

Abstract
Asthma is a chronic disease most commonly associated with allergy and type 2 inflammation. However, the mechanisms that link airway inflammation to the structural changes that define asthma are incompletely understood. Using a human model of allergen-induced asthma exacerbation, we compared the lower airway mucosa in allergic asthmatics and allergic non-asthmatic controls using single-cell RNA sequencing. In response to allergen, the asthmatic airway epithelium was highly dynamic and up-regulated genes involved in matrix degradation, mucus metaplasia, and glycolysis while failing to induce injury-repair and antioxidant pathways observed in controls. IL9-expressing pathogenic TH2 cells were specific to asthmatic airways and were only observed after allergen challenge. Additionally, conventional type 2 dendritic cells (DC2 that express CD1C) and CCR2-expressing monocyte-derived cells (MCs) were uniquely enriched in asthmatics after allergen, with up-regulation of genes that sustain type 2 inflammation and promote pathologic airway remodeling. In contrast, allergic controls were enriched for macrophage-like MCs that up-regulated tissue repair programs after allergen challenge, suggesting that these populations may protect against asthmatic airway remodeling. Cellular interaction analyses revealed a TH2–mononuclear phagocyte–basal cell interactome unique to asthmatics. These pathogenic cellular circuits were characterized by type 2 programming of immune and structural cells and additional pathways that may sustain and amplify type 2 signals, including TNF family signaling, altered cellular metabolism, failure to engage antioxidant responses, and loss of growth factor signaling. Our findings therefore suggest that pathogenic effector circuits and the absence of proresolution programs drive structural airway disease in response to type 2 inflammation.
INTRODUCTION
Asthma affects 1 in 13 people in the United States and is increasing in prevalence (1). Most asthma is characterized by allergic inflammation to environmental antigens (2). However, not all allergic individuals sensitized to inhaled allergens develop asthma, a disease of the lower airways defined by airway inflammation, bronchial hyper-responsiveness, and mucus hypersecretion (2, 3). We therefore hypothesized that identifying key differences in the lower airway response to allergen between allergic asthmatics (AAs) and allergic individuals without asthma would provide fundamental insights into the mechanisms that drive asthma.
It is increasingly recognized that cellular cross-talk within the airway microenvironment is important in asthma pathogenesis (2–4). Consistent with this, the airway mucosa in asthma is characterized by innate and adaptive type 2 immune cells, including eosinophils, mast cells, and T helper 2 (TH2) cells, that localize in proximity to the airway epithelium. Moreover, murine and human studies have established that understanding epithelial cell activation and immune cell enrichment in the airway mucosa in response to allergen challenge is essential to elucidate asthma pathobiology (3–11). Single-cell transcriptional approaches have facilitated the discovery of tissue-specific cell subsets associated with allergic disease, including a recent steady-state analysis of human airways that showed that pathogenic effector TH2 cells, mast cells, and secretory airway epithelial cells (AECs) are enriched in asthmatics compared with healthy controls (12–16). However, high-dimensional analyses of the dynamic lower airway mucosal response to allergen challenge have not been reported.
Bronchoscopic segmental allergen challenge (SAC) is a powerful research tool that recapitulates the features of an allergen-induced asthma exacerbation in a single airway segment and has been used to identify asthma-relevant pathways and successfully predict the clinical efficacy of new therapeutics in asthma (9, 17, 18). We have previously shown that SAC induces type 2 airway inflammation in both AAs and allergic non-asthmatic controls (ACs) (9). However, asthmatics have increased measures of type 2 inflammation and uniquely demonstrate airway structural changes, including increases in mucin production, epithelial and subepithelial thickness, and airway smooth muscle thickness (9, 19). Thus, we hypothesize that asthmatics have distinct cellular and transcriptional programs in the airway microenvironment that amplify type 2 inflammation and link mucosal inflammation to the airway structural changes that define asthma.
We leveraged bronchoscopic SAC and single-cell RNA sequencing (scRNA-seq) to define the airway mucosal microenvironment and identified transcriptional programs that were only observed after allergen exposure. By comparing AAs with ACs, we distinguished cellular and molecular pathways that define asthma from those associated with allergy alone. These findings included a robust transcriptional response to allergen challenge in basal and secretory AECs from asthmatics that was greater than in ACs and characterized by up-regulation of genes involved in mucus metaplasia and matrix remodeling. In addition, an AEC glycolytic gene signature was associated with asthma, whereas antioxidant and growth factor signaling pathways were identified as potentially protective. IL9-expressing pathogenic TH2 cells were only found in asthmatic airways after SAC, along with enrichment of conventional type 2 dendritic cells (DC2) and pathogenic monocyte-derived cells (MCs) that up-regulated inflammatory mediators and metalloproteinases. Cell-cell interaction analyses highlighted immune-epithelial interactions specific to asthma and were dominated by a TH2–mononuclear phagocyte (MNP)–basal cell interactome, suggesting that cross-talk between these key cell subsets is critical to the pathogenesis of allergic asthma.
RESULTS
A human model of allergen-induced asthma exacerbation transformed the airway mucosal cellular landscape
To test the hypothesis that differences in immune or structural cells in the airway mucosa distinguish asthma from allergy alone, we enrolled 61 adults with allergy to house dust mite (HDM) or cat with or without asthma, as well as 5 nonallergic healthy controls, to undergo SAC (Fig. 1A, data file S1, and Supplementary Methods) (9, 19). Allergy was defined as a clinical history of allergic rhinitis and/or conjunctivitis to HDM or cat, which was confirmed by skin prick testing (SPT). Quantitative SPT was used to standardize the dose of allergen administered according to each participant’s level of allergy (fig. S1A and Supplementary Methods). ACs had no history of asthma, normal forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC), and a negative methacholine challenge test. AAs had a history of mild asthma, FEV1 ≥ 75% predicted, and bronchial hyperresponsiveness confirmed by positive methacholine challenge testing. Bronchoalveolar lavage (BAL) and endobronchial brush samples of third- to fourth-generation airway segments were collected at baseline and 24 hours after administration of allergen and diluent, which controlled for the effects of the bronchoscopy procedure (Fig. 1B). Similar to prior reports (9), SAC induced robust eosinophilic inflammation in both allergic groups that was restricted to the allergen-challenged segment and was not observed in healthy controls (Fig. 1C).
Fig. 1. A human model of allergen-induced asthma exacerbation using SAC.
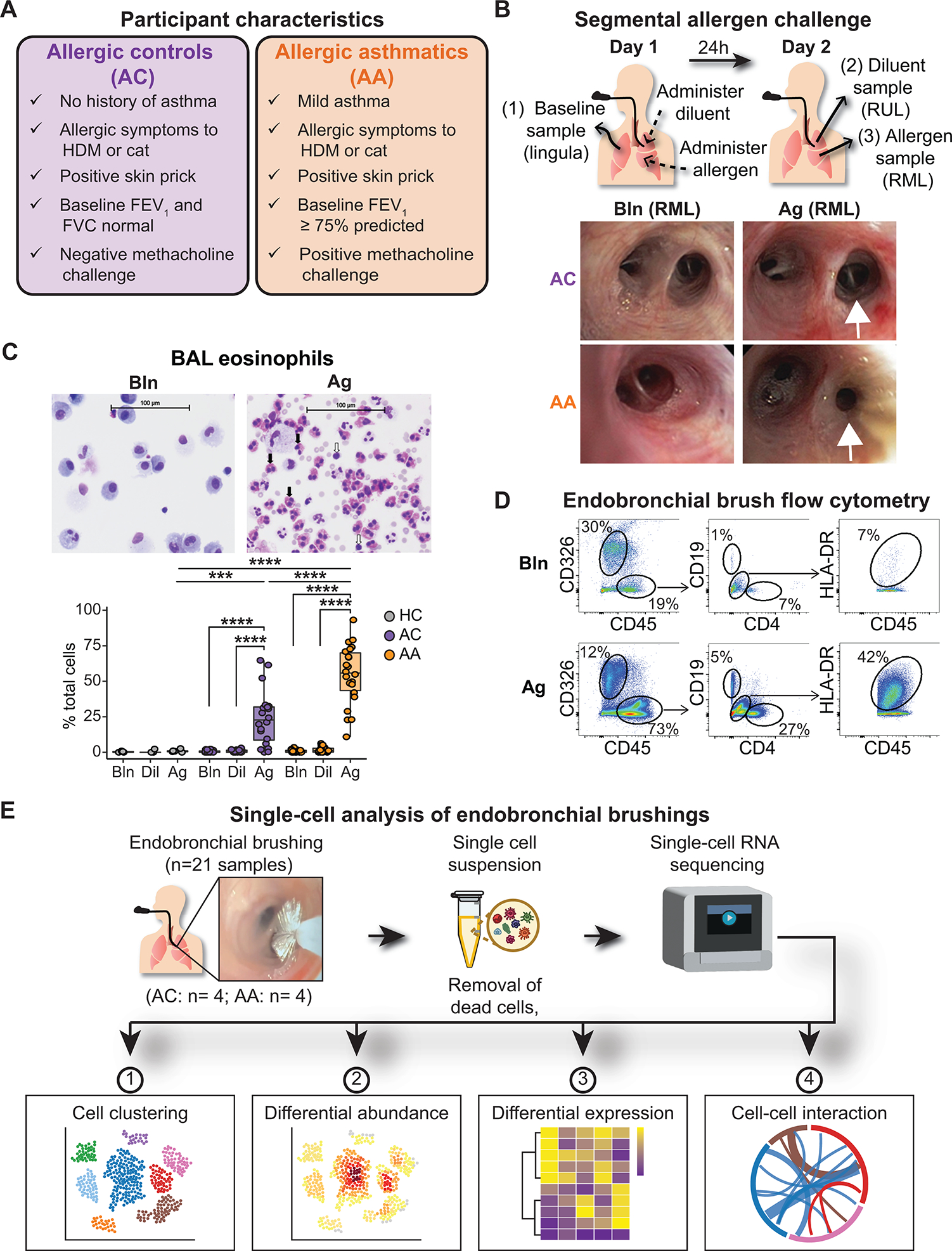
(A) Characteristics of AC and AA participants. (B) Design of the bronchoscopic SAC procedure. BAL and endobronchial brush samples were obtained at baseline (from the lingula) and 24 hours after administration of diluent to the right upper lobe (RUL) and allergen to the right middle lobe (RML). Representative images of RML airway segments from one AC participant (top row) and one AA participant (bottom row) at baseline (left) and after SAC (right). In the AA participant, the allergen-challenged segmental airway is narrowed, and mucus is present after SAC. (C) Representative images of cytospin preparations from BAL from one AA participant and quantification of BAL eosinophils in nonallergic healthy controls (HCs; n = 5), ACs (n = 20), and AAs (n = 22). Black arrows indicate eosinophils; white arrows indicate lymphocytes. (D) Representative flow cytometry of endobronchial brush samples at baseline (Bln; top row) and after allergen challenge (Ag; bottom row) from one AA participant. Epithelial cells were identified as CD326+CD45−. After gating on CD45+ cells, CD19+ B cells and CD4+ T cells were identified, as were CD19−CD4−HLA-DR+ antigen-presenting cells. (E) scRNA-seq was performed on endobronchial brushing samples (n = 21) collected from segmental airways, and downstream analysis was performed to identify differences between ACs (n = 4) and AAs (n = 4). In (C), boxes represent the median (line) and IQR, with whiskers extending to the remainder of the distribution no more than 1.5× IQR, with dots representing individual samples. P values were generated using a mixed-effects model with Šidák correction to adjust for multiple comparisons. ***P < 0.001 and ****P < 0.0001.
To characterize the lower airway mucosa during allergic inflammation, we performed flow cytometry of endobronchial brush samples and found enrichment of CD45+ immune cells after SAC, including CD4+ T cells, CD19+ B cells, and HLA-DR+ antigen-presenting cells (Fig. 1D). Given the limited number of cells recovered with this approach and difficulty identifying specific cell subsets due to overlapping expression of cell surface markers, we leveraged scRNA-seq to define cellular populations in the lower airway mucosa using an unbiased analytical framework. We generated high-quality scRNA-seq data from 21 lower airway brushings (totaling 52,152 cells) collected from four AAs (25,397 cells) and four ACs (26,755 cells) (Fig. 1E, fig. S1, B and C, and data files S2 and S3). Brushings were collected at baseline (Bln; 22,406 cells) and 24 hours after exposure to allergen (Ag; 17,462 cells) or diluent (Dil; 12,284 cells). Diluent samples could not be collected in two AAs and one AC and were thus excluded from downstream statistical comparisons with the other experimental conditions. We systematically performed iterative cell clustering to define distinct global and lineage-specific cellular populations in the lower airway mucosa. We subsequently used differential abundance, differential gene expression (DGE), and cell-cell interaction analyses to identify populations, transcriptional programs, and cellular networks that distinguish asthma from allergy alone (Fig. 1E and Materials and Methods).
Principal components analysis revealed variability driven by both disease group and experimental condition (fig. S1D). After data integration and unsupervised clustering analysis (Materials and Methods), we identified seven global cell lineages, including AECs (EPCAM), CD4 T cells (CD3D, CD4, and IL7R), CD8 T cells (CD3D and CD8A), MNPs (LYZ), B cells (MS4A1), mast cells (CPA3), and natural killer cells (GNLY) (Fig. 2, A to C, fig. S1E, and data file S4). All cell lineages were captured across all participants and experimental conditions (fig. S1E). At baseline, the airway mucosal landscape was dominated by AECs and CD8 T cells in both groups (Fig. 2D), with a similar cellular composition observed after diluent administration (fig. S1, F and G). Mixed-effects logistic regression was used to identify differences in cellular abundance that were associated with each disease group [ACs: odds ratio (OR) < 1, AAs: OR > 1; Fig. 2E, fig. S1H, and Materials and Methods] (20). Both B cells (OR 4.48, 95% confidence interval [1.73 to 11.61]) and mast cells (OR 6.68, [3.20 to 13.94]) were associated with asthma at baseline, and mast cells expressed genes (CD38 and KIT) consistent with a subset that proliferates in situ during allergic disease (fig. S1I and data file S5) (13). We did not observe DGE [log2FC (fold change) > 0.5 and false discovery rate (FDR) < 0.1; Materials and Methods] in B cells or mast cells between groups, but mast cells in AAs responded to allergen challenge by up-regulating genes involved in mast cell growth and survival (IL3RA, IL2RA, BIRC3, and BCL2), chemotaxis (CXCR4 and C5AR1), and KIT expression (RUNX1), suggesting that these pathways may promote mast cell enrichment in asthmatic airways (fig. S1, J and K, and data file S6). After allergen challenge, immune cells dominated airway mucosal samples (Bln: 9658 cells, 43.1%; Ag: 13,459 cells, 77.1%) (Fig. 2F). The number of MNPs increased the most after allergen challenge in both groups (ACs: 6.7% versus 33.4%, P = 0.009; AAs: 5.9% versus 28.1%, P = 0.02), whereas CD4 T cells only increased in asthmatics (7.1% versus 20.6%, P = 0.03).
Fig. 2. Allergen-induced immune cell enrichment in the airway mucosa.
