

Summary
EGFR-specific tyrosine kinase inhibitors (TKIs), especially osimertinib, have changed lung cancer therapy, but secondary mutations confer drug resistance. Because other EGFR mutations promote dimerization-independent active conformations but L858R strictly depends on receptor dimerization, we herein evaluate the therapeutic potential of dimerization-inhibitory monoclonal antibodies (mAbs), including cetuximab. This mAb reduces viability of cells expressing L858R-EGFR and blocks the FOXM1-aurora survival pathway, but other mutants show no responses. Unlike TKI-treated patient-derived xenografts, which relapse post osimertinib treatment, cetuximab completely prevents relapses of L858R+ tumors. We report that osimertinib’s inferiority associates with induction of mutagenic reactive oxygen species, whereas cetuximab’s superiority is due to downregulation of adaptive survival pathways (e.g., HER2) and avoidance of mutation-prone mechanisms that engage AXL, RAD18, and the proliferating cell nuclear antigen. These results identify L858R as a predictive biomarker, which may pave the way for relapse-free mAb monotherapy relevant to a large fraction of patients with lung cancer.
Keywords: adaptive mutability, EGFR, kinase inhibitors, lung cancer, monoclonal antibody, receptor tyrosine kinases, cetuximab, NSCLC, osimertinib
Graphical abstract
Highlights
-
•
EGFR+ lung tumors respond to kinase inhibitors but evolve new mutations and resistance
-
•
Unlike other EGFR mutations, L858R needs dimerization that is inhibitable by cetuximab
-
•
Cetuximab and kinase inhibitors inhibit L858R tumors; only cetuximab prevents relapses
-
•
When singly expressed in xenograft models, L858R-EGFR predicts response to cetuximab
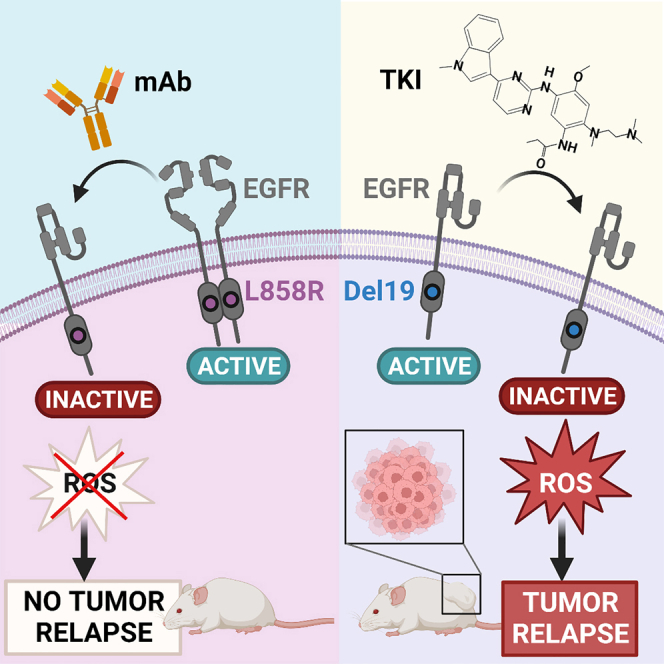
Marrocco et al. report that, among the most common EGFR mutations, the dimerization-dependent L858R can be inhibited by an antibody, such as cetuximab. Accordingly, xenograft models of lung cancer singly expressing L858R can be persistently inhibited by cetuximab, thus preventing tumor relapses, which often follow treatments with kinase inhibitors.
Introduction
Genome-based, personalized cancer medicine is rapidly becoming the standard of medical oncology1: the patient tumor’s genome is firstly characterized using state-of-the-art technologies. Later, the genomic data are filtered to identify driver aberrations and biomarkers and, finally, through a database of anticancer drugs the treating oncologist can select the most effective drugs. Treatment of non-small cell lung cancer (NSCLC) offers a suitable example: activating mutations in KRAS, BRAF, and the epidermal growth factor receptor (EGFR) gene, rearrangements of ALK, NTRK, RET, and ROS1, as well as amplifications of MET and ERBB2/HER2, are considered actionable candidates.2 Unfortunately, although many different EGFR mutations exist and several lines of evidence indicate biological differences among the mutations, currently all patients with EGFR+ lung tumors are treated in the same way. Moreover, despite high efficacy and acceptable safety of EGFR-specific tyrosine kinase inhibitors (TKIs), most TKI-treated NSCLC patients will eventually develop resistance due to new mutations or bypass pathways.3,4,5
Depending on ethnicity and gender, somatic EGFR mutations are present in 15%–40% of lung adenocarcinomas. The single point mutation, L858R, in exon 21, and variable length deletions in exon 19 (Del19) represent 85%–90% of all known EGFR mutations in lung cancer.6 These two highly common mutations confer sensitivity to EGFR-specific TKIs, but a third, less common mutation, T790M, typically emerges when patients are treated with TKIs since it confers resistance to the first- and second-generation drugs.3,7 Rare mutations account for 10%–15% of all EGFR mutations in NSCLC and include G719X (X denotes A, S, C, or other amino acids), Del18, E709K, exon 19 insertions (Ins19), S768I, L861Q, and exon 20 insertions (Ins20). The latter are resistant to clinically approved EGFR inhibitors due to unique mechanisms of kinase activation.8
Although Del19 includes more than 20 variants, all these mutations, along with L858R, have been classified into the “classical” (TKI sensitive) group. Interestingly, meta-analyses of multiple randomized trials that compared TKIs and chemotherapy confirmed the superiority of TKIs and, unexpectedly, revealed that the hazard ratio of progression-free survival for tumors with Del19 was 50% greater than the ratio calculated for tumors with L858R.9 Another interesting difference relates to the mechanism of kinase activation: while it is well known that wild-type EGFR requires dimerization for proper signaling,10,11 most kinase-activating mutations induce an active conformation of the enzyme that is independent of ligand-induced dimerization.12 For example, exon 19 deletions, exon 20 insertions, and the dual L858R/T790M EGFR mutant do not require dimerization. This contrasts with the L858R mutant, which depends on dimerization.13 Importantly, crystal structures of a complex comprising the extracellular region of EGFR and the antigen binding fragment of cetuximab, an anti-EGFR antibody that is clinically approved for patients with colorectal, head, and neck tumors, revealed that the antibody partially occludes the ligand binding region and sterically prevents the receptor from adopting the extended conformation required for dimerization.14
The reviewed observations predicted that cetuximab would inhibit tumors carrying the L858R mutation, but lung tumors expressing other EGFR mutants will show no responses. Reminiscent of this prediction, a second-generation TKI, afatinib, cannot overcome resistance when used alone, but a phase Ib trial of patients who acquired TKI resistance found that the combination of afatinib and cetuximab resulted in a response rate of 29%.15 Along this line, combining cetuximab and chemotherapy yielded statistically significant, albeit small effects on patients with lung cancer.16 In aggregate, these observations raise the possibility that L858R might serve as a biomarker able to predict responses to cetuximab. However, such responses are likely diluted because clinical trials do not regularly stratify patients on the basis of their L858 status. To examine this prediction, we employed patient-derived xenografts (PDXs) and other models. As predicted, cetuximab monotherapy completely inhibited relapses of L858R models, but tumors harboring other mutations rapidly relapsed post treatment with either cetuximab or TKIs. Further assays supported the in vivo observations: cetuximab and certain other anti-EGFR monoclonal antibodies (mAbs) inhibited growth of L858R-EGFR-expressing cells but Del19- and T790M-expressing cells were not affected by cetuximab. Interestingly, unlike TKIs, which elevated reactive oxygen species (ROS) and induced robust cell death, mAb treatments only moderately associated with apoptosis, but they accelerated the rate of EGFR degradation and downregulated multiple receptor tyrosine kinases (RTKs) that have previously been implicated in drug resistance. In addition, cetuximab uncoupled EGFR from a mutation-prone DNA replication pathway involving AXL, RAD18, and the mono-ubiquitinated form of PCNA. Taken together, our results warrant clinical tests aimed at genome-based immunotherapeutic treatments that minimize the use of TKIs and avoid engagement of mutagenic DNA replication.
Results
Certain anti-EGFR mAbs decrease viability of cells expressing L858R-EGFR by inhibiting the FOXM1-aurora kinase pathway, but cells expressing other EGFR mutants are unaffected
Because kinase-activating mutations other than L858R promote a dimerization-independent active conformation of EGFR’s kinase domain12 but the L858R mutant depends on receptor dimerization,13 we tested the effect of a dimerization-inhibitory antibody, cetuximab,14 on NSCLC cells expressing EGFR-L858R (Figures 1A and S1A). As predicted, a colorimetric test revealed that cetuximab inhibited two L858R-expressing cells (H3255 and 11-18), but it did not affect five other NSCLC cell lines: PC9, HCC4006, HCC2935 (EGFR-E746_A750), PC9ER (E746_A750 and T790M), and H1975 cells (L858R and T790M). Interestingly, HCC827 cells (Del19-EGFR) represent an exception17 that might be explained by receptor overexpression (Figure S1B) due to high gene copy number.18 As control, we used trastuzumab, an anti HER2 mAb, which was ineffective, but a combination of cetuximab and trastuzumab (2XmAbs) was inhibitory. Next, we counted surviving cells post treatment with increasing doses of cetuximab. This experiment confirmed that cetuximab inhibits L858R-expressing cells (Figure S1C). In addition, we asked if other anti-EGFR antibodies might inhibit growth of L858R+ cells. Three additional mAbs were tested: panitumumab, a clinically approved mAb that shares antigenic specificity with cetuximab, and two murine mAbs, 111 and 565. Previous analyses reported that mAb111 binds with EGFR without significantly inhibiting the binding of cetuximab, but mAb565 can compete with cetuximab for EGFR.19 In line with these observations, panitumumab and mAb565 inhibited growth of H3255 and 11-18 cells but mAb111 only weakly retarded growth of these cell lines (Figure 1B). As expected, neither antibody inhibited L858R-negative cells, with the exception of HCC827 cells that responded weakly to panitumumab (Figure S1D). Conceivably, mAb565 and panitumumab, like cetuximab, better block EGFR dimerization, hence can retard cell growth, but mAb111 cannot strongly impede growth because this antibody engages an EGFR site distinct from the dimerization site.
