

Abstract
Prostate cancer remains a major cause of mortality and morbidity, affecting millions of men, with a large percentage expected to develop the disease as they reach advanced ages. Treatment and management advances have been dramatic over the past fifty years or so, and one aspect of these improvements is reflected in the multiple advances in diagnostic imaging techniques. Much attention has been focused on molecular imaging techniques that offer high sensitivity and specificity and can now more accurately assess disease status and detect recurrence earlier. During development of molecular imaging probes, single-photon emission computed tomography (SPECT) and positron emission tomography (PET) must be evaluated in preclinical models of the disease. If such agents are to be translated to the clinic, where patients undergoing these imaging modalities are injected with a molecular imaging probe, these agents must first be approved by the FDA and other regulatory agencies prior to their adoption in clinical practice. Scientists have worked assiduously to develop preclinical models of prostate cancer that are relevant to the human disease to enable testing of these probes and related targeted drugs. Challenges in developing reproducible and robust models of human disease in animals are beset with practical issues such as the lack of natural occurrence of prostate cancer in mature male animals, the difficulty of initiating disease in immune-competent animals and the sheer size differences between humans and conveniently smaller animals such as rodents. Thus, compromises in what is ideal and what can be achieved have had to be made. The workhorse of preclinical animal models has been, and remains, the investigation of human xenograft tumor models in athymic immunocompromised mice. Later models have used other immunocompromised models as they have been found and developed, including the use of directly derived patient tumor tissues, completely immunocompromised mice, orthotopic methods for inducing prostate cancer within the mouse prostate itself and metastatic models of advanced disease. These models have been developed in close parallel with advances in imaging agent chemistries, radionuclide developments, computer electronics advances, radiometric dosimetry, biotechnologies, organoid technologies, advances in in vitro diagnostics, and overall deeper understandings of disease initiation, development, immunology, and genetics. The combination of molecular models of prostatic disease with radiometric-based studies in small animals will always remain spatially limited due to the inherent resolution sensitivity limits of PET and SPECT decay processes, fundamentally set at around a 0.5 cm resolution limit. Nevertheless, it is central to researcher’s efforts and to successful clinical translation that the best animal models are adopted, accepted, and scientifically verified as part of this truly interdisciplinary approach to addressing this important disease.
Graphical Abstract
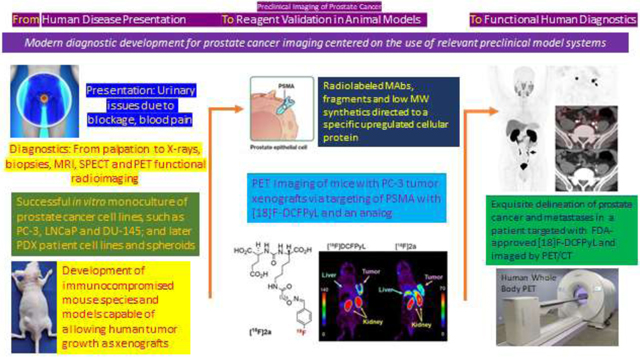
INTRODUCTION
In a general context, preclinical studies on prostate cancer imaging represents one example of a broader challenge related to the need to establish relevant preclinical models for human cancers as a requisite prelude to clinical testing. Historically, the first animal model considerations applied to drug testing and concentrated typically on in vitro binding studies followed by in vivo testing of drug candidates in animal survival studies.1,2 As such, model systems were an essential step in the development, testing and validation of new therapy agents eventually meant for use in patients. With the recent shift of making cancer therapeutics more molecularly targeted to the tumors being treated, there has been an increasing trend of linking diagnostics to therapeutics. Typically done with pairs of drugs that are chemically similar, such agent pairs are most often termed ‘theranostics’ (or theragnostics) and their use aims to improve patient management by confirming disease prior to treatment decisions or improve the safety and performance of the therapies.3 Clearly, the topic of this review forms part of a much larger picture of preclinical research and development in modern oncology. Prostate cancer, however, represents a leading example of the successful development of modern research efforts against human cancer in that the disease history and efforts to ameliorate it are relatively long, the disease is very common making new treatments highly impactful, the progress of the disease is fairly well understood, and its biology and biochemical progress has been somewhat elucidated. Thus, there are now clearly addressable antigens, receptors and mechanisms that can be logically targeted. These include those molecules that are specific to prostate cancer biology and also those that are seen more generally in other tumor types.
BACKGROUND AND HISTORICAL PROSTATE CANCER CLINICAL TREATMENTS
Historically, most of the therapeutic agents currently used for prostate and other cancers were discovered and applied to patients before the development of sophisticated imaging agents.4 Early and localized disease is typically treated with surgery or external beam radiation, sometimes supplemented by adjuvant hormonal or radiotherapies, while more advanced disease had been treated with systemic treatments such as chemotherapies.5 Diagnostic radiology typically plays a supportive role in diagnosis as well as treatment planning/response evaluation and has benefited from numerous advances in multiple technologies to bring the field to its current state of knowledge and competence. The broader history of the field’s development lies far beyond the scope of this review, but the reader is referred to excellent discussions of a scientific and a more general nature regarding cancer and its related treatment and diagnoses.6,7
Prostate cancer is considered a disease of older men although it can occur in younger men who carry one or more risk factors. Clinically, patients can present with difficulties in urination due to urinary obstruction at the urethra from an enlarged prostate, but many patients are completely asymptomatic. Together with the patient’s history, the first indication of prostate cancer is often from abnormalities noted on screening exams such as a digital rectal examination performed by physicians, or blood tumor markers such as the prostate specific antigen (PSA). However, a benign growth patten, termed benign prostate hyperplasia (BPH) may also be the cause of the anatomic growth and symptoms, and patients are often subjected to invasive procedures such as needle biopsies performed on multiple parts of the prostate gland to differentiate the diagnosis with tissues sent for pathological work-up for a definitive positive diagnosis. More recently, magnetic resonance imaging (MRI) may be added to this general paradigm, when available, and is excellent in defining the extent of tumor growth more accurately into and through the surrounding tissues, and to identify specific targets for biopsy. This information may be crucial to decisions regarding individual patient management. While this multiplicity of tests strongly assists the physicians in their courses of action it should be understood that all are anatomic tests, backed up with the definitive results from the pathologist experienced in detecting differences in the normal and non-normal appearance of cellular structures. None of these tests address the modern molecular medicine concept of testing that relies on identifying biochemical differences between normal and cancerous cells, and that forms the basis of the detailed discussions below.
Historically, detection of prostate cancer would direct the patient to a surgeon to remove the prostate gland or a radiation oncologist to apply radiation to the prostate gland and most common areas of spread within the pelvis. In advanced cases this could be combined with reduction in androgens, also known as Androgen Deprivation Therapy (ADT). These treatments, in most cases, thwart the disease’s progress, slowing or eliminating metastatic spread, and increasing survival. However, given the slow pace of prostate cancer progression, many men die of other causes than prostate cancer and so not all treatments are considered necessary or beneficial to the patient. For this reason, some men with low-risk disease are directed to active surveillance or watchful waiting rather than surgery or radiation. While many men with prostate cancer die of other causes, a significant percentage will die from metastatic disease often after considerable suffering accounting for over 34,500 deaths per year in the United States. The big questions then are which men will have progressive disease and which will not, who among the diagnosed population needs further treatment, and what treatment is best?
Historically, drug treatments directed to testosterone reduction represented a major effort to combat the disease, based on the seminal findings of Charles Brenton Huggins, who won the 1966 Nobel Prize for Physiology or Medicine for showing testosterone reduction strategies could slow prostate cancer growth.9 The later discovery that the hypothalamus controls hormone production and release by the pituitary gland leading to the production of gonadotropin releasing hormone (GnRH) and luteinizing hormone releasing hormone (LH-RH) won Andrew Schally the Nobel Prize in Physiology or Medicine in 1977 and led to the expanded further studies of these hormones in reproductive processes.10
Particularly for this discussion, the role of LH-RH in suppressing testosterone production was studied and shown to have essentially the same effects as orchiectomy in suppressing the male hormone. Representing what is essentially a ‘chemical castration’ it proved more acceptable to patients since no disfiguring surgery was involved, despite the loss of sexual function. These research efforts on LH-RH are historically important because they required the development of animal models which endure to this day. Experiments relied on the use of normal rats, typically Copenhagen strain (injected with squamous cell prostate tumor 11095) or Fisher (injected with R-3327 Dunning rat prostate carcinoma) and treated with LH-RH antagonist analogs.11,12 Such models required tumor growth of from 140 days post-inoculation to as much as six months post-inoculation.13 These slow growth rates are clearly not conducive to performing multiple, efficient imaging experiments because of the times and animal sizes involved and despite the advantage of being a syngeneic model (rat tumor implanted in rats), modern imaging experiments required the development of better animal models, most notably derived from the independent de novo development of immunocompromised mice that could accept and allow growth of mammalian tumor xenografts.14–19
A separate independent series of developments involved novel radionuclide imaging technologies and, the development of the Anger camera may be considered as a historical starting point.20–22 Dr. Anger is a giant in the history of nuclear medicine as he also invented that other stalwart of preclinical radioactive animal biodistribution experiments, the scintillation well counter.23 Roëntgen’s demonstration of the use of X-rays had led over time to their widespread use as an anatomic technique for detection of tumors and became, and still often remain, a definitive test for the presence of tumors.24–25 Radiography is a mature technology, cheap, easily applied by technical staff, and simply read and interpreted by general radiologists. Of course, a major breakthrough in imaging technology came in the 1970s with the invention of x-ray computed tomography that allowed radiologists to see anatomy in cross section. In its simplest form, as a technique, the analysis of penetrating X-rays is complemented by the injection of a radioactive substance that has specificity for the disease under suspicion. Originally, the Anger camera was used but this has given way to single photon emission tomography (SPECT), and later positron emission tomography (PET). Not only multiple advances in specific technologies but also the worldwide revolution in computing power were also requirements for successful studies in modern radioimaging techniques.
PROSTATE ANATOMY, FUNCTION AND CARCINOGENESIS
A schematic drawing of the prostate general anatomy and cellular structure is shown in FIGURE 1.26 Briefly, the healthy gland, normally the size of a walnut, surrounds the urethra and the ejaculatory ducts leading from the seminal vesicles and its major function is to add multiple components to formulate and protect the viability of the sperm just prior to ejaculation. Clinically, the gland is divided into separate zones, with the central, transitional, and peripheral zones being most cited. The bulk of prostate cancers originate in the peripheral zone. Functionally, the zones contain ducts and acini that produce the ejaculatory secretions, and these are supported and confounded with various other structures and cells such as the extracellular matrix, muscle cells, fibroblasts, and neuroendocrine cells. Cancers arise when the ductal cells undergo changes due to inheritable mutations that eventually lead to aggressive adenocarcinomas. These early prostate tumors are usually dependent on androgens for their growth. Tumors may appear in multiple parts of the prostate and eventually, tumors may break through basement membranes and invade surrounding tissues and in some patients early metastatic spread to distant organs may occur. Aside from cited diagnostic methods physicians have relied on the serum test of rising levels of prostate specific antigen (PSA; a serine protease, MW 33kD) secreted by the prostate to indicate the presence of cancer, both for initial diagnosis and in follow-up diagnostics during patient management.27 However, PSA is also secreted into serum in non-cancerous conditions such as benign prostatic hyperplasia or BPH. Notably, PSA is not elaborated in rodents and can therefore not be followed in preclinical models as a disease marker, although a mouse homologue of a human prostate secretory protein of 94 amino acids (PSP94) has been suggested as a marker in murine models.28
FIGURE 1.
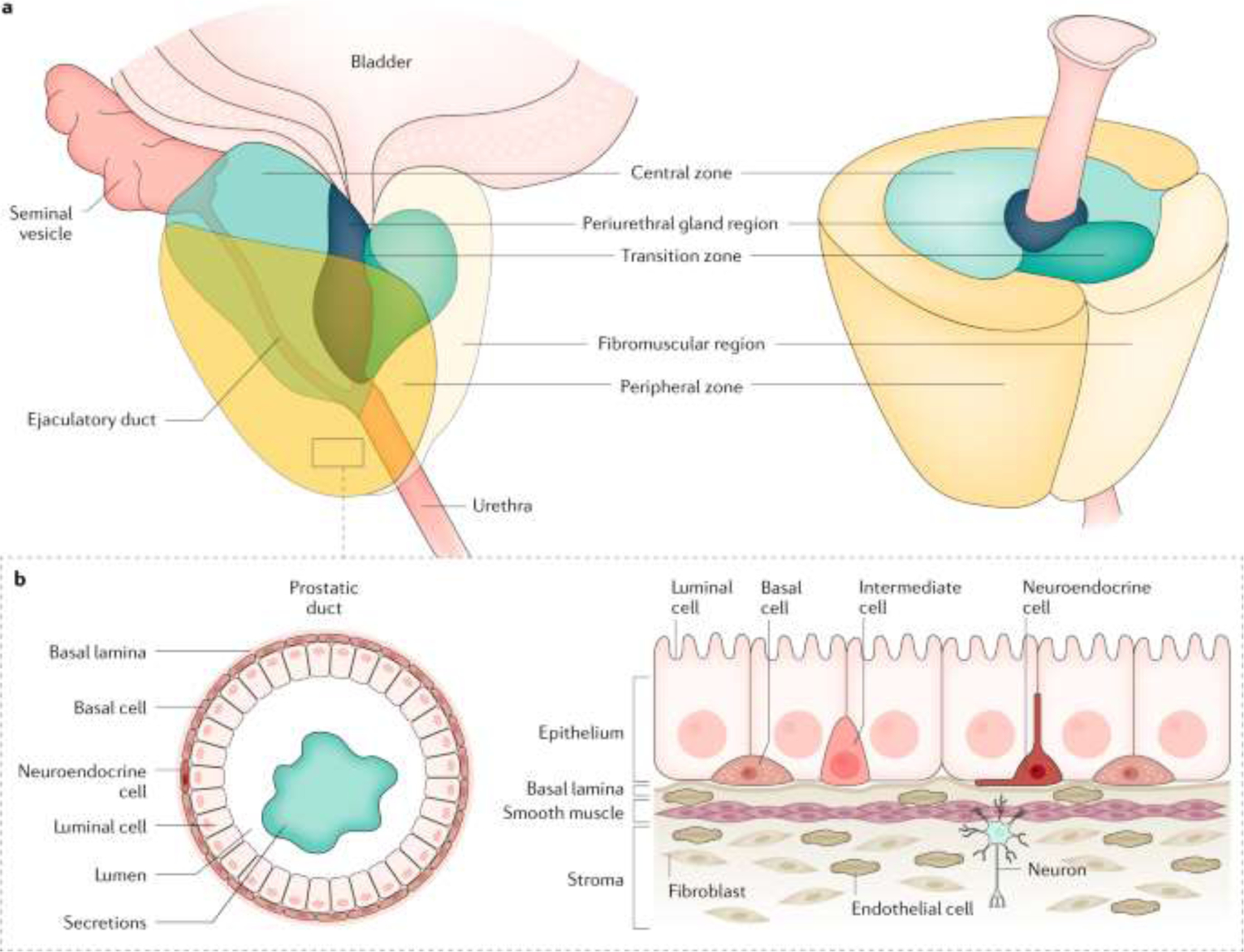
Anatomy of the human prostate and surrounding tissues.