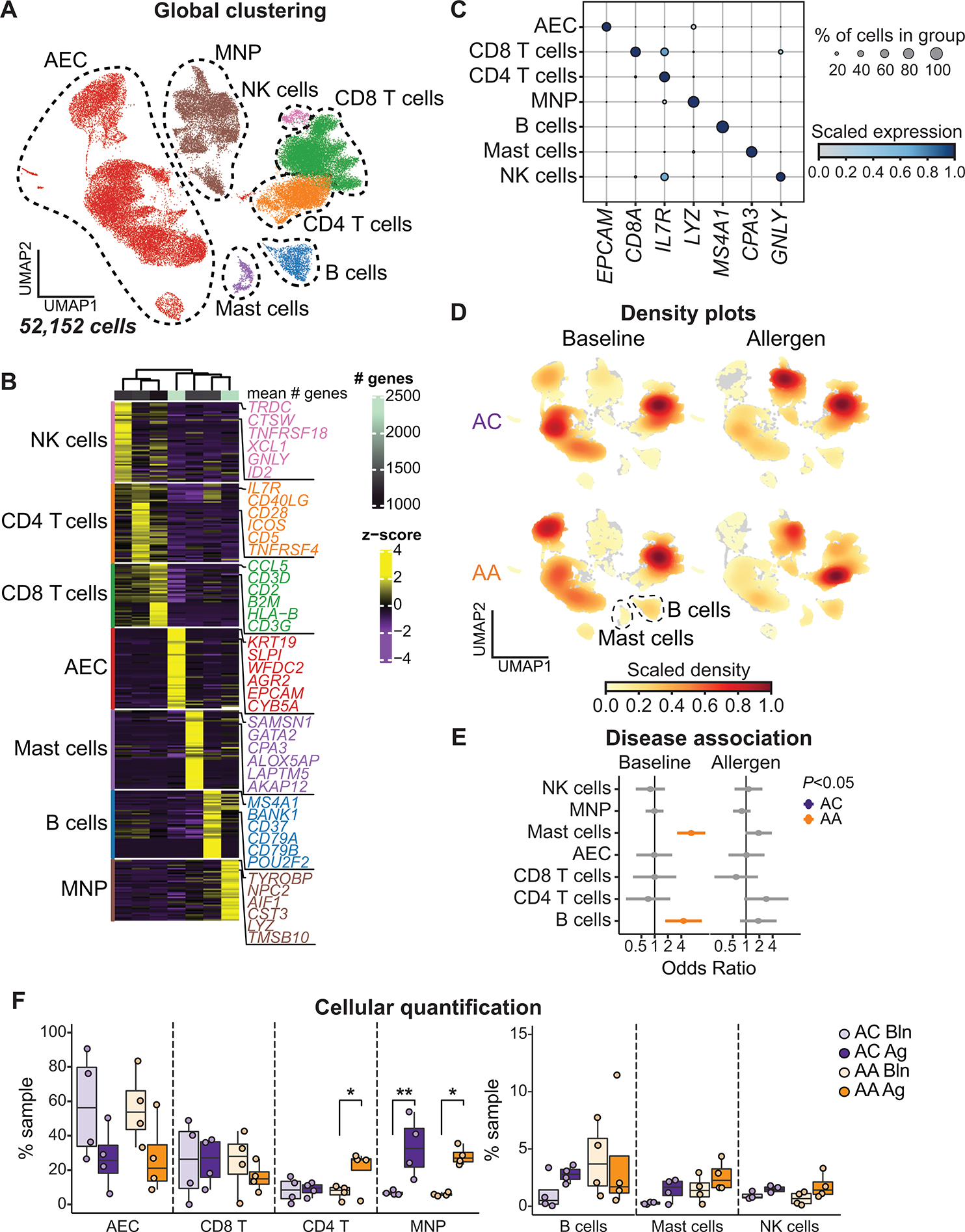
(A) UMAP embedding of 52,152 high-quality single cells color-coded by predicted cell lineage. NK, natural killer. (B) Heatmap showing the top discriminative gene sets for each cell lineage compared with every other lineage. Color scales denote the normalized gene expression (mean zero, unit variance) for each cluster and the mean number of genes per cluster (top bar). (C) Dot plot showing the percentage and expression level of marker genes defining each cell lineage, with the size of the dot representing the percentage of cells within each lineage expressing each marker and the color intensity indicating the scaled expression level. (D) UMAP embedding of cell density displaying the proportion of each cell lineage at baseline (left) and after allergen challenge (right) compared with every other cell lineage, faceted by disease group (ACs: top, AAs: bottom). (E) OR of disease association by cell lineage at baseline and after allergen challenge. Color-coding denotes significant associations with ACs (OR < 1, purple) or AAs (OR > 1, gold). (F) Contribution of each cell lineage defined in (B), shown as percentage (%) of total sample at baseline (Bln) and after allergen challenge (Ag). In (E), dots and whiskers represent OR with 95% confidence interval, calculated using a mixed-effects association logistic regression model (P < 0.05 corrected for multiple comparisons using the Tukey method). In (F), boxes represent the median (line) and IQR, with whiskers extending to the remainder of the distribution no more than 1.5× IQR and dots representing individual samples. P values generated using a mixed-effects model with Šidák correction to adjust for multiple comparisons. *P < 0.05 and **P < 0.01.
Basal and secretory epithelial cells in asthmatics extensively altered their transcriptional profiles in response to allergen
Our prior work suggests that AECs in asthmatics are uniquely sensitive to type 2 inflammation (9). Subclustering of the 20,410 AECs enabled detailed characterization of the transcriptional programs characterizing AECs at baseline and in the setting of allergic inflammation. We identified 14 distinct AEC subsets [area under the curve (AUC) ≥ 0.75 and pseudo-bulk DGE FDR < 0.05; Materials and Methods], annotated by cross-referencing top marker genes with published transcriptional data (Fig. 3, A to C, fig. S2, A and B, and data file S4) (4, 16, 21–25). Three ciliated clusters (FOXJI and PIFO) were observed, including mucous-ciliated cells (SCGB1A1 and MUC5AC). A prior study identified mucous-ciliated cells in asthmatics but not healthy controls (16). However, we found similar numbers of mucous-ciliated cells in both asthmatics and ACs, indicating that these cells do not distinguish asthma from allergy alone. Four basal cell clusters (KRT5 and S100A2) were also identified. Suprabasal cells were distinguished from basal cells by lower TP63 and KRT5 expression, cycling basal cells expressed markers of proliferation (PCLAF and MKI67), and a cluster that was distinguished primarily by lower overall gene expression was termed quiescent (quiesBasal). Three secretory clusters had high expression of secretoglobin genes (SCGB1A1 and SCGB3A1). Goblet and quiescent goblet (quiesGoblet) cells had high expression of the tethered mucin genes (MUC5AC and MUC5B), whereas club cells were distinguished by lower expression of mucin and goblet cell differentiation genes (FOXA3 and SPDEF) (4, 16, 21, 23, 26). A small serous cluster expressed markers of both serous (LYZ, PRB3) and mucous cells (AZGP1, BPIFB2, and MUC5B), suggesting sampling of submucosal glands. Moreover, we identified the recently described deuterosomal (CCNO, CDC20B), hillock (KRT13 and SPRR3), and ionocyte (FOXI1 and CFTR) populations (Fig. 3, A and B, and fig. S2C) (4, 16, 21–25). We observed AEC subset–specific expression of asthma-relevant mediators (Fig. 3C). Basal cells were enriched for IL33 and uniquely expressed TSLP. Periostin (POSTN) was expressed in both basal and ionocyte subsets, and ionocytes also expressed genes involved in prostaglandin signaling (PTGS2 and PTGER3).
Fig. 3. Dynamic transcriptional response in basal and secretory epithelial cells after allergen challenge.
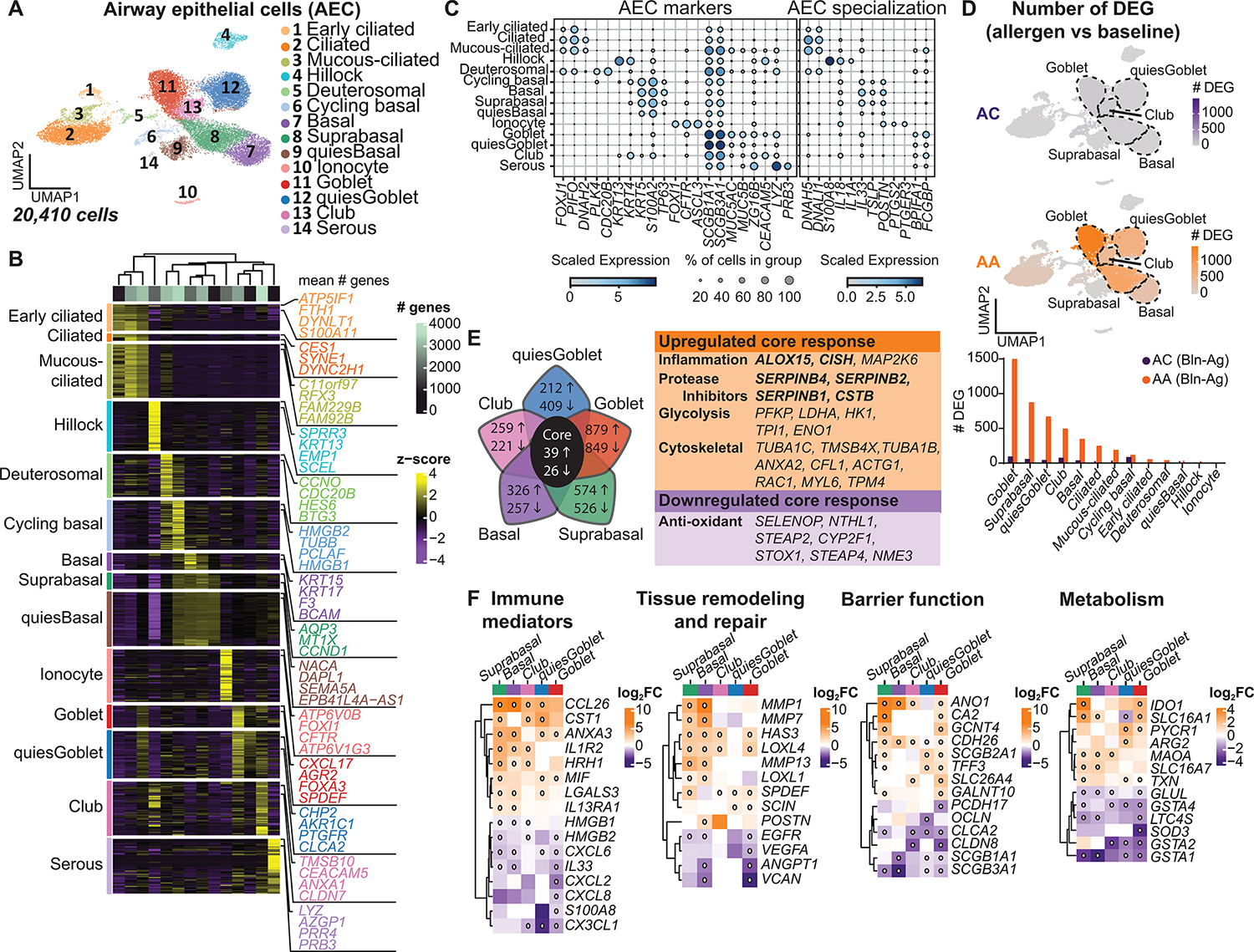
(A) UMAP embedding derived from subclustering of 20,410 AECs. (B) Heatmap showing the top discriminative gene sets for each cluster compared with every other AEC cluster. Color scales denote the normalized gene expression (mean zero, unit variance) for each cluster and the mean number of genes per cluster (top bar). (C) Dot plot depicting gene expression levels and percentage of cells expressing genes across AECs. (D) UMAP plots with color intensity (top) indicating the number of DEGs induced by SAC in ACs and AAs compared with baseline. The number of DEGs induced by SAC, quantified by cluster for AAs and ACs (bottom). (E) Venn diagram depicting the top five AEC clusters with the most DEGs after SAC, with the number of genes up-regulated (up arrow) or down-regulated (down arrow) in AAs compared with ACs using an interaction term for disease state and experimental condition. The core transcriptional response is denoted in the table (right). Bolded genes are induced by IL-13. (F) Heatmap of selected DEGs. Color scale indicates FC differences between AAs and ACs after SAC. White dot indicates FDR < 0.1. (D) DEG based on FDR < 0.1 and log2FC > 0.5 using the Wald test on pseudo-bulk count matrix. (E and F) DEGs based on FDR < 0.1 and log2FC > 0.5 using a likelihood ratio test on pseudo-bulk count matrix.
Hillock cells expressed genes involved in squamous differentiation (KRT13 and KRT4), barrier integrity (SPRR3 and ECM1), and immune responses (IL1A, S100A8, and ALOX15) (Fig. 3, B and C, and fig. S2C). Hillock cells have been described as a highly proliferative transitional population between basal and secretory cells (4, 23, 25). In our dataset, hillock cells did not express proliferation markers, nor did they cluster between basal and secretory cells in Uniform Manifold Approximation and Projection (UMAP) space. However, we identified a discrete region within the club cell cluster that expressed hillock cell markers and aligned with transitional “hillock club” cells previously only described in mouse trachea (fig. S2, C and D, and data file S7) (23). In humans, hillock cells have only been reported in the nasal mucosa (25). To validate our scRNA-seq findings, we performed immunofluorescence staining of explanted asthmatic lung and observed contiguous groups of KRT13+ cells lining the airway without overlying ciliated cells (fig. S2E), consistent with prior descriptions of hillock cells (23, 25). In atopic dermatitis and eosinophilic esophagitis, keratinized squamous cells can sense and potentiate type 2 inflammation and induce barrier dysfunction (27). Consistent with this, hillock cells in asthmatics expressed interleukin-13 (IL-13)–responsive genes, including calpain 14 (CAPN14), which was highly enriched in this subset, has been linked to asthma, and induces epithelial barrier dysfunction (fig. S2F) (28, 29). These findings suggest that hillock cells may play a previously unappreciated role in directing the lower airway response to allergen.
Although quantification of AEC subsets did not reveal significant differences between groups (fig. S2G and data file S3), mixed-effects logistic regression identified an association between ACs and quiesGoblet (OR 0.39, [0.50 to 1.51]) at baseline and deuterosomal cells (OR 0.35, [0.17 to 0.75]) after allergen challenge (fig. S2, H and I). Ciliated cells (OR 2.10, [1.25 to 3.54]) and ionocytes (OR 2.59, [1.34 to 5.02]) were associated with asthmatics at baseline. To assess the response of AECs to allergen challenge, we quantified the number of differentially expressed genes (DEGs; log2FC > 0.5 and FDR < 0.1; Materials and Methods) after allergen compared with baseline (Fig. 3D and data file S6). AECs in asthmatics markedly altered their transcriptional profile after allergen challenge compared with ACs (total DEGs = 4286 in AAs versus 269 in ACs). Goblet cells in asthmatics had the greatest transcriptional response to allergen challenge (DEGs = 1454), followed by suprabasal (DEGs = 850), quiesGoblet (DEGs = 645), club (DEGs = 472), and basal cells (DEGs = 323), suggesting that these AEC subsets play a central role during allergic inflammation. At baseline, we found minimal transcriptional differences between groups (data file S6). Given the large number of DEGs induced by allergen challenge, we created an interaction term for disease group and experimental condition to identify DEGs uniquely associated with asthma after allergen challenge (FDR < 0.1; Materials and Methods and data file S6). Using the interaction term, we identified 65 shared DEGs across basal and secretory clusters specific to asthma (Fig. 3E). This core response was characterized by up-regulation of IL-13–induced genes, along with genes involved in inflammatory signaling, tissue remodeling, glycolysis, and down-regulation of antioxidant genes.