Figure 1.

An anti-EGFR antibody decreases viability of lung cancer cell lines expressing L858R-EGFR but it does not affect cells expressing the E746_A750 deletion mutant (Del19) or T790M-EGFR
(A) H3255 cells (2 × 104; L858R), along with 11-18 (5 × 103; L858R), PC9 (3 × 103; Del19), PC9ER (3 × 103; Del19 and T790M), and H1975 cells (5 × 103; L858R and T790M), were seeded in 96-well plates and treated for 72 h with cetuximab, trastuzumab (anti-HER2), or the combination of antibodies (2XmAbs) at 5, 10, or 20 μg/mL. Cell viability was assessed using the MTT assay. Data are presented as means ± SEM of three biological replicates.
(B) The indicated NSCLC cell lines (H3255, 2 × 104 cells; 11-18, 5 × 103; PC9 and PC9ER, each at 3 × 103) were seeded in 96-well plates and later treated for 72 h with the indicated anti-EGFR antibodies (each at 10 μg/mL). Cell viability was assessed using the MTT assay. Results are presented as means + SEM of three biological replicates. Significance was assessed using one-way ANOVA followed by Dunnett’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant.
(C) H3255, 11-18 and PC9 cells were treated for 48 h with cetuximab (Cetux., 10 μg/mL), trastuzumab (Trast., 10 μg/mL), or 2XmAbs (cetuximab + trastuzumab, each at 5 μg/mL). Protein extracts were resolved, blotted and probed with antibodies specific to the indicated apoptosis and cell-cycle markers, including an antibody specific to the cleaved form of caspase-3 (Cl.Casp.3). Vinculin and GAPDH were used as gel loading controls. See also Figures S1 and S2.
To uncover mechanisms that underlie the inhibitory effect of cetuximab, we applied immunoblotting on extracts derived from antibody-treated lung cancer cells. This experiment detected moderate upregulation of several markers of apoptosis in cells treated with either cetuximab alone or a mixture of cetuximab and trastuzumab (Figures 1C and S1E). Alongside, we observed downregulation of survivin, an anti-apoptosis protein, and reduced abundance of the intact form of caspase-3. Next, we referred to our previous study,20 in which we showed that prevention of tumor relapses may be achieved by inhibiting a well-characterized pathway comprising FOXM1, aurora kinase A (AURKA), and a few regulators of mitosis and cytokinesis.21 Along this line, we observed antibody-induced downregulation of FOXM1, AURKA, and survivin in both H3255 and 11-18 cells (Figure 1C), but L858R-negative cells displayed no consistent effects of cetuximab on AURKA, survivin, and FOXM1. However, treatment of HCC827 cells with cetuximab inhibited the FOXM1 pathway and upregulated BIM and pH2AX (also called γH2AX; Figure S1E). It is interesting to note that both AURKA22 and FOXM123 have previously been implicated in resistance to TKIs, implying that by inhibiting the FOXM1-AURKA survival pathway, cetuximab can inhibit NSCLC cells expressing L858R-EGFR. Altogether, our data confirmed that certain anti-EGFR antibodies can effectively inhibit L858R-EGFR-expressing cells, but they are not inhibitory toward all exon 19 deletion mutants of EGFR that were available to us, with the exception of HCC827 cells.
Cetuximab elicits nuclear translocation of FOXO3A in L858R-expressing cells, but osimertinib induces the translocation in a mutation type-independent manner
FOXM1 is suppressed mainly by another forkhead box transcription factor, FOXO3A, that can initiate apoptosis and cell-cycle arrest by activating BIM and p27Kip1.24 Because oxidative damage and other stressors stabilize and translocate FOXO3A into the nucleus, which permits suppression of FOXM1, we used immunofluorescence. As demonstrated in Figure S2 (upper part), under resting conditions FOXO3A was mostly cytoplasmic, but treatment with either cetuximab or osimertinib promoted its nuclear translocation. Importantly, analysis of cells that do not respond to cetuximab detected osimertinib-induced nuclear localization of the FOXO3A tumor suppressor, but cetuximab treatment failed translocating FOXO3A (lower part of Figure S2). Taken together, these observations indicated that treatment with cetuximab promotes nucleocytoplasmic transport of FOXO3A to inhibit FOXM1 and cell growth. However, this mechanism appears relevant only to H3255 (L858R-EGFR) cells, since it was not detectable in PC9 cells (Exon19Del-EGFR).
Treatment with either osimertinib or cetuximab reduces KI67 staining but the TKI initially decreases and later increases the S-phase fraction of L858R+ cells
Next, we performed colony formation assays, which employed the L858R-expressing H3255 and 11-18 cells. As expected, incubation with cetuximab partly inhibited the ability of H3255 (Figure S3A) and 11-18 cells (Figure 2A) to form colonies, but osimertinib almost completely prevented colony formation and cetuximab did not affect PC9 cells. Consistent with these observations, both L858R+ cell lines displayed lower KI67 staining, which marks proliferating cells, following treatment with cetuximab (Figure 2B), but the antibody did not change KI67 levels in PC9 cells (Figure S3B).
Figure 2.

Both cetuximab and osimertinib reduce proliferation and induce apoptosis of cells driven by L858R-EGFR, but only osimertinib inhibits cells expressing Del19-EGFR
(A) 11-18 (L858R-EGFR) and PC9 (Del19-EGFR) cells were seeded on six-well plates and on the next day they were treated for 48 h with cetuximab (Cetux., 10 μg/mL) or osimertinib (Osim., 50 nM for PC9 or 500 nM for 11-18 cells). Thereafter cells were treated with trypsin and counted. Five thousand (11-18) or 150 (PC9) cells were seeded in six-well plates to allow colony formation. Media (without drugs) were refreshed once every 3 days. After 14 days, cells were fixed and stained with crystal violet. For image quantification, five different fields were quantified per sample using ImageJ. Signals were normalized to the control wells. Values represent mean + SEM of three biological replicates. Significance was assessed using one-way ANOVA followed by Dunnett’s multiple comparison test. ∗∗∗∗p < 0.0001; ns, not significant.
(B) H3255 and 11-18 cells were seeded on coverslips and treated for 48 or 72 h, respectively, with cetuximab (Cetux., 10 μg/mL) or osimertinib (Osim., 50 nM for H3255 and 500 nM for 11-18 cells). Cells were fixed in paraformaldehyde (4%) and incubated with an anti-KI67 antibody, followed by an Alexa Fluor 555-conjugated secondary antibody. DAPI (blue) was used to stain nuclei. Images were captured using confocal microscopy (40× magnification). The number of KI67-positive cells was normalized to the total number of nuclei. The signals shown in the graph bars are relative to Control. Scale bars, 20 μm. Values represent mean + SEM of three biological replicates. Significance was assessed using one-way ANOVA followed by Dunnett’s multiple comparison test. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(C) H3255, 11-18 and PC9 cells were treated for 48 h with cetuximab (Cetux., 10 μg/mL), erlotinib or osimertinib (Erlot. or Osim., 500 nM for 11-18, or 50 nM for the other cell lines). Protein extracts were blotted and probed with specific antibodies. Vinculin was used as the gel loading control. See also Figure S3.
In contrast with the partial effects of cetuximab on L858R+ cell lines, osimertinib treatment almost completely eliminated KI67 expression in all cell lines. Congruent with the partial vs. nearly complete anti-proliferation effects of cetuximab and osimertinib, respectively, we observed similar effects when analyzing three markers of apoptosis: BIM, a predictor of clinical benefit from TKIs,25 cleaved caspase-3, and survivin (Figure 2C). Immunoblotting extracts prepared after treatment of the L858R+ cell lines with either osimertinib or erlotinib confirmed that the apoptosis markers were strongly elevated, whereas survivin was clearly downregulated (Figure 2C). Still, consistently weaker effects on caspase-3 cleavage were observed following treatments with cetuximab. Furthermore, in line with the other results presented in Figure 2, we observed no effects of cetuximab on PC9 cells (Figure 2C). In conclusion, both cetuximab and TKIs can inhibit proliferation of and colony formation by L858R-expressing cells by means of inducing apoptosis, but in general the effects of the TKIs were stronger and extended to cells expressing other mutant alleles of EGFR.
To unravel longer-term effects of cetuximab and osimertinib, we made use of Fucci, a fluorescent cell-cycle indicator.26 Lentiviruses were used to infect 11-18 cells that were treated for as long as 9 days. Next, we used high-content imaging to measure fluorescence intensity of green and red signals and determine the proliferating fraction of cells (S-phase), arrested cells (G1-phase), and other fractions (G2-M). Figure S3C presents the population averages obtained by using CellProfiler.27 Note that we stopped taking measurements once confluence was reached. As shown, osimertinib initially induced growth arrest (lower S-phase fraction and higher G1 fraction) of L858R+ cells, but this was followed by up- and downregulation of the S and G1 fractions, respectively. This pattern likely reflects emergence of osimertinib-tolerant cells, but no similar phenotype was induced by the antibody.
