

Abstract
Regulated cell death occurs in many forms, including apoptosis, pyroptosis, necroptosis, and NETosis. Most obviously, the purpose of these pathways is to kill the cell. However, many cells need to complete a set of effector programs before they die, which we define as a cellular “bucket list”. These effector programs are specific to the cell type, the mode of death, and the circumstances of death. For example, intestinal epithelial cells need to complete the process of extrusion before they die. In order to temporarily prolong their life, cells use regulatory mechanisms to temporarily prolong their life, including ESCRT- and acid sphingomyelinase-driven membrane repair. These allow cells to complete their bucket lists before they die.
Keywords: Gasdermin, Membrane repair, ESCRT, Acid sphingomyelinase, Caspase-7, Cell death
Cellular “bucket lists” during pyroptosis
Pyroptosis is a lytic form of regulated cell death. After the discovery of gasdermin D as an executioner of pyroptosis in 2015 [1,2], how this pore-forming protein and the subsequent lytic cell death is regulated has been an active area of research. Accumulating evidence showed that there are regulatory mechanisms governing lytic death downstream of gasdermin D cleavage – gasdermin activation, gasdermin pore formation, and plasma membrane rupture can all be regulated. Interestingly, recent reports showed that after gasdermin D pore opening, cells can remain alive for a substantial duration of time. Eventually these cells may undergo plasma membrane rupture and die, but before that occurs, the cells have time to embark into effector pathways, and to complete those tasks before they die. This can manifest differently in different cell types. An example of an effector pathway in macrophages and neutrophils during pyroptosis is the cleavage of IL-1β and IL-18 to their mature forms. Living dendritic cells (DCs) also activate, release IL-1β, and potentiate T cell responses [3]. A second example of an effector pathway is the activation of a cell extrusion pathway by intestinal epithelial cells (IECs). Remarkably, IECs open the gasdermin pore and simultaneously activate a pore-repair pathway, and without this repair IEC extrusion fails, suggesting that cell extrusion must be completed before the cells die [4]. These findings lead us to propose that each cell type has its own specific set of effector programs the cell needs to complete before it dies (Figure 1).
Figure 1. The cellular bucket list.
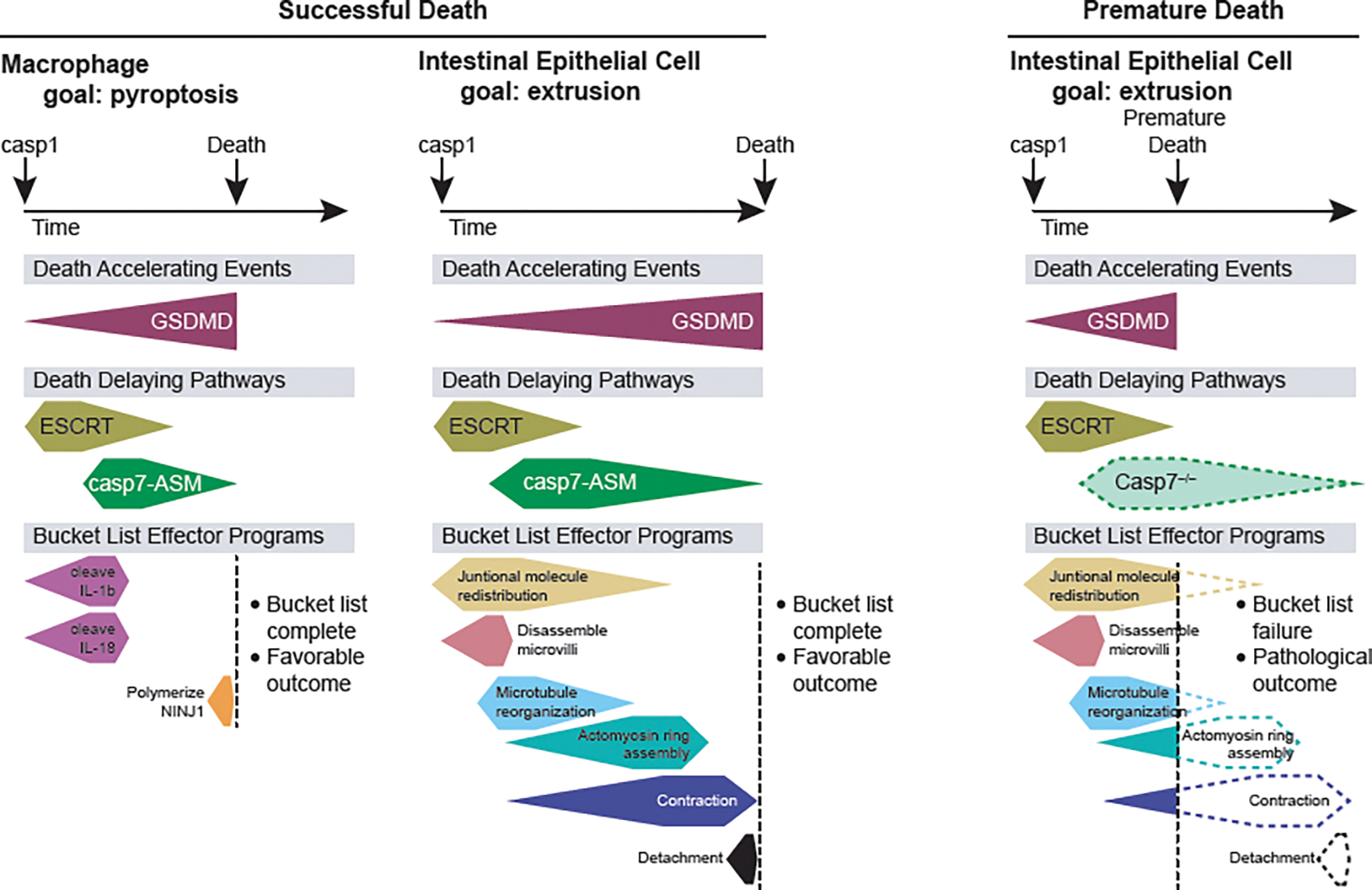
After a death trigger, events occur in three categories: 1) death-accelerating pathways, 2) death-delaying pathways, and 3) bucket list effector programs. Different cell types and even different death triggers in the same cell type will have distinct survival durations of the cell and different bucket list tasks to complete. We depict specific examples of events in each category after caspase-1 activates in either a macrophage or an intestinal epithelial cell. In both, opening of the gasdermin D pore accelerates the progression towards death of the cell. In both, membrane repair pathways should delay the death of the cell, however because caspase-7 expression is higher in intestinal epithelial cells, the delay may be slightly longer. Simultaneously, the bucket list effector pathways are initiated. In macrophages, this includes cleaving IL-1β and IL-18 to their mature forms and activating NINJ1 to rupture membranes. In intestinal epithelial cells the bucket list includes a series of tasks that, once performed in order, accomplish the extrusion of the cell into the gut lumen. When premature death occurs (depicted at right), for example in a caspase-7-deficient cell, the bucket lists remain incomplete and pathologic outcomes can occur.