Reprinted with Permission from Nature, Rebello et al 2021. adapted from Verze et al. (2016), Springer Nature Limited.
PSA targeting strategies were among the earliest tried molecular diagnostic approaches both preclinically and clinically. The concept of population-based screening PSA tests is controversial. Advocates claim that it can lead to earlier diagnosis preventing bad outcomes whereas detractors argue that no statistical effect from PSA screening efforts can be discerned.29 Everyone agrees PSA testing is less than ideal but it continues as a mainstay of patient management.
CELL LINES AND MODEL SYSTEMS FOR ANIMAL RESEARCH
In the modern age, and most particularly for preclinical research on imaging methods, cell lines taken from patients and shown to grow in laboratory culture and as xenografts in immunocompromised rodents, particularly mice, have formed the basis of most preclinical research on new imaging and therapy agents. TABLE 1 shows a list of the more common cell lines used in preclinical models. The well-known and still widely used LNCaP cell line was described as long ago as 1983.30 It was obtained from a patient with a metastatic lymph node caused by prostatic adenocarcinoma. It was found to grow well in vitro and as subcutaneous xenografts (86-hour growth doubling time) in athymic [nude] mice. These attributes as well as its sensitivity to androgen receptors are appealing to preclinical researchers needing reproducible, predictable, and reasonably fast-growing xenograft lines, although perhaps one should be cautioned regarding how well findings obtained with this system might relate to primary glandular prostate cancer or to prostate cancer with bony metastases. An androgen-dependent cell line isolated at about the same time as LNCaP, PC 82, was obtained from a primary prostate cancer and so may more accurately model primary disease.31 PC 82 was obtained from a patient’s primary tumor that had breached the prostate and invaded surrounding tissues. However, in modelling work, after multiple inoculations of PC 82 tumor tissue into nude mice it still displayed a [xenograft size] doubling time of 4 weeks, considerably slower than LNCaP and therefore, less desirable as a tumor model.
TABLE 1.
Cell lines commonly used in xenograft mouse models of prostate cancer notably with radionuclides
| Cell Line | Type | Androgen dependence for xenograft growth | Source | Reference |
|---|---|---|---|---|
| LNCaP | Metastatic | − | Lymph node | 30 |
| PC-82 | Primary | + | Prostate | 31 |
| PC-3 | Metastatic | − | Bone | 34 |
| DU 145 | Metastatic | − | Brain | 36 |
| PC346 | Primary | + | Prostate | 37 |
| RM-9/hPSCA | Murine | Prostate | ||
| 22Rv1 | Primary | + | Prostate | 83 |
Later development of the severe combined immunodeficient mouse (SCID) model32 allowed further extension of LNCaP preclinical models into a more immunocompromised murine model system.33 This further enabled the study of metastatic spread of LNCaP either from subcutaneous xenografts or from orthotopic implantations directly into mouse prostate. PC-334,35 and DU14536 are other cell lines that, like LNCaP, are widely used in animal models, because they are rapidly growing in androgen-independent media and are easy to culture and transplant. But, also like LNCaP they more accurately represent metastatic rather than primary disease, again, as the latter type are much more difficult to grow reasonably quickly and reproducibly either in culture or as serial xenograft transplantations.35 Injected intravenously into SCID mice, PC3, DU145 and LNCaP cells are all able to localize to bone and to establish tumors in mouse lungs, further emphasizing their metastatic origin and their further potential applications.36
The PC346 line and a series of its derivative cell lines were created from a patient with primary prostate cancer. The derivative cell lines are thought to reflect different stages of tumor development.37 The model begins as an androgen-dependent tumor that transitions to a more aggressive and more metastatic, androgen-independent tumor. The originally developed PC-3 and DU145 show little or no androgen growth sensitivity due to low receptor expression while the LNCaP line shows robust androgen growth sensitivity.38 While the original PC346 line displayed limited mutations, repeat culture or xenografting led to ‘daughter’ lines. These then proved more similar to cell lines from metastatic disease, and reliably show apparently increasing levels of mutations with xenograft disease progression. It has been long hypothesized that primary prostate tumors are composed of mixtures of cells with differing androgen sensitivity, and the observed patient transition to dominant androgen-independent lines involves clonal expansion of independent cells already present in tumor before androgen ablation treatments are undertaken.39 The natural selection process toward a more aggressive form often seen in patient subjects is almost certainly expedited during establishment of monoclonal cell lines for laboratory and animal uses. Genomically, the point mutation T877A was noted in the LNCaP line and is seen again in the PC346 line series.40 More recently other point mutations have been noted in cultured cell lines41 and such findings have been associated with tumor aggressiveness.
It has been recognized for some time that prostate cancer research is hindered by a lack of preclinical models and diversity in those preclinical models that better reflect the true diversity of prostate cancer found in humans.42 The Prostate Cancer Foundation has held working groups to try to address this issue.2,43 Topics included limitations of available and developed cell lines, xenograft models and their application, and newer genetically engineered mouse models.44 Aside from the practical development issue of models, a major overriding issue is the relevance of these model systems to human disease. The number of models is insufficient to meet researcher’s needs and does not truly reflect the broad spectrum of human prostate cancer. Moreover, molecular events that define tumorigenesis such as point mutations are poorly understood, while the study of tumor and host interactions in preclinical xenograft models which do not have an intact immune system probably represents a profoundly inadequate model for studying the human/tumor system.
Most recently, the establishment of animal models for prostate cancer preclinical research has morphed in multiple different directions as scientists have addressed the need for better model systems with diverse new approaches. Transgenic mice that develop prostate cancers on their own have been established, however the incubation time is long and generally uncertain.44 More sophisticated applications of LNCaP has extended the system to metastatic systems, reflecting its original isolation as metastatic disease,45 and to the use of advanced injection techniques for orthotopic models.46 A significant advance and alternative to murine models may be establishment of a canine prostate cancer cell line (Ace-1) into the prostates of immunosuppressed, intact, adult male dogs after transabdominal injection.47 An orthotopic model has also been established in immunocompromised rabbits, perhaps opening another new avenue to more relevant patient models.48 Meanwhile, significant attempts have been made to ‘naturalize’ the growth and progress of prostate tumors, by direct injection of cells into the prostate,49 and using electroporation/transposon techniques.50 Considerable work at multiple institutions has been directed toward the development of patient derived xenograft (PDX) models of prostate cancer, lately leading to the establishment of the Melbourne Urological Research Alliance (MURAL) collection of prostate cancer cell lines for preclinical research.51 This collection includes cell lines from the treatment-naïve primary tumors to castration-resistant metastases and embraces inter- and intra-tumor heterogeneity in both adenocarcinoma and neuroendocrine phenotypes. However, not all PDX tumors grow ex-vivo so there is some selection bias. Moreover, PDX tumors lack a normal tumor microenvironment because they must be injected into immune-compromised animals.
SPECT and PET
Despite all the issues with preclinical models of disease, SPECT and PET imaging of targeted molecular imaging probes have proven to reliably predict functionality in humans. Radiolabeled targeting moieties simply need to demonstrate the ability to bind to representative prostate cancer xenografts well enough to justify an initial clinical investigation, more fundamental disease questions aside.
A variety of radionuclides have been proposed and investigated for both SPECT and PET nuclear imaging (TABLE 2). All have relatively short physical and biologic half-lives consistent with their intended purposes. Radionuclides vary in the energy of the gamma ray or positron they produce. PET isotopes uniformly produce two 511 keV gamma ray emissions but differ in the energy levels for the positron emission energy with higher energy positrons tending to give less sharp images. Before PET became more widely available, most initial research work focused on SPECT agents although once PET was developed to an acceptable technical level its superior inherent sensitivity and resolution, relating to its two co-incident 511 keV gamma rays emitted 180° apart, that eliminates the need for collimation made PET the more desirable radionuclide type.
TABLE 2.
Radionuclides used for SPECT and PET preclinical imaging and intended for clinical development. (Emissions: γ, gamma; β− beta; β+ positron), energy in keV (% abundance).
| Radionuclide | Half-Life | Major Energy Emissions γ, Positron & Negatron | Vector Attachment |
|---|---|---|---|
| Indium-111 | 2.8 d | γ 173 & 245 keV (>90%) | Chelate |
| Technetium-99m | 6 h | γ 140 keV (98%) | Chelate |
| Gallium-67 | 3.3 d | γ 93 & 185 keV (38 & 20%) | Chelate |
| Gallium-68 | 68 min | B+ 511 keV (89%) γ 1899 keV (89%) | Chelate |
| Copper-64 | 13 h | B+ (18%) γ, β− (43 & 39%) | Chelate |
| Copper-67 | 62 h | γ 184 keV (48%) β-(392, 483, 577; 57, 22, 20 %) | Chelate |
| Carbon-11 | 20 min | B+ 960 keV | Covalent |
| Fluorine-18 | 6 h | B + 511 keV (96%) | Covalent |
| Iodine-123 | 13 h | γ 159 keV (83%) | Covalent |
| Iodine-124 | 4.2 d | B + > 1500 keV (22%) | Covalent |
| Iodine-131 | 8 d | γ 364 keV (82%) β - 192 keV (90%) | Covalent |
| Zirconium-89 | 3.3 d | γ 908 keV (100%) β + 902 keV (23%) | Chelate |
A relatively recent innovation is the dual use of targeted radionuclides as either diagnostic or therapeutic agents. Molecularly targeted radiotherapy can result from substituting a non-therapeutic isotope for one that emits either an alpha or beta particle while maintaining the same targeting ligand (as much as possible). Extension of diagnostic findings to therapeutic potential will always remain an attractive option for clinicians and the demonstration of binding of a detectable and target-specific radiolabeled vector naturally leads to considerations of therapeutic analogs. Most recently, scientists have brought this idea to fruition by the development of multiple radionuclide-based targeting agents directed to different tumor sites that provide molecularly specific diagnostics, which are then rapidly being extended to their therapeutic radio-analogs. The preclinical development of selected novel agents is discussed below.
ESTABLISHING TARGETS FOR MOLECULAR IMAGING
Metabolic Imaging
The era of molecular medicine for prostate cancer targeting has been ushered in with a variety of basic laboratory advances including the landmark discovery of monoclonal antibodies (MAbs),52 subsequent protein engineering discoveries,53 increasing knowledge of cancer growth mechanisms54, and development of drug discovery screening chemistries.55 However in the realm of radioimmunodiagnostic imaging the development of PET as a superseding technology to SPECT in turn led to the seminal breakthrough of the discovery of 18F-fluorodeoxyglucose (18F-FDG) as a preferred molecular imaging agent for cancers, and this is not based on molecular recognition but rather on inherent metabolic processes.56 18F-FDG localizes to cancers at higher levels than it does to normal tissues, often resulting in enhanced uptake in cancer cells due to their greater metabolic uptake of glucose.57 Mechanistically, at a basic biochemical level, the 18F-FDG is internalized into cells and broken down into 18F-tagged molecular fragments that are then accumulated within the cell, enabling sufficient time to complete imaging studies. Unfortunately, in prostate cancer several factors make 18F-FDG less useful than in other tumor types including high excretion through the urinary tract where the prostate is located, and lower avidity of the glucose analog for relatively indolent primary prostate cancers. As the disease progresses and prostate cancers become more aggressive, 18F-FDG uptake increases. Animal xenograft studies using comparative localizing agents and differing tumor xenograft models confirm this result.58 Further metabolic targeting agent options, relying on the concept of increased uptake by more active tumor cells, have been explored including 11C- and 18F- analogs of choline (lipid synthesis), acetate (acetyl coenzyme A mediated metabolic processes), methionine and glutamate (protein synthesis), and amino acid analogs such as FLT and FACBC.59 Many of these agents have also been proposed for niche applications such as radiation therapy planning and following the progress of external beam therapies, and one might assume that the same comments directed to 18F-FDG PET metabolic imaging above may be applicable to these agents with respect to the indolent growth profiles generally seen in primary prostate cancer. In general, this metabolic approach is less sensitive and less specific than more receptor targeted approaches.
Monoclonal Antibodies and Derivatives
Originally, PSA, a serum protease, was explored as a specific antigen for monoclonal antibody targeting,60 but its use was discontinued due to the high amounts secreted into serum, both in patients and in animal xenografts, as well as the binding of many such PSA antibodies to the related kallikrein proteins, which are also serine proteases. More recently this approach has returned with targeted agents against kallikreins, and early results are promising. A better antibody target was identified as prostate specific membrane antigen (PSMA) and the comparison to PSA is shown in TABLE 3.61 Also known as glutamate carboxypeptidase G2 (N-acetyl-L-aspartyl-L-glutamate peptidase I; folate hydrolase), it is present as a transmembrane protein in all types of prostate tissue but often increased in tumor tissue. An original monoclonal [7E11] was first developed with the LNCaP cell line and shown to bind an intracellular/membrane epitope of the PSMA protein.61 Eventually work on 7E11 led to the first FDA approved SPECT agent for prostate cancer diagnosis [ProstaScint® scan (Cytogen Corp., Princeton, NJ).62 However, due to its high background signal and low sensitivity clinical performance related to prostate recurrence, lymph and bone metastases, possibly due to recognizing an internal antigen binding site, and its inferior radiolabeling protocol producing an unstable 111In-MAb linkage, it was later withdrawn from commercial use.
TABLE 3.
