

Summary
Signal regulatory protein (SIRPα) is an immune inhibitory receptor expressed by myeloid cells to inhibit immune cell phagocytosis, migration, and activation. Despite the progress of SIRPα and CD47 antagonist antibodies to promote anti-cancer immunity, it is not yet known whether SIRPα receptor agonism could restrain excessive autoimmune tissue inflammation. Here, we report that neutrophil- and monocyte-associated genes including SIRPA are increased in inflamed tissue biopsies from patients with rheumatoid arthritis and inflammatory bowel diseases, and elevated SIRPA is associated with treatment-refractory ulcerative colitis. We next identify an agonistic anti-SIRPα antibody that exhibits potent anti-inflammatory effects in reducing neutrophil and monocyte chemotaxis and tissue infiltration. In preclinical models of arthritis and colitis, anti-SIRPα agonistic antibody ameliorates autoimmune joint inflammation and inflammatory colitis by reducing neutrophils and monocytes in tissues. Our work provides a proof of concept for SIRPα receptor agonism for suppressing excessive innate immune activation and chronic inflammatory disease treatment.
Keywords: SIRPα, neutrophil, monocyte, arthritis, colitis, autoimmune inflammation, agonistic antibody
Graphical abstract
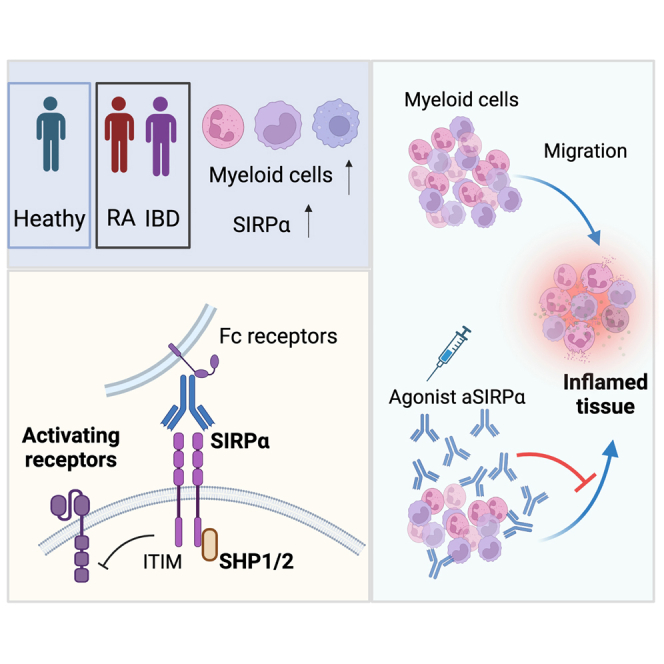
Highlights
-
•
Increased SIRPα+ monocytes and neutrophils in inflamed biopsies of RA and IBD patients
-
•
Agonistic SIRPα antibody inhibits neutrophils and monocyte migration to inflamed tissues
-
•
Agonistic SIRPα treatment ameliorates experimental arthritis and colitis
Xie et al. report an agonistic anti-SIRPα antibody that reduces neutrophils, monocytes, and cytokines in inflamed tissues. Anti-SIRPα agonistic antibody treatment inhibits neutrophil and monocyte migration and ameliorates tissue injury in experimental arthritis and colitis.
Introduction
Human immune responses evolved through an exquisitely regulated process integrating activating and inhibitory receptors on the immune cell surface. The best examples for activating and inhibitory immune receptor families are immunoreceptor-tyrosine-based activation motif (ITAM)-containing receptors and immunoreceptor-tyrosine-based inhibitory motif (ITIM)-containing receptors.1,2 Complex interactions and fully integrated signal inputs from ITAM- or ITIM-containing receptors regulate the quality and magnitude of immune responses and are widely targeted for immune-based therapies.1 Blockade of ITIM-containing inhibitory receptors, such as programmed death 1 (PD1) and lymphocyte activation gene 3 (LAG3), to amplify activating signals and promote anti-tumor immunity, has been clinically beneficial, but such activation is often accompanied by a hyper-immune response and autoimmunity, a common side effect of checkpoint blockade.3,4 In the context of immune suppression, blockade of activating receptors remains challenging due to the redundancy between this class of immune receptors. Successful application of inhibitory receptor agonism remains limited for immune regulation.5
SIRPα is an immunoinhibitory receptor primarily expressed by myeloid lineage of immune cells, including neutrophils, monocytes, macrophages, and dendritic cells (DCs).6 CD47 (also known as integrin-associated protein, IPA) is the only known endogenous ligand for SIRPα, and SIRPα-CD47 interaction leads to recruitment of protein-tyrosine phosphatases (SHP1 and SHP2) that counteract activating signals through dephosphorylation of its substrates in proximity, thereby transducing inhibitory signals that restrict immune cell function.6,7 Temporal blockade of CD47 and/or SIRPα leads to integrin-mediated DC activation,8 macrophage phagocytosis,9,10 and increased immune cell migrations.11,12 Furthermore, anti-CD47 and anti-SIRPα antibodies are under clinical investigation as a cancer immunotherapy for blood and solid tumor types.13,14 Despite the success of CD47 and SIRPα blockades in cancer immunity, agonism of SIRPα to control inflammation has not yet been achieved.
Here, we reported that SIRPα was elevated in inflamed human tissues, and antibody-mediated agonism inhibited neutrophil and monocyte tissue infiltration. In preclinical animal models of arthritis and colitis, the agonistic anti-SIRPα ameliorated diseases through reduction of injurious neutrophil and monocyte tissue infiltration. Our work provides a proof-of-concept study demonstrating the therapeutic benefit of agonizing ITIM-containing inhibitory receptors for innate immune suppression in inflammatory diseases.
Results
Elevated SIRPα+ myeloid cells in inflammatory tissues and associated with treatment non-responsiveness
We first investigated SIRPA expression in synovial biopsies from healthy, osteoarthritis (OA), and rheumatoid arthritis (RA) patients and found SIRPA was significantly elevated in RA patients as compared with healthy and OA patients (Figure 1A). In addition, SIRPA transcripts were also upregulated in inflamed colon tissues of ulcerative colitis (UC) and Crohn disease (CD) patients (Figure 1B). Enhanced SIRPA expression in inflamed UC/CD colonic biopsies was correlated with other neutrophil and inflammatory monocyte-associated genes (e.g., S100A8, S100A9, FCGR2A, VNN2, NCF2) (Figure 1C and Table S1). Consistent with prior publications revealing elevated neutrophil and monocyte gene signatures are associated with inflammatory bowel disease (IBD) patients with adalimumab and vedolizumab non-responsiveness,15,16 SIRPA, as well as other neutrophil- and monocyte-associated genes, were most highly increased in baseline colonic biopsies of patients who failed with anti-tumor necrosis factor (TNF) (infliximab) or anti-α4β7 (vedolizumab) inhibitory antibody (Figure 1D and Table S2). Highly correlated upregulation of SIRPA and other neutrophil/monocyte-associated genes in inflamed tissues suggests an increased frequency SIRPα+ neutrophils/monocytes rather than a cellular increase in SIRPα receptor expression. To confirm this hypothesis, we performed anti-SIRPα immunohistochemistry staining and found increased frequency of SIRPα+ mononuclear cells in the RA synovium and CD-derived inflamed colon biopsies (Figure 1E). Taken together, we observed increased SIRPα+ neutrophils/monocytes in inflamed biopsies of RA and IBD patients.
Figure 1.

Elevated SIRPα+ myeloid cells in inflammatory tissues and associated with treatment non-responsiveness
(A) Transcriptional data of SIRPA expression in synovial biopsies from healthy control, OA, and RA patients (n = 69; each dot represents an individual patient biopsy).
(B) Transcriptional data of SIRPA expression in colon biopsies from healthy control, CD, and UC patients (n = 254; each dot represents an individual patient biopsy).
(C) Transcriptional heatmap of SIRPA and other neutrophil-/monocyte-associated genes in colonic biopsies from healthy control (HC), inflamed, and uninflamed UC/CD colonic biopsies. Data are presented as log2 relative expression.
(D) Transcriptional heatmap of SIRPA and other neutrophil-/monocyte-associated genes in baseline colonic biopsies of patients who responded (R) or non-responded (NR) with infliximab or vedolizumab. Data are presented as log2 relative expression.