We also identified subset-specific DEGs between asthmatics and ACs, highlighting new aspects of AEC biology associated with asthma. Basal and suprabasal cells in allergic controls were characterized by an injury-repair response after allergen challenge, with increased expression of alarmins (IL33 and HMGB1) and neutrophil chemoattractants (CXCL6) (Fig. 3F). In contrast, these cells in asthmatics up-regulated genes involved in type 2 inflammatory cell recruitment and signaling (CCL26 and IL13RA1), mucus metaplasia (SPDEF), and genes that promote extracellular matrix (ECM) degradation (MMP1 and MMP7) and allow for connective tissue regeneration and biogenesis (POSTN and LOXL4). We then performed Ingenuity Pathway Analysis (IPA) of DEGs identified by the interaction term to identify potential upstream regulators of transcriptional changes in suprabasal cells in each group, with |z score| > 2 considered significant (Materials and Methods and data file S8). In asthmatics, the strongest predicted upstream regulators included IL-13 and fibroblast growth factor 2 (FGF2) (|z scores| = 2.45), both involved in airway inflammation and remodeling (fig. S2J).
ACs also up-regulated stress response genes (S100A8 and HMGB2) in goblet cells, along with genes that orchestrate tissue integrity and angiogenesis (VEGFA) and antioxidant defense (GSTA1, GSTA2, and GSTA4) (Fig. 3F). Although all secretory clusters in asthmatics up-regulated SPDEF, a signal transducer and activator of transcription 6 (STAT6)–induced transcription factor that directs goblet cell differentiation (26), goblet cells in asthmatics additionally up-regulated genes that control mucin glycosylation and hydration (GCNT4 and GALNT10) and regulate airway surface liquid pH (CA2 and SLC26A4) (30). In asthmatics, the strongest predicted upstream regulator of goblet cells was the epidermal growth factor receptor (EGFR; |z score| = 2.0; fig. S2J), which can promote goblet cell hyperplasia and mucin production (21, 31, 32). Collectively, these results identify airway basal and secretory cells as highly dynamic cells during allergic inflammation and reveal mechanisms by which they may drive asthma pathogenesis.
IL9-expressing pathogenic TH2 cells are specific to asthmatic airways after allergen challenge
TH2 cells play a central role in the inflammatory response to allergen in the airways (2, 6, 9). Subclustering of 18,714 T cells identified 10 distinct subsets annotated by cross-referencing top marker genes (AUC ≥ 0.75 and pseudo-bulk DGE FDR < 0.05; Materials and Methods and data file S4) with published transcriptional data (16, 33–39) and included four CD8 T cell subsets, five CD4 T cell subsets, and a small γδ T cell (TRDC) population (Fig. 4, A and B, and fig. S3, A and B). Quiescent CD8 T cells were distinguished by low overall gene expression. CD8 T (GZMK) cells were enriched for late effector genes (EOMES and KLRG1), whereas CD8 T (CLIC3) and CD8 T (EGR2) cells were both enriched for tissue-resident memory markers (ITGAE, ITGA1, and CD69). These findings were supported by gene set enrichment analysis (GSEA) (fig. S3C, Materials and Methods, and data file S7) (36). CD4 T cell subsets included CD4 regulatory T (Treg) (FOXP3), CD4 TH2 (GATA3), CD4 T (CD40LG), CD4 TH17 (RORA), and a recently described interferon (IFN)–responsive CD4 THIFNR (ISG15) population (33, 37, 38). The identities of TH2, TH17, and THIFNR clusters were confirmed by GSEA (Fig. 4C, fig. S3C, and data file S7) (33, 39). CD8 T cells represented the highest proportion of T cells in both groups at baseline and remained the largest proportion of T cells in ACs after allergen challenge (Fig. 4D, fig. S3D, and data file S3). In contrast, the proportion of CD4 T (CD40LG), Treg, and TH2 cells increased in asthmatics after allergen challenge. TH2 abundance was associated with asthma both at baseline (OR 3.57, [1.71 to 7.45]) and after allergen challenge (OR 3.43, [1.87 to 6.29]) (Fig. 4E, fig. S3E, and data file S5). CD4 T (CD40LG) (OR 2.12, [1.23 to 3.68]) and Treg (OR 3.47, [2.20 to 5.47]) were also associated with asthma after allergen challenge. Further interrogation of the TH2 population identified a subset of cells expressing genes associated with enhanced pathogenic function (PTGDR2 and HPGDS) and receptors for TH2-polarizing cytokines (IL1RL1 and IL17RB) (Fig. 4F and fig. S3F) (33–35).
Fig. 4. IL9-expressing pathogenic TH2 cells specific to asthmatic airways.
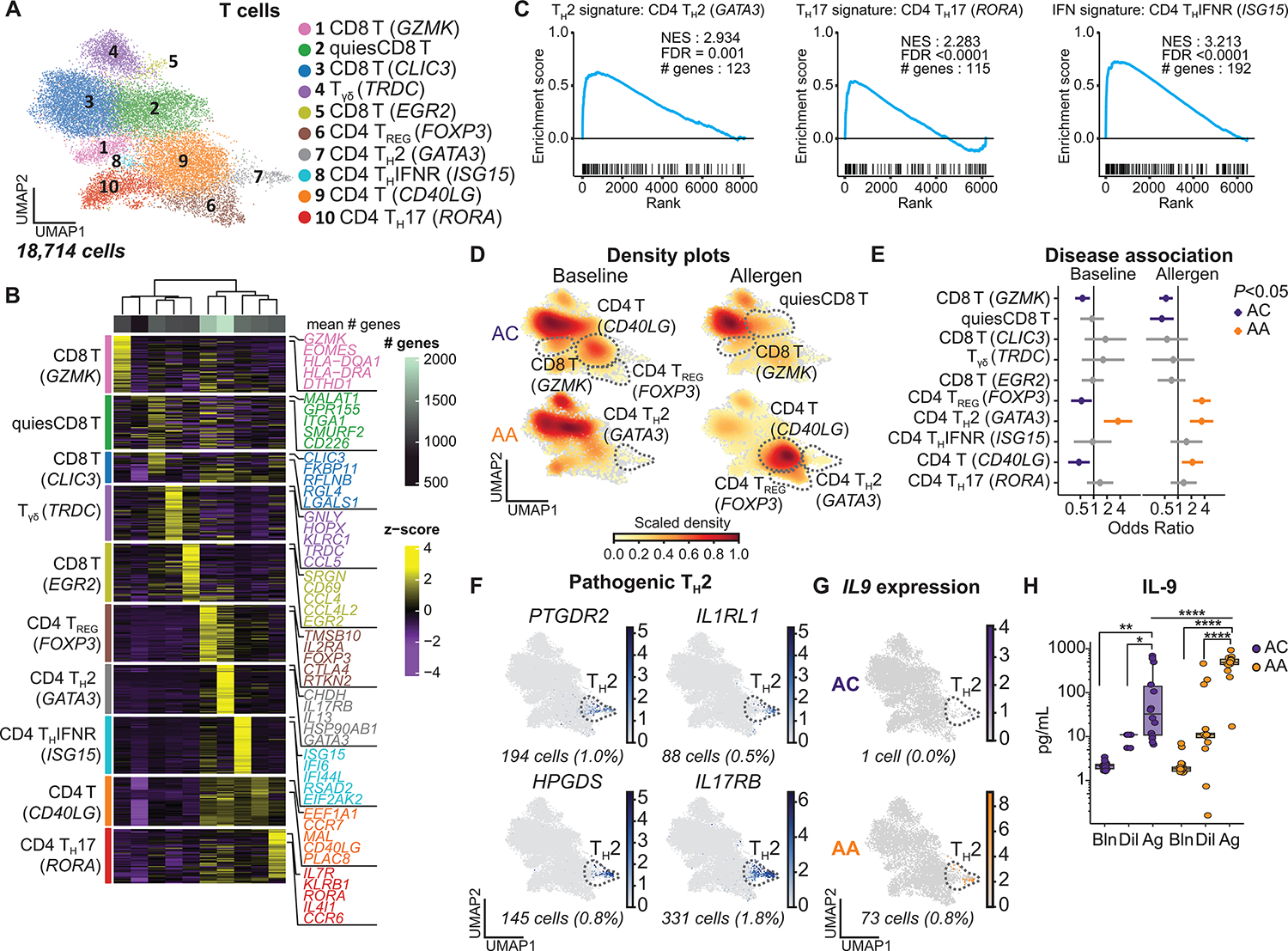
(A) UMAP embedding derived from subclustering of 18,714 T cells. (B) Heatmap showing the top discriminative gene sets for each cluster, compared with every other T cell cluster and the mean number of genes per cluster (top bar). (C) GSEA of CD4 TH2, TH17, and THIFNR clusters comparing the marker genes for these clusters with published T cell gene sets from Seumois et al. (33) (data file S7). (D) UMAP embedding of cell density displaying the proportion of each T cell subset at baseline (left) and after allergen challenge (right) compared with every other T cell subset, faceted by disease group (ACs: top, AAs: bottom). (E) OR of disease association by cluster at baseline and after allergen challenge. (F) Feature plots of pathogenic TH2 genes using pseudo-coloring to indicate gene expression. (G) Feature plot of IL9 expression using pseudo-coloring to indicate gene expression, faceted by group. (H) BAL concentration of IL-9 (ACs, n =14; AAs, n = 18). In (E), dots and whiskers represent OR with 95% confidence interval, calculated using a mixed-effects association logistic regression model (P < 0.05 corrected for multiple comparisons using the Tukey method). In (F) and (G), cell number and percentages (%) represent gene expression across all T cell subsets. Scaled gene expression in log(CPM). In (H), boxes represent the median (line) and IQR, with whiskers extending to the remainder of the distribution no more than 1.5× IQR. P values were generated using a mixed-effects model with Šidák correction to adjust for multiple comparisons. *P < 0.05, **P < 0.01, and ****P < 0.0001.
To assess the response of T cells to allergen challenge, we quantified the number of DEGs (log2FC > 0.5 and FDR < 0.1; Materials and Methods) after allergen compared with baseline and found the greatest number of DEGs in TH2 (DEGs = 76) and CD4 T (CD40LG) (DEGs = 54) in asthmatics (fig. S3G and data file S6). There were few DEGs identified between groups after allergen challenge (fig. S3, H and I, and data file S6). IL9 was the most up-regulated gene in asthmatic TH2 cells in response to allergen (logFC 8.3, FDR = 2.8 × 10−8). IL9-expressing pathogenic TH2 cells have previously been described in the blood of AAs (33), but they have not been reported in the airway. In our model, IL9-expressing TH2 cells were only observed after allergen challenge and were highly specific to asthmatic airways (Fig. 4G and fig. S3J). These cells were also enriched for inducers of IL-9 production (PPARG and IRF4) (fig. S3F) (33, 40, 41). Moreover, IL-9 protein levels in BAL samples increased in response to allergen challenge and were higher in asthmatics compared with ACs (median [interquartile range, IQR]: 489.9 [429.9 to 583.5] versus 33.5 [10.2 to 238.9] pg/ml, P < 0.0001) (Fig. 4H). IL-9 directly promotes mast cell activation and expression of profibrotic mediators, induces autocrine production of type 2 cytokines by TH2 cells, and can indirectly increase AEC mucin production via induction of IL-13 (40, 42–44). IL9R expression was identified in mast cells and pathogenic TH2, similar to previous reports (figs. S1I and S3F) (16, 45). These results demonstrate that pathogenic TH2 cells in the airway, and in particular their expression of IL9, distinguish asthma from allergy alone. Furthermore, they suggest that allergen-induced IL-9 expression may amplify type 2 inflammation and promote pathologic airway remodeling.
DC2 and CCR2-expressing MCs are enriched in asthmatics after allergen challenge, whereas macrophage-like MCs are enriched in ACs
MNPs are necessary to sustain type 2 inflammation in murine models of asthma (46); however, the precise role of MNP subsets and their functional specialization in the context of human allergic airway inflammation are incompletely understood. MNPs are recruited to the lung during allergic inflammation (Figs. 1D and 2, D and F) and include dendritic cells (DCs), macrophages (Macs), and MCs (47–49). Subclustering of 8510 MNP cells identified 14 distinct subsets that were annotated by cross-referencing top marker genes (AUC ≥ 0.75 and pseudo-bulk DGE FDR < 0.05; Materials and Methods and data file S4) with published transcriptional data (Fig. 5, A to C, and fig. S4, A and B) (49–58). DC subsets included DC1 (CLEC9A), DC2 (CD1C), migratory DCs (migDCs; CCR7), plasmacytoid DCs (pDCs; TCF4), and Axl-Siglec6 DCs (AXL). Three clusters were annotated as Macs on the basis of their abundance in baseline samples, indicative of their tissue residency. Mac1 (FABP4) and quiesMacs (defined by overall lower gene expression) were enriched for canonical Macs (MARCO, MSR1, and VSIG4) and lipid metabolism genes (FABP4 and APOE), consistent with previously described luminal Macs (16, 25, 51). Mac2 (A2M) had high expression of complement family genes (C1QA, C1QB and C1QC) and lower relative expression of canonical Mac markers, reminiscent of airway-associated Macs (AAMs) recently characterized in mice (57, 58).
Fig. 5. Distinct MNP profiles in asthmatics and ACs.