Comparison of prostate-specific antigen (PSA) and prostate-specific membrane antigen (PSMA) properties.
| PSA | PSMA |
|---|---|
| Secretory Protein | Membrane Protein |
| Serine Protease | Carboxypeptidase |
| Function: Liquefaction of semen | Cleaves C-terminal protein glutamate residues |
| Serum cancer marker | Expression correlated with cancer aggressiveness |
| Level falls with androgen blockage | Upregulated with androgen blockage |
Other antibodies raised against human PSMA were discovered and developed63 and in comprehensive testing in xenografted mice bearing prostate carcinoma cell lines LNCaP, DU145, and PC-3 MAbs radiolabeled with both 131I and 111In were shown to both strongly target and rapidly internalize into cells via the PSMA antigen.64 Of this Mab series, the J591 clone was chosen for further theranostic development, and it was later radiolabeled with 89Zr for PET imaging.65 Preclinically, the 89Zr-J591 was compared in LNCaP (PSMA positive) and PC-3 (PSMA negative) s.c. xenograft models (FIGURE 3). With separate animals having xenografts in left (PSMA+) or right (PSMA−) shoulder regions the efficacy of the positive targeting agent was shown clearly, with the striking images for the LNCaP animals reflecting 8-, 29- and 18- to 1 tumor to blood ratios at 48-, 96- and 144-hours post-injection, respectively. Interestingly, the relatively high bone uptakes with carrier-free 89Zr-J591 could be partially blocked by excess injected cold J591 MAb suggesting that relatively high bone uptakes often seen will all metallic radiolabeled MAbs was not due entirely to dissociated 89Zr localization, but partially due to recognition of the PSMA antigen in bone by the 89Zr-J591 targeting MAb.
FIGURE 3.
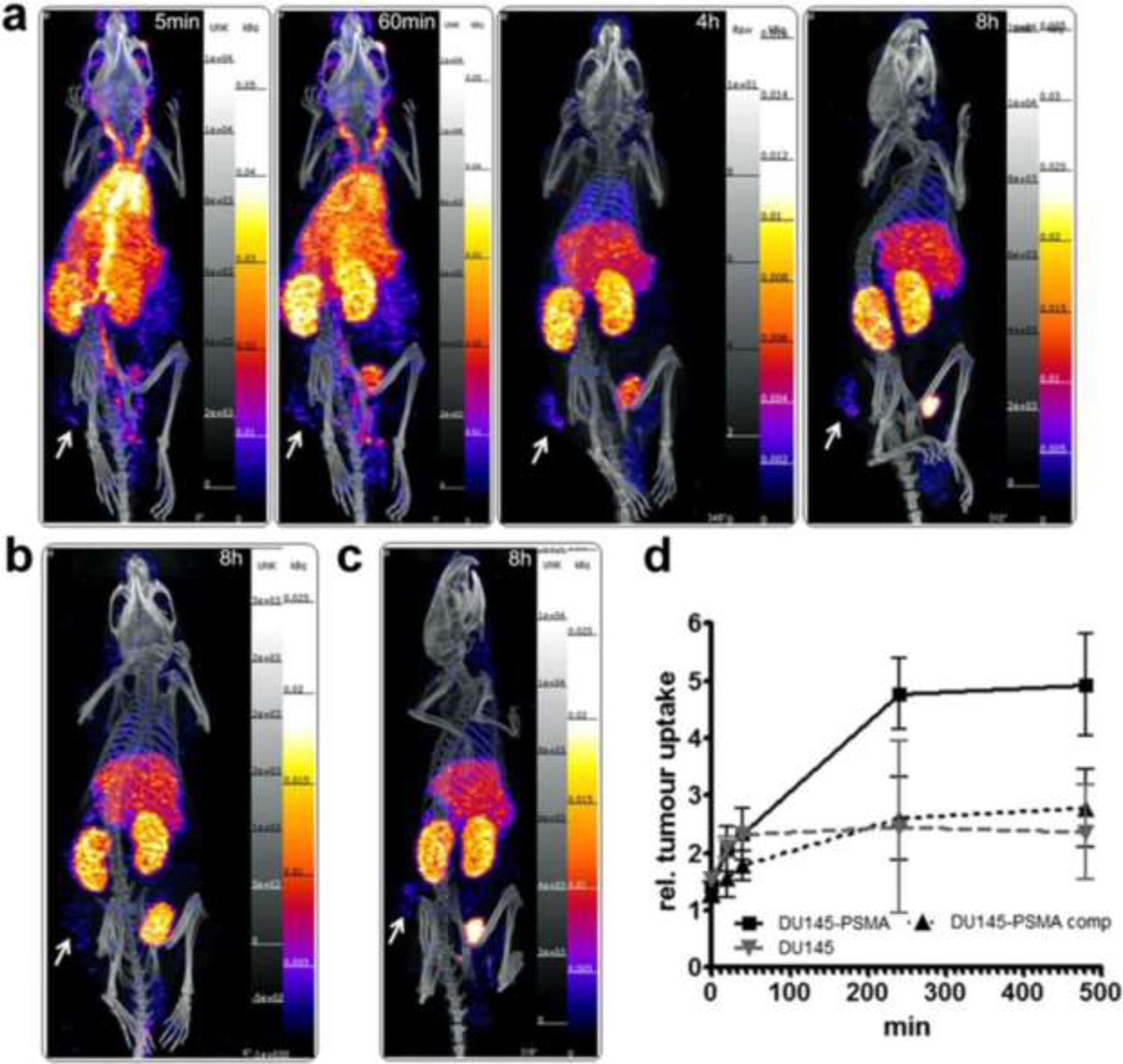
SPECT imaging of 99mTc-J591 diabody in mice bearing s.c. xenografts of the native DU145 PSMA− cell line or the DU145 line expressing PSMA as positive target. a. Serial images of PSMA-DU145 taken from 5 min to 8 h post-injection. b. Mouse with PSMA negative tumor line at 8 h post-injection. c. Mouse with PSMA-DU145 cell line given blocking dose of Tc99m-J591 diabody. Tumor shown by arrows.
Reprinted under Creative Commons from: Florian Kampmeier et al. Design and preclinical evaluation of a 99mTc-labelled diabody of mAb J591 for SPECT imaging of prostate-specific membrane antigen (PSMA), March 7, 2014. EJNMMI Research. Springer Nature
The images from FIGURE 2 illustrate the utility of these xenograft models in predicting likely human performance. The tumor xenografts are well delineated and clearance and biodistribution accurately quantified for both non-targeting and targeting models. Normal tissue and tumor uptake of the radionuclide and its retention over time clearly shows the tumor, not only indicating optimal imaging times but giving an accurate estimation of the dosimetry to be expected to tumor and normal tissues. This data can be extended to human predictions of dosimetry and forms an essential part of the preparation of materials for potential clinical applications. Dosimetry is an important aspect of the reagent under development as the expected patient radiation doses (a negative aspect from inherent doses to patients) must be counter-balanced, or more than counter-balanced by the utility of information being obtained (positive aspect from the imaging). Even more importantly, with theranostic extension under consideration anticipated patient doses from an analogous therapeutic conjugate, such as 177Lu-J591, can be gleaned from this imaging study. So, while the images are strongly positive, the study also cautions that high non-target tissue uptakes, principally in liver and bone are to be expected, and perhaps further predicts that attempts to use this agent for identification of bone metastases may be problematic. As mentioned above, radiometal labeling of proteins can lead to dissociation of the two components in vivo often leading to unwanted tissue uptakes, principally in liver and bone, and this concern is present when using all radiolabeled MAbs with nuclides listed in TABLE 2. Xenograft studies over several decades have been crucial in identifying this problem and, more importantly, quantifying the extent of the problem for every candidate radiometal labeled targeting vector under consideration for development. The J591 methodology and techniques applied here result in a superior conjugate to many others also outlined here and are built upon numerous previous preclinical investigations with xenograft models developed to identify and select the most stable radiometal-MAb combinations for clinical development.66–72
FIGURE 2.
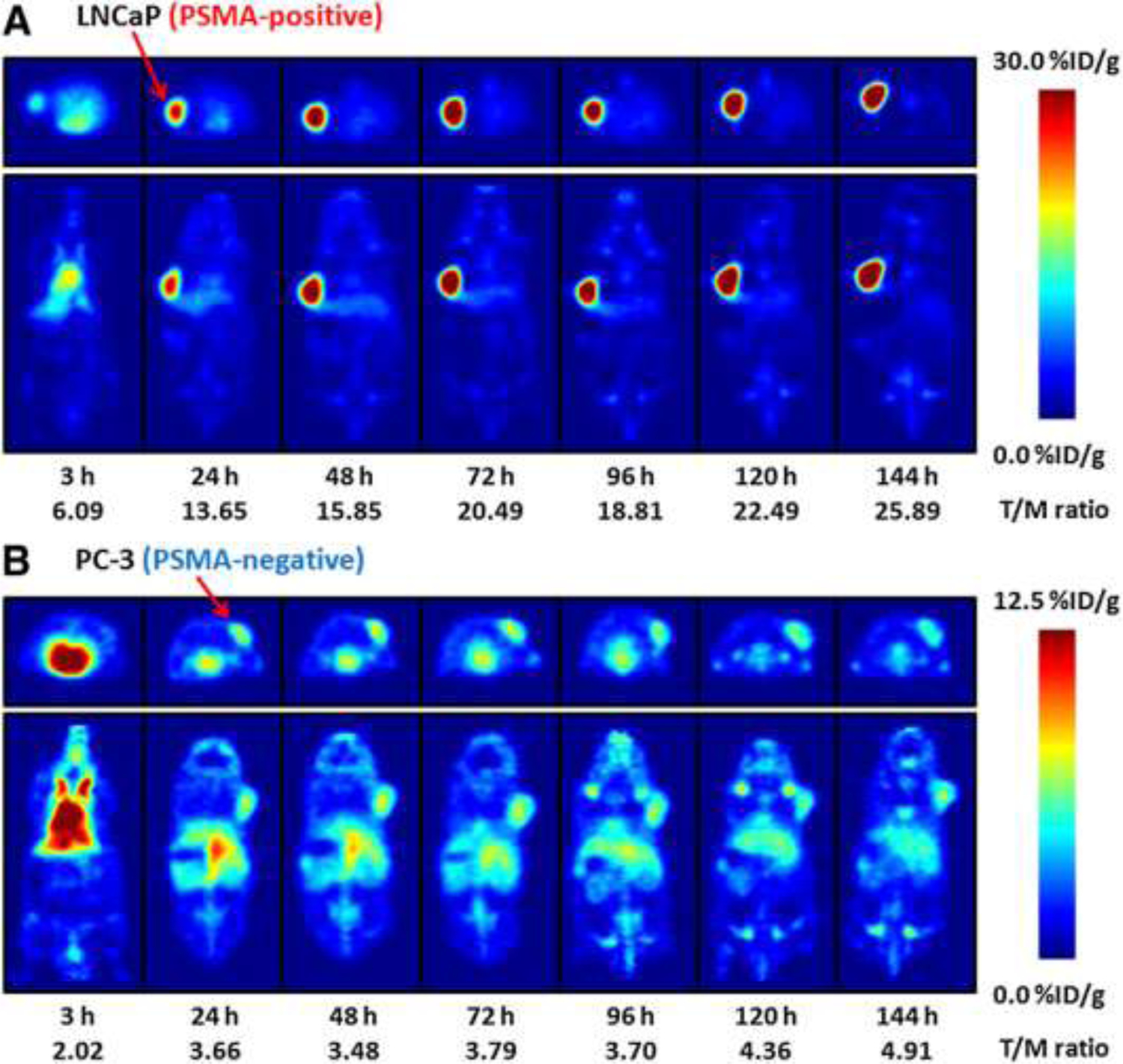
PET imaging of nude mice bearing LNCaP (top) or PC-3 (bottom) xenografts from 3 to 144 h post-injection of 89Zr-J591 with transverse images shown above coronal images in each case and the strong and specific uptake in the PSMA positive lesions.
Reprinted with permission. This research was originally published in JNM. Holland JP et al. 89Zr-DFO-J591 for ImmunoPET of Prostate-Specific Membrane Antigen Expression In Vivo. J Nucl Med. 2010;51:1293–1300. © SNMMI.
Mouse models, however, have inherent disadvantages. If the mouse does not express PSMA in normal tissue (and it doesn’t) an inherently optimistic biodistribution will be obtained since the injected targeting vector does not have to deal with competition from normal tissue expression of the same antigen, as it will in humans. Effects may be further exacerbated by the antigen expression in normal tissues in patients being more naturally available in normal tissue than in tumors, which often have compromised blood flow and aberrant lymphatic drainage, characteristics not seen in mouse xenografts. Furthermore, human MAbs often clear rodent circulation faster giving the false impression of a shorter biological serum half-life than one may later see in a human subject. For imaging, one wants maximum specific tumor uptake combined with the most rapid normal all-tissue clearance, as an ideal situation. The radiolabeled IgG localizes exceptionally well in the xenograft model, but the model indicates an extended circulation time, under even the best of conditions. While imaging performance and dosimetry are considerations, a more mundane factor might be patient acceptance of imaging procedures that take multiple days to complete. Because of this point, extended research has investigated potential improvements in MAb imaging by using MAbs with shorter serum half-lives, such as antibody fragments or smaller re-engineered versions of the originally developed intact MAbs.73
As an example of this, a 99mTc radiolabeled diabody construct of the J591 antibody was developed for SPECT imaging (FIGURE 3). Despite use of this 55K Dalton diabody ensuring rapid serum clearance and the well- engineered xenograft model system, the test agent shows limited positive uptake in PSMA+ DU145 target cells and considerable background uptake in normal tissues. While the results may indicate that this particular agent may not have a clinical future, the imaging model performed its experimental function superbly thanks to the exquisite and quantifiable mouse images obtained. The recognition that a radiolabeled antibody’s long serum presence is a disadvantage has been continually challenged with experiments on smaller and smaller radiolabeled recombinant proteins, with molecular weights down to and including peptides. Mirroring prior work when intact IgG MAbs were enzymatically converted to F(ab’)2, Fab’ and similar species reflecting molecular weights of approximately 150,000 Dalton, 100,000 Dalton and 50,000 Dalton respectively, recombinant constructs of minibodies (80,000 Dalton), diabodies (50,000 Dalton) and scFv fragments (27 Dalton) have been generated, radiolabeled, and tested in various xenograft models. In the prostate cancer xenograft models and continuing with the J591 model exemplar, 89Zr-J591-IgG -minibody and -diabody conjugates were compared in LNCaP models.74
The specific comparisons in the animal xenograft model system are again highly instructive in a general manner (FIGURE 4). All three agents show strong uptake on planar PET images of xenografts at 12 h post-injection, but the IgG still shows considerable circulating 89Zr-MAb, making it more difficult to pick out tumors whereas the smaller fragments have essentially cleared the blood. However, the 89Zr-IgG can still accumulate into tumor at extended time periods, while the two fragments have maximized their uptake at the earlier time-point and tumor retention of 89Zr in xenografts begins to fall at later time-points, indicating early time-point PET must be done since some the radiolabeled agents are surface bound and will dissociate from their binding sites when uptakes are not supported by constantly circulating levels of unbound agent. Despite the similarity in the images, biodistributions in %ID/g terms indicate much higher uptakes from the 89Zr-IgG than the fragments and the fragments show high uptakes in kidney, with the lower MW diabody clearing circulation faster than the minibody. In essence, early time-point PET will offer superior contrast against a general background, except perhaps in the pelvic area, which is of most concern in primary prostate cancer.
