancestor (swIAV lineages in Cambodia) | Time to most recent common ancestor (stem) | ||||||
|---|---|---|---|---|---|---|---|
| Lineage | Mean | 95% lower HPD | 95% upper HPD | Mean | 95% lower HPD | 95% upper HPD | Undetected years |
| H1 CS | 27 Jun 2019 | 06 Nov 2018 | 18 Jan 2020 | 21 Jan 2015 | 06 Mar 2014 | 09 Dec 2015 | 4.43 |
| H1 EA | 24 Jun 2021 | 01 Apr 2021 | 29 Aug 2021 | 04 Oct 2014 | 20 Feb 2014 | 02 Dec 2014 | 6.72 |
| H1 pdm09 | 16 Feb 2020 | 16 Dec 2019 | 16 Mar 2020 | 21 Aug 2017 | 22 May 2017 | 26 Oct 2017 | 2.49 |
| H3 seasonal group 1 | 09 Feb 2021 | 19 Apr 2019 | 10 Nov 2021 | 17 Nov 2010 | 20 Dec 2009 | 24 Jun 2011 | 10.23 |
| H3 seasonal group 2 | 01 Jul 2017 | 17 Mar 2015 | 23 Apr 2019 | 25 Jan 2012 | 25 Mar 2010 | 26 Apr 2013 | 5.43 |
| H3 seasonal group 3 | 14 Mar 2015 | 18 Oct 2012 | 02 Dec 2017 | 07 May 2011 | 26 Apr 2010 | 25 Jan 2012 | 3.85 |
| N1 EA swine | 06 Nov 2018 | 09 Dec 2017 | 03 Aug 2019 | 12 Dec 2014 | 08 Oct 2013 | 05 Dec 2015 | 3.90 |
| N1 pdm09 | 25 Jan 2020 | 30 Oct 2019 | 16 Mar 2020 | 21 Nov 2017 | 30 Jul 2017 | 27 Feb 2018 | 2.18 |
| N2 seasonal (EA H1)* | 24 Jun 2021 | 25 Mar 2021 | 07 Aug 2021 | 28 Nov 2005 | 21 May 2004 | 26 May 2007 | 15.57 |
| N2 seasonal (CS H1) | 09 Aug 2012 | 13 Jun 2010 | 10 Aug 2014 | 10 Jan 2008 | 17 Mar 2006 | 08 Jul 2009 | 4.58 |
| N2 seasonal (human H3) | 16 Dec 2021 | 03 Aug 2021 | 06 Mar 2022 | 06 May 2008 | 07 Aug 2006 | 06 Nov 2009 | 13.61 |
*The corresponding HA of the N2 seasonal genes are indicated in Fig. 4E. Abbreviations: CS, classical swine; EA, European avian-like swine; HPD, highest posterior density; pdm09, human H1N1/2009 pandemic.
The EA H1-HA viruses have acquired mutations at a mean rate of 3.77 × 10−3 substitutions per site per year (s/s/y), which was similar to the rates estimated for the H1 of H1N1/pdm09 and CS viruses (mean rates of 3.90 × 10−3 s/s/y and 3.37 × 10−3 s/s/y, respectively). Notably, the G4 EA H1-HA lineage acquired sequential HA amino acid mutations since 2014. The HA mutation A135S defines the 2021 to 2022 Cambodian and related Liaoning EA viruses that circulated in 2014 to 2016 (Fig. 3A). Liaoning EA viruses from 2020 displayed extensive HA mutations (K47Q, K66E, T107A, K171R, I370T, R433K, K458R and I533V) than earlier strains (Fig. 3A), indicative of EA lineage diversification in Asia. In addition, a comparison of Cambodian EA H1N2 HA with other distantly related G4 (e.g. A/swine/Jiangsu/J004/2018) and G5 (e.g. A/Hunan/42443/2015) viruses showed they were identical at HA-binding residues (190-Thr and 225-Gly) and differed by only a single Ala-135-Ser (H1 numbering) mutation at HA antigenic sites (SI Appendix, Fig. S9).
The N2 genes of Cambodian EA H1N2 viruses were ultimately derived from an established swine lineage and a distinct human seasonal N2 lineage (PP = 1.00, Fig. 4E). Of note, the mean TMRCA was estimated around June 2021 (95% HPD intervals: March 2021 to August 2021, Table 2), which overlaps with the TMRCA of EA H1 gene lineage in pigs from Cambodia. These N1 sequences are most closely related to ancestral swine H3N2 viruses circulated in 2012–2016 from Canada (e.g., A/swine/Manitoba/D0208/2013) and North America (e.g., A/swine/Iowa/13E045/2013), but with a combined TMRCA of 2005 (Fig. 4E). Our data suggest that the North American swine N2 segment may have been introduced into Asia over 15 B.P., highlighting decades of unsampled diversity due to a lack of systematic surveillance of swine influenza viruses in Southeast Asia.
Fig. 4.