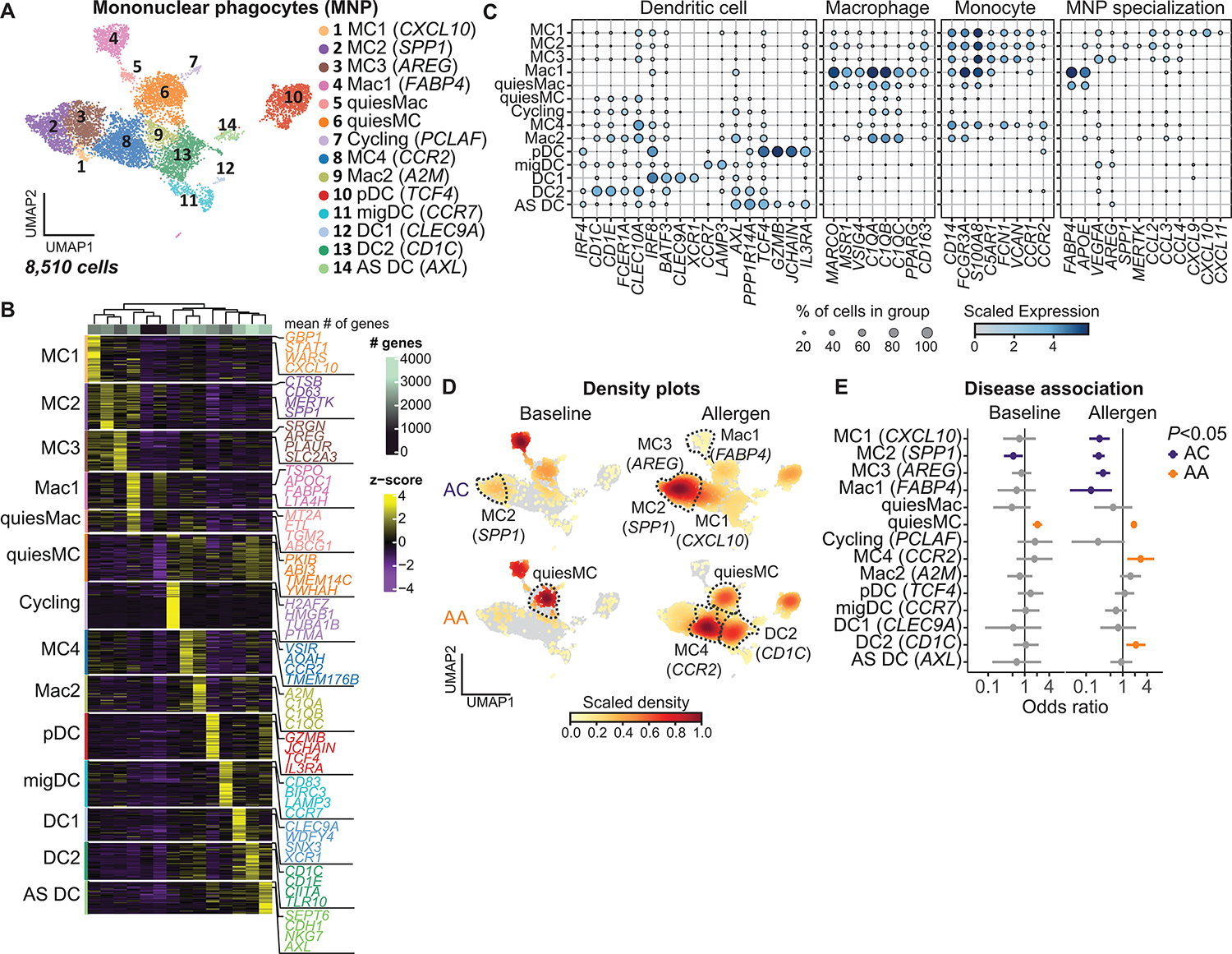
(A) UMAP embedding derived from subclustering of 8510 MNPs. (B) Heatmap showing the top discriminative gene sets for each cluster compared with every other MNP cluster and the mean number of genes per cluster (top bar). (C) Dot plot depicting gene expression levels and percentage of cells expressing genes across MNP clusters. (D) UMAP embedding of cell density displaying the proportion of each MNP subset at baseline (left) and after allergen challenge (right) compared with every other MNP subset, faceted by disease group (ACs: top, AAs: bottom). (E) OR of disease association by cluster at baseline and after allergen challenge. In (E), dots and whiskers represent OR with 95% confidence interval, calculated using a mixed-effects association logistic regression model (P < 0.05 corrected for multiple comparisons using the Tukey method).
Five clusters were annotated as MCs on the basis of high expression of canonical monocyte genes (CD14, VCAN, and FCN1) and varying expression of Mac and DC markers, consistent with the well-described plasticity of MCs (47, 48, 59). Quiescent MCs were characterized by markers of monocyte plasticity and had lower overall gene expression levels than the remaining MC clusters. MC1 expressed high levels of IFN-responsive genes (GBP1, STAT1, and WARS) and uniquely expressed CXCR3 ligands (CXCL9, CXCL10, and CXCL11) (Fig. 5C and fig. S4C). MC2 were enriched for matrix-interacting genes (SPP1, MERTK, and LGMN), consistent with a previously described lung resident population (50–52). MC3 distinctively expressed genes involved in tissue repair (AREG, VEGFA, and HBEGF) and NR4A1, a transcription factor necessary for monocytes to restrict inflammation and differentiate into regenerative Macs (60, 61). MC4, in contrast, had low expression of Mac markers, had high expression of genes associated with undifferentiated monocytes (TMEM176B, AIF1, and CSF1R), and were enriched for the monocyte chemotactic receptor CCR2, consistent with a recently recruited immature MC.
Tissue MCs and Macs play an important role in lung homeostasis, and imbalance in their proportions has been associated with pulmonary disease (50, 62). Mac1 (FABP4) was abundant in both groups at baseline, whereas DC subsets were rare (Fig. 5D, fig. S4D, and data file S3). Using mixed effects logistic regression, MC2 (SPP1) abundance was associated with ACs at baseline (OR 0.52, [0.31 to 0.89]), whereas quiescent MCs were associated with asthmatics (OR 2.05, [1.59 to 2.66]); Fig. 5, D and E, fig. S4E, and data file S5). Allergen challenge induced a dramatic shift in the MNP profile that was distinct between ACs and asthmatics. The abundance of Mac1 and Mac-like MCs (MC1, MC2, and MC3) was associated with ACs after allergen challenge, whereas the abundance of MC4 (OR 2.77, [1.29 to 5.92]) and DC2 (OR 2.11, [1.23 to 3.61]) was associated with asthmatics. Flow cytometry of endobronchial brush samples confirmed the enrichment of DC2 (CD45+HLA-DRhiCD1c+) in asthmatics compared with ACs after SAC (6.6% [2.6 to 9.2%] versus 2.9% [2.4 to 3.2%], P = 0.02; fig. S4F).
To identify genes associated with enrichment of MNP subsets in each group after allergen challenge, we used least absolute shrinkage and selection operation (LASSO) modeling (empirical P < 0.01 cutoff; fig. S4, G to I, Materials and Methods, and data file S9). MC2 (SPP1) and MC3 (AREG) enrichment in ACs was associated with EGFR signaling (AREG and EGFR). Lymphotoxin genes (LTB and LTA) were positively associated with DC2 (CD1C) enrichment in asthmatics but negatively associated with MC2 (SPP1) and MC3 (AREG) enrichment in ACs. Lymphotoxin is a tumor necrosis factor (TNF) family member whose signaling orchestrates immune cell aggregation at mucosal surfaces and effector immune responses in the tissue, including DC maturation and DC–T cell interactions (63). Secreted LTα forms a heterotrimer with membrane-bound LTβ to signal via LTβR, and we identified LTBR as primarily expressed in MNPs and AECs (fig. S4J). These data suggest that lymphotoxin signaling in the airway may play an important role in regulating MNPs in asthma.
CCR2-expressing MCs persist in an immature and pathogenic state in asthmatic airways
Monocytes are highly plastic cells that are recruited to the tissue during inflammation and are programmed by the tissue microenvironment (47, 48, 59). Murine models suggest that these cells are transiently pro-inflammatory but subsequently differentiate into Macs that resolve inflammation (48, 59, 62). To determine whether there is any transition between monocyte-derived subsets in the airway mucosa, cellular trajectory was inferred using RNA velocity and latent time analyses and predicted a transition from MC4 (CCR2) to MC2 (SPP1) and MC3 (AREG) with high confidence (Fig. 6A, and fig. S5, A and B, and Materials and Methods). Velocity analysis identified MC2 (SPP1) as the terminal population, suggesting that MC3 (AREG) is an intermediate state. Cells along this differentiation trajectory sequentially acquired expression of genes associated with Mac identity and survival (MARCO, PPARG, and CSF1), phagolysosomal function (LAMP1, CTSD, and CTSL), and lipid metabolism (APOC1, MGLL, and FABP5) (Fig. 6B and data file S10).
Fig. 6. A pathogenic transcriptional program in airway MCs in asthmatics after SAC.
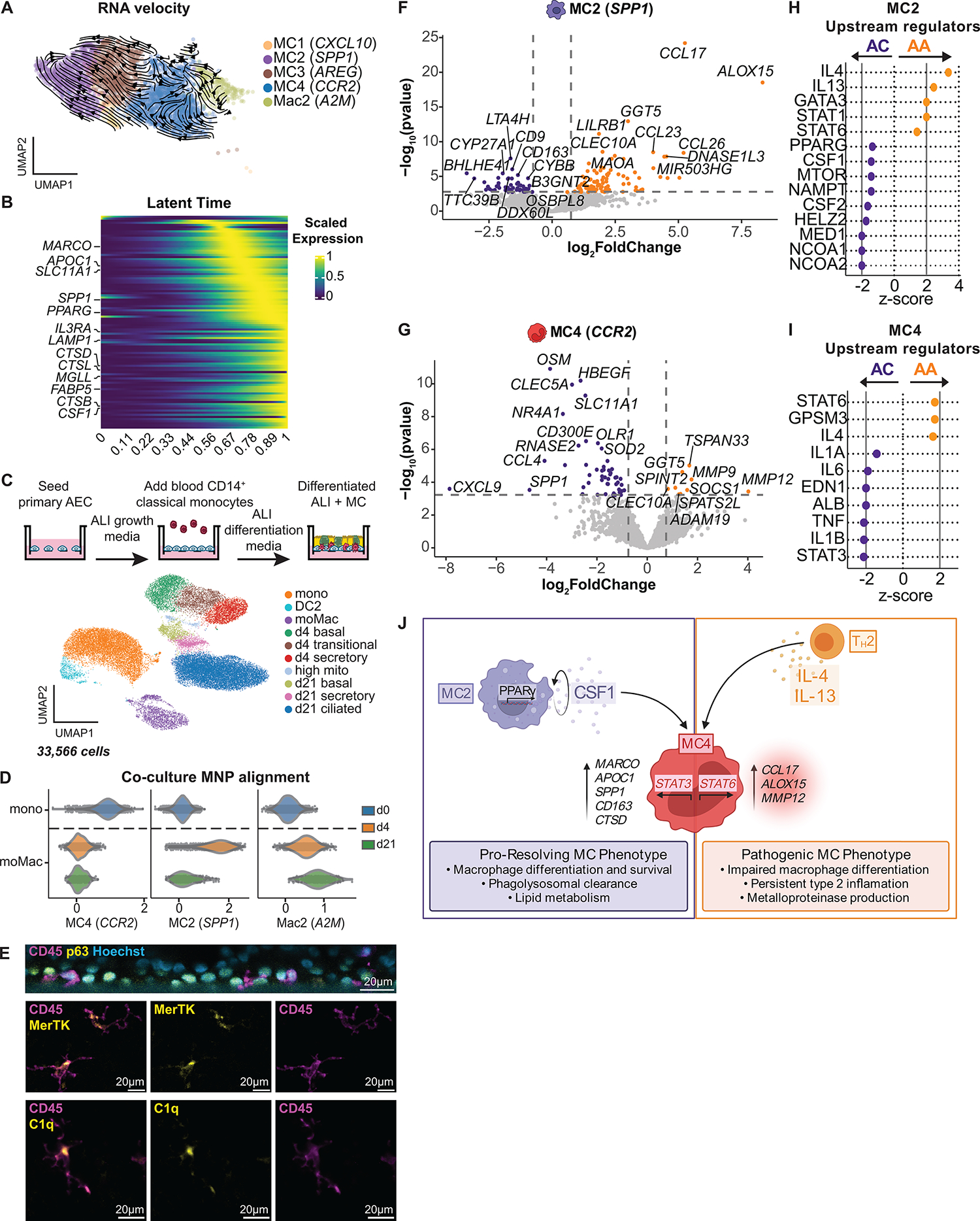
(A) RNAvelocities of a subset of MNP clusters visualized as streamlines projected on UMAP embedding. (B) Heatmap of gene expression pattern for the top 100 lineage driver genes correlated along the inferred differentiation trajectory. (C) Coculture model for blood CD14+ monocytes and AECs at ALI. UMAP embedding derived from clustering of 33,566 cells consisting of baseline blood CD14+ monocytes and cocultured cells collected at day 4 (d4) and day 21 (d21). Mono, monocyte; moMac, monocyte-derived Mac; mito, mitochondrial. (D) Alignment of blood CD14+ monocytes (day 0; d0) and moMacs from d4 and d21 coculture, using gene set scores (x axis) of airway MNP subclusters (Fig. 5A). (E) Immunofluorescence staining with orthogonal reconstruction at d21 of coculture demonstrates CD45+ immune cells (magenta) integrated into the p63+ basal cell layer (yellow) with Hoechst nuclear counterstain (cyan). CD45+ immune cells (magenta) exhibit prominent cytoplasmic projections and expression of MERTK and C1q (yellow). (F and G) Volcano plots representing DEGs in MC2 (F) and MC4 (G), comparing ACs (purple) with AAs (gold) after SAC. Vertical dotted lines represent cutoff of |log2FC| = 0.5, and horizontal dotted lines represent FDR cutoff = 0.1. (H and I) Predicted upstream regulators of MC2 (H) and MC4 (I) based on DEGs identified in (F) and (G) (ACs: purple, AAs: gold). Vertical solid lines represent z-score cutoff of |2|. (J) Summary figure of MC maturation sequence. (F and G) DEGs based on FDR < 0.1 and log2FC > 0.5 using the Wald test on pseudo-bulk count matrix.
We further interrogated this transitional relationship by performing scRNA-seq of human CD14+ classical monocytes isolated from peripheral blood (9668 cells) and of cocultures of monocytes and primary AECs at air-liquid interface (ALI) after 4 days (8759 cells) and 21 days (15,139 cells) (Fig. 6C, fig. S5, C and D, Materials and Methods, and data files S2 and S4). We identified 10 distinct populations, including 3 MNP and 7 AEC clusters (AUC ≥ 0.75 and pseudo-bulk DGE FDR < 0.05; Materials and Methods). MNP clusters included classical monocytes (mono; CD14) and a small population of DC2 (CD1C) from day 0 peripheral blood, as well as monocyte-derived Macs (moMacs; C1QA) identified in both day 4 and 21 cocultures (Fig. 6C and fig. S5C). Using gene set scores derived from our airway mucosal MNP marker genes (Fig. 5B and data file S4), day 0 monocytes from blood aligned with MC4 (CCR2), whereas moMacs after 4 days in coculture aligned most closely with MC2 (SPP1), consistent with the differentiation continuum predicted by RNA velocity analyses (Fig. 6D and fig. S5E). moMacs after 21 days in coculture acquired a transcriptional profile similar to that of Mac2 (A2M) and were highly enriched for complement genes (C1QA, C1QB, and C1QC) and MERTK. Murine AAMs, which are transcriptionally similar to Mac2 (A2M), are replenished by CCR2+ monocytes, integrate in the airway epithelium, and exhibit dendriform morphology (57, 58). We therefore performed immunofluorescence staining of day 21 cocultures and confirmed that these cells colocalize with the epithelial basal layer, acquire dendritic projections, and express C1Q and MERTK (Fig. 6E).