FIGURE 4.
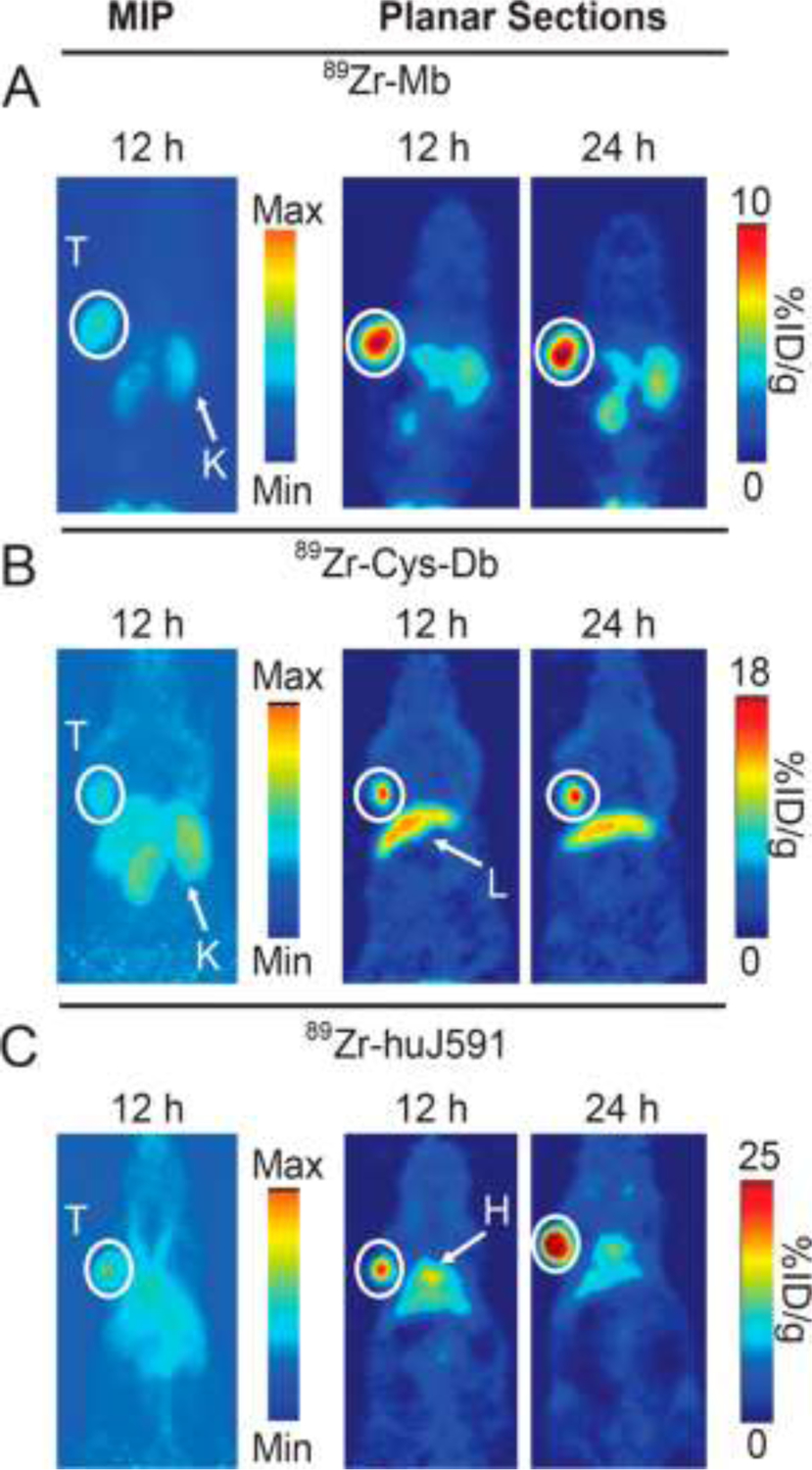
PET images of 89Zr-minibody (A), 89Zr-diabody (B) and 89Zr-IgG (C) of the J591 targeting MAb in the LNCaP xenograft system. Animals have tumors (T) in their shoulder regions and all three targeting agents clearly show specific targeting.
Reprinted with permission from Molecular Pharmaceutics, 11:3965–3973, 2014. https://pubs.acs.org/doi/10.1021/mp500164r American Chemical Society. Further permissions related to the material excerpted should be directed to the ACS.
Further molecular weight reduction in targeting proteins beyond what is achievable with antibody fragmentation was achieved with antibody analogs rather than recombinant antibody fragments with the development of Affibody® molecules which are about 6,000 Dalton MW and comprised of alpha helices of 58 amino acids that are obtainable by library screening methods and raised with exceptionally high, picomolar, affinity against investigator-selected antigen targets. Notable efforts with these agents in prostate cancer have been directed against more general cancer antigens than PSMA, such as epidermal growth factor (EGF). In preclinical testing of an 111In-labeled affibody, ZEGFR:2377 for binding in all three standard xenograft lines, LNCaP, PC-3 and DU-145, all of which express EGF receptors, at average levels of 43,000, 99,000 and 207,000 receptors/cell, respectively high specific binding levels were seen.75 In extension of the work into a DU-145 preclinical xenograft model a similar anti-HER2 affibody construct radiolabeled with 57Co was demonstrated to show positive uptake in xenografts implanted in the animal’s hind legs. Again, the models provided good general guidance on what to expect with further development. Tumor uptakes seen with larger vectors were again considerably reduced with 1–2 %ID/g uptakes in the xenografts, counterbalanced by 3–7 %ID/g uptakes in liver, and 247–300 %ID/g uptakes in kidney. The general trend of reduced target uptakes and increased renal uptakes seen with smaller antibody fragments is strongly reinforced here with a 6,000 MW vector. The data suggests ever- faster clearance with ever-smaller proteins/peptides, lower target uptakes resulting from the faster pharmacokinetics, but enhanced and, importantly, retained renal uptake of radionuclide. Most simply interpreted, smaller protein vectors do not apparently have the circulatory time adequate to obtain substantial tumor uptakes, and ominously this effect is seen with vectors having target affinities in the picomolar range, suggesting clearance is a more important factor than affinity.
In passing, other targeting constructs tested included camelids (camel VHH, variable region heavy chains) recombinant non-antibody targeting vectors such as ankyrins and DARPins are being developed as imaging and therapy agents and although it may be too early to gauge successes in these areas one preliminary animal study using a 57Co-labeled DARPin raised against epithelial cell adhesion molecule (EpCAM) has been published.76 Very high uptake and retention was seen in kidney when using radiometal labeled constructs, with > 50-fold lower uptakes in DU145 xenografts, echoing prior results, using different radionuclides, labeling methods, protein vectors, and antigen targets.
Recently, more novel targets have been investigated with radiolabeled protein constructs such as diabodies targeting prostate stem cell antigens (PSCA).77 This work used two 124I-labeled diabodies for targeting that showed very low uptakes in LAPC-9 xenografts at early time-points post-injection, with modest renal uptake since 124I clears rapidly from renal tissues, but also clears rapidly from tumor target cells since it is not retained internally in cells. Such efforts have continued against this target using 131I and 177Lu minibodies78 and a more sophisticated syngeneic model using ‘KNOCK-IN’ transfused versions of original RM-9 murine cells elaborating enhanced levels of hPSCA.79 The latter used 89Zr-diabodies and therefore had the same general distribution seen with such radiolabels, although the establishment of a syngeneic model in immunocompetent mice may have significant importance in studies of the effects of the immune system in prostate cancer models.
Small Molecule Imaging Agents
Low MW agents based on specific targeting of receptors have been suggested and investigated in some detail.80 A considerable effort has been expended on bombesin-like peptides, principally gastrin-releasing peptide (GRP), a naturally occurring 27-amino acid length peptide, that can be subjected to multiple synthetic and peptide modifications which, along with multi-radionuclide radiolabeling options, leads to hundreds (theoretically limitless) of analogs for investigations.81 Only one or two highlights can be discussed here. A GRP dimer was produced to give two peptide recognition units on the targeting moiety.82 SPECT and optical images (agent also had a fluorescent dye attached) obtained with this In-111-labeled agent are shown in FIGURE 5. Analogs containing 68Ga, 177Lu, 89Zr and other similar radiometals are, similar in biodistribution. Shoulder PC-3 tumor xenografts are clearly delineated but the great mass of the injectate is present in the bowels, particularly pancreas, as expected given the role of gastrin in digestive processes. Interestingly, considering possible prostate cancer imaging in the pelvis, little or no activity is seen in the kidneys or urethra and future placement of xenografts in or around this area would be interesting, as might application of the newer orthotopic model approaches mentioned elsewhere in this review. Radiolabeled bombesin analogs have been compared with RGD, 18F-FDG, PSMA and neurotensin analogs for prostate cancer xenograft imaging in an exploratory study in the 22Rv1 (androgen positive) and PC-3 (androgen negative) xenograft cell lines, with the intent of identifying a peptide vector that might we used in PSMA negative patients.83
FIGURE 5.
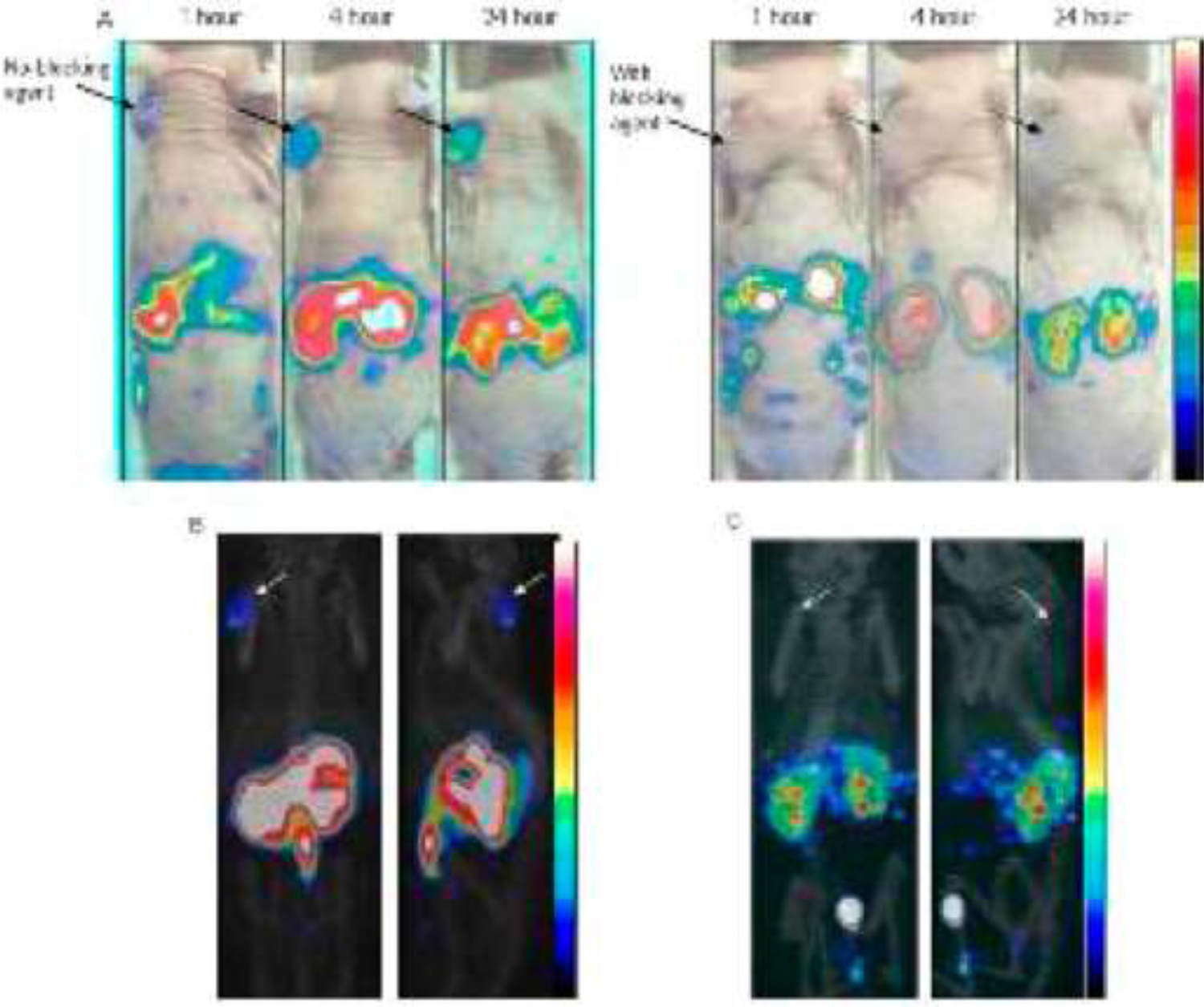
(A) Static coronal microSPECT images of Bombesin targeting bivalent peptide 111In-DOTA-[Aca-BN(7−14)]2 on PC-3 tumor bearing athymic mice at 1, 4, and 24 h p.i. (top) with and without blocking agent and combined with the optical photograph. Below: Static coronal microSPECT/CT images acquired at 24 h p.i. without (left) and with blocking agent (right). Arrows indicate tumor sites.
“Reprinted (adapted) with permission from Molecular Pharmaceutics 10:1716–1724. Copyright 2013. American Chemical Society.”
The most fundamental and practical advance in prostate cancer diagnostic imaging was made with the discovery of glutamate carboxypeptidase G non-natural binding agents, that could be radiolabeled with imaging radionuclides.84,85 The glutamate carboxypeptidase II enzyme, one of a larger family of carboxypeptidase enzymes, cleaves the amino acid glutamate from the N-acetyl-L-aspartyl-L-glutamate (NAAG) dipeptide and is also active in cleaving glutamate for other C-terminal glutamate substrates including peptidyl-, aminoacyl-, benzoyl-, benzyloxycarbonyl-, folyl- and pteroyl- groups. It is also termed folate hydrolase and for our purposes, prostate specific membrane antigen (PSMA). Early work in neurosciences led Pomper et al. to recognize the enzyme’s ability to cleave glutamate from multiple natural and non-natural structures including the urea derivatives shown (FIGURE 6). This work led to the discovery and development of multiple radiolabeled analogs including 18F-DCFBC,86 images of which in nude mice bearing PC-3 xenografts are shown in FIGURE 7. The PC-3 cells modified with PIP to express PSMA clearly show xenograft uptake while the original PC-3 line, which is PSMA negative shows no uptake. Tumors are placed in the shoulders, remote from the renal and bladder areas which show the typically high radioactivity uptakes and radioactivity retentions seen with almost all low molecular weight targeting vectors. In structural development to the DCFBC analog the central urea framework, is flanked by the required glutamate function on one nitrogen and an aromatic function on the other urea nitrogen. As such, the central urea replacement of the original peptide bonds present in natural NAAG produces a binding but enzymatically resistant variant while the aromatic group was shown to offer a sub-structure type that results in a molecule with stronger affinity to the target enzyme, and also offered the aromatic ring for convenient radiofluorination producing an agent resistant to in vivo defluorination reactions.
