

Abstract
Introduction
BAY1128688 is a selective inhibitor of aldo-keto reductase family 1 member C3 (AKR1C3), an enzyme implicated in the pathology of endometriosis and other disorders. In vivo animal studies suggested a potential therapeutic application of BAY1128688 in treating endometriosis. Early clinical studies in healthy volunteers supported the start of phase IIa.
Objective
This manuscript reports the results of a clinical trial (AKRENDO1) assessing the effects of BAY1128688 in adult premenopausal women with endometriosis-related pain symptoms over a 12-week treatment period.
Methods
Participants in this placebo-controlled, multicenter phase IIa clinical trial (NCT03373422) were randomized into one of five BAY1128688 treatment groups: 3 mg once daily (OD), 10 mg OD, 30 mg OD, 30 mg twice daily (BID), 60 mg BID; or a placebo group. The efficacy, safety, and tolerability of BAY1128688 were investigated.
Results
Dose-/exposure-dependent hepatotoxicity was observed following BAY1128688 treatment, characterized by elevations in serum alanine transferase (ALT) occurring at around 12 weeks of treatment and prompting premature trial termination. The reduced number of valid trial completers precludes conclusions regarding treatment efficacy. The pharmacokinetics and pharmacodynamics of BAY1128688 among participants with endometriosis were comparable with those previously found in healthy volunteers and were not predictive of the subsequent ALT elevations observed.
Conclusions
The hepatotoxicity of BAY1128688 observed in AKRENDO1 was not predicted by animal studies nor by studies in healthy volunteers. However, in vitro interactions of BAY1128688 with bile salt transporters indicated a potential risk factor for hepatotoxicity at higher doses. This highlights the importance of in vitro mechanistic and transporter interaction studies in the assessment of hepatoxicity risk and suggests further mechanistic understanding is required.
Clinical Trial Registration
NCT03373422 (date registered: November 23, 2017)
Supplementary Information
The online version contains supplementary material available at 10.1007/s40268-023-00427-5.
Key Points
| The doses of the AKR1C3 inhibitor BAY1128688 tested in this trial reached the anticipated plasma exposure range and the pharmacodynamics (such as increased androsterone) in the patients with endometriosis were comparable with those observed in healthy volunteers over a period of 4 weeks. |
| Dose-/exposure-dependent hepatotoxicity was observed following treatment with the AKR1C3 inhibitor BAY1128688 with characteristic clustering around the 12-week time point, although no hepatotoxicity had been observed in animal toxicology studies. |
| The absence of predictivity by the animal toxicology studies and by the short-term clinical studies in healthy volunteers in identifying potential effects in the human liver emphasizes the additional value of in vitro mechanistic and transporter interaction studies for the assessment of risk of hepatoxicity with continuous treatment (such as bile acid transporter interactions). |
Introduction
Hypothesized Role of the Enzyme AKR1C3 in Disease Pathology
The aldo-keto reductase family 1 member C3 (AKR1C3; also known as 17β-hydroxysteroid dehydrogenase type 5) is a steroidogenic enzyme that plays a central role in intra-tissue steroid and prostaglandin synthesis, both of which are of high relevance in many pathological processes [1, 2]. AKR1C3 is therefore implicated in the pathophysiology of a number of malignancies and endocrine disorders, such as castration-resistant prostate cancer, polycystic ovarian syndrome, and endometriosis, making it a potential target for the treatment of these diseases [2].
Rationale for Testing AKR1C3 Inhibitors as Treatments for Patients with Endometriosis
Endometriosis is a chronic, benign, estrogen-dependent, gynecological inflammatory disease affecting women of reproductive age [3–6]. Pathologically, the disease is defined by the presence of ectopic endometrial tissue. It is characterized by chronic, non-menstrual pelvic pain, dysmenorrhea, and dyspareunia; is often associated with subfertility or infertility; and may substantially impact quality of life [3, 4, 6–8].
AKR1C3 is expressed in the human endometrium and has been found to be upregulated in endometriotic tissue [9]. Increased AKR1C3 expression has been postulated to trigger the estrogen-dependent proliferation of lesions through steroid biosynthesis and to induce local inflammation through prostaglandin synthase activity [1]. Preclinical pharmacology studies in the marmoset monkey (Callithrix jacchus) endometriosis model showed that treatment with ARK1C3 inhibitor BAY1128688 reduced endometriotic lesions when compared with the vehicle group [10]. Furthermore, data from the animal study showed that ovarian cyclicity remained unchanged after treatment with BAY1128688 [10]. Inhibition of AKR1C3 may therefore be a promising treatment option for endometriosis in women that need to or want to avoid the suppression of ovarian function that accompanies other endometriosis treatments.
Selectivity of the AKR1C3 Inhibitor BAY1128688
BAY1128688 is a potent AKR1C3 (in vitro 50% inhibitory concentration [IC50] < 2 nM in a biochemical assay) inhibitor identified in a compound optimization program. BAY1128688 exhibited no relevant agonism or antagonism at the following nuclear hormone receptors (estrogen, androgen, progesterone receptor) [10]. In addition, BAY1128688 was screened preclinically and found to have no relevant activity against a range of related enzymes, namely recombinant human AKR1C enzymes (1C1, 1C2, 1C4) and AKR1D1. Specificity was also tested in a panel towards 134 human targets including G protein-coupled and nuclear receptors, ion channels, transporters, and enzymes, and no relevant interactions were identified. However, interactions of BAY1128688 with transporters involved in bile acid metabolism have been detected. Analyses performed prior to conducting the clinical study are reported in this manuscript. Additional investigations performed after study conduct had been completed, including modeling using DILIsym® (Simulations Plus, Durham, NC, USA), are reported separately [11].
In Vitro Characterization of the Interaction Potential of the AKR1C3 Inhibitor BAY1128688 Relevant to Liver Function
In vitro data generated prior to start of the clinical trials in patients showed that BAY1128688 could potentially inhibit the activity of bile salt transporters (e.g., bile salt export pump [BSEP]) as well as bilirubin-metabolizing enzymes and transporters (e.g., UDP-glucuronosyltransferase 1A1 [UGT1A1], multidrug resistance-associated protein 2 [MRP2]) and organic anion-transporting polypeptides 1B1/3 [OATP1B1/3]) in a dose-dependent manner (Online Resource 1, Online Resource 2, see electronic supplementary material [ESM]).
Toxicological Characterization of the AKR1C3 Inhibitor BAY1128688 in Rats and Cynomolgus Monkeys
The initial characterization of the safety profile of BAY1128688, including determination of exposure levels, dose–response relationship, target organs of toxicity, and reversibility of effects, was carried out in repeat-dose in vivo toxicity studies according to ICH guidelines [Bayer data on file]. Rats (Rattus norvegicus, Wistar strain) and female cynomolgus macaque monkeys (Macaca fascicularis) were dosed for 4 and 13 weeks. No hepatotoxicity was observed up to the highest tested dose of 70 mg/kg BAY1128688 in female rats and 40 mg/kg BAY1128688 in female monkeys dosed over 13 weeks. These doses correspond to an approximately 21-fold (rat) and 16-fold (monkey) higher systemic exposure (by the area under the unbound concentration–time curve at steady state over 24 h, AUC[0–24]ss,u) than the anticipated human exposure at the maximum clinical dose of 60 mg twice daily (BID). Some adaptive changes, including increases in total serum bilirubin, increased liver weight, and microscopic findings in the liver (e.g., prominent bile ducts, hepatocellular hypertrophy) were observed in both primate and rat studies. However, all changes were reversible and occurred at exposures that were higher than the maximum expected human exposure. Thus, the in vivo preclinical investigations provided no evidence for potential drug-induced liver toxicity with BAY1128688 at the selected doses for human clinical trials.
Hepatic Safety of the AKR1C3 Inhibitor BAY1128688, Pharmacodynamic Biomarker, and Ovarian Function in a Multiple-Dose Phase I Study
In a multiple-dose phase I study conducted and published prior to the one reported here, healthy pre- and postmenopausal women were exposed to doses of up to 60 mg BID BAY1128688 for up to 4 weeks [12]. Increases in mean and median total serum bilirubin were observed in postmenopausal women after 2 weeks of treatment at doses of 90 mg once daily (OD) and 60 mg BID, but not at 30 mg and 3 mg OD. These increases in total serum bilirubin were not associated with symptoms and were isolated, i.e., no increases in alanine aminotransferase (ALT) or aspartate transaminase (AST) were observed. Maximum median total serum bilirubin increases of 2.2-fold at day 6 of treatment relative to baseline values were observed in the 90 mg OD dose group (individual maximum increase around 2.3-fold upper limit of normal [ULN] at day 6). In the 60 mg BID dose group, maximum median increases of 2.8-fold at day 6 of treatment relative to baseline values were observed (individual maximum increase around 1.2-fold ULN at day 13). This dataset was used to derive a population-based pharmacokinetic/pharmacodynamic (PK/PD) model which predicted that increases in total serum bilirubin would not exceed 2.5-fold ULN.
In order to assess whether BAY1128688 had an effect on bile acid metabolism, an exploratory evaluation of bile acids was conducted, which did not indicate any influence of BAY1128688 on bile acid metabolism.
AKR1C3 has been described to convert various substrates in androgen metabolism, including androsterone [13]. A dose-dependent increase in serum androsterone was observed during the administration of multiple doses of BAY1128688 in the phase I study. Accordingly, these increases in serum androsterone have been postulated as a pharmacodynamic biomarker of target engagement with AKR1C3 [12].
In a small cohort of healthy premenopausal women exposed to up to 60 mg BID of BAY1128688 for 28 days, clinical data also demonstrated that normal ovarian cycles were maintained [12].