We subsequently quantified the number of DEGs (log2FC > 0.5 and FDR < 0.1; Materials and Methods) in MNP subsets after allergen challenge compared with baseline (fig. S5F and data file S6) and found that the transcriptional response was greater in asthmatics compared with ACs (total DEGs = 671 in AAs versus 89 in ACs). MC4 in asthmatics had the greatest transcriptional response to allergen (DEGs = 289), followed by MC2 (DEGs = 122), DC2 (DEG = 94), and Mac2 (DEGs = 48). To identify transcriptional changes in MNPs specific to allergic asthma, we next identified DEGs between groups after allergen challenge (fig. S5G and data file S6). MC2 (SPP1) and MC4 (CCR2) had the most DEGs between groups in response to allergen challenge. In ACs, MC2 (SPP1) up-regulated genes involved in monocyte and Mac chemotaxis and survival (CCL2 and CSF1), phagolysosomal function (MARCO, CD163, and CTSD), and lipid transport and metabolism (ACSL1 and ABCA1) (Fig. 6F). In contrast, MC2 in asthmatics up-regulated genes involved in chemotaxis of immune cells during type 2 inflammation (CCL17 and CCL26), antigen presentation (HLADRB1, HLADPA1, and CIITA), and eicosanoid synthesis (ALOX15 and GGT5). MC4 (CCR2) in ACs also up-regulated genes involved in monocyte and Mac chemotaxis and survival (OSM and CCL4), along with genes that orchestrate tissue repair (HBEGF and VEGFA) and reactive oxidative species clearance (NAMPT and SOD2), whereas asthmatics up-regulated genes implicated in ECM degradation and pathologic remodeling (MMP9, MMP12, and ADAM19) (Fig. 6G).
IPA and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of the DEGs between groups after allergen challenge were used to predict signaling pathways (pORA < 0.1) and upstream regulators that may be driving transcriptional differences between groups, with |z score| > 2 considered significant (Materials and Methods and data file S8). Distinct predicted signaling pathways were identified in MC2 (SPP1) in ACs [phagolysosome, mitogen-activated protein kinase (MAPK), and peroxisome proliferator–activated receptor (PPAR)] compared with asthmatics (antigen processing and presentation, C-type lectin receptor, and Wnt) (fig. S5H). The predicted upstream regulators of MC2 (SPP1) in ACs direct Mac survival and identity and included regulators of CSF1 (NCOA1) and PPARγ signaling (MED1), along with NCOA2, which encodes GRIP1 and promotes an anti-inflammatory Mac phenotype (|z score| = 2.0) (Fig. 6H) (64). In contrast, IL-4 (| z score| = 3.36) and IL-13 (|z score| = 2.45) were the strongest predicted drivers of transcriptional changes in MC2 (SPP1) in asthmatics.
Distinct predicted signaling pathways were also identified in MC4 (CCR2) in ACs [complement cascade, nuclear factor κB (NF-κB), and PPAR] compared with asthmatics [arachidonic acid metabolism and Janus kinase (JAK)–STAT] (fig. S5H). STAT3 (|z score| = 2.12) was the strongest predicted upstream regulator of transcriptional changes in MC4 (CCR2) in ACs, whereas a nonsignificant trend was observed in asthmatics for STAT6 (|z score| = 1.73), which mediates IL-4/IL-13 signaling (Fig. 6I). Many STAT6-induced genes (ALOX15, CCL17, and MMP12) were enriched across MNP clusters in asthmatics after allergen challenge (fig. S5I). These data suggest that, in ACs, the MC phenotype is characterized by autocrine production of factors important in endocytic clearance, Mac differentiation and survival, and expression of trophic factors promoting angiogenesis and tissue repair. In contrast, IL-4/IL-13 signaling via STAT6 in asthmatics may prevent or arrest Mac differentiation and instead direct a pathogenic MC phenotype characterized by up-regulation of genes involved in inflammatory signaling, antigen presentation, and pathologic airway remodeling (summarized in Fig. 6J).
Asthmatic airways are characterized by a pathogenic TH2–MNP–basal cell interactome
Cellular cross-talk between airway epithelial and immune cells is critical to the initiation and resolution phases of allergic inflammation (2–4, 65, 66). We therefore used CellPhoneDB (67) to predict receptor-ligand pairs specific to ACs and asthmatics after allergen challenge (empirical P < 0.001 cutoff; Materials and Methods and data file S11). The greatest number of unique predicted interactions in both groups was between AEC and MNP subsets (Fig. 7A and fig. S6A). In asthmatics, the remaining interactions were dominated by TH2 cells.
Fig. 7. A pathogenic cellular interactome in asthmatic airways after allergen challenge.
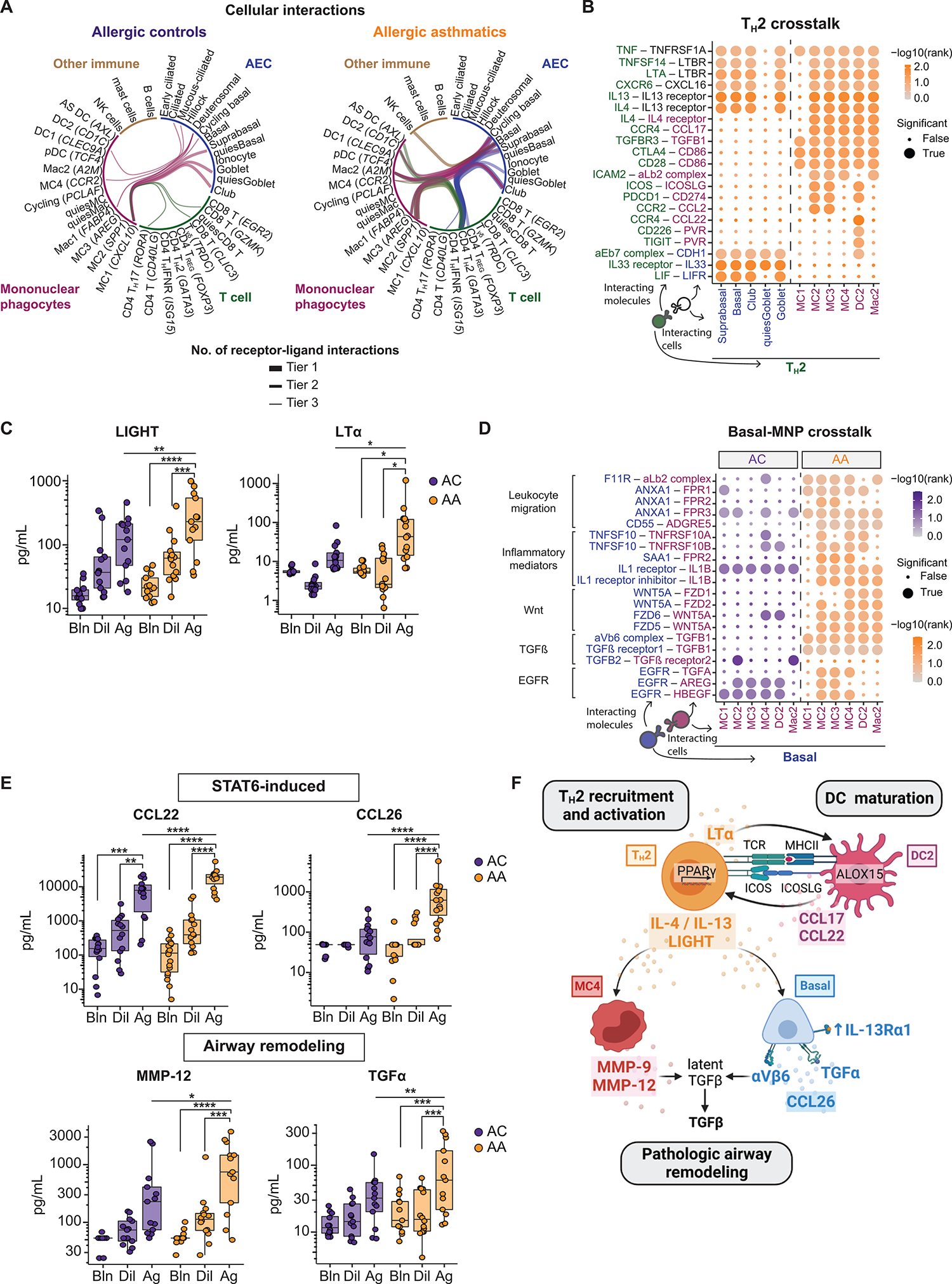
(A) Top 25 cell-cell pairs with the most unique receptor-ligand interactions in ACs and AAs, restricted to interactions between distinct cell lineages. Tiers represent the number of interactions (tier 1: ≥50, tier 2: 40 to 49, tier 3: 30 to 39). (B) Dot plots showing predicted interactions between TH2-AECs and TH2-MNPs after SAC in AAs. (C) BAL concentration of LIGHT (TNFSF14; ACs: n = 13, AAs: n = 14) and LTα (ACs: n = 13, AAs: n = 16). (D) Dot plots showing predicted interactions between AEC-MNP after SAC in AAs and ACs. (E) BAL concentration of CCL22, CCL26, MMP-12, and TGFα (ACs: n = 13, AAs: n = 14). (F) Summary figure of a pathogenic TH2–MNP–basal cell interactome in allergic asthma. In (B) and (D), dot size indicates significance (true: empirical P < 0.001), and color intensity indicates specificity of interaction to disease group [−log10(rank)]. In (C) and (F), boxes represent the median (line) and IQR, with whiskers extending to the remainder of the distribution no more than 1.5× IQR. In (C) and (F), P values were generated using a mixed-effects model with Šidák correction to adjust for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
In asthmatics, TNF family members (TNF, LTA, and TNFSF14) and type 2 cytokines (IL4 and IL13) from TH2 were predicted to interact across AEC and MNP subsets (Fig. 7B and fig. S6B). Protein validation in BAL demonstrated an increase in LIGHT (TNFSF14; 229.1 [84.8 to 584.6] versus 119.6 [37.1 to 211.6] pg/ml, P = 0.006) and LTα (43.3 [12.8 to 124.3] versus 11.7 [7.2 to 26.8] pg/ml, P = 0.03) in asthmatics compared with ACs after allergen challenge (Fig. 7C). TH2 were also predicted to interact with AEC subsets via IL33R/IL1RL1-IL33 and LIFR-LIF. TH2 and MNPs were predicted to interact via receptor-ligand pairs involved in T cell localization (CCR4-CCL17), immune synapse formation (ICAM2–integrin αLβ2 complex), costimulation (CTLA4-CD86 and ICOS-ICOSLG), and T cell polarization (TGFBR3-TGFB1 and CCR2-CCL2) (40, 68). We additionally observed specific predicted interactions between TH2 and DC2, including those important in T cell chemotaxis (CCR4-CCL22) and immunomodulation (CD226-PVR and TIGIT-PVR).
MNPs were predicted to interact with multiple AEC subsets in ACs, whereas basal cells were the primary interacting AEC subset in asthmatics (Fig. 7A). Interactions favoring IL-1 signaling were identified in ACs via CellPhoneDB analysis, as well as by performing linear modeling of the receptor-ligand pairs (FDR < 0.1; Fig. 7D, fig. S6, C to E, Materials and Methods, and data files S11 and S12). Whereas IL1B from MNPs was predicted to interact with its cognate receptor IL1R1 on basal cells in both groups, this cytokine was only predicted to interact with the decoy receptor IL1R2 in asthmatics. IL-1R2 is induced by IL-4/IL-13 and antagonizes the effects of IL-1β (69, 70). In asthmatics, predicted AEC-MNP interactions involved inflammatory leukocyte migration, along with noncanonical Wnt and transforming growth factor β (TGFβ) signaling. TGFβ was predicted to interact with epithelial-specific integrin αvβ6, which mediates TGFβ activation and drives airway structural remodeling (71). TRAIL (TNFSF10) signaling was also enriched in asthmatics and, in addition to its inflammatory effects, can up-regulate metalloproteinase production and promote airway remodeling in allergic asthma (72). EGFR signaling was predicted to occur between basal cells and distinct MNP-derived ligands in asthmatics compared with ACs. Specifically, EGFR ligands known to promote tissue repair (AREG and HBEGF) characterized ACs (73, 74), whereas asthmatics uniquely interacted via TGFA, which promotes epithelial mucin production (31, 32).
To further characterize AEC-MNP cross-talk, we performed NicheNet analysis (75) to identify the regulatory networks between these cell types that distinguish allergy from asthma (Materials and Methods and data file S13). Thus, we defined ligands and their downstream targets as genes that were differentially expressed between asthmatics and ACs after allergen challenge (data file S6). Cellular communication pathways in ACs were characterized by growth factor signaling and the injury-repair response across subsets (fig. S6, F and G). In contrast, asthmatics were dominated by basal-MNP interactions that included STAT6-induced signaling and mediators of airway remodeling. To validate these findings, we quantified BAL protein levels in asthmatics compared with ACs (Fig. 7E and fig. S6H). Protein levels of STAT6-induced chemokines were higher in asthmatics compared with ACs after allergen challenge (CCL22: 19,080.6 [8876.2 to 22,692.5] versus 7927.6 [1301.3 to 13,504.2] pg/ml, P < 0.0001; CCL26: 627.5 [176.1 to 1276.9] versus 76.9 [20.9 to 126.5] pg/ml, P < 0.0001), as were levels of proteins involved in mucin production and pathologic airway remodeling (TGFα: 59.7 [20.1 to 211.4] versus 32.2 [15.6 to 55.2] pg/ml, P = 0.003; matrix metalloproteinase-12 (MMP-12): 745.7 [176.7 to 1957.7] versus 228.6 [69.7 to 493.5] pg/ml, P = 0.02). Overall, these data support a dominant TH2–MNP–basal cell interactome that may override a protective injury-repair response observed in ACs, thereby driving asthma pathobiology (summarized in Fig. 7F).
DISCUSSION
This study directly compared allergic asthmatics with allergic non-asthmatic controls to identify cell subsets and transcriptional effector programs in the lower airway relevant to asthma pathogenesis. In response to allergen challenge, basal and secretory epithelial cells in asthmatics up-regulated genes involved in matrix degradation, mucus metaplasia, and glycolysis and failed to induce antioxidant pathways observed in ACs. Allergen challenge also induced IL9-expressing pathogenic TH2 cells and led to enrichment of DC2 (CD1C) and immature pro-inflammatory MCs in asthmatic airways. Cross-talk between these cell types may be critical to the persistence and amplification of airway inflammation as well as the pathologic structural remodeling that defines allergic asthma.
Whereas SAC leads to increased eosinophils and type 2 cytokine levels in the airways of both AAs and ACs (9), our current study demonstrates that tissue reprogramming in response to type 2 inflammation is a defining feature of asthma (summarized in Fig. 8). This finding is most clearly observed in airway-resident basal and goblet cells. Basal and secretory epithelial cells in asthmatics up-regulated genes involved in type 2 immune cell chemotaxis, mucus metaplasia, and ECM remodeling after SAC. Asthmatic basal cells up-regulated IL13RA1 after allergen challenge, suggesting a possible mechanism of heightened responsiveness of these cells to IL-13. In contrast, allergen challenge induced genes involved in epithelial repair and cytoprotection in ACs. Thus, our data suggest that type 2 programming overrides protective and repair pathways in the asthmatic airway epithelium. It remains unknown whether this programming is induced by persistent type 2 airway inflammation or results from an intrinsic susceptibility of AECs in allergic asthma.
Fig. 8. Transcriptional reprogramming in response to type 2 airway inflammation as a defining feature of allergic asthma.