FIGURE 6.
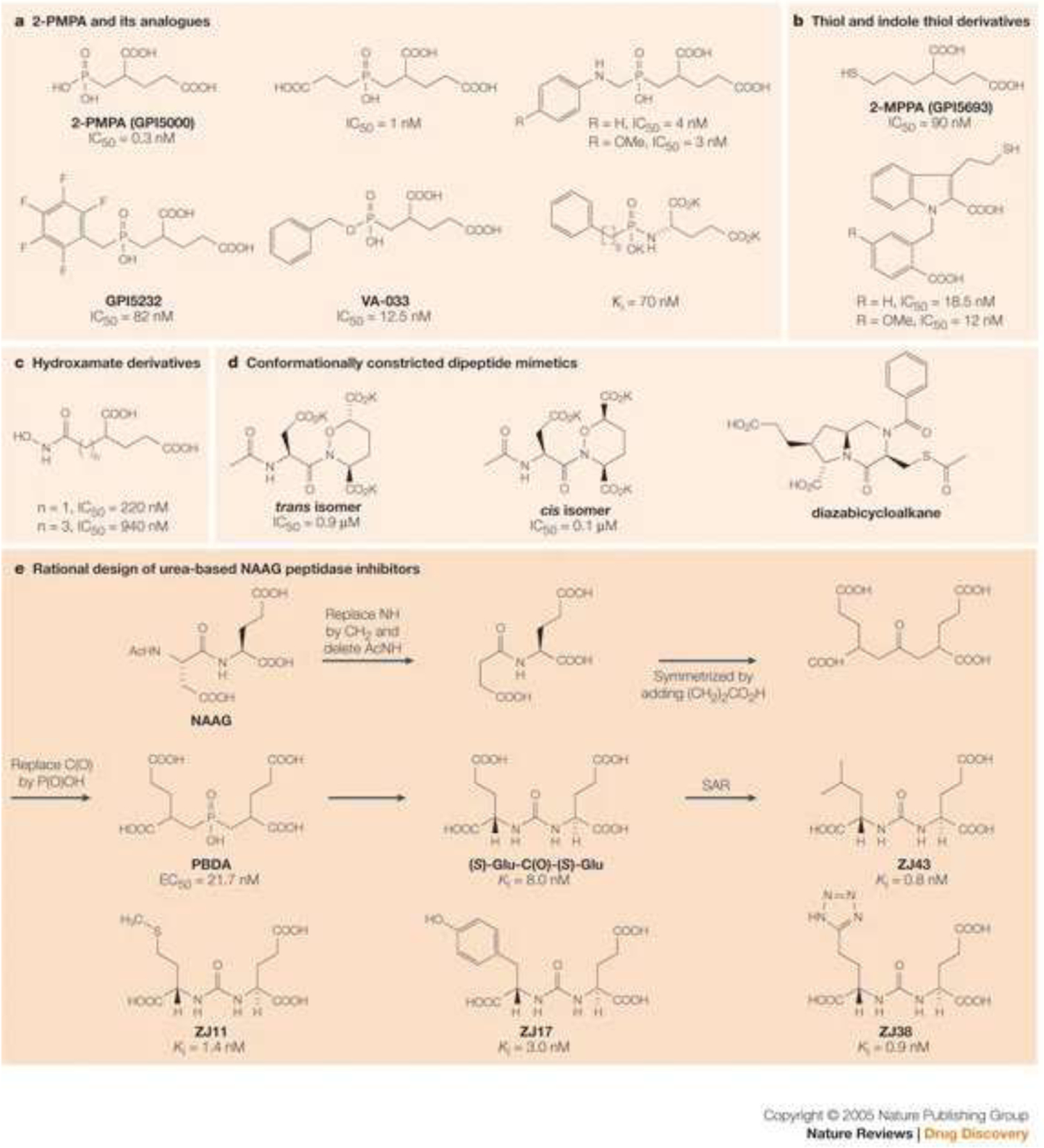
Structures with C-terminal glutamic acid units cleaved by the NAAG peptidase (PSMA) enzyme. The bottom panel reflects the discovery of the urea-based series of non-natural derivatives and the ability of the enzyme to remain active against a large and diverse number of such structures.
Reprinted with permission from Zhou et al. Nature Reviews Drug Discovery. Nature Publishing Group.
FIGURE 7.
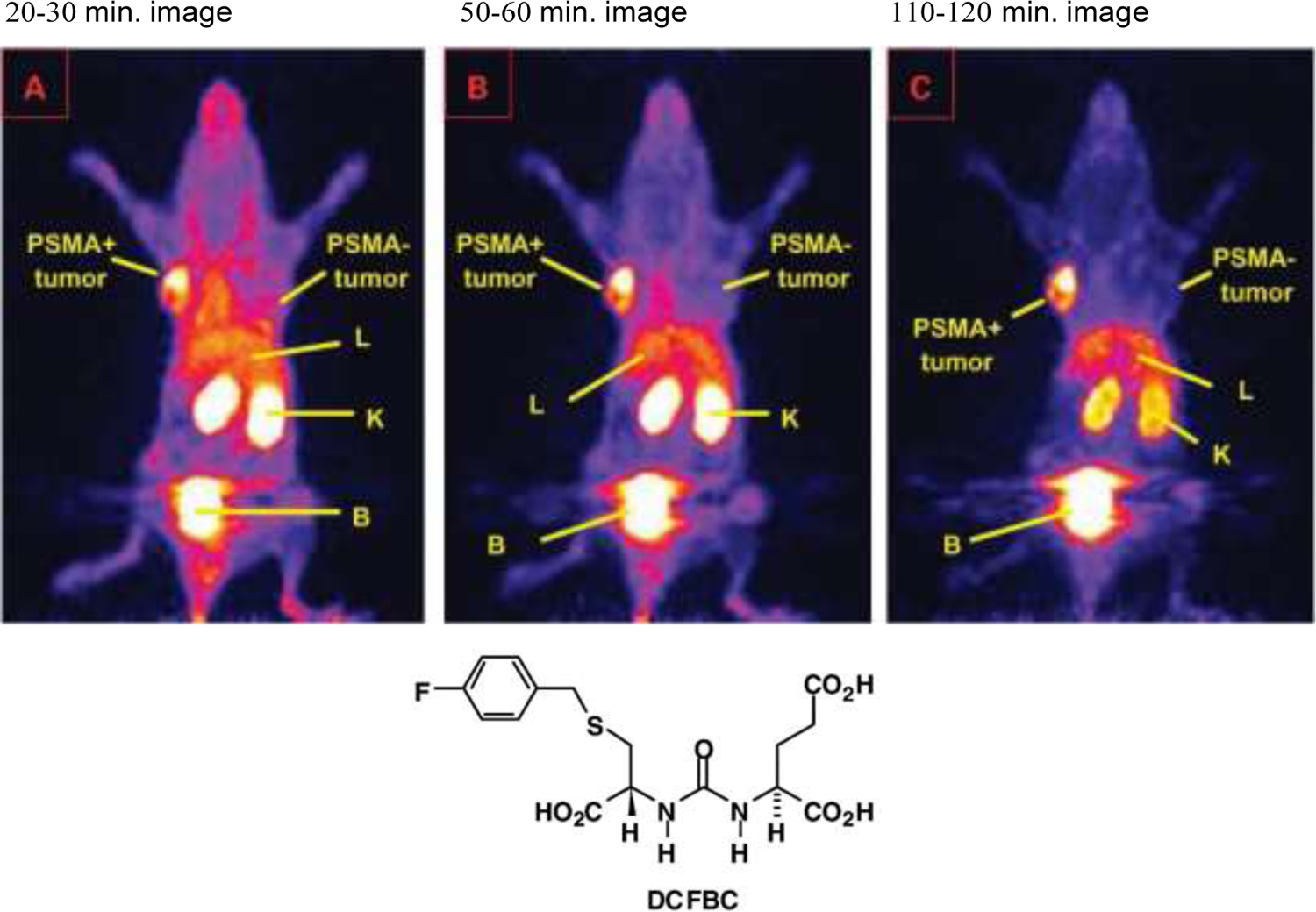
PET images of animals with PSMA+ PC-3 PIP tumors in their left shoulders and PSMA− PC-3 tumors in their right shoulders after injection with [18F]DCFBC at times shown. Accumulation is only seen in the PSMA + tumor which also shows better retention than initial renal uptakes indicating slow urine wash-out and high bladder activity.
Reprinted with permission from Mease et al. Clinical Cancer Research, AACR journals.
The later-generation agent, 18F-DCFPyL was developed and then tested in the same PC-3 (PSMA+/PSMA−) model system.87 As seen in FIGURE 8, the model showed more rapid and compete clearance through the renal system than the earlier analog. Structurally, DCFPyL contained an α-substituted amino lysyl- residue whereas the DCFBC contained an α-substituted cysteinyl- residue on one of the ureal-functionalities with 18F-aroyl substitutions on the respective amino acid side chains. Additionally, the DCFPyL derivative was substituted with an 18F-nicotinyl- side chain whereas the DCFBC had an 18F-benzyl derivative making the former radiolabel easier to produce, and the final product more water-soluble. In human dosimetry estimates from this study, the effective dose based on ICRP 60 tissue weighting factors was 13.6 mSv/MBq, indicating a maximum of 331 MBq (9mCi) could be administered without exceeding the maximum 50 mGy critical organ dose limit (to the urinary bladder wall). The improved properties directly resulted in the superior xenograft imaging results seen in FIGURE 9, which was crucial in deciding to proceed with further development of a clinical product. Piflufolastat F-18 injection (Pylarify, Lantheus, N. Billerica MA) became the first radiofluorinated PSMA agent approved by the FDA for PET imaging in May 2021 after two pivotal clinical trials (CONDOR and OSPREY studies). Updated clinical investigations suggest that 18F-DCFPyL PSMA imaging in combination with advanced magnetic resonance imaging may change the paradigm for prostate cancer patient diagnoses and management.88
FIGURE 8.
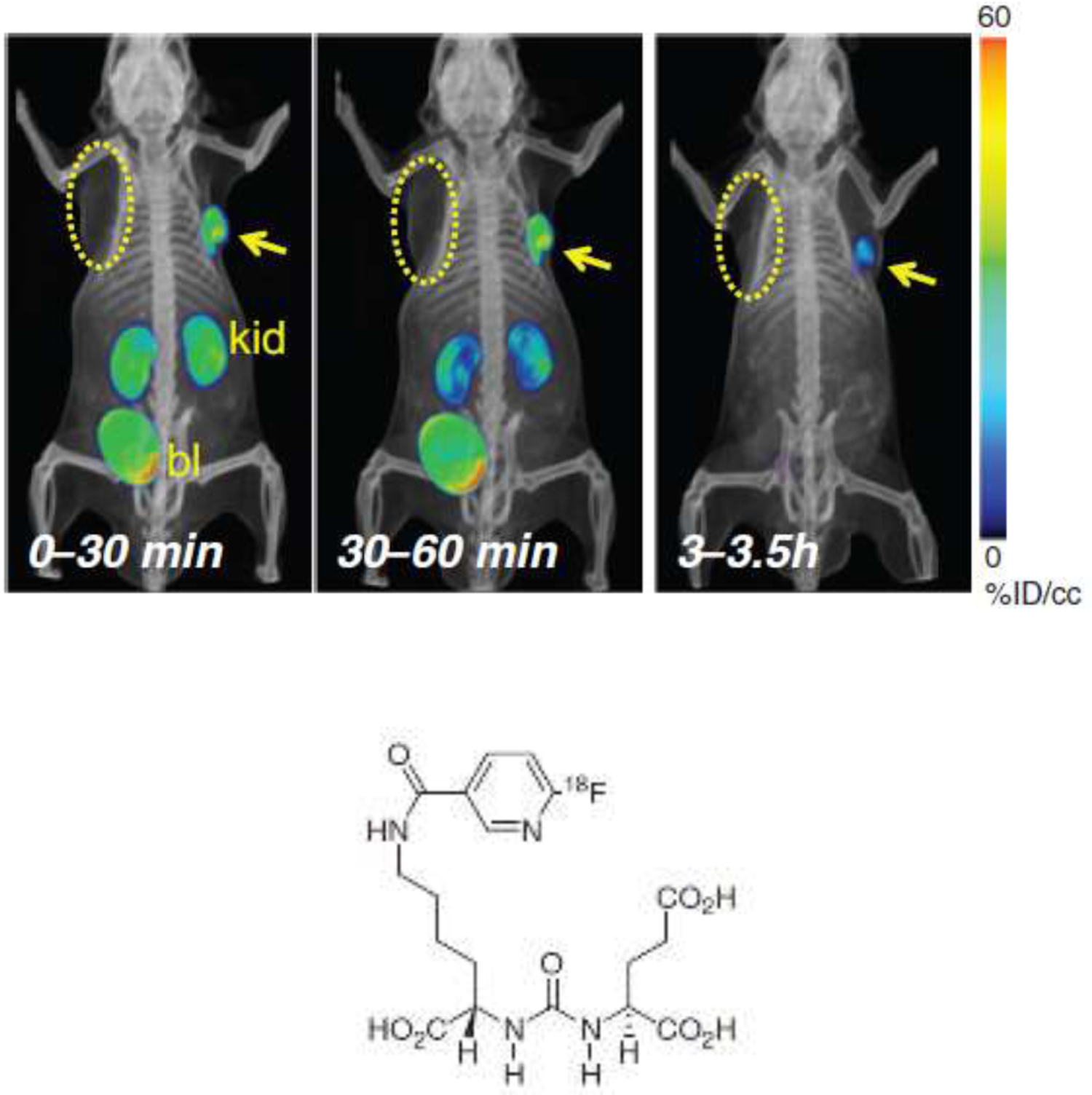
PET-CT volume-rendered composite images of the biodistribution of [18F]-DCFPyL in PC3 PIP (PSMA+) and PSMA− PC3 flu (PSMA−) with tumor xenografts in opposite forearms of NOD-SCID mice. At 30 minutes post-injection radionuclide uptake was evident within the PSMA+ tumor and the kidneys, and uptake receded from kidneys with bladder excretion faster than from the tumor and was not evident at 3.5 hours. Compare with FIGURE 7 images.
Reprinted with permission from Chen et al. Clinical Cancer Research, AACR journals.