Summary
Results from preclinical pharmacology studies suggested that AKR1C3 inhibition by BAY1128688 may be an effective treatment for endometriosis that does not affect systemic endogenous sex hormones or ovulation (the latter also confirmed clinically). Initial in vitro data showed that there was a potential for BAY1128688 to inhibit bile salt transport through hepatocyte and bilirubin metabolism and transport in a dose-dependent manner. However, there were no signs of liver toxicity based on animal toxicology studies after up to 13 weeks of treatment and no clinical signs during 4 weeks of treatment in humans (see Online Resource 3 in the ESM for a full summary of risk factors for drug-induced liver injury [DILI] from preclinical studies).
Here we report findings from AKRENDO1, an exploratory phase IIa trial that investigated the dose–response relationship, safety, and tolerability of BAY1128688 in adult premenopausal women with endometriosis-related pain symptoms over a 12-week treatment period.
Methods
Study Design
AKRENDO1 was a randomized, double-blind, parallel group, placebo-controlled, multicenter phase IIa clinical trial (ClinicalTrials.gov identifier: NCT03373422) conducted from November 30, 2017 to October 22, 2018, at 69 sites in 13 European countries.
The primary objective was to explore the dose–response relationship of different doses of BAY1128688 relative to a placebo in the treatment of endometriosis-related pain symptoms over a 12-week treatment period. The secondary objective was to assess the safety and tolerability of BAY1128688 over this period. Exploratory objectives included the assessment of menstrual bleeding patterns and health-related quality of life, as well as further investigation of the pharmacokinetics and biomarker endpoints of BAY1128688.
The inclusion criteria were adult (≥18 years) women with confirmed endometriosis (confirmed either visually during surgical procedures within 10 years or confirmed via imaging within 12 months prior to screening) who met defined pain criteria but were otherwise in good general health. The defined pain criteria were a visual analog scale (VAS) score of at least 40 mm on a scale between 0 and 100 mm for participants not on hormonal treatment and at least 30 for participants on hormonal treatment at visit 1 (pre-screening); at least 24 daily diary entries in the endometriosis symptoms diary (ESD) [14], a multi-item patient-related outcome (PRO) instrument designed to assess the patient’s experience of endometriosis symptoms by rating symptoms on a scale from 0 to 10; and a total ‘worst pain’ score of at least 98 using the ESD’s numerical rating scale (NRS) over the screening period (pre-randomization). Exclusion criteria included pregnant or lactating women and those with risks related to bilirubin metabolism, liver function, and hyperandrogenemia.
It was planned to randomize 120 participants, with a planned 84 participants valid for primary efficacy analysis. Eligible participants were randomized (via an interactive voice/web-response system) in an imbalanced fashion into one of six treatment groups: 3 mg OD (n = 10 planned participants for randomization/n = 7 planned participants valid for primary efficacy analysis), 10 mg OD (n = 30/n = 21), 30 mg OD (n = 10/n = 7), 30 mg BID (n = 10/n = 7), 60 mg BID (n = 30/n = 21), and placebo (n = 30/n = 21).
The screening visit was carried out at visit 1, with randomization to the treatment groups at visit 2 and start of treatment at visit 3. Monitoring during the treatment period was carried out at visit 4 on day 10 of treatment ± 4 days, visit 5 on day 28 ± 4 days, and visit 6 on day 56 ± 4 days. Visit 7 was the end-of-treatment (EoT) visit, scheduled at 12 weeks or day 84 ± 4 days of treatment, or as soon as possible after the last dose of the study drug if a subject discontinued the study drug prematurely. An additional visit 7a was introduced as a result of study termination for safety monitoring and occurred 14 days ± 4 days after EoT. The end of study took place at visit 8, 6 weeks (42 days ± 4 days) after treatment completion. The overall study design and visit scheduling are shown in Fig. 1.
Fig. 1.
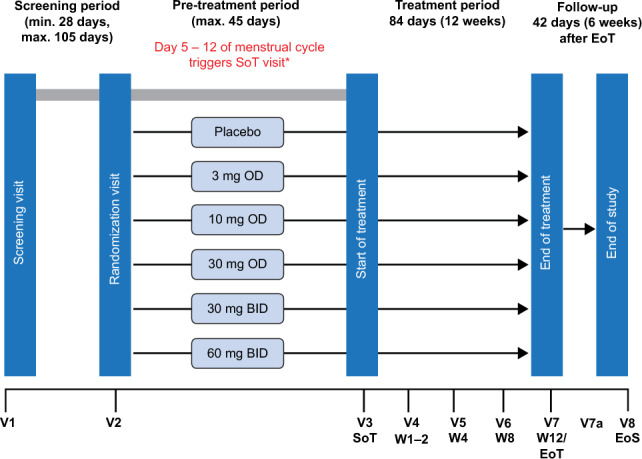
Study design and visit schedule. *Menstrual bleeding is defined as 2 consecutive days with more than spotting. Day 1 of the menstrual cycle is defined as the first of the 2 consecutive days with more than spotting. NOTE that additional visit 7a 2 weeks after end of treatment was introduced as a result of study termination for safety monitoring. BID twice daily, EoT end of treatment, EoS end of study, OD once daily, SoT start of treatment, V visit, W week
Based on preclinical in vivo and clinical phase I data dosages, 3 mg OD and 10 mg OD were not expected to be associated with increases in total serum bilirubin concentrations and were judged to have the potential to be efficacious [10, 12]. Higher dose groups (30 mg OD, 30 mg BID, and 60 mg BID) were selected to cover the in vitro 50% inhibitory concentration (IC50) and 80% inhibitory concentration (IC80) of BAY1128688 for the inhibition of the AKR1C3 enzyme, using plasma-free trough concentrations (Ctrough, unbound). To maximize Ctrough and reduce maximum concentration (Cmax), doses of 120 mg and 60 mg were split into two administrations (2 doses of 60 mg and 30 mg, respectively).
Efficacy and Safety
Efficacy was assessed using PRO measures and responses to electronic diary questionnaires and included an ESD for the daily assessment of symptoms, including endometriosis pain (NRS scale from 0 to 10), vaginal bleeding, and trial drug and other pain medication use; an endometriosis impact scale for the weekly assessment of the effects of primary endometriosis symptoms on participants’ lives (i.e., pain); and VAS (0–100 mm) for endometriosis-associated pelvic pain (EAPP) with a 4-week recall. The primary efficacy variable was absolute change in the mean weekly pain, comparing the 7 days with the worst EAPP from baseline (the last 28 days before visit 3) with the last 28 days before visit 7. This was measured on the NRS of the ESD. Safety was assessed by monitoring the incidence of adverse events, serious adverse events, and treatment-emergent adverse events (TEAEs). Adverse events of special interest were the worsening of EAPP, increases in total serum bilirubin, and increases in liver laboratory values.
The work-up of all cases of suspected liver injury was performed in accordance with FDA 2009 Drug-Induced Liver Injury (DILI) Guidelines [15]. The severity of liver injury was defined using the DILI Expert Working Group Severity Criteria [16]. The causality of any liver injury was assessed according to the Roussel Uclaf Causality Assessment Method (RUCAM) criteria described by Danan and Benichou in 1993 [17]. The nature of any liver injury (hepatocellular, cholestatic, or mixed) was determined based on the ratio of liver test abnormalities (r-value, defined as serum ALT/ULN divided by serum alkaline phosphatase [AP]/ULN) [18].
Population Pharmacokinetic and Exposure–Response Analyses
An exploratory population pharmacokinetic (popPK) analysis of BAY1128688 and an exposure–response (ER) analysis were conducted. PopPK modeling was performed via non-linear mixed-effects modeling methods applied in NONMEM® (ICON plc, Dublin, Ireland; version 7.3). For the ER analysis, treatment-emergent increases in ALT concentration > 2 × ULN were analyzed as binary events; no relevant increase in ALT concentrations versus ALT concentrations elevated above 2 × ULN for at least one time point after baseline. ER modeling was performed using the software R (version 3.2.5).
Pharmacodynamics
Pharmacodynamic biomarker serum androsterone was measured at all visits except visit 2 throughout the study. Steroid analyses, including the measuring of androsterone levels, were conducted using 500-µL serum samples and the AbsoluteIDQ® Stero 17 kit (Biocrates, Innsbruck, Austria) [19]. The kinetics of the pharmacodynamic response to AKR1C3 inhibition was assessed over time in all participants as well as the subset of 50 completers with complete datasets for the 12 weeks of treatment.
Biomarker Sub-Study
This exploratory biomarker sub-study was conducted using the dataset of patients who had completed 12 weeks of treatment (‘completers’, n = 50). Standard liver safety laboratory parameters (ALT, AST, total serum bilirubin, and the international normalized ratio [INR]), a panel of 22 biomarkers (including 15 plasma bile acids, total bile acids, and 6 biomarkers described to predict DILI events, see Online Resource 4 in the ESM), and the pharmacodynamic biomarker androsterone were measured at all visits except visit 2.
The analyses aimed to determine:
if there were associations between standard liver safety laboratory parameters measured during the study with the panel of 22 biomarkers and the pharmacodynamic biomarker androsterone. Specifically, it was determined whether the concentration of these biomarkers was correlated (Spearman’s rank correlation) with liver safety parameters measured at time points from visit 3 to visit 8.
the predictive value of a panel of the 22 biomarkers described above and androsterone (measured prior to the ALT increase occurrence) in identifying patients who experienced increases in ALT > 2 × ULN after 12 weeks of treatment or thereafter. Specifically, differences in biomarker concentrations between completers who experienced increases in ALT > 2 × ULN at visit 7 or thereafter and completers who did not experience an increase in ALT were assessed (using equal variance t-tests). Hence, the time points prior to increases in ALT > 2 × ULN (visit 3–6) were used in this analysis. Biomarker data from visits 7 and 8 were also assessed, despite not being of predictive value.