When tested on L858R+ cells, both cetuximab and kinase inhibitors downregulate compensatory RTKs and inactivate AKT and ERK
To effectively inhibit NSCLC harboring EGFR mutations, anti-EGFR drugs must block not only EGFR but also a set of compensatory RTKs that converge on activation of the AKT and ERK survival pathways. In addition to two family members of EGFR, HER2 and HER3,28,29 two other RTKs, MET30,31 and AXL,32 are able to mediate survival when EGFRs are blocked. Western blot analyses of cetuximab-treated H3255 cells (L858R+) confirmed reduced levels of the active forms of ERK and AKT (Figure 3A). Likewise, we observed simultaneous downregulation of EGFR, HER2, HER3, MET, and AXL. Importantly, treatment with either erlotinib (50 nM) or osimertinib (50 nM) yielded similar inhibitory effects. While analyses of the less-characterized L858R-EGFR+ cell line, 11-18, similarly confirmed drug-induced decreases in pERK and pAKT, we observed variable responses at the RTK level (Figure 3B). In contrast, our control cells, PC9 (E746_A750), did not respond to cetuximab, although they responded to the TKIs in terms of pEGFR, pAKT, pERK, and the compensatory RTKs (Figure 3C). In summary, congruent with the ability of both cetuximab and TKIs to inhibit L858R+ cells, these two different classes of drugs intercept in vitro the same survival pathways while inactivating RTK-mediated evasion routes.
Figure 3.

Unlike cells expressing other EGFR mutants, the FOXM1 pathway and compensatory RTKs are inhibited in L858R expressors following cetuximab treatment
(A–C) H3255 (L858R-EGFR), 11-18 (L858R-EGFR), and PC9 cells (Del19-EGFR) were treated for 24 h with cetuximab (10 μg/mL), or erlotinib or osimertinib (500 nM for 11-18; 50 nM for the other cell lines). Protein extracts were resolved and probed with antibodies specific to the indicated receptors and downstream pathways. GAPDH was used as the loading control.
(D–F) The indicated cell lines, including PC9ER (Del19 and T790M EGFR), were treated for 48 h with cetuximab (10 μg/mL), erlotinib, or osimertinib (each at 50 nM). Protein extracts were resolved, blotted, and probed with antibodies specific to components of the FOXM1 and other pathways. GAPDH and vinculin were used to ensure equal gel loading.
Cetuximab shares with TKIs the ability to intercept mitosis by inhibiting KIF4A and upregulating p27Kip1, but this occurs only in L858R-EGFR+ cells
FOXM1 and FOXO3 integrate AKT and ERK signals to regulate transcription of aurora kinases, cyclins, and kinesins such as KIF4A, which has been implicated in chromosome segregation33 and cell-cycle control.24 Accordingly, resistance to a wide variety of anti-cancer drugs is linked to deregulated FOXM1 and aurora signaling.24,34 To analyze the ability of cetuximab and TKIs to harness this pathway, we employed H3255 (L858R; sensitive to cetuximab, erlotinib, and osimertinib), PC9 (E746_A750; sensitive to erlotinib and osimertinib), and PC9ER cells (T790M and E746_A750; sensitive to osimertinib, see Figures 3D–3F). Along with downregulation of FOXM1 by all drugs, we observed decreased abundance of three of FOXM1’s targets, AURKA, cyclinB1, and KIF4A, in H3255 cells. Concurrently, the cyclin-dependent kinase inhibitor p27Kip1 was strongly induced, in line with a previous report.35 Curiously, we also observed drug-inducible putative cleavage of p27Kip1. Interestingly, loss of sensitivity to cetuximab associated with disappearance of the lower form (Figure 3E). Furthermore, this form characterized PC9ER cells treated with osimertinib, but it was lost when cells were treated with drugs to which they acquired resistance (Figure 3F). Importantly, PC9ER cells displayed no mAb- (or erlotinib)-induced downregulation of FOXM1 and its three direct targets, and the phosphorylated form of histone H2A (gamma H2A.X) was induced only by osimertinib. In conclusion, the data we presented reinforce the ability of cetuximab to partly mimic TKIs in terms of activating growth inhibitory pathways as long as L858R-EGFR exists in the absence of other EGFR mutations.
Production of ROS and emergence of drug-tolerant persister cells differentiate between treatments using osimertinib or cetuximab
Newly acquired resistance to TKIs is frequently due to the emergence of on-target secondary mutations, but resistance to mAbs often associates with non-mutational adaptations.36 Because endogenous production of ROS is considered a major driver of stress-induced mutations, we assayed hydrogen peroxide (H2O2) production in cells that were treated for 8 h with either cetuximab or osimertinib. The assay was based on a cell-permeant fluorogenic dye, 2′,7′-dichlorofluorescin diacetate (DCFDA), that measures hydroxyl, peroxyl, and other ROS activities. Following cellular uptake, DCFDA is deacetylated by cellular esterases to a non-fluorescent compound, which is oxidized by ROS into 2′,7′-dichlorofluorescein (DCF). While treatment with cetuximab did not detectably alter the basal level of ROS, we observed significantly elevated ROS levels following treatments of either L858R- (H3255 and 11-18) or Del19-expressing cells (PC9 and HCC827) with the TKI (Figures 4A and 4B). Next, we extended the analysis to the highly reactive superoxide anion (O2−), which was assayed using DHE (dihydroethidium). Similar to the results obtained with DCF, in TKI-treated cells we observed approximately 2- to 3-fold increases in the DHE signal (Figures S4A and S4B). As with DCFDA, cetuximab induced no detectable superoxide anions and the effect of osimertinib was independent from EGFR’s mutant alleles. Taken together, the absence of detectable mAb-induced effect on ROS production raised the possibility that, in contrast to TKIs, antibody treatments might not be followed by mutagenesis and drug tolerance.
Figure 4.

In contrast to treatments with TKIs, cetuximab induces no ROS production or emergence of persister cells
(A) PC9 (Del19-EGFR), H3255 (L858R-EGFR), HCC827 (Del19-EGFR), and 11-18 (L858R-EGFR) cells were treated for 8 h with cetuximab (Cetux., 10 μg/mL) or osimertinib (Osim., 500 nM for 11-18; 50 nM for the other cell lines). NAC (N-acetyl-L-cysteine; 10 mM) was used as a ROS scavenger. DCFDA (2ʹ,7ʹ-dichlorofluorescin diacetate) was employed for determining the intracellular content of hydrogen peroxide. Representative images of epifluorescence microscopy (original magnification, ×100) are shown. Scale bar, 200 μm
(B) Shown are quantifications of the hydrogen peroxide fluorescence signals from (A) (mean +SEM of three biological replicates). Significance was assessed using one-way ANOVA followed by Dunnett’s multiple comparison test. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(C) H3255, PC9, and HCC827 cells were seeded on six-well plates at high confluence and on the next day they were treated for 9 days with either cetuximab (Cetux., 10 μg/mL) or osimertinib (Osim., 300 nM). Media and drugs were refreshed once every 3 days. After 9 days, cells were fixed and stained with crystal violet. Images corresponding to five different fields were quantified using ImageJ. The experiment was repeated thrice. Representative images and the respective histograms are shown. Signals were normalized to the control. Values represent mean + SEM of three biological replicates. Significance was assessed using one-way ANOVA followed by Dunnett’s multiple comparison test. Scale bars, 200 μm. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant. See also Figure S4.
As a prelude to in vivo tests of this prediction, we employed a previously described drug tolerance assay.37 In analogy to drug-tolerant persister (DTP) bacteria,38 a small subpopulation of cancer cells demonstrates recurring tolerance, which might precede de novo mutagenesis.39,40,41 Consistent with such models, prolonged treatments with osimertinib almost completely eradicated all H3255, PC9, and HCC827 cells, but very small fractions survived the treatment (Figure 4C). In contrast, approximately 40% of H3255 cells survived similar treatments that made use of cetuximab. As expected, PC9 cells were not affected by the antibody. Interestingly, HCC827 cells displayed an intermediary response, probably due to high expression of EGFR. The observed different outcomes reinforced the possibility that DTP cells will repopulate TKI-treated tumors, but antibody treatment, which associates with no oxidative stress, might involve only limited relapses.
Cetuximab and osimertinib similarly inhibit L858R+ spheroids and xenografts but the effect of cetuximab on Del19-EGFR models is heterogeneous
To test the prediction that treatments with cetuximab might inhibit tumorigenic growth of H3255 cells, we firstly attempted xenograft studies. However, due to the very slow rate of in vitro growth of these cells, our experiments were underpowered. As an alternative, we established a 3D model (spheroids) of H3255 cells, an experimental strategy that phenocopies some in vivo effects of anti-cancer drugs.42 As shown in Figures S5A and S5B, when grown under low attachment conditions, H3255 cells developed well-rounded spheroids, but both cetuximab and osimertinib strongly inhibited spheroid formation. Because PC9 cells were unable to form spheroids, we assessed the growth of this cell line under low attachment conditions. As expected, osimertinib impaired formation of PC9’s irregular 3D structures, which were not affected by cetuximab (Figure S5C). In conclusion, in similarity to 2D monolayers, both cetuximab and osimertinib can inhibit 3D growth of H3255 cells (L858R+), but only osimertinib can inhibit 3D growth of PC9 cells (Del19+).
Next, we implanted 11-18 cells in mice and, after tumors became palpable, randomized all animals in groups that were either untreated or treated with cetuximab or osimertinib. The results presented in Figures 5A and S6A confirmed that cetuximab, like osimertinib, caused rapid shrinkage of all tumors, followed by prolonged inhibition of tumor regrowth post treatment ending. Note that neither drug significantly affected body weight (Figure S6B). These observations supported the in vitro data and proposed that cetuximab and osimertinib are equally effective when applied on L858R+ cells. The above-described animal studies were next replicated using two non-L858R animal models: PC9 and HCC827 (Figures 5B and 5C; see curves of survival and body weight in Figures S6A and S6B). Although treatments lasted only 3–5 weeks, tumor volumes were monitored for several additional weeks. As expected, the results obtained with the PC9 xenografts confirmed that cetuximab treatment falls short of preventing relapses in this model. Nevertheless, relative to osimertinib, cetuximab better delayed onset of resistance. Surprisingly, but in accordance with Figure S1A, HCC827 xenografts responded to both treatments (Figure S6A). This implies that the antibody is especially effective when EGFR is highly expressed due to gene amplification.18
Figure 5.

