

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic kidney disease and is characterized by the formation of renal cysts and the eventual development of end-stage kidney disease. One approach to treating ADPKD is through inhibition of the mammalian target of rapamycin (mTOR) pathway, which has been implicated in cell overproliferation, contributing to renal cyst expansion. However, mTOR inhibitors, including rapamycin, everolimus, and RapaLink-1, have off-target side effects including immunosuppression. Thus, we hypothesized that the encapsulation of mTOR inhibitors in drug delivery carriers that target the kidneys would provide a strategy that would enable therapeutic efficacy while minimizing off-target accumulation and associated toxicity. Toward eventual in vivo application, we synthesized cortical collecting duct (CCD) targeted peptide amphiphile micelle (PAM) nanoparticles and show high drug encapsulation efficiency (>92.6%). In vitro analysis indicated that drug encapsulation into PAMs enhanced the anti-proliferative effect of all three drugs in human CCD cells. Analysis of in vitro biomarkers of the mTOR pathway via western blotting confirmed that PAM encapsulation of mTOR inhibitors did not reduce their efficacy. These results indicate that PAM encapsulation is a promising way to deliver mTOR inhibitors to CCD cells and potentially treat ADPKD. Future studies will evaluate the therapeutic effect of PAM-drug formulations and ability to prevent off-target side effects associated with mTOR inhibitors in mouse models of ADPKD.
Keywords: Micelles, ADPKD, Targeted, Rapalogs, mTOR
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a chronic kidney condition that affects approximately 600,000 individuals in the United States, making it the most common inherited kidney disorder [1]. ADPKD is caused by mutations in the PKD1 or PKD2 genes, encoding the proteins polycystin 1 (PC1) and polycystin 2 (PC2) that are involved in the regulation of cell growth, cell movement, and cell-to-cell interactions [2]. ADPKD is characterized by renal cell overproliferation and cyst formation throughout the nephron, and the eventual decline in kidney function [3]. Approximately half of ADPKD patients progress to end-stage kidney disease (ESKD) by the time they reach their 50’s and will require dialysis or kidney transplantation [4]. To date, there is no cure for ADPKD, and the vasopressin V2-receptor antagonist tolvaptan is the only treatment that has been approved by the U.S. Food and Drug Administration (FDA) to slow disease progression. However, tolvaptan has modest therapeutic effects in delaying ESKD [5] and can cause undesirable reactions, including polyuria, excessive thirst, dizziness, and severe liver damage. Thus, finding alternative therapeutic options is crucial for the treatment of ADPKD.
A potential target for ADPKD treatment is the mammalian target of rapamycin (mTOR) pathway. mTOR pathway dysregulation is associated with diseases characterized by cell overproliferation, e.g., colorectal cancer [6], breast cancer [7], and renal cell carcinoma (RCC) [8]. In ADPKD, the mTOR pathway is activated by decreased functional expression of PC1 or PC22. Subsequent activation of mTOR complex 1 (mTORC1) leads to increased cell proliferation, which contributes to cyst formation [9].
Considering the role of mTOR activation in cell proliferation, multiple mTOR inhibitors have been investigated, including the hydrophobic small molecule drug rapamycin and its chemical analogs (rapalogs). Rapamycin inhibits cell proliferation by binding to the FK506 binding protein-12 (FKBP12) binding pocket and the FKBP–rapamycin binding (FRB) pocket on mTORC1 [10]. Everolimus, a rapalog that mimics the structure of rapamycin with the addition of a hydroxyethyl ester at the C40 position [11], was developed to combat the low oral bioavailability seen with rapamycin (16% for everolimus vs. 10% for rapamycin in animal models) [12]. Both drugs are already FDA approved: rapamycin for induction of immunosuppression in renal transplant patients and to treat lymphangioleiomyomatosis [13], and everolimus to treat advanced RCC, renal angiomyolipoma, and tuberous sclerosis complex (TSC), and have undergone clinical trials to treat ADPKD. However, a phase III trial in 2014 evaluating rapamycin treatment for ADPKD (NCT00346918) failed due to off-target side effects, such as immunosuppression. Similarly, everolimus underwent phase IV clinical trials in 2010 to treat ADPKD (NCT00414440). Although everolimus was shown to slow the rate of renal cyst growth, it did not reduce the progression of renal impairment, potentially due to insufficient accumulation in the kidneys [14]. Furthermore, many patients experienced adverse, off-target side effects, including gastrointestinal disorders (e.g., stomach pain, development of stomach ulcers) and blood disorders (e.g., anemia) [14].
Targeted drug delivery to the kidneys using nanoparticles may combat these disadvantages by facilitating specific uptake to diseased cells and limiting off-target accumulation and associated adverse effects [15]. Our lab has previously developed peptide amphiphile micelles (PAMs) [16] that incorporate kidney targeting peptides [17] and showed preferential accumulation in the kidneys [18]. PAMs consist of peptide amphiphiles capable of self-assembling into micelle nanoparticles with a hydrophobic core, suitable for encapsulating hydrophobic drugs such as rapamycin and rapalogs [19].
In this study, we developed PAMs using the CKDSPKSSKSIRFIPVST (CKD) peptide as our targeting moiety for the first time, which was previously reported to accumulate in the cortical collecting duct (CCD) of the kidneys, a region of therapeutic relevance as cyst formation is most prevalent in the CCD in ADPKD [20]. We encapsulated rapamycin, everolimus, or RapaLink-1, which is rapamycin chemically linked with MLN0128 [21] that can simultaneously bind to and inhibit both the FKBP12 region (via rapamycin) and the TOR kinase inhibitor (TORki)-binding region (via MLN0128) of mTORC1 [22]. We characterized the nanoparticle-drug formulations and evaluated their ability to inhibit cell proliferation in vitro in human CCD cells compared to free drug. Furthermore, we assessed their effect on markers of mTOR activation. This work aims to address the unmet clinical need for ADPKD treatment and provides insight for the delivery of drugs for kidney disease.