The six biomarkers described as carrying potential in detecting DILI risk prior to observed ALT/AST increases were selected based on the recommendations listed in corresponding letters of support issued by the FDA and EMA in 2016 resulting from the IMI SAFE-T consortium (Online Resource 4, see ESM) [20, 21].
Analysis of the above-mentioned six biomarkers described to predict DILI events was conducted at SYNLAB using 500-µL serum samples (SYNLAB, Berlin, Germany).
Plasma bile acids and total bile acids were analyzed in 200-µL plasma samples using liquid chromatography with tandem mass spectrometry (Biocrates, Innsbruck, Austria) [22].
For further methodology details, refer to Online Resource 5 in the ESM.
Results
Of the 230 total participants screened, 141 were randomized into one of the six treatment groups. Twenty participants did not receive BAY1128688 or the placebo due to early termination. Thus, 121 participants underwent treatment, of whom 90 were exposed to BAY1128688 and 31 received the placebo (Fig. 2). Demographics and baseline characteristics were similar between treatment groups (Table 1).
Fig. 2.
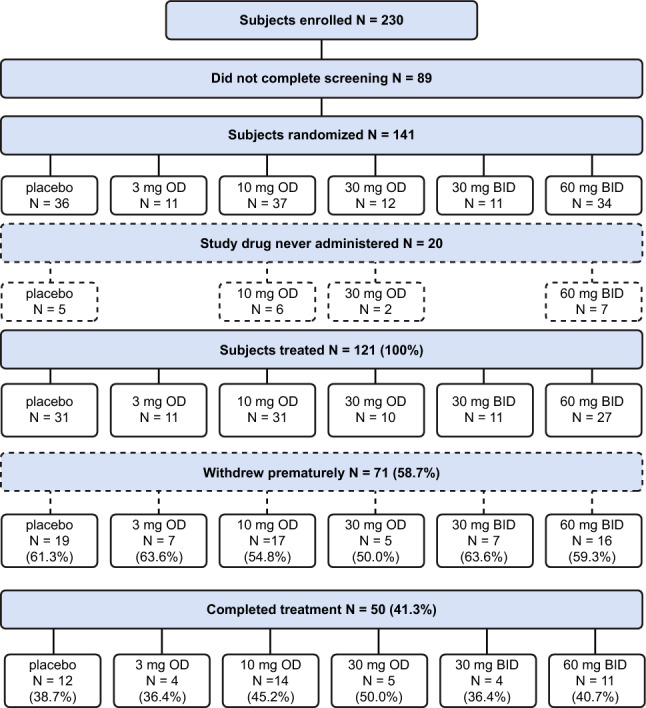
Participant flowchart. 3 mg OD, 10 mg OD, 30 mg OD, 30 mg BID, and 60 mg BID refer to active dose groups of BAY1128688. BID twice daily, OD once daily
Table 1.
Baseline demographics
| Participants | Placebo n = 31 |
BAY1128688 | Total N = 121 |
||||
|---|---|---|---|---|---|---|---|
| 3 mg OD n = 11 |
10 mg OD n = 31 |
30 mg OD n = 10 |
30 mg BID n = 11 |
60 mg BID n = 27 |
|||
| Age at baseline (years) | |||||||
| Mean (SD) | 31.4 (5.7) | 32.5 (9.2) | 32.7 (6.1) | 32.9 (4.9) | 33.5 (5.4) | 31.3 (7.3) | 32.1 (6.4) |
| Race, n (%) | |||||||
| White | 31 (100) | 11 (100) | 30 (96.8) | 10 (100) | 11 (100) | 26 (96.3) | 119 (98.3) |
| Multiple or other | 0 | 0 | 1 (3.2) | 0 | 0 | 1 (3.7) | 2 (1.6) |
| Baseline BMI (kg/m2) | |||||||
| Mean (SD) | 25.1 (4.0) | 23.4 (5.4) | 23.3 (2.5) | 23.2 (4.0) | 24.2 (4.0) | 22.9 (3.5) | 23.8 (3.9) |
| Alcohol use, n (%) | |||||||
| Abstinent | 10 (32.3) | 2 (18.2) | 6 (19.4) | 1 (10.0) | 4 (36.4) | 9 (33.3) | 32 (26.4) |
| Light | 17 (54.8) | 9 (81.8) | 24 (77.4) | 6 (60.0) | 7 (63.6) | 17 (63.0) | 80 (66.1) |
| Moderate | 4 (12.9) | 0 | 1 (3.2) | 3 (30.0) | 0 | 1 (3.7) | 9 (7.4) |
| Tobacco use, n (%) | |||||||
| Never | 16 (51.6) | 8 (72.7) | 20 (64.5) | 4 (40.0) | 10 (90.9) | 14 (51.9) | 72 (59.5) |
| Former | 7 (22.6) | 1 (9.1) | 3 (9.7) | 2 (20.0) | 0 | 3 (11.1) | 16 (13.2) |
| Current | 8 (25.8) | 2 (18.2) | 8 (25.8) | 4 (40.0) | 1 (9.1) | 10 (37.0) | 33 (27.3) |
BID twice daily, BMI body mass index, OD once daily, SD standard deviation
The trial was terminated prematurely due to clinical indications of hepatotoxicity. Of the 121 participants who received BAY1128688 or the placebo, 71 (58.7%) discontinued treatment prematurely, primarily (60/71 patients) due to the sponsor’s decision to terminate the trial (Fig. 2). Seven participants discontinued due to TEAEs (placebo: worsening of EAPP in 1 participant; BAY1128688: depression or mood changes in 3 participants, gastrointestinal AEs, vertigo, and ocular icterus in 1 participant each). There was one discontinuation due to lack of efficacy, one due to protocol deviation, and two participants withdrew from the study. Of the 50 participants (41.3%) who completed 12 weeks of treatment, 45 were valid for primary efficacy analysis.
Efficacy
Because of the early termination, the AKRENDO1 study has insufficient power to assess the efficacy of BAY1128688, that is, the ability of BAY1128688 to reduce or prevent EAPP could not be properly assessed, as a minimum of 84 evaluable participants were required for efficacy evaluations. Accordingly, no inferential statistical analyses were performed and therefore no final conclusions can be derived regarding the efficacy of the AKR1C3 inhibitor BAY1128688 in patients with endometriosis. Changes in EAPP from baseline to the end of treatment are shown in Online Resource 6 (see ESM).
Overall Safety
Safety analyses were conducted using data from all participants who received at least one dose of BAY1128688.
Overall, at least one TEAE was reported in 71 of the 121 participants (58.7%). In the dose groups receiving BAY1128688, TEAEs were reported in 32.3% (10 mg OD) to 90.0% (30 mg OD) of participants. There was no apparent dose dependency in the number of TEAEs reported (Table 2). In the placebo group, TEAEs were reported in 64.5% of participants. TEAEs that were considered by the investigator to be study-drug–related were reported in 9.7% (10 mg OD) to 51.9% (60 mg BID) of participants in the BAY1128688 treatment groups (with no apparent dose dependency) and in 29.0% for participants receiving the placebo. Overall, seven participants (5.8%) discontinued treatment because of TEAEs, with five of the seven having received the highest or second-highest dose of BAY1128688 (dose-dependency). Across all treatment groups, the majority of participants had TEAEs of either mild or moderate intensity (as rated by the investigator). Severe TEAEs were reported in 12 participants (9.9%), six of whom were in the 60 mg BID group, none in the 30 mg BID group, three in the 30 mg OD group, one in the 10 mg OD group, one in the 3 mg OD group, and two in the placebo group.
Table 2.
Summary of adverse events
| Number (%) of participants with AEs | Placebo n = 31 (100%) |
BAY1128688 | Total N = 121 (100%) |
||||
|---|---|---|---|---|---|---|---|
| 3 mg OD n = 11 (100%) |
10 mg OD n = 31 (100%) |
30 mg OD n = 10 (100%) |
30 mg BID n = 11 (100%) |
60 mg BID n = 27 (100%) |
|||
| Any AE | 24 (77.4) | 8 (72.7) | 16 (51.6) | 9 (90.0) | 7 (63.6) | 23 (85.2) | 87 (71.9) |
| Any TEAE | 20 (64.5) | 5 (45.5) | 10 (32.3) | 9 (90.0) | 6 (54.5) | 21 (77.8) | 71 (58.7) |
| Study-drug-related TEAE | 9 (29.0) | 4 (36.4) | 3 (9.7) | 4 (40.0) | 2 (18.2) | 14 (51.9) | 36 (29.8) |
| Procedure-related TEAE | 0 | 0 | 0 | 0 | 0 | 2 (7.4) | 2 (1.7) |
| Any SAE | 1 (3.2) | 1 (9.1) | 0 | 2 (20.0) | 0 | 1 (3.7) | 5 (4.1) |
| Any TESAE | 1 (3.2) | 1 (9.1) | 0 | 2 (20.0) | 0 | 1 (3.7) | 5 (4.1) |
| Study-drug-related SAE | 1 (3.2) | 0 | 0 | 1 (10.0) | 0 | 1 (3.7) | 3 (2.5) |
| SAE related to procedure | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Discontinuation due to TEAE | 1 (3.2) | 0 | 1 (3.2) | 0 | 2 (18.2) | 3 (11.1) | 7 (5.8) |
| Discontinuation due to TESAE | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
AE adverse event, BID twice daily, OD once daily, SAE serious adverse event, TEAE treatment-emergent adverse event, TESAE treatment-emergent serious adverse event
A total of six treatment-emergent serious adverse events (SAEs) were reported in five participants, all of which occurred after cessation of treatment. There were two events of surgical endometriosis ablation (n = 1 in the placebo group; n = 1 in the 3 mg OD group), three events related to liver disorders in two patients (events ‘DILI’ and ‘Hepatic enzyme increased’ in the same patient in the 30 mg OD group, and event ‘Hepatitis’ in the 60 mg BID group), and one event of peritoneal hemorrhage (in the 30 mg OD group). Four out of the six SAEs were considered trial-drug–related by the investigator (one event of endometriosis ablation and the three events related to liver disorders).