Cetuximab inhibits relapses of animal models carrying either L858R-EGFR (11-18) or an overexpressed Del19-EGFR (HCC827), but a model expressing normal levels of Del19-EGFR (PC9) does not respond to cetuximab
11-18 (A, 1 × 107 cells/mouse), PC9 (B, 3 × 106/mouse), or HCC827 cells (C, 4 × 106/mouse) were subcutaneously implanted in the flanks of CD1-nu/nu mice. When tumors became palpable, mice were randomized in groups of 5–7 animals (each) that were treated (gray areas) for 58 days (A) or 21 days (B and C) with cetuximab (0.2 mg/mouse/injection) once every 3 days, or daily with either osimertinib (10 mg/kg/day in A or 5 mg/kg in C) or erlotinib (50 mg/kg/day in B). Shown are tumor volumes of individual mice from each group. Animal numbers per group are indicated (N). See also Figures S5 and S6.
Patient-derived L858R+ tumors displayed no relapses post cetuximab treatment, but Del19+ tumors did not respond to the antibody
In the next step, we analyzed PDXs, which represent tumor heterogeneity better than other xenografts.43 Three models were selected: TM00199 and TM00253, both expressing L858R-EGFR, and TM00193, which expresses E746_A750 Del19-EGFR. Mice bearing pre-established tumors were randomized in three groups: untreated, cetuximab treated, and TKI treated. Note that we employed NSG mice, which have no active T cells, B cells, NK cells, and dendritic cells, and applied a drug holiday to examine early relapses. In addition, we kept following both body weight and tumor volume after ending all treatments. The individual tumor growth curves are presented in Figures 6A–6C, and graphs depicting animal survival and body weight are shown in Figures S7A and S7B. In sharp contrast to the relapses experienced by all TKI-treated mice and the cetuximab-treated Del19-EGFR group, no mAb-treated L858R+ mouse displayed post-treatment relapses, or lost body weight, although animals were followed for 49-120 additional days. Importantly, and in keeping with the other lines of evidence, relatively short mAb treatments were sufficient for complete eradication of the L858R+ models. In summary, despite their intrinsic heterogeneity, the PDX models indicated that cetuximab can rapidly inhibit pre-established tumors harboring L858R-EGFR, as well as durably block relapses. This contrasts with the uniform relapses we observed post TKI treatments, which underscores the superiority of cetuximab and the specificity of its inhibitory effect to the L858R mutation.
Figure 6.

Cetuximab treatment prevents relapses of two PDX models expressing L858R-EGFR but a third model expressing Del19-EGFR does not respond to the antibody
NSG mice were pre-implanted with tumor fragments derived from three different PDX models: TM00199 (PDX1, L858R-EGFR) (A), TM00253 (PDX2, L858R-EGFR) (B), and TM00193 (E746_A750 Del19-EGFR) (C). Once tumors reached approximately 500 mm3, mice were treated for 5 weeks (A and B) or 3 weeks (C), as indicated by the gray areas. Shown are tumor volumes corresponding to individual mice. The drugs were administered as follows: cetuximab (0.2 mg/intraperitoneal injection), twice a week, or TKIs, either erlotinib (50 mg/kg) or osimertinib (10 mg/kg), which were orally administered daily. Animal numbers per group are indicated (N). Note that each color represents one animal. See also Figure S7.
Molecular analyses of drug-treated PDXs unveils the molecular mechanisms that likely underlie cetuximab’s therapeutic superiority
To address mechanisms underlying cetuximab’s superiority in terms of preventing relapses, we repeated the TM00199 (L858R+) experiment with four animals per experimental arm. Mice were treated, or not, for only 7 days before tumor extraction. Figures 7A and 7B present the growth curves and relative tumor volumes at the time of excision. While they were shrinking under drugs, both cetuximab- and TKI-treated tumors exhibited nearly complete inhibition of EGFR phosphorylation (Figure 7C). However, in the cetuximab group this was due to mAb-induced EGFR degradation, whereas in the TKI group this was due primarily to kinase inhibition. Importantly, all five receptors previously implicated in resistance to TKIs, namely AXL, HER3, MET, HER2, and IGF1R, underwent more extensive downregulation when mice were treated with cetuximab. In line with this, cetuximab treatment associated with stronger inhibition of pERK. In addition, both drugs decreased survivin and increased BIM (Figure 7D), as well as downregulated FOXM1 and its transcriptional targets, AURKA, KIF4A, and cyclinB1 (Figure 7E).
Figure 7.

Immunoblot analyses of extracts derived from a PDX model (L858R-EGFR) uncover differences between the modes of action of cetuximab and osimertinib
(A and B) NSG mice were pre-implanted with tumor fragments derived from the PDX model TM00199 (L858R-EGFR). Once tumors reached approximately 500 mm3, mice were randomized into groups of four animals each, which were untreated (Control), or treated for 1 week only with either cetuximab (0.2 mg/injection, twice a week), or osimertinib (10 mg/kg, daily). Thereafter, all tumors were removed and their volumes were determined (A). Also shown are growth curves of individual tumors (B). The timing of tumor excisions is marked by red arrows.
(C–F) Cleared whole extracts of individual tumors were resolved and probed with the indicated antibodies, with the aim of evaluating signaling pathways (C), apoptosis markers (D), cell-cycle markers (E), and specific components of the DNA damage response (F). Vinculin and GAPDH were used as gel loading controls.
Of the RTKs that undergo compensatory upregulation, the relevance of AXL to drug resistance is well understood. AXL’s ligand, GAS6, serves as a sensor of cell death, and although we observed no effects on GAS6, in all cetuximab-treated mice we noted strong downregulation of RAD18, another AXL binder44 (Figure 7F). Notably, AXL activates RAD18,41 which enhances ubiquitination of proliferating cell nuclear antigen (PCNA) and consequent recruitment of mutagenic DNA polymerases to stalled replication forks.45 In accord, treatment of mice with cetuximab decreased both Ub1-PCNA and total PCNA, as well as downregulated the error-prone DNA polymerase kappa (Figure 7F). In addition, both cetuximab- and TKI-treated tumors displayed downregulation of several DNA repair proteins, including mismatch repair (MMR) enzymes. Analogously, it has been reported that a combination of cetuximab and a kinase inhibitor downregulated several MMR genes.40 Taken together, the results presented in Figure 7 propose that cetuximab might prevent tumor relapses due to its ability to inhibit the AXL-to-RAD18/PCNA mutagenic pathway.
In summary, our study identified L858R as a potential predictive biomarker and uncovered a clear advantage of cetuximab over TKIs in terms of preventing relapses of L858R-EGFR tumors. In addition, among all other cell lines tested, carrying EGFR mutations other than L858R, we confirmed that HCC827 cells (Del19-EGFR) respond to EGFR mAbs, but the mechanism underlying the sensitivity of this model remains unknown. Although cetuximab and TKIs share several functional features, as shown herein, they employ remarkably different mechanisms: accelerated EGFR downregulation, inhibition of receptor dimerization, and blocking compensatory pathways in the case of cetuximab, vs. kinase inhibition and increased apoptosis due to elevated ROS levels in the case of osimertinib. In addition, while both classes of drugs inhibit the FOXM1-AURKA axis, analysis of tumor extracts attributed to cetuximab a stronger downstream effect, at the level of pERK. It is also important to note that all six RTKs that are trans-regulated by the two drugs (i.e., EGFR, HER2, HER3, IGF1R, MET, and AXL) undergo stronger in vivo downregulation by cetuximab compared with osimertinib. These differences extend to RAD18, which interacts with AXL and coordinates mutagenic DNA polymerases. Thus, beyond the identification of L858R as a predictive biomarker, our study sheds light on the basic mechanisms of drug resistance.
Discussion
Identification of new biomarkers able to predict responses to molecular targeted drugs is the essence of contemporary cancer therapy research.46 By identifying L858R as a potential biomarker in NSCLC, the experiments we describe herein offer an alternative to the current standard of care of patients with lung cancer. Stated differently, treatment of L858R+ tumors with cetuximab, rather than osimertinib, might prevent or delay the relapses that characterize TKI-treated L858R+ patients. A recent meta-analysis of data from 21 clinical trials, including approximately 5,800 patients with lung cancer,47 seems to support our conclusion. According to the recent report, the best first-line therapeutic outcome for L858R-mutated NSCLC was achieved by a combination of a second-generation TKI, afatinib, plus cetuximab. However, this was not the case for patients with tumors harboring deletions in exon 19 of EGFR. In addition to the identification of L858R as a putative biomarker, our study invokes several interesting lessons in drug resistance. For instance, while acquisition of new on-target mutations frequently heralds patient resistance to anti-cancer TKIs (e.g., imatinib and osimertinib), acquired resistance to antibodies such as rituximab and cetuximab rarely involves emergence of new mutations.48,49 Below we focus on the medical and other implications of our results and deal with their molecular bases.