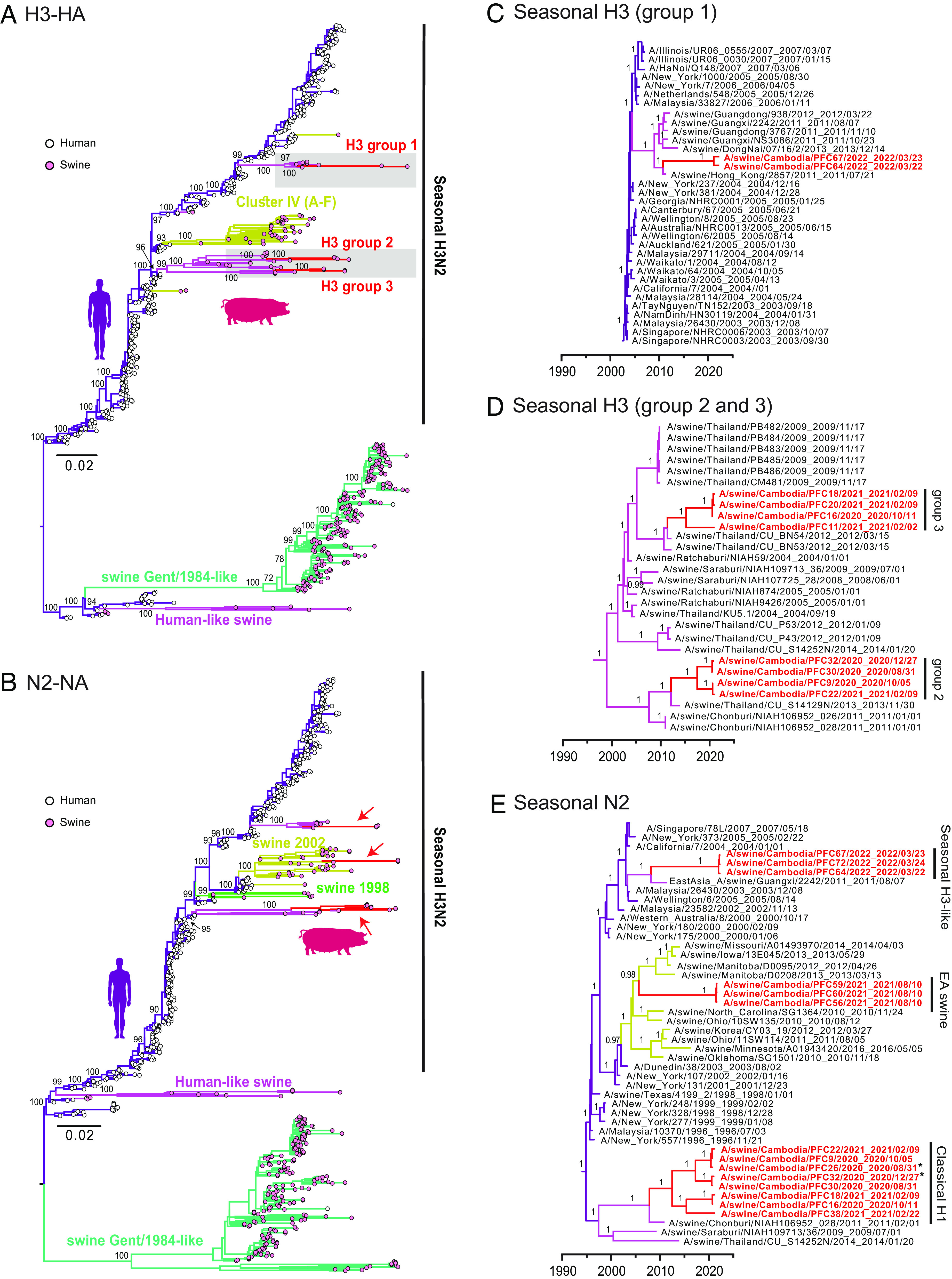
Phylogenies of H3-HA and N2-NA gene segments. (A and B) Evolutionary relationships of the H3-HA (A) and N2-NA (B) genes of influenza viruses inferred by maximum likelihood method in RAxML. Human and swine influenza viruses are denoted by pink and white tip circles, respectively. Colored branches represent different IAV lineages. Bootstrap support (≥50) are indicated at major nodes. (C and D) Time-scaled phylogenies of H3-HA genes. (E) Seasonal H3 avian-like H1N1 swine viruses. Red branches and arrows represent swIAV sequences generated from this study. Bayesian PP values (≥0.95) are shown at major nodes. *The corresponding HA gene of A/swine/Cambodia/PFC26/2020 is H1N1/pdm09, whereas that of A/swine/Cambodia/PFC32/2020 contained both H1N1/pdm09 and H3.
Most internal genes (PB2, PB1, PA, NP, MP, and NS) of EA H1N2 virus from Cambodia pigs were found to have descended from North American TRIG lineage (Fig. 5). Phylogenetic positions of these internal genes of Cambodian EA viruses were either basal or nested within the CS H1N1 viral lineage. Of note, the NS gene of Cambodian EA viruses were related to the genes of G4 viruses from pigs in China, including recent Liaoning strains (e.g., A/swine/Liaoning/HLD1795/2020); these viruses collectively clustered within the TRIG lineage. In contrast, the MP gene of Cambodian EA viruses were most related to swine H1N1 viruses from China and North America, and nested within the human H1N1/pdm09 lineage. Interestingly, the H1, N1, most TRIG internal genes and MP gene of the three Cambodia EA swine exhibited overlapping TMRCA dates, estimated around October 2020 to August 2021 (SI Appendix, Fig. S10). However, the associated PB2 showed an earlier TMRCA date in late 2017, indicative of independent introduction of the gene segment.
Fig. 5.
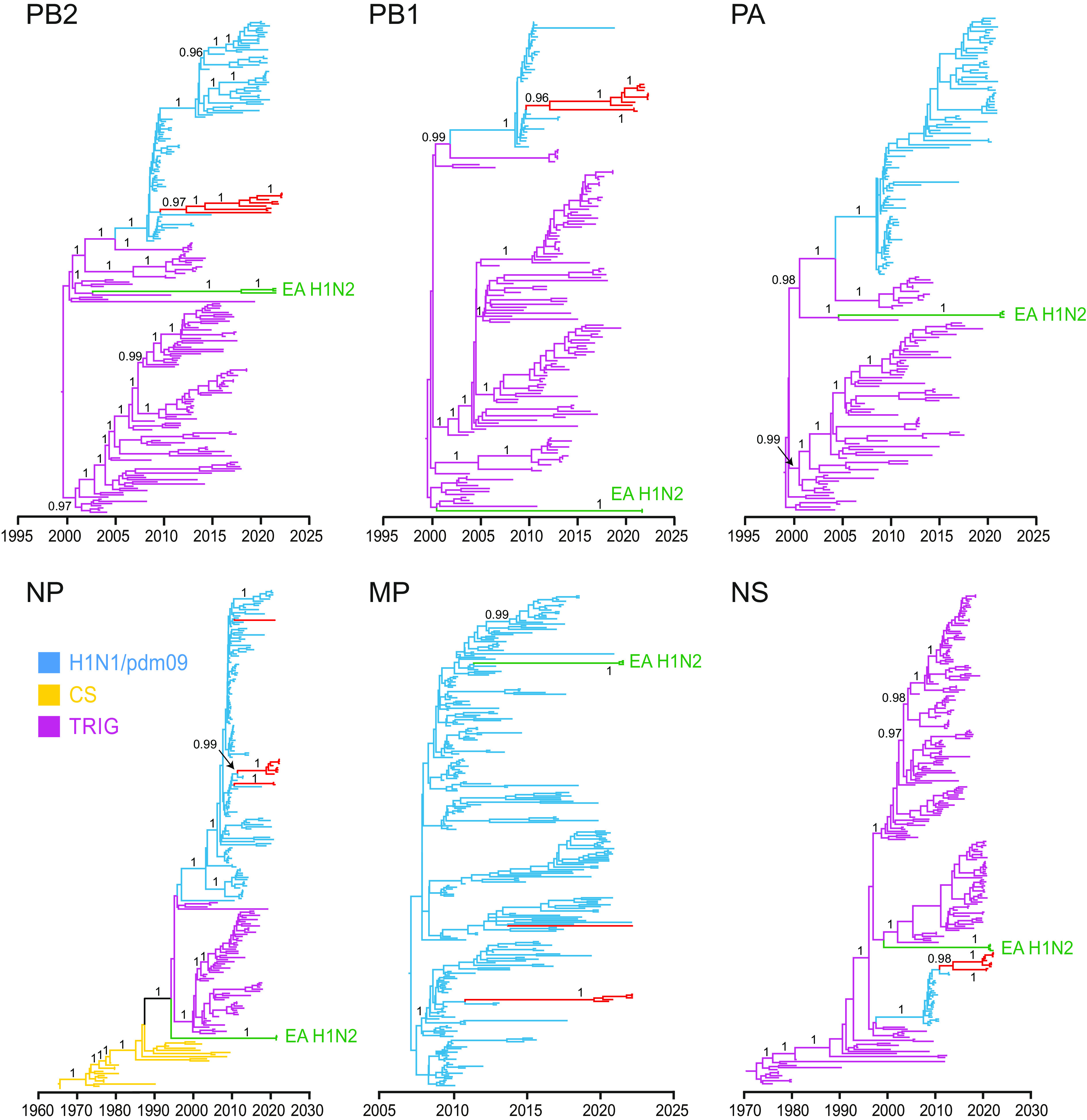
Temporal phylogenetic trees highlighting the origin of EA H1N2 internal genes. Purple, blue, and yellow branches represent TRIG, H1N1/pdm09 and CS virus lineages, respectively. Red and green branches represent swIAV and EA H1N2 sequences generated from this study, respectively. Bayesian PP values (≥0.95) are shown at major nodes.