Hepatic Safety
A structured analysis of the number of participants with treatment-emergent laboratory abnormalities for ALT, AST, bilirubin, and INR over time was conducted (Table 3). Ten participants had increased ALT > 2 × ULN and eight of these had increased ALT > 3 × ULN. In seven of these eight cases, the increase in ALT was first observed at visit 7 or thereafter, that is, there was a characteristic clustering of increases in ALT detected at the 12-week time point and in follow-up visits (Fig. 3). The initial increases in ALT were typically followed by further increases in transaminases over approximately 4 weeks following the end of treatment. Restoration of normal ALT levels took up to 3 months (Fig. 3 and Online Resource 7 and 8, see ESM).
Table 3.
Participants with treatment-emergent laboratory abnormalities relating to ALT, AST, bilirubin, and INR
| Laboratory variable | Placebo n = 31 (100%) |
BAY1128688 | Total N = 121 (100%) |
||||
|---|---|---|---|---|---|---|---|
| 3 mg OD n = 11 (100%) |
10 mg OD n = 31 (100%) |
30 mg OD n = 10 (100%) |
30 mg BID n = 11 (100%) |
60 mg BID n = 27 (100%) |
|||
| ALT, U/L; n (%)a | |||||||
| Total number of participants with > 2 × ULN | 0 | 0 | 1 (3.2) | 3 (30.0) | 0 | 6 (22.2) | 10 (8.3) |
| > 2 × ULN and ≤3 × ULNb | 0 | 0 | 1 (3.2) | 0 | 0 | 1 (3.7) | 2 (1.7) |
| > 3 × ULN and ≤5 × ULNb | 0 | 0 | 0 | 2 (20.0) | 0 | 3 (11.1) | 5 (4.1) |
| > 5 × ULN and ≤8 × ULNb | 0 | 0 | 0 | 0 | 0 | 1 (3.7) | 1 (0.8) |
| > 8 × ULNb | 0 | 0 | 0 | 1 (10.0) | 0 | 1 (3.7) | 2 (1.7) |
| AST, U/L; n (%)a | |||||||
| Total number of participants with > 2 × ULN | 0 | 0 | 0 | 1 (10.0) | 0 | 3 (11.1) | 4 (3.3) |
| > 3 × ULN and ≤5 × ULNb | 0 | 0 | 0 | 0 | 0 | 2 (7.4) | 2 (1.7) |
| > 8 × ULNb | 0 | 0 | 0 | 1 (10.0) | 0 | 1 (3.7) | 2 (1.7) |
| Bilirubin in serum, mg/dL; n (%)a | |||||||
| > 2 × ULN | 0 | 0 | 0 | 1 (10.0) | 0 | 2 (7.4) | 3 (2.5) |
ALT alanine aminotransferase, AST aspartate aminotransferase, BID twice daily, INR international normalized ratio, OD once daily, ULN upper limit of normal
aPercentages refer to the total sample size per treatment group (i.e., n)
bOnly the largest value per participant was considered, such that a participant was only counted once, in the highest category. All individuals with AST > 2 × ULN were also observed to have ALT > 2 × ULN. The case of bilirubin > 2 × ULN in the 30 mg OD group refers to the participant described as ‘case 1’ (see Section 3.3) and was found to also have > 2 × ULN for ALT and AST. In contrast, the two participants in the 60 mg BID group with bilirubin > 2 × ULN demonstrated isolated increases of bilirubin only (i.e., no increase in ALT or AST)
Fig. 3.

ALT values in participants with ALT > 3-fold the ULN. Only participants whose maximum value between the start of treatment and the end of the study exceeds 3 × ULN are included here. Lower and upper black horizontal lines correspond to 2 × ULN and 3 × ULN, respectively. Gray vertical dotted line corresponds to planned EoT visit at day 84 ± 4 days of treatment. ALT alanine aminotransferase, EoT end-of-treatment visit, ULN upper limit of normal
Serum total bilirubin increased in a dose-dependent manner, with initial onset during the first 4 weeks of treatment (Fig. 4). The mean and median total, direct, and indirect bilirubin levels increased to a similar extent (approximately 2- to 3-fold increase relative to baseline) (Online Resource 9, see ESM). These findings were in line with the population-based PK/PD model derived from results of the multiple-dose phase I study in healthy volunteers, which had predicted increases in total serum bilirubin not exceeding 2.5 × ULN (see Sect. 1.6). Values started to normalize during treatment towards week 12.
Fig. 4.

Total serum bilirubin change from baseline at each visit in completers of 12 weeks of treatment. Placebo: N = 12; 3 mg OD: N = 4; 10 mg OD: N = 14; 30 mg OD: N = 5; 30 mg BID: N = 4; 60 mg BID: N = 11 Day 1 was the SoT visit (visit 3). The EoT visit (visit 7) was scheduled after 84 days of treatment ± 4 days. The visits at days 10, 28, and 56 also had a leeway of ± 4 days. aBaseline is the last available measurement before the start of treatment (either the measurement taken at visit 3 [labelled ‘Day 1 SoT’ in this figure] or, if that is not available, the measurement taken at visit 1[screening]). BID twice daily, EoT end of treatment, OD once daily, SoT start of treatment
In three participants, the increase in total bilirubin during treatment exceeded the level of 2 × ULN. In one of these cases, total bilirubin increased concurrently with an increase in ALT >3 × ULN around the 12-week time point, therefore fulfilling Hy’s law criteria [15, 23] (patient on 30 mg OD, see case 1 described below). In the other two cases (both in the 60 mg BID group), no concurrent increases in ALT above the threshold of 2 × ULN were observed. One of these two participants had normal baseline ALT values, with the increase occurring from day 12 of treatment and remaining at an increased level for 54 days of ongoing treatment. ALT values returned to normal after the end of treatment. The other participant exhibited borderline high normal baseline values, and her total bilirubin levels increased rapidly after the start of treatment, reaching 4.3 × ULN on day 34. A concomitant ocular icterus led to the discontinuation of treatment in this case. Total bilirubin normalized thereafter.
Increases in serum bilirubin exhibited dynamic characteristics different from those seen in the ALT increases described above, and hence may be facilitated by a different mechanism (see Table 4 and Discussion Sect. 4).
Table 4.
Observations from the AKRENDO1 study, supporting the hypothesis that differing mechanisms lead to isolated increases in serum bilirubin and hepatotoxicity in humans
| Observation | Timing | Dose-dependency | Predictive value of animal toxicology studies | Hypothesis |
|---|---|---|---|---|
| Isolated increase in serum bilirubin (total, direct, and indirect) | Within days/2 weeks of exposure to BAY1128688 with tendency towards normalization at visit after 12 weeks of treatment | Directly correlated with exposure | Observed in cynomolgus monkey animal toxicology study with BAY1128688 | Result of BAY1128688-mediated inhibition of transporters facilitating uptake and efflux of bilirubin to/from hepatocytes (e.g., via OATP1B1 and MRP2) |
| Increase in liver transaminases up to liver injury | >8 weeks of exposure to BAY1128688 | Risk increased with dose, but extent of ALT increase/liver injury not directly correlated with exposure | Not observed in cynomolgus monkey animal toxicology study with BAY1128688 | Result of BAY1128688-mediated inhibition of transporters facilitating exit/export of bile acids/salts from the hepatocyte (e.g., via BSEP) |
ALT alanine aminotransferase, BSEP bile salt export pump, MRP2 multidrug resistance-associated protein 2, OATP organic anion-transporting polypeptides
Three participants had an ALT increase > 5 × ULN, meeting international criteria for liver injury [16] (Table 3).
Case 1: The participant was randomized into the 30 mg OD dose group and exhibited the following serious adverse events: ‘drug-induced liver injury’ and ‘hepatic enzyme increased’ (MedDRA-preferred terms). Assessment revealed a probable severe DILI (according to the RUCAM diagnostic scale [17]), with increased INR and increased total serum bilirubin > 2 × ULN, in combination with up to 76 × ULN increases in ALT (Online Resource 7, see ESM). This was detected at the regular EoT visit following 12 weeks of treatment. Case 1 was identified as hepatocellular liver injury [18] and fulfilled the criteria of Hy’s law [15, 23]. ALT and AST increased between weeks 8 and 12 and continued to increase after the end of treatment, peaking at around 5 weeks post-treatment, ALT and AST levels then normalized (Fig. 3, Online Resource 7, see ESM). Total serum bilirubin did not increase until after 12 weeks of exposure, also peaking around 5 weeks post-treatment. The patient developed additional clinical symptoms (malaise, nausea, vomiting, pain below the right side of the rib cage, fatigue, constipation, scleral icterus, and weight loss), requiring hospitalization. The patient recovered from the event. Alternative explanations for liver injury were excluded. The serious adverse event was assessed by the investigator as severe and related to the study drug. The sponsor assessed the event as related to the study drug.