2. Methods
2.1. Nanoparticle formulation
2.1.1. Kidney-targeting peptide synthesis and purification
The CCD-targeting peptide, CKDSPKSSKSIRFIPVST (CKD), was synthesized using standard Fmoc-mediated solid phase peptide synthesis with rink amide resin (Gyros Protein Technologies, SE) using an automatic peptide synthesizer (PS3, Protein Technologies, Tucson, AZ) [18]. Peptides were cleaved from the rink amide resin using a 94:2.5:2.5:1 by volume solution of trifluoroacetic acid, Milli-Q water, triisopropylsilane, and ethanediol, and then precipitated and washed twice in ice-cold diethyl ether, dissolved in Milli-Q water, and lyophilized. The resultant powder was stored at − 20 °C until further use.
Crude peptide was filtered using a 0.22 μm PES syringe filter and further purified with reverse-phase high performance liquid chromatography (HPLC) on a C18 column at 55 °C using 0.1% formic acid in acetonitrile and water mixtures. The molecular weight of the purified peptide was confirmed using matrix-assisted laser desorption ionization time-of-flight mass spectral analysis (MALDI-TOF) or electrospray ionization (ESI). The fraction containing the peptide of interest was lyophilized and stored at −20 °C until further use.
2.1.2. Amphiphile synthesis, purification, and characterization
Amphiphiles were produced by carrying out thioester conjugation between the CKD peptide and DSPE-PEG(2000)-maleimide (Avanti Polar Lipids, AL, USA). Equimolar amounts of the lipid and pure peptide were mixed in Milli-Q water (pH 7.2) and gently agitated for at least 24 h at room temperature (RT). The conjugate mixture was filtered with a 0.22 μm PES filter and purified using HPLC on a C4 column and characterized using MALDI-TOF as described above.
2.1.3. Peptide Amphiphile Micelle (PAM) formation
PAMs were prepared by thin lipid film hydration. Non-targeting micelles (NTM) consisted of DSPE-PEG(2000)-methoxy (Avanti Polar Lipids). CCD-targeting micelles were synthesized using a 45:55 molar ratio of DSPE-PEG(2000)-CKD:DSPE-PEG(2000)-methoxy. The appropriate ratios of lipids and lipid-peptide conjugates were dissolved in methanol and evaporated under a steady flow of nitrogen to form a thin film. The film was further dried under vacuum overnight at RT and hydrated in syringe-filtered Milli-Q water or phosphate buffered saline (PBS) to form PAMs. After sonication for 10 min, PAMs were incubated at 80 °C for 30 min. For encapsulation studies, mTOR inhibitors rapamycin, everolimus, or RapaLink-1 were added to the methanol solution before thin film formation. Different molar ratios of lipid to drug (20:1, 10:1, 5:1, and 2:1) were assessed to determine the lipid-to-drug ratio that resulted in the highest drug loading and encapsulation efficiency.
2.2. PAM characterization
2.2.1. Hydrodynamic diameter, zeta potential, and morphology of PAMs
The hydrodynamic diameter in Milli-Q water and zeta potential in 1 mM NaCl of PAMs (100 μM) were measured using Zetasizer Ultra (Malvern Instruments, Malvern, UK). Size and morphology were further confirmed by transmission electron microscopy (TEM). PAMs (7 μL in Milli-Q water) were loaded onto 400 mesh lacey carbon grids (Ted Pella, Redding, CA, USA) for 5 min. Excess moisture was wicked away with KimWipes, then the grids were incubated with 7 μL of 2% wt. uranyl acetate solution at RT in the dark for 8 min. Excess moisture was wicked away once more, and the grids were left to dry for at least 1 h. Then, samples were imaged on a JEOL JEM-2100F TEM (JEOL, Ltd., Tokyo, Japan).
2.2.2. Calculation of drug loading and encapsulation efficiency
Following hydration, PAMs were syringe filtered using a 0.22 μm PES filter in order to remove excess drug that had not been encapsulated within the PAMs. Then, PAMs were disassembled using a 10:1 volume ratio of dimethyl sulfoxide (DMSO), releasing the encapsulated drug into solution. After agitation for 30 s and sonication for 5 min at RT, the absorbance of the solution was read at 278 nm using a plate reader. A standard curve was created using the absorbance values of different concentrations of free drug (25, 50, 100, and 150 μg/mL), and encapsulation efficiency (EE) and drug loading were calculated using the following equations:
The EE and drug loading was calculated for non-targeted PAMs at 4 lipid-to-drug ratios (2:1, 5:1, 10:1, and 20:1) for rapamycin (Santa Cruz Biotechnology, Dallas TX, USA), everolimus (Sigma Aldrich, St. Louis, MO, USA), and RapaLink-1 (MedChemExpress, Monmouth Junction, NJ, USA). The highest ratio of lipid-to-drug in terms of EE was used for further studies using CCD-targeted PAMs (10:1 for both rapamycin and everolimus and 20:1 for RapaLink-1).