Reverse Zoonosis of H1N1/pdm09-Like Virus in Pigs from Cambodia.
Most of the H1 and N1 sequences from pigs in Cambodia belonged to human H1N1/pdm09 lineage, forming a distinct monophyletic sublineage (PP = 1.00, H1 and N1 in Fig. 3B and SI Appendix, Fig. S11A, respectively) that nested within the H1N1/pdm09 HA and NA lineages. The H1 and N1 gene segments had an estimated mean TMRCAs of approximately February 2020 and January 2020, respectively (both PP=1.00, denoted by red branches in Fig. 3B, SI Appendix, Fig. S11A, and Table 2). In the HA phylogeny, the most closely related strains are human H1N1/pdm09 viruses from Thailand collected in November 2017, and dating suggests that human-to-swine transmission occurred as early as August 2017, followed by enzootic spread of a genetically distinct H1N1/pdm09 sublineage among pigs in Cambodia. During this period, the swine H1N1/pdm09 viruses in Cambodia acquired six H1-HA amino acid substitutions (K120E, N162S, M227I, T232I, K308E and N451S) (Fig. 3B) and eight N1-NA substitutions (T9A, N50S, Q78K, P126H, I163V, T332I, I374V and V394I) (SI Appendix, Fig. S11A). These HA and NA mutations may be due to adaptation to pigs following reverse zoonosis; however, it is not known if they may have arisen through virus circulation in Cambodian pigs or spread from other countries. The remaining internal genes (PB2, PB1, PA, NP, MP, and NS) were predominantly derived from human H1N1/pdm09 viruses (SI Appendix, Figs. S12–S17), except for A/swine/Cambodia/PFC32/2020 where the MP gene was from an EA H1N1 virus.
Cocirculation of Multiple Classical Swine H1N1 and H3N2 Lineages in Pig Populations.
Two enzootic swIAV viruses, CS H1 and H3N2, were also detected in pigs sampled from Kandal, Phnom Penh, and Takeo provinces. The CS H1 subtype was detected in 7 swine samples, all HA sequences grouped into a monophyletic clade, which nested within the classical swine H1 alpha 1A.1.2 lineage (PP =1 .00, Fig. 3C). The accompanying N1 gene is derived from EA viruses and the internal genes are from H1N1/pdm09 virus. The mean TMRCA of the Cambodian CS H1 sequences was estimated around June 2019 (Fig. 3C and Table 2) and they grouped with CS H1 viruses from Thailand detected from 2009 to 2018. The introduction of the CS H1 gene from Thailand to Cambodia likely occurred as early as January 2015. The EA N1 sequences of these samples (with an additional NA gene from A/swine/Cambodia/PFC52/2021) were most closely related to Thailand swine 2017 viruses and had a TMRCA of November 2018 (PP = 1.00, SI Appendix, Fig. S11B and Table 2), suggesting independent introduction into Cambodia than the slightly older CS H1 gene. These CS/EA H1N1 reassortants have also acquired amino acid mutations near the HA receptor-binding sites (K130R, Y230H and Y261H) and in the NA protein (I53M, V62L and Q308H). The multiple origins of gene segments in these viruses highlights the frequent reassortment between diverse virus lineages that occurs in the region.
A high diversity of A/H3N2 subtype virus was found in 10 swine samples in Cambodia, with three distinct groups of H3N2 viruses circulating that all clustered with swine H3N2 viruses that were ultimately derived from human seasonal H3N2 (Fig. 4 A and B). Interestingly, H3N2 virus was codetected with H1N1/pdm09 HA genes in eight samples. Group 1 H3 contained two samples (A/swine/Cambodia/PFC64/2022 and A/swine/Cambodia/PFC67/2022), with the mean TMRCA estimated in February 2021 (Table 2). Both viruses were closely related to other swine H3 2011 to 2013 viruses from southern China (Guangdong and Guangxi) and southern Vietnam (Fig. 4C). Group 2 and 3 H3 each contained four viruses with estimated mean TMRCAs of July 2017 and March 2015, respectively (Fig. 4D and Table 2). Both groups were independently derived from Thailand swine viruses detected in swine since 2004. Prior to detection in swine, this H3N2 lineage was detected in humans as early as 1996, indicating there were multiple reverse-zoonoses of human H3N2 to swine that occurred between 1996 to 2004. The currently circulating swine N2 sequences from Cambodia also originated from human seasonal H3N2 viruses (Fig. 4E). Eight Cambodian swine N2 sequences formed a clade with an estimated TMRCA in August 2012 (Table 2) and are related to Thailand swIAVs from 2009 to 2014 (Fig. 4E). These N2 sequences may have been introduced from Thailand as early as January 2008.