In the movie “The Bucket List”, two main characters are patients diagnosed with terminal cancer. They decide to make a list of activities they want to accomplish before they “kick the bucket” and die. The characters then set out to complete their “bucket list”. In cell biology, regulated cell death pathways initiate a signaling cascade that results in the death of a cell. There is a discrete amount of time between initiation and total loss of cellular function. Different cell types have distinct tasks, “cellular bucket lists”, that they must accomplish before they die. Successful completion of the bucket list is beneficial to an organ returning to homeostasis, whereas failure to complete the bucket list can result in tissue pathology or a failure to eliminate an infection.
Bucket list can be initiated by caspase signaling, by the cellular response to gasdermin pores, or by other regulated cell death signaling pathways. Completion of the bucket list may be at odds with imminent cell death, and thus a cell may simultaneously trigger mechanism to postpone its death. In this review, we will summarize recently identified mechanisms of how gasdermin D pores are regulated to alter the timing of pyroptosis, and also discuss newly reported programs of bucket list and how these can be specific to distinct cell types including macrophages, neutrophils, dendritic cells, intestinal epithelial cells, and hepatocytes.
Death-delaying regulations of the GSDMD pore
Pyroptosis is initiated by inflammasome sensors, including NOD-like Receptors (NLRs), AIM2, and pyrin. These respond to patterns of pathogenesis, which trigger their oligomerization to form multi-protein complexes called inflammasomes that activate caspase-1 [5]. Regulation of inflammasome activation and the caspase itself are reviewed elsewhere [5–7]. In parallel, caspase-4/5/11 activates in response to cytosolic LPS [8–10]. Activated caspase-1/4/5/11 attain full proteolytic activity necessary for targeting and cleaving gasdermin D [11]. Gasdermins consists of a N-terminal pore-forming domain (GSDMDNT), a liner region, and a C-terminal autoinhibitory domain (GSDMDCT). Upon cleavage, the N-terminal pore forming domain of gasdermin D oligomerizes and subsequently inserts 18–20 nm pores into the plasma membrane. Subsequently, osmotic swelling leads to membrane rupture through a regulated process [12]. The timing of pyroptosis is regulated by various steps: alternative gasdermin D cleavage events, gasdermin D oligomerization and insertion, membrane repair pathways, regulatory volume decrease, and regulation of plasma membrane rupture (Figure 2).
Figure 2. Mechanisms regulating gasdermin D-mediated cell death.
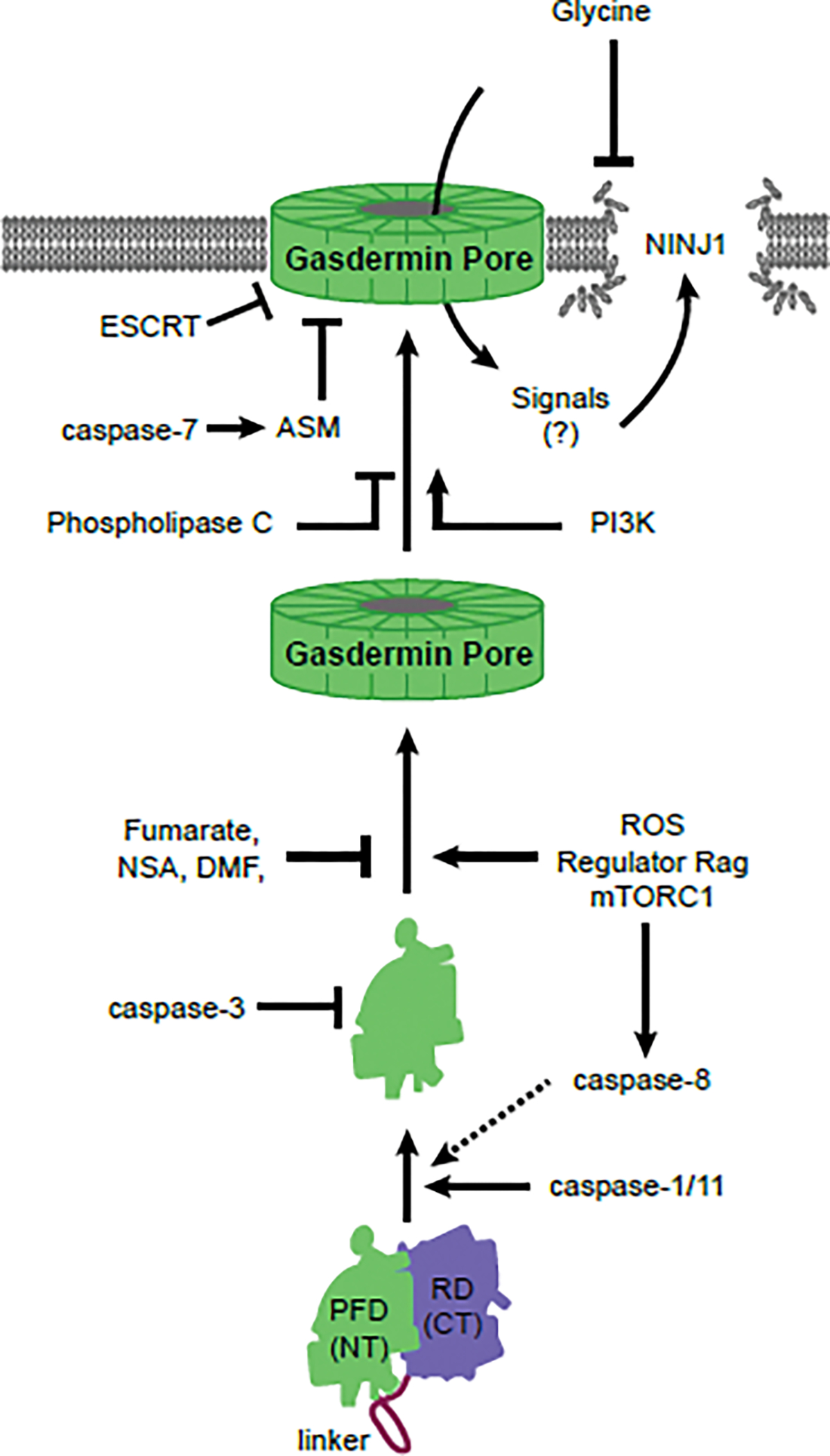
The process of gasdermin D-mediated pyroptosis is strictly regulated. Before activation, the C-terminal (CT) regulatory domain (RD) of gasdermin D seal and inhibit the N-terminal (NT) pore forming domain (PFD). Caspase-1/11 (or caspase-8 slowly) can cleave gasdermin D, releasing the active PFD that oligomerizes into large pore complexes. In parallel, gasdermin D can be cleaved by caspase-3 into an inactive form. During oligomerization, active gasdermin D PFDs require a critical cysteine to remain unmodified to permit oligomerization via ROS generating activation of Regulator Rag complex-mTORC1. Several drugs (fumarate, NSA, DMF) or intrinsic metabolites such as succinate derivative inhibit the availability of this cysteine. After pore formation, ESCRT and/or ASM-driven membrane repair pathways can remove the pores form the plasma membrane. Phosphoinositide-3 kinase (PI3K) and phospholipase C can modulate membrane phosphoinositide composition to alter the pore-opening/closing dynamics. After gasdermin D pores open, ninjurin-1 (NINJ1) causes plamsa membrane rupture. Extracellular glycine applied to cells can inhibit plasma membrane rupture in a manner that is similar to the phenotype of Ninj1-deficient cells.