2.3. Comparison of anti-proliferative effect of free vs. encapsulated drugs in vitro
Cell proliferation was evaluated using an MTS cell proliferation colorimetric assay according to the manufacturer’s specifications (BioVision Incorporated, San Francisco, CA, USA). Human CCD (hCCD) cells were gifted by Remi Piedagnel (INSERM, Paris, France). hCCD cells were grown in medium consisting of a 1:1 ratio of DMEM:Ham’s F-12, 5 μg/ml of transferrin, 30 nM of sodium selenite, 2 mM of glutamine, 50 nM of dexamethasone, 5 μg/ml of insulin, 20 mM HEPES (pH 7.4), and 2% FBS. Medium for all cells was changed every 2–3 days and cells were maintained at 37 °C and 5% CO2. For MTS assay, cells (5 × 103/well) were seeded in a 96-well plate and grown for 72 h. Cell medium was then removed and 50, 100, or 200 μM of drug-PAMs or the equivalent amount of free drug suspended in 100 μL cell medium was added to the cells and incubated for 24 or 72 h before conducting MTS assays. Empty CCD-targeting PAMs were used as a control. Cell proliferation was calculated as a percentage of the proliferation measured in cells incubated with cell medium only.
2.4. Electrophoresis and immunoblotting analysis
Human CCD cells were cultured as described in Section 2.3. After treatment, cells were washed with ice-cold PBS and lysed in lysis buffer containing 20 mM Tris– HCl pH 7.4, 50 mM NaCl, 50 mM NaF, 5 mM Na pyrophosphate, 250 mM sucrose, 1% Triton X-100, 1 mM DTT, 1 mM PMSF, and complete protease inhibitor cocktail (Roche, Basel, Switzerland). After incubation on ice for 15 min, lysate was centrifuged at 16,000 × g for 15 min at 4 °C, and the protein concentration in the supernatants was measured using the Bradford method (Bio-Rad Laboratories, Hercules, CA, USA). Samples were separated by 4–12% gradient Gel (Nu-PAGE, Invitrogen Waltham, MA, USA) and electrically transferred to a nitrocellulose membrane. The membrane was stained with Revert 700 total protein stain solution for 5 min, then the total protein stain solution was decanted thoroughly and rinsed using Revert 700 wash solution two times. Immediately, membranes were imaged (700 nm channel) using an Odyssey® Fc Imaging System (LI-COR Biosciences, Lincoln, NE, USA) for total protein quantification. After destaining with Revert destaining solution, the membrane was blocked in LI-COR Odyssey Blocking Buffer for 1 h and then incubated in primary antibody overnight at 4° C with gentle shaking. After one wash in TBS-0.1% Tween 20, the membrane was incubated for 1 h with secondary antibody in blocking buffer with 0.2% Tween 20. After one wash in TBS-0.1% Tween 20, proteins were detected by an Odyssey® Fc Imaging System. Membranes probed with another primary antibody were stripped by incubation in NewBlot™ IR stripping solution for 15 min. Quantification of Western blots was performed by densitometry with analysis using Image Studio Lite Ver 5.2 software. A full list of antibodies used is provided in Table S1.
2.5. Statistical analysis
All statistical analyses were performed using GraphPad Prism 9 (San Diego, CA, USA). Two-tailed Student’s t-test was used to compare means of two groups. Analysis of variance (ANOVA) with a Tukey’s test for post-hoc analysis was used to determine statistical significance between 3 or more groups. p < 0.05 was considered statistically significant.
3. Results and discussion
3.1. Peptide and amphiphile synthesis
CCD-targeting peptides were synthesized, purified using HPLC, and characterized by MALDI-TOF spectrophotometry (Fig. S1). DSPE-PEG(2000)-peptide amphiphiles were also purified by HPLC and ESI was used to confirm successful conjugation (Fig. S2). A yield of 38.0 ± 3.4% of DSPE-PEG(2000)-peptide amphiphiles was obtained after HPLC and purified conjugates were used to form nanoparticles in this study..
3.2. PAM characterization
Initially, non-targeted PAMs (100 μM) were used to determine the optimal PAM:drug molar ratio in terms of maximum encapsulation efficiency (EE) and drug loading (Fig. 1). Rapamycin and everolimus showed similar results, likely due to their similar structure and molecular weight (Fig. S3). The highest drug loading for rapamycin (23.3%wt/wt) and everolimus (26.0%wt/wt) was found at the lipid:drug ratio of 5:1. However, since the EE was not significantly different at the 10:1 ratio, future experiments used the lowest effective dose (10:1 ratio) for rapamycin and everolimus. RapaLink-1 had equal drug loading (13–15%wt/wt) at the 5:1, 10:1, and 20:1 ratios, but EE increased with increasing PAM:drug ratio. Thus, the 20:1 ratio was chosen for further studies.
Fig. 1.
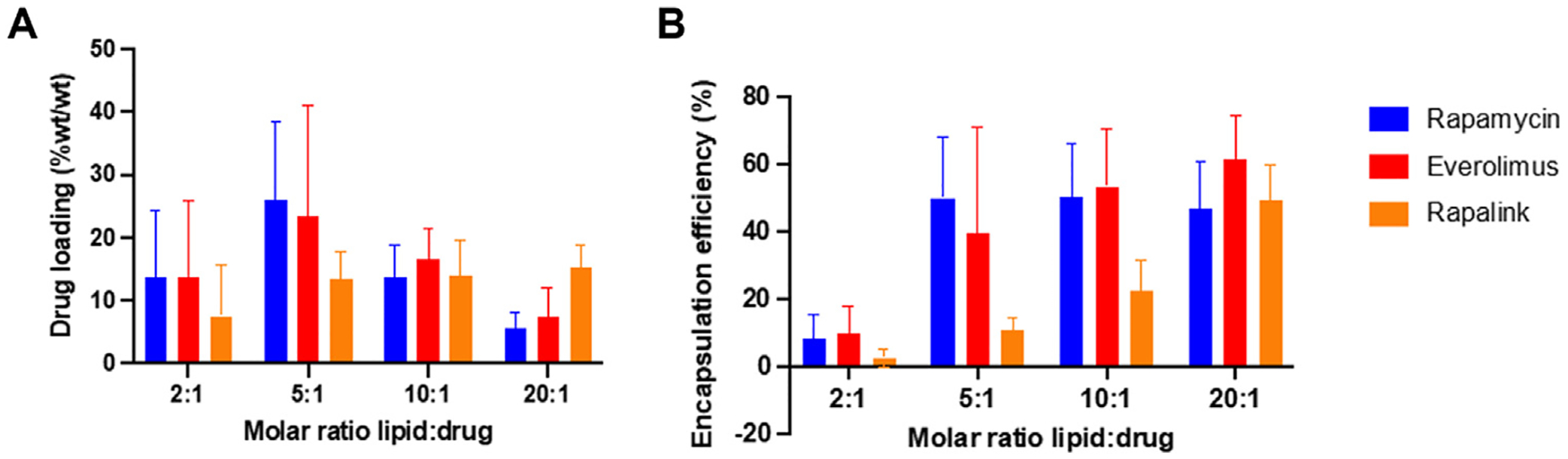
(A) Drug loading (%wt/wt) and (B) encapsulation efficiency (EE,%) of rapamycin, everolimus and RapaLink-1 in micelles. Varying molar ratios of lipid: drug were used (2:1, 5:1, 10:1, 20:1, n = 3).