Spatiotemporal Diffusion Patterns of EA Swine Viruses.
To determine the migration processes of EA swine H1-HA viruses, we reconstructed ancestral geographical locations and inferred diffusion patterns between locations using a discrete phylogeographic model. Phylogeographic reconstruction based on 1,073 sequences revealed that EA swine viruses circulated for decades among pigs in Europe until the early 2000s when we observed sporadic introductions into south central China and Southeast Asia (Fig. 6 A and B). Since 2010, south central China became the dominant source for EA swine virus transmission, displaying strong diffusion links from south central China to east China, with strong migration support (BF > 10,000, SI Appendix, Table S2). Significant migration pathways with higher diffusion rates (2.11 to 3.83, SI Appendix, Table S3) were also present from east China to other parts of China, including north China, northeast China, south central China (Fig. 6B). After 2016, there was an increased circulation of EA swine viruses between different regions of China, leading to a marked expansion of EA swine lineage diversity. During this period, strong migration links were also evident from south central China to northeast China, and from northeast China to Southeast Asia (SI Appendix, Table S2), including Cambodia.
Fig. 6.
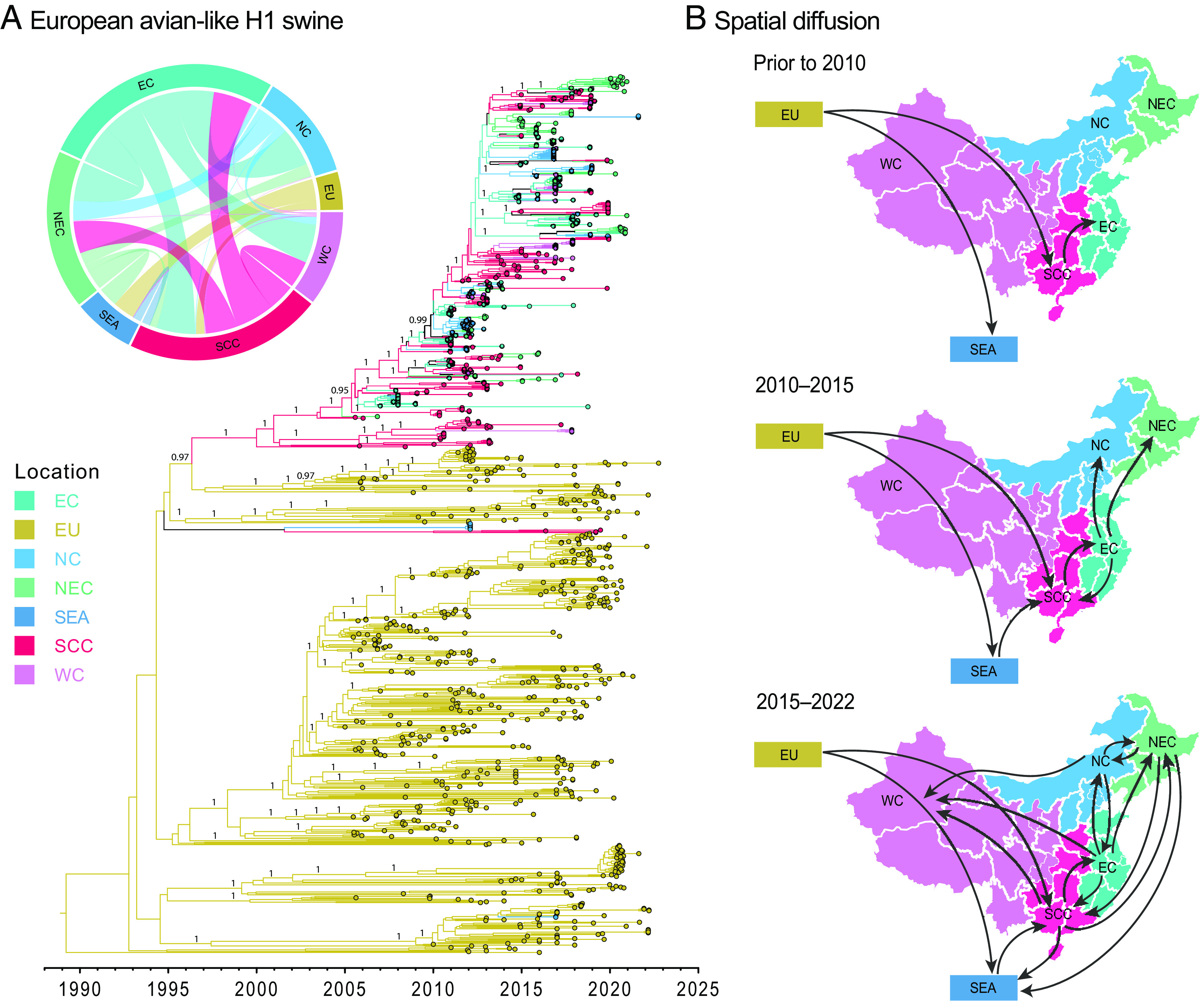
Discrete phylogeographic reconstruction of EA-like swine H1 diffusion dynamics. (A) Time-scaled maximum clade credibility tree of the Bayesian phylogeographic inference. Branch color corresponds to different geographic locations. The insert displays the circular migration plot based on log Bayes factor support between pairwise locations (see SI Appendix, Table S2 for support values). Abbreviations: EC, east China; EU, Europe; NC, north China; NEC, northeast China; SEA, Southeast Asia; SCC, south central China; WC, west China. Bayesian PP values (≥0.95) are shown at major nodes only. (B) Spatial migration pathways of EA swine viruses through time.
It is of note that there is an uneven sampling across geographical regions due to the limited number of all publicly available EA H1 sequences from 2005 to 2022 (n = 1,584 in total) (SI Appendix, Fig. S18). An abundance of EA H1 sequences can be found in Europe, although fewer exist from Southeast Asia and regions of China like west China. This could possibly be due to a scarcity or absence of active surveillance for swine viruses in these areas per se. To better understand the effect of sampling bias, we downsampled the large dataset to a smaller subset (n = 534) to achieve a relatively even sampling among locations (SI Appendix, Fig. S19) and compared the two sets of sequences. Our phylogeographic reconstructions indicate that the smaller subset inferred 15 strongly supported dispersal pathways, whereas the larger dataset yielded 16 significant pathways. Both datasets displayed relatively similar dispersal patterns, for instance, strongly supported diffusion pathways were inferred from east China to north China, northeast China, south central China and west China, and from south central China to east China and northeast China (SI Appendix, Fig. S19). In addition, the two datasets consistently showed that the highest migration rate was inferred from east China to south central China, at 3.83 and 3.43, respectively (SI Appendix, Table S4). The impact of sampling bias can inevitably lead to some level of discrepancies in the reconstruction of spatial patterns, for instance, the large dataset displayed strong diffusion links from south central China to west China, with a moderately higher diffusion rate (0.68), in contrast to the absence of statistical support from south central China to west China and a lower diffusion rate (0.21) as inferred by the smaller subset of sequences.
