

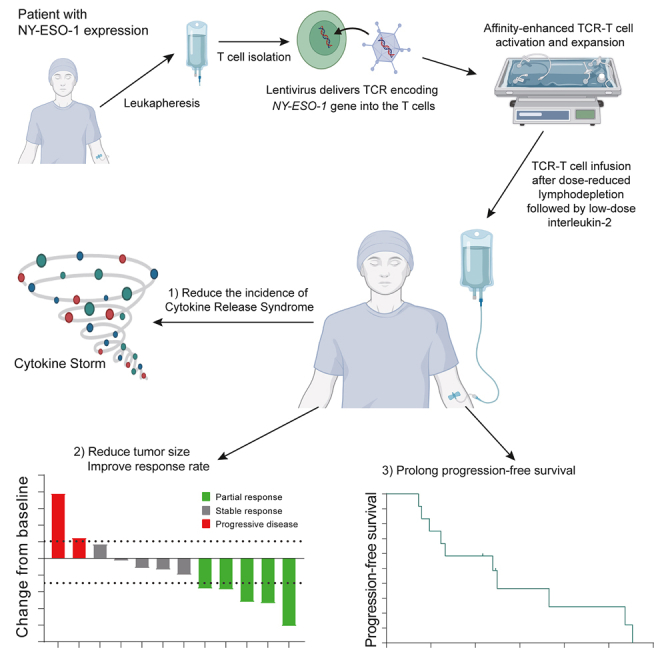
Summary
New York esophageal squamous cell carcinoma-1 (NY-ESO-1)-specific T cell receptor (TCR) T cell therapy is effective in tumors with NY-ESO-1 expression, but a safe and effective TCR-T cell therapeutic protocol remains to be improved. Here, we report a phase 1 investigational new drug clinical trial with TCR affinity-enhanced specific T cell therapy (TAEST16001) for targeting NY-ESO-1. Enrolled patients receive TAEST16001 cell infusion after dose-reduced lymphodepletion with cyclophosphamide (15 mg/kg/day × 3 days) combined with fludarabine (20 mg/m2/day × 3 days), and the TCR-T cells are maintained with low doses of interleukin-2 injection post-adoptive transfer. Analysis of 12 patients treated with the regimen demonstrates no treatment-related serious adverse events. The overall response rate is 41.7%. The median progression-free survival is 7.2 months, and the median duration of response is 13.1 months. The protocol of TAEST16001 cells delivers a safe and highly effective treatment for patients with advanced soft tissue sarcoma (ClinicalTrials.gov: NCT04318964).
Graphical abstract
Highlights
-
•
TAEST16001 cells are high-affinity NY-ESO-1-specific TCR-T cells
-
•
TAEST16001 cell therapy is well tolerated in advanced soft tissue sarcoma
-
•
TAEST16001 cell therapy has promising anti-tumor activity and durable response
-
•
Several prespecified biomarkers are associated with patient response
Pan et al. demonstrate the safety, efficacy, and survival of TAEST16001 cells in HLA-A∗02:01 patients with advanced soft tissue sarcoma. They show that TAEST16001 cell therapy is well tolerated and has promising anti-tumor activities for advanced soft tissue sarcoma expressing the NY-ESO-1 antigen.
Introduction
Soft tissue sarcomas (STSs) are a group of heterogeneous mesenchymal cancers that comprise more than 50 different histological types.1 About 60% of patients with STS present with localized disease at the time of diagnosis, and 40% of patients develop metastases within 5 years.2 Treatment options for advanced, unresectable, or metastatic STS are scarce, both in number and efficacy. Anthracycline (e.g., doxorubicin), either as monotherapy or in combination with ifosfamide, is the most conventional first-line treatment regimen.3 After progression on an anthracycline regimen, second-line treatment options include dacarbazine, ifosfamide, gemcitabine, docetaxel, trabectedin, eribulin, or pazopanib.1,4 However, survival benefits with these drugs are generally unsatisfactory because they are correlated with low overall response rates (5%–16%), short duration of disease control, and risk of treatment-related side effects.5 Thus, there is an urgent need for more effective and tolerable treatment for patients with advanced STS, and immunotherapy may offer attractive solutions.
Immunotherapy with immune checkpoint inhibitors represents a landscape for the management of many solid tumors. However, the total objective response rate (ORR) of anti-PD-1 antibody monotherapy in sarcomas was only 5%–18%,6,7 which is far from fulfilling clinical demand. Moreover, the progression-free survival (PFS) and overall survival (OS) data of the current immune checkpoint inhibitor trials were similar to that of traditional chemotherapy trials in sarcomas.8 Poor T cell infiltration, marginal PD-L1 expression, and lower levels of nonsynonymous somatic mutation burden in sarcoma may at least partially explain the poor clinical benefit of immune checkpoint inhibitors.9,10 Therefore, other immunotherapy approaches, such as adoptive T cell immunotherapy, deserve important exploration in sarcomas.
New York esophageal squamous cell carcinoma-1 (NY-ESO-1) is a cancer-germline antigen (CGA) or cancer-testis antigen that is expressed in a wide range of tumor types.11 NY-ESO-1 has been considered a potential target of immunotherapy in sarcomas12 and is reported to be expressed in 88% of myxoid liposarcomas, 49% of synovial sarcomas, and 35% of myxofibrosarcomas by immunohistochemical staining.13 Although several previous studies have demonstrated that adoptively transferred patient-derived T cell receptor (TCR)-engineered T cells (TCR-T) specific for NY-ESO-1 are preliminarily effective in NY-ESO-1+ synovial sarcomas,14,15,16,17,18 some important questions of TCR-T cell therapy such as the consensus on optimal affinity, cell dose, predictive biomarkers, and minimized toxicity still needed to be explored.
T cell infiltration is required for effective T cell therapy. TCR-T cell tumor infiltration can be improved by optimizing the TCR-pHLA interation.19 This improvement can be achieved by generating high-affinity TCR through mutations in complementarity-determining regions (CDRs).19 Using previously published approaches,20,21 we enhanced the affinity of a TCR, which was isolated with previously published methods22 from peripheral blood mononuclear cells (PBMCs) of a healthy donor, by phage display for the development of TCR-engineered T cells (TAEST16001 cells) specific for the NY-ESO-1157–165 epitope in complex with HLA-A∗02:01. Our preclinical investigation indicated a good safety and efficacy for TAEST16001 cells (data not shown). Here, we report results from an open-label, dose-escalating phase 1 investigational new drug clinical trial evaluating safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary efficacy of TAEST16001 cells maintained with low-dose interleukin-2 (IL-2) in patients after dose-reduced lymphodepletion for the treatment of advanced soft tissue sarcoma. We also investigated the correlations between responses and selected prespecified biomarkers.
Results
Manufacturing of TAEST16001 cells
We developed a manufacturing procedure for producing TCR-T cells under good manufacturing practice (GMP) conditions using closed and semi-closed modular systems (Figure S1A). To validate the procedure, TAEST16001 cells were produced using PMBCs from three healthy donors. The final products showed a predominant memory phenotype (Figure S1B) and mediated interferon γ (INF-γ) release and specific lysis of NY-ESO-1+ tumor cell lines or T2 cells loaded with the NY-ESO-1157–165 peptide (Figures S1C–S1G). The manufacturing procedure was used for all the patients in this clinical study.
Patient and treatment characteristics
Between March 23, 2020, and December 31, 2021, 436 patients with sarcoma were screened for HLA-A∗02:01 and NY-ESO-1 expression. A total of 12 HLA-A∗02:01+ patients with NY-ESO-1+ advanced, unresectable sarcoma were included (Figure S2). Of the 12 enrolled patients, 10 had synovial sarcomas and 2 had liposarcomas (1 myxoid liposarcoma, 1 dedifferentiated liposarcoma). The median age was 33 (25–67) years, and 58.3% (7/12) of patients were men. 83.3% of patients had received at least 2 lines of prior chemotherapy. The median time from enrollment to TAEST16001 cell infusion was 43 days (interquartile range [IQR], 35.5–46.5 days). All 12 patients received modified lymphodepletion, which was a lower dose of precondition than previously reported (Table S1),14,15,17 followed by a median of 3.24 × 109 TAEST16001 cells (range, 0.51–11.96 × 109) plus low-dose systemic IL-2 (500,000 IU per time, twice daily for 14 days). Characteristics of the patients and the administered TCR-T cells are shown in Tables 1 and S2, respectively.