(E) Immunohistochemical (IHC) staining of SIRPα in normal or RA synovium (top row) and normal or CD colons (bottom row). Scale bar, 100 μm (one representative image was selected from each group with 3–5 biopsy samples). Data were analyzed using a Kruskal-Wallis test with Dunn’s multiple comparisons test. p values (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) were calculated using a one-way ANOVA and accounting for multiple testing.
Agonistic anti-SIRPα antibody inhibits neutrophil and inflammatory monocyte tissue infiltration
Given the abundant SIRPα+ neutrophils/monocytes in inflamed tissues, we hypothesized that the SIRPα agonistic antibodies could suppress innate immune activation during inflammation through transducing inhibitory signals. Accordingly, we immunized hamsters with a murine recombinant SIRPα extracellular domain protein and identified a novel SIRPα activating antibody clone (termed agonistic anti-SIRPα). Agonistic anti-SIRPα recognized mouse SIRPα in the cell surface (Figure S1A) and induced rapid SIRPα receptor phosphorylation (Figure 2A). To validate the activating function of the agonistic anti-SIRPα antibody, we added murine immunoglobulin (Ig)G2a to the mouse macrophage cell line RAW 264.7 cells with or without anti-SIRPα, and subsequently added protein A beads to crosslink the IgG2a and the agonistic anti-SIRPα immune complex. Using this system, we found that the agonistic SIRPα antibody potently inhibited immune complex induced RAW 264.7 cell secretion of TNFα and granulocyte colony-stimulating factor (G-CSF) (Figure S1B). CD47-Fc fusion protein, which mimics endogenous SIRPα activation, also suppressed TNFα and G-CSF secretion, albeit only at the high concentrations (1 μg/mL) and was significantly less potent at lower protein concentrations (10–100 ng/mL) as compared with the agonistic anti-SIRPα antibody (Figure 2B). Suboptimal agonistic activity of CD47-Fc likely resulted from 200-fold lower affinity of CD47-Fc (2 μM binding Kd for SIRPa/CD47)17 as compared with our agonistic antibody (9 nM binding Kd). ITIM-containing inhibitory receptors need to be in proximity to the activating receptors to recruit SHP1 and dephosphorylate activating receptors.1,10 To test whether the agonistic anti-SIRPα could inhibit immune complex activation without protein A crosslinking of anti-SIRPα and IgG2a together, we added fragment antigen binding 2 (F(ab’)2) to RAW 264.7 cells stimulated with immune complex. We did not observe inhibition of TNFα and G-CSF with anti-SIRPα (F(ab’)2 (Figure S1C). Thus, these in vitro data demonstrate the anti-inflammatory effects of the agonistic SIRPα antibody requires crosslinking with activating receptors.
Figure 2.

The agonistic anti-SIRPα antibody limits neutrophil and inflammatory monocyte immune infiltration
(A) Immunoblot analysis of phosphorylated SIRPα. RAW264.7 cells were stimulated with no treatment, isotype control, or agonistic anti-SIRPα for 5 min.
(B) TNFα and G-CSF in supernatant from RAW 264.7 cells stimulated by mIgG2a-protein A beads immune complex (IC) in the presence of isotype control, agonistic anti-SIRPα, or CD47-Fc.
(C) Flow cytometric quantification of mouse neutrophil (Ly6G+CD11b+) and monocyte (Ly6C+CD11b+) cell number from peritoneal lavage collected 6 h after zymosan injection.
(D) IL-1β, TNFα, and G-CSF in peritoneal lavage collected 6 h after zymosan injection with indicated treatment.
(E and F) Mice were treated with isotype control, agonistic anti-SIRPα, and control anti-SIRPα antibodies. Overnight after antibody treatment, neutrophil and monocyte cell number were quantified by flow cytometry quantification in peritoneal cavity 4 h post intraperitoneal injection of recombinant CXCL1. In (F), indicated groups of mice were pre-treated with blocking antibodies against LFA-1 and MAC-1 16 h before CXCL1 injection.
(G) Mice were treated isotype control, agonistic anti-SIRPα, or control anti-SIRPα antibody. Overnight, after the treatment, migration of bone marrow neutrophils from indicated mouse groups in response to CXCL1 (2 ng/mL) was quantified by a transwell migration assay. Data are from one representative experiment of three independent experiments with at least three biological replicates per group. Each symbol represents one individual mouse. Bar graph is shown as mean ± standard error. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001, by ordinary one-way ANOVA with Tukey’s multiple comparisons test.
We next tested our agonistic anti-SIRPα antibody in vivo in a zymosan-induced peritonitis model, which is commonly used to quantify the recruitment of monocytes and neutrophils into the peritoneal cavity.18 The agonistic anti-SIRPα, but not a commercial anti-SIRPα antagonist antibody,8 significantly attenuated the frequency and number of neutrophils and monocytes in the peritoneal lavage (Figures 2C and S1D). Reduction of neutrophils and monocytes was associated with reduced TNFα, interleukin (IL)-1β, and G-CSF in peritoneal lavage (Figure 2D). Treatment with the CD47-Fc fusion protein was associated with a trend in inhibiting neutrophil/monocyte infiltration and cytokine production (Figures 2C and 2D). We observed a similar effect of the agonistic anti-SIRPα antibody on neutrophil recruitment using the thioglycollate induced peritonitis model (Figures S1E and S1F), which indicates that agonistic SIRPα antibody reduces neutrophil and inflammatory monocyte tissue infiltration in two independent innate stimuli.
In order to test whether the agonistic SIRPα antibody could directly affect immune cell migration, we injected a neutrophil and monocyte chemokine CXCL1 into the peritoneal cavity, and quantified CXCL1-mediated neutrophil and monocyte chemotaxis. The agonistic anti-SIRPα antibody, but not control SIRPα antibody, reduced the frequency and number of peritoneal neutrophils and monocytes (Figures 2E and S1G). Blockade of LFA-1 and MAC-1-dependent endothelial cell adhesion abrogated the difference between isotype control or anti-SIRPα antibody-treated mice (Figure 2F). There was no effect on neutrophils and monocytes in the spleen (Figure S1H), indicating the agonistic anti-SIRPα inhibited integrin-dependent trans-endothelial tissue migration. Consistent with our in vitro data that antibody crosslinking is necessary for inhibition, no effect was seen using an SIRPα Fc mutant antibody (D265A and N297A, dubbed DANA), which lacks the ability to bind to Fc receptors19 (Figure S1I). These data indicate that Fc receptor binding by agonistic anti-SIRPα antibody is critical for in vivo crosslinking of SIRPα to inhibit cell migration. SIRPα was reported to inhibit integrin-dependent cell adhesion and immune cell migration through activation of RhoA and cytoskeletal rearrangement.12,20 Supporting the anti-chemotactic effects of anti-SIRPα treatment, bone marrow neutrophils isolated from the agonistic anti-SIRPα antibody-treated mice had reduced CXCL1-induced ex vivo chemotaxis in a transwell assay (Figure 2G). Reduced neutrophil chemotaxis by anti-SIRPα antibody is SIRPα dependent, as no reduction was observed in SIRPα-deficient mice (Figure S1J). Taken together, these data reveal that the agonistic anti-SIRPα antibody inhibits neutrophil and monocyte migration to tissues during inflammation.
The anti-SIRPα agonist antibody ameliorates experimental arthritis through inhibiting neutrophil and monocyte infiltration
Given the elevated expression of SIRPα expression in human RA biopsies, and that the agonistic SIRPα antibody inhibited monocytes and neutrophil tissue migration, we next sought to investigate the effect of the agonistic anti-SIRPα antibody in a K/BxN serum-induced arthritis preclinical model. In this model, inflammation and associated joint damage is driven by serum autoantibodies against ubiquitously expressed self-antigen, glucose-6-phosphate isomerase (G6PI), which leads to the formation and deposition of immune complexes, followed by infiltration of pathogenic neutrophils and inflammatory monocytes, leading to joint injury.21,22 The agonistic anti-SIRPα antibody significantly diminished the paw and joint erythema and edema clinical arthritis scores as compared with isotype control and CD47-Fc fusion protein (Figures 3A and 3D). Histological analysis revealed reduced arthritis severity, characterized by reduced synovial and intra-articular inflammation, with attenuation of articular cartilage erosion and bone remodeling in the animals treated with the agonistic anti-SIRPα antibody (Figures 3B and 3C).