Gasdermin D inactivation by alternative cleavage
Gasdermin D has another cleaved site within the GSDMDNT that is efficiently cleaved by caspase-3, ablates pore-forming activity during apoptosis [14,15]. However, during caspase-1 signaling, pore formation and pyroptosis occur faster than the slower shunt signaling to caspase-3. Thus, in the only case of slower gasdermin D activation, for example by caspase-8, will utilize caspase-3 to inactivate Gasdermin D faster than its activation, thus preventing rapid lysis (unless it is inhibited by a pathogen) [15–17].
Gasdermin oligomerization and insertion dynamics
After cleavage, GSDMDNT should use its positively-charged helix to interact with negatively-charged membrane phospholipids, especially such as phosphatidylinositol (PI) phosphates (PIPs) and weakly to phosphatidylserine, all of which are on the cytosol-facing leaflets of the plasma membrane [18–20].
To form the pores on the membrane, GSDMDNT must first oligomerize [18–24]. The Regulator-Rag complex recruits and activates mTORC, which is required for GSDMD oligomerization after caspase-1 cleaves it [25,26]. mTORC1 activation generates ROS production in mitochondria, which is essential and sufficient for the GSDMDNT oligomerization [25]. However, another report suggests that the Regulator-Rag complex only regulates GSDMDNT at the level of activation of caspase-8 when this caspase activates gasdermin D [26]. Therefore, in different pathways, the Regulator-Rag complex can control gasdermin D pore formation by different mechanisms.
A cysteine residue (C192 in mice, C191 in humans) within the GSDMDNT is important for gasdermin D oligomerization [19]. The sensitivity of this cysteine was demonstrated by using three recently-identified gasdermin D-inhibiting drugs (necrosulfamide (NSA) [27], dimethyl-fumarate (DMF) [28], disulfiram [29]), all of which covalently modify this cysteine residue. This is also illustrated in the setting of endogenous metabolites fumarate, which succinates C191 and inhibits caspase-1 binding as well as GSDMDNT oligomerization [28]. Another immunomodulating molecule, itaconate, also modifies C192 [30]. Consistently, using prediction analysis for the cysteine, this C191 is the most reactive for oxidation among all cysteine residues of gasdermin D in human [29].
After oligomerization, the dynamics of opening/closing states of gasdermin D pores are regulated by phosphoinositide phosphate (PIP) modification [31]. Gasdermin D prefers to insert into PIP2-rich membranes, where they are highly dynamic, having repeated opening and closing states [31]. Once open pores permit Ca2+ influx, phospholipase C (PLC) cleaves PIP2 and generates diacylglycerol, which closes the pores. In contrast, PI3K phosphorylates PIP2 into PIP3, which keeps the pores predominantly in open states. Therefore, the lipid composition of the cytosolic leaflet of the membrane can be used to either promote or impede gasdermin D pores.
Gasdermin D N-terminus inhibits caspase-1/11
After cleavage, liberated N-terminal gasdermin D can still bind to caspase-1/11 and prevent its activation by utilizing its RFWK motif, serving as a negative feedback system [31].
Membrane repair – Exocytic
Eukaryotic cells are frequently exposed to the threat of plasma membrane damage. This can occur due to physical damage, or be caused by bacterial pore-forming toxins. To counteract these harmful events, cells have evolved several membrane repair pathways: ESCRT-induced shedding, acid sphingomyelinase (ASM)-driven endocytosis, constriction, and patching [32].