Table 1.
Baseline characteristics of patients
| Patient | Age (years) | Gender | Sites of disease | Prior chemotherapy regimensa | Pathological subtype | NY-ESO-1 expression (%) | Cells (×109) |
|---|---|---|---|---|---|---|---|
| T01 | 67 | M | leg | 2: doxorubicin+ifosfamide+dacarbazine, apatinib | synovial sarcoma | 70 | 0.53 |
| T02 | 27 | M | leg | 1: doxorubicin+ifosfamide | myxoid liposarcoma | 90 | 0.51 |
| T03 | 40 | M | arm, pleura, mediastinum | 3: doxorubicin+ifosfamide, anlotinib+TQB2450 (clinical trial), apatinib | synovial sarcoma | 100 | 0.51 |
| T04 | 28 | F | arm, pleura, lung | 3: cyclophosphamide+epirubicin+vindesine+dacarbazine, apatinib, pazopanib | synovial sarcoma | 100 | 1.94 |
| T05 | 25 | F | lung, vertebra, mediastinum | 2: doxorubicin+ifosfamide, anlotinib | synovial sarcoma | 90 | 1.70 |
| T06 | 42 | M | arm, lung | 2: doxorubicin+ifosfamide, anlotinib | synovial sarcoma | 30 | 1.86 |
| T07 | 33 | M | leg, lung | 2: doxorubicin+ifosfamide, ifosfamide | synovial sarcoma | 95 | 4.65 |
| T08 | 37 | F | arm, lung, pleura | 2: doxorubicin+ifosfamide, ifosfamide | synovial sarcoma | 95 | 4.71 |
| T09 | 33 | M | arm, lung | 2: doxorubicin+ifosfamide, ifosfamide | synovial sarcoma | 35 | 4.55 |
| T10 | 32 | M | leg, lung, pleura, bone | 3: doxorubicin+ifosfamide+dacarbazine+endostar, doxorubicin+dacarbazine+endostar+anti-PD-1 antibody, anlotinib | synovial sarcoma | 40 | 11.09 |
| T11 | 64 | F | arm | 1: doxorubicin+ifosfamide | dedifferentiated liposarcoma | 40 | 11.96 |
| T12 | 27 | F | subcutaneous, liver, peritoneum, pelvis | 2: gemcitabine+docetaxel, doxorubicin+ifosfamide | synovial sarcoma | 95 | 9.56 |
F, female; M, male.
Lines of therapy: names of agents.
Safety and tolerability of TAEST16001 cells
No predefined dose-limiting toxicities (DLTs) within 28 days after TAEST16001 cell infusion were observed during the dose-escalation phase. The most common adverse events of grade 3 or higher among the 12 treated subjects were lymphopenia (100%), neutropenia (92%), leukopenia (83%), and anemia (33%), which were primarily attributable to the precondition of lymphodepletion (Table 2). Other relevant grade 3–4 toxicities included fever (8%), thrombocytopenia (8%), hypokalemia (8%), increased alanine aminotransferase (8%), proteinuria (8%), and hypertriglyceridemia (8%). Two patients in the second cell-dose level experienced grade 2 cytokine release syndrome (CRS) and developed fever on the second day after the cell infusion. Hypotension and hypoxia occurred in the two patients on the third and fourth days, respectively. One patient was treated with tocilizumab, the other with oxygen therapy, and both recovered from CRS within 1 week with no remaining complications. None of the patients had neurotoxicity or serious adverse events related to cell infusion.
Table 2.
Adverse events
| Adverse event | Grade 1 (%) | Grade 2 (%) | Grade 3 (%) | Grade 4 (%) | Total (%) |
|---|---|---|---|---|---|
| Hematological | |||||
| Neutropenia | 0 (0) | 1 (8) | 5 (42) | 6 (50) | 12 (100) |
| Lymphopenia | 0 (0) | 0 (0) | 1 (8) | 11 (92) | 12 (100) |
| Leukopenia | 0 (0) | 2 (17) | 4 (33) | 6 (50) | 12 (100) |
| Anemia | 3 (25) | 3 (25) | 4 (33) | 0 (0) | 10 (83) |
| Thrombocytopenia | 2 (17) | 0 (0) | 1 (8) | 0 (0) | 3 (25) |
| Metabolism and nutrition disorders | |||||
| Hypoalbuminemia | 10 (83) | 2 (17) | 0 (0) | 0 (0) | 12 (100) |
| Hypocalcemia | 5 (42) | 6 (50) | 0 (0) | 0 (0) | 11 (92) |
| Hyponatremia | 8 (67) | 1 (8) | 0 (0) | 0 (0) | 9 (75) |
| Hyperglycemia | 7 (58) | 1 (8) | 0 (0) | 0 (0) | 8 (67) |
| Hypokalemia | 5 (42) | 1 (8) | 1 (8) | 0 (0) | 7 (58) |
| Decreased appetite | 5 (42) | 2 (17) | 0 (0) | 0 (0) | 7 (58) |
| Gastrointestinal | |||||
| Constipate | 3 (25) | 3 (25) | 0 (0) | 0 (0) | 6 (50) |
| Diarrhea | 3 (25) | 3 (25) | 0 (0) | 0 (0) | 6 (50) |
| Nausea | 4 (33) | 2 (17) | 0 (0) | 0 (0) | 6 (50) |
| Vomit | 4 (33) | 2 (17) | 0 (0) | 0 (0) | 6 (50) |
| Other | |||||
| Fever | 4 (33) | 7 (58) | 1 (8) | 0 (0) | 12 (100) |
| Increased ALT | 7 (58) | 0 (0) | 1 (8) | 0 (0) | 8 (67) |
| Proteinuria | 5 (42) | 1 (8) | 1 (8) | 0 (0) | 7 (58) |
| Fatigue | 7 (58) | 0 (0) | 0 (0) | 0 (0) | 7 (58) |
| Hypertriglyceridemia | 4 (33) | 1 (8) | 1 (8) | 0 (0) | 6 (50) |
| Increased AST | 3 (25) | 2 (17) | 0 (0) | 0 (0) | 5 (42) |
| Chills | 4 (33) | 1 (8) | 0 (0) | 0 (0) | 5 (42) |
| Dizziness | 4 (33) | 0 (0) | 0 (0) | 0 (0) | 4 (33) |
| Cytokine release syndrome | 0 (0) | 2 (17) | 0 (0) | 0 (0) | 2 (17) |
| Neurotoxicities | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Efficacy of TAEST16001 cells
All 12 patients that received the prescribed cell dose of TAEST16001 were eligible for efficacy analysis, and 9 (75%) patients showed tumor regression (Figure 1A). Patients infused with TAEST16001 cells in this trial demonstrated a median time to response (TTR) of 1.9 months (range, 0.9–3 months) and a median duration of response of 13.1 months (range, 5–14.3 months; Figures 1B and S3). Six (50%) patients experienced continued decreases in tumor burden following the first radiological assessment (Figure 1C). At the time of the primary analysis, the best response was partial response in 5 of 12 patients for an ORR of 41.7% (95% confidence interval [CI], 15.2–72.3; Figure 1A; Table S3). Five (41.7% [95% CI, 15.2–72.3]) patients achieved stable disease for a disease control rate of 83.3% (95% CI, 51.6–97.9). Four of five (80%) patients without lung metastases had partial response. The median PFS was 7.2 months (95% CI, 2.5–11.8) (Figure 1D). At the time of data cutoff (April 15, 2022), 10 patients had disease progression, and one of them had died from disease. Therefore, the proportion censored at the time was 11/12 (91.7%), and the median OS was immature.
Figure 1.

Clinical response and progression-free survival outcomes in 12 patients who were treated with TAEST16001 cells
(A) Waterfall plot of the best clinical response for each evaluable patient.
(B) Swimlane plot shows clinical outcomes of treated patients.
(C) Spider plot shows the change in the sum of the diameters of each patient’s target lesions over time.
(D) Kaplan-Meier estimates of the progression-free survival. Tick marks indicate the time of data censoring at their last date of contact.
Notably, patient T06 (42-year-old male), who had received two prior lines of therapy and experienced grade 2 CRS after TAEST16001 treatment, exhibited a partial response of upper arm and multiple lung metastatic lesions after 4 weeks of cell infusion, maintaining more than 14 months following treatment (Figure 2A). As the lung metastatic lesions remained a great partial response even at the time of progression, the patient underwent surgical resection of a lesion in the upper arm. Another patient, T02 (27-year-old male), who had recurrent myxoid liposarcoma in the right leg after receiving five surgeries, also exhibited a partial response and sustained more than 1 year following treatment (Figure 2B).