Because early studies reported that lung cancer tissue shows significantly increased EGFR staining compared with normal lung,50 and animal xenografts of lung and other cancer cell lines showed sensitivity to anti-EGFR antibodies,51 later studies tested cetuximab in lung and other cancer trials that applied mono- and combination therapies.52 These early efforts heralded the approval of cetuximab, in combination with either chemotherapy or radiotherapy, for colorectal cancer (CRC) and head/neck cancer, respectively.53,54 However, the lung cancer efforts have remained fruitless. Subsequent reports found that high expression of EGFR ligands, especially amphiregulin, might serve as a biomarker of response of CRC patients to antibody plus chemotherapy regimens.55 However, unlike CRC, in which such autocrine mechanisms expose vulnerability to anti-EGFR strategies, combining cetuximab and chemotherapy yielded relatively small, albeit consistent, effects on patients with lung cancer.16 Hence, activating EGFR mutations, which mimic autocrine mechanisms, have emerged as the most critical vulnerability of lung cancer. For example, applying a combination of a TKI and cetuximab on first-line patients, or patients who already acquired resistance to the first-generation TKIs, reported either no efficacy56 or weak but still significant clinical activity.15
Importantly, analysis of the latter clinical trial reveals differences between the mechanism we uncovered herein and the mode of action of the cetuximab + TKI combination.15 Firstly, while their results reported no efficacy differences between patients without and with the T790M mutation, our data predict that patients with mutations other than L858R would be resistant to the antibody. Secondly, all our L858R-EGFR animal models completely responded to cetuximab alone, such that the addition of a TKI might not improve efficacy. Practically, these differences translate to a requirement to perform ultra-deep DNA sequencing to ensure absence of T790M or any mutation other than L858R, in order for cetuximab monotherapy to be effective. As detailed below, we attribute the efficacy of cetuximab treatment to suppression of adaptive mechanisms and evasion of intrinsic mutagenic processes. Yet another mode of cetuximab action was revealed by a recent study that applied brigatinib. According to this report, the novel TKI collaborates with cetuximab because the antibody can decrease both surface and total EGFR expression.57 Thus, in addition to the ability of cetuximab to inhibit EGFR dimerization, this antibody might collaborate with TKIs in various different ways that have previously been investigated.28
The remarkably different effects of osimertinib and cetuximab monotherapies on relapses of L858R-EGFR tumors have not been reported before. Beyond the potential clinical implications, the ability of cetuximab to durably prevent relapses offers unexpected glimpses of the mechanisms underlying progression of tumors while they are being treated with a TKI or a mAb. The in vitro assays and animal comparative analyses portray the following scenarios: although both treatments can inhibit the FOXM1 pathway, cetuximab more strongly inhibited ERK. Likewise, mAb treatment more effectively reduced expression of all five well-characterized compensatory RTKs, while causing more extensive degradation of EGFR. Equally significant, unlike the robust induction of ROS and apoptosis by TKIs, cetuximab induced no ROS and only moderately elevated markers of apoptosis. The following lines of evidence suggest that these two highly different pharmacological scenarios are causally linked to the induction of relapses in the case of TKIs and to the avoidance of relapses in the case of cetuximab: (1) one of the five compensatory RTKs that were more strongly suppressed by cetuximab is AXL. When upregulated, AXL binds with RAD18 and increases two endogenous mutators, a previously described purine mutational bias58 and low-fidelity DNA polymerases.41 (2) According to another report, when treated with a combination of cetuximab and a BRAF kinase inhibitor, human CRC cells downregulate DNA repair genes and concomitantly upregulate error-prone DNA polymerases, a mechanism that increases adaptive mutability and evades therapeutic pressure.40 (3) According to a recent study, stress-induced mutagenesis (SIM) was identified in multiple models of human cancer.59 Furthermore, the cited study identified the mechanistic target of rapamycin as the molecule that mediates SIM.
In summary, the observations we made while contrasting the responses of different EGFR mutants to TKIs and mAbs have far-reaching clinical and mechanistic implications. The superiority of cetuximab monotherapy over treatments using TKIs might pave the way for relapse-free treatments of a large fraction of patients with EGFR+ lung cancer. Similarly important, our results provide unexpected insights into drug resistance. Specifically, many observations from various oncology indications have shown that emergence of secondary mutations characterizes acquired resistance to TKIs rather than to antibodies.48,49 Our observations might explain this duality: ROS generation and the associated extensive cell death, which typically accompany treatments with TKIs, inevitably evoke mutagenesis and drug resistance. In contrast, antibody treatments frequently involve milder stress and only limited apoptosis, which translates to evasion of adaptive mutability and no emergence of drug-resistant clones. As evidence accumulates in support of this concept, minimizing exposure of EGFR+ tumors to TKIs and application of mAb treatments might become major goals of future research.
Limitations of the study
Herein, we are reporting our efforts to approach pre-treatment selection of EGFR+ patients with NSCLC, based on the identity of their specific EGFR mutations. Our approach made use of in vitro and in vivo models (i.e., immunocompromised mice), rather than humanized animals or human patients. While we demonstrated antibody sensitivity of multiple cell lines and tumors expressing L858R-EGFR, as well as attributed the underlying mechanism to the dependency of L858R-EGFR on receptor dimerization, one model, HCC827 cells, stood out from all other Del19-EGFR models we tested. These cells displayed sensitivity to cetuximab, although Del19-EGFR is known to be dimerization independent. Presumably, the unexpected antibody sensitivity of HCC827 cells is related to the extremely high overexpression of EGFR in HCC827 cells. Notably, this knowledge gap must be filled in if the current results are going to be translated in the clinic. Nevertheless, if we are correct, the herein provided information offers a promising path forward for targeted therapy for subsets of EGFR mutated NSCLC.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EGFR, Rabbit monoclonal | Cell Signaling Technology | Cat#4267; RRID:AB_2246311 |
| pEGFR (Y1068), Rabbit polyclonal | Cell Signaling Technology | Cat#2234; RRID:AB_331701 |
| HER2, Rabbit monoclonal | Cell Signaling Technology | Cat#4290; RRID:AB_10557104 |
| pHER2 (Y1221/1222), Rabbit polyclonal | Cell Signaling Technology | Cat#2249; RRID:AB_2099241 |
| HER3, Rabbit monoclonal | Cell Signaling Technology | Cat#12708; RRID:AB_2721919 |
| pHER3(Y1289), Rabbit monoclonal | Cell Signaling Technology | Cat#4791; RRID:AB_2099709 |
| IGF1Rβ, Rabbit monoclonal | Cell Signaling Technology | Cat#9750; RRID:AB_10950969 |
| AXL, Rabbit monoclonal | Cell Signaling Technology | Cat#8661; RRID:AB_11217435 |
| MET, Rabbit monoclonal | Cell Signaling Technology | Cat#8198; RRID:AB_10858224 |
| ERK1/2, Rabbit monoclonal | Cell Signaling Technology | Cat#4695; RRID:AB_390779 |
| pERK1/2 (Y202/204), Rabbit polyclonal | Cell Signaling Technology | Cat#9101; RRID:AB_331646 |
| AKT1, Rabbit monoclonal | Cell Signaling Technology | Cat#2938; RRID:AB_915788 |
| pAKT (S473), Rabbit monoclonal | Cell Signaling Technology | Cat#4060; RRID:AB_2315049 |
| Caspase 3, Rabbit polyclonal | Cell Signaling Technology | Cat#9662; RRID:AB_331439 |
| Cleaved Caspase 3 (D175), Rabbit polyclonal | Cell Signaling Technology | Cat#9661; RRID:AB_2341188 |
| BIM, Rabbit monoclonal | Cell Signaling Technology | Cat#2933; RRID:AB_1030947 |
| Survivin, Rabbit polyclonal | Cell Signaling Technology | Cat#2803; RRID:AB_490807 |
| p27, Rabbit monoclonal | Cell Signaling Technology | Cat#3686; RRID:AB_2077850 |
| γH2AX (S139), Rabbit | Cell Signaling Technology | Cat#2577; RRID:AB_2118010 |
| AURKA, Rabbit monoclonal | Cell Signaling Technology | Cat#4718; RRID:AB_2061482 |
| FOXM1, Rabbit monoclonal | Cell Signaling Technology | Cat#5436; RRID:AB_10692483 |
| Cyclin B1, Rabbit monoclonal | Cell Signaling Technology | Cat#12231; RRID:AB_2783553 |
| RAD18, Rabbit monoclonal | Cell Signaling Technology | Cat#9040; RRID:AB_2756446 |
| GAS6, Rabbit monoclonal | Cell Signaling Technology | Cat#67202; RRID:AB_2799720 |
| RAD51, Rabbit monoclonal | Cell Signaling Technology | Cat#8875; RRID:AB_2721109 |
| Ub1-PCNA (K164), Rabbit monoclonal | Cell Signaling Technology | Cat#13439; RRID:AB_2798219 |
| PCNA, Mouse monoclonal | Cell Signaling Technology | Cat#2586; RRID:AB_2160343 |
| FOXO3A, Rabbit monoclonal | Cell Signaling Technology | Cat#2497; RRID:AB_836876 |
| KI67, Mouse monoclonal | Cell Signaling Technology | Cat#9449; RRID:AB_2797703 |
| β-Actin, Rabbit polyclonal | Cell Signaling Technology | Cat#4967; RRID:AB_330288 |
| GAPDH, Mouse monoclonal | Millipore | Cat#MAB374; RRID:AB_2107445 |
| Vinculin, Mouse monoclonal | Sigma-Aldrich | Cat#V9264; RRID:AB_10603627 |
| Pol η, Rabbit polyclonal | Abcam | Cat#ab234855 |
| Pol κ, Rabbit polyclonal | Abcam | Cat#ab86076; RRID:AB_1925334 |