Case 2: Another participant (in the 60 mg BID dose group) with ALT > 5 × ULN exhibited the serious adverse event ‘hepatitis’ and was assessed as having possible (according to the RUCAM diagnostic scale) mild hepatocellular DILI (Fig. 3, Online Resource 8, see ESM). Total bilirubin increased during the first 8 weeks to 1.6 × ULN without accompanying increases in ALT or AST. Between weeks 8–12 of treatment, ALT and AST increased but total bilirubin decreased. As with Case 1, ALT and AST continued to increase after cessation of treatment, peaking around 2 weeks post-treatment (ALT: 32 × ULN). Alternative explanations for liver injury were excluded. The patient remained asymptomatic, and the event resolved. The investigator and the sponsor assessed the serious adverse event of hepatitis as related to the study drug.
Case 3: A third participant (in the 60 mg BID dose group) exhibited ALT > 5 × ULN and the non-serious adverse event ‘transaminases increased’ after 12 weeks of treatment. Mild liver injury (peak ALT 7.1 × ULN) was detected after early termination. However, according to RUCAM criteria, this was classified as not related to the study drug (mainly based on negative dechallenge, i.e., ALT remained mildly elevated (< 3 × ULN) for 2.5 months after termination of the study drug). Alternative explanations for liver injury were partly excluded. The patient remained asymptomatic, and the event resolved.
Taking the above-described findings together, the sponsor considered the likelihood that the observed liver injuries and ALT increases were related to the study drug to be “near certain”; this assessment was confirmed by an external hepatologist with special expertise in the field of DILI [Watkins P, personal communication, 2018]. The continued increases in ALT long after treatment termination were interpreted by the external hepatologist to most likely reflect an adaptive immune response [Watkins P, personal communication, 2018].
In light of case 1 and case 2, the sponsor concluded that the benefit–risk balance was no longer enough to justify the continuation of AKRENDO1 and decided to terminate the study early. After trial termination, participants were closely monitored for emerging increases in various liver parameters. Unscheduled visits took place for those participants under close liver monitoring after findings of increased transaminases until normalization or at least a substantial decrease in liver enzymes was observed.
Population Pharmacokinetic Modeling and Exposure–Response Analysis for Increases in ALT
Pharmacokinetic analyses were conducted using data from all participants who received at least one dose of BAY1128688 and had at least one measurable plasma concentration post-dose.
BAY1128688 pharmacokinetic data from this study could be adequately described using a one compartment model with interindividual variability quantified on oral clearance and on relative bioavailability. Differences in relative bioavailability between 3 mg OD and other BAY1128688 doses were identified by an increase of 35% (95% confidence interval, 11–59) relative to all doses >3 mg.
The studied doses reached the anticipated BAY1128688 plasma exposure range, with the average being approximately 2-fold higher than expected based on previous population pharmacokinetic modeling using pharmacokinetic data from the healthy volunteer phase I studies [12].
In addition, ER modeling was undertaken to assess the probability of ALT levels exceeding 2 × ULN during BAY1128688 treatment (Fig. 5). The individual area values under the BAY1128688 concentration–time curve at a steady state over 24 h (AUC[0–24]ss) were estimated based on the popPK model for the 10 participants with increased ALT concentrations > 2 × ULN and 80 participants with normal ALT concentrations. The ER of ALT concentrations was characterized by fitting a binary response variable (increased ALT > 2 × ULN vs normal ALT concentration on treatment) versus the BAY1128688 AUC[0–24]ss by means of an Emax-model, parametrized by the maximum effect that can be attributed to the drug (Emax) model, exposure AUC[0–24]ss that produces half of Emax (EAUC50). A baseline rate (E0) of 3.2% was derived from the placebo arm (although one out of 31 participants had increased ALT > 2 × ULN at baseline).
Fig. 5.

Exposure–response analysis for increase in ALT (ALT > 2-fold ULN). Exposure–response relationship of daily steady-state AUC [AUC(0–24)ss] and increased ALT level > 2 × ULN during the course of treatment. Single dots represent each participant of the study with (symbols above ‘1.00’, top end of graph) or without (symbols below ‘0.00’, at lower part of graph) increased ALT level > 2 × ULN versus its model-predicted daily steady-state AUC. The black squares and horizontal bars represent observed ‘increased ALT (> 2 × ULN) rate’ and range of exposure in the different exposure quartiles. Solid red line (red shaded area) represents Emax-model fit (95% confidence band). Dots and horizontal bars at bottom of graph indicate median and range of exposure within respective treatment group. Color of symbols and bars represent the treatment group. Note, in the placebo group, 1 out of 31 participants included in this analysis had increased ALT level > 2 × ULN (at baseline). ALT alanine aminotransferase, AUC(0–24)ss steady-state area under the concentration–time curve for 24 h, BID twice daily, Emax maximum effect that can be attributed to the drug, ER exposure–response, OD once daily, ULN upper limit of normal
A significant relationship, indicating increasing risk for increased ALT > 2 × ULN with increasing BAY1128688 plasma exposure, was identified (p-value < 0.01). However, EAUC50 and Emax could not be identified reliably based on the limited number of subjects in the study and the low number of observed events, leading to a relative high uncertainty in risk prediction (Fig 5).
Pharmacodynamics
To perform pharmacodynamic analyses, data from the 50 completers were used. Additionally, data for all participants who received at least one dose of BAY1128688 or placebo and had at least one measurable serum androsterone were included in analyses.
Serum concentrations of the pharmacodynamic biomarker androsterone increased dose-dependently from visit 4 onwards in all completers, yielding a complete data set covering 12 weeks of treatment (Fig. 6, Online Resource 10, see ESM). No changes in the cyclicity of sex hormones were detected among the completers. Both findings confirm prior observations in a previously conducted healthy volunteer phase I study [12].
Fig. 6.
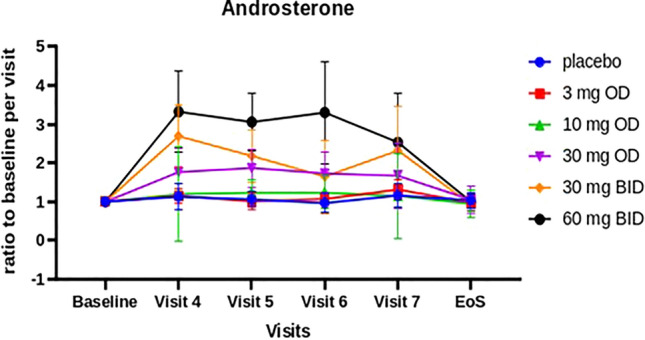
Ratio of mean androsterone values from baseline at each visit (for the subset of 50 participants who completed the study). Baseline is the last available measurement before the start of treatment (either the measurement taken at visit 3 (start of treatment) or, if that is not available, the measurement taken at visit 1 (screening). BID twice daily, OD once daily
Biomarker Sub-Study
Twenty-two exploratory biomarkers (15 plasma bile acids, total bile acids, and 6 biomarkers) previously described as potential predictors of DILI events (see Online Resource 4 in the ESM), as well as androsterone, were tested to determine whether they a) were associated with liver safety parameters (ALT, AST, total serum bilirubin, and INR) or b) could predict increases of ALT > 2 × ULN observed at visit 7 or thereafter.
Associations of 22 Exploratory Biomarkers and Androsterone with Liver Safety Parameters from Visit 3 to Visit 8
The subset of completers (n = 50) was used for this analysis.
This biomarker sub-study investigated whether serum/plasma concentrations of the 22 exploratory biomarkers and androsterone at visits 3–8 were associated with liver safety parameters (ALT, AST, total bilirubin, and INR) at visit 7 or thereafter. In summary, few of the biomarkers investigated were significantly associated with these liver safety parameters at visit 7 or thereafter. Of the 1560 association tests performed, only six were significant after correction for multiple testing (false discovery rate [FDR]-adjusted p-value < 0.05), described in Online Resource 5, see ESM. Specifically, macrophage colony-stimulating factor receptor 1 (MCSFR) at visit 3 (2 h post-dose) and at visit 5 was significantly and positively correlated with ALT at visit 8 (FDR-adjusted p-value = 3.209e-2, ρ = 0.469, FDR-adjusted p-value = 1.358e–2, ρ = 0.470, respectively). Glutamate dehydrogenase (GLDH) at visit 5 was positively correlated with both AST and ALT at visit 8 (FDR-adjusted p-value = 1.145e–2, ρ = 0.744, and FDR-adjusted p-value = 1.145e–2, ρ = 0.733, respectively). Androsterone at visit 6 was positively correlated with total bilirubin at visits 7 and 8 (FDR-adjusted p-value = 3.184e-2 and ρ = 0.454 and FDR-adjusted p-value = 7.231e–3, ρ = 0.523, respectively).
The results were subsequently explored at a relaxed significance threshold of unadjusted p-value < 0.05. While few results were statistically significant even at this threshold, some trends were observed. MCSFR was positively correlated with ALT and/or AST in almost all time point comparisons. These correlations were significant (unadjusted p-value < 0.05) at several time points, with the highest observed correlation coefficient being ρ = 0.47. Of the bile acids, very few significant correlations were identified. Taurolithocholic acid was significantly and positively correlated with AST, ALT, and total bilirubin (unadjusted p-value < 0.05, ρ > 0.3) at visit 7 and visit 8 at multiple time points; this was most notable in correlation tests involving data from visit 3. Glycocholic acid was positively correlated with AST in all time point comparisons and this relationship was significant (unadjusted p-value < 0.05) in three of these comparisons.
In conclusion, associations between selected biomarkers and liver safety parameters could be confirmed, but none were consistently related among all time point comparisons and/or did not reach statistical significance after correction for multiple testing.