ESCRTs are involved in a variety of the membrane dynamics, including endosomal sorting and biogenesis of intraluminal vesicles (ILVs), vesicular budding, membrane scission, cytoskeletal abscission, autophagy, and membrane repair [33]. ESCRTs are subdivided into several functional complexes; ESCRT-0, ESCRT-I, ESCRT-II, ESCRT-III. After plasma membrane damage, local Ca2+ entry rapidly recruits ALG2, a Ca2+ binding protein that bridges a Bro1 family protein, ALIX and a ESCRT-I protein, TSG101, thereby further recruiting the ESCRT-III machinery, which exerts membrane scission and completes shedding of the damaged membrane by exocytosis (Figure 3a) [34–36]. ESCRT-III can repair mixed lineage kinase domain-like pseudokinase (MLKL) pores during necroptosis [37], gasdermin D pores during pyroptosis [38], and perforin pores downstream of CTL attack [39]. In the context of gasdermin D pores, the inhibitory effect for pyroptosis was dependent on Ca2+ entry, however, ALG2 and ALIX were surprisingly not required [38].
Figure 3. Membrane repair pathways antagonize gasdermin D pores.
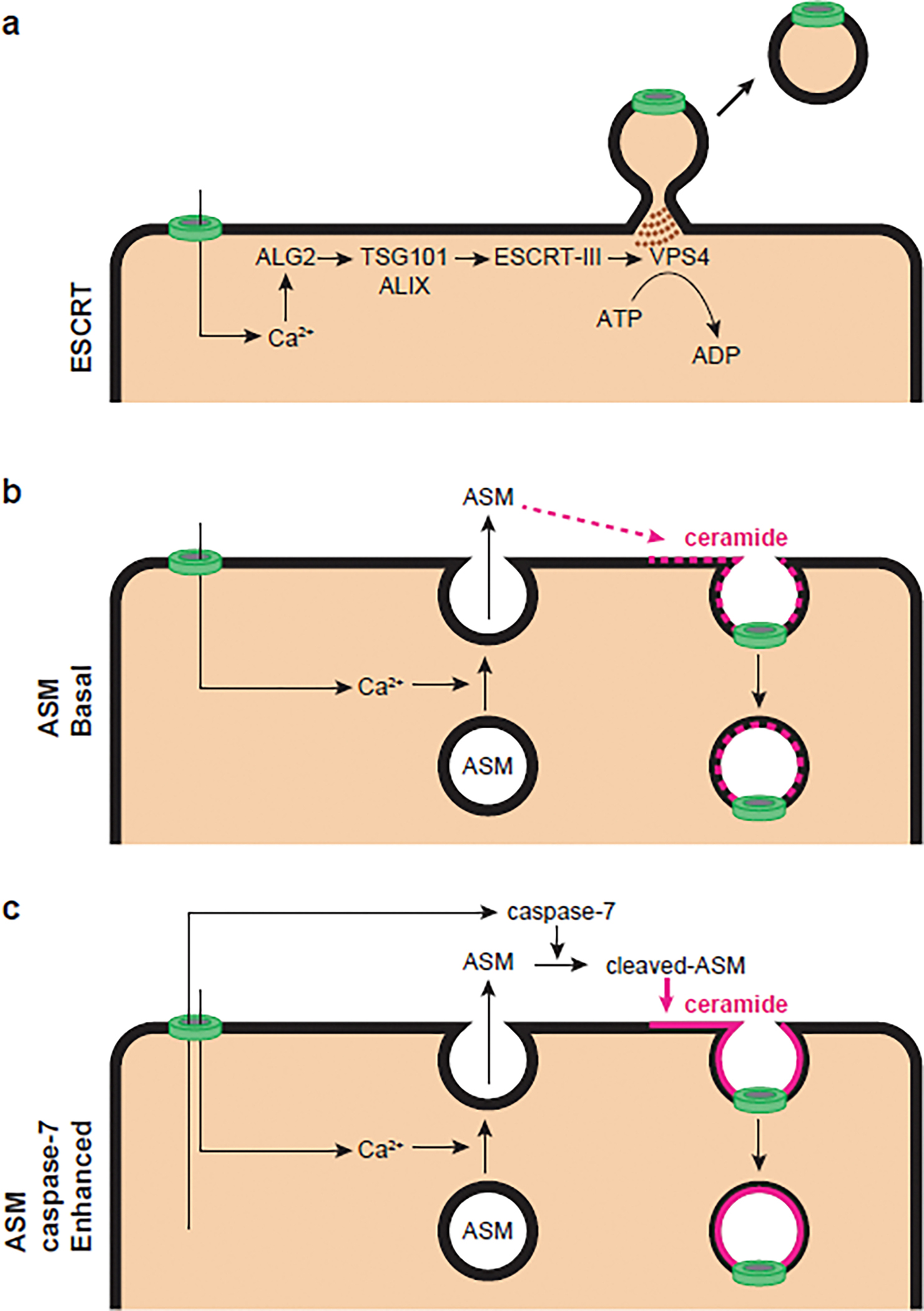
Gasdermin D pores (green) allow Ca2+ influx into the cytosol, which initiates ESCRTs/ASM-mediated repair pathways. In ESCRT repair (a), local increases of Ca2+ rapidly recruit ALG2, a Ca2+ sensor that bridges ALIX and TSG101. This further recruits the ESCRT-III machinery to drive repair by exocytosis. VPS4 completes the shedding by a scissors function in an ATP-dependent manner. In basal ASM repair (b), Ca2+ entry though pores rapidly drives lysosomal exocytosis, which releases lysosomal ASM onto the cell surface. ASM cleaves off the head group of sphingomyelin to generate ceramide within the outer leaflet of the plasma membrane. This ceramide drives spontaneous membrane repair by endocytosis that does not require ATP. In caspase-7-enhanced ASM repair (c), caspase-7 cleaves and activates ASM to enhance its enzymatic activity, resulting in the generation of more ceramide, thereby enhancing the endocytic repair process.