Figure 2.

Computed tomography (CT) scans demonstrating tumor regression
(A) CT scans from patient T06, who had large synovial sarcoma in the right arm with numerous lung metastatic lesions.
(B) CT scans from patient T02, who had recurrent myxoliposarcoma in the right leg. Red arrows and ellipse point to tumors.
TAEST16001 cell expansion and persistence
All 12 patients were assessed for pharmacokinetics. The TCR gene copies per μg genomic DNA (gDNA) at prescribed points before and after infusion are shown in Figure 3A. The median Cmax value after cell infusion was 52,582 copies per μg gDNA (range, 11,156–838,655), and the median Tmax value was 6 days (range, 0.08–20). The Cmax value did not show significant differences among the four dosing groups (Figure 3B). There was a trend toward increased expansion of the TCR-T cells as assessed by qPCR in the responders (median, 103,806 copies per μg gDNA; range, 26,260–838,655) compared with the nonresponders (median, 25,160 copies per μg gDNA; range, 11,156–447,847; Figure 3C, p = 0.1061). Circulating TCR-expressing T cells were detected in the 5 patients for whom monitoring continued beyond 25 weeks (Figure 3A).
Figure 3.

Peak TCR-engineered T cell expansion and association with response
(A) TCR copies as measured by vector transgene copies per μg genomic DNA in peripheral blood, according to clinical response. LLOQ, lower limit of quantitation.
(B) Peak TCR-engineered T cells levels by dose level.
(C) Association between peak TCR-engineered T cell expansion and the occurrence of tumor response. The horizontal lines within each box represent the median, the lower and upper borders of each box represent the interquartile range, and the bars show the range.
Phenotypic evolution of TAEST16001 cells over time
The memory cell phenotype of the TAEST16001 cells was assessed in the manufactured product and following infusion in a subset of patients. We evaluated the following four T cell subsets, naive T (TN; CD45RO−CCR7+) cells, central memory T (TCM; CD45RO+CCR7+) cells, effector memory T (TEM; CD45RO+CCR7−) cells, and terminally differentiated effector T (TTE; CD45RO−CCR7−) cells and found that TN was the lowest proportions in the four T cell subsets in the manufactured product (p = 0.0335; Figure 4A). After adoptive transfer, we consistently observed an increase in the frequency of TCM and TEM subsets within the circulating Tetramer+ CD3+ TCR-T cells (Figures 4A and 4B). The increase of the frequency of TCM (p = 0.0365) and TEM (p = 0.0268) subsets was more significantly obvious on days 180–270 after cell infusion (Figure 4A). We also analyzed the correlation between frequencies of T cell subsets in the manufactured product and the peak of TCR gene copies per μg gDNA after cell infusion. The results revealed that the frequency of CD45RO+CCR7− (TEM) cells was positively correlated with the peak of TCR gene copies per μg gDNA (Figure 4C).
Figure 4.

Phenotypic evolution of TAEST16001 cells and their correlation with the peak of TCR gene copies per μg gDNA after cell infusion
(A) The compositional evolution of TAEST16001 cell pool in peripheral blood. Naive T (TN; CD45RO−CCR7+) cells, central memory T (TCM; CD45RO+CCR7+) cells, effector memory T (TEM; CD45RO+CCR7−) cells, and terminally differentiated effector T (TTE; CD45RO−CCR7−) cells were detected by flow cytometry in the manufactured product (MP) and at the time points indicated in each graph. The horizontal lines within each box represent the median, the lower and upper borders of each box represent the interquartile range, and the bars show the range. We calculated the p values using the Kruskal-Wallis (KW) test. ∗p < 0.05.
(B) Flow cytometry of tetramer-binding TAEST 16001 cells expressing CD45RO and CCR7 from patient T02 within the manufactured cell product (MP) and at the time points indicated in each graph after cell infusion. TN cells, TCM cells, TEM cells, and TTE cells were detected by flow cytometry.
(C) Correlation between frequencies of T cell subsets in the MP and the peak of TCR gene copies per μg gDNA after cell infusion. Spearman correlation analysis of the peak of TCR gene copies per μg gDNA after cell infusion and the frequencies of, TCM (CD45RO+CCR7+) cells, TEM (CD45RO+CCR7−) cells, TN (CD45RO−CCR7+) cells, and TTE(CD45RO−CCR7−) cells in TAEST16001 cell product.
Correlation between peripheral blood biomarkers and response
Several kinds of inflammatory cytokines, including IL-2, IL-6, IL-10, IFN-γ, serum amyloid A (SAA), C-reactive protein (CRP), and ferritin, as well as the phenotype of peripheral blood T cells, were detected after cell infusion. We found that the median (range) times of the peak value for IL-2, IL-6, IL-10, IFN-γ, SAA, CRP, and ferritin were 2 (1−19), 3 (2−6), 3.5 (2−9), 3 (1−7), 5 (3−20), 4.5 (3−6), and 6 days (3−9), respectively. No statistically significantly correlation was found between response and peak value of IL-2, IL-6, IL-10, SAA, and ferritin. However, the peak values of IFN-γ (p = 0.0051) as well as CRP (p = 0.0303) were statistically significantly higher in responders than those in nonresponders (Figure 5). Accordingly, the percentage of CXCR3+CD3+ T cells or CXCR3+CD3+CD8+ T cells on day 28 after cell infusion were significantly positively correlated with patient responses (Figure S4). Other T cell subsets, such as CD3+CD4+, CD3+CD8+, TN, TCM, TEM, TTE, CD3+CD25+Foxp3+ (regulatory T [Treg] cells), CD3+CXCR6+, CD3+PD-1+, CD3+LAG-3+, CD3+TIM-3+, CD3+CTLA4+, CD3+CD8+CXCR6+, CD3+CD8+PD-1+, CD3+CD8+LAG-3+, CD3+CD8+TIM-3+, and CD3+CD8+CTLA4+, were unrelated to patient responses (Figure S4).
Figure 5.

Associations between peak values of inflammatory cytokines and tumor response
(A–G) Relationship between (A) post-infusion interleukin-2 (IL-2) peak and treatment response; (B) post-infusion IL-6 peak and treatment response; (C) post-infusion IL-10 peak and treatment response; (D) post-infusion interferon-γ (IFN-γ) peak and treatment response; (E) post-infusion serum amyloid A (SAA) peak and treatment response; (F) post-infusion C-reactive protein (CRP) peak and treatment response; and (G) post-infusion ferritin peak and treatment response. Data are presented as mean ± SD. We calculated the p values using the two-sided Wilcoxon rank-sum test.
Discussion
Adoptive T cell therapy with HLA-A∗02:01-restricted NY-ESO-1-transduced T cells has shown promising results in patients with NY-ESO-1 antigen expression.14,15,16,17,18 However, the safety and effectiveness of NY-ESO-1-specific TCR-T cell therapy remain to be further optimized. In our previous study, we developed NY-ESO-1-specific TCR-T cells (TAEST16001 cells), of which the TCR was affinity enhanced by phage display from a parental TCR isolated from PBMCs of an HLA-matched healthy donor (data not shown), differing from the popular TCR that was affinity enhanced by random mutations from a 1G4 TCR isolated from a patient with cancer.17,23 TAEST16001 cells showed good specificity and safety in previous investigator-initiated trial (data not shown; ClinicalTrials.gov: NCT03462316). Therefore, in the current phase 1 study, we further evaluated the safety and clinical activity of TAEST16001 cells in HLA-A∗02:01-positive patients with advanced soft tissue sarcomas expressing the NY-ESO-1 antigen.
By this rigorous dose-escalation study, we further demonstrated that TAEST16001 cell treatment was well tolerated in terms of no serious adverse events related to cell infusion and no treatment-related deaths or adverse events leading to study withdrawal, although the maximum escalating dose of TAEST16001 cells in the study was lower than the dose already used in the National Cancer Institute studies.15 Besides, our results suggested that the maximum escalating dose in this study, which tended to correlate with increased expansion level of TCR-T cells in vivo and have similar safety with the low-level dose, could be selected for dose confirmation in phase 2 study. Once the maximum escalating dose was determined to be safe and effective, a higher cell dose beyond the maximum escalating dose may be unnecessary in patient treatment for cost-effective reasons.