Predictive Value of a Panel of 22 Biomarkers and Androsterone for Increases in ALT > 2 × ULN After 12 Weeks of Treatment and Thereafter
This biomarker sub-study investigated whether serum concentrations of the 22 exploratory biomarkers at visits 3–6 could predict events of ALT > 2 × ULN identified at visit 7 and thereafter. Biomarker data at visits 7 and 8 were also assessed, despite not being of predictive value.
Data from completers with ALT increases > 2 × ULN at visit 7 or thereafter were compared with those of completers that did not exhibit such ALT increases. One participant who completed 12 weeks of treatment exhibited an increase in ALT before visit 7 and hence was assigned to the non-affected group of completers. There were five participants with increases in ALT > 2 × ULN at visit 7 and a further two participants with increases in ALT > 2 × ULN detected after visit 7. Please note that two participants listed in Table 3 with treatment-emergent increases in ALT > 2 × ULN were excluded from these analyses because they did not complete the 12 weeks of treatment. The five participants were compared with those 45 completers who did not experience an increase in ALT > 2 × ULN at visit 7 (n = 45), and then all seven participants were compared with the 43 completers who did not experience an increase in ALT > 2 × ULN at visit 7 or thereafter (n = 43).
Serum androsterone concentration did not significantly vary between the seven participants with ALT > 2 × ULN at visit 7 or thereafter and the 43 other participants at any time point except for a single instance (visit 6, p-value < 0.05).
None of the six potentially predictive biomarkers investigated would have been able to predict the increases in ALT detected after 12 weeks of treatment or thereafter prior to week 12 (visit 7). Further details can be found in Online Resource 5, see ESM.
Micro-RNA (miR)-122, caspase-cleaved cytokeratin 18 (ccCK 18), GLDH, cytokeratin 18 (CK 18), and MCSFR levels observed at visits 7 and 8 were significantly different between the seven participants with ALT > 2 × ULN at visit 7 or thereafter and the other 43 participants (FDR-adjusted p-value < 0.05). These differences between the two participant groups were expected considering the increases in ALT concentration at these time points. Full details are given in Online Resource 5, see ESM.
There were no significant associations with ALT > 2 × ULN involving bile acids at FDR-adjusted p-value < 0.05. At a less stringent significance threshold (unadjusted p-value < 0.05), higher plasma concentrations of selected bile acids (glycocholic acid, taurolithocholic acid, and hyodeoxycholic acid) were observed among the seven participants with ALT > 2 × ULN at visit 7 or thereafter when compared with the other 43 participants.
Discussion
The AKRENDO1 trial was designed to assess the dose–response relationship of five different oral doses of the AKR1C3 inhibitor BAY1128688 relative to a placebo in the treatment of endometriosis-related pain. AKRENDO1 was prematurely terminated following the detection of hepatocellular drug-induced liver injury in two participants, one of which fulfilled Hy’s law criteria [15, 23]. The detection of these liver injuries indicated a safety profile that precludes further clinical development of BAY1128688. The limited number of completers reduced the statistical power of any analyses using data from AKRENDO1, thus preventing the application of any inferential statistics. Hence, no conclusions around the efficacy of BAY1128688 could be drawn.
The comparatively high incidence of observations of ALT increase >3 × ULN in eight of the 90 participants treated with BAY1128688, in combination with the latency at which ALT increases, were observed relative to termination of treatment (this pattern was observed in seven of the eight participants that exhibited ALT >3 × ULN) and led to the assumption that BAY1128688 has the potential to be hepatotoxic. No hepatotoxicity was observed until after 8 weeks of exposure in all dose groups, in line with the previously performed 4-week multiple-dose phase I trial [12].
Two—believed to be separate—effects on liver function were observed and are described below and in Table 4.
(1) Early elevations in total serum bilirubin as well as direct and indirect bilirubin were dose-related. They were not associated with elevations in serum aminotransferases (Fig. 3) and were accordingly described as ‘isolated’. All three measures of bilirubin appeared to peak between weeks 1 and 8 before declining towards baseline at 12 weeks of treatment (see Fig. 4 and Online Resource 7 in the ESM). Bilirubin levels had not reached baseline by the end of the 12-week period.
In the prior preclinical 13-week monkey study, the observed elevations in serum bilirubin (total, conjugated, and unconjugated) were reversible, trending towards normalization as treatment progressed (see Sect. 1.5). Early isolated elevations in serum bilirubin had also been witnessed in a preceding healthy volunteer study (see Sect. 1.6). A population PK/PD model using data from the healthy volunteer study predicted concentration-dependent increases in serum total bilirubin up to approximately 2 × ULN, similar to the results observed in this study.
(2) Late elevations in serum ALT occurred after more than 8 weeks of treatment (Fig. 3) and peaked several weeks post-treatment. Late ALT elevation was less clearly dose-/exposure-related than early elevations in serum total bilirubin, but the probability of elevated ALT increased significantly with increased exposure to BAY1128688. ALT increases have been observed in the 30 mg OD (n = 10) and the 60 mg BID (n = 27) groups, but not in the dose group in between (30 mg BID; n = 11). The fact that, despite the otherwise apparent dose–response relationship, no cases have been detected in the 30 mg twice daily group is considered a chance finding due to small sample size and exposures overlapping between dose groups.
These increases in ALT did not coincide with increases in bilirubin levels, except in the most severe case (case 1, the Hy’s law case described in Sect. 3.3) of delayed increases in serum aminotransferases, in which liver injury was associated with an accompanying rise in serum total bilirubin. Increases in serum ALT were not observed in the animal toxicology study (see Sect. 1.5).
In conclusion, the nature of serum bilirubin increases differed from that seen in ALT increases, and hence may be underpinned by a different mechanism.
The inhibition of the human transporter OATP1B1 has been described as a potential cause of mild hyperbilirubinemia via the blocking of bilirubin uptake into hepatocytes [24]. MRP2 function has also been implicated as a potential cause of mild hyperbilirubinemia as it facilitates the excretion of bilirubin from hepatocytes into the bile [25]. Human genetic evidence exists (OATP1B1/1B3 deficiency—Rotor syndrome; MRP2 deficiency—Dubin-Johnson syndrome) that supports both hypotheses. There were no signs of increased bilirubin production (hemolysis) in this study, nor are we aware of any literature describing a role of AKR1C3 in the metabolism of bilirubin. Accordingly, it is hypothesized that the potential of BAY1128688 to inhibit OATP1B1 and MRP2 (see Online Resource 1 in the ESM and section 1.4) plays a key role in the isolated cases of hyperbilirubinemia and increases direct and indirect serum bilirubin levels, as observed in participants within the first 8 weeks of AKRENDO1. In addition, the inhibitory potency of BAY1128688 to UGT1A1 (see Online Resource 2 in the ESM), which is also involved in bilirubin metabolism, could also contribute but is considered of minor clinical relevance. No other human UGTs involved in bile salt metabolism are inhibited by BAY128688 in vitro.
BAY1128688 also has the potential to inhibit the bile salt export pump (BSEP; Online Resource 2, see ESM), an adenosine triphosphate (ATP) binding cassette (ABC)-transporter claimed to be responsible for the excretion of bile salts from hepatocytes into the bile. Inhibition of bile salt transport is thought to increase hepatotoxicity risk and hence may play a role in the ALT increases observed in AKRENDO1 [26]. This has also been incorporated into computational models used in the prediction of hepatotoxicity [27].
DILI caused by intracellular hepatic bile salt accumulation may be clinically represented as hepatocellular injury, resulting in a liver chemistry profile reflecting substantial hepatocyte death (i.e., prominent elevations in serum aminotransferases with minimal elevation in serum AP and bilirubin) [28]. The findings described below support but cannot ultimately prove the hypothesis that the hepatotoxicity observed in AKRENDO1 was the result of the intrahepatic accumulation of bile salts/bile acids due to the inhibition of transporters involved in hepatocyte bile salts/bile acid secretion by the AKR1C3 inhibitor BAY1128688 (for an overview see Online Resource 1 in the ESM).
The pattern of hepatotoxicity associated with intrahepatic bile salt/bile acid accumulation accompanied by minimal increases in serum AP observed in this trial (cases 1 and 2) is consistent with hepatocellular liver injury [28].
Higher plasma concentrations of selected bile acids (glycocholic acid, taurolithocholic acid, hyodeoxycholic acid) were observed among patients with ALT > 2 × ULN after 12 weeks, compared with patients not showing ALT > 2 × ULN. In addition, increases in selected (e.g., tauro-conjugated) plasma bile acids at the start of treatment that lasted until week 8 were found to be associated with increases in serum ALT and/or AST at week 12 and post-treatment (see Sect. 3.6 and Online Resource 10 in the ESM for details). However, both findings lacked statistical strength (e.g., the associations were not statistically significant after correcting for multiple testing). Taken together, these findings may indicate a delay before bile salts-/bile acid-mediated toxicity (due to elevated bile salts/bile acid concentrations) becomes apparent in hepatocytes.
Signs of hepatotoxicity were absent in animal toxicology studies, which are typically known for their weak accuracy in predicting hepatotoxicity in humans, particularly relating to intrahepatic bile salt/bile acid accumulation. This absence reflects the variable sensitivity of different animal species to the modulation of bile salt content in hepatocytes [28].
Taking into consideration the possible risk factors for DILI according to the relevant compound characteristics (potential to inhibit transporters including bile salt transporters and other mediators of bile acid metabolism such as BSEP, see ESM, Online Resources 1–3) [24, 28, 29], hepatotoxicity observed after >8 weeks of treatment suggests an ‘off-target’ effect of the AKR1C3 inhibitor BAY1128688. The most likely cause is the inhibition of intrahepatic bile acids/bile salt transporters, resulting in the observed DILI [30].