Figure 3.

Anti-SIRPα agonistic antibody ameliorates experimental arthritis through reducing neutrophil and monocyte infiltration
(A–G) Mice received indicated treatment agents following K/BxN serum transfer. (A) Daily arthritis score. (B and C) Representative joint histopathology image and paw/joint histological scores of mice 8 days post K/BxN serum transfer. Scale bar, 500 μm. (D) Area under the curve (AUC) of total clinical arthritis scores. (E and F) Flow cytometric quantification of neutrophils and monocytes (F) from knee joint (E) and spleen (F) on 8 days post K/BxN serum transfer. (G) RNA sequencing of total paw tissues (each from four individual mice) of naive, isotype control, and agonistic anti-SIRPα treated mice 7 days post K/BxN serum transfer. Heat maps showing differential expression of genes related to myeloid cells (left) and genes of cytokines and chemokines (right).
(H and J) Clinical arthritis scores of naive or collagen-induced arthritis (CIA) mice with treatments of isotype control, agonistic anti-SIRPα, and CD47-Fc.
(I) Representative joint histopathology of naive or CIA mice that received indicated treatment. Scale bar, 500 μm. Data are from one representative experiment of two independent experiments with at least three technical replicates per group. Each symbol represents one mouse. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001, by ordinary one-way ANOVA with Tukey’s multiple comparisons test.
In this KBxN serum transfer arthritis model, neutrophils and monocytes are the primary cellular drivers of disease pathogenesis.21,22 Treatment with depleting antibodies to Ly6G+ neutrophils significantly ameliorated arthritis preclinical manifestations, and furthermore, depleting Ly6G+/Ly6C+ neutrophils and monocytes together led to an even greater effect on inflammation, completely abrogating disease (Figures 3A–3D). Depletion of neutrophils and monocytes in the blood and spleen by anti-Ly6G (neutrophil depletion) and anti-Ly6C/G antibodies was confirmed by flow cytometry; however, depletion was not seen in mice treated with the agonistic anti-SIRPα antibody (Figures S2A and S2B). This indicates disease amelioration by agonistic SIRPα antibody is not through an antibody-dependent cell cytotoxicity (ADCC) mechanism. Corroborating the effect of the anti-SIRPα agonistic antibody in dampening neutrophil and monocyte migration to inflamed tissues, the agonistic anti-SIRPα antibody reduced more than 80% neutrophils and inflammatory monocytes in joint synovial fluids (Figures 3E and S2C), but increased the number of neutrophils and monocytes in systemic lymphoid organ spleen (Figures 3F and S2D). The redistribution of neutrophils in spleen and joint corroborates our findings that anti-SIRPα antibody affects trans-endothelial neutrophil/monocyte tissue migration (Figures 2E–2G). Furthermore, we collected joint tissues from mice treated with isotype control and anti-SIRPα antibodies for RNA-sequencing and observed a reduction of genes associated with neutrophils/monocytes as well as proinflammatory chemokines and cytokines (Figure 3G and Table S3). Last, to test whether the agonistic anti-SIRPα antibody could be efficacious in a preclinical arthritis model initiated from more complex antigen-specific T cell activation, cytokine activation, and subsequent innate cell activation, we tested the efficacy of our agonistic anti-SIRPα antibody and CD47-Fc fusion protein in a collagen-induced arthritis model. We observed a significant amelioration of joint swelling, edema, and erythema in mice treated with the agonistic anti-SIRPα antibody and a reduction in arthritis severity on histopathology (Figures 3H, 3I, and 3J). Taken together, our data indicate that the agonistic anti-SIRPα antibody ameliorates joint inflammation and arthritis via inhibiting detrimental neutrophil and monocyte infiltration in inflamed joints.
The agonistic anti-SIRPα ameliorates T cell transfer colitis through reducing neutrophil and inflammatory monocyte in tissues
During T cell-mediated autoimmunity, CD4+ T cell-derived cytokines, such IL-17 and IL-22, promote tissue inflammation through recruiting neutrophils and monocytes.23,24 Neutrophil infiltration is a hallmark of histological manifestations seen in active UC.25,26 We asked whether our agonistic anti-SIRPα could inhibit neutrophil and monocyte recruitment in a T cell transfer colitis model. In this model, adoptive transfer of sorted CD45RBhighCD4+ T cells into C.B.17 SCID mice initially causes the activation of autoreactive Th1/Th17 T cells, followed by neutrophil and monocyte infiltration and tissue destruction.27,28,29 We investigated the therapeutic efficacy of anti-SIRPα agonistic antibody in a T cell transfer colitis and initiated the treatment right after T cell transfer (Figure S3A). Anti-SIRPα agonistic antibody treatment significantly reduced body weight loss (Figure S3B) and reduced visual colon and histopathology score (Figure S3C). To test whether anti-SIRPα agonistic antibody is efficacious in established colitis, we treated mice with an isotype control, agonistic anti-SIRPα antibody, or anti-Integrin β7 blocking antibody 6 weeks after T cell adoptive transfer when animals had exhibited significant body weight loss and diarrhea, indicative of disease (Figure 4A). Therapeutic treatment with the agonistic anti-SIRPα antibody led to a trend in improvement in the body weight (Figure S3D), improved visual colon score measuring colon edema and thickness (Figure 4B), and decreased histopathological scores (Figures 4B and 4C). Fewer foci of epithelial inflammation, decreased crypt loss, and reduced reactive epithelial hyperplasia was observed in animals treated with the agonistic anti-SIRPα (Figure 4C). Treatment of the anti-Integrin β7 antibody (FIB504) showed a trend of improvement in visual colon and histopathology score (Figures 4B and 4C). Furthermore, we performed automated image analysis on the entire colon tissue sections that were immunohistochemically stained with anti-CD4, anti-F4/80, and anti-GR1. We found that treatment with the SIRPα agonist antibody significantly reduced the GR1+ neutrophil/monocytes in mucosa and lamina propria regions (Figure 4F). The agonist anti-SIRPα antibody had no effect on the frequency of CD4+ T cells and F4/80+ macrophages (Figures 4D and 4E). As a control, a trend in the reduction of CD4+ T frequency in lamina propria and intestinal mucosa was observed for mice treated with anti-Integrin β7 antibody (Figure 4D). Taken together, our data indicate that the efficacy of the agonistic anti-SIRPα antibody in the T cell-mediated transfer colitis preclinical model is through the inhibition of neutrophil and/or monocyte recruitment into inflamed colon tissues.
Figure 4.

The agonistic anti-SIRPα ameliorates T cell transfer colitis through reducing neutrophils and inflammatory monocytes in tissues
(A) Schema of the CD45RBhighCD4+ T cell transfer colitis therapeutic model. C.B17 SCID mice were transferred with unsorted splenocytes or sorted CD45RBhighCD4+ T cells. Six weeks post cell transfer, all mice were re-randomized and started with indicated treatments every other day. All mice were euthanized 12 weeks post cell transfer for tissue collection and histopathology.
(B and C) Visual colon scores and histological scores of colons. (C) Hematoxylin and eosin staining of colon. Asterisks indicate increased lamina propria inflammation, epithelial hyperplasia, and areas of crypt loss. Scale bar, 400 μm.
(D–F) IHC staining of colons was conducted per standard protocols on an autostainer. Sections were stained with antibodies for CD4, F4/80, and GR1(Ly6G/C). Scale bar, 200 μm; inset scale bar, 100 μm. Data are from one representative experiment of two independent experiments with at least three biological replicates per group. Each symbol represents one mouse. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001, by ordinary one-way ANOVA with Tukey’s multiple comparisons test.
Discussion
Despite the successful application of anti-CD47 and anti-SIRPα blocking antibodies in cancer immunotherapy, agonism of inhibitory SIRPα to restrain excessive immune activation in the context of autoimmune inflammation has not been attempted. Here, we provide a proof of concept for an agonistic anti-SIRPα antibody to inhibit innate immune activation and therapeutic intervention in experimental autoimmune arthritis and colitis conditions. There were elevated SIRPα+ neutrophils and monocytes in inflamed tissues from RA and IBD patients. The agonistic anti-SIRPα antibody attenuated inflammation through inhibiting detrimental neutrophil and monocyte tissue infiltration. In preclinical animal models of autoimmune arthritis and colitis, the agonistic anti-SIRPα antibody was highly efficacious in dampening inflammation, dramatically improving tissue pathology. Our study provides a therapeutic approach to agonize inhibitory immune receptors and curb excessive inflammation and autoimmunity.