We further explore the potential factors driving the spread of EA H1 viruses, six predictors that related to pigs were selected, including the number of pigs, pig production, and pork consumption. We employed a linear regression method using a generalized least squares model (GLS) and a Bayesian inference using a generalized linear model (GLM) to evaluate the association between predictors and diffusion rates. The GLS model suggests pork production at origin was positively correlated (P-value < 0.1) to the spatial spread of EA H1N1 swIAV (SI Appendix, Fig. S20). In contrast, the consumption of pork in both origin and destination regions was negatively correlated with virus spread. These results were corroborated with the GLM model, inferring that pork production at origin was strongly related to viral migration (PP > 0.9, SI Appendix, Fig. S20). Phylogeographic inference is a powerful tool to assess potential factors that drive virus spread; however, inference with predictors is sensitive to the availability and reliability of predictor data, and therefore should be interpreted with caution.
Discussion
Influenza A viruses are an integral part of a complex ecosystem that results in viral emergence and zoonotic diseases. Pigs are natural drivers for the emergence of novel influenza virus strains that cross species boundaries by means of shuffling gene segments between avian, swine, and human hosts. While much is known about the genomic diversity of swIAVs in Europe and North America (12), the evolutionary dynamics of swIAV populations in Southeast Asia remains largely unknown.
Despite the difficulties posed by COVID-19, we conducted an extensive longitudinal study of influenza surveillance in pigs from slaughterhouses across four major provinces in Cambodia—spanning March 2020 through July 2022. Our study provides a detailed picture of virus circulation and dynamics in this region; showing diverse groups of swIAV viruses in pig populations and tracing the evolutionary footprints of their ancestral origins.
Our study highlights that circulating swIAV lineages in Southeast Asia acquired reassorted gene segments from diverse geographical origins, facilitated by intercontinental and intracontinental spread of previously segregated swIAV lineages, the same process that led to the emergence of the H1N1/pdm09 virus in Mexico (10). This process also gave rise to the novel EA H1N2 subtype virus that likely emerged in late 2014, approximately 7 y before its first detection in Cambodia in 2021. This novel EA H1N2 virus has a unique gene constellation comprised of a swine-origin EA G4-like H1-HA, an N2-HA originating from human viruses and internal genes from TRIG viruses. In contrast, the ancestral EA G4 H1N1 virus that emerged from pigs in Henan, China in 2011 (13), contained EA virus H1 and N1 genes, human H1N1/pdm09-derived internal genes and a TRIG-derived NS gene, and has since spread to other parts of China including Jilin, Liaoning and Tianjin (37). EA G4 H1N1 viruses bind preferentially to alpha 2,6-linked sialic acid receptors, they replicate efficiently in human airway epithelial cells and are highly transmissible among ferrets, and they demonstrate low serological cross-reactivity with human H1N1/pdm09 viruses, all of which highlights their zoonotic potential (12, 13). While the molecular characterization of EA H1N2 viruses shows identical HA receptor–binding residues and suggests a similar antigenic profile to EA G4 H1N1 viruses, virus isolation restrictions in the present study have limited our ability to characterize these viruses. Further studies are needed to understand the zoonotic potential and pandemic threat of the EA G4-like H1N2 viruses, including determining serological cross-reactivity with human viruses and assessing replicative fitness and transmissibility in animal models.
Our study emphasizes the importance of early identification of novel swIAV lineages and highlights concerns surrounding interregional spread of swIAVs as a key driver in the emergence of new virus lineages, particularly in Southeast Asia where surveillance is sporadic. We also found multiple instances of CS H1 coupled with EA N1. Ancestral sequences to this viral lineage are primarily present in swine populations from Asia (China and Thailand) and North America, providing more evidence of global intermixing of previously distinct virus populations. Given the lack of systematic pig surveillance, it is likely that this classical swine virus had circulated undetected in Cambodia since its introduction in 2017.
Pig value chains play a critical role in maintaining swine-to-swine virus transmission, regional and global migration, and the reassortment events that result from these activities. These prompt the establishment of diverse swIAV lineages among pig populations, including human influenza viruses that pose a constant threat for the reintroduction of antigenically and genetically distinct swIAVs being transmitted into humans (11, 12, 38, 39). We found that H1N1/pdm09 virus is circulating enzootically in Cambodian swine herds. Since the H1N1/2009 pandemic, multiple H1N1/pdm09 virus spillovers back to pigs have occurred, seeding novel H1N1 reassortant viruses across the world (40–42). We also detected multiple reverse zoonotic H3 lineages in Cambodian pigs that had circulated undetected for about 10 y. Follow-up experiments are needed to determine if the Cambodian H3 clades are antigenically distinct. The independent evolutionary trajectories of these viruses in pigs are recognized as a future pandemic threat (43). Evidence of little to no antigenic cross-reactivity to currently circulating human influenza A viruses would indicate the potential for reintroduction of these viruses to susceptible human populations.
Pork is a major component of nutrition in many parts of Asia. The rapid growth in meat consumption is driven by increasing incomes, population sizes, and rapid urbanisation in developing countries (44). However, a number of pig diseases such as influenza, porcine reproductive and respiratory syndrome and African swine fever viruses have caused significant economic loss in swine industries (45). As swine-rearing operations in most countries continue to expand and intensify, routine and sustained surveillance in pigs is indispensable in identifying new viruses so that their zoonotic risk can be assessed. The current methods of disease surveillance are not fit for this purpose, primarily because sampling individual animals is expensive and time consuming. As most of the funding for this work in Southeast Asia comes from research grants, the efforts are also sporadic, which is reflected in the sparse swIAV genomic data available from the region. It is therefore critical that more efficient and continuous surveillance methods such as metagenomic surveillance (46) of air and wastewater samples in farms and slaughterhouses are integrated with automated analytical tools to rapidly provide information on changes in the spatiotemporal occurrence of a broad range of human and animal pathogens. Such a system would serve to improve animal health through selection of efficacious vaccines, and aid in human health by monitoring viruses with the potential for zoonotic transmission.