Another aspect of good tolerance is manifested by the grade 4 adverse events and adverse events of special interest (CRS and neurotoxicity). No grade 4 adverse events other than hematological toxicity occurred. The incidence and grade of CRS reported in our study (16.7%, 2/12) were both lower than that observed in a previous study of NY-ESO-1-specific TCR-T cell therapy (41.6%, 5/12).14 The differences in the incidence and severity of CRS between ours and others may be partially related to lymphodepletion intensity and IL-2 dose (Table S1). Previous TCR-T trials administered a lymphodepletion regimen containing a high dose of fludarabine with cyclophosphamide to support TCR-T cell engraftment and improve clinical efficacy (Table S1).14,15,16,17 Our lymphodepleting regimen utilized dose-reduced fludarabine (60 mg/m2) and cyclophosphamide (45 mg/kg). Besides, instead of high-dose IL-2, such as 720,000 IU/kg three times daily,15,17 low-dose IL-2 (500,000 IU, twice daily for 14 days) was used after cell infusion in our study, which is even lower than the low-dose IL-2 (500,000 IU/m2 twice daily for 14–28 days) used in another clinical study.24 Modified (dose-reduced) lymphodepletion with fludarabine and cyclophosphamide and low-dose IL-2 usage may contribute to the moderate adverse events observed in our clinical study. In addition, the affinity of TCR is considered to be related to the toxicity of TCR-T cell therapy.25,26 Severe lung injury or lethal cardiotoxicity has been reported in several studies related to affinity-enhanced TCR-T cells.18,24,27 However, our NY-ESO-1-specific TCR with optimal affinity and specificity was selected with phage display libraries, which were constructed on templates of a TCR from a health donor, and the affinity-enhancement approaches were verified to maintain good antigen specificity and potentially enlarge therapeutic windows.28 Unlike the high incidence of immune effector cell-associated neurotoxicity syndrome (ICANS) in chimeric antigen receptor (CAR) T cells treatment,29 no neurotoxicity was observed in our study. This may relate to the TCR-T and CAR-T forming different immunologic synapses.30 Collectively, these data provided evidence that TAEST16001 cell treatment was safe when used with modified lymphodepletion and low-dose IL-2.
The impressive responses to TAEST16001 cells included a median TTR of 1.9 months and 41.7% ORR, indicating highly efficient anti-tumor activity by TAEST16001 cells. The median PFS was 7.2 months, and a median OS was not reached. As observed in previous studies,14,17 not all patients respond to TCR-T cell therapy. Low expansion peak of TCR-T cells in peripheral blood, downregulation of HLA, and defects in the antigen-presenting machinery may explain the primary resistance to TCR-T cell therapy.31 Although cross-study comparisons have limitations, the response rate, time, and durability are comparable to the previous NY-ESO-1-specific TCR-T cell trial reported by D'Angelo et al., in which the ORR was 50%, the TTR was 6.2 weeks, and the median PFS was 15 weeks.14 It is reported that T cells equipped with a high-affinity TCR exhibited increased anti-tumor effects because of their improvement of intratumor infiltration23 and faster and better response to antigen recognition.17,32 Another two clinical trials also reported a high ORR in patients with synovial sarcoma after NY-ESO-1-specific TCR-T cell infusion.15,17 As stated above, a high dose of fludarabine with cyclophosphamide and high-dose IL-2 were used in their studies,15,17 which might be related to the high ORR. In fact, a majority of patients eventually exhibited progressive disease after a period of remission in all these studies. A possible reason for patients’ resistance to TCR-T cell therapy may be related to T cell exhaustion after overactivation of high-affinity TCR-T cells,25,26,33 as exhausted T cells show progressive loss of anti-tumor function and upregulate the expression of inhibitory receptors.34 Further studies are needed for a better understanding of the mechanisms underlying acquired resistance against TCR-T cells.
Excitedly, two patients with liposarcoma had objective responses in the study, with one lasting for more than one year and the other exhibiting an ongoing PFS for 6 months. Generally, liposarcoma, especially well-differentiated and dedifferentiated liposarcoma, is insensitive to systemic chemotherapy, and no standard treatment was recommended for recurrent liposarcoma after failure of doxorubicin-based chemotherapy.35 It is reported that liposarcoma has some unique immune profile features, such as a higher proportion of tumor-infiltrating T cells and PD-L1 expression and more macrophage infiltration, compared with other STSs,36 suggesting that immunotherapy could be an alternative therapeutic option for liposarcoma. Indeed, liposarcoma was one of the sarcoma subtypes that responded to anti-PD-1 antibody immunotherapy as reported in the SARC028 study.6 Collectively, these studies suggest that liposarcoma may be another dominant sarcoma subtype in the clinical trial of TCR-T cell immunotherapy.
Clinical efficacy was corroborated by swift and robust expansions of TAEST16001 cells, as indicated by pharmacokinetic data from PCR. Moreover, continued expression of the TCR was observed up to 9 months post-infusion in some of the patients, suggesting the long-term persistence of TAEST16001 cells in vivo. In consistent with the long-term persistence of TAEST16001 cells in vivo, the duration of response in some patients last long as well in the current study. Similar to observations by D'Angelo et al.,14 the frequency of TCM subsets within the circulating TCR-T cells was increased with the extension of cell infusion, and this subset became the predominant cell subset on day 180–270 after cell infusion, which also in part explained the long persistence of TCR-T cells in the peripheral blood in our study. Although there was no statistically significantly difference in the peak of TCR gene copies among the four dosage levels, a relatively poor tumor response rate was observed at dose levels 2 and 3, which could be associated with a slow expansion of TCR in vivo. A special example was observed in patient T04, for whom the time to reach peak TCR-T cells was 1 h after cell infusion, suggesting that TCR-T cells did not proliferate in vivo. Correspondingly, a short stable disease was observed in patient T04. Our results did not show a positive correlation between the peak expansion of the TCR-T cells and tumor response, which may be due to the small sample size, and further investigation is warranted in the phase 2 study. All these results reveal that the magnitude and persistence of TCR-T cell expansion may be related to clinical efficacy.
The clinical efficacy of TAEST16001 cells was further verified by the higher amount of CRP and IFN-γ as observed in responders compared with that in nonresponders. Their increase suggested T cell activation, in line with previous reports.37,38 IFN-γ has always been regarded as a key role in the activation of cellular immunity and the stimulation of anti-tumor response. Nowadays, pro- and anti-tumor effects of IFN-γ have been found; however, patients treated with high levels of IFN-γ had tumor regression.39 Interestingly, the percentage of CXCR3+CD3+ or CXCR3+CD3+CD8+ T cells was statistically significantly increased in the responder group in our study, as IFN-γ is an essential factor associated with the induction of CXCR3 on T cells.40 These results suggest that more CXCR3+CD8+ T cells in peripheral blood means more T cells trafficking to the tumor microenvironment and exhibiting stronger anti-tumor immune responses,40 but this could not be evaluated in this study due to lack of available tumor samples.
In conclusion, data from this phase 1 clinical trial showed that autologous T cells engineered to express a high-affinity NY-ESO-1-specific TCR derived from PBMCs of an HLA-matched healthy donor are safe and highly active in the HLA-A∗02:01 population with advanced STS expressing NY-ESO-1 antigens. A dose-reduced lymphodepletion with fludarabine and cyclophosphamide and low-dose IL-2 usage could minimize toxicity without decreasing efficacy. An expansion and phase 2 study of TAEST16001 cell treatment is ongoing to further assess the safety and clinical activity of the maximum escalating dose in patients with advanced soft tissue sarcoma. In addition, further investigation is needed to fully understand the long persistence of TAEST16001 cells and its association with patient responses.