CCD-targeted PAMs were used for all further studies and were characterized following encapsulation of all three mTOR inhibitors (Table 1). Size and surface charge of drug-PAMs was determined, as well as the EE and drug loading (Table 1). All PAMs were < 15 nm, and TEM analysis indicated that PAMs were spherical and monodisperse (Fig. 2). Zeta potential for PAMs was negative and similar to previously published results [18]. PAM characteristics showed slight changes upon drug encapsulation, including decreased size and increased zeta potential, similar to previous reports with rapamycin encapsulation in hyaluronan-streptomycin micelles [23]. EE was high for all mTOR inhibitors, likely due to their hydrophobic nature which facilitates effective encapsulation within the water-protected hydrophobic core of nanoparticles, as previously shown in hyaluronan-streptomycin micelles [23], PLGA-carbopol nanoparticles [24], and polysulfide micelles [25].
Table 1.
Characterization of drug-loaded, CCD-targeted PAMs.
| PAM:drug molar ratio | Encapsulation efficiency (%) | Loading capacity (%wt/wt) | Hydrodynamic diameter (nm) | Zeta potential (mV) | |
|---|---|---|---|---|---|
| Empty | – | – | – | 13.5 ± 4.6 | −17.5 ± 0.6 |
| Rapamycin | 10:1 | 105.2 ± 24.3 | 2.5 ± 0.6 | 12.4 ± 1.6 | −11.9 ± 0.6 |
| Everolimus | 10:1 | 92.6 ± 20.9 | 2.3 ± 0.5 | 11.7 ± 1.2 | −11.4 ± 2.3 |
| RapaLink-1 | 20:1 | 113.1 ± 6.4 | 2.8 ± 0.2 | 10.5 ± 0.9 | −10.8 ± 3.7 |
Values are the mean of 3 measurements ± SD.
Fig. 2.

Representative TEM images show empty and drug loaded-PAMs were spherical and monodisperse.
3.3. Inhibition of cell proliferation in vitro
Rapamycin, everolimus, and RapaLink-1 have been reported to inhibit cell proliferation [26] and the impact of encapsulating these drugs into CCD targeted micelles on their anti-proliferative efficacy was assessed via MTS assay. hCCD cells were incubated with free or drug-PAMs, or with control empty PAMs, for 24 h (Fig. 3) and 72 h (Fig. 4). Low (50 μM), intermediate (100 μM), and high (200 μM) doses of PAMs were used and concentrations of free drug matched the amount of drug that was encapsulated in PAMs at these doses.
Fig. 3.
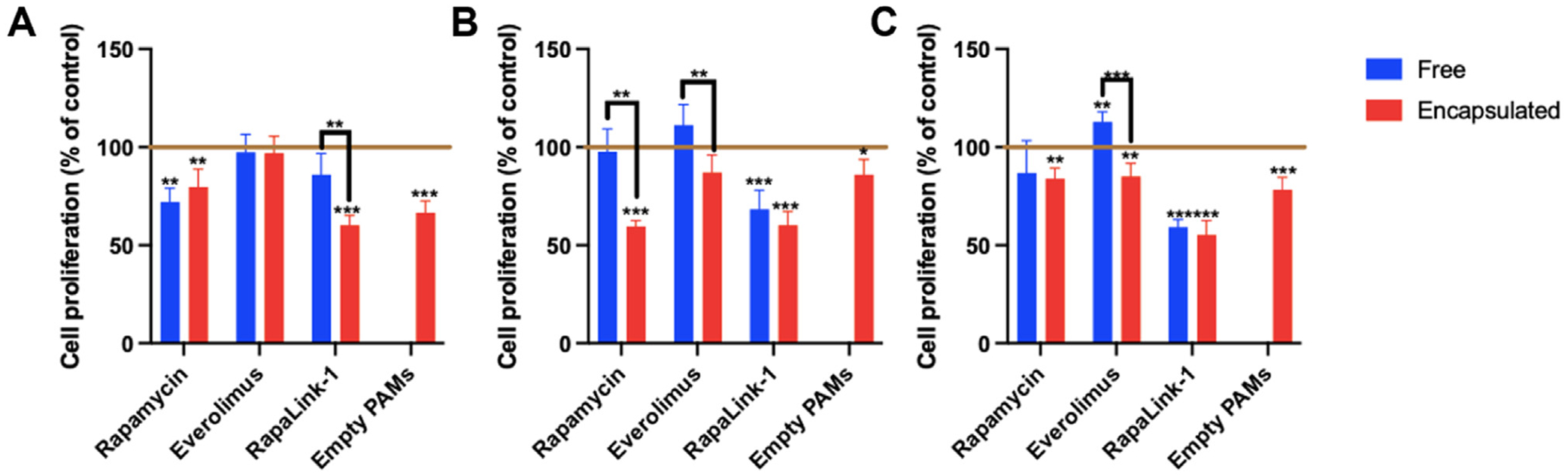
hCCD cell proliferation after 24 h incubation with mTOR inhibitors, either free or encapsulated in PAMs at (A) 50 μM, (B) 100 μM, or (C) 200 μM, as assessed by MTS assay (n = 6, * p < 0.05, ** p < 0.01, *** p < 0.001, comparing values to control with cell medium alone, indicated by horizontal line).