SIRPα is an important modulator regulating innate immune activation. SIRPα has a long intracellular domain that contains four tyrosine residues to form two ITIM motifs, which are highly evolutionarily conserved across mouse, rat, and human.1 Binding of SIRPα with its extracellular ligand CD47 results in phosphorylation of ITIM and phosphorylated ITIMs serve as recruitment sites for SHP1 and SHP2 phosphatase.6 This leads to an inhibition of cell signaling events, negatively impacting phagocytosis, TNFα production, integrin-dependent adhesion, and in vitro transmigration.10,11,12,30 Engagement of SIRPα with CD47 leads to dysregulated Rho activation, cell skeleton rearrangement, and inhibition of cell migration.12 Consistent with earlier reports of migration inhibition by SIRPα and SHP1,11,12,20 we observed that the agonistic anti-SIRPα antibody was highly effective in reducing neutrophil and monocyte infiltration in peritoneal lavage, arthritic joints, and inflamed colon tissues. Neutrophils from the agonistic anti-SIRPα-treated mice showed reduced ability for chemotaxis. Furthermore, reduction of neutrophils and monocytes in the joint tissues was associated with an increase of these cells in the spleen, indicating inhibition of tissue infiltration likely happens at the step of trans-endothelial migration. Indeed, the difference in cell infiltration diminished after blockade of LFA-1/MAC-1 and integrin-dependent adhesion. SIRPα agonism serves as an effective approach to inhibit neutrophil and monocyte-mediated tissue damage.
SIRPα location was reported as one of the key determinants of receptor activity.10 Without CD47, SIRPα is relegated to the phosphatase-rich zone outside of immune synapse.10,31 This localization prevents SIRPα activation as well as excluding its interaction with activating receptors.10 During CD47 engagement, SIRPα is recruited to the Src-kinase immune synapse,10 where it is activated and suppresses engulfment, cell skeleton rearrangement, and cytokine secretion. Such spatial segregation of inhibitory and activating receptors is similar between SIRPα for myeloid cells and CD45 phosphatase for T cell receptors.10,32 Therefore, the regulation of SIRPα inhibitory receptor requires the temporal recruitment to the immunological synapse to enable activating receptor inactivation. Corroborating the critical role of spatial localization of SIRPα for inhibitory signaling transduction, we found that crosslinking SIRPα with the activating receptor was required for in vitro inhibition of macrophage Fc receptor-induced cytokine secretion (Figure 2). In vivo efficacy of the agonistic anti-SIRPα required an Fc effector function and we observed a reduced efficacy and activity of anti-SIRPα antibody in effectorless Fc format (Figure S3E). We could not see any evidence of ADCC for our agonistic anti-SIRPα and instead we hypothesize that in vivo Fc receptor binding may facilitate trans- or cis-SIRPα clustering to bring SIRPα into the immune synapse during the neutrophil/monocyte adhesion and migration process. Such a requirement for Fc receptor binding and/or activating receptor crosslinking is also seen with other inhibitory agonistic antibodies, including PD-1, BTLA, and CD200R1 agonistic antibodies.5,33,34,35 It is not yet known which Fc receptors (e.g., FCGRIIA, FCGRIIB, FCGRIIIA) and which cells crosslink SIRPα receptor to enable in vivo inhibitory receptor agonism. More than one type of Fc receptors could be involved. Our data suggest that multi-specific protein scaffold with higher magnitude of inhibitory receptor clustering and crosslinking between inhibitory and activating receptors may deliver stronger inhibitory signal transduction and immune cascade suppression.
Despite the advance in effective therapeutics against inflammatory cytokines (e.g., TNFα, IL-23), cytokine signaling (e.g., JAK1, TYK2), and integrin-dependent adaptive immune trafficking (e.g., integrin α4 and α4β7), there remains high number of inflammatory disease patients who are refractory to current treatment, representing a tremendous unmet medical need. There are limited therapeutic options for targeting innate immune cell infiltrations. Our data suggest that agonism of inhibitory receptors on myeloid cells by anti-SIRPα may represent an approach to limit pathogenic innate immune infiltration and chronic tissue inflammation.
Limitations of the study
Our current study focused on investigating how anti-SIRPα agonistic antibody regulates cellular response in the context of preclinical inflammatory conditions. It remains unclear the molecular mechanism by which SIRPα agonism regulates cell migration. In addition, Fc effector function is required for agonistic activity, but it is not yet known which Fc receptors and which cells crosslink SIRPα receptor to enable in vivo inhibitory receptor agonism. Further experiments to investigate SIRPα signaling in the context of leukocyte trafficking could provide additional mechanistic information. Finally, clinical studies of SIRPα or other ITIM-containing receptor agonistic antibodies will be necessary to fully evaluate the therapeutic translatability of our findings.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC/Cy7 anti-mouse SIRPα (clone P84) | BioLegend | Catalog #: 144018; RRID:AB_2629558 |
| APC/Cy7 anti-mouse GR-1 (clone: RB6-8C5) | BioLegend | Catalog #: 108424; RRID:AB_2137485 |
| APC/Cy7 anti-mouse F4/80 (clone: BM8) | BioLegend | Catalog #: 123118; RRID:AB_893477 |
| PE/Cy7 anti-mouse Ly6G (clone: 1A8) | BioLegend | Catalog #: 127618; RRID:AB_1877261 |
| PE/Cy7 anti-mouse CD8a (clone: 53-6.7) | BioLegend | Catalog #: 100722; RRID:AB_312761 |
| PE/Cy7 anti-mouse SIRPα (clone: P84) | BioLegend | Catalog #: 144008; RRID:AB_2563546 |
| PE/Cy7 anti-mouse CD11c (clone: N418) | BioLegend | Catalog #: 117318; RRID:AB_493568 |
| PE/Cy7 anti-mouse CD86 (clone: GL-1) | BioLegend | Catalog #: 105014; RRID:AB_439783 |
| BUV395 anti-mouse CD11b (clone: M1/70) | BD Biosciences | Catalog #: 563553; RRID:AB_2738276 |
| BV421 anti-mouse CD4 (clone: RM4-4) | BioLegend | Catalog #: 116023; RRID:AB_2800579 |
| BV421 anti-mouse Ly6C (clone: HK1.4) | BioLegend | Catalog #: 128014; RRID:AB_1732079 |
| BV421 anti-mouse F4/80 (clone: BM8) | BioLegend | Catalog #:123132; RRID:AB_11203717 |
| BV421 anti-mouse I-A/I-E (clone: M5/114.15.2) | BioLegend | Catalog #: 107632; RRID:AB_2650896 |
| PE anti-mouse F4/80 (clone: BM8) | BioLegend | Catalog #: 123110; RRID:AB_893486 |
| PE anti-mouse Ly6C (clone: HK1.4) | BioLegend | Catalog #: 128008; RRID:AB_1186132 |
| PERCP/Cy5.5 F4/80 (clone: BM8) | BioLegend | Catalog #: 123128; RRID:AB_893484 |
| PERCP/Cy5.5 Ly6C (clone: HK1.4) | BioLegend | Catalog #: 128012; RRID:AB_1659241 |
| PERCP/Cy5.5 CD4 (clone: RM4-5) | BioLegend | Catalog #: 100540; RRID:AB_893326 |
| PERCP/Cy5.5 Ly6G (clone: 1A8) | BioLegend | Catalog #: 127616; RRID:AB_1877271 |