Membrane repair – Endocytic
Another major plasma membrane repair pathway is ASM-mediated endocytosis [40]. When a membrane pore opens, Ca2+ enters at the damaged site, driving lysosomal exocytosis within seconds (Figure 3b). This localizes the lysosomal enzyme ASM to the extracellular space, where ASM can access sphingomyelin, a major lipid component of the outer leaflet of the plasma membrane. Sphingomyelin consists of a phosphocholine headgroup attached to two lipids (a sphingosine and a fatty acid) [32]. ASM removes the headgroup, generating the strongly hydrophobic lipid ceramide, which spontaneously self-aggregates. The resulting ceramide-enriched microdomains in the lipid bilayer spontaneously invaginate inward without the need for cellular ATP, resulting in spontaneous endocytosis that internalizes the pore [41,42].
Recently it was reported that this ASM membrane repair pathway is hyperactivated by caspase-7 during regulated cell death when membrane pores are present (Figure 3c) [4]. Caspase-7 had been considered as a weak back-up for caspase-3 [43,44], however, caspase-7 was also shown to be activated by the inflammatory caspase-1 for previously unknown reasons [45]. Caspase-7 was also reported to cleave and hyperactivate the enzymatic activity of ASM, but again a physiologically relevant role was unknown [46]. In IEC organoids, after NAIP/NLRC4 inflammasome-dependent caspase-1 activation, we found that propidium iodide entry through these gasdermin D pores was faster and ceramide generation was lost in IECs that were deficient in caspase-7 or ASM, or when the caspase-7 cleavage site in ASM was mutated. Therefore, caspase-1 activates both gasdermin D pores and simultaneously a negative feedback mechanism via caspase-7/ASM-driven membrane repair [4]. This mechanism can be applicable to the similar membrane-protective phenotype of caspase-7 observed in macrophages infected with Listeria monocytogenes in vitro [47] or infected with Legionella pneumophila [48]. Of note, Gsdmd−/− organoids had no ASM cleavage after caspase-1 activation, implying that the gasdermin D pore itself serves as a conduit for caspase-7 to exit the cytosol and meet ASM in extracellular space (Figure 3c). The caspase-7 dimer is around 6 nm in diameter, which can pass through the 18–20 nm diameter gasdermin D pore [18,22].
Membrane repair – a model for non-redundancy
Whether the ESCRT and ASM pathways act cooperatively or redundantly has not been investigated. Both pathways are triggered by Ca2+ influx into the cytosol. A recent report found that during candidalysin pores from Candida albicans, Ca2+ influx first activated ESCRT-repair, followed by lysosomal exocytosis at later timepoints [49]. We speculate that lower calcium concentrations trigger ESCRT-repair, and only if damage overwhelms ESCRT-repair does the ASM-repair pathway activate in response to even greater Ca2+ influx. If this tiered repair system is correct, it would be consistent with the expected energy state of the cell; if pores persist without repair, cellar energy stores may become depleted not only by cytosolic leakage, but also by energy-consuming pathways such as effector pathways in a bucket list, cell volume regulation, and membrane repair pathways. Indeed, ESCRT-repair pathway requires ATP hydrolysis by Vps4 in the membrane scission cycle [50]. In contrast, synaptotagmins that act during exocytosis of ASM only require forces generated by Ca2+-dependent SNARE proteins conformational changes [51], and ceramide-dependent endocytosis also does not require ATP. This speculative model would explain why both of ESCRTs and ASM-dependent membrane repair can be observed to be required instead of redundant, depending on different experimental settings (see Outstanding questions).
Outstanding questions.
Bucket lists are determined in part by the cell type, the initiating death trigger, and the infection status. Can other factors such as environmental stress, position in embryonic development, cellular age, or other specific cell conditions add new effector programs to the bucket list?
In pyroptosis, individual known death-delaying mechanisms cannot explain the long-term cell survival after gasdermin D activation. There may be additional death-delaying mechanisms yet to be discovered. How do multiple death-delaying mechanisms in DCs/macrophages/neutrophils allow a cell to survive for days after caspase-1/11 activation?
The ESCRT repair pathway delays death in various pore-induced cell death pathways. However, ESCRT machinery has a broad spectrum of functions other than plasma membrane repair and is essential in embryonic development. How can we analyze the in vivo relevance of ESCRT membrane repair in pore-induced death in isolation from its other functions?
The apoptotic program activates a multitude of downstream pathways (e.g., DNA laddering, bleb formation, etc). Which pathways are required to clear intracellular infection in hepatocytes? Are different apoptotic bucket lists required in defense against different pathogens infecting diverse cell types?
In necroptosis, the ESCRT pathway repairs the MLKL pores. Do additional death-delaying pathways also impede necroptosis? Are specific bucket lists activated in a cell undergoing necroptosis?
Ferroptosis occurs when cells experience severe oxidative damage resulting in lipid peroxidation. If such a form of cell death occurs without the cell activating a regulated pathway, does a cell still have bucket list tasks to accomplish before it dies?
What kind of bucket lists exist in other cell types not discussed herein?
Do the failures to complete bucket lists contribute to the tissue pathology that occurs in many diseases?
Regulatory volume decrease
Cells constantly keep their osmolality by activating ion transporters such as Na+/K+ ATPase [52]. Membrane pores lead to osmotic water influx. This osmotic swelling activates Na+/K+ pumps and others, and release non-essential osmolytes such as K+ and Cl−, and water to return the normal cell volume, called regulatory volume decrease [52]. Different cell types may have diverse capacity to use regulatory volume mechanisms. Therefore, regulatory volume decrease could be a death-delaying mechanism by preventing membrane rupture. This process also could contribute to depletion of cellular energy.
Regulating plasma membrane rupture by NINJ1 and glycine
When accumulated membrane pores overwhelm membrane repair, plasma membrane rupture occurs. Cell death and lysis were recently shown to be separate events, as ninjurin1 (NINJ1), a cell surface molecule with two transmembrane domains, multimerizes and causes plasma membrane rupture [12]. Gasdermin D pore opening causes macrophage swelling, but in Ninj1−/− macrophages death occurs without plasma membrane rupture. Ninj1−/− cells still released IL-1β, indicating that gasdermin D pores opened, but failed to release larger molecules including HMGB1 and LDH [12].