FIGURE 9.
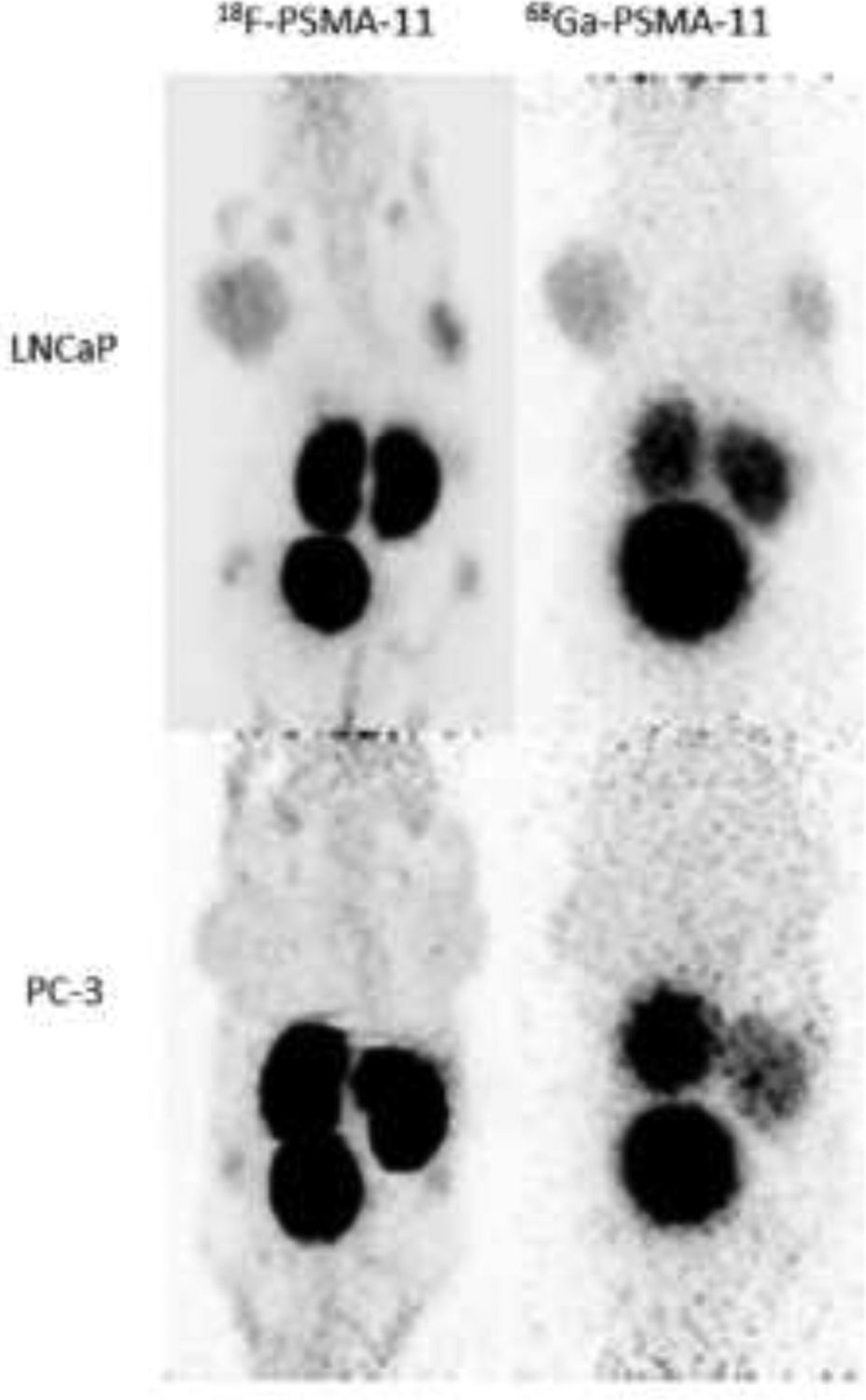
68Ga- and 18F-PSMA 11 analogs. Maximum intensity projection (MIP) PET images at 1 h p.i. of two mice with either PSMA positive tumors (LNCaP, top) or PSMA negative tumors (PC3, bottom). Images show high kidney and bladder uptake with tumor implanted in the animal’s shoulder regions.
Reprinted under Creative Commons from Piron et al. Intra-individual dynamic comparison of 18F-PSMA-11 and 68Ga-PSMA-11 in LNCaP xenograft bearing mice. Scientific Reports, 2020, Springer Nature.
Pylarify was not the first anti-PSMA targeting agent to gain regulatory approval. It was preceded by a 68Ga radiolabeled agent, Ga68 PSMA 11 (UCLA and UC San Francisco). 68Ga-PSMA 11 contains the same urea recognition unit as DCFPyL but has a gallium-68 chelation sub-structure termed HBED on its lysyl-epsilon amino residue. However, despite excellent imaging results, the dependency of this agent on 68Ga produced from an in-house 68Ga/68Ge generator system and its short half-life and high energy positron emission mitigate against its widespread use, and investigators are actively looking for 18F analogs of the agent.89 Preclinically, in LNCaP xenograft models 68Ga- and 18F-PSMA 11 analogs behave quite similarly with very high kidney and bladder radioactivity uptakes at 1-hour post-injection (FIGURE 9). The 18F PSMA 11 analog was structurally much different from the 68Ga-PSMA 11 original version, with the HBED 68Ga chelate discarded in favor of a polyazamacrocyclic structure, making the comparison somewhat contrived with regard to being ‘PSMA 11’, as it is really between two quite different compositions-of-matter. However, this study demonstrates further the remarkable ability of the PSMA enzyme to bind to multiple diverse structural derivatives of the basic urea-glutamate structural template, and this has led to numerous other analogs targeting PSMA with diagnostic and therapeutic structural variations of that basic structural sub-unit.
Examples of the diverse agents based on the ureal-glutamate subunit include 123/124/131I-MIP-1072/−1095, 99mTc-MIP-1404/−1405, 68Ga-HBED-CC-PSMA, 68Ga/177Lu-PSMA I&T, 68Ga/177Lu-PSMA-617 and 18F-PSMA-1007, with a clear thrust toward theranostic agents.90 Driven mostly by the needs of radiotherapeutic agents the most significant efforts are now directed toward reduction of renal and salivary gland uptakes of agents by blocking agents91 or by (there is no salivary gland uptake via PSMA targeting in mice) by continuing modifications of chelates, isotopes, linkers and PSMA-targeting sub-structures, as well as use of imaging vectors in xenograft models to establish dosimetry for planned future therapeutics.92 Additionally, with respect to anti-PSMA agents, attempts to modify biodistributions of PSMA targeting vectors have explored the temporary binding of such agents to albumin by addition of a hydrophobic (usually aromatic group) to extend serum half-life, as a much greater appreciation of the role of protein binding in final deployment of administered drugs and radiolabeled agents is appreciated.93 Initial explorations in this field have already led to modification of well-known anti-PSMA targeting agents such as PSMA-617 with serum-half-life extenders such as the dye Evans blue.94 FIGURE 10 shows the effect of the attachment of Evans blue dye to PSMA-617, by two methods, their radiolabeling with 86Y- and the PET imaging biodistribution in PSMA+ xenografts. Enhanced xenograft retention of the radiolabel out to 24–48 h post-injection is demonstrated. Multiple other serum-extending agents based on lipophilic attachments, such as poly(ethylene)glycols, can be envisaged and work is very active in formulating many compositions designed to ameliorate the too-rapid washout of low molecular weight, peptide and small protein tumor targeting agents.95
FIGURE 10.
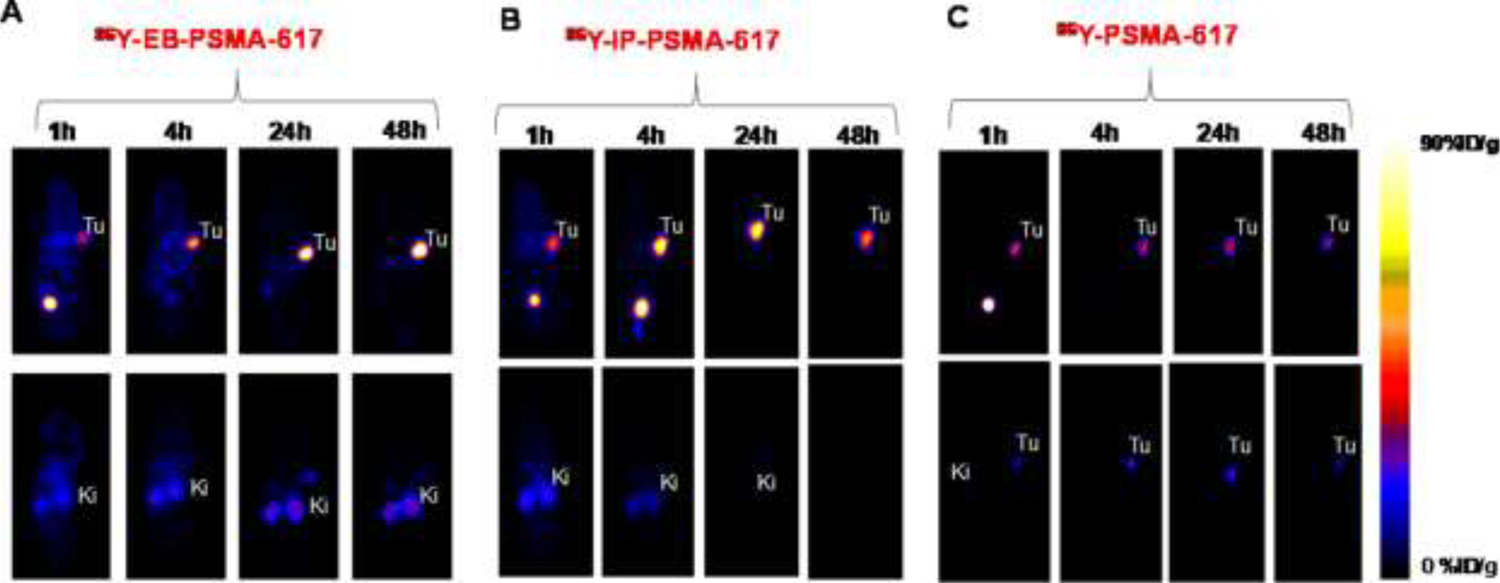
Albumin binding. PET imaging of 86Y-PSMA-617 [90Y therapeutic analog radionuclide] derivatives with and without attachment of the albumin-binding dye Evans Blue, out to 48 hours post-injection in mice with xenografts of PC3-PIP (PSMA+) or PC-3 (PSMA−) cells. Upper ventral, lower dorsal, slices.
“Reprinted (adapted) with permission from Bioconjugate Chemistry 29:3213–3221. Copyright 2018 American Chemical Society.”
Some of the most recent work dedicated to low MW anti-PSMA agents have focused on bivalent targeting strategies, for example combined PSMA and GRPR targets.96 Using PC-3 PIP subtype tumor models with PC-3 cells transfected to express PSMA (FIGURE 11) both protein targets could be identified with a low MW agent that had a chelating group for 111In and 177Lu, together with the ureayl- glutamate sub-unit for PSMA and a bombesin analog peptide for GRPR, with selective receptor blockage with 2-PMPA and bombesin, respectively. Further, similar work extended the concept to bispecific targeting of two epitopes, one to PSMA and the other to fibroblast activation protein (FAP) using 64Cu labeling. While showing multiple cancers, high background radioactivity was also seen.97
FIGURE 11.
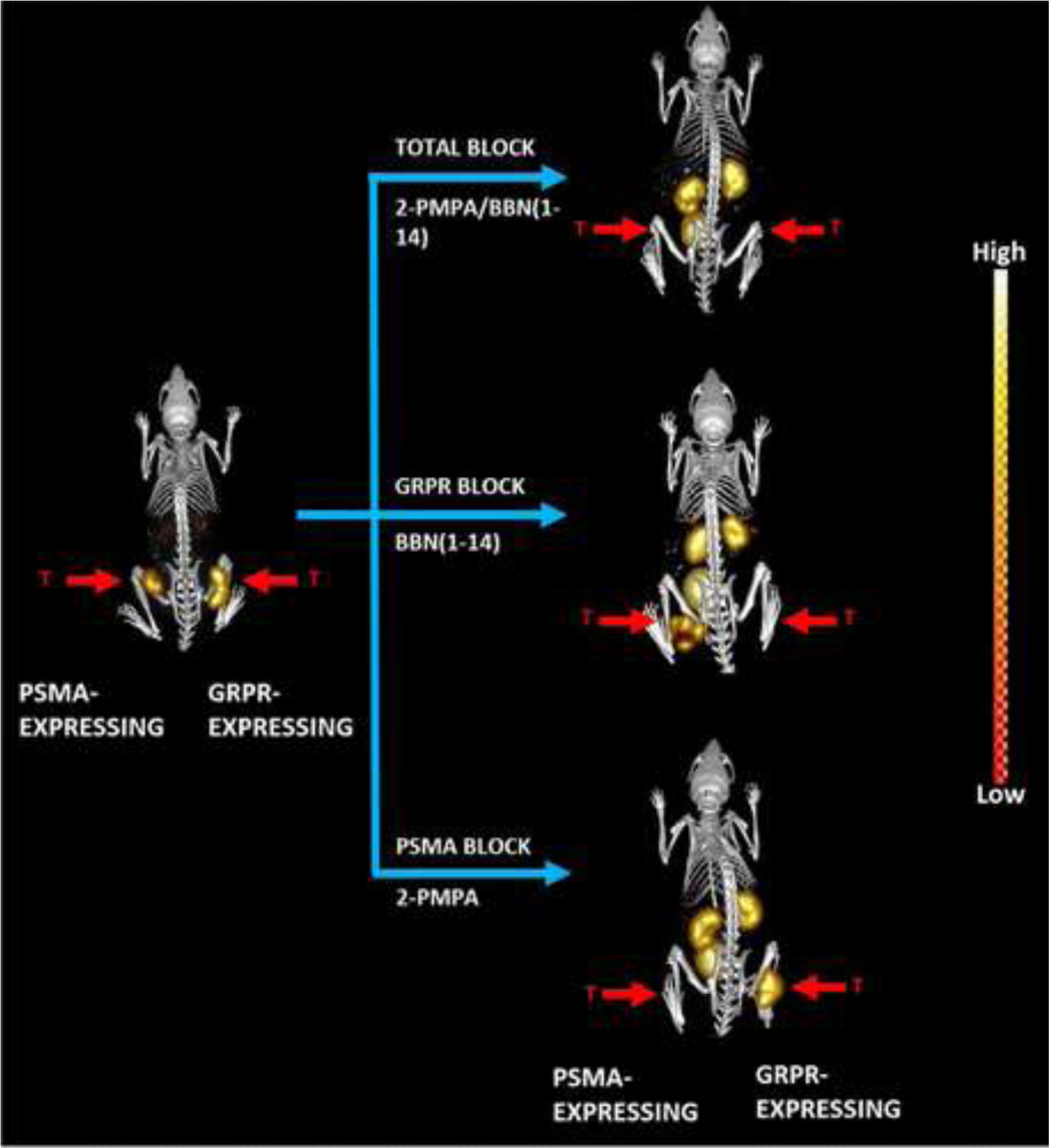
Bispecific Targeting. Maximum-intensity microSPECT tumor and microCT skeletal fusion coronal, whole-body images with and without blocking of PC-3 and PC-PIP tumor-bearing SCID mice at 4 h post-tail vein injection of [DUPA-6-Ahx-([111In]In-DO3A)-8-Aoc-BBN ANT]. PSMA blocked with excess 2-PMPA, and GRPR blocked with cold bombesin. Bilateral xenografted tumors in hind legs are indicated by red arrows.