Materials and Methods
Ethics Statement.
This study was approved by the ethics committees at LSHTM’s Institutional Review Board (approval number 16635) and Animal Welfare and Ethical Research Board (ref 2019-12), the National Ethics Committee for Health Research in Cambodia (ref 105), and the Human Research Protection Office (HRPO, ref A-21055) and Animal Care and Use Review Office of the USAMRDC Office of Research Protections.
Sample Collection and Sequencing.
From March 2020 to July 2022, we collected 4,089 nasal swab samples from pigs in 18 slaughterhouses located in four neighboring provinces (Kampong Speu, Kandal, Phnom Penh, and Takeo) of Cambodia (Fig. 2 and SI Appendix, Fig. S21). Viral RNA was extracted from samples using the QIAmp Viral RNA Mini Kit (Qiagen) and real-time RT-PCR was performed to detect the presence of influenza A virus (M gene) using AgPath-ID™ One-Step RT-PCR Kit (Thermo Fisher Scientific). Positive influenza samples were then selected for whole-genome sequencing using next generation sequencing (NGS) methods following previously published protocols (47). Amplified cDNA was quantified using the Qubit dsDNA HS Assay kits (Thermo Fisher Scientific) and subsequently diluted to 1 ng/µL. Libraries were prepared using the Nextera XT DNA library preparation kits (Illumina) and pooled. The pooled libraries were sequenced using an Illumina MiSeq sequencing platform at the Duke-NUS Genome Biology Facility, generating 250-bp paired-end reads. The NGS reads were checked with FastQC (48) in Unipro UGENE v40.1 (49) and were processed by trimming adaptors via Trimmomatic v0.39 (50). For each sample, reads were de novo assembled using SPAdes v3.15.3 (51), and individual gene segment was determined by BLASTn v2.2.18 (52). Reads were subsequently mapped to most similar segments using the map to reference tool in UGENE v40.1, and consensus sequences were extracted.
Evolutionary Analysis.
All available global H1N1 IAV genomes (as of 08 September 2022) collected from human and swine hosts were downloaded from NCBI GenBank and the Global Initiative on Sharing All Influenza Data (GISAID) (53) databases during 1957 to 2022 (n > 15,000). The datasets were subsampled to 1,008 sequences for H1 and 766 sequences for H3 using SMOT (54). Maximum likelihood (ML) phylogenies for H1, H3, N1, N2, and six internal genes (PB2, PB1, PA, NP, MP, and NS) were individually reconstructed using RAxML-NG v1.1.0 (55) with the following number of sequences H1-HA (n = 1,009), N1-NA (n = 986), H3-HA (n = 766), N2-NA (n = 773), PB2 (n = 924), PB1 (n = 923), PA (n = 915), NP (n = 927), MP (n = 927) and NS (n = 927). Branch support was assessed using 1,000 bootstrap replicates and bootstrap values greater than 50% are shown at major nodes. Classification designations for H1 sequences were assigned using the Swine H1 Clade Classification tool provided by Influenza Research Database (56). H3 classifications were assigned based on phylogenetic inference (57, 58). Lineages for internal genes were assigned using the OctoFLU pipeline (59) supplemented with A/swine/Arnsberg/1/1979 to indicate the EA-like lineage (60). Comparisons of nucleotide and amino acid sequences were aligned and visualized using Geneious PRIME 2022.1.1 (Biomatters Ltd., New Zealand).
To estimate the date of introduction for each genomic segment into Cambodian pigs, we utilized the ML phylogenies reconstructed above to extract the Cambodian swine viruses and representative viruses that pertain to the corresponding lineages. Bayesian time-scaled trees were inferred for the HA and NA genes using BEAST v1.10.4 (61) with BEAGLE library v3.1.0 (62). Posterior trees were sampled using an uncorrelated relaxed clock with lognormal distribution (63), under a generalized time-reversible substitution model (64) with four rate categories of gamma-distributed site heterogeneity (65), and a Gaussian Markov random field tree prior with time-aware smoothing (66). Two runs of Markov chain Monte Carlo sampling were performed, each with 100 million iterations and sampled every 10,000 trees. Convergence metrics were checked using Tracer v1.7.2 (67). Time-scaled maximum clade credibility (MCC) trees were summarized after the removal of 10% burn-in using TreeAnnotator v1.10.4 (61). The resulting MCC trees were visualized using FigTree v.1.4.2. Amino acid substitutions of the HA and NA were mapped using treesub (https://github.com/tamuri/treesub).
Phylogeographic Analysis.
To examine the spatial patterns of EA-swine H1 viruses, we reconstructed the spatiotemporal pathways between locations using discrete phylogeographic analyses in BEAST v.10.4. All available H1 sequences of European avian-like swine viruses were downloaded from NCBI and GISAID databases from 2005 to 2022 (accessed 20 December 2022), and seven discrete geographical regions were coded (Europe, east China, north China, northeast China, south central China, west China, and Southeast Asia). The HA dataset was subsampled using SMOT (54), and the final dataset consisted of 1,073 taxa. Asymmetric diffusion rates between discrete locations were inferred using Bayesian Stochastic Search Variable Selection (68), strict molecular clock model, the HKY85 nucleotide substitution model with four rate categories of gamma-distributed site heterogeneity, and a coalescent exponential growth tree prior. At least four independent runs of 100 million generations were performed and sampled every 10,000 generations. The runs were combined after removal of burn-in, with effective sample size of >200. Nonzero diffusion rates were calculated from the resulting log files. Bayes factor (BF) values were determined by SpreaD3 (69), and the circular migration diagram was plotted in R using the circlize package (70). BF ≥ 1,000 indicates decisive support, 100 ≤ BF < 1,000 as very strong support, 10 ≤ BF < 100 as strong support, and 3 ≤ BF < 100 as supported (SI Appendix, Table S2). To understand the effect of sampling bias, we subsampled the larger dataset to obtain a more even sampling of sequences among locations (n = 534). Phylogeographic analyses were performed using the same criteria as stated above, and asymmetric diffusion rates between discrete locations were estimated.