Interestingly, this phenotype is similar to the cytoprotective effect of glycine treatment [53]. Concomitantly, intracellular glycine inhibits NINJ1 clustering [54], suggesting that NINJ1 may only cause plasma membrane rupture when the cytosolic concentration of glycine falls. It is important to address the unanswered questions about how NINJ1 oligomerization causes plasma membrane rupture in the future.
Bucket lists in pore-induced death
These death-delaying pathways to inhibit or repair gasdermin D pores could allow cells to survive long enough to complete their bucket lists (Figure 1). To be clear, these inhibitory or repair mechanisms are not included in the bucket list, rather they facilitate the completion of the bucket list. We propose that at least two events can independently initiate bucket lists during regulated cell death: the death signaling pathway (e.g. caspases cleaving cytokines such as IL-1β for processing before cell death), and the opening of pores (e.g. gasdermin pores initiating Ca2+ signaling for supporting NET formation in neutrophils).
Interestingly, these two bucket list initiators are identical to the pathways that initiate membrane repair. Apical caspase-1 activates the caspase-7/ASM repair pathway. Similarly, gasdermin pores permit Ca2+ surge that initiates membrane repair. The cell then furiously attempts to accomplish the bucket lists before it experiences total cellular dysfunction. Depending on the different bucket lists they have, distinct cell types or distinct death pathways will lead to different durations of cellular survival. Some specific bucket lists may take so long to complete, perhaps several days, that it becomes more efficient for the cell to simply not die. Thus, after initiating cell death signaling and embarking on bucket list completion, some cells have strong enough death inhibitory mechanisms that they survive long term. Next, we will discuss how bucket lists and the death-delaying mechanisms can be different in distinct cell types (Table 1).
Table 1.
Cellular bucket lista
| Death trigger | Death pathway | Bucket lists | Death-delaying mechanism | Living duration | Consequence of missing execution | Refs | |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Neutrophils | Intracellular rod, needle, flagellin | NAIP/NLRC4/Caspase-1/GSDMD | IL-1β processing/release, phagocytosis, ROS production | ND | 5 hours and more | Reduced bacterial killing | [55–57] |
| Neutrophils | Intracellular LPS, PMA | Caspase-11/GSDMD, Neutrophil Elastase/GSDMD | NET formation pathway | ND | 30–60 min | Failure to form NETs and reduced bacterial killing | [63,64] |
| Macrophages | Activators for NLRP1, NLRP3, NLRC4, Aim2, Pyrin | Caspase-1/GSDMD | IL-1β processing | ESCRT repair? | 30–60 min | Failure to release mature IL-1β | |
| Macrophages DCs | oxPAPCs bacterial peptidoglycan fragments, and mutant Staphylococcus aureus | CD14/Caspase-11/NLRP3/Caspase-1/GSDMD | Both macrophages and DCs; IL-1β processing/release, DCs only; Antigen presentation | ND | Macroph ages; 16 hours or more DCs; A few days | Failure to release mature IL-1β, Less antigen-specific T cell responses | [3, 66–68] |
| IECs | Intracellular rod, needle, flagellin | NAIP/NLRC4/Caspase-1/GSDMD | Cell extrusion pathway; | Caspase-7/ASM-mem brane repair | 30–60 min | Failure to extrude IECs, More tissue damage | [4] |
| Hepatocytes | NK/CTL attack | Perforin/granzymes/Caspase-3 | Apoptosis-rel ated bacterial clearance | Caspase-7/ASM-mem brane repair | ND | Failure to clear intracellular pathogens | [4] |
Cellular bucket list table
ND; not determined
Neutrophil cytokine release and NET formation
Neutrophils have at least two different programs in their bucket lists after death initiation by caspase-1/11. Several papers reported that cytoplasmic LPS-driven caspase-11 activation trigger pyroptosis while NLRC4- and NLRP3-driven caspase-1 activation promotes IL-1β release without plasma membrane rupture in neutrophils even after gasdermin D cleavage [55–57]. The investigators speculated that this non-lytic activation may allow neutrophils to retain their antimicrobial activity and simultaneously release IL-1β [57]. GSDMDNT in neutrophils preferentially trafficked to organelle membranes such as azurophilic granules rather than the plasma membrane [57]. The regulatory switch between pyroptosis and survival is determined by the route of entry of the agonist; those which cross the plasma membrane cause pyroptosis, whereas agonists that cross the phagosomal membrane trigger caspase-1 activation that foregoes pyroptosis [58].
Another program of a bucket list that is specific to neutrophils is the formation of neutrophil extracellular traps (NETs) [59]. The regulatory mechanism of NET formation and cell death (NETosis) have been reviewed elsewhere [60]. This process requires a substantial process, typically including activation of myeloperoxidase (MPO) and NADPH oxidase, neutrophil elastase, and protein-arginine deiminase type 4 (PAD4) [61]. PAD4 is a Ca2+ activated nuclear enzyme that removes the positive charge from histones by converting arginine residues to citrulline, resulting in chromatin decondensation followed by nuclear envelop rupture [62]. Gasdermin D initiates NET formation [63,64], and plasma membrane rupture occurs only late in the process [65], implying that controlling plasma membrane rupture timing could be quite important for NET formation. Most likely the gasdermin pore-mediated Ca2+ surge activates PAD4, which would be an example of pore-initiation of a bucket list. In this case, the NET program is the bucket list, which upon completion simultaneously completes the bucket list and kills the cell – the event of DNA extrusion typically kills the neutrophil. However, if the goal of the cell death program is NETs to more efficiently kill bacteria, and instead the cell dies of lysis without extruding NETs, then this would be an example of a failed bucket list. And that neutrophil would have failed to fight the infection.