Fig. 4.
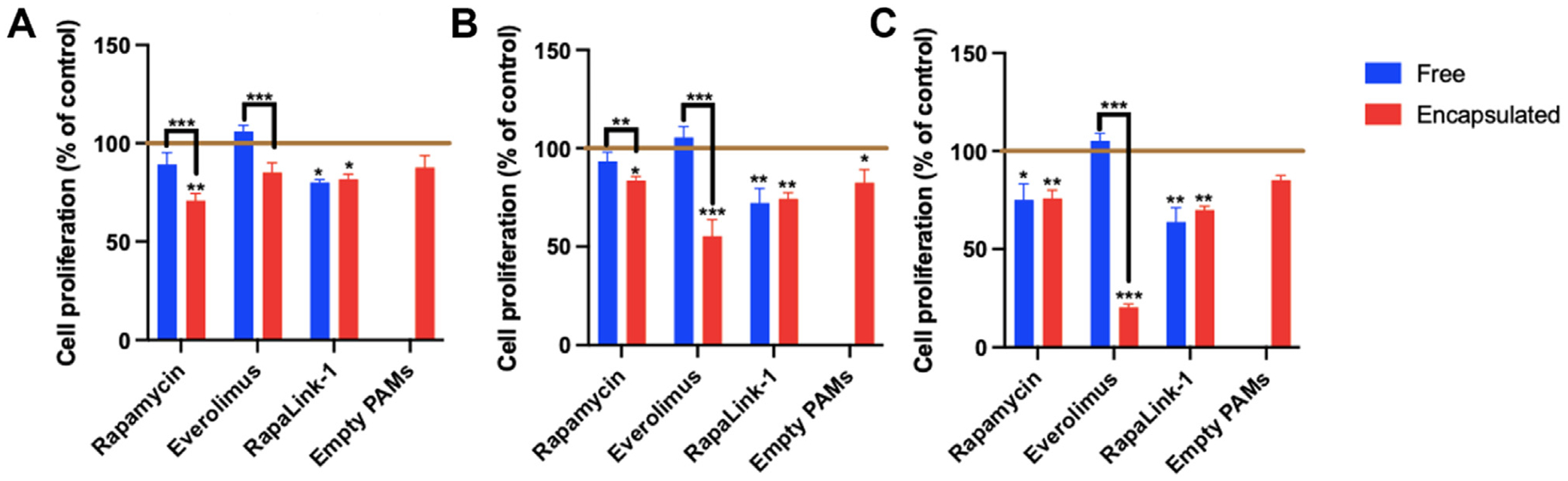
hCCD cell proliferation after 72 h incubation with mTOR inhibitors, either free or encapsulated in PAMs at (A) 50 μM, (B) 100 μM, or (C) 200 μM, as assessed by MTS assay (n = 6, * p < 0.05, ** p < 0.01, *** p < 0.001, comparing values to control with cell medium alone, indicated by horizontal line).
After 24 h of incubation, Fig. 3 shows that encapsulation did not affect the anti-proliferative effect of low dose rapamycin. Everolimus did not have any significant effect on cell proliferation at low dose when administered free or encapsulated. However, the anti-proliferative effect of low dose RapaLink-1 was enhanced when the drug was encapsulated within CCD-targeted PAMs (60.0 ± 5.2% of control respectively) vs. when administered as a free drug (85.9 ± 10.9% of control, p = 0.003) at this timepoint. At intermediate doses incubated for 24 h, rapamycin and everolimus both showed enhanced anti-proliferative effects when encapsulated in PAMs. Similarly, encapsulated RapaLink-1 induced a slightly stronger decrease in cell proliferation vs. free drug, although this change was not significant. Finally, at high doses, everolimus showed inhibition of cell proliferation only when encapsulated within PAMs (85.1 ± 6.6% vs. 112.8 ± 5.1% free; p < 0.001). Interestingly, RapaLink-1-PAMs were the most effective in inhibiting cell proliferation at all three doses, demonstrating this nanoformulation may be a promising option for further development.
An extended incubation period at 72 h was also used to assess longer term effects of the varying doses of mTOR inhibitors on hCCD cell proliferation (Fig. 4). At low dose (50 μM), encapsulated rapamycin showed higher inhibition of cell proliferation (70.8 ± 3.6%) vs. free rapamycin (89.2 ± 6.0%; p = 0.0006). Strikingly, everolimus caused significant inhibition of cell proliferation when encapsulated (85.0 ± 5.1%) but not when administered as free drug (105.9 ± 3.1%, p < 0.001), suggesting nanocarriers promote cellular uptake and enhance the therapeutic effect of the drug. At the intermediate dose (100 μM), encapsulation increased the efficacy of both rapamycin and everolimus in hCCD cells. Encapsulated rapamycin (59.5 ± 3.1%) had a greater anti-proliferative effect than free rapamycin (97.7 ± 11.6%), with the same trend for encapsulated everolimus (87.1 ± 8.9%) vs. free everolimus (111.2 ± 10.5%; p = 0.009). At a high dose (200 μM), encapsulated everolimus showed high inhibition of cell proliferation (20.4 ± 1.7%) vs. free everolimus (105.2 ± 3.8%).