We further evaluated the effect of potential predictors for the spatial migration of EA H1 swine (SI Appendix, Table S5). Six predictors were selected: number of pigs at destination, number of pigs at origin, pork production at destination, pork production at origin, pork consumption at destination, and pork consumption at origin. The pig data were obtained from a published study (71) and online resources from Ministry of Agriculture and Rural Affairs of the People’s Republic of China (72) and Our World in Data (73). We employed a GLS model using the nlme v3.1 package in R and applied a linear function between migration rates and six potential predictors. A P-value <0.1 indicates statistical significance for predictor support. We used the same dataset (n = 1,073 taxa) to test the predictors, and the GLS results were plotted in R. In addition, we also used a Bayesian GLM to assess the potential predictors while simultaneously estimating the spatiotemporal patterns of virus diffusion (74). The predictors were incorporated in BEAST and run using the same parameters as stated above, using 100 million generations. The plots were generated using in-house scripts in R.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
The study was supported by the United States Department of Defense Threat Reduction Agency Cooperative Biological Research project: PigFluCam+ (HDTRA11810051), contracts HHSN272201400006C and 75N93021C00016 from the National Institute of Allergy and Infectious Diseases, NIH, Department of Health and Human Services, USA, and by the Duke-NUS Signature Research Programme funded by the Ministry of Health, Singapore. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. We thank Dr. Rong Zhang at Duke-NUS for providing assistance on generalized linear models. We thank GISAID and all submitters for access to their databases and all submitters of data and sequences to these databases.
Author contributions
S.T., S.S., A.H., H.H., J.W.R., G.J.D.S., and Y.C.F.S. designed research; A.H., H.H., T.C., S.S., D.K., B.S., S.C., G.G.K.N., and Z.Y. performed research; M.A.Z. contributed new analytic tools; M.A.Z., J.M., F.Y.W., and Y.C.F.S. analyzed data; S.T., A.H., H.H., S.S., T.C., and M.C. coordinated data and sample collection; J.W.R., G.J.D.S., and Y.C.F.S. conceived the study; and M.A.Z., J.W.R., G.J.D.S., and Y.C.F.S. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Although PNAS asks authors to adhere to United Nations naming conventions for maps (https://www.un.org/geospatial/mapsgeo), our policy is to publish maps as provided by the authors.
Contributor Information
Gavin J. D. Smith, Email: gavin.smith@duke-nus.edu.sg.
Yvonne C. F. Su, Email: yvonne.su@duke-nus.edu.sg.
Data, Materials, and Software Availability
Genome sequences data have been deposited in GenBank [OQ459860 to OQ460225 (SI Appendix, Table S6)] (75).
Supporting Information
References
- 1.Garten R. J., et al. , Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans. Science 325, 197–201 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith G. J., et al. , Dating the emergence of pandemic influenza viruses. Proc. Natl. Acad. Sci. U.S.A. 106, 11709–11712 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vijaykrishna D., et al. , Long-term evolution and transmission dynamics of swine influenza A virus. Nature 473, 519–522 (2011). [DOI] [PubMed] [Google Scholar]
- 4.VanderWaal K., Deen J., Global trends in infectious diseases of swine. Proc. Natl. Acad. Sci. U.S.A. 115, 11495–11500 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies P. R., Intensive swine production and pork safety. Foodborne Pathogens Dis. 8, 189–201 (2011). [DOI] [PubMed] [Google Scholar]
- 6.FAO Food, Food and Agriculture Organization of the United Nations. Rome (2022). http://faostat.fao.org. Accessed 10 April 2023.
- 7.Tep B., "Swine production and disease status in Cambodia" in Third Regional Workshop on Swine Disease Control in Asia (General Directorate of Animal Health and Production Ministry of Agriculture, Forestry and Fisheries Cebu, Philippines, 2018). [Google Scholar]
- 8.Smith G. J., et al. , Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Fraser C., et al. , Pandemic potential of a strain of influenza A (H1N1): Early findings. Science 324, 1557–1561 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mena I., et al. , Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. Elife 5, e16777 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson M. I., et al. , Global migration of influenza A viruses in swine. Nat. Commun. 6, 6696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henritzi D., et al. , Surveillance of european domestic pig populations identifies an emerging reservoir of potentially zoonotic swine influenza A viruses. Cell Host. Microbe. 28, 614–627.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Sun H., et al. , Prevalent Eurasian avian-like H1N1 swine influenza virus with 2009 pandemic viral genes facilitating human infection. Proc. Natl. Acad. Sci. U.S.A. 117, 17204–17210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nasamran C., et al. , Persistence of pdm2009-H1N1 internal genes of swine influenza in pigs, Thailand. Sci. Rep. 10, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charoenvisal N., et al. , Genetic characterization of Thai swine influenza viruses after the introduction of pandemic H1N1 2009. Virus Genes 47, 75–85 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Nonthabenjawan N., et al. , Genetic diversity of swine influenza viruses in Thai swine farms, 2011–2014. Virus Genes 50, 221–230 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Takemae N., et al. , Antigenic variation of H1N1, H1N2 and H3N2 swine influenza viruses in Japan and Vietnam. Arch. Virol. 158, 859–876 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Ngo L. T., et al. , Isolation of novel triple-reassortant swine H3N2 influenza viruses possessing the hemagglutinin and neuraminidase genes of a seasonal influenza virus in Vietnam in 2010. Influenza Other Respir. Viruses 6, 6–10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baudon E., et al. , Detection of novel reassortant Influenza A (H3N2) and H1N1 2009 pandemic viruses in swine in Hanoi, Vietnam. Zoonoses Public Health 62, 429–434 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Mon P. P., et al. , Swine influenza viruses and pandemic H1N1-2009 infection in pigs, Myanmar. Transboundary Emerging Dis. 67, 2653–2666 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Long J. S., et al. , Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 17, 67–81 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Beigel J. H., et al. , Avian influenza A (H5N1) infection in humans. N. Engl. J. Med. 353, 1374–85 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Chen Y., et al. , Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: Clinical analysis and characterisation of viral genome. Lancet 381, 1916–1925 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Reeth K., Avian and swine influenza viruses: Our current understanding of the zoonotic risk. Veterinary Res. 38, 243–260 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Myers K. P., Olsen C. W., Gray G. C., Cases of swine influenza in humans: A review of the literature. Clin. Infect. Dis. 44, 1084–1088 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeller M. A., et al. , Complete genome sequences of two novel human-like H3N2 influenza A viruses, A/swine/Oklahoma/65980/2017 (H3N2) and A/Swine/Oklahoma/65260/2017 (H3N2), detected in swine in the United States. Microbiol. Resour. Announc. 7, e01203-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajao D. S., et al. , Novel reassortant human-like H3N2 and H3N1 influenza A Viruses detected in pigs are virulent and antigenically distinct from swine viruses endemic to the United States. J. Virol. 89, 11213–11222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vincent A. L., et al. , Characterization of a newly emerged genetic cluster of H1N1 and H1N2 swine influenza virus in the United States. Virus Genes 39, 176–185 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Zhou N. N., et al. , Genetic reassortment of avian, swine, and human influenza A viruses in American pigs. J. Virol. 73, 8851–8856 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webby R. J., et al. , Evolution of swine H3N2 influenza viruses in the United States. J. Virol. 74, 8243–8251 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chea B., et al. , Assessment of pig disease prevention of smallholder farmers and village animal health workers in rural and peri-urban Cambodia. Open J. Animal Sci. 10, 572–591 (2020). [Google Scholar]