| MLH1, Rabbit monoclonal | Abcam | Cat#ab92312; RRID:AB_2049968 |
| MSH2, Rabbit monoclonal | Abcam | Cat#ab92473; RRID:AB_10585291 |
| KIF4A, Rabbit polyclonal | Aviva Systems Biology | Cat#OAGA05174; RRID:AB_2861368 |
| BRCA1, Rabbit polyclonal | Santa Cruz Biotechnology | Cat#sc-642; RRID:AB_630944 |
| Anti-mouse Alexa Fluor 555-conjugated secondary antibody | Thermo Fisher Scientific | Cat#A-31570; RRID:AB_2536180 |
| Anti-rabbit Alexa Fluor 555-conjugated secondary antibody | Thermo Fisher Scientific | Cat#A-31572; RRID:AB_162543 |
| Biological samples | ||
| Patient derived xenograft (PDX) TM00199 | The Jackson Laboratory | TM00199 |
| Patient derived xenograft (PDX) TM00193 | The Jackson Laboratory | TM00193 |
| Patient derived xenograft (PDX) TM00253 | The Jackson Laboratory | TM00253 |
| Chemicals, peptides, and recombinant proteins | ||
| Erlotinib | LC Laboratories | Cat#E−4997 |
| Osimertinib | Gift from Astrazeneca | N/A |
| Cetuximab (Erbitux®) | Merck | N/A |
| Trastuzumab (Herceptin®) | Roche | N/A |
| Panitumumab (Vectibix®) | Amgen | N/A |
| mAb565 | Generated in our lab19 | N/A |
| mAb111 | Generated in our lab19 | N/A |
| NAC (N-acetyl-L-cysteine) | Sigma-Aldrich | Cat# A9165; CAS: 616-91-1 |
| DHE (Dihydroethidium) | Thermo Fisher Scientific | Cat# D11347 |
| DCFDA (2ʹ,7ʹ-Dichlorofluorescin Diacetate) | Thermo Fisher Scientific | Cat#D399 |
| Triton X-100 | Sigma-Aldrich | Cat#T8787; CAS: 9036-19-5 |
| MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) | Sigma-Aldrich | Cat#M5655; CAS: 298-93-1 |
| FITC-labeled phalloidin | Sigma-Aldrich | Cat#P5282 |
| Critical commercial assays | ||
| FuGENE HD Transfection Reagent | Promega | Cat#E2311 |
| Experimental models: Cell lines | ||
| H3255 | National Cancer Institute (NCI) | RRID:CVCL_6831 |
| 11–18 | Riken BRC | RRID:CVCL_6659 |
| PC9 | Gift from Julian Downward, Francis Crick Institute, London | RRID:CVCL_B260 |
| PC9ER | Gift from Julian Downward, Francis Crick Institute, London | N/A |
| HCC4006 | ATCC | Cat# CRL-2871; RRID:CVCL_1269 |
| HCC2935 | ATCC | Cat# CRL-2869; RRID:CVCL_1265 |
| HCC827 | Gift from Ravid Straussman, Weizmann Institute of Science, Israel | RRID:CVCL_2063 |
| H1975 | ATCC | Cat# CRL-5908; RRID:CVCL_1511 |
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| Experimental models: Organisms/strains | ||
| CD1 nude mice (HsdHli:CD1-Foxn1nu) | Envigo Israel | N/A |
| NSG mice | The Jackson Laboratory | RRID:IMSR_JAX:005557 |
| Recombinant DNA | ||
| Fucci(CA)2/pCSII-EF | RIKEN BRC | RDB15446 |
| psPAX2 | Addgene | Plasmid #12260 |
| pMD2.G | Addgene | Plasmid #12259 |
| Software and algorithms | ||
| GraphPad Prism v8.0.2 | GraphPad Software | RRID:SCR_002798 |
| ImageJ v1.53t | National Institute of Health (NIH) | RRID:SCR_003070 |
| Cell Profiler v4.2.1 | Broad Institute | RRID:SCR_007358 |
| Image Lab v6.0.1 | Bio-Rad | RRID:SCR_014210 |
| ReViSP v2.3 | Piccinini et al.60 Piccinini et al.61 |
N/A |
| IncuCyte S3 software | Sartorius | RRID:SCR_023147 |
| R v4.2.1 | The R Foundation | RRID:SCR_001905 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Yosef Yarden (yosef.yarden@weizmann.ac.il).
Materials availability
This study generated no unique reagents.
Experimental models and subjects details
Cell cultures
Human NSCLC (HCC4006, HCC2935, H1975) and HEK293T cells were obtained from the American Type Tissue Culture Collection (ATCC, Bethesda, MD), the National Institutes of Health (NCI; H3255 cells) and from Riken BioResource Research Center (Riken BRC; 11–18 cells, also called II-18). PC9/PC9ER and HCC827 were a gift from Julian Downward (Francis Crick Institute, London, UK) and Ravid Straussman (Weizmann Institute of science, Rehovot, Israel), respectively. Cells were maintained in RPMI-1640 or DME media (for HEK293T) containing 10% fetal bovine serum (FBS) and antibiotics. Cells were treated with the following drugs: erlotinib (LC Laboratories), osimertinib (a gift from AstraZeneca), cetuximab (Merck), trastuzumab (Roche), panitumumab (Amgen), mAb565 and mAb111 (generated in our lab19).
Animal studies
All animal studies were approved by our institutional board and they were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC). Mice were housed and handled in a pathogen-free, temperature-controlled (22°C ± 1°C) facility on a 12 h light/dark cycle. Animals were fed a regular chow diet (2018 Teklad Global 18% Protein Rodent Diet) and given ad libitum access to food and water. PC9 cells (3X106 cells per mouse), 11–18 cells (10X106 per mouse) or HCC827 cells (4X106 per mouse) were subcutaneously injected in the right flanks of 6-weeks old CD1 female nude mice (Envigo Israel). Once tumors reached a volume of approximately 500 mm3, mice were randomized into different groups and treated as indicated. Kinase inhibitors were daily administered using oral gavage. Antibodies were administered twice a week using intraperitoneal injection at a final dose of 0.2 mg/mouse/injection. The TM00193 (exon 19 deletion), TM00199 and TM00253 (L858R mutation) PDX models were purchased from the Jackson Laboratory and implanted in 8–10 weeks old male and female NSG mice (The Jackson Laboratory). Following euthanasia, tumors were removed from donor mice and cut into small fragments. A small pouch was made in the lower back of each mouse and one fragment was later inserted into the pouch. The wound was closed using a surgical clip. Clips were removed 4–5 days after surgery. Mice were labeled with RF identification chips (from Troven). Tumors were measured twice a week with a caliper, and body weight was measured once a week. Tumor volume was calculated using the formula 3.14Xshortest diameterX(longest diameter)2X1/6. Mice were euthanized when tumors reached approximately 1500 mm3 (PC9, 11–18 and HCC827 xenografts) or 1000 mm3 (PDX models).
Method details
Immunofluorescence analysis
Cells were grown on coverslips in 12-well plates. Following treatments, cells were fixed for 15 min at room temperature in paraformaldehyde (4%). This step was followed by extensive washes and permeabilization for 10 min in saline containing Triton X-100 (0.2%). Blocking was carried out for 60 min using bovine serum albumin (1%), followed by an overnight incubation at 4°C with a primary antibody (FOXO3A and KI67 antibodies). Thereafter, cells were washed, stained for 60 min in dark with an anti-mouse or anti-rabbit Alexa Fluor 555- conjugated secondary antibody, and with DAPI (without or with FITC-labeled phalloidin). Images were captured using a Zeiss Spinning disk confocal microscope and processed using the Zeiss ZEN 3.1 software.
Cell viability assays
Cell viability was assessed using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). PC9 (3 × 103 cells/well), 11–18 (5 × 103/well), H3255 (2 × 104/well), PC9ER (3 × 103 cells/well), H1975 cells (5 × 103/well), HCC827 (8X103 cells), HCC4006 (1X104) and HCC2935 (1.5X104) were seeded in 96-well plates. On the next day, cells were treated for 72 h with the indicated drugs. Afterward, cells were incubated for 3 h at 37°C with the MTT solution (0.5 mg/mL). The formazan crystals formed by metabolically active cells were dissolved in DMSO and the absorbance was determined at 570 nm.
Colony formation assays
Following 48-h long treatments, cells were seeded in 6-well plates at a density of 150–5000 cells per well, depending on the cell line, to permit colony formation. Media (without drugs) were refreshed once every 3 days. After 14 days, cells were fixed for 20 min in ice-cold methanol, followed by staining for 15 min at room temperature with 2% crystal violet. Full well photos were captured using an EPSON PERFECTION 4870 Photo Scanner (Long Beach, CA, USA). For signal quantification, images corresponding to 5 non-overlapping fields were taken with a light microscope (Olympus Corporation, Tokyo, Japan) and quantified using ImageJ.
Cell proliferation assays
Cells were seeded in 96-well plates at a density of 1000 cells per well, and on the following day they were treated for 1, 3 or 7 days with increasing concentrations of antibodies. At the indicated time points, cells were fixed in ice-cold methanol for 20 min at room temperature, followed by staining for 15 min (at room temperature) with 2% crystal violet. Cell growth was quantified by dissolving the cells in a detergent solution (2% SDS) and determining light absorbance (590 nm) using a microplate reader.
Cell cycle distribution using Fucci
The ubiquitination-based cell cycle indicator (Fucci),26 which enables real-time monitoring of interphase and cell-cycle progression, was employed using 11–18 cells. Lentiviruses were packaged by co-transfecting HEK293T cells with Fucci(CA)2/pCSII-EF (RIKEN BRC, Tsukuba, Japan), psPAX2 (Addgene, #12260) and pMD2.G (Addgene #12259), along with the Fugene HD reagent (from Promega). Fluorescent, virus transduced 11–18 cells were treated for 1–9 days in the absence or presence of drugs, which were refreshed once every three days. A high-content imaging system (CV8000, from Yokogawa) was used to capture cellular images. The fluorescence intensity of green and red signals was used to determine the proliferating fraction of cells (S-phase), arrested cells (G1-phase) and other fractions (G2-M). Analysis was performed using CellProfiler27 and the R software.
Spheroid assays
In Suspension: H3255 (3 × 104) or PC9 (1 × 104) cells were grown in suspension over a 0.6% agar layer diluted in full medium supplemented with the indicated drugs. Non-overlapping fields of each well were photographed after 8–10 days using the OpTech IB4 microscope. The volume of H3255 spheroids was calculated using the ReViSP software.60,61 Flat cell layers were analyzed for the percentage of covered area using the ImageJ software.