Comparing short and long incubation times, although there was a slight decrease in cell proliferation upon treatment with empty PAMs at 24 h, this effect was ameliorated by 72 h. For drug-PAM formulations, encapsulated everolimus induces a greater decrease in cell proliferation after 72 h vs. 24 h incubation at all doses, whereas free everolimus shows no significant decrease at both timepoints. Rapamycin follows a similar, although less pronounced trend, with increased inhibition of cell proliferation at longer time points. In contrast, the extent of RapaLink-1 cell proliferation inhibition remained constant over time.
3.4. Activation of the mTOR pathway
In order to assess the effects of drug encapsulation into PAMs on the mTOR pathway, Western blotting was carried out to quantify the phosphorylation of P70S6K and acetyl-CoA carboxylase (ACC), and the autophagy protein microtubule-associated protein light chain 3 (LC3) following incubation of free drug or drug-PAMs (Fig. 5). P70S6K is a mitogen-activated Ser/Thr protein kinase and lies on a mitogen-activated signaling pathway downstream of mTORC1 [27]. Phosphorylation of P70S6K at Thr389 has been used as a key marker of mTORC1 activity. Rapalogs have been shown to decrease the phosphorylated P70S6K to total P70S6K ratio, which indicates mTORC1 inhibition and the subsequent reduction in cellular proliferation [28]. As found in Fig. 5, our results show that encapsulation of mTOR inhibitors does not blunt the significant decrease in mTORC1 activity in human CCD cells after 24 h of incubation.
Fig. 5.
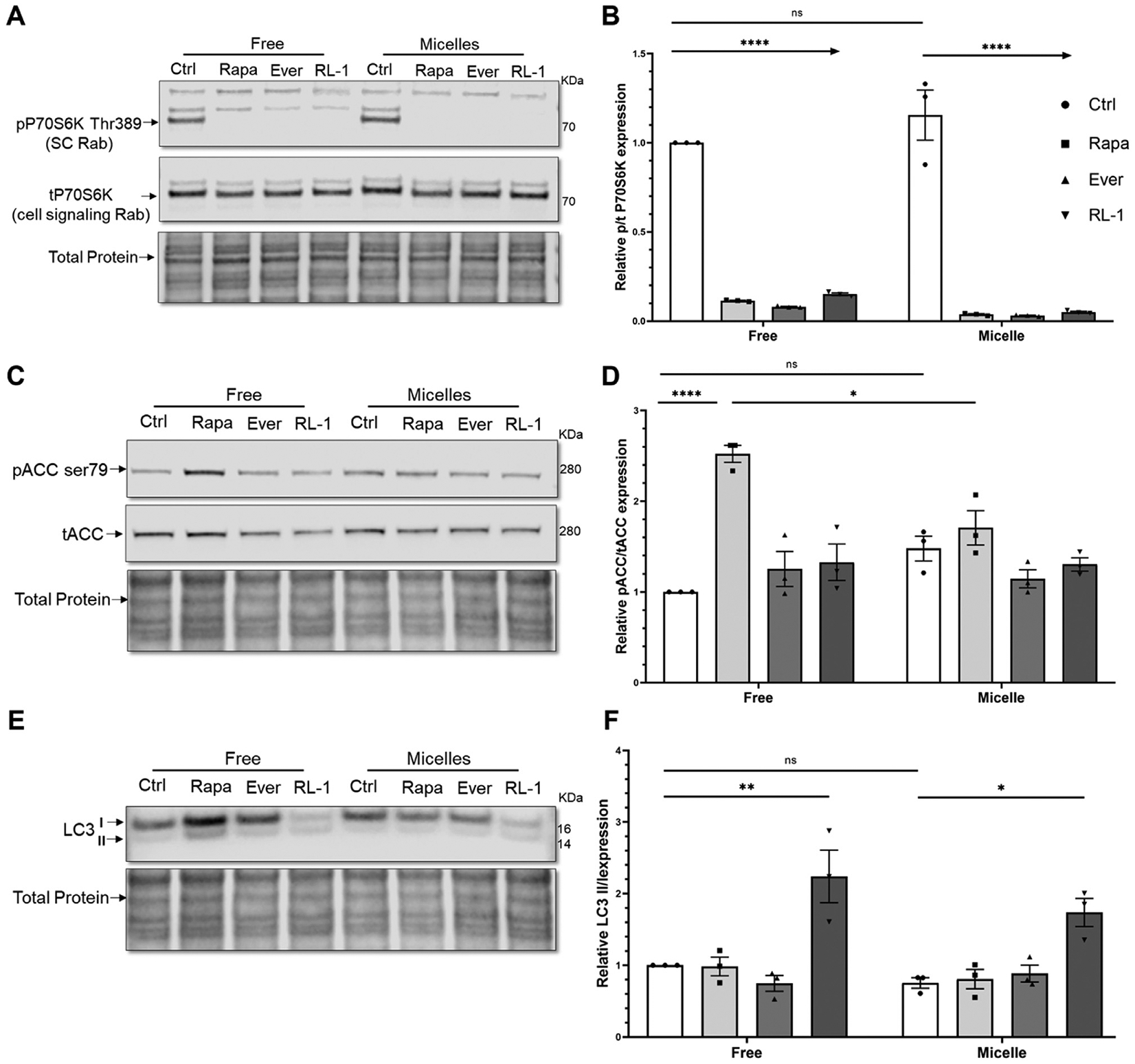
Western blot and quantification of the activation of the mTOR pathway by measuring phosphorylation of (A, B) P70S6K and (C, D) ACC, and (E, F) LC3 II/I expression after 24 h incubation of free or encapsulated drugs in PAMs with hCCD cells (n = 3, * p < 0.05, ** p <0.01, **** p < 0.0001).