- 32.Scholtissek C., Pigs as ‘mixing vessels’ for the creation of new pandemic influenza A viruses. Med. Princ. Pract. 2, 65–71 (1990). [Google Scholar]
- 33.Pensaert M., et al. , Evidence for the natural transmission of influenza A virus from wild ducks to swine and its potential importance for man. Bull. World Health Organ. 59, 75 (1981). [PMC free article] [PubMed] [Google Scholar]
- 34.Chauhan R. P., Gordon M. L., A systematic review of influenza A virus prevalence and transmission dynamics in backyard swine populations globally. Porcine Health Manag. 8, 10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osbjer K., et al. , Influenza A virus in backyard pigs and poultry in rural Cambodia. Transbound Emerg. Dis. 64, 1557–1568 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Takemae N., et al. , Influenza A viruses of swine (IAV-S) in Vietnam from 2010 to 2015: Multiple introductions of A(H1N1)pdm09 viruses into the pig population and diversifying genetic constellations of enzootic IAV-S. J. Virol. 91, e01490-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H., et al. , Prevalence, genetics and evolutionary properties of eurasian avian-like H1N1 swine influenza viruses in Liaoning. Viruses 14, 643 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson M. I., et al. , Global transmission of influenza viruses from humans to swine. J. Gen. Virol. 93, 2195–2203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson M. I., et al. , Spatial dynamics of human-origin H1 influenza A virus in North American swine. PLoS Pathog. 7, e1002077 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danilenko D. M., et al. , Antigenic and genetic characterization of swine influenza viruses identified in the European Region of Russia, 2014–2020. Front. Microbiol. 12, 662028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryt-Hansen P., et al. , Co-circulation of multiple influenza A reassortants in swine harboring genes from seasonal human and swine influenza viruses. Elife 10, e60940 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mon P. P., et al. , Swine influenza viruses and pandemic H1N1-2009 infection in pigs, Myanmar. Transbound Emerg. Dis. 67, 2653–2666 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Wong F. Y. K., et al. , Divergent human-origin influenza viruses detected in Australian swine populations. J Virol. 92, e00316-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mottet A., Tempio G., Global poultry production: Current state and future outlook and challenges. World’s Poultry Sci. J. 73, 245–256 (2017). [Google Scholar]
- 45.Woonwong Y., Do Tien D., Thanawongnuwech R., The future of the pig industry after the introduction of African swine fever into Asia. Anim. Front. 10, 30–37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holmes E. C., The ecology of viral emergence. Annu. Rev. Virol. 9, 173–192 (2022). [DOI] [PubMed] [Google Scholar]
- 47.Ali M., et al. , Genetic characterization of highly pathogenic avian influenza A (H5N8) virus in Pakistani live bird markets reveals rapid diversification of clade 2.3. 4.4 b viruses. Viruses 13, 1633 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andrews S., FastQC: A quality control tool for high throughput sequence data (2010). https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 01 Mar 2022.
- 49.Okonechnikov K., Golosova O., Fursov M., Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 28, 1166–7 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bankevich A., et al. , SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altschul S. F., et al. , Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shu Y., McCauley J., GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 22, 30494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arendsee Z. W., V. Baker A. L., Anderson T. K., smot: A python package and CLI tool for contextual phylogenetic subsampling. J. Open Source Softw. 7, 4193 (2022). [Google Scholar]
- 55.Kozlov A. M., et al. , RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson T. K., et al. , A phylogeny-based global nomenclature system and automated annotation tool for H1 hemagglutinin genes from swine influenza A viruses. mSphere 1, e00275-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anderson T. K., et al. , Swine influenza A viruses and the tangled relationship with humans. Cold Spring Harb. Perspect. Med. 11, a038737 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watson S. J., et al. , Molecular epidemiology and evolution of influenza viruses circulating within European swine between 2009 and 2013. J. Virol. 89, 9920–9931 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang J., et al. , octoFLU: Automated classification for the evolutionary origin of influenza A virus gene sequences detected in US swine. Microbiol. Resour. Announc. 8, e00673-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zell R., Scholtissek C., Ludwig S., Genetics, evolution, and the zoonotic capacity of European Swine influenza viruses. Curr. Top. Microbiol. Immunol. 370, 29–55 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Drummond A. J., et al. , Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ayres D. L., et al. , BEAGLE: An application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 61, 170–173 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drummond A. J., et al. , Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tavaré S., Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 17, 57–86 (1986). [Google Scholar]
- 65.Yang Z., Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 39, 306–314 (1994). [DOI] [PubMed] [Google Scholar]
- 66.Minin V. N., Bloomquist E. W., Suchard M. A., Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 25, 1459–1471 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rambaut A., Drummond A. J., Tracer v1. 6, Tracer (2007). http://beast.bio.ed.ac.uk. Accessed 01 March 2022.
- 68.Lemey P., et al. , Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5, e1000520 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bielejec F., et al. , Sprea D3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 33, 2167–2169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gu Z., et al. , circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Zhao Q., et al. , Distribution and intensification of pig production in China 2007–2017. Environ. Res. Lett. 17, 124001 (2022). [Google Scholar]
- 72.National Bureau of Statistics of China. China Statistical Yearbook (2018). http://www.stats.gov.cn/sj/ndsj/2018/indexeh.htm. Accessed 10 April 2023.
- 73.Roser M., Ortiz-Ospina E., Pigmeat production (2021). https://ourworldindata.org/grapher/pigmeat-production-tonnes. Accessed 10 April 2023.
- 74.Lemey P., et al. , Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog. 10, e1003932 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeller M. A., et al. , Accession numbers for The National Center for Biotechnology Information (NCBI), GenBank. https://www.ncbi.nlm.nih.gov/nuccore/?term=OQ459860:OQ460225[accn]. Deposited 26 June 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Genome sequences data have been deposited in GenBank [OQ459860 to OQ460225 (SI Appendix, Table S6)] (75).