Limitations of the study
Our study has several limitations. First, the major limitation was small sample size, so the association between peak TCR-T cell expansion in vivo and the infused cell dose should be further investigated. Second, biomarkers related to the response of the TCR-T cell treatment were not comprehensively understood, and more clinical validation is required.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD3 APC | Biolegend | Cat# 317318; RRID: AB_1937212 |
| Anti-CD3 BV510 | BD biosciences | Cat# 564713; RRID: AB_2738909 |
| Anti-CD4 BUV496 | BD biosciences | Cat# 564651; RRID: AB_2744422 |
| Anti-CD8 APC-Cy7 | Biolegend | Cat# 300926; RRID: AB_10613636 |
| Anti-CD45 AF700 | Biolegend | Cat# 304024; RRID: AB_493761 |
| Anti-CCR7 BV421 | Biolegend | Cat# 353208; RRID: AB_11203894 |
| CCR7-FITC | Biolegend | Cat# 353216; RRID: AB_10916386 |
| CD45RO-BV421 | Biolegend | Cat# 304224; RRID: AB_2563817 |
| Anti-CD45RO BV605 | BD biosciences | Cat# 562791; RRID: AB_2744411 |
| Anti-CD25 BUV737 | BD biosciences | Cat# 612806; RRID: AB_2870132 |
| Anti-CXCR6 BV421 | BD biosciences | Cat# 566007; RRID: AB_2744472 |
| Anti-CXCR3 BV605 | Biolegend | Cat# 353728; RRID: AB_2563157 |
| Anti-Tim-3 BV711 | BD biosciences | Cat# 565566; RRID: AB_2744370 |
| Anti-CTLA-4 PE | Biolegend | Cat# 349906; RRID: AB_10641842 |
| Anti-LAG-3 PE-Cy7 | eBioscience | Cat# 25-2239-42; RRID: AB_2573430 |
| Anti-PD-1 PerCP-Cy5.5 | Biolegend | Cat# 329914; RRID: AB_1595461 |
| Anti-Foxp3 AF488 | Biolegend | Cat# 320112; RRID: AB_430883 |
| Biological samples | ||
| Leukopak | Milestone Biotechnologies | Cat# PBLP-F-3 |
| Chemicals, peptides, and recombinant proteins | ||
| Peptides | Genscript | Cat# SC1208 |
| Tetramer | This manuscript | N/A |
| Recombinant Human Il-2 | Beijing Four Rings Biopharmaceuticals Co., Ltd. | Cat# S20040018 |
| DEPC-H2O | Invitrogen | Cat # 750023 |
| Buffer TE | Invitrogen | Cat # AM9849 |
| Ethanol (96–100%) | Sinopharm | Cat # 10009259 |
| HIV-Psi F | GENEWIZ | NA |
| HIV-Psi R | GENEWIZ | NA |
| HIV-Psi Probe | GENEWIZ | NA |
| XL-1901 | Provided by sponsor | Cat # XL-1901 ps190820 |
| RCLVSVG F | GENEWIZ | NA |
| RCLVSVG R | GENEWIZ | NA |
| RCLVSVG Probe | GENEWIZ | NA |
| pMD2.G | Provided by sponsor | Cat # ps19010901 |
| Human Fc block | BD | Cat#564220 |
| Staining Buffer | Biolegend | Cat#420201 |
| Brilliant Stain Buffer | BD | Cat#566349 |
| IC fixation buffer | BD | Cat#554655 |
| RBC lysing solution | BD | Cat#555899 |
| PIC | Genescript | customized |
| P72A | Genescript | 7195038–1 |
| RPMI-1640 | Gibco | Cat # A1049101 |
| FBS | ExCell | FSP500 |
| Critical commercial assays | ||
| CytoTox 96® non-radioactive cytotoxicity assay kit | Promega | Cat# G1780 |
| Human IFN-γ ELISpot Set | BD biosciences | Cat# 551849 |
| V-PLEX Custom Human Cytokine Panel | MSD | Cat# K151A0H-1 |
| Proinflammatory panel 1 (human) control pack | MSD | Cat# C4049-1 |
| Chemokine panel 1(human) control pack | MSD | Cat# C4047-1 |
| Cytokine panel 1(human) control pack | MSD | Cat# C4050-1 |
| QIAamp DNA Blood Midi Kit | Qiagen | Cat# 51183 or 51185 |
| Faststart Universal Probe Master | Roche | Cat# 04914058001 |
| CD14 Microbeads | Miltenyi | Cat# 272-01 |
| CD25 Microbeads | Miltenyi | Cat# 274-01 |
| Dynabeads CD3/CD28 CTS | ThermoFisher | Cat# 40203D |
| QIAamp DNA Blood Midi Kit | Qiagen | Cat # 51185 |
| FastStart Universal Probe Master | Roche | Cat # 04914058001 |
| V-PLEX Custom Human Cytokine panel | Meso Scale Discovery | Cat # MSD-K151A9H-1 |
| Proinflammatory panel 1(human) control pack | Meso Scale Discovery | Cat #C4049-1 |
| Chemokine panel 1 (human) control pack | Meso Scale Discovery | Cat #C4047-1 |
| Cytokine panel 1 (human) control pack | Meso Scale Discovery | Cat #C4050-1 |
| LIVE/DEAD™ Fixable Red Dead Cell Stain Kit | eBioscience | Cat#L34972 |
| Foxp3/Transcription Factor Staining Buffer Set (contain Fix/Perm buffer, Perm/Wash buffer) | eBioscience | Cat#00-5523-00 |
| Human IFN-γ ELISpot PLUS kit (ALP) | Mabtech | Cat # 3420-4AST-10 |
| Experimental models: Cell lines | ||
| A375 | ATCC | Cat# CRL-1619 |
| IM9 | ATCC | Cat# CCL-159 |
| U266B1 | ATCC | Cat# TIB-196 |
| T2 | ATCC | Cat# CRL-1992 |
| NCI-H1650 | ATCC | Cat# CRL-5883 |
| NCI-H1703 | ATCC | Cat# CRL-5889 |
| NCI-H1299 | ATCC | Cat# CRL-5803 |
| SW480 | ATCC | Cat# CCL-228 |
| Software and algorithms | ||
| Statistical Package for the Social Science, version 22.0 | IBM Corp | N/A |
| GraphPad Prism 9 | GraphPad Software, Inc. | N/A |
| cytExpert, version 2.0 | Beckman Coulter | N/A |
| FlowJo software, Version 10 | BD | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xing Zhang (zhangxing@sysucc.org.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and study particpant details
Study design
This study was an open-label, single-arm phase 1 study, with a conventional 3 + 3 dose escalation/expansion design. Four dose levels were planned: dose level 1, 5 × 108 ± 30%; dose level 2, 2 × 109 ± 30%; dose level 3, 5 × 109 ± 30%; and dose level 4, 1.2 × 1010 ± 30%. Starting doses were based on previous studies,15,17 in which the number of infused TCR-T cells was between 3.5×108 and 2.9×1010. At least three patients per dose cohort were recruited and no dose-limiting toxic (DLT) effect was observed during a 28-day observation period before dose escalation. If a DLT was recorded, three more patients were recruited to the cohort in which the DLT occurred. Intrapatient dose escalation was not allowed. The MTD was defined as the highest dose at which one or fewer of six patients had a DLT after cell infusion.
Trial oversight
The study was approved by the Institutional Review Board of Sun Yat-sen University Cancer Center, conducted according to the principles of the Declaration of Helsinki, and was registered on ClinicalTrials.gov (NCT04318964). All patients provided written informed consent before enrollment.
Eligibility
Patients were eligible if they were aged 18 through 70 years; had HLA-A∗02:01 allele and ≥20% of tumor cells expressing the NY-ESO-1 tumor antigen by immunohistochemistry; had histologically confirmed advanced soft tissue sarcoma that was unresectable, metastatic, or refractory to standard treatments; have at least 1 measurable disease by Computed Tomography (CT) or Magnetic Resonance (MR) scans (per Response Evaluation Criteria In Solid Tumors [RECIST] v1.1); had an Eastern Cooperative Oncology Group performance status score of 0 or 1; had a life expectancy of at least 3 months; had left ventricular ejection fraction of ≥50%; and had adequate liver, kidney, and bone marrow function.
Key exclusion criteria included: anticancer therapy within 4 weeks of cell infusion; history of NY-ESO-1-targeted therapy; concomitant corticosteroids, or immunomodulatory drugs within 4 weeks of cell infusion; history of brain metastases or active infection or other uncontrolled significant medical or psychiatric illness; and history of autoimmune diseases.
Treatment
After TAEST16001 cells were successfully manufactured and met the quality control, patients received a modified lymphodepleting regimen of intravenous fludarabine 20 mg/m2 per day and cyclophosphamide 15 mg/kg per day for 3 consecutive days beginning on day −7 before cell infusion. On day 1, the full dosage of TAEST16001 cells was administered intravenously over 30 min once (at dose level 1 or 2) or twice in two consecutive days (at dose level 3 or 4). IL-2 was subcutaneous injection within 30 min after the first cell infusion at a dose of 500,000 IU each time, twice daily for 14 days.