Additionally, pACC to total ACC (tACC) was measured as phosphorylation of acetyl-CoA carboxylase (ACC) at Ser79 [29] is another indicator of AMPK activation [30]. Active AMPK assists in the inhibition of the mTORC1 kinase complex, and the pACC to tACC ratio has previously been shown to increase upon rapalog treatment [31]. As shown in Fig. 5, all three mTOR inhibitors induce an increase in AMPK activation vs. non-treated control, although only free rapamycin treatment induced a significant increase. PAM-encapsulated rapamycin also significantly increased this ratio, confirming the ability of nanoparticle-mediated drug delivery to retain therapeutic effects.
Finally, relative LC3 II/I expression was explored as a marker of autophagy, which is decreased in PKD and is induced by rapalogs [32]. LC3 is processed from cytosolic, pre-autophagic LC3-I and subsequently proceeds to autophagosome membrane-bound LC3-II upon activation of autophagy. An increase in the ratio of autophagosomal membrane-bound LC3-II to cytosolic LC3-I is an established indicator of autophagic activity [33]. In our study, only RapaLink-1 induced autophagy following 24 h of incubation, indicating its superiority over first generation mTOR inhibitors rapamycin and everolimus. Furthermore, encapsulation did not significantly hinder the effect of RapaLink-1. Overall, these results indicate that drug encapsulation into micelles does not inhibit drug activity and its effect on the mTOR pathway and may be beneficial for future in vivo studies aimed to target the kidney and diseased tissue while minimizing delivery to off-target sites.
3.5. Conclusions
Here, we created PAMs that can efficiently encapsulate rapamycin, everolimus, and RapaLink-1, accumulate in hCCD cells, and exert anti-proliferative effects via the mTOR pathway. PAM encapsulation enhanced the anti-proliferative effects of these mTOR inhibitors at multiple doses in hCCD cells following 24 and 72 h of incubation. Specifically, PAM encapsulation enhanced the antiproliferative effect of (1) rapamycin at low and intermediate doses for up to 72 h, (2) everolimus at low and intermediate doses at 24 and 72 h, and at high dose at 24 h, and (3) RapaLink-1 at a low dose after 24 h and high dose after 72 h.
In addition to the development of dose-optimized nanotherapeutics for specific drugs, the applications of this technology are potentially diverse and promising, although further studies are required. Although this study used healthy hCCD cells for evaluation, PAM-encapsulated drugs should be evaluated on 3D ADPKD cystic models to validate our results on an in vitro model that further mimics the disease and to identify an ideal nanoformulation for future, in vivo application. Moreover, multiple PAM-encapsulated drugs should be tested simultaneously to evaluate the potential benefits of combination therapy. Notably, the mTOR pathway, targeted in this study, is highly conserved and implicated in the progression of many chronic, proliferative diseases, including a variety of cancers and cystic disorders. While our PAMs were specifically designed for renal accumulation, potential future applications could include incorporation of different targeting moieties to deliver mTOR inhibitors directly to diseased tissues in a variety of proliferation disorders, including cancer.
Supplementary Material
Acknowledgements
This work was supported by the University of Southern California, New Innovator Award (NIH, DP2-DK121328) and WISE Major Support Award granted to E.J.C, and by the PKD Foundation postdoctoral fellowship 839636 to A.C. TEM images were taken in the USC Center of Excellence in Nano Imaging. The authors declare no competing interests.
Footnotes
Declaration of competing interests
The authors declare no financial interests/personal relationships which may be considered as potential competing interests.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.slast.2023.02.001.
References
- [1].Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008;359(14):1477–85. doi: 10.1056/NEJMcp0804458 From NLM. [DOI] [PubMed] [Google Scholar]
- [2].Al-Bhalal L, Akhtar M. Molecular basis of autosomal dominant polycystic kidney disease. Adv Anat Pathol 2005;12(3):126–33. doi: 10.1097/01.pap.0000163959.29032.1f. [DOI] [PubMed] [Google Scholar]
- [3].Cabrita I, Kraus A, Scholz JK, Skoczynski K, Schreiber R, Kunzelmann K, Buchholz B. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat Commun 2020;11(1):4320. doi: 10.1038/s41467-020-18104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Calvet JP. Strategies to inhibit cyst formation in ADPKD. Clin J Am Soc Nephrol 2008;3(4):1205–11. doi: 10.2215/CJN.05651207. [DOI] [PubMed] [Google Scholar]
- [5].van Gastel MDA, Torres VE. Polycystic kidney disease and the vasopressin pathway. Ann Nutr Metab 2017;70(Suppl 1):43–50. doi: 10.1159/000463063. [DOI] [PubMed] [Google Scholar]
- [6].Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget 2014;5(1):49–66. doi: 10.18632/oncotarget.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Costa RLB, Han HS, Gradishar WJ. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: a review. Breast Cancer Res Treat 2018;169(3):397–406. doi: 10.1007/s10549-018-4697-y. [DOI] [PubMed] [Google Scholar]
- [8].Chen H, Zhu D, Zheng Z, Cai Y, Chen Z, Xie W. CEP55 promotes epithelial-mesenchymal transition in renal cell carcinoma through PI3K/AKT/mTOR pathway. Clin Transl Oncol 2019;21(7):939–49. doi: 10.1007/s12094-018-02012-8. [DOI] [PubMed] [Google Scholar]