Increasing exposure to the AKR1C3 inhibitor BAY1128688 was found to be associated with increased risk of elevated ALT and DILI. While increasing BAY1128688 exposure results in a greater inhibition of AKR1C3, as indicated by increases in the pharmacodynamic biomarker androsterone, it also results in the greater inhibition of bile acid/bile salt transporters. Hence, information regarding BAY1128688 exposure alone does not allow the separation of on- and off-target effects.
The inhibition of human steroid 5β-reductase AKR1D1 has been discussed as a potential off-target effect of BAY1128688 that could contribute to or cause the hepatotoxicity observed in AKRENDO1 by interfering with bile acid synthesis [9]. The AKR1D1 enzyme generates all 5β-reduced dihydrosteroid metabolites for C19-, C21-, and C27-steroids (i.e., androgens, glucocorticoids, and bile acids or oxysterols) and accordingly is involved in steroid hormone inactivation and bile acid synthesis. BAY1128688 was screened preclinically for activity not only against a range of related enzymes including recombinant human AKR1C enzymes (1C1, 1C2, 1C4) but also against human AKR1D1. Using cortisone as a model substrate, BAY1128688 was found to inhibit human AKR1D1 activity by 11% at a concentration of 100 µM (Bayer AG, data on file). Accordingly, it is unlikely that the observed hepatotoxicity of BAY1128688 is related to an off-target effect on AKR1D1.
The inhibitory effect of BAY1128688 on bile salt transporters could lead to alterations in bile acid homeostasis and the accumulation of toxic bile acids in the hepatocytes [30]. Increased concentrations of conjugated bile acids in plasma are potentially monitorable mechanism-based biomarkers of transport protein inhibition in the liver (in particular, BSEP inhibition) in vivo [28, 31]. Although the fraction unbound of BAY1128688 in human plasma is very low, leading to an unbound maximum steady-state concentration in human plasma of < 0.1 µM after 60 mg BID application, the total concentration in human plasma is approximately 10 µM [12]. Therefore, the increase of selected bile acids (known to be substrates of BSEP [32]) may have been caused by total BAY1128688 concentrations above the in vitro IC50 value of 3.6 µM for BSEP inhibition [Online Resource 1, see ESM]. In our AKRENDO1 trial, individual bile acid levels were increased at isolated ‘early’ time points suggesting a mechanistic link, however, the strength of the association was insufficient to allow consistent prediction of DILI events based on bile acid increases (total and/or individual). While in this specific case bile acid measurements were of limited value for prediction of increases in ALT, we continue to consider bile acid measurements as generally useful for monitoring and mechanistic understanding.
Multiple serum biomarkers were suggested to have prognostic value in predicting DILI or severe outcomes [21, 33]. However, none of the six potentially predictive biomarkers nor any of the bile acids investigated would have been able to consistently predict the increases in ALT detected after 12 weeks of treatment or thereafter prior to week 12 of treatment (visit 7). Moreover, increases in androsterone, the pharmacodynamic biomarker of AKR1C3 inhibition, were not associated with increases in ALT.
The observed trends in circulating MCSFR (macrophage colony-stimulating factor receptor 1) are of particular note. MCSFR has been described as a predictive biomarker in the context of analyses of serum ALT increases observed in patients exposed to the anticoagulant ximelagatran [34]. Another result of note in the development of BAY1128688 is the lack of hepatotoxicity signals in animal toxicology studies. MCSFR is the receptor for CSF-1, a cytokine that controls the proliferation, differentiation, and function of macrophages. Increases in (shed) plasma-derived MCSFR have been proposed as an indicator of macrophage activation. Hence, a role of macrophages in the inflammatory response associated with drug-induced liver injury has been hypothesized [35]. However, while associations between increased systemic MCSFR and ALT were detected consistently across a range of time points (visits), these associations were not statistically significant after correction for multiple testing.
This clinical trial highlights the limitations of in vivo testing in predicting DILI in long-term toxicity studies, as an interaction of BAY1128688 with bile salt transport proteins (including BSEP) was observed in vitro without any clear evidence of hepatotoxicity having been observed in preclinical animal studies. In the 13-week animal study, total, conjugated, and unconjugated serum bilirubin levels were transiently increased, but concomitant increases in liver enzymes or microscopic liver findings were not seen. Preclinical in vivo testing cannot accurately predict DILI resulting from bile acid homeostasis alterations as animal studies do not specifically investigate the mechanism of transport inhibition-related hepatotoxicity [36–38]. In addition, differences in bile salt metabolism and the tolerability of bile salt accumulation between humans and animals means liver toxicity risk can be difficult to predict [39, 40].
Conclusion
The AKRENDO1 trial, which aimed to assess the efficacy and safety of the AKR1C3 inhibitor BAY1128688 as a potential treatment for endometriosis-related pain symptoms, was prematurely terminated by the sponsor due to cases of hepatotoxicity. The pharmacodynamics of BAY1128688 observed in AKRENDO1 (such as increases in androsterone and bilirubin) were comparable with those found in a previously conducted, healthy volunteer, 4-week study. However, as indicated by preclinical studies [9, 10], AKR1C3 inhibition remains a potentially valid therapeutic approach to treating hormone-dependent malignancies and endocrine disorders characterized by AKR1C3 overexpression, including endometriosis and polycystic ovary syndrome [2]. The development of other drugs that target AKR1C3 and are not associated with risks of hepatotoxicity may allow for treatment of diseases proposed to benefit from AKR1C3 inhibition.
The absence of hepatotoxicity indicators in prior animal toxicity studies highlights the importance of in vitro mechanistic studies in identifying potential DILI risks related to drug exposure in humans. Further investigations into the mechanisms behind the hepatotoxicity observed in this study have been performed using a Quantitative Systems Toxicology modeling tool, including DILIsym® (Simulations Plus, Durham, NC, USA) [41], to evaluate the safety of BAY1128688 relative to drug exposure and the activity of hepatotoxic mechanisms including mitochondrial dysfunction, oxidative stress, and bile acid accumulation [11].
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgments
This study has been funded by Bayer AG, Berlin, Germany. The authors would like to thank the participants for their cooperation and the investigators and study team, led by the study manager Tea Vedenkannas and co-study manager Anu Sulamaa, for their excellent operational conduct of this trial and their continued commitment to the study. The authors would like to acknowledge Andreas Kaiser (Bayer AG, Berlin, Germany) and Franco Mendolia (Bayer AG, Wuppertal, Germany) for their assistance with statistical analyses, Beate Rohde (Bayer AG, Berlin, Germany) for their critical review of the manuscript, and Udo Oppermann (Botnar Research Centre, University of Oxford) and Michaele Peters (Bayer AG, Berlin, Germany) for their critical review of the discussion section and assistance in describing selectivity against other human AKR enzymes, including AKR1D1. The authors would also like to acknowledge Highfield, Oxford, UK for providing medical writing assistance with funding from Bayer AG and Heike Gimeson (medical writing at Bayer AG, Berlin, Germany) for support during the revision process.
Declarations
Funding
The study was funded by Bayer AG, Berlin, Germany. Bayer AG was responsible for the study design, data collection and analysis, and overall conduct of the study.
Conflicts of Interest
Jan Hilpert, Esther Groettrup-Wolfers, Kerstin Gude, Stefanie Reif, Thomas Steger-Hartmann, Christian Scheerans, Alexander Solms, Antje Rottmann, and Guangping Mao are full-time employees of Bayer AG, Berlin, Germany. Isabella Gashaw was a full-time employee of Bayer AG, Berlin, Germany, at the time of study conduct. Hristiyan Kosturski is a full-time employee of Svelte scientific GmbH, Berlin, Germany. Svelte scientific was responsible for medical monitoring during the study and received funding for this from Bayer AG, Berlin, Germany. Laura Bennett and Catriona Barnes are full-time employees of Fios Genomics Ltd, Edinburgh, United Kingdom. Fios Genomics was responsible for the statistical analyses in the biomarker sub-study and received funding for this from Bayer AG, Berlin, Germany. Professor Charles Chapron acted as a consultant and has received honorarium from Abbvie, Besins, Bayer, Gedeon Richter, and Ipsen.
Ethics Approval
Before the start of the study, the protocol and protocol amendments were reviewed and approved by each study site’s independent ethics committee/institutional review board (see Online Resource 11 in the ESM for a full list). The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use’s Good Clinical Practice Guideline (E6).
Consent to Participate
Written informed consent was obtained from all study participants.
Consent for Publication
Written consent for publication was obtained from the sponsor (Bayer AG).
Availability of Data and Material
The availability of the data reported in this publication is determined according to Bayer’s commitment to the European Federation of Pharmaceutical Industries and Associations and Pharmaceutical Research and Manufacturers of America principles for responsible clinical trial data sharing, pertaining to scope, time points, and the process of data access. Bayer commits to sharing patient-level and study-level clinical trial data and protocols, upon request from qualified scientific and medical researchers as necessary for doing legitimate research. This commitment applies to data relating to new medicines and indications that have been approved by the European Union and US regulatory agencies on or after January 1, 2014. Researchers can request access to anonymized patient-level data and supporting documents from clinical studies via www.clinicalstudydatarequest.com in order to perform further research to advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the ‘study sponsors section’ of this portal. Data access to anonymized patient-level data, protocols, and clinical study reports is granted after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
Code Availability
Not applicable.