Clinical assessments
Peripheral blood samples were taken for safety, pharmacokinetic, and pharmacodynamic analyses during the study period at predefined visit. Safety was assessed by means of adverse event monitoring, graded according to the NCI Common Terminology Criteria for Adverse Events v5.0. Cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS) were graded according to the ASTCT 2018 guideline.41 DLTs were defined as adverse events occurred within 28 days of the TAEST16001 cell infusion judged possibly related to the cell infusion. Non-haematological DLTs were any toxicity of grade 3 persisted for seven or more days or any toxicity of grade 4 regardless of duration, with exceptions detailed in the study protocol (Methods S1). Haematological DLTs were grade 4 toxicity (except lymphopenia) lasting more than 28 days if not due to underlying disease. Grade 3 CRS lasting more than 7 days or grade 4 CRS was also considered as DLT. The parameters related with pharmacokinetic and pharmacodynamic analyses were provided by Immunology Center WuXi AppTec in Shanghai, China.
Tumors were assessed by CT or MR scans performed at baseline and on day 28, day 60, day 90, day 180, and day 270 after TAEST16001 cell infusion. Tumor response was evaluated by the RECIST v1.1. Patients who were evaluated as progressive disease (PD) by RECIST v1.1 could be re-evaluated as immune unconfirmed PD (iUPD) according to the immune Response Evaluation Criteria In Solid Tumors (iRECIST) and re-assessed after 4–6 weeks until immune confirmed progressive disease (iCPD) or initiating other anti-tumor treatment, whichever occur first.
The primary objectives were to investigate the safety, tolerability, and DLTs of TAEST16001 cells to identify the MTD. Safety endpoints included adverse events, serious adverse events, adverse events of special interest (CRS and ICANS), laboratory tests, physical examination assessments, electrocardiogram, and vital sign measurements.
The secondary objectives were pharmacokinetics, pharmacodynamics, objective response rate (ORR, the proportion of patients who achieved a RECIST 1.1 confirmed complete response or partial response), disease control rate (DCR, the proportion of patients who reached an objective response or stable disease), and PFS (the number of days between patients’ enrollment and either disease progression or death after cell infusion). For pharmacokinetics analyses, TAEST16001 cells as well as the TCR gene copies per μg genomic DNA (gDNA) were detected by flow cytometry and qPCR, respectively, to calculate the maximum value of TCR-T cells in peripheral blood (Cmax) and the time to reach peak concentration (Tmax). For pharmacodynamics analyses, T cell subsets and antigen-specific cytotoxic T lymphocytes in peripheral blood were detected.
We also assessed correlations between response and selected prespecified biomarkers: TCR gene copies per μg gDNA, inflammatory cytokines (IL-2, IL-6, IL-10, interferon-γ (IFN-γ), Serum amyloid A (SAA), C-reactive protein (CRP), and ferritin).
Method details
NY-ESO-1 expression
NY-ESO-1 antigen was tested by immunostaining at the Sun Yat-sen University Cancer Center. Immunohistochemistry (IHC) was carried out using primary antibody against NY-ESO-1 on 3∼5-mm-thick sections of formalin-fixed, paraffin-embedded tumor tissue sections. Then, the sections were visualized by DAB and counterstained with hematoxylin as per the manufacturer’s instructions. More than 20% of tumor cells expressing the NY-ESO-1 tumor antigen by IHC were defined as NY-ESO-1 positive expression in the study.
The full TCR sequence
atggagacactgctgggcgtgagcctggtcatcctgtggctccagctggcccgggtgaacagccagcagggagaggaggacccccaggccctgagcatccaggagggcgagaatgccaccatgaactgctcctacaagacatccatcaacaacctccagtggtatcggcagaacagcggccggggcctggtgcacctgatcctgatcaggtctaacgagagggagaagcacagcggccggctgagagtgaccctggacacaagcaagaagagctcctctctgctgatcaccgcctccagagccgccgatacagcctcttacttttgcatgtatgaccagaacggcaagatcatcttcggcaagggcaccaggctgcacatcctgcctaatatccagaacccagatcccgccgtgtaccagctgcgcgacagcaagagctccgataagagcgtgtgcctgttcaccgactttgattcccagacaaacgtgagccagtctaaggactctgacgtgtacatcaccgacaagacagtgctggatatgaggagcatggactttaagagcaattccgccgtggcctggtctaacaagagcgacttcgcctgcgccaacgcctttaacaatagcatcatcccagaggataccttcttttgctccccagagtctagctgtgacgtgaagctggtggagaagagcttcgagacagatacaaatctgaactttcagaatctgtccgtgatcggcttcagaatcctgctgctgaaggtggccggctttaacctgctgatgaccctgagactgtggtcctctggctctcgggccaagagatctggcagcggcgccacaaatttcagcctgctgaagcaggcaggcgatgtggaggagaacccaggacctagagacagctggaccctgtgctgcgtgagcctgtgcatcctggtggccaagcacacagacgccggcgtgatccagtctccacggcacgaggtgaccgagatgggccaggaggtgacactgaggtgtaagcccatcagcggccacgattacctgttttggtataggcagaccatgatgcgcggcctggagctgctgatctacttcaacaataacgtgcccatcgacgattctggcatgcctgaggaccggtttagcgccaagatgccaaatgcctccttctctaccctgaagatccagccttctgagccaagagatagcgccgtgtacttttgcgccagctccctgggctccaatgagcagtatttcggccccggcacaaggctgaccgtgacagaggacctgaagaacgtgttcccccctgaggtggccgtgtttgagccttccgagtgcgagatctctcacacccagaaggccaccctggtgtgcctggcaaccggcttctatccagatcacgtggagctgagctggtgggtgaatggcaaggaggtgcactccggcgtgtctacagacccacagcccctgaaggagcagcccgccctgaacgattcccgctactgcctgtctagcaggctgcgcgtgtctgccaccttttggcagaatcctcggaaccacttcagatgtcaggtgcagttttatggcctgagcgagaacgatgagtggacccaggacagggccaagcctgtgacacagatcgtgtccgccgaggcctggggaagggcagactgtggcttcacaagcgagtcctaccagcagggcgtgctgtccgccaccatcctgtacgagatcctgctgggcaaggccacactgtatgccgtgctggtgtccgccctggtgctgatggccatggtgaagaggaaggattctcgcggctga.
Generation of TAEST16001 cells
Patients underwent leukapheresis at enrollment to collect peripheral blood mononuclear cells (PBMCs). High-affinity NY-ESO-1-specific TCR-T cells (TAEST16001 cells) were manufactured at Xiangxue Life Science Technology (Guangdong) Co., Ltd. TAEST16001 cells were generated from CD14−and CD25-depleted PBMCs by Sepax C-pro and CliniMACS plus. CD14 and CD25 negative cells were activated and expanded using αCD3/αCD28 antibody-conjugated beads in the presence of 200 IU/mL of recombinant IL-2, transduced with a lentivirus at a multiplicity of infection of 1 transducing unit per cell. Cells were expanded for 9–13 days by Wave Bioreactor and then harvested by Sefia and formulated by Sepax C-pro. TCR-T cell product would be frozen for release testing, which took an additional 7–10 days. So far, the manufacture process works well and no manufacturing failures occurred.
qPCR analysis of TAEST16001 cell expansion and persistence
TCR gene copies per microgram of gDNA were determined at the Immunology Center WuXi AppTec by quantification of the WPRE region of the lentivirus backbone by qPCR. 6 mL peripheral blood samples were taken at baseline and after infusion. Genomic DNA was extracted from PBMCs using the QIAamp DNA Blood Midi Kit (Qiagen). A series dilution of a plasmid was used to prepare a standard curve with a high, middle and low controls. 5 μL gDNA and standard were amplified with Quant Studio 7 Flex Real-Time PCR System (Thermo Fisher Applied Biosystems) using the respective primers, probes and FastStart Universal Probe Master Mix. The number of TCR gene copies per microgram of gDNA was determined triplicated for each sample. The limit of detection for this assay was 20 copies per microgram of gDNA.