| PERCP/Cy5.5 I-A/I-E (clone: M5/114.15.2) | BioLegend | Catalog #: 107626; RRID:AB_2191071 |
| APC anti-mouse Ly6G (clone: 1A8) | BioLegend | Catalog #: 127614; RRID:AB_2227348 |
| APC anti-mouse F4/80 (clone: BM8) | BioLegend | Catalog #: 123116; RRID:AB_893481 |
| APC anti-mouse CD11c (clone: N418) | BioLegend | Catalog #: 117310; RRID:AB_313779 |
| APC anti-mouse CD4 (clone: RM4-5) | BioLegend | Catalog #: 100516; RRID:AB_312719 |
| APC anti-mouse CD8a (clone: 53-6.7) | BioLegend | Catalog #: 100766; RRID:AB_2572113 |
| FITC anti-mouse CD86 (clone: GL-1) | BioLegend | Catalog #: 105005; RRID:AB_313148 |
| FITC anti-mouse Ly6C (clone: HK1.4) | BioLegend | Catalog #: 128006; RRID:AB_1186135 |
| Anti-human SIRPα (Mouse IgG2b Clone #602411) | R&D systems | Catalog #: MAB4546; RRID:AB_10718553 |
| Anti-human CD4 (Polyclonal Goat IgG) | R&D systems | Catalog #: AF-379-NA; RRID:AB_354469 |
| Anti-mouse F4/80 (clone: T45-2342) | BD Pharmingen | Catalog #: BDB565410 |
| Anti-mouse GR-1 (clone: T45-2342) | BD Pharmingen | Catalog #: BDB553123 |
| Fc block- anti-mouse CD16/32 | BioLegend | Catalog #: 101302; RRID:AB_312801 |
| Anti-mouse β7 (clone FIB504) | This paper | Antibody ID: AB_2892125 |
| Agonist anti-mouse SIRPα (clone 6F2) see below | This paper | this manuscript |
| Agonist anti-mouse SIRPα (clone 6F2) Heavy Chain sequence: | EVQLVESGGGLVKPGGSLKLSCAASGFSFSTYWM TWVRQAPGKGLEWVGEINEGGSTTNYAPSVK GRFTISRDNARNTLFLQMNSVKSEDTAAYYCARDY WDTPYYFDYWGQGTMVTVSSAKTTAPSVYPLAPVC GDTTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGV HTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAH PASSTKVDKKIEPRGPTIKPCPPCKCPAPNLLGGPSV FIFPPKIKDVLMISLSPIVTCVVVDVSEDDPDVQISWFV NNVEVHTAQTQTHREDYNSTLRVVSALPIQHQDW MSGKEFKCKVNNKDLPAPIERTISKPKGSVRAPQVY VLPPPEEEMTKKQVTLTCMVTDFMPEDIYVEWTNN GKTELNYKNTEPVLDSDGSYFMYSKLRVEKK NWVERNSYSCSVVHEGLHNHHTTKSFSRTPGK |
this manuscript |
| Agonist anti-mouse SIRPα (clone 6F2) Light Chain sequence: | DIVMTQSPSSLAVSVGEKVTIGCKSSQSLLFN KDQKNYLSWYLQKPGQSPKLLIYYASTRH TGVPDRFIGSGSGTDFTLTIDSVQSEDLADY YCLQTYSAPRTFGPGTKLEIKRADAAPTV SIFPPSSEQLTSGGASVVCFLNNFYPKDIN VKWKIDGSERQNGVLNSWTDQDSKDSTY SMSSTLTLTKDEYERHNSYTCEATHKTST SPIVKSFNRNEC |
this manuscript |
| Isotype Control (anti-GP120) | This paper | this manuscript |
| InVivoMAb anti-mouse Ly6G (clone: 1A8) | BioXCell | Catalog #: BE0075-1; Antibody ID: AB_1107721 |
| InVivoMAb anti-mouse Ly6G/Ly6C GR-1 (clone: RB6-8C5) | BioXCell | Catalog #: BE0075; Antibody ID: AB_10312146 |
| Anti-rat IgG HRP for IP | R&D systems | Catalog #: HAF005; RRID:AB_1512258 |
| Anti-mouse SIRPa (clone: P84) for IP | BD Biosciences | Catalog #: 552371; RRID:AB_394371 |
| Anti-phosphotyrosine (clone: 4G10) | EMD Millipore | Catalog #: 05-321X; RRID:AB_568858 |
| Biological samples | ||
| Healthy or IBD patient intestinal tissue biopsies | Genentech | GEO: GSE179285 |
| Healthy or arthritis patient synovial tissue biopsies | University of Michigan | GEO: GSE48780 and GSE236924 |
| Chemicals, peptides, and recombinant proteins | ||
| Complete Freund’s Adjuvant | InvivoGen | Catalog #: vac-cfa-60 |
| Incomplete Freund’s Adjuvant | InvivoGen | Catalog #: vac-ifa-60 |
| Zymosan | InvivoGen | Catalog #: tlrl-zyn |
| Recombinant Mouse CXCL1 | R&D systems | Catalog #: 453-KC-050 |
| Recombinant Mouse CXCL1 | PeproTECH | Catalog #: 250-11-250UG |
| Thioglycollate Medium | Sigma-Aldrich | SKU: 1462200100 |
| Protein A beads/DYNABEADS | Thermo fisher | Catalog #: 10002D |
| Red Blood Cell Lysing Buffer Hybri-MaxTM | Sigma-Aldrich | SKU: R7757-100ML |
| Protein A-agarose | Roche | Discontinued N/A |
| Critical commercial assays | ||
| RNeasy Fibrous Tissue Mini Kit | QIAGEN | Cat. No./ID: 74704 |
| CD4+ T cell Isolation Kit, mouse | Miltenyi Biotec | Order no. 130-104-454 |
| Lipofectamine 2000 kit | ThermoFisher Scientific | Catalog #: 11668019 |
| HTS Transwell®-24-well Permeable Support with 5.0 μm Pore Polycarbonate Membrane and 6.5 mm Inserts, Sterile | Corning | Discontinued N/A |
| High Sensitivity D1000 ScreenTape and reagents | Agilent Technologies | Catalog #: 5067-5584 |
| IdeZ Protease | Promega | Catalog #: 8341 |
| SMARTer Stranded Total RNA-Seq Kit | Takara | Catalog #: 634412 |
| Qubit™ dsDNA HS and BR Assay Kits | ThermoFisher Scientific | Catalog #: Q32851 |
| Deposited data | ||
| Bulk RNA-sequencing of mouse paw samples | This paper | GEO: GSE235400 |
| Microarray of arthritis patient tissue samples | University of Michigan | GEO: GSE48780 and GSE236924 |
| Experimental models: Cell lines | ||
| RAW 264.7 cells | ATCC | TIB-71 |
| HEK 293T | ATCC | CRL-1573 |
| Experimental models: Organisms/strains | ||
| C57BL/6 | Charles River | Strain code: 027 |
| SIRPa-flox/flox Lyzm-Cre | This paper | this manuscript |
| BALB/cAnNCrl | Charles River | Strain code: 028 |
| C.B17 SCID | Taconic | Model #: CB17SC-F |
| Software and algorithms | ||
| R (v 3.5.1) | The R Project | http://www.r-project.org |
| GraphPad Prism v.8 | GraphPad Software | https://www.graphpad.com/ |
| FlowJo v.10 | FlowJo | https://www.flowjo.com/ |
| EndNote 20 | EndNote | https://endnote.com/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/ |
| BioRender | BioRender | https://www.biorender.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tangsheng Yi (tangshengy@gmail.com).
Materials availability
All unique/stable reagents generated in this study are available from the corresponding author Tangsheng Yi (tangshengy@gmail.com) with a completed Materials Transfer Agreement (https://www.gene.com/scientists/mta).
Experimental model and study participant details
Mice
Female C57BL/6, Balb/c and C.B17 SCID mice were from Charles River Laboratories and were used at age 6–10 weeks for studies.
The generation of Sirpa conditional Knockout mouse (cKO): the construct for targeting the C57BL/6N Sirpa locus in ES cells was made using a combination of recombineering and standard molecular cloning techniques. Exons 1 and 2 were floxed by inserting a loxP site upstream of exon 1 and a loxP-Frt-Pgk1-Neo-Frt cassette downstream of exon 2. The final vector was confirmed by DNA sequencing. The Sirpa cKO vector was linearized and C57BL/6N C2 ES cells were targeted using standard methods (G418 positive and gancyclovir negative selection). Positive clones were identified using PCR and taqman analysis, and confirmed by sequencing. Correctly targeted ES cells were transfected with a Flpe plasmid to remove Neo and create the Sirpa conditional knock-out allele (cKO). Sirpa cKO ES cells were then injected into blastocysts using standard techniques, and germline transmission was obtained after crossing resulting chimeras with C57BL/6N females. Sirpa flox mice were further crossed with LyzM-Cre (The Jackson Laboratory) to obtain LyzM-cre+Sirpa flox/flox cKO mice.
All mice used in this study were kept at Genentech under specific pathogen-free conditions. All protocols were reviewed and approved by the Genentech Institutional Animal Care and Use Committee.