Authors’ Contributions
Jan Hilpert: conception, design, and conduct of the study including safety surveillance; interpretation of results; critical review and revision of the manuscript; and approval of the final manuscript for submission. Esther Groettrup-Wolfers: conception and design of the study; safety surveillance; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Hristiyan Kosturski: conduct of the study including safety surveillance; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Laura Bennett: biomarker sub-study statistical analyses; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Catriona Barnes: biomarker sub-study statistical analyses; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Kerstin Gude: safety surveillance; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Isabella Gashaw: conception, design, and conduct of the study; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Stefanie Reif: contributions to preparation and conduct of the study; evaluation and interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Thomas Steger-Hartmann: evaluation and interpretation of toxicity data; critical review and revision of toxicology sections; approval of the final manuscript for submission. Christian Scheerans: study design; sparse pharmacokinetic sampling strategy; pharmacometric data analysis strategy; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Alexander Solms: pharmacometric data analysis; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Antje Rottmann: preparation of the study; evaluation of preclinical transporter and enzyme interaction data; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Guangping Mao: conception, design, and conduct of the study including safety surveillance; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission. Charles Chapron: design and conduct of the study; interpretation of results; critical review and revision of the manuscript; approval of the final manuscript for submission.
References
- 1.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol. 2011;125(1–2):95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Penning TM. AKR1C3 (type 5 17beta-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol Cell Endocrinol. 2019;489:82–91. doi: 10.1016/j.mce.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zondervan KT, et al. Endometriosis. Nat Rev Dis Primers. 2018;4(1):9. doi: 10.1038/s41572-018-0008-5. [DOI] [PubMed] [Google Scholar]
- 4.Bulun SE, et al. Endometriosis. Endocr Rev. 2019;40(4):1048–1079. doi: 10.1210/er.2018-00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marquardt RM, et al. Progesterone and estrogen signaling in the endometrium: what goes wrong in endometriosis? Int J Mol Sci. 2019;20(15):3822. doi: 10.3390/ijms20153822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapron C, et al. Rethinking mechanisms, diagnosis and management of endometriosis. Nat Rev Endocrinol. 2019;15(11):666–682. doi: 10.1038/s41574-019-0245-z. [DOI] [PubMed] [Google Scholar]
- 7.Marinho MCP, et al. Quality of life in women with endometriosis: An integrative review. J Womens Health (Larchmt) 2018;27(3):399–408. doi: 10.1089/jwh.2017.6397. [DOI] [PubMed] [Google Scholar]
- 8.La Rosa VL, et al. Quality of life in women with endometriosis: a narrative overview. Minerva Med. 2020;111(1):68–78. doi: 10.23736/S0026-4806.19.06298-0. [DOI] [PubMed] [Google Scholar]
- 9.Rizner TL, Penning TM. Aldo-keto reductase 1C3-Assessment as a new target for the treatment of endometriosis. Pharmacol Res. 2020;152:104446. doi: 10.1016/j.phrs.2019.104446. [DOI] [PubMed] [Google Scholar]
- 10.Peters MEA, Bothe U, Sohler F, Fischer OM, Zollner TM. A novel AKR1C3 inhibitor demonstrates strong efficacy in a marmoset monkey endometriosis model. 2016: Poster presented at the Society for Reproductive Investigation, 63rd Annual Scientific Meeting, March 16–19, 2016, Montreal, Canada.
- 11.Battista C, et al. Quantitative Systems Toxicology identifies independent mechanisms for hepatotoxicity and bilirubin elevations due to AKR1C3 Inhibitor BAY1128688. Paper submitted to Journal: Clinical Pharmacology & Therapeutics on 19 May 2023 (submission ID: 2023-0318). [DOI] [PubMed]
- 12.Gashaw I, et al. Novel AKR1C3 inhibitor affects androgen metabolism but not ovarian function in healthy women: A phase 1 study. Accepted for publication: 11-Apr-2023. Online ahead of print as Eur J Endocrinol. 2023 Jun 12;lvad063 / EJE-22-1077.R2. [DOI] [PMC free article] [PubMed]
- 13.O'Reilly MW, et al. AKR1C3-mediated adipose androgen generation drives lipotoxicity in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2017;102(9):3327–3339. doi: 10.1210/jc.2017-00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gater A, et al. Development and content validation of two new patient-reported outcome measures for endometriosis: the Endometriosis Symptom Diary (ESD) and Endometriosis Impact Scale (EIS) J Patient Rep Outcomes. 2020;4(1):13. doi: 10.1186/s41687-020-0177-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Food and Drug Administration. Guidance for Industry Drug-induced liver injury: Premarketing clinical evaluation. 2009 [cited Nov 17, 2020.
- 16.Aithal GP, et al. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther. 2011;89(6):806–815. doi: 10.1038/clpt.2011.58. [DOI] [PubMed] [Google Scholar]
- 17.Danan G, Benichou C. Causality assessment of adverse reactions to drugs–I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol. 1993;46(11):1323–1330. doi: 10.1016/0895-4356(93)90101-6. [DOI] [PubMed] [Google Scholar]
- 18.Chalasani NP, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950–966. doi: 10.1038/ajg.2014.131. [DOI] [PubMed] [Google Scholar]
- 19.Koal T, et al. Standardized LC-MS/MS based steroid hormone profile-analysis. J Steroid Biochem Mol Biol. 2012;129(3–5):129–138. doi: 10.1016/j.jsbmb.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Food and Drug Administration. Letter of Support (LOS) Initiative: Letter of support for drug-induced liver injury (DILI) biomarker(s). 2016 [cited Nov 17, 2020].
- 21.Church RJ, Watkins PB. Serum biomarkers of drug-induced liver injury: Current status and future directions. J Dig Dis. 2019;20(1):2–10. doi: 10.1111/1751-2980.12684. [DOI] [PubMed] [Google Scholar]
- 22.Pham HT, et al. Inter-laboratory robustness of next-generation bile acid study in mice and humans: International ring trial involving 12 laboratories. J Appl Lab Med. 2016;1(2):129–142. doi: 10.1373/jalm.2016.020537. [DOI] [PubMed] [Google Scholar]
- 23.Reuben A. Hy's law. Hepatology. 2004;39(2):574–578. doi: 10.1002/hep.20081. [DOI] [PubMed] [Google Scholar]
- 24.Kotsampasakou E, Escher SE, Ecker GF. Linking organic anion transporting polypeptide 1B1 and 1B3 (OATP1B1 and OATP1B3) interaction profiles to hepatotoxicity—the hyperbilirubinemia use case. Eur J Pharm Sci. 2017;100:9–16. doi: 10.1016/j.ejps.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Keppler D. The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia. Drug Metab Dispos. 2014;42(4):561–565. doi: 10.1124/dmd.113.055772. [DOI] [PubMed] [Google Scholar]
- 26.Kock K, et al. Risk factors for development of cholestatic drug-induced liver injury: inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab Dispos. 2014;42(4):665–674. doi: 10.1124/dmd.113.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Longo DM, et al. Quantitative systems toxicology analysis of in vitro mechanistic assays reveals importance of bile acid accumulation and mitochondrial dysfunction in TAK-875-induced liver injury. Toxicol Sci. 2019;167(2):458–467. doi: 10.1093/toxsci/kfy253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgan RE, et al. A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci. 2013;136(1):216–241. doi: 10.1093/toxsci/kft176. [DOI] [PubMed] [Google Scholar]
- 29.Strassburg CP, et al. Developmental aspects of human hepatic drug glucuronidation in young children and adults. Gut. 2002;50(2):259–265. doi: 10.1136/gut.50.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mosedale M, Watkins PB. Drug-induced liver injury: Advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther. 2017;101(4):469–480. doi: 10.1002/cpt.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dawson S, et al. In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab Dispos. 2012;40(1):130–138. doi: 10.1124/dmd.111.040758. [DOI] [PubMed] [Google Scholar]
- 32.Byrne J, et al. The human bile salt export pump: Characterization of substrate specificity and identification of inhibitors. Gastroenterology. 2002;123(5):1649–1658. doi: 10.1053/gast.2002.36591. [DOI] [PubMed] [Google Scholar]
- 33.Church RJ, et al. Candidate biomarkers for the diagnosis and prognosis of drug-induced liver injury: An international collaborative effort. Hepatology. 2019;69(2):760–773. doi: 10.1002/hep.29802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersson U, et al. A systems biology approach to understanding elevated serum alanine transaminase levels in a clinical trial with ximelagatran. Biomarkers. 2009;14(8):572–586. doi: 10.3109/13547500903261354. [DOI] [PubMed] [Google Scholar]
- 35.Kullak-Ublick GA, et al. Drug-induced liver injury: recent advances in diagnosis and risk assessment. Gut. 2017;66(6):1154–1164. doi: 10.1136/gutjnl-2016-313369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen M, et al. Toward predictive models for drug-induced liver injury in humans: are we there yet? Biomark Med. 2014;8(2):201–213. doi: 10.2217/bmm.13.146. [DOI] [PubMed] [Google Scholar]
- 37.Kuna L, et al. Models of drug induced liver injury (DILI) - Current issues and future perspectives. Curr Drug Metab. 2018;19(10):830–838. doi: 10.2174/1389200219666180523095355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Albrecht W, et al. Prediction of human drug-induced liver injury (DILI) in relation to oral doses and blood concentrations. Arch Toxicol. 2019;93(6):1609–1637. doi: 10.1007/s00204-019-02492-9. [DOI] [PubMed] [Google Scholar]
- 39.Thakare R, et al. Species differences in bile acids II. Bile acid metabolism. J Appl Toxicol. 2018;38(10):1336–1352. doi: 10.1002/jat.3645. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Dawson PA. Animal models to study bile acid metabolism. Biochim Biophys Acta Mol Basis Dis. 2019;1865(5):895–911. doi: 10.1016/j.bbadis.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woodhead JL, et al. Application of a mechanistic model to evaluate putative mechanisms of tolvaptan drug-induced liver injury and identify patient susceptibility factors. Toxicol Sci. 2017;155(1):61–74. doi: 10.1093/toxsci/kfw193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.