RCL discovery by using TaqMan Real-time PCR
TaqMan qPCR was used to determine the RCL. A dilution series of a plasmid was used to prepare standard curve with high, middle and low controls. gDNA were extracted from ∼2 mL whole blood using QIAamp DNA Blood Midi Kit. 5 μL gDNA and standard were amplified with Quant Studio 7 Flex Real-Time PCR System (Thermo Fisher Applied Biosystems) using the respective primers, probes and FastStart Universal Probe Master Mix.
Cytokine measurement
Human serum was used to detect human cytokine/chemokine/proinflammatory. The MSD V-PLEX Plus Custom Human Biomarkers Kit from MSD was used to measure IL-2, IL-6, IL-10, and IFN-γ. MSD plates were analyzed on the MESO SECTOR S600. The assay was performed following the manufacturer’s instructions. All standards, controls and samples were measured in duplicate.
Flow cytometry
Whole blood samples were collected with heparin vacuum tube (BD) and processed with RBC lysis buffer to remove red blood cell. Multi-parametric immune-phenotyping for peripheral blood was performed using approximately 1.0 × 106 total cells per condition (depending on cell yield in samples), and using isotype stains in PK analysis. Cells were stained in staining buffer for 45 min at room temperature using antibody and reagent concentrations recommended by the manufacturer. PK samples were stained with A2P1B, anti-CD45, CD3, CD4 and CD8 antibodies, PD samples were stained with A2P1B, anti-CD45, CD3, CD4, CD8, CCR7, and CD45RO antibodies for Tet+ T cell subsets analysis; and were stained with A2P1B, anti-CD45, CD3, CD4, CD8, CD25, CXCR3, CXCR6, TIM-3, PD-1, CTLA-4 and LAG-3 antibodies, permeablized with Fix/Perm buffer for 30 min, and stained with anti-Foxp3 antibody for Treg analysis, respectively. Cells were then washed, and re-suspended in staining buffer and acquired using an BD Fortessa cytometer equipped with an Ultraviolet (355 nm), Violet (405 nm), Blue (488 nm), Yellow (561 nm) and Red (633 nm) laser. Flow cytometry files were exported in.fcs file format and analyzed using FlowJo software (Version 10, BD).
Peptides and tetramer
Peptides were synthesized (>95% purity) by GenScript and verified using mass spectroscopy. MHC tetramers were produced in-house.
Human IFN-γ ELISpot assay
IFN-γ ELISpot were determined using the Human IFN-γ ELISpot Set (BD Biosciences). Human PBMCs were used to detect effector cell activity. IFN-γ ELISpot assay was performed using 96-well ELISpot strip plates (8 wells × 12 strips) pre-coated with monoclonal antibody. The assay was performed following the manufacturer’s instructions. Plates were blocked with RPMI-1640 medium (0.2 mL/well) containing 10% FBS for at least 30 min at room temperature after washing 4 times with sterile DPBS (0.2 mL/well). 100 μL Peptide pools were added to the plate. Plate were incubated the at 37°C 5% CO2 for 16 h without moving after adding the same volume cells. Plate was washed 5 times using PBS with a volume of 0.2 mL per well. Diluted detection antibody (R4-6A2-biotin) was added with a volume of 0.1 mL per well for 2 h at room temperature. After washing the plate 5 times with DPBS, diluted Streptavidin-ALP was added with a volume of 0.1 mL per well for 1 h. Plates were washed 5 times with PBS, and then substrate solution (BCIP/NBT-plus) were added with a volume of 0.1 mL per well. Plates were developed until distinct spots emerge and stopped color development by washing extensively in tap water. After the plates were dried, read the plates and analyzed the results by using AID ELISpot Software 7.0.
Quantification and statistical analysis
The sample size was based on the conventional 3 + 3 dose escalation/expansion design and the number of DLTs. We summarized the number of cases, median, interquartile range, minimum and maximum values for continuous variables and frequency distributions for categorical variables with descriptive statistics. We assessed the association between response and biomarkers as stated above with a Mann-Whitney U test. We evaluated the difference in the phenotype of TCR-T cells among different time with a Kruskal-Wallis (KW) test. We reported estimates of ORR and DCR with the corresponding two-sided exact binomial 95% CI, calculated by means of the Clopper-Pearson method. Kaplan-Meier method was used to estimate progression-free survival (PFS). We used SPSS software (Statistical Package for the Social Science, version 22.0, IBM Corp. Armonk, NY, USA) and GraphPad Prism 9 (Version 9.3.1, GraphPad Software, Inc.) for all analyses.
Additional resources
This trial is registered with ClinicalTrials.gov, NCT04318964.
Acknowledgments
We thank patients and their families, as well as their caregivers, for taking part in this trial. This study was sponsored by Xiangxue Life Science Technology (Guangdong) Co., Ltd., the National Key Research and Development Program (2021YFC2400601), and the National Natural Science Foundation of China (82072958).
Author contributions
X.Z., Z.F., and Y. Li conceived and designed the study. D.W., Q.P., J.L., B.X., R.P., Y.Q., X.W., J.Y., Z.F., and X.Z. enrolled and treated patients. Y.O. was responsible for TCR-T cell product manufacturing. H.G. was responsible for TCR-T cell product quality. Y. Lin and K.M. did the immunological assay for cell product. Q.P., Y. Li, Z.H., S.Z., and X.Z. did exploratory immunological studies. Q.P. did the statistical analyses. Q.P., D.W., Z.H., L.Z., A.C., J.C., J.Y.N.L., J.L., Y. Li, Z.F., and X.Z. analyzed and interpreted the data. Q.P., Y. Li, and X.Z. wrote the manuscript. All authors reviewed the data, participated in the development of the manuscript, and approved the final version for publication.
Declaration of interests
Z.H., Y.O., L.Z., H.G., A.C., J.C., S.Z., Y. Lin, Y. Li, and K.M. are employees of Xiangxue Life Science Technology (Guangdong) Co., Ltd.
Published: August 15, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101133.
Contributor Information
Yi Li, Email: li_yi@gibh.ac.cn.
Zhengfu Fan, Email: zhengfufan@126.com.
Xing Zhang, Email: zhangxing@sysucc.org.cn.