Human clinical cohorts
Expression in colon samples from healthy control, Crohn disease, or ulcerative colitis patient samples was measured by using microarrays (Agilent). 254 samples used for this analysis were from the internal Embark study (SPR1001)36 and were obtained from Crohn’s disease patients, Ulcerative Colitis patients, and healthy controls by ileocolonoscopy. Specifically, biopsies were taken in the sigmoid colon (n = 21) and ascending/descending colon of healthy controls; in the ascending/descending colon (n = 107) in uninflamed areas in all patients with Crohn’s disease, as well as additional biopsies were taken in inflamed regions of the ascending/descending colon (n = 35) in patients with mucosal lesions. Paired uninflamed sigmoid (n = 48) and inflamed sigmoid biopsies (n = 46) were taken in Ulcerative Colitis patients. Extracted RNA was quantified using an Agilent human 4x44kv1 array.
Expression in two separate Rheumatoid Arthritis patient collections (Two sequential cohorts, n = 49 and n = 20) from the University of Michigan was measured by using Affymetrix microarrays. Detailed study information was published earlier.37 All patients provided consentin accordance with Institutional Review Board guidance for each institution as noted above.
Method details
Agonistic monoclonal anti-SIRPα generation
Monoclonal antibodies were generated by immunizing hamsters with murine SIRPα extracellular domain fused to a human IgG1 Fc. Hybridomas were generated using traditional methods and supernatants were screened for binding to murine SIRPα by ELISA. Positive hybridoma clones were scaled up and purified for further characterization. Variable regions of top binders were molecularly cloned into murine IgG2a Fcs with and without DANA (D265A and N297A) mutations, recombinantly expressed and purified for further applications. To generate F(ab’)2 fragment of the antibody, IdeZ kit (Promega) was used. One unit of IdeZ protease was added per 1 μg of the antibody and incubated at 37°C for 30 min before application for further studies.
K/BxN serum transfer arthritis model
Mice were treated with indicated treatment group (250 μg per mouse) every other day starting one day before serum transfer until the termination. Subsequently, mice were intravenously injected once with 100 μl of arthrogenic K/BxN serum. Mice were checked and monitored for joint and paw clinical scoring every other day. Briefly the animals were scored as follows: 0, no evidence of erythema and swelling; 1, erythema and mild swelling confined to the mid-foot (tarsal) or ankle; 2, erythema and mild swelling extending from the ankle to the mid-foot; 3, erythema and moderate swelling extending from the ankle to the metatarsal joints; 4, erythema and severe swelling encompass the ankle, foot and digits. Total score was the sum of the 4 paw scores.
Collagen induced arthritis (CIA) model
Mice were immunized with 100 μg type II chicken collagen in 100 μl Complete Freunds Adjuvant (CFA). The collagen type II in CFA were injected intradermally (i.d.) on the side of the back, with the dose divided into 2, 50 μL injections. At Day 21, a second immunization with 100 μg chicken collagen type II in 100 μL of incomplete Freunds adjuvant was given i.d. on the side of the back, with the dose divided into 2, 50 μL injections. Mice were treated with indicated antibodies (250 μg/mouse) every other day starting one day before CIA recall responses on Day 21 post the first immunization. Paw and joint arthritic inflammation were scored on indicated dates. On Day 38, all paws were harvested for histopathological analysis. Mice were checked and monitored for joint and paw clinical scoring regularly. Briefly the animals were scored as follows: 0, no evidence of erythema and swelling; 1, erythema and mild swelling confined to the mid-foot (tarsal) or ankle; 2, erythema and mild swelling extending from the ankle to the mid-foot; 3, erythema and moderate swelling extending from the ankle to the metatarsal joints; 4, erythema and severe swelling encompass the ankle, foot and digits. Total score was the sum of the 4 paw scores.
Peritonitis models
To elicit sterile peritoneal inflammation, mice pre-treated (16 h) with indicated antibodies (250 μg/mouse/time) were intraperitoneally injected with 2 mg of Zymosan (Invivogene) suspended in 200 μL PBS. Peritoneal lavage was done 6 h after zymosan injection. Briefly, 3 mL PBS was injected via i.p. and 2 mL of lavage was aspirated out using a syringe. To elicit thioglycollate-induced peritonitis mAbs pre-administered mice were injected with 5 mL of thioglycollate broth (Sigma-Aldrich) i.p. Peritoneal lavage was collected 6 h after thioglycollate injection. Cell pellets were collected after centrifuge for flow cytometric analysis while suspensions were used for cytokine Luminex assay.
CD45RBhighCD4+ T cell transfer colitis model
On Week 0, mice received 3 x105 CD45RBhighCD4+ T cells or unsorted T cells in 200 μl PBS via i.v. For the preventive model, treatments were started the next day of T cell transfer (250 μg/mouse/time). Mice were treated 3 times per week via subcutaneous (s.c.) route in 200 μL sterile PBS until the end of experiment. For the therapeutic model, all mice were bled by retro-orbital (RO) route under anesthesia for 200 μL whole blood on heparin for flow cytometric analysis 6 weeks post adoptive T cell transfer. Animals were re-randomized and grouped out according to percentage and number of CD4 cells and body weight (BW). Treatments were started on the same day of re-randomization (250 μg/mouse/time). Mice were treated 2 times per week via s.c. route in 200 μL sterile PBS until end of experiment. Weights will be taken prior to cell transfer (baseline), week 6 and the termination of the study. Beginning at week 4, mice will be monitored daily for signs of IBD. The study will be terminated at week 12. At the end of the study mouse colons were harvested. A gross colon score and colon weight were determined after flushing the tissues with cold saline.38 Colons were stored in formalin for histopathology analysis.
CXCL1-mediated cell recruitment assay
Mice were injected intraperitoneally with 250 μg mAbs in 200 μl PBS 16 h prior to CXCL1 injection. To elicit CXCL1 induced cell recruitment, 4 μg CXCL1 (R & D) suspended in 200μL PBS was injected intraperitoneally. After 4 h, peritoneal lavage was carried out by injecting 3 mL PBS i.p. and 2 mL of lavage was aspirated out using a syringe. Cell pellets were collected after centrifuge for flow cytometric analysis.
Histology
In arthritis models, the left and right fore- and hindlimbs were immersion fixed in 10% neutral buffered formalin for 24 h, then hemi-sectioned, paraffin embedded and routinely processed. Forelimb sections comprised the distal radius, carpi, metacarpi and P1-3. Hindlimb sections included the distal tibia, occasionally the distal fibula, talus, tarsal, metatarsal bones, and P1-3. Duplicate sections of each limb were examined and scored on a scale of 0–5 based on the number of joints affected, and the severity of inflammation, spindle cell proliferation, cartilage erosion, and bone lysis and remodeling. A total arthritis severity score was calculated from the average of these scores across each limb.39
In the T cell transferred colitis model, colons were flushed with saline and fixed in 10% formalin overnight and processed into paraffin. 4-μm sections were stained with hematoxylin and eosin and were scored for colitis severity by a pathologist. Each of 4 anatomical segments were scored on a scale of 0–5 based on extent of inflammation, epithelial hyperplasia, and epithelial loss, resulting in a summed score ranging from 0 to 20.
Immunohistochemistry
IHC staining was conducted per standard protocols on an autostainer. In brief, sections were deparaffinized, subjected to antigen retrieval, and incubated with primary antibodies (SIRPα, CD4 - R&D Systems; F4/80, GR1 - Pharmingen), ABC-Peroxidase Elite secondary antibody system, and detection with 3,3′-Diaminobenzidine Chromogen.
Flow cytometry/FACS analysis
Cell suspension was washed twice in staining buffer (PBS with 2% of fetal bovine serum), blocked with Fc block (anti-mouse CD16/32, Biolegend) for 5 min at room temperature, and stained with indicated fluorescence conjugated antibodies below for 20 min. Fluorochrome-conjugated anti-mouse antibodies used are as follows: SIRPα (P84), GR1 (RB6-8C5), F4/80 (BM8), Ly6G (1A8), CD11b (M1/70), MHCII (M5/114.15.2), Ly6C (HK1.4). All samples were acquired on a BD FACSymphony flow cytometer (Becton Dickinson) and analyzed with FlowJo software.