Macrophages surviving pores and PIT formation
In macrophages, several cell death initiation pathways trigger programs of the bucket lists. One of the important function of macrophages is sensing bacterial invasion and releasing inflammatory cytokines such as IL-1β. For all inflammasome activating stimuli, cleavage of IL-1β and IL-18 are on the bucket list. These cytokines can be released first through gasdermin D pores and later by plasma membrane rupture. In murine bone marrow derived macrophages stimulated in vitro with strong inflammasome agonists the gap between gasdermin D pores and plasma membrane rupture can be around 15 minutes. A 15 minute delay in cytokine release might not impact the final outcome of the immune response. However, other inflammasome agonists might activate gasdermin D pores under slower kinetics, thus creating a greater gap between the pore opening and plasma membrane rupture. In such a case, the faster cytokine release through the gasdermin D pore could have great biological significance, allowing faster immune responses than would be accomplished by plasma membrane rupture alone. Indeed, recent reports suggested that macrophages stimulated with a subset of inflammasome agonists (e.g. oxPAPCs, bacterial peptidoglycan fragments, and mutant Staphylococcus aureus) can live for at least 16 hours and all the while can release IL-1β via gasdermin D pores [66]. This IL-1β-releasing phenotype without pyroptosis could help initiate a local inflammatory response, meanwhile the macrophage can continue to fight the infection. Thus, in these cases, gasdermin D pore-mediated IL-1β release is included in the bucket list. There are also the cases where cell death signaling is inactivated while bucket lists are completed, resulting in long term survival (which applies to DCs).
It is interesting to also consider the process of forming pore-induced intracellular traps (PITs), which is the end state of the corpse of a pyroptotic macrophage [67]. PITs trap intracellular bacteria, and neutrophil efferocytosis and thereby killing of the bacteria [59,67]. PITs have been thought to be the inevitable structure of a macrophage that died via pyroptosis. However, considering that NET formation is an active process, and the new concept of cellular bucket lists, it may be that macrophages must complete certain programs to form functional PITs in an active way to efficiently trap the intracellular bacteria by caspase-1/gasdermin D-dependent PIT formation.
Dendritic cells releasing IL-1β and surviving
DCs are professional antigen presenting cells. DCs stimulated with specific agonists (e.g., oxPAPCs) enhance antigen presentation and simultaneously release IL-1β without cell death [3]. 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC) is a membrane lipid, that when exposed to ROS are converted to oxidized PAPCs (oxPAPCs). These oxPAPCs trigger innate immune sensors such as CD14 that normally respond to LPS, and thus are danger associate molecular patterns (DAMPs) [68]. oxPAPCs bind and activate caspase-11, resulting in the processing and release of IL-1β, and the potentiation of T cell responses. These bucket lists are initiated by the caspase-11 cell death signaling pathway, which normally kill the cell instantly, are performed for several days but without signs of cell death under oxPAPCs. Presumably, in order to enhance its own bucket list, DC evolved the aforementioned death-delaying pathways, perhaps in conjunction with autophagic removal of the activated caspase-11 [7], fully inactivate cell death signaling. Thus, we think this can still be within the concept of cellular bucket lists even the death-delaying pathways finally overcome death-accelerating pathways. However, we acknowledge that cells that use cell death pathways to accomplish a task, but are destined to not die under normal circumstances, may be an exception that could be discussed further and possibly excluded from the bucket list concept.
Intestinal epithelial cells and extrusion
IECs cover the intestinal luminal surface and are a physical barrier against microbial invasion. Among the ways of maintaining barrier functions, cell extrusion is an effector mechanism: ejecting only the dying or target cell out of the epithelial layer and resealing the monolayer, meanwhile preventing permeability [69]. Extrusion can also remove infected or pre-cancerous IECs [69,70]. The IEC extrusion program is activated during homeostasis to turn over old IECs as well as during apoptotic and pyroptotic cell death. Apoptotic extrusion occurs in several phases: initiation, progression, and completion. Various effector molecules govern each step, redistributing junctional molecules and remodeling cytoskeletal molecules to eject the extruding IEC into the lumen [71–73]. Caspase-1 and gasdermin D also initiate IEC extrusion, and as is typical for extrusion, IECs extrude as single cells [74,75]. It was recently demonstrated that this extrusion fails in the absence of the caspase-7/ASM-repair pathway, resulting in abnormal extrusion clusters. Organoids from Casp7−/− mice or ASM cleavage-resistant mice undergoing caspase-1 initiated extrusion showed earlier initiation of extrusion and defective completion of extrusion [4]. This suggests that the caspase7/ASM membrane repair pathway provides enough time for dying IECs to complete their bucket list of molecular programs that must be completed during extrusion.
Hepatocytes and apoptosis
Perforin pores are similar in size to gasdermin D pores [19,76]. Natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) use perforin pores to deliver granzymes to target cells identified for elimination during infection. A recent paper showed a role for the bucket list in hepatocytes that have been targeted by perforin attack. Hepatocytes are the major parenchymal cells in the liver, and are often infected by viruses and bacteria. During Chromobacterium and Listeria infection, perforin attack from NK cells and CTLs, respectively, clears the bacteria from infected hepatocytes [77]. Perforin attack delivers granzyme B to the cytosol of target cells, which activates caspase-3, -6, and -7 and promotes apoptosis [78], however, excessive numbers of perforin pores also could cause lysis [79]. Repair of perforin pores was demonstrated to facilitate perforin attack-induced apoptosis [80–83]. We found that such repair during Chromobacterium violaceum and Listeria monocytogenes infection required caspase-7 and ASM for bacterial clearance in the infected hepatocytes [4]. The caspase-7/ASM membrane repair pathway buys the time for the completion of the process of apoptosis by inhibiting perforin-driven rapid lysis. In this case, only the parts of these pathways specifically responsible for the pathogen clearance during apoptosis are included as the bucket list that must be completed before the cell ceased to function.
Of note, bacterial clearance in hepatocytes has a clear contrast to that of macrophages which expresses caspase-1/gasdermin D and forms PITs during pyroptosis. It seems that lytic cell death is sufficient to clear bacteria that are inside macrophages, whereas apoptosis is only required to clear bacteria that reside within hepatocytes, at least during Chromobacterium and Listeria infection. One possible mechanism to explain this could be if hepatocytes are unable to form PITs that trap bacteria. This could occur if hepatocytes expressed very high levels of NINJ1 compared to macrophages. This might result in excessive membrane ruptures that release bacteria instead of trapping them.