In Matrigel: 96-well plates were pre-coated with basement membrane extract (BME; 80%) and incubated for 30 min at 37°. Thereafter, H3255 (5 × 104) and PC9 (1 × 103) cells were layered on top of the gelled BME, and cultured in 5% of BME diluted in drug-containing medium. IncuCyte S3 Live-Cell was used to follow the multiple H3255 spheroids using time-lapse. Analyses were performed using the IncuCyte S3 Software (v2018C). PC9 spheroids were analyzed using ImageJ.
Immunoblotting analyses
Protein extracts were prepared either from cell lines or from tumors extracted from mice. Tumors collected from mice were smashed using the gentleMACS Dissociator in RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 1 mM EDTA, 1 mM Na3VO4 and a protease inhibitor cocktail). Cells were washed in saline and then extracted in RIPA buffer. Proteins were separated using gel electrophoresis and transferred to nitrocellulose membranes. After blocking, membranes were incubated overnight with the indicated primary antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (1 h), and treatment with Clarity Western ECL Blotting Substrates (Bio-Rad). ECL signals were detected using the ChemiDoc Imaging System (Bio-Rad) and images were acquired using the ImageLab Software. The antibodies used for immunoblotting were purchased from Cell Signaling (anti- EGFR, pEGFR Y1068, HER2, pHER2 (Y1221/1222), HER3, pHER3 Y1289, IGF1Rβ, AXL, MET, ERK1/2, pERK1/2, AKT1, pAKT S473, caspase3, cleaved caspase3, BIM, survivin, p27, γH2AX, AURKA, FOXM1, cyclin B1, RAD18, GAS6, RAD51, Ub1-PCNA, PCNA and β-actin). An anti-GAPDH antibody was obtained from Millipore and the anti-vinculin antibody (V9264) was obtained from Sigma. The following antibodies were purchased from Abcam: anti- Pol η (ab234855), Pol κ (ab86076), MLH 1 (ab92312) and MSH 2 (ab92473). A KIF4A antibody (#OAGA05174) was obtained from Aviva Systems Biology and the anti-BRCA1 (sc-642) antibody was from Santa Cruz Biotechnology.
ROS production assays
Cells were seeded in 6-well plates (70% confluency) and drugs were added and incubated for 8 h on the following day. Hydrogen peroxide and anion superoxide levels were respectively determined using2ʹ,7ʹ-dichlorofluorescin diacetate (DCFDA) or dihydroethidium (DHE) at a final concentration of 10 mM (diluted in Krebs-Ringer phosphate buffer). Cells were incubated for 30 min in the dark at 37°C and under 5% CO2. After 30 min, the cells were washed twice in fresh Krebs-Ringer phosphate buffer. Finally, the cellular fluorescence signal was recorded using epifluorescence microscopy (Olympus Corporation, Japan) at a wavelength of 500 nm (excitation), and 580 nm emission for DCFDA, or 495/525 nm for DHE. Signals were quantified using ImageJ.
Quantification and statistical analysis
The GraphPad Prism (version 8.0.2) software was used to perform statistical analyses. Sample numbers and other information (mean ± SEM or SD, number of replicates and specific statistical tests) are indicated in the respective figure legends. Differences were considered statistically significant if p < 0.05. The ImageJ, Cell Profiler, R, Image Lab, ReViSP and IncuCyte S3 software packages were used to perform data analysis.
Acknowledgments
We thank all members of our laboratories for their kind help and insightful comments. I.M. would like to thank the Lombroso Foundation for their fellowship. This work was performed in the Marvin Tanner Laboratory for Research on Cancer. Y.Y. is the incumbent of the Harold and Zelda Goldenberg Professorial Chair in Molecular Cell Biology. Our studies were supported by the Israel Science Foundation, European Research Council (ERC), the Israel Cancer Research Fund (ICRF), and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. In addition, this study was partially supported by the Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research; BINDS) from AMED under grant no. JP22ama121054. M. Lauriola was supported by MIUR PRIN2017, grant no. 2017TATYMP_002, and Fondazione del Monte 2021. The graphical abstract accompanying this manuscript was created with BioRender.com.
Author contributions
Conceptualization, I.M. and Y.Y.; methodology, I.M., Y.H., D.R., and R.O.; formal analysis, I.M., S.G., A.S.-N., N.G., Y.H., and D.R.; investigation, I.M., S.G., A.S.-N., N.G., A.R., Y.H., D.R., A.S., M.Z., R.O., M. Lindzen, and D.F.; resources, Y.T., M. Lauriola, and Y.Y.; writing – original draft, I.M. and Y.Y.; writing – review & editing, I.M. and Y.Y.; supervision, Y.T., M. Lauriola, L.T., and Y.Y.; project administration, I.M. and Y.Y.; funding acquisition, Y.Y.
Declaration of interests
The authors declare no competing interests. Yeda, the technology transfer arm of the Weizmann Institute, has filed a patent covering in part the findings reported in this article.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 8, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101142.
Supplemental information
Data and code availability
-
•
All data reported in this article will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to re-analyse the data reported in this paper is available from the lead contact upon request.
References
- 1.Garraway L.A., Verweij J., Ballman K.V. Precision oncology: an overview. J. Clin. Oncol. 2013;31:1803–1805. doi: 10.1200/JCO.2013.49.4799. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pao W., Miller V.A., Politi K.A., Riely G.J., Somwar R., Zakowski M.F., Kris M.G., Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi S., Boggon T.J., Dayaram T., Jänne P.A., Kocher O., Meyerson M., Johnson B.E., Eck M.J., Tenen D.G., Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 5.Oxnard G.R., Arcila M.E., Chmielecki J., Ladanyi M., Miller V.A., Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 2011;17:5530–5537. doi: 10.1158/1078-0432.Ccr-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gazdar A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28:S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sequist L.V., Waltman B.A., Dias-Santagata D., Digumarthy S., Turke A.B., Fidias P., Bergethon K., Shaw A.T., Gettinger S., Cosper A.K., et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vyse S., Huang P.H. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct. Target. Ther. 2019;4:5. doi: 10.1038/s41392-019-0038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee C.K., Wu Y.L., Ding P.N., Lord S.J., Inoue A., Zhou C., Mitsudomi T., Rosell R., Pavlakis N., Links M., et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J. Clin. Oncol. 2015;33:1958–1965. doi: 10.1200/jco.2014.58.1736. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X., Gureasko J., Shen K., Cole P.A., Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 11.Yarden Y., Schlessinger J. Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry. 1987;26:1434–1442. doi: 10.1021/bi00379a034. [DOI] [PubMed] [Google Scholar]
- 12.Papakyriakou A., Vourloumis D., Tzortzatou-Stathopoulou F., Karpusas M. Conformational dynamics of the EGFR kinase domain reveals structural features involved in activation. Proteins. 2009;76:375–386. doi: 10.1002/prot.22353. [DOI] [PubMed] [Google Scholar]
- 13.Cho J., Chen L., Sangji N., Okabe T., Yonesaka K., Francis J.M., Flavin R.J., Johnson W., Kwon J., Yu S., et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013;73:6770–6779. doi: 10.1158/0008-5472.CAN-13-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S., Schmitz K.R., Jeffrey P.D., Wiltzius J.J.W., Kussie P., Ferguson K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Janjigian Y.Y., Smit E.F., Groen H.J.M., Horn L., Gettinger S., Camidge D.R., Riely G.J., Wang B., Fu Y., Chand V.K., et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4:1036–1045. doi: 10.1158/2159-8290.Cd-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pujol J.L., Pirker R., Lynch T.J., Butts C.A., Rosell R., Shepherd F.A., Vansteenkiste J., O'Byrne K.J., de Blas B., Heighway J., et al. Meta-analysis of individual patient data from randomized trials of chemotherapy plus cetuximab as first-line treatment for advanced non-small cell lung cancer. Lung Cancer. 2014;83:211–218. doi: 10.1016/j.lungcan.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Mukohara T., Engelman J.A., Hanna N.H., Yeap B.Y., Kobayashi S., Lindeman N., Halmos B., Pearlberg J., Tsuchihashi Z., Cantley L.C., et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J. Natl. Cancer Inst. 2005;97:1185–1194. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 18.Amann J., Kalyankrishna S., Massion P.P., Ohm J.E., Girard L., Shigematsu H., Peyton M., Juroske D., Huang Y., Stuart Salmon J., et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–235. [PubMed] [Google Scholar]