- [9].Zou Z, Tao T, Li H. Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci 2020;10:31. doi: 10.1186/s13578-020-00396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zheng Y, Jiang Y. mTOR Inhibitors at a Glance. Mol Cell Pharmacol 2015;7(2):15–20 From NLM.. [PMC free article] [PubMed] [Google Scholar]
- [11].Li A, Fan S, Xu Y, Meng J, Shen X, Mao J, Zhang L, Zhang X, Moeckel G, Wu D, et al. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J Cell Mol Med 2017;21(8):1619–35. doi: 10.1111/jcmm.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].MacKeigan JP, Krueger DA. Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro Oncol 2015;17(12):1550–9. doi: 10.1093/neuonc/nov152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Meng LH, Zheng XF. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol Sin 2015;36(10):1163–9. doi: 10.1038/aps.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Klawitter J, Nashan B. Christians U. Everolimus and sirolimus in transplantation-related but different. Expert Opin Drug Saf 2015;14(7):1055–70. doi: 10.1517/14740338.2015.1040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Koul PA, Mehfooz N. Sirolimus in lymphangioleiomyomatosis: a case in point for research in ‘orphan’ diseases. Lung India 2019;36(4):353–5. doi: 10.4103/lungindia.lungindia_280_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, Kunzen-dorf U, Banas B, Hörl WH, Obermüller N, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2010;363(9):830–40 note = PMID20581392. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- [17].Cox A, Lim SA, Chung EJ. Strategies to deliver RNA by nanoparticles for therapeutic potential. Mol Aspect Med 2021:100991. doi: 10.1016/j.mam.2021.100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jiang K, Huang Y, Chung EJ. Combining metformin and drug-loaded kidney-targeting micelles for polycystic kidney disease. Cell Mol Bioeng 2023;16(1):55–67. doi: 10.1007/s12195-022-00753-9 From NLM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huang Y, Jiang K, Zhang X, Chung EJ. The effect of size, charge, and peptide lig- and length on kidney targeting by small, organic nanoparticles. Bioeng Transl Med 2020;5(3):e10173. doi: 10.1002/btm2.10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang J, Poon C, Chin D, Milkowski S, Lu V, Hallows KR, Chung EJ. Design and in vivo characterization of kidney-targeting multimodal micelles for renal drug delivery. Nano Res 2018;11(10):5584–95. doi: 10.1007/s12274-018-2100-2. [DOI] [Google Scholar]
- [21].Chin D, Poon C, Wang J, Joo J, Ong V, Jiang Z, Cheng K, Plotkin A, Magee GA, Chung EJ. Nanoparticle-mediated microRNA-145 delivery for vascular smooth muscle cell phenotype modulation and atherosclerosis treatment. bioRxiv 2020. [Google Scholar]
- [22].Chung EJ, Mlinar LB, Sugimoto MJ, Nord K, Roman BB, Tirrell M. In vivo biodistribution and clearance of peptide amphiphile micelles. Nanomedicine 2015;11(2):479–87. doi: 10.1016/j.nano.2014.08.006. [DOI] [PubMed] [Google Scholar]
- [23].Joo J, Poon C, Yoo SP, Chung EJ. Shape effects of peptide amphiphile micelles for targeting monocytes. Molecules 2018(11):23. doi: 10.3390/molecules23112786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Giesecke T, Himmerkus N, Leipziger J, Bleich M, Koshimizu TA, Fähling M, Smorod-chenko A, Shpak J, Knappe C, Isermann J, et al. Vasopressin increases urinary acidification. J Am Soc Nephrol 2019;30(6):946–61. doi: 10.1681/ASN.2018080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guo Y, Kwiatkowski DJ. Equivalent benefit of rapamycin and a potent mTOR ATP-competitive inhibitor, MLN0128 (INK128), in a mouse model of tuberous sclerosis. Mol Cancer Res 2013;11(5):467–73. doi: 10.1158/1541-7786.MCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, Won H, Wong W, Berger M, de Stanchina E, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016;534(7606):272–6. doi: 10.1038/nature17963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Qiu Y, Lu C, Chen P, Sun F, Wang D, Wang Z, Hou C, Mu H, Duan J. Synergistic clearance of intracellular pathogens by hyaluronan-streptomycin micelles encapsulated with rapamycin. Carbohydr Polym 2019;210:364–71. doi: 10.1016/j.carbpol.2019.01.068. [DOI] [PubMed] [Google Scholar]
- [28].Zou W, Cao G, Xi Y, Zhang N. New approach for local delivery of rapamycin by bioadhesive PLGA-carbopol nanoparticles. Drug Deliv 2009;16(1):15–23. doi: 10.1080/10717540802481307. [DOI] [PubMed] [Google Scholar]
- [29].El Mohtadi F, d’Arcy R, Burke J, Rios De La Rosa JM, Gennari A, Marotta R, Francini N, Donno R, Tirelli N. “Tandem” nanomedicine approach against osteoclastogenesis: polysulfide micelles synergically scavenge ROS and release rapamycin. Biomacromolecules 2020;21(2):305–18. doi: 10.1021/acs.biomac.9b01348. [DOI] [PubMed] [Google Scholar]
- [30].La Manna F, De Menna M, Patel N, Karkampouna S, De Filippo MR, Klima I, Kloen P, Beimers L, Thalmann GN, Pelger RCM;, et al. Dual-mTOR inhibitor Rapalink-1 reduces prostate cancer patient-derived xenograft growth and alters tumor heterogeneity. Front Oncol 2020;10:1012. doi: 10.3389/fonc.2020.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373–9. doi: 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kirchner GI, Meier-Wiedenbach I, Manns MP. Clinical pharmacokinetics of everolimus. Clin Pharmacokinet 2004;43(2):83–95. doi: 10.2165/00003088-200443020-00002. [DOI] [PubMed] [Google Scholar]
- [33].Pullen N, Thomas G. The modular phosphorylation and activation of p70s6k. FEBS Lett 1997;410(1):78–82. doi: 10.1016/s0014-5793(97)00323-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.