“Reprinted (adapted) with permission from Journal of Medicinal Chemistry, 64:2151–2166, Copyright 2021, American Chemical Society.”
In a study of growth patterns in response to androgen deprivation therapies (ADT) in LNCaP xenografted mice, an 18F-anti-PSMA PET agent showed great variability in tumor uptake values over the time courses of androgen blockade, indicating highly variable PSMA expression in tumors at different periods of tumor treatments.98 Since ADT is such an important aspect of prostate cancer therapy it is important to know its effects on PSMA uptake and animal models are useful to accumulate this data.
Testosterone and Steroids
Testosterone and related steroids are extremely hydrophobic and tend to circulate bound to sex hormone globulin or other ‘carrier’ proteins such as serum albumin. To this day testosterone activity and biology is poorly understood with controversies over the role of free testosterone still unresolved.99 Typically, only 2% of serum testosterone is in free form with 44% and 50% bound to sex hormone binding globulin (SHBG) and serum albumin, respectively. A PET radionuclide such as 18F- bound to a steroidal testosterone analog displays extremely fast and near-complete deposition into the liver soon after injection, making efforts to image animal xenograft models with testosterone derivatives largely futile, besides which rodents do not express SHBG. Nevertheless, a radiofluorinated testosterone analog was prebound in vitro to SHBG and injected into LNCaP xenografted mice as a strategy to overcome the albumin binding problems, however, poor xenograft uptakes were still seen in the face of overwhelming radionuclide uptake in the liver.100 In 1992 a series of fluorinated testosterone analogs were prepared and studied to provide guidance with regard to a useful 18F-labeled testosterone analog and showed that fluorination did not markedly affect receptor binding or interfere with other steroid binding if the C-3 and C17 substitution positions were avoided,101 and subsequently fluorinated analogs were synthesized as prospective PET agents.102
[18F]16β-fluoro-5α-dihydrotestosterone (DHT) was studied in an LNCaP animal model to delineate testosterone biology and link PET imaging observations to biological reactions, rather than using it to merely highlight a tumor’s presence.103 In this study the role of glucuronidation as a detoxification for drugs was examined, with the 17-oxy position of dihydrotestosterone being the point (vide supra) where the mice effectively can glucuronidate and remove excess dihydrotestosterone from its system. Loss of this ability to glucuronidate the steroid enabled the system to survive with extra-gonadal testosterone and steroid production and showed a parallel finding to that seen in the clinic with treatments involving an extended steroid depletion strategy.104 The availability of new androgen blocking drugs, opens the door to the preparation of radiofluorinated analogs with superior imaging pharmacokinetics to testosterone analogs. Such agents may be used to target androgen receptors in the prostate, and the first radiofluorinated analog of enzalutamide was recently investigated in the LNCaP xenograft model for this purpose.105
SUMMARY
Molecular medicine, and its diagnostic partner, molecular imaging, have made great advances over the last two decades. SPECT and PET are essential to carefully evaluate potential advances in this area using animal models. Xenograft models of prostate cancer are well established and proven to be trusted standards in predictions of clinical utility, and just as importantly, indicators of potential clinical problems. The limitations of such models and their relevance to the clinical situation always needs to be kept in mind, from the simplest comparisons of size and species to the equally important aspects of biochemistry and natural and xenograft disease distinctions.
Newer animal models using advanced transplantation techniques, genetic engineering, better patient disease samples, improved understanding of prostate cancer growth in mice, and the use of other animals in modeling, promise further improvements in the relevance of tumor models to clinical disease. Clinically, prostate cancer remains notoriously difficult to detect on a patient-by patient basis both regarding its initial presence and for its potential in any specific individual to progress to metastatic or more aggressive growth. It is in these areas that in vivo modeling systems may best serve future patient needs.
Highlights:
Immunocompromised mice allow controlled tumor xenograft growth.
Prostate and metastatic prostate cancer cell lines isolated and grown represent various stages of the disease.
Advances in PET technology allows highly accurate imaging in xenografted mice.
Biodistribution of radiolabeled reagents can be fully quantified.
Any type of targeting vector can be radiolabeled and accurately studied.
This methodology has formed the basis of extension to human studies.
Results strongly support the regulatory approvals for human use.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. 75N91019D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.Kallman RF: Animal experiments in radiotherapy I - small animals. J Can Assoc Radiol 26:15–24, 1975. [PubMed] [Google Scholar]
- 2.Handelsman H: The limitations of model systems in prostatic cancer. Oncology 34:96–99, 1977. [DOI] [PubMed] [Google Scholar]
- 3.Farolfi A, Mei R, Ali S, et al. : Theragnostics in prostate cancer. Q J Nucl Med Mol Imaging 65:333–341, 2021. [DOI] [PubMed] [Google Scholar]
- 4.Murphy GP, Merrin CE: Chemotherapy of advanced prostatic cancer today. Prog Clin Biol Res 6:285–299, 1976. [PubMed] [Google Scholar]
- 5.Heller JR: Cancer chemotherapy, history, and present status. Bull N Y Acad Med 38:348–363, 1962. [PMC free article] [PubMed] [Google Scholar]
- 6.De Vita VT, Lawrence TS, Rosenberg SA (eds): Cancer: Principles and Practice of Oncology (ed 11). Philadelphia, PA, Wolters-Kluwer, 2022. [Google Scholar]
- 7.Mukherjee S, The Emperor of All Maladies: A Biography of Cancer. New York, NY, Scribner, 2011. [Google Scholar]
- 8.Merseberger AS, Roesch MC: Advanced delivery of leuprorelin acetate for the treatment of prostatic cancer. Expert Rev Anticancer Ther 22: 703–715, 2022. [DOI] [PubMed] [Google Scholar]
- 9.Huggins C, Hodges CV: Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin 22:232–240, 1972. [DOI] [PubMed] [Google Scholar]
- 10.Redding TW, Schally AV: Inhibition of prostate tumor growth in two rat models by chronic administration of D-Trp6 analogue of luteinizing hormone-releasing hormone. Proc Natl Acad Sci USA 78:6509–6512, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schally AV, Redding TW, Comaru-Schally AM: Potential use of analogs of luteinizing hormone-releasing hormones in the treatment of hormone-sensitive neoplasms. Clinical Trial Cancer Treat Rep 68:81–89, 1984. [PubMed] [Google Scholar]
- 12.Schally AV, Redding TW: Combination of long-acting microcapsules of the D-tryptophan-6 analog of luteinizing hormone-releasing hormone with chemotherapy: investigation in the rat prostate cancer model. Proc Natl Acad Sci U S A 82:2498–2502, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jungwirth A, Schally A, Nagy A, et al. : Regression of rat Dunning R-3327-H prostate carcinoma by treatment with targeted cytotoxic analog of luteinizing hormone-releasing hormone AN-207 containing 2-pyrrolinodoxorubicin. Int J Oncol 10:877–884, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Flanagan SP: ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res Camb 8:295–309, 1966. [DOI] [PubMed] [Google Scholar]
- 15.Pantelouris EM: Absence of thymus in a mouse mutant. Nature 217:370–371, 1968. [DOI] [PubMed] [Google Scholar]
- 16.Povlsen CO, Rygaard J: Heterotransplantation of human adenocarcinomas of the colon and rectum to the mouse mutant Nude. A study of nine consecutive transplantations. Acta Pathol Microbiol Scand A 79:159–69, 1971. [DOI] [PubMed] [Google Scholar]
- 17.Artzt K: Breeding and husbandry of “nude” mice. Transplantation 13:547–549, 1972. [DOI] [PubMed] [Google Scholar]
- 18.Giovanella BC, Stehlin JS: Heterotransplantation of human malignant tumors in “nude” thymusless mice. I. Breeding and maintenance of “nude” mice. J Natl Cancer Inst 51:615–619 1973. [PubMed] [Google Scholar]
- 19.Schroeder FH, Okada K, Jellinghaus W, et al. : Human prostatic adenoma and carcinoma. Transplantation of cultured cells and primary tissue fragments in “nude” mice. Invest Urol 13:395–403, 1976. [PubMed] [Google Scholar]
- 20.Gottschalk A, Anger HO: Sensitivity of the positron scintillation camera for detecting simulated brain tumors UCRL-11033. UCRL US At Energy Comm 86:126–129 1963. [PubMed] [Google Scholar]
- 21.Mallard JR, Myers MJ: The performance of a gamma camera for the visualization of radioactive isotope in vivo. Phys Med Biol 8:165–182, 1963. [DOI] [PubMed] [Google Scholar]
- 22.Anger HO, Gottschalk A: Localization of brain tumors with the positron scintillation counter. J Nucl Med 4:326–30, 1963. [PubMed] [Google Scholar]
- 23.Anger HO: Radio-activity distribution detector. US Patent 2,779,876 Filed March 3, 1953, Issued January 29, 1957.
- 24.Roentgen W: [Diagnostic imaging 100 years ago]. Servir. 44:207–10, 1996. [PubMed] [Google Scholar]
- 25.Walsh JM, Amendola MA, Konerding KF, et al. : Computed tomographic detection of pelvic and inguinal lymph-node metastases from primary and recurrent pelvic malignant disease. Radiology 137:157–166, 1980. [DOI] [PubMed] [Google Scholar]
- 26.Rebello RJ, Oing C, Knudsen KE, et al. : Nat Rev Dis Primers 7, 9, 2021. 10.1038/s41572-020-00243-0. [DOI] [PubMed] [Google Scholar]
- 27.Bostwick DG, Meiers I: Prostate Chapter 32. pp 1121–1180. Weidner N, Cote RJ, Suster S, Weiss LM (Eds) In: Modern Surgical Pathology, (ed 2). Philadelphia, PA, Saunders, 2009. [Google Scholar]
- 28.Van Huizen I, Wu G, Moussa M, et al. : Establishment of a serum tumor marker for preclinical trials of mouse prostate cancer models. Clin Cancer Res 11:7911–7919, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Ilic D, Neuberger MM, Djulbegovic M, et al. : Screening for prostate cancer. Cochrane Database Syst. Rev 1, CD004720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horoszewicz JS, Leong SS, Kawinski E, et al. : LNCaP model of human prostatic carcinoma. Cancer Res 43:1809–1818, 1983. [PubMed] [Google Scholar]
- 31.Hoehn W, Schroeder FH, Reimann JF, et al. : Human prostatic adenocarcinoma: some characteristics of a serially transplantable line in nude mice (PC 82). The Prostate 1:95–104, 1980. [DOI] [PubMed] [Google Scholar]
- 32.Bosma GC, Custer RP, Bosma MJ: A severe combined immunodeficiency mutation in the mouse. Nature 301:527–530, 1983. [DOI] [PubMed] [Google Scholar]
- 33.Sato N, Gleave NE, Bruchovsky N, et al. : A metastatic and androgen-sensitive human prostate cancer model using intraprostatic inoculation of LNCaP cells in SCID mice. Cancer Res 57:1584–1589, 1997. [PubMed] [Google Scholar]
- 34.Shevrin DH, Gorny KI, Kukreja SC: Patterns of metastasis by the human prostate cancer cell line PC-3 in athymic nude mice. The Prostate 15:187–194, 1989. [DOI] [PubMed] [Google Scholar]
- 35.Wang M, Stearns ME: Isolation and characterization of PC-3 human prostatic tumor sublines which preferentially metastasize to select organs in S.C.I.D. mice. Differentiation 48:115–125, 1991. [DOI] [PubMed] [Google Scholar]
- 36.Paine-Murrieta GD, Taylor CW, Curtis RA, et al. : Human tumor models in the severe combined immune deficient (scid) mouse. Cancer Chemother Pharmacol 40:209–214, 1997. [DOI] [PubMed] [Google Scholar]
- 37.Marques RB, van Werden WM, Erkens-Schulze S, et al. : The human PC346 xenograft and cell line panel: A model system for prostate cancer progression. Eur Urol 49: 245–257, 2006. [DOI] [PubMed] [Google Scholar]
- 38.van Bokhoven A, Caires A, Maria MD, et al. : Spectral karyotype (SKY) analysis of human prostate carcinoma cell lines. The Prostate 57: 226–244, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Isaacs JT, Coffey DS: Adaptation versus selection as the mechanism responsible for the relapse of prostatic cancer to androgen ablation therapy as studied in the Dunning R-3327-H adenocarcinoma. Cancer Res 41:5070–5075, 1981. [PubMed] [Google Scholar]
- 40.Veldscholte J, Ris-Stalpers C, Kuiper GG, et al. : A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun 173: 534–540, 1990. [DOI] [PubMed] [Google Scholar]
- 41.Bladou F, Gleave ME, Penault-Llorca F, et al. : [In vitro and in vivo models developed from human prostatic cancer] [Article in French] Prog Urol 7:384–396, 1997. [PubMed] [Google Scholar]
- 42.Nemeth JA, Harb JF, Barroso U Jr, et al. : Severe combined immunodeficient-hu model of human prostate cancer metastasis to human bone. Cancer Res 59:1987–1993, 1999. [PubMed] [Google Scholar]
- 43.Pienta KJ, Abate-Shen C, Agus DB, et al. : The current state of preclinical prostate cancer animal models. The Prostate 68:629–639, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belloli S, Jachetti E, Moresco RM, et al. : Characterization of preclinical models of prostate cancer using PET-based molecular imaging. EJNMMI 36:1245–1255, 2009. [DOI] [PubMed] [Google Scholar]
- 45.Thalmann GN, Sikes RA, Wu TT, et al. : LNCaP progression model of human prostate cancer: androgen-independence and osseous metastasis. The Prostate 44:91–103, 2000. [DOI] [PubMed] [Google Scholar]