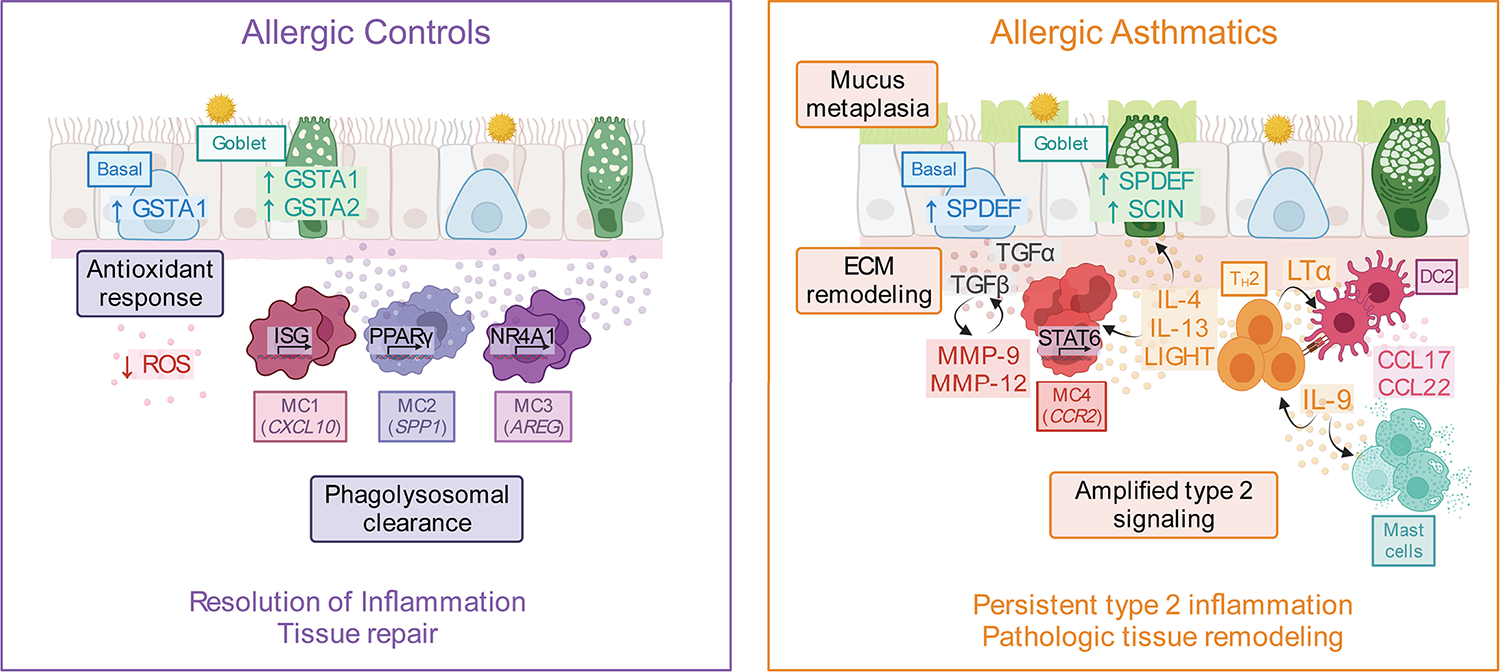
After SAC, the airway landscape in allergic non-asthmatic controls was characterized by enrichment of macrophage-like MCs expressing growth factors associated with tissue repair as well as up-regulation of antioxidant genes in basal and goblet cells. In contrast, asthmatics failed to up-regulate these pathways and instead had enrichment of DC2 (CD1C), pro-inflammatory MC4 (CCR2), and IL9-expressing pathogenic TH2 cells. Type 2 cytokines and other mediators, including TNF family members, produced by TH2 cells may promote a pathogenic MC4 phenotype in asthmatics by interrupting the default MC maturation sequence to proresolution macrophages. These TH2 mediators may also override the epithelial injury response observed in ACs and instead promote ECM degradation, subepithelial fibrosis, and mucus metaplasia in asthma. Figure was created using biorender.com.
Type 2 programming was also a defining feature of MCs recruited to asthmatic airways after SAC. Specifically, asthmatics were enriched for immature MC4 (CCR2) that up-regulated IL-13–induced inflammatory mediators and metalloproteinases. In marked contrast, macrophage-like MCs (MC1, MC2, and MC3) dominated the airway mucosal immune landscape in ACs 24 hours after allergen challenge. The proresolving phenotype observed in MCs from ACs included genes encoding scavenger receptors and phagocytic machinery, trophic mediators that promote epithelial repair, and regulators of lipid metabolism. Monocytes are highly plastic cells that are recruited to the tissue during inflammation and acquire functional roles based on environmental signals. Murine models suggest that these cells are transiently pro-inflammatory but subsequently differentiate into macrophages that resolve inflammation (48, 59, 62). Consistent with this, coculture of blood monocytes with AECs predicted a continuum of differentiation from monocytes to macrophage-like MCs. Together, these results suggest that IL-13 may arrest recently recruited MC4 (CCR2) in a pro-inflammatory state, thereby preventing differentiation into macrophage-like MCs that restore airway homeostasis.
Our study also identified signaling pathways that may amplify type 2 inflammation and critically link airway inflammation to the structural remodeling observed in asthma. IL9-expressing pathogenic TH2 cells were highly specific to asthmatic airways after allergen challenge. IL-9 is known to amplify type 2 inflammation, promote mast cell activation and survival, and drive tissue pathology (40–44). Whereas three previous small trials targeting IL-9 in asthma had mixed results, they included heterogeneous asthma cohorts followed over short time periods (76, 77). Our findings, along with recent murine studies (78), suggest that IL-9 may be specific for the allergen recall response and thus may be an important disease mediator in a targeted subset of patients. Murine models of asthma demonstrate that T cell receptor activation by antigen-presenting DCs after allergen challenge is required for IL9 induction (41). Airway mucosal DC2 were enriched in AAs after SAC and highly expressed ALOX15, a lipoxygenase induced by type 2 cytokines that generates arachidonic acid metabolites. These metabolites can in turn bind to and activate PPARγ, a lipid-activated transcription factor that drives pathogenic TH2 effector responses in the airway (79). Cell-cell interaction analysis highlighted additional DC2-TH2 interactions comprising a feedforward loop of immunotaxis, DC maturation, and TH2 costimulation specific to asthmatic airways. This suggests that local DC2-TH2 cross-talk may establish TH2 residence in the airway, license the pathogenic TH2 phenotype, and promote production of IL-9 via PPARγ activation. A similar DC2-TH2 pathogenic circuit was recently described in atopic dermatitis and persisted despite IL-4Rα blockade (12). Thus, targeting pathways that recruit, activate, and facilitate the persistence of airway mucosal DC2 and TH2 may be necessary to induce remission in allergic disease.
Our data additionally highlight a critical role for TGFα and TNF family members in linking airway inflammation to pathologic tissue remodeling in asthma. TGFα is an IL-13–induced EGFR ligand that promotes epithelial mucin production (31, 32), and TGFA-EGFR signaling was predicted to be unique to asthmatics in our interaction analysis. Interaction analyses also predicted that TNF family members LIGHT and TRAIL interact with MC4 (CCR2) to induce MMP9 expression in asthma. Together with MMP-12, which is induced by type 2 cytokines, these MMPs increase collagen synthesis, degrade ECM, and activate TGFβ (80–83). In addition to being a potent elastase, MMP-12 has important immunoregulatory roles in the lung, including amplification of IL-13 signaling, promotion of eosinophil and MNP chemotaxis, and B cell follicle formation (80, 84). TNF family member lymphotoxin also induces immune cell aggregation at mucosal sites and promotes DC maturation, amplifying local inflammation (63). LTA was highly enriched in pathogenic TH2 cells and associated with DC2 enrichment in asthmatic airways. BAL protein levels of TGFα, LIGHT, MMP-12, and LTα increased in asthmatics but not controls after SAC, further supporting their relevance in asthma. These data reveal a TH2–MNP–epithelial cellular interactome that may drive asthma pathogenesis and suggest that targeting cross-talk between these cell types may be necessary to induce disease remission.
Our study focused on mild asthmatics in the setting of allergen-induced inflammation, and tissue sampling was restricted to the airway mucosa. Thus, our results may not be generalizable to the full spectrum of asthma, and additional studies that include other asthma endotypes and severities and alternative challenge models are warranted. Although the number of participants was limited, we observed highly significant differences in cluster abundances and DGE between groups that were represented across all participants, supporting the relevance of these findings to allergic asthma. We have not previously observed differences in the airway response to SAC due to participant sex or the allergen administered. However, we cannot rule out that some of our findings are attributable to these factors. Cellular origin and function cannot be definitively ascribed on the basis of gene expression alone, and follow-up studies are needed. Last, a single time point after allergen exposure cannot interrogate the full kinetics of airway inflammation and resolution, and analysis of later time points may identify additional pathologic processes in asthma.
Our data indicate that type 2 cytokines may override the “default” epithelial injury-repair program and proresolution macrophage maturation sequence observed in ACs, thereby programming pathogenic niche circuits that amplify and link airway inflammation to remodeling in asthma. Our findings also support the involvement of additional pathways that act in concert with type 2 inflammation to promote asthma, including TNF family signaling, altered cellular metabolism, failure to engage antioxidant responses, loss of growth factor signaling, or a complex interplay between these mechanisms. In sum, we present an integrated analysis of the airway cellular ecosystem in the context of allergen-induced inflammation and identify cell subsets and transcriptional effector programs specific to asthma, thereby advancing the current understanding of asthma pathogenesis and identifying potential targets to achieve sustained therapeutic responses.
MATERIALS AND METHODS
Study design
Participant recruitment and study protocol
This study was a nonrandomized, nonblinded mechanistic study with the primary aim of comparing the airway mucosal response to allergen challenge in AAs and ACs. Volunteers were screened for eligibility with a full medical history, baseline spirometry and methacholine challenge, and allergen skin testing. Detailed inclusion and exclusion criteria can be found in Supplementary Methods, as previously published (9). Briefly, male and female participants between 18 and 50 years old with a clinical history of allergic rhinitis and/or conjunctivitis to HDM and/or cat hair and a positive skin prick test to the same allergen were enrolled. AAs additionally had a clinical history of mild asthma, baseline FEV1 ≥ 75% of the predicted value, and positive methacholine challenge testing defined as a provocative concentration causing a 20% drop in FEV1 (PC20) <16 mg/ml. Participants could not be on systemic steroids for 6 weeks before the study, and inhaled corticosteroids were held for 2 weeks before the study procedure. All participants provided written informed consent before testing and sample collection. The study protocol was approved by the Massachusetts General Brigham Institutional Review Board (IRB# 2007P001050) and registered at clinicaltrials.gov (NCT00595491). A full list of participants is compiled in data file S1.
Allergen skin testing
AAs and ACs had positive SPT to standardized HDM [Dermatophagoides pteronyssinus; Greer Laboratories, 10,000 allergy units (AU) per ml] and/or cat hair extract [Felis catis; Greer Laboratories, 10,000 bioequivalent allergy units (BAU) per ml]. The threshold level of allergen sensitivity was determined by quantitative SPT using serial threefold dilutions of extract. The lowest concentration of extract eliciting a positive skin prick test (3-mm wheal diameter) was used as the allergen dose administered during SAC (9). The same stock of allergen was used for both SPT and SAC.
Segmental allergen challenge
Briefly, baseline BAL samples were obtained by administering 120 ml of normal saline to the lingula. After clearing luminal contents, baseline endobronchial brushings were collected from the lingula using a 4-mm sterile nylon cytology brush under direct visualization (85). Diluent (2 ml) was then administered to the anterior segment of the right upper lobe (RUL) followed by allergen (2 ml) to the lateral segment of the right middle lobe (RML). Twenty-four hours later, BAL and endobronchial brush samples were obtained in a similar manner from the diluent- and allergen-challenged segmental airways.
Cytospins
Cell differential counts for BAL were determined by enumerating eosinophils on cytocentrifuge preparations (9).
Flow cytometry
Endobronchial brushings were obtained as above and stained with 4′,6-diamidino-2-phenylindole (DAPI) for dead cell exclusion and fluorescently conjugated antibodies against CD326, CD45, CD3, CD19, HLA-DR, and CD1c after Fc receptor blockade. Detailed information is provided regarding the antibodies used for flow cytometry staining in table S1. Samples were run on a BD FACSAria Fusion flow cytometer (BD Biosciences) and analyzed with FlowJo v10.7 (Tree Star).
Cytokine quantification
Cytokines were analyzed by MilliplexMAP kit (EMD Millipore) using 30-fold concentrated BAL supernatant (9). BAL was concentrated in Amicon Ultra-15 centrifugal filter units (EMD Millipore, catalog no. UFC9100) or Sartorius Vivaspin 15R centrifugal concentrators (Thermo Fisher Scientific, catalog no. 14-558-499) according to the manufacturer’s instructions. Cytokines were measured on Luminex 200 and analyzed using xPONENT v3.1 (Luminex Corp.).
Human classical monocyte purification
Whole blood was collected from healthy volunteers (IRB# 2002P001658), and peripheral blood mononuclear cells (PBMCs) were separated using Ficoll gradient centrifugation. CD14+ classical monocytes were isolated from PBMCs using magnetic bead isolation (Miltenyi Biotec, catalog no. 130-117-337) in accordance with the manufacturer’s protocol. Excess cells were frozen in CryoStor CS10 (BioLife Solutions, catalog no. 210102) and later thawed and serially diluted in phosphate-buffered saline (PBS) + 2 mM EDTA + 0.5% bovine serum albumin (BSA) solution. Dead cells were magnetically removed (BioLegend, catalog no. 480159), and the remaining cells were resuspended at a concentration of 1000 cells per μl in preparation for loading on the 10X Chromium instrument (10X Genomics).
Human monocyte and airway epithelial coculture and scRNA-seq
Human primary small AECs (Lonza, catalog no. CC-2547) were cultured at ALI on laminin-coated Transwell inserts (Corning, catalog no. 3470) using PneumaCult Ex-Plus followed by ALI medium (STEMCELL Technologies, catalog nos. 05040 and 05001) as previously described (86). CD14+ classical monocytes were added to the apical chamber at a density of 9 × 104 cells per cm2 after an intact ALI was formed. Before dissociation of cocultures at days 4 and 21 for scRNA-seq, excess mucus was removed by adding 75 μl of 10 mM dithiothreitol (MilliporeSigma, catalog no. 43819) in PBS to the apical chamber for 5 min followed by PBS wash (×2). Cocultures were then dissociated with TrypLE Select (Gibco, catalog no. 12563011) for 15 min. Magnetic CD45+ enrichment (STEMCELL Technologies, catalog no. 1000105) was used to increase the fraction of recovered MNPs from dissociated cocultures. All samples underwent magnetic dead cell removal (BioLegend, catalog no. 480159) and were resuspended at a target concentration of 1000 cells per μl in preparation for loading on the 10X Chromium instrument (10X Genomics).
Immunofluorescence staining
Detailed information is provided regarding the antibodies used for immunofluorescence staining in table S1. Staining for MERTK was performed on live cells by first adding Fc receptor blockade for 1 hour before adding anti-human CD45 and anti-human MERTK for 2 hours at 37°C. All other coculture staining was performed after fixation using 4% paraformaldehyde (PFA). Cells were permeabilized with 0.1% Triton X-100 and blocked with 1% BSA–0.3% Triton X-100. Cells were stained with mouse anti-human CD45 (2 hours at 37°C), rat anti-human C1q (overnight at 4°C), and rabbit anti-human p63 (overnight at 4°C). Secondary antibodies were added for 2 hours at 37°C along with Hoechst 33258. Fluorescent images were obtained using an inverted Zeiss LSM780 confocal laser-scanning microscope.