Supplemental information
Data and code availability
Data from this study can be made available upon request and approval by the study management committee and subject to appropriate data transfer agreements. Requests should be directed to X.Z. No custom code was used for statistical analysis for this study. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.von Mehren M., Randall R.L., Benjamin R.S., Boles S., Bui M.M., Ganjoo K.N., George S., Gonzalez R.J., Heslin M.J., Kane J.M., et al. Soft Tissue Sarcoma, Version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018;16:536–563. doi: 10.6004/jnccn.2018.0025. [DOI] [PubMed] [Google Scholar]
- 2.Li R.H., Zhou Q., Li A.B., Zhang H.Z., Lin Z.Q. A nomogram to predict metastasis of soft tissue sarcoma of the extremities. Medicine. 2020;99 doi: 10.1097/MD.0000000000020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sleijfer S., Ouali M., van Glabbeke M., Krarup-Hansen A., Rodenhuis S., Le Cesne A., Hogendoorn P.C.W., Verweij J., Blay J.Y. Prognostic and predictive factors for outcome to first-line ifosfamide-containing chemotherapy for adult patients with advanced soft tissue sarcomas: an exploratory, retrospective analysis on large series from the European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC-STBSG) Eur. J. Cancer. 2010;46:72–83. doi: 10.1016/j.ejca.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Gronchi A., Miah A.B., Dei Tos A.P., Abecassis N., Bajpai J., Bauer S., Biagini R., Bielack S., Blay J.Y., Bolle S., et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021;32:1348–1365. doi: 10.1016/j.annonc.2021.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Schöffski P., Cornillie J., Wozniak A., Li H., Hompes D. Soft tissue sarcoma: an update on systemic treatment options for patients with advanced disease. Oncol. Res. Treat. 2014;37:355–362. doi: 10.1159/000362631. [DOI] [PubMed] [Google Scholar]
- 6.Tawbi H.A., Burgess M., Bolejack V., Van Tine B.A., Schuetze S.M., Hu J., D'Angelo S., Attia S., Riedel R.F., Priebat D.A., et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18:1493–1501. doi: 10.1016/S1470-2045(17)30624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Angelo S.P., Mahoney M.R., Van Tine B.A., Atkins J., Milhem M.M., Jahagirdar B.N., Antonescu C.R., Horvath E., Tap W.D., Schwartz G.K., Streicher H. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018;19:416–426. doi: 10.1016/S1470-2045(18)30006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer C.F. Immunotherapy for Sarcoma: A Work in Progress. J. Clin. Oncol. 2022;40:1267–1270. doi: 10.1200/JCO.21.01338. [DOI] [PubMed] [Google Scholar]
- 9.Rizvi H., Sanchez-Vega F., La K., Chatila W., Jonsson P., Halpenny D., Plodkowski A., Long N., Sauter J.L., Rekhtman N., et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018;36:633–641. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollack S.M., He Q., Yearley J.H., Emerson R., Vignali M., Zhang Y., Redman M.W., Baker K.K., Cooper S., Donahue B., et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer. 2017;123:3291–3304. doi: 10.1002/cncr.30726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caballero O.L., Chen Y.T. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100:2014–2021. doi: 10.1111/j.1349-7006.2009.01303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas R., Al-Khadairi G., Roelands J., Hendrickx W., Dermime S., Bedognetti D., Decock J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018;9:947. doi: 10.3389/fimmu.2018.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Endo M., de Graaff M.A., Ingram D.R., Lim S., Lev D.C., Briaire-de Bruijn I.H., Somaiah N., Bovée J.V.M.G., Lazar A.J., Nielsen T.O. NY-ESO-1 (CTAG1B) expression in mesenchymal tumors. Mod. Pathol. 2015;28:587–595. doi: 10.1038/modpathol.2014.155. [DOI] [PubMed] [Google Scholar]
- 14.D'Angelo S.P., Melchiori L., Merchant M.S., Bernstein D., Glod J., Kaplan R., Grupp S., Tap W.D., Chagin K., Binder G.K., et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018;8:944–957. doi: 10.1158/2159-8290.CD-17-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robbins P.F., Kassim S.H., Tran T.L.N., Crystal J.S., Morgan R.A., Feldman S.A., Yang J.C., Dudley M.E., Wunderlich J.R., Sherry R.M., et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin. Cancer Res. 2015;21:1019–1027. doi: 10.1158/1078-0432.CCR-14-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramachandran I., Lowther D.E., Dryer-Minnerly R., Wang R., Fayngerts S., Nunez D., Betts G., Bath N., Tipping A.J., Melchiori L., et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer. 2019;7:276. doi: 10.1186/s40425-019-0762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robbins P.F., Morgan R.A., Feldman S.A., Yang J.C., Sherry R.M., Dudley M.E., Wunderlich J.R., Nahvi A.V., Helman L.J., Mackall C.L., et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishihara M., Kitano S., Kageyama S., Miyahara Y., Yamamoto N., Kato H., Mishima H., Hattori H., Funakoshi T., Kojima T., et al. NY-ESO-1-specific redirected T cells with endogenous TCR knockdown mediate tumor response and cytokine release syndrome. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q., Tian Y., Li Y., Zhang W., Cai W., Liu Y., Ren Y., Liang Z., Zhou P., Zhang Y., et al. In vivo therapeutic effects of affinity-improved-TCR engineered T-cells on HBV-related hepatocellular carcinoma. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-001748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y., Moysey R., Molloy P.E., Vuidepot A.L., Mahon T., Baston E., Dunn S., Liddy N., Jacob J., Jakobsen B.K., Boulter J.M. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H., Zhang J., Chen L., Weng Z., Tian Y., Zhao H., Li Y., Chen L., Liang Z., Zheng H., et al. Targeting naturally occurring epitope variants of hepatitis C virus with high-affinity T-cell receptors. J. Gen. Virol. 2017;98:374–384. doi: 10.1099/jgv.0.000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z., Gong H., Liu Q., Wu W., Cheng J., Mei Y., Chen Y., Zheng H., Yu X., Zhong S., Li Y. Identification of an HLA-A∗24:02-restricted alpha-fetoprotein signal peptide-derived antigen and its specific T-cell receptor for T-cell immunotherapy. Immunology. 2020;159:384–392. doi: 10.1111/imm.13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robbins P.F., Li Y.F., El-Gamil M., Zhao Y., Wargo J.A., Zheng Z., Xu H., Morgan R.A., Feldman S.A., Johnson L.A., et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J. Immunol. 2008;180:6116–6131. doi: 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowicki T.S., Berent-Maoz B., Cheung-Lau G., Huang R.R., Wang X., Tsoi J., Kaplan-Lefko P., Cabrera P., Tran J., Pang J., et al. A Pilot Trial of the Combination of Transgenic NY-ESO-1-reactive Adoptive Cellular Therapy with Dendritic Cell Vaccination with or without Ipilimumab. Clin. Cancer Res. 2019;25:2096–2108. doi: 10.1158/1078-0432.CCR-18-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones H.F., Molvi Z., Klatt M.G., Dao T., Scheinberg D.A. Empirical and Rational Design of T Cell Receptor-Based Immunotherapies. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.585385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Ippolito E., Schober K., Nauerth M., Busch D.H. T cell engineering for adoptive T cell therapy: safety and receptor avidity. Cancer immunology, immunotherapy. 2019;68:1701–1712. doi: 10.1007/s00262-019-02395-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linette G.P., Stadtmauer E.A., Maus M.V., Rapoport A.P., Levine B.L., Emery L., Litzky L., Bagg A., Carreno B.M., Cimino P.J., et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.June C.H. Adoptive T cell therapy for cancer in the clinic. J. Clin. Invest. 2007;117:1466–1476. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schubert M.L., Schmitt M., Wang L., Ramos C.A., Jordan K., Müller-Tidow C., Dreger P. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol. 2021;32:34–48. doi: 10.1016/j.annonc.2020.10.478. [DOI] [PubMed] [Google Scholar]
- 30.Carrasco-Padilla C., Hernaiz-Esteban A., Álvarez-Vallina L., Aguilar-Sopeña O., Roda-Navarro P. Bispecific Antibody Format and the Organization of Immunological Synapses in T Cell-Redirecting Strategies for Cancer Immunotherapy. Pharmaceutics. 2022;15 doi: 10.3390/pharmaceutics15010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parkhurst M.R., Yang J.C., Langan R.C., Dudley M.E., Nathan D.A.N., Feldman S.A., Davis J.L., Morgan R.A., Merino M.J., Sherry R.M., et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tucker C.G., Mitchell J.S., Martinov T., Burbach B.J., Beura L.K., Wilson J.C., Dwyer A.J., Singh L.M., Mescher M.F., Fife B.T. Adoptive T Cell Therapy with IL-12-Preconditioned Low-Avidity T Cells Prevents Exhaustion and Results in Enhanced T Cell Activation, Enhanced Tumor Clearance, and Decreased Risk for Autoimmunity. J. Immunol. 2020;205:1449–1460. doi: 10.4049/jimmunol.2000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLane L.M., Abdel-Hakeem M.S., Wherry E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019;37:457–495. doi: 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- 35.Crago A.M., Dickson M.A. Liposarcoma: Multimodality Management and Future Targeted Therapies. Surg. Oncol. Clin. 2016;25:761–773. doi: 10.1016/j.soc.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roulleaux Dugage M., Nassif E.F., Italiano A., Bahleda R. Improving Immunotherapy Efficacy in Soft-Tissue Sarcomas: A Biomarker Driven and Histotype Tailored Review. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.775761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qi C., Gong J., Li J., Liu D., Qin Y., Ge S., Zhang M., Peng Z., Zhou J., Cao Y., et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat. Med. 2022;28:1189–1198. doi: 10.1038/s41591-022-01800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Z.Q., Zhao J.J., Pan Q.Z., Chen C.L., Liu Y., Tang Y., Zhu Q., Weng D.S., Xia J.C. PD-L1 expression is a predictive biomarker for CIK cell-based immunotherapy in postoperative patients with breast cancer. J. Immunother. Cancer. 2019;7:228. doi: 10.1186/s40425-019-0696-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jorgovanovic D., Song M., Wang L., Zhang Y. Roles of IFN-gamma in tumor progression and regression: a review. Biomark. Res. 2020;8:49. doi: 10.1186/s40364-020-00228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groom J.R., Luster A.D. CXCR3 in T cell function. Exp. Cell Res. 2011;317:620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee D.W., Santomasso B.D., Locke F.L., Ghobadi A., Turtle C.J., Brudno J.N., Maus M.V., Park J.H., Mead E., Pavletic S., et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019;25:625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from this study can be made available upon request and approval by the study management committee and subject to appropriate data transfer agreements. Requests should be directed to X.Z. No custom code was used for statistical analysis for this study. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.