In vitro cell culture
RAW 264.7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum and 100 mg/mL penicillin +100 μg/mL streptomycin in a humidified 37°C incubator with 5% CO2 and atmospheric oxygen (21% O2). For immune complex assay, RAW 264.7 cells were cultured at 4 × 105 cells/well in 1mL medium in 24-well flat bottom tissue culture plates. The cells were allowed to adhere overnight and the following day the medium was supplemented with indicated antibodies at 10 μg/mL in medium for 1 h. After 1 h, 10 μL DYNABEADS (PROTEIN A). After another 24 h’ culture, 150 μL medium supernatant from each well was collected for cytokine Luminex assay.
HEK 293T cells were grown to 80–90% confluency in 5 mL of DMEM with 10% FBS and 1% antibiotics in a 6 well plate. Cells were transfected with SIRPα expressing vectors by using Lipofectamine 2000 (ThermoFisher Scientific). 2.5 μg of vector were mixed in 150 μl Opti-MEMTM I reduced serum medium (ThermoFisher Scientific), and 15 μL Lipofectamine 2000 was mixed in another 150 μl Opti-MEMTM I reduced serum medium. After combining, this mixture was incubated at room temperature for 15 min. The combined mixture was gently added to the HEK293T cells culture plate. After 72 h, the cells were harvested for FACS analysis.
Western blot/Immunoprecipitation
Raw 264.7 cells were seeded in a 6 well plate and treated at 90% confluence with 100 ng/mL antibody for 5 min. Cells treated with media alone serves as a no treatment control. After 5 min media was aspirated and cells were lysed with 400 μl IP lysis buffer (Pierce) containing a phosphatase inhibitor cocktail (Sigma) and Complete protease inhibitor (Roche) and kept on ice. Lysates were immunoprecipitated with 2 μg anti-phosphotyrosine clone 4G10 (EMD Millipore) for 1 h at 4C. Immune complexes were precipitated with Protein A-agarose (Roche), washed three times in lysis buffered prior to running on SDS-PAGE. Proteins were transferred to nitrocellulose (Invitrogen) and blocked with 5% BSA in TRIS-buffered saline tween 20 and blotted using anti-SIRPα clone P84 (BD) and anti-rat IgG HRP (R&D). Proteins were visualized by chemiluminescence (Amersham).
Bone marrow cell isolation and transwell migration assay
Mice were injected intraperitoneally with 250 μg indicated antibodies in 200 μl PBS 20 h prior to bone marrow collection. Bone marrow cells were collected from two femurs and red blood cells were lysed using the lysis buffer (Red Blood Cell Lysing Buffer Hybri-Max, Sigma-ALDRICH). Bone marrow cells were washed in migration medium (RPMI containing 0.5% fatty acid-free BSA, 10 mM HEPES and 50 IU/L penicillin/streptomycin) and resuspended in migration medium at 2 × 106 cells/ml. Cells were resensitized for 10 min in a 37°C water bath in migration medium. To set up the migration assay, 600 μl CXCL1 containing migration medium was added into the transwell bottom. Transwell filters (6 mm insert, 5 μm pore size, Corning) were placed on top of each well, and 100 μl containing 2 × 105 cells of each group was added to the transwell insert. The cells were allowed to migrate for 3 h, after which the cells in the bottom well were counted by flow cytometry. As described in,40 a percentage of input migration was plotted as cell migration.
RNAseq and bioinformatics analysis
Mouse hind paws from one KB/xN study were collected and unskinned for RNA isolation. RNA was isolated from tissues using the QIAGEN RNeasy Fibrous Tissue Mini Kit (QIAGEN) according to the manufacturer’s instructions. Total RNA was quantified with Qubit RNA HS Assay Kit (Thermo Fisher Scientific) and quality was assessed using RNA ScreenTape on 4200 TapeStation (Agilent Technologies). For sequencing library generation, the SMARTer Stranded Total RNA-Seq Kit v2 – Pico Input Mammalian kit (Takara) was used with an input of 1–2 ng of total RNA. Libraries were quantified with Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) and the average library size was determined using High Sensitivity D1000 ScreenTape on 4200 TapeStation (Agilent Technologies). Libraries were pooled and sequenced on NovaSeq 6000 (Illumina) to generate 30 million single-end 50-base pair reads for each sample. For RNA-seq analysis, we used custom scripts written in the R programming language and packages from the Bioconductor project as described before.41
Quantification and statistical analysis
GraphPad Prism software was used for statistical analysis. Graph bars represent the mean ± SEM. Unless otherwise stated, One-Way ANOVA with Tukey’s post hoc analysis was used to determine significance. Significant differences (p < 0.05) are indicated in the figures.
Acknowledgments
We thank Saiyu Hang for critically reviewing this manuscript. We thank Genentech Research Pathology Core Laboratories (Necropsy, Histology, and Immunohistochemistry), FACS Core Laboratory, and Laboratory Animal Systems and Reports staff for their assistance. This research was supported and funded by Genentech, Inc., USA.
Author contributions
M.M.X. performed in vitro and in vivo experiments, analyzed data, and wrote the manuscript; J.A.H. analyzed data; M.J.T. and D.A.F. contributed to the acquisition of human data; B.D., S.T., J.Z., A.S., J.K.J., S.J., R.A.I.C., Y.L., Y.F., H.B., E.R.S., M.E.K., J.B., G.N., W.P.L., and P.J.G. provided support with data analysis and designed and executed experiments. M.M.X. and J.A.H. performed statistical analyses. P.G. initiated the project and supervised SIRPα antibody generation. M.M.X., F.M., R.P., and T.Y. designed experiments and wrote the manuscript; all authors were involved in manuscript editing and finalization.
Declaration of interests
All authors except D.A.F. are current or past employees of Genentech, a member of the Roche group, and may hold Roche stock or stock options.
Published: July 24, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101130.
Contributor Information
Paul J. Godowski, Email: godowski89@gmail.com.
Rajita Pappu, Email: Pappu.Rajita@gene.com.
Tangsheng Yi, Email: tangshengy@gmail.com.
Supplemental information
Data and code availability
RNA-seq and gene expression data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This work does not contain any custom software for the data analysis.