Concluding remarks
There are many ways to delay cell death, including membrane repair and mechanisms to control pore formation. These are not included in our definition of the bucket list, rather, these mechanisms buy the cell time to complete the bucket list. Such pathways may be complex beyond what is discussed here, and may prevent death by circuitous routes. For example, we have speculated that autophagy/xenophagy against cytosolic bacteria, if successful, could eliminate the bacteria and thereby abort the need for cell death and allow a cell to survive long term [7]. Future studies should seek to understand whether all death delaying mechanisms act simultaneously, whether they act in sequence, and whether they are functionally redundant. Carefully controlled experiments studying fine kinetic detail will likely be required to understand how a single initiating signal such as Ca2+ influx acts both to drive exocytic and endocytic membrane repair. The ESCRT pathway is quite complex, which will raise challenges in isolating its role in membrane repair from its other functions. It may be that use of Alg2−/− cells, lacking the Ca2+ binding protein, may allow study of cells that lack ESCRT-dependent membrane repair without affecting other ESCRT functions.
Every cell type is diverse in its identity and is included into the whole animal’s physiology in distinct ways. Naturally, such diversity between cell’s “personalities” will lead each to have a different bucket list that they must complete before they die (see Outstanding Questions). Nevertheless, when one sensor initiates cell death in different cell types, it is likely that their different bucket lists actually work together to accomplish a common goal. For example, caspase-1-activated macrophages release IL-18, which can drive innate production of IFN-γ that primes cell-autonomous immunity to prevent intracellular infection. Meanwhile caspase-1-activated IECs extrude to prevent intracellular infection. Thus, these diverse bucket lists work together towards a common goal.
Highlights.
During regulated cell death, apparently contradictory events occur: cells simultaneously activate death-accelerating and death-delaying pathways. Meanwhile, dying cells are undertaking other activities, specific effector programs (e.g. epithelial cell extrusion, cytokine processing) that are not directly related to living or dying.
Here, we propose that specific effector programs are “bucket lists” that need to be accomplished before the cell dies.
For example, intestinal epithelial cells need to complete the extrusion bucket lists before they die. Likewise, macrophages that activate caspase-1 must process IL-1β/IL-18 before they undergo pyroptosis.
In order to complete bucket lists, cells activate death-delaying pathways that repair membrane pores.
If a cell dies prematurely then it will fail to complete these bucket lists, resulting in pathological consequences.
Acknowledgements
This work was supported by the following NIH grants: AI133236, AI139304, AI136920, and AR072694.
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Shi J et al. (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 [DOI] [PubMed] [Google Scholar]
- 2.Kayagaki N et al. (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 [DOI] [PubMed] [Google Scholar]
- 3.Zanoni I et al. (2016) An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nozaki K et al. (2022) Caspase-7 activates ASM to repair gasdermin and perforin pores. Nature 606, 960–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nozaki K et al. (2022) Innate Sensors Trigger Regulated Cell Death to Combat Intracellular Infection. Annu. Rev. Immunol. 40, 469–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross C et al. (2022) Inflammatory Caspases: Toward a Unified Model for Caspase Activation by Inflammasomes. Annu. Rev. Immunol. 40, 249–269 [DOI] [PubMed] [Google Scholar]
- 7.Harvest CK and Miao EA (2022) Autophagy may allow a cell to forbear pyroptosis when confronted with cytosol-invasive bacteria. Front. Immunol. 13, 871190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagar JA et al. (2013) Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayagaki N et al. (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 [DOI] [PubMed] [Google Scholar]
- 10.Shi J et al. (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 [DOI] [PubMed] [Google Scholar]
- 11.Wang K et al. (2020) Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 180, 941–955.e20 [DOI] [PubMed] [Google Scholar]
- 12.Kayagaki N et al. (2021) NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 [DOI] [PubMed] [Google Scholar]
- 13.Taabazuing CY et al. (2017) Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol 24, 507–514.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demarco B et al. (2020) Caspase-8–dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Science Advances 6, eabc3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orning P et al. (2018) Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarhan J et al. (2018) Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. U. S. A. 115, E10888–E10897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding J et al. (2016) Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 [DOI] [PubMed] [Google Scholar]
- 18.Liu X et al. (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruan J et al. (2018) Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aglietti RA et al. (2016) GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proceedings of the National Academy of Sciences 113, 7858–7863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sborgi L et al. (2016) GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia S et al. (2021) Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593, 607–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X et al. (2016) Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26, 1007–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evavold CL et al. (2021) Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 184, 4495–4511.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Z et al. (2021) The lysosomal Rag-Ragulator complex licenses RIPK1– and caspase-8–mediated pyroptosis by Yersinia. Science 372, eabg0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rathkey JK et al. (2018) Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Humphries F et al. (2020) Succination inactivates gasdermin D and blocks pyroptosis. Science 369, 1633–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu JJ et al. (2020) FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 21, 736–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin W et al. (2020) Chemoproteomic Profiling of Itaconation by Bioorthogonal Probes in Inflammatory Macrophages. J. Am. Chem. Soc. 142, 10894–10898 [DOI] [PubMed] [Google Scholar]
- 30.Santa Cruz Garcia AB et al. (2022) Gasdermin D pores are dynamically regulated by local phosphoinositide circuitry. Nat. Commun. 13, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu Y et al. (2022) The Gasdermin D N-terminal fragment acts as a negative feedback system to inhibit inflammasome-mediated activation of Caspase-1/11. Proceedings of the National Academy of Sciences 119, e2210809119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrews NW et al. (2014) Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. 24, 734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hurley JH (2015) ESCRTs are everywhere. EMBO J. 34, 2398–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheffer LL et al. (2014) Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 5, 5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jimenez AJ et al. (2014) ESCRT machinery is required for plasma membrane repair. Science 343, 1247136. [DOI] [PubMed] [Google Scholar]
- 36.Okumura M et al. (2009) Penta-EF-hand protein ALG-2 functions as a Ca2+-dependent adaptor that bridges Alix and TSG101. Biochem. Biophys