- 19.Ferraro D.A., Gaborit N., Maron R., Cohen-Dvashi H., Porat Z., Pareja F., Lavi S., Lindzen M., Ben-Chetrit N., Sela M., Yarden Y. Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc. Natl. Acad. Sci. USA. 2013;110:1815–1820. doi: 10.1073/pnas.1220763110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marrocco I., Romaniello D., Vaknin I., Drago-Garcia D., Oren R., Uribe M.L., Belugali Nataraj N., Ghosh S., Eilam R., Salame T.M., et al. Upfront admixing antibodies and EGFR inhibitors preempts sequential treatments in lung cancer models. EMBO Mol. Med. 2021;13:e13144. doi: 10.15252/emmm.202013144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du R., Huang C., Liu K., Li X., Dong Z. Targeting AURKA in cancer: molecular mechanisms and opportunities for cancer therapy. Mol. Cancer. 2021;20:15. doi: 10.1186/s12943-020-01305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah K.N., Bhatt R., Rotow J., Rohrberg J., Olivas V., Wang V.E., Hemmati G., Martins M.M., Maynard A., Kuhn J., et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019;25:111–118. doi: 10.1038/s41591-018-0264-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y., Zhang W., Wen L., Yang H., Wen M., Yun Y., Zhao L., Zhu X., Tian L., Luo E., et al. FOXM1 confers resistance to gefitinib in lung adenocarcinoma via a MET/AKT-dependent positive feedback loop. Oncotarget. 2016;7:59245–59259. doi: 10.18632/oncotarget.11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao S., Fan L.Y.N., Lam E.W.F. The FOXO3-FOXM1 axis: a key cancer drug target and a modulator of cancer drug resistance. Semin. Cancer Biol. 2018;50:77–89. doi: 10.1016/j.semcancer.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faber A.C., Corcoran R.B., Ebi H., Sequist L.V., Waltman B.A., Chung E., Incio J., Digumarthy S.R., Pollack S.F., Song Y., et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1:352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakaue-Sawano A., Yo M., Komatsu N., Hiratsuka T., Kogure T., Hoshida T., Goshima N., Matsuda M., Miyoshi H., Miyawaki A. Genetically encoded tools for optical dissection of the mammalian cell cycle. Mol. Cell. 2017;68:626–640.e5. doi: 10.1016/j.molcel.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Stirling D.R., Swain-Bowden M.J., Lucas A.M., Carpenter A.E., Cimini B.A., Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics. 2021;22:433. doi: 10.1186/s12859-021-04344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romaniello D., Mazzeo L., Mancini M., Marrocco I., Noronha A., Kreitman M., Srivastava S., Ghosh S., Lindzen M., Salame T.M., et al. A combination of approved antibodies overcomes resistance of lung cancer to osimertinib by blocking bypass pathways. Clin. Cancer Res. 2018;24:5610–5621. doi: 10.1158/1078-0432.CCR-18-0450. [DOI] [PubMed] [Google Scholar]
- 29.Romaniello D., Marrocco I., Belugali Nataraj N., Ferrer I., Drago-Garcia D., Vaknin I., Oren R., Lindzen M., Ghosh S., Kreitman M., et al. Targeting HER3, a catalytically defective receptor tyrosine kinase, prevents resistance of lung cancer to a third-generation egfr kinase inhibitor. Cancers. 2020;12:2394. doi: 10.3390/cancers12092394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turke A.B., Zejnullahu K., Wu Y.-L., Song Y., Dias-Santagata D., Lifshits E., Toschi L., Rogers A., Mok T., Sequist L., et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelman J.A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J.O., Lindeman N., Gale C.-M., Zhao X., Christensen J., et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 32.Taniguchi H., Yamada T., Wang R., Tanimura K., Adachi Y., Nishiyama A., Tanimoto A., Takeuchi S., Araujo L.H., Boroni M., et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019;10:259. doi: 10.1038/s41467-018-08074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazumdar M., Sundareshan S., Misteli T. Human chromokinesin KIF4A functions in chromosome condensation and segregation. J. Cell Biol. 2004;166:613–620. doi: 10.1083/jcb.200401142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka K., Yu H.A., Yang S., Han S., Selcuklu S.D., Kim K., Ramani S., Ganesan Y.T., Moyer A., Sinha S., et al. Targeting Aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis. Cancer Cell. 2021;39:1245–1261.e6. doi: 10.1016/j.ccell.2021.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yabuuchi S., Katayose Y., Oda A., Mizuma M., Shirasou S., Sasaki T., Yamamoto K., Oikawa M., Rikiyama T., Onogawa T., et al. ZD1839 (IRESSA) stabilizes p27Kip1 and enhances radiosensitivity in cholangiocarcinoma cell lines. Anticancer Res. 2009;29:1169–1180. [PubMed] [Google Scholar]
- 36.Garraway L.A., Jänne P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- 37.Sharma S.V., Lee D.Y., Li B., Quinlan M.P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M.A., et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaldalu N., Hauryliuk V., Tenson T. Persisters-as elusive as ever. Appl. Microbiol. Biotechnol. 2016;100:6545–6553. doi: 10.1007/s00253-016-7648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Recasens A., Munoz L. Targeting cancer cell dormancy. Trends Pharmacol. Sci. 2019;40:128–141. doi: 10.1016/j.tips.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Russo M., Crisafulli G., Sogari A., Reilly N.M., Arena S., Lamba S., Bartolini A., Amodio V., Magrì A., Novara L., et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science. 2019;366:1473–1480. doi: 10.1126/science.aav4474. [DOI] [PubMed] [Google Scholar]
- 41.Noronha A., Belugali Nataraj N., Lee J.S., Zhitomirsky B., Oren Y., Oster S., Lindzen M., Mukherjee S., Will R., Ghosh S., et al. AXL and error-prone DNA replication confer drug resistance and offer strategies to treat EGFR-mutant lung cancer. Cancer Discov. 2022;12:2666–2683. doi: 10.1158/2159-8290.CD-22-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boucherit N., Gorvel L., Olive D. 3D tumor models and their use for the testing of immunotherapies. Front. Immunol. 2020;11:603640. doi: 10.3389/fimmu.2020.603640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siolas D., Hannon G.J. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao Y., Mutter-Rottmayer E., Greenwalt A.M., Goldfarb D., Yan F., Yang Y., Martinez-Chacin R.C., Pearce K.H., Tateishi S., Major M.B., Vaziri C. A neomorphic cancer cell-specific role of MAGE-A4 in trans-lesion synthesis. Nat. Commun. 2016;7:12105. doi: 10.1038/ncomms12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bienko M., Green C.M., Crosetto N., Rudolf F., Zapart G., Coull B., Kannouche P., Wider G., Peter M., Lehmann A.R., et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- 46.Tsimberidou A.M., Fountzilas E., Nikanjam M., Kurzrock R. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev. 2020;86:102019. doi: 10.1016/j.ctrv.2020.102019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen C., Zhang C., Lin H., Liu Q., Wu L., Zhou C., Zhang J. First-line therapeutic strategy for patients with advanced non-small cell lung cancer with Leu858Arg epidermal growth factor receptor mutations: a Bayesian network meta-analysis. Ther. Adv. Chronic Dis. 2022;13 doi: 10.1177/20406223221125706. 20406223221125706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konieczkowski D.J., Johannessen C.M., Garraway L.A. A convergence-based framework for cancer drug resistance. Cancer Cell. 2018;33:801–815. doi: 10.1016/j.ccell.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marrocco I., Romaniello D., Yarden Y. Cancer immunotherapy: the dawn of antibody cocktails. Methods Mol. Biol. 2019;1904:11–51. doi: 10.1007/978-1-4939-8958-4_2. [DOI] [PubMed] [Google Scholar]
- 50.Veale D., Ashcroft T., Marsh C., Gibson G.J., Harris A.L. Epidermal growth factor receptors in non-small cell lung cancer. Br. J. Cancer. 1987;55:513–516. doi: 10.1038/bjc.1987.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masui H., Kawamoto T., Sato J.D., Wolf B., Sato G., Mendelsohn J. Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 1984;44:1002–1007. [PubMed] [Google Scholar]
- 52.Baselga J., Pfister D., Cooper M.R., Cohen R., Burtness B., Bos M., D'Andrea G., Seidman A., Norton L., Gunnett K., et al. Phase I studies of anti-epidermal growth factor receptor chimeric antibody C225 alone and in combination with cisplatin. J. Clin. Oncol. 2000;18:904–914. doi: 10.1200/JCO.2000.18.4.904. [DOI] [PubMed] [Google Scholar]
- 53.Cunningham D., Humblet Y., Siena S., Khayat D., Bleiberg H., Santoro A., Bets D., Mueser M., Harstrick A., Verslype C., et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 54.Bonner J.A., Harari P.M., Giralt J., Azarnia N., Shin D.M., Cohen R.B., Jones C.U., Sur R., Raben D., Jassem J., et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 55.Stahler A., Stintzing S., Modest D.P., Ricard I., Giessen-Jung C., Kapaun C., Ivanova B., Kaiser F., Fischer von Weikersthal L., Moosmann N., et al. Amphiregulin expression is a predictive biomarker for EGFR inhibition in metastatic colorectal cancer: combined analysis of three randomized trials. Clin. Cancer Res. 2020;26:6559–6567. doi: 10.1158/1078-0432.CCR-20-2748. [DOI] [PubMed] [Google Scholar]
- 56.Goldberg S.B., Redman M.W., Lilenbaum R., Politi K., Stinchcombe T.E., Horn L., Chen E.H., Mashru S.H., Gettinger S.N., Melnick M.A., et al. Randomized trial of afatinib plus cetuximab versus afatinib alone for first-line treatment of EGFR-mutant non-small-cell lung cancer: final results from SWOG S1403. J. Clin. Oncol. 2020;38:4076–4085. doi: 10.1200/jco.20.01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uchibori K., Inase N., Araki M., Kamada M., Sato S., Okuno Y., Fujita N., Katayama R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017;8:14768. doi: 10.1038/ncomms14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keshet R., Lee J.S., Adler L., Iraqi M., Ariav Y., Lim L.Q.J., Lerner S., Rabinovich S., Oren R., Katzir R., et al. Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nat. Cancer. 2020;1:894–908. doi: 10.1038/s43018-020-0106-7. [DOI] [PubMed] [Google Scholar]
- 59.Cipponi A., Goode D.L., Bedo J., McCabe M.J., Pajic M., Croucher D.R., Rajal A.G., Junankar S.R., Saunders D.N., Lobachevsky P., et al. MTOR signaling orchestrates stress-induced mutagenesis, facilitating adaptive evolution in cancer. Science. 2020;368:1127–1131. doi: 10.1126/science.aau8768. [DOI] [PubMed] [Google Scholar]
- 60.Piccinini F., Tesei A., Arienti C., Bevilacqua A. Cancer multicellular spheroids: volume assessment from a single 2D projection. Comput. Methods Programs Biomed. 2015;118:95–106. doi: 10.1016/j.cmpb.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 61.Piccinini F., Tesei A., Bevilacqua A. Single-image based methods used for non-invasive volume estimation of cancer spheroids: a practical assessing approach based on entry-level equipment. Comput. Methods Programs Biomed. 2016;135:51–60. doi: 10.1016/j.cmpb.2016.07.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this article will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to re-analyse the data reported in this paper is available from the lead contact upon request.