References
- 1.Daëron M., Jaeger S., Du Pasquier L., Vivier E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol. Rev. 2008;224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x. [DOI] [PubMed] [Google Scholar]
- 2.Getahun A., Cambier J.C. Of ITIMs, ITAMs, and ITAMis: revisiting immunoglobulin Fc receptor signaling. Immunol. Rev. 2015;268:66–73. doi: 10.1111/imr.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young A., Quandt Z., Bluestone J.A. The Balancing Act between Cancer Immunity and Autoimmunity in Response to Immunotherapy. Cancer Immunol. Res. 2018;6:1445–1452. doi: 10.1158/2326-6066.CIR-18-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chao M.P., Takimoto C.H., Feng D.D., McKenna K., Gip P., Liu J., Volkmer J.P., Weissman I.L., Majeti R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2019;9:1380. doi: 10.3389/fonc.2019.01380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paluch C., Santos A.M., Anzilotti C., Cornall R.J., Davis S.J. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front. Immunol. 2018;9:2306. doi: 10.3389/fimmu.2018.02306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barclay A.N., Van den Berg T.K. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu. Rev. Immunol. 2014;32:25–50. doi: 10.1146/annurev-immunol-032713-120142. [DOI] [PubMed] [Google Scholar]
- 7.Myers D.R., Abram C.L., Wildes D., Belwafa A., Welsh A.M.N., Schulze C.J., Choy T.J., Nguyen T., Omaque N., Hu Y., et al. Shp1 Loss Enhances Macrophage Effector Function and Promotes Anti-Tumor Immunity. Front. Immunol. 2020;11:576310. doi: 10.3389/fimmu.2020.576310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yi T., Li J., Chen H., Wu J., An J., Xu Y., Hu Y., Lowell C.A., Cyster J.G. Splenic Dendritic Cells Survey Red Blood Cells for Missing Self-CD47 to Trigger Adaptive Immune Responses. Immunity. 2015;43:764–775. doi: 10.1016/j.immuni.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oldenborg P.A., Zheleznyak A., Fang Y.F., Lagenaur C.F., Gresham H.D., Lindberg F.P. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 10.Morrissey M.A., Kern N., Vale R.D. CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity. 2020;53:290–302.e6. doi: 10.1016/j.immuni.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu D.Q., Li L.M., Guo Y.L., Bai R., Wang C., Bian Z., Zhang C.Y., Zen K. Signal regulatory protein alpha negatively regulates beta2 integrin-mediated monocyte adhesion, transendothelial migration and phagocytosis. PLoS One. 2008;3:e3291. doi: 10.1371/journal.pone.0003291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Motegi S.I., Okazawa H., Ohnishi H., Sato R., Kaneko Y., Kobayashi H., Tomizawa K., Ito T., Honma N., Bühring H.J., et al. Role of the CD47-SHPS-1 system in regulation of cell migration. EMBO J. 2003;22:2634–2644. doi: 10.1093/emboj/cdg278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willingham S.B., Volkmer J.P., Gentles A.J., Sahoo D., Dalerba P., Mitra S.S., Wang J., Contreras-Trujillo H., Martin R., Cohen J.D., et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng M., Jiang W., Kim B.Y.S., Zhang C.C., Fu Y.X., Weissman I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer. 2019;19:568–586. doi: 10.1038/s41568-019-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedrich M., Pohin M., Jackson M.A., Korsunsky I., Bullers S.J., Rue-Albrecht K., Christoforidou Z., Sathananthan D., Thomas T., Ravindran R., et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med. 2021;27:1970–1981. doi: 10.1038/s41591-021-01520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arijs I., De Hertogh G., Lemmens B., Van Lommel L., de Bruyn M., Vanhove W., Cleynen I., Machiels K., Ferrante M., Schuit F., et al. Effect of vedolizumab (anti-alpha4beta7-integrin) therapy on histological healing and mucosal gene expression in patients with UC. Gut. 2018;67:43–52. doi: 10.1136/gutjnl-2016-312293. [DOI] [PubMed] [Google Scholar]
- 17.Logtenberg M.E.W., Scheeren F.A., Schumacher T.N. The CD47-SIRPalpha Immune Checkpoint. Immunity. 2020;52:742–752. doi: 10.1016/j.immuni.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louwe P.A., Badiola Gomez L., Webster H., Perona-Wright G., Bain C.C., Forbes S.J., Jenkins S.J. Recruited macrophages that colonize the post-inflammatory peritoneal niche convert into functionally divergent resident cells. Nat. Commun. 2021;12:1770. doi: 10.1038/s41467-021-21778-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shields R.L., Namenuk A.K., Hong K., Meng Y.G., Rae J., Briggs J., Xie D., Lai J., Stadlen A., Li B., et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 20.Inagaki K., Yamao T., Noguchi T., Matozaki T., Fukunaga K., Takada T., Hosooka T., Akira S., Kasuga M. SHPS-1 regulates integrin-mediated cytoskeletal reorganization and cell motility. EMBO J. 2000;19:6721–6731. doi: 10.1093/emboj/19.24.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen A.D., Haase C., Cook A.D., Hamilton J.A. K/BxN Serum-Transfer Arthritis as a Model for Human Inflammatory Arthritis. Front. Immunol. 2016;7:213. doi: 10.3389/fimmu.2016.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wipke B.T., Allen P.M. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 23.Kvedaraite E. Neutrophil-T cell crosstalk in inflammatory bowel disease. Immunology. 2021;164:657–664. doi: 10.1111/imm.13391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernshtein B., Curato C., Ioannou M., Thaiss C.A., Gross-Vered M., Kolesnikov M., Wang Q., David E., Chappell-Maor L., Harmelin A., et al. IL-23-producing IL-10Ralpha-deficient gut macrophages elicit an IL-22-driven proinflammatory epithelial cell response. Sci. Immunol. 2019;4:eaau6571. doi: 10.1126/sciimmunol.aau6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kucharzik T., Walsh S.V., Chen J., Parkos C.A., Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am. J. Pathol. 2001;159:2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muthas D., Reznichenko A., Balendran C.A., Böttcher G., Clausen I.G., Kärrman Mårdh C., Ottosson T., Uddin M., MacDonald T.T., Danese S., Berner Hansen M. Neutrophils in ulcerative colitis: a review of selected biomarkers and their potential therapeutic implications. Scand. J. Gastroenterol. 2017;52:125–135. doi: 10.1080/00365521.2016.1235224. [DOI] [PubMed] [Google Scholar]
- 27.Yen D., Cheung J., Scheerens H., Poulet F., McClanahan T., McKenzie B., Kleinschek M.A., Owyang A., Mattson J., Blumenschein W., et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ostanin D.V., Bao J., Koboziev I., Gray L., Robinson-Jackson S.A., Kosloski-Davidson M., Price V.H., Grisham M.B. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G135–G146. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fournier B.M., Parkos C.A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5:354–366. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- 30.Abram C.L., Lowell C.A. Shp1 function in myeloid cells. J. Leukoc. Biol. 2017;102:657–675. doi: 10.1189/jlb.2MR0317-105R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodridge H.S., Reyes C.N., Becker C.A., Katsumoto T.R., Ma J., Wolf A.J., Bose N., Chan A.S.H., Magee A.S., Danielson M.E., et al. Activation of the innate immune receptor Dectin-1 upon formation of a 'phagocytic synapse. Nature. 2011;472:471–475. doi: 10.1038/nature10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James J.R., Vale R.D. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature. 2012;487:64–69. doi: 10.1038/nature11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cherwinski H.M., Murphy C.A., Joyce B.L., Bigler M.E., Song Y.S., Zurawski S.M., Moshrefi M.M., Gorman D.M., Miller K.L., Zhang S., et al. The CD200 receptor is a novel and potent regulator of murine and human mast cell function. J. Immunol. 2005;174:1348–1356. doi: 10.4049/jimmunol.174.3.1348. [DOI] [PubMed] [Google Scholar]
- 34.Shane Krummen Atwell A.C.V., Obungu V.H. BTLA agonist antibodies and uses thereof. patent US. 2020;10:604. patent application 15/977,003 Mar. 31. [Google Scholar]
- 35.Suzuki K., Tajima M., Tokumaru Y., Oshiro Y., Nagata S., Kamada H., Kihara M., Nakano K., Honjo T., Ohta A. Anti-PD-1 antibodies recognizing the membrane-proximal region are PD-1 agonists that can down-regulate inflammatory diseases. Sci. Immunol. 2023;8:eadd4947. doi: 10.1126/sciimmunol.add4947. [DOI] [PubMed] [Google Scholar]
- 36.Faubion W.A., Jr., Fletcher J.G., O'Byrne S., Feagan B.G., de Villiers W.J., Salzberg B., Plevy S., Proctor D.D., Valentine J.F., Higgins P.D., et al. EMerging BiomARKers in Inflammatory Bowel Disease (EMBARK) study identifies fecal calprotectin, serum MMP9, and serum IL-22 as a novel combination of biomarkers for Crohn's disease activity: role of cross-sectional imaging. Am. J. Gastroenterol. 2013;108:1891–1900. doi: 10.1038/ajg.2013.354. [DOI] [PubMed] [Google Scholar]
- 37.Dennis G., Jr., Holweg C.T.J., Kummerfeld S.K., Choy D.F., Setiadi A.F., Hackney J.A., Haverty P.M., Gilbert H., Lin W.Y., Diehl L., et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res. Ther. 2014;16:R90. doi: 10.1186/ar4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erben U., Loddenkemper C., Doerfel K., Spieckermann S., Haller D., Heimesaat M.M., Zeitz M., Siegmund B., Kühl A.A. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014;7:4557–4576. [PMC free article] [PubMed] [Google Scholar]
- 39.Patrick Caplazi L.D. Molecular Histopathology and Tissue Biomarkers in Drug and Diagnostic Development. Humana Press; 2014. Histopathology in Mouse Models of Rheumatoid Arthritis; pp. 65–78. [DOI] [Google Scholar]
- 40.De Giovanni M., Tam H., Valet C., Xu Y., Looney M.R., Cyster J.G. GPR35 promotes neutrophil recruitment in response to serotonin metabolite 5-HIAA. Cell. 2022;185:815–830.e19. doi: 10.1016/j.cell.2022.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai B., Hackney J.A., Ichikawa R., Nguyen A., Elstrott J., Orozco L.D., Sun K.H., Modrusan Z., Gogineni A., Scherl A., et al. Dual targeting of lymphocyte homing and retention through alpha4beta7 and alphaEbeta7 inhibition in inflammatory bowel disease. Cell Rep. Med. 2021;2:100381. doi: 10.1016/j.xcrm.2021.100381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and gene expression data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This work does not contain any custom software for the data analysis.