- 46.Liu W, Zhu Y, Ye L, et al. : Establishment of an orthotopic prostate cancer xenograft mouse model using microscope-guided orthotopic injection of LNCaP cells into the dorsal lobe of the mouse prostate. BMC Cancer 22:173, 2022. doi: 10.1186/s12885-022-09266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keller JM, Schade GR, Ives K, et al. : A novel canine model for prostate cancer. The Prostate. 73:952–959, 2013. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Abenojar EC, Wang J, et al. : Development of a novel castration-resistant orthotopic prostate cancer model in New Zealand White rabbit. The Prostate 82:695–705, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linxweiler J, Körbel C, Müller A, et al. : A novel mouse model of human prostate cancer to study intraprostatic tumor growth and the development of lymph node metastases. The Prostate 78:664–675, 2018. [DOI] [PubMed] [Google Scholar]
- 50.Choi H-J, Lee H-B, Jung S, et al. : Development of a mouse model of prostate cancer using the sleeping beauty transposon and electroporation. Molecules 23:1360, 2018. doi: 10.3390/molecules23061360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Risbridger GP, Clark AK, Porter LH, et al. : The MURAL collection of prostate cancer patient derived xenografts enables discovery through preclinical models of uro-oncology. Nat Commun 12:5049, 2021. doi: 10.1038/s41467-021-25175-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Köhler G, Milstein C: Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 236:495–497, 1975. [DOI] [PubMed] [Google Scholar]
- 53.Luo R, Liu H, Cheng Z: Protein scaffolds: antibody alternatives for cancer diagnosis and therapy. RSC Chem Biol 3:830–847, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merkens L, Sailer V, Lessel D, et al. : Aggressive variants of prostate cancer: underlying mechanisms of neuroendocrine transdifferentiation. J Exp Clin Cancer Res 41:46. 2022. doi: 10.1186/s13046-022-02255-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fredolini C, Liotta LA, Petricoin EF: Application of proteomic technologies for prostate cancer detection, prognosis, and tailored therapy. Crit Rev Clin Lab Sci 47:125–138, 2010. [DOI] [PubMed] [Google Scholar]
- 56.Conti PS, Lilien DL, Hawley K, et al. : PET and [18F]-FDG in oncology: a clinical update. Nucl Med Biol 23:717–735, 1996. [DOI] [PubMed] [Google Scholar]
- 57.Salas JR, Clark PM: Signaling pathways that drive 18F-FDG accumulation in cancer. J Nucl Med 63:659–663, 2022. [DOI] [PubMed] [Google Scholar]
- 58.Kukuk D, Reischl G, Raguin O, et al. : Assessment of PET tracer uptake in hormone-independent and hormone-dependent xenograft prostate cancer mouse models. J Nucl Med 52:1654–1663, 2011. [DOI] [PubMed] [Google Scholar]
- 59.Zhu A, Marcus DM, Shu H-KG, et al. : Application of metabolic PET imaging in radiation oncology. Rad Res 177:436–448, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stenman UH, Paus E, Allard WJ, et al. : Summary report of the TD-3 workshop: characterization of 83 antibodies against prostate-specific antigen. Tumor Biol (Suppl 1) 20:1–12, 1999. [DOI] [PubMed] [Google Scholar]
- 61.Chang SS: Overview of prostate-specific membrane antigen. Rev Urol. (suppl 10) 6:S13–18, 2004. [PMC free article] [PubMed] [Google Scholar]
- 62.Hinkle GH, Burgers JK, Neal CE, et al. : Multicenter radioimmunoscintigraphic evaluation of patients with prostate carcinoma using indium-111 capromab pendetide. Cancer 83:739–747, 1998. [PubMed] [Google Scholar]
- 63.Chang SS, Reuter VE, Heston WD, et al. : Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res 59:3192–3198, 1999. [PubMed] [Google Scholar]
- 64.Smith-Jones PM, Vallabhajosula S, Navarro V, et al. : Radiolabeled monoclonal antibodies specific to the extracellular domain of prostate-specific membrane antigen: preclinical studies in nude mice bearing LNCaP human prostate tumor. J Nucl Med 44:610–617, 2003. [PubMed] [Google Scholar]
- 65.Holland JP, Divilov V, Bander NH, et al. : 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J Nucl Med 51:1293–1300, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Esteban JM, Schlom J, Gansow OA, et al. : New method for the chelation of indium-111 to monoclonal antibodies: biodistribution and imaging of athymic mice bearing human colon carcinoma xenografts. J Nucl Med 28:861–870, 1987. [PubMed] [Google Scholar]
- 67.DeNardo GL, Kroger LA, DeNardo SJ, et al. : Comparative toxicity studies of yttrium-90 MX-DTPA and 2-IT-BAD conjugated monoclonal antibody (BrE-3). Cancer (3 suppl) 73:1012–1022, 1994. [DOI] [PubMed] [Google Scholar]
- 68.Govindan SV, Shih LB, Goldenberg DM, et al. : 90Yttrium-labeled complementarity-determining-region-grafted monoclonal antibodies for radioimmunotherapy: radiolabeling and animal biodistribution studies. Bioconjug Chem 9:773–782, 1998. [DOI] [PubMed] [Google Scholar]
- 69.Kang CS, Song HA, Milenic DE, et al. : Preclinical evaluation of NETA-based bifunctional ligand for radioimmunotherapy applications using 212Bi and 213Bi: radiolabeling, serum stability, and biodistribution and tumor uptake studies. Nucl Med Biol 40:600–605, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mastren T, Marquez BV, Sultan DE, et al. : Cyclotron production of high-specific activity 55Co and in vivo evaluation of the stability of 55Co metal-chelate-peptide complexes. Mol Imaging 14:526–533, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feiner IJV, Brandt M, Cowell J, et al. : The race for hydroxamate-based zirconium-89 chelators. Cancers (Basel) 13:4466, 2021. doi: 10.3390/cancers13174466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cho H, Al-Saden N, Lam H, et al. : A comparison of DFO and DFO* conjugated to trastuzumab-DM1 for complexing 89Zr - In vitro stability and in vivo microPET/CT imaging studies in NOD/SCID mice with HER2-positive SK-OV-3 human ovarian cancer xenografts. Nucl Med Biol 84–85:11–19, 2020. [DOI] [PubMed] [Google Scholar]
- 73.Kampmeier F, Williams JD, Maher J, et al. : Design and preclinical evaluation of a 99mTc-labelled diabody of mAb J591 for SPECT imaging of prostate-specific membrane antigen (PSMA). EJNMMI 4:13 2014. http://www.ejnmmires.com/content/4/1/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Viola-Villegas NT, Sevak KK, Carlin SD, et al. : Noninvasive imaging of PSMA in prostate tumors with (89)Zr-labeled huJ591 engineered antibody fragments: the faster alternatives. Mol Pharm 11:3965–3973, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Malmberg J, Tolmachev V, Orlova A: Imaging agents for in vivo molecular profiling of disseminated prostate cancer - targeting EGFR receptors in prostate cancer: comparison of cellular processing of [111In]-labeled affibody molecule ZEGFR:2377 and cetuximab. Int J Oncol 38:1137–1143, 2011. [DOI] [PubMed] [Google Scholar]
- 76.Mitran B, Andersson KG, Lindström E, et al. : Affibody-mediated imaging of EGFR expression in prostate cancer using radiocobalt-labeled DOTA-ZEGFR:2377. Oncol Rep 41:534–542, 2019. [DOI] [PubMed] [Google Scholar]
- 77.Leyton JV, Olafsen T, Sherman MA, et al. : Engineered humanized diabodies for microPET imaging of prostate stem cell antigen-expressing tumors. Protein Eng Des Sel 22:209–216, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsai W-TK, Zettlitz KA, Dahlbom M, et al. : Evaluation of [131I]I- and [177Lu]Lu-DTPA-A11 minibody for radioimmunotherapy in a preclinical model of PSCA-expressing prostate cancer. Mol Imaging Biol 22:1380–1391, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zettlitz KA, Tsai W-TK, Knowles SM, et al. : [89Zr]A2cDb immuno-PET of prostate cancer in a human prostate stem cell antigen knock-in (hPSCA KI) syngeneic model. Mol Imaging Biol 22:367–376, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Apolo AB, Pandit-Taskar N, Morris MJ: Novel tracers and their development for the imaging of metastatic prostate cancer. J Nucl Med 49:2031–2041, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ananias HJK, de Jong IJ, Dierckx RA, et al. : Nuclear imaging of prostate cancer with gastrin-releasing-peptide-receptor targeted radiopharmaceuticals. Review Curr Pharm Des 14:3033–3047, 2008. [DOI] [PubMed] [Google Scholar]
- 82.Carlucci G, Ananias HJK, Yu Z, et al. : Preclinical evaluation of a novel 111In-labeled bombesin homodimer for improved imaging of GRPR-positive prostate cancer. Mol Pharm 10:1716–1724, 2013. [DOI] [PubMed] [Google Scholar]
- 83.Zhang-Yin J, Provost C, Cancel-Tassin G, et al. : A comparative study of peptide-based imaging agents [68Ga]Ga-PSMA11, [68Ga]Ga-AMBA, [68Ga]Ga-NODAGA-RGD and [68Ga]Ga-DOTA-NT20.3 in preclinical prostate tumour models. Nucl Med Biol 84–85:88–95, 2020. [DOI] [PubMed] [Google Scholar]
- 84.Zhou J, Neale JH, Pomper MG, et al. : NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nat Rev Drug Discov 4:1015–1026, 2005. [DOI] [PubMed] [Google Scholar]
- 85.Rowe SP, Gorin MA, Pomper MG: Imaging of prostate-specific membrane antigen with small molecule PET radiotracers: from the bench to advanced clinical applications. Ann Rev Med 70:461–477, 2019. [DOI] [PubMed] [Google Scholar]
- 86.Mease RC, Dusich CL, Foss CA, et al. : N-[N-[(S)-1,3-dicarboxypropyl]carbamoyl]-4-[18F]fluorobenzyl-L-cysteine, [18F]DCFBC: A new imaging probe for prostate cancer. Clin Cancer Res 14:3036–3043, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Y, Pullambhatla M, Foss CA, et al. : 2-(3-{1-Carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pentanedioic acid, [18F]DCFPyL, a PSMA-based PET imaging agent for prostate cancer. Clin Cancer Res 17:7645–7653, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gaur S, Mena E, Harmon SA, et al. : Prospective evaluation of 18F-DCFPyL PET/CT in detection of high-risk localized prostate cancer: comparison with mpMRI. AJR Am J Roentgenol 215:652–659, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Piron S, Verhoeven J, Descamps B, et al. : Intra-individual dynamic comparison of 18F-PSMA-11 and 68Ga-PSMA-11 in LNCaP xenograft bearing mice. Sci Reports 10.1038/s41598-020-78273-7 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lütje S, Slavik R, Fendler W, et al. : PSMA ligands in prostate cancer - probe optimization and theranostic applications. Methods 130:42–50, 2017. [DOI] [PubMed] [Google Scholar]
- 91.Kalidindi TM, Lee S-G, Jou K, et al. : A simple strategy to reduce the salivary gland and kidney uptake of PSMA-targeting small molecule radiopharmaceuticals. EJNMMI 48:2642–2651, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim SB, Song IH, Song YS, et al. : Biodistribution and internal radiation dosimetry of a companion diagnostic radiopharmaceutical, [68Ga]PSMA-11, in subcutaneous prostate cancer xenograft model mice. Sci Reports 10.1038/s41598-021-94684-6 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hu H, Quintana J, Weissleder R, et al. : Deciphering albumin-directed drug delivery by imaging. Adv Drug Del Rev 185:114237, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Z, Tian R, Niu G, et al. : Single low-dose injection of Evans Blue modified PSMA-617 radioligand therapy eliminates prostate-specific membrane antigen positive tumors. Bioconjug Chem 29:3213–3221, 2018. [DOI] [PubMed] [Google Scholar]
- 95.Xu M, Zhang P, Ding J, et al. : Albumin binder-conjugated fibroblast activation protein inhibitor radiopharmaceuticals for cancer therapy. J Nucl Med 63:952–958, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bandari RP, Carmack TL, Malhotra A, et al. : Development of heterobivalent theranostic probes having high affinity/selectivity for the GRPR/PSMA. J Med Chem 64:2151–2166, 2021. [DOI] [PubMed] [Google Scholar]
- 97.Bionapally S, Lisok A, Lofand G, et al. ; Hetero-bivalent agents targeting FAP and PSMA. EJNMMl. 10.1007/s00259-022-05933-3. [DOI] [PubMed] [Google Scholar]
- 98.Simaeys GV, Doumont G, De Maeseneire C, et al. [18F]-JK-PSMA-7 and -FDG tumour PET uptake in treated xenograft human prostate cancer model in mice. EJNMMI 48:1773–1784, 2021. [DOI] [PubMed] [Google Scholar]
- 99.Narinx N, David K, Walravens J, et al. : Role of sex hormone-binding globulin in the free hormone hypothesis and the relevance of free testosterone in androgen physiology. Cell Mol Life Sci 79:543, 2022. 10.1007/s00018-022-04562-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Larimer BM, Dubois F, Bloch E, et al. : Specific 18F-FDHT accumulation in human prostate cancer xenograft murine models is facilitated by prebinding to sex hormone-binding globulin. J Nucl Med 59:1538–1543, 2018. [DOI] [PubMed] [Google Scholar]
- 101.Liu A, Carlson KE, Katzenellenbogen JA. Synthesis of high affinity fluorine-substituted ligands for the androgen receptor. Potential agents for imaging prostatic cancer by positron emission tomography. J Med Chem 35:2113–2129, 1992. [DOI] [PubMed] [Google Scholar]
- 102.Parent EE, Carlson KE, Katzenellenbogen JA. Synthesis of 7alpha-(fluoromethyl)dihydrotestosterone and 7alpha-(fluoromethyl)nortestosterone, structurally paired androgens designed to probe the role of sex hormone binding globulin in imaging androgen receptors in prostate tumors by positron emission tomography. J Org Chem 72:5546–5554, 2007. [DOI] [PubMed] [Google Scholar]
- 103.Zhu Z, Chung Y-M, Sergeeva O, et al. : Loss of dihydrotestosterone-inactivation activity promotes prostate cancer castration resistance detectable by functional imaging. J Biol Chem 293:17829–17837, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.James ND, de Bono JS, Spears MR, et al. : Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med 377:338–351, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Antunes IF, Dost RJ, Hoving HD, et al. : Synthesis and evaluation of 18F-enzalutamide, a new radioligand for PET imaging of androgen receptors: A comparison with 16β18F-fluoro-5α-dihydrotestosterone. J Nucl Med 62:1140–1145, 2021. [DOI] [PubMed] [Google Scholar]