Lung tissue was collected from a lobectomy specimen of a patient with a clinical history of asthma (IRB# 2013P002332). Tissue was fixed overnight in PFA, washed three times, dehydrated in 30% sucrose, embedded in optimal cutting temperature (OCT) (Sakura), and cryosectioned. Slides were stained with antibodies against cytokeratin 13 (KRT13) and acetylated tubulin, and coverslips were mounted in antifade medium containing DAPI. Images were obtained on a ZEISS Axio Imager M2 widefield fluorescent microscope. All images were processed using Fiji or Zen Blue (Zeiss).
Single-cell RNA sequencing and computational data analysis
Preparation of single-cell suspensions from endobronchial brushings
Endobronchial brush samples were collected in phenol-free RPMI 1640 (Thermo Fisher Scientific) with 2% human AB serum and ROCK inhibitor (Y-27632, Tocris Bioscience, catalog no. 1254) on ice and processed within 60 min of collection. Cells were gently removed from cytology brushes and resuspended in phenol-free RPMI 1640. Single-cell suspensions were depleted of dead cells using annexin V–conjugated beads (STEMCELL Technologies, catalog no. 17899) and of red blood cells using antibodies directed against glycophorin A (STEMCELL Technologies, catalog no. 18170). Cell viability was assessed via trypan blue (0.4%) staining and manual cell counting using a hemocytometer both before and after red blood cell and dead cell removal to achieve >95% viability after dead cell removal. Viable cells were resuspended at a concentration of 800 to 1000 cells per μl in preparation for loading on the 10X Chromium controller instrument (10X Genomics).
Single-cell RNA sequencing
About 12,000 single cells were loaded to each 10X channel with a recovery goal of 6000 single cells. Cell suspensions were loaded along with reverse transcriptase reagents, 3′ gel beads, and emulsification oil onto separate channels of a 10X Single Cell B Chip, which was loaded into the 10X Chromium instrument to generate emulsions. Emulsions were transferred to polymerase chain reaction (PCR) strip tubes for immediate processing and reverse transcription. Library preparation was performed according to the manufacturer’s recommendations. Expression libraries were generated either using Chromium Single Cell 3′V3 chemistry (10X Genomics PN-120262; endobronchial brush samples) or Next GEM Single Cell 3′V3.1 chemistry (10X Genomics PN-1000268; coculture samples). DNA and library quality was evaluated using an Agilent 2100 Bioanalyzer, and concentration was quantified using the Qubit dsDNA high-sensitivity reagents (Thermo Fisher Scientific). Gene expression libraries were sequenced on an Illumina NextSeq instrument. Endobronchial brush samples were sequenced on the Illumina NextSeq 500/550 system using the following single-index sequencing configuration: read 1 = 28 base pairs (bp) [cell barcode, unique molecular identifier (UMI)], read 2 = 56 bp (insert), index 1 = 8 bp (sample index), and index 2 = 0 bp. Coculture samples were sequenced on the Illumina NextSeq 2000 system using the following dual-index sequencing configuration: read 1 = 28 bp (cell barcode, UMI), read 2 = 96 bp (insert), index 1 = 10 bp (sample index), and index 2 = 10 bp (sample index). All sequencing statistics are compiled in data file S2. Detailed methods for read alignment and quantification, cell clustering, marker gene identification, DGE analysis, and all other downstream computational analyses can be found in Supplementary Methods.
Statistical analysis
Statistical approaches for computational analyses of scRNA-seq data are detailed in Supplementary Methods. Mixed-effects modeling with repeated measures was used to compare median differences in cellular proportions and protein levels between groups unless otherwise specified, where experimental condition and disease group were considered fixed effects and each participant was treated as a random effect. Adjustment for multiple comparisons was performed using Šidák’s correction. A two-sided P < 0.05 was considered significant.
Supplementary Material
Acknowledgments:
We thank the staff of the Massachusetts General Hospital Pulmonary Special Procedures Unit and Translational and Clinical Research Centers, clinical research coordinators (M. Kone, N. Yang, A. N. Dartley, C. Fromson, G. Lopes, and E. White), D. L. Hamilos, and C. Leary.
Funding:
Funding for this study was provided by NIH grants 5KL2TR002542-04 (to J.A.), T32HL116275 (J.A. and T.K.), T32AR007258 (to K.S.), K08HL140173 (to R.A.R.), R01AI040618 and R01AI168131 (to A.D.L.), DP2CA247831 (to A.-C.V.), and UH2AI44434-01 (to J.L.C.); Massachusetts General Hospital Transformative Scholar in Medicine Award (to A.-C.V.); Department of Defense PR150903/W81XWH-16-1-0493 (to B.D.M.); and Sanofi iAward (to B.D.M.). A portion of the data presented here was obtained at the Bioinformatics Division of the Iowa Institute of Human Genetics, which is supported, in part, by the University of Iowa Carver College of Medicine. This work was conducted with support (KL2, Harvard Catalyst Clinical Research Center) from Harvard Catalyst | the Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, NIH Award grants KL2TR002542 and UL1TR002541). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, or the NIH. None of these funders had a role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Competing interests: L.P.H. reports grants from Boehringer Ingelheim and has received personal consulting fees from Boehringer Ingelheim, Pliant Therapeutics, Biogen Idec, and Bioclinica. R.J.X. is a cofounder of Celsius Therapeutics and Jnana Therapeutics, a director at MoonLake Immunotherapeutics, and a scientific advisory board member at Nestle. B.D.M. receives research funding from and served as a consultant for Sanofi and Regeneron. The other authors declare that they have no competing interests.
Data and materials availability:
scRNA-seq count matrices and related data are deposited in the GEO database (under accession number GSE193816), and raw human sequencing data are available in the controlled access repository dbGaP (www.ncbi.nlm.nih.gov/gap/) under accession number phs003101.v1.p1. Source code for data analysis in each main figure is available on GitHub (https://github.com/villani-lab/airway_allergic_asthma) and has been archived to Zenodo (https://zenodo.org/badge/latestdoi/412260708). A user-friendly portal is available to browse the single-cell data generated in this manuscript. Users can query the single-cell clustering solutions, visualize gene expression, and browse differential expression of genes. Users may navigate the portal by visiting https://villani.mgh.harvard.edu/allergy-asthma.
REFERENCES AND NOTES
- 1.Centers for Disease Control and Prevention, CDC Virtal Signs—Asthma in the US (Centers for Disease Control and Prevention, 2011); www.cdc.gov/vitalsigns/asthma/index.html. [Google Scholar]
- 2.Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD, Asthma. Nat. Rev. Dis. Primers 1, 15025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammad H, Lambrecht BN, The basic immunology of asthma. Cell 184, 1469–1485 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Hewitt RJ, Lloyd CM, Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 21, 347–362 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MAM, Kool M, Muskens F, Lambrecht BN, Inflammatory dendritic cells—not basophils—are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J. Exp. Med. 207, 2097–2111 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rahimi RA, Nepal K, Cetinbas M, Sadreyev RI, Luster AD, Distinct functions of tissue-resident and circulating memory Th2 cells in allergic airway disease. J. Exp. Med. 217, e20190865 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jahnsen FL, Moloney ED, Hogan T, Upham JW, Burke CM, Holt PG, Rapid dendritic cell recruitment to the bronchial mucosa of patients with atopic asthma in response to local allergen challenge. Thorax 56, 823–826 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Djukanović R, Roche WR, Wilson JW, Beasley CR, Twentyman OP, Howarth RH, Holgate ST, Mucosal inflammation in asthma. Am. Rev. Respir. Dis. 142, 434–457 (1990). [DOI] [PubMed] [Google Scholar]
- 9.Cho JL, Ling MF, Adams DC, Faustino L, Islam SA, Afshar R, Griffith JW, Harris RS, Ng A, Radicioni G, Ford AA, Han AK, Xavier R, Kwok WW, Boucher R, Moon JJ, Hamilos DL, Kesimer M, Suter MJ, Medoff BD, Luster AD, Allergic asthma is distinguished by sensitivity of allergen-specific CD4+ T cells and airway structural cells to type 2 inflammation. Sci. Transl. Med. 8, 359ra132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thornton EE, Looney MR, Bose O, Sen D, Sheppard D, Locksley R, Huang X, Krummel MF, Spatiotemporally separated antigen uptake by alveolar dendritic cells and airway presentation to T cells in the lung. J. Exp. Med. 209, 1183–1199 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lilly CM, Tateno H, Oguma T, Israel E, Sonna LA, Effects of allergen challenge on airway epithelial cell gene expression. Am. J. Respir. Crit. Care Med. 171, 579–586 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Bangert C, Rindler K, Krausgruber T, Alkon N, Thaler FM, Kurz H, Ayub T, Demirtas D, Fortelny N, Vorstandlechner V, Bauer WM, Quint T, Mildner M, Jonak C, Elbe-Bürger A, Griss J, Bock C, Brunner PM, Persistence of mature dendritic cells, TH2A, and Tc2 cells characterize clinically resolved atopic dermatitis under IL-4Rα blockade. Sci. Immunol. 6, eabe2749 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Dwyer DF, Ordovas-Montanes J, Allon SJ, Buchheit KM, Vukovic M, Derakhshan T, Feng C, Lai J, Hughes TK, Nyquist SK, Giannetti MP, Berger B, Bhattacharyya N, Roditi RE, Katz HR, Nawijn MC, Berg M, van den Berge M, Laidlaw TM, Shalek AK, Barrett NA, Boyce JA, Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation. Sci. Immunol. 6, eabb7221 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, Wadsworth MH, Hughes TK, Kazer SW, Yoshimoto E, Cahill KN, Bhattacharyya N, Katz HR, Berger B, Laidlaw TM, Boyce JA, Barrett NA, Shalek AK, Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 560, 649–654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan DM, Ruiter B, Smith NP, Tu AA, Monian B, Stone BE, Virk-Hundal N, Yuan Q, Shreffler WG, Love JC, Clonally expanded, GPR15-expressing pathogenic effector TH2 cells are associated with eosinophilic esophagitis. Sci. Immunol. 6, eabi5586 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, Brouwer S, Gomes T, Hesse L, Jiang J, Fasouli ES, Efremova M, Vento-Tormo R, Talavera-López C, Jonker MR, Affleck K, Palit S, Strzelecka PM, Firth HV, Mahbubani KT, Cvejic A, Meyer KB, Saeb-Parsy K, Luinge M, Brandsma C-A, Timens W, Angelidis I, Strunz M, Koppelman GH, van Oosterhout AJ, Schiller HB, Theis FJ, van den Berge M, Nawijn MC, Teichmann SA, A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 25, 1153–1163 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Gauvreau GM, Davis BE, Scadding G, Boulet L-P, Bjermer L, Chaker A, Cockcroft DW, Dahlén B, Fokkens W, Hellings P, Lazarinis N, O’Byrne PM, Tufvesson E, Quirce S, Van Maaren M, de Jongh FH, Diamant Z, Allergen provocation tests in respiratory research: Building on 50 years of experience. Eur. Respir. J. 60, 2102782, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diamant Z, Gauvreau GM, Cockcroft DW, Boulet L-P, Sterk PJ, de Jongh FHC, Dahlén B, O’Byrne PM, Inhaled allergen bronchoprovocation tests. J. Allergy Clin. Immunol. 132, 1045–1055.e6 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Adams DC, Miller AJ, Applegate MB, Cho JL, Hamilos DL, Chee A, Holz JA, Szabari MV, Hariri LP, Harris RS, Griffith JW, Luster AD, Medoff BD, Suter MJ, Quantitative assessment of airway remodelling and response to allergen in asthma. Respirology 24, 1073–1080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fonseka CY, Rao DA, Teslovich NC, Korsunsky I, Hannes SK, Slowikowski K, Gurish MF, Donlin LT, Lederer JA, Weinblatt ME, Massarotti EM, Coblyn JS, Helfgott SM, Todd DJ, Bykerk VP, Karlson EW, Ermann J, Lee YC, Brenner MB, Raychaudhuri S, Mixed-effects association of single cells identifies an expanded effector CD4+ T cell subset in rheumatoid arthritis. Sci. Transl. Med. 10, eaaq0305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitsett JA, Kalin TV, Xu Y, Kalinichenko VV, Building and regenerating the lung cell by cell. Physiol. Rev. 99, 513–554 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis JD, Wypych TP, Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol. 14, 978–990 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket S, Yuan F, Chen S, Leung HM, Villoria J, Rogel N, Burgin G, Tsankov A, Waghray A, Slyper M, Waldmann J, Nguyen L, Dionne D, Rozenblatt-Rosen O, Tata PR, Mou H, Shivaraju M, Bihler H, Mense M, Tearney GJ, Rowe SM, Engelhardt JF, Regev A, Rajagopal J, A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, Klein AM, Jaffe AB, A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deprez M, Zaragosi L-E, Truchi M, Becavin C, Ruiz García S, Arguel M-J, Plaisant M, Magnone V, Lebrigand K, Abelanet S, Brau F, Paquet A, Pe’er D, Marquette C-H, Leroy S, Barbry P, A single-cell atlas of the human healthy airways. Am. J. Respir. Crit. Care Med. 202, 1636–1645 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Park K-S, Korfhagen TR, Bruno MD, Kitzmiller JA, Wan H, Wert SE, Khurana Hershey GK, Chen G, Whitsett JA, SPDEF regulates goblet cell hyperplasia in the airway epithelium. J. Clin. Invest. 117, 978–988 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goleva E, Berdyshev E, Leung DY, Epithelial barrier repair and prevention of allergy. J. Clin. Invest. 129, 1463–1474 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Litosh VA, Rochman M, Rymer JK, Porollo A, Kottyan LC, Rothenberg ME, Calpain-14 and its association with eosinophilic esophagitis. J. Allergy Clin. Immunol. 139, 1762–1771.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang IV, Pedersen BS, Liu AH, O’Connor GT, Pillai D, Kattan M, Misiak RT, Gruchalla R, Szefler SJ, Khurana Hershey GK, Kercsmar C, Richards A, Stevens AD, Kolakowski CA, Makhija M, Sorkness CA, Krouse RZ, Visness C, Davidson EJ, Hennessy CE, Martin RJ, Togias A, Busse WW, Schwartz DA, The nasal methylome and childhood atopic asthma. J. Allergy Clin. Immunol. 139, 1478–1488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagami Y, Favoreto S, Zhen G, Park S-W, Nguyenvu LT, Kuperman DA, Dolganov GM, Huang X, Boushey HA, Avila PC, Erle DJ, The epithelial anion transporter pendrin is induced by allergy and rhinovirus infection, regulates airway surface liquid, and increases airway reactivity and inflammation in an asthma model. J. Immunol. 181, 2203–2210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burgel P, Nadel J, Roles of epidermal growth factor receptor activation in epithelial cell repair and mucin production in airway epithelium. Thorax 59, 992–996 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeyama K, Dabbagh K, Lee HM, Agustí C, Lausier JA, Ueki IF, Grattan KM, Nadel JA, Epidermal growth factor system regulates mucin production in airways. Proc. Natl. Acad. Sci. U.S.A. 96, 3081–3086 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seumois G, Ramírez-Suástegui C, Schmiedel BJ, Liang S, Peters B, Sette A, Vijayanand P, Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci. Immunol. 5, eaba6087 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wambre E, Bajzik V, DeLong JH, O’Brien K, Nguyen Q-A, Speake C, Gersuk VH, DeBerg HA, Whalen E, Ni C, Farrington M, Jeong D, Robinson D, Linsley PS, Vickery BP, Kwok WW, A phenotypically and functionally distinct human TH2 cell subpopulation is associated with allergic disorders. Sci. Transl. Med. 9, eaam9171 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruiter B, Smith NP, Monian B, Tu AA, Fleming E, Virkud YV, Patil SU, Whittaker CA, Love JC, Shreffler WG, Expansion of the CD4+ effector T-cell repertoire characterizes peanut-allergic patients with heightened clinical sensitivity. J. Allergy Clin. Immunol. 145, 270–282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho S-H, Lerner H, Friedman AL, Shen Y, Farber DL, Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tibbitt CA, Stark JM, Martens L, Ma J, Mold JE, Deswarte K, Oliynyk G, Feng X, Lambrecht BN, De Bleser P, Nylén S, Hammad H, Arsenian Henriksson M, Saeys Y, M J. Coquet, Single-cell RNA sequencing of the T helper cell response to house dust mites defines a distinct gene expression signature in airway Th2 cells. Immunity 51, 169–184.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Kiner E, Willie E, Vijaykumar B, Chowdhary K, Schmutz H, Chandler J, Schnell A, Thakore PI, LeGros G, Mostafavi S, Mathis D, Benoist C; Immunological Genome Project Consortium, Gut CD4+ T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nat. Immunol. 22, 216–228 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cano-Gamez E, Soskic B, Roumeliotis TI, So E, Smyth DJ, Baldrighi M, Willé D, Nakic N, Esparza-Gordillo J, Larminie CGC, Bronson PG, Tough DF, Rowan WC, Choudhary JS, Trynka G, Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4+ T cells to cytokines. Nat. Commun. 11, 1801 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Angkasekwinai P, Dong C, IL-9-producing T cells: Potential players in allergy and cancer. Nat. Rev. Immunol. 21, 37–48 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Micossé C, von Meyenn L, Steck O, Kipfer E, Adam C, Simillion C, Seyed Jafari SM, Olah P, Yawlkar N, Simon D, Borradori L, Kuchen S, Yerly D, Homey B, Conrad C, Snijder B, Schmidt M, Schlapbach C, Human “TH9” cells are a subpopulation of PPAR-γ+ TH2 cells. Sci. Immunol. 4, eaat5943 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Kearley J, Erjefalt JS, Andersson C, Benjamin E, Jones CP, Robichaud A, Pegorier S, Brewah Y, Burwell TJ, Bjermer L, Kiener PA, Kolbeck R, Lloyd CM, Coyle AJ, Humbles AA, IL-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. Am. J. Respir. Crit. Care Med. 183, 865–875 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steenwinckel V, Louahed J, Lemaire MM, Sommereyns C, Warnier G, McKenzie A, Brombacher F, Van Snick J, Renauld J-C, IL-9 promotes IL-13-dependent paneth cell hyperplasia and up-regulation of innate immunity mediators in intestinal mucosa. J. Immunol. 182, 4737–4743 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Temann U-A, Ray P, Flavell RA, Pulmonary overexpression of IL-9 induces Th2 cytokine expression, leading to immune pathology. J. Clin. Invest. 109, 29–39 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andersson CK, Mori M, Bjermer L, Löfdahl C-G, Erjefält JS, Novel site-specific mast cell subpopulations in the human lung. Thorax 64, 297–305 (2009). [DOI] [PubMed] [Google Scholar]
- 46.van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, Hoogsteden HC, Lambrecht BN, In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 201, 981–991 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K, Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jakubzick CV, Randolph GJ, Henson PM, Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 17, 349–362 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S, Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 14, 571–578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, Jiang Y, Kass DJ, Gibson K, Chen W, Mora A, Benos PV, Rojas M, Lafyatis R, Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 54, 1802441 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mould KJ, Moore CM, McManus SA, McCubbrey AL, McClendon JD, Griesmer CL, Henson PM, Janssen WJ, Airspace macrophages and monocytes exist in transcriptionally distinct subsets in healthy adults. Am. J. Respir. Crit. Care Med. 203, 946–956 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen C-I, Ren Z, Verma R, Abdala-Valencia H, Nam K, Chi M, Han S, Gonzalez-Gonzalez FJ, Soberanes S, Watanabe S, Williams KJN, Flozak AS, Nicholson TT, Morgan VK, Winter DR, Hinchcliff M, Hrusch CL, Guzy RD, Bonham CA, Sperling AI, Bag R, Hamanaka RB, Mutlu GM, Yeldandi AV, Marshall SA, Shilatifard A, Amaral LAN, Perlman H, Sznajder JI, Argento AC, Gillespie CT, Dematte J, Jain M, Singer BD, Ridge KM, Lam AP, Bharat A, Bhorade SM, Gottardi CJ, Budinger GRS, Misharin AV, Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 199, 1517–1536 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villar J, Segura E, Decoding the heterogeneity of human dendritic cell subsets. Trends Immunol. 41, 1062–1071 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Bourdely P, Anselmi G, Vaivode K, Ramos RN, Missolo-Koussou Y, Hidalgo S, Tosselo J, Nuñez N, Richer W, Vincent-Salomon A, Saxena A, Wood K, Lladser A, Piaggio E, Helft J, Guermonprez P, Transcriptional and functional analysis of CD1c+ human dendritic cells identifies a CD163+ subset priming CD8+CD103+ T cells. Immunity 53, 335–352.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dutertre C-A, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, Ng PY, van den Hoogen LL, Leong JY, Lee B, Chevrier M, Zhang XM, Yong PJA, Koh G, Lum J, Howland SW, Mok E, Chen J, Larbi A, Tan HKK, Lim TKH, Karagianni P, Tzioufas AG, Malleret B, Brody J, Albani S, van Roon J, Radstake T, Newell EW, Ginhoux F, Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity 51, 573–589.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Engler AE, Ysasi AB, Pihl RMF, Villacorta-Martin C, Heston HM, Richardson HMK, Thapa BR, Moniz NR, Belkina AC, Mazzilli SA, Rock JR, Airway-associated macrophages in homeostasis and repair. Cell Rep. 33, 108553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang X-Z, Kreuk LSM, Cho C, Metzger RJ, Allen CDC, Bronchus-associated macrophages efficiently capture and present soluble inhaled antigens and are capable of local Th2 cell activation. eLife 11, e63296 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guilliams M, Mildner A, Yona S, Developmental and functional heterogeneity of monocytes. Immunity 49, 595–613 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Hilgendorf I, Gerhardt LMS, Tan TC, Winter C, Holderried TAW, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK, Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 114, 1611–1622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koenis DS, Medzikovic L, van Loenen PB, van Weeghel M, Huveneers S, Vos M, Evers-van Gogh IJ, Van den Bossche J, Speijer D, Kim Y, Wessels L, Zelcer N, Zwart W, Kalkhoven E, de Vries CJ, Nuclear receptor Nur77 limits the macrophage inflammatory response through transcriptional reprogramming of mitochondrial metabolism. Cell Rep. 24, 2127–2140.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puttur F, Gregory LG, Lloyd CM, Airway macrophages as the guardians of tissue repair in the lung. Immunol. Cell Biol. 97, 246–257 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Upadhyay V, Fu Y-X, Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat. Rev. Immunol. 13, 270–279 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coppo M, Chinenov Y, Sacta MA, Rogatsky I, The transcriptional coregulator GRIP1 controls macrophage polarization and metabolic homeostasis. Nat. Commun. 7, 12254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holgate ST, Lackie P, Wilson S, Roche W, Davies D, Bronchial epithelium as a key regulator of airway allergen sensitization and remodeling in asthma. Am. J. Respir. Crit. Care Med. 162, S113–S117 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Lambrecht BN, Hammad H, Allergens and the airway epithelium response: Gateway to allergic sensitization. J. Allergy Clin. Immunol. 134, 499–507 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R, CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ, Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 404, 407–411 (2000). [DOI] [PubMed] [Google Scholar]
- 69.Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Giri JG, Dower SK, Sims JE, Mantovani A, Interleukin-1 type II receptor: A decoy target for IL-1 that is regulated by IL-4. Science 261, 472–475 (1993). [DOI] [PubMed] [Google Scholar]
- 70.Colotta F, Re F, Muzio M, Polentarutti N, Minty A, Caput D, Ferrara P, Mantovani A, Interleukin-13 induces expression and release of interleukin-1 decoy receptor in human polymorphonuclear cells. J. Biol. Chem. 269, 12403–12406 (1994). [PubMed] [Google Scholar]
- 71.Makinde T, Murphy RF, Agrawal DK, The regulatory role of TGF-beta in airway remodeling in asthma. Immunol. Cell Biol. 85, 348–356 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Collison A, Li J, Pereira de Siqueira A, Zhang J, Toop HD, Morris JC, Foster PS, Mattes J, Tumor necrosis factor-related apoptosis-inducing ligand regulates hallmark features of airways remodeling in allergic airways disease. Am. J. Respir. Cell Mol. Biol. 51, 86–93 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Dao DT, Anez-Bustillos L, Adam RM, Puder M, Bielenberg DR, Heparin-binding epidermal growth factor–like growth factor as a critical mediator of tissue repair and regeneration. Am. J. Pathol. 188, 2446–2456 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zaiss DMW, Gause WC, Osborne LC, Artis D, Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42, 216–226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Browaeys R, Saelens W, Saeys Y, NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Oh CK, Leigh R, McLaurin KK, Kim K, Hultquist M, Molfino NA, A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir. Res. 14, 93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parker JM, Oh CK, LaForce C, Miller SD, Pearlman DS, Le C, Robbie GJ, White WI, White B, Molfino NA; MEDI-528 Clinical Trials Group, Safety profile and clinical activity of multiple subcutaneous doses of MEDI-528, a humanized anti-interleukin-9 monoclonal antibody, in two randomized phase 2a studies in subjects with asthma. BMC Pulm. Med. 11, 14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ulrich BJ, Kharwadkar R, Chu M, Pajulas A, Muralidharan C, Koh B, Fu Y, Gao H, Hayes TA, Zhou H-M, Goplen NP, Nelson AS, Liu Y, Linnemann AK, Turner MJ, Licona-Limón P, Flavell RA, Sun J, Kaplan MH, Allergic airway recall responses require IL-9 from resident memory CD4+ T cells. Sci. Immunol. 7, eabg9296 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nobs SP, Natali S, Pohlmeier L, Okreglicka K, Schneider C, Kurrer M, Sallusto F, Kopf M, PPARγ in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J. Exp. Med. 214, 3015–3035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, Rabach ME, Shipley JM, Shapiro SD, Senior RM, Elias JA, Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and −12 in IL-13-induced inflammation and remodeling. J. Clin. Invest. 110, 463–474 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doherty TA, Soroosh P, Khorram N, Fukuyama S, Rosenthal P, Cho JY, Norris PS, Choi H, Scheu S, Pfeffer K, Zuraw BL, Ware CF, Broide DH, Croft M, The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nat. Med. 17, 596–603 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weckmann M, Collison A, Simpson JL, Kopp MV, Wark PAB, Smyth MJ, Yagita H, Matthaei KI, Hansbro N, Whitehead B, Gibson PG, Foster PS, Mattes J, Critical link between TRAIL and CCL20 for the activation of TH2 cells and the expression of allergic airway disease. Nat. Med. 13, 1308–1315 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Atkinson JJ, Senior RM, Matrix metalloproteinase-9 in lung remodeling. Am. J. Respir. Cell Mol. Biol. 28, 12–24 (2003). [DOI] [PubMed] [Google Scholar]
- 84.Pyle CJ, Patel DF, Peiró T, Joulia R, Grabiec AM, Hussell T, Tavernier G, Simpson A, Pease J, Harker JA, Lloyd CM, Snelgrove RJ, Matrix metalloproteinase-12 supports pulmonary B cell follicle formation and local antibody responses during asthma. Am. J. Respir. Crit. Care Med. 206, 1424–1428 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Corleis B, Cho JL, Gates SJ, Linder AH, Dickey A, Lisanti-Park AC, Schiff AE, Ghebremichael M, Kohli P, Winkler T, Harris RS, Medoff BD, Kwon DS, Smoking and human immunodeficiency virus 1 infection promote retention of CD8+ T cells in the airway mucosa. Am. J. Respir. Cell Mol. Biol. 65, 513–520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Medoff BD, Seung E, Hong S, Thomas SY, Sandall BP, Duffield JS, Kuperman DA, Erle DJ, Luster AD, CD11b+ myeloid cells are the key mediators of Th2 cell homing into the airway in allergic inflammation. J. Immunol. 182, 623–635 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH, Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fleming SJ, Chaffin MD, Arduini A, Akkad A-D, Banks E, Marioni JC, Philippakis AA, Ellinor PT, Babadi M, Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. bioRxiv 791699 [Preprint]. 12 July 2022. 10.1101/791699. [DOI] [PubMed] [Google Scholar]
- 89.Li B, Gould J, Yang Y, Sarkizova S, Tabaka M, Ashenberg O, Rosen Y, Slyper M, Kowalczyk MS, Villani A-C, Tickle T, Hacohen N, Rozenblatt-Rosen O, Regev A, Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq. Nat. Methods 17, 793–798 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wolock SL, Lopez R, Klein AM, Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, Raychaudhuri S, Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Traag VA, Waltman L, van Eck NJ, From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McInnes L, Healy J, Melville J, UMAP: Uniform Manifold Approximation and Projection for dimension reduction. arXiv:1802.03426 [stat.ML] (18 September 2020). [Google Scholar]
- 94.Reyes M, Filbin MR, Bhattacharyya RP, Billman K, Eisenhaure T, Hung DT, Levy BD, Baron RM, Blainey PC, Goldberg MB, Hacohen N, An immune-cell signature of bacterial sepsis. Nat. Med. 26, 333–340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lun ATL, Bach K, Marioni JC, Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gu Z, Eils R, Schlesner M, Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, Sud M, Andrews E, Velonias G, Haber AL, Jagadeesh K, Vickovic S, Yao J, Stevens C, Dionne D, Nguyen LT, Villani A-C, Hofree M, Creasey EA, Huang H, Rozenblatt-Rosen O, Garber JJ, Khalili H, Desch AN, Daly MJ, Ananthakrishnan AN, Shalek AK, Xavier RJ, Regev A, Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, Fan J, Borm LE, Liu Z, van Bruggen D, Guo J, He X, Barker R, Sundström E, Castelo-Branco G, Cramer P, Adameyko I, Linnarsson S, Kharchenko PV, RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gu Z, Gu L, Eils R, Schlesner M, Brors B, Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scRNA-seq count matrices and related data are deposited in the GEO database (under accession number GSE193816), and raw human sequencing data are available in the controlled access repository dbGaP (www.ncbi.nlm.nih.gov/gap/) under accession number phs003101.v1.p1. Source code for data analysis in each main figure is available on GitHub (https://github.com/villani-lab/airway_allergic_asthma) and has been archived to Zenodo (https://zenodo.org/badge/latestdoi/412260708). A user-friendly portal is available to browse the single-cell data generated in this manuscript. Users can query the single-cell clustering solutions, visualize gene expression, and browse differential expression of genes. Users may navigate the portal by visiting https://villani.mgh.harvard.edu/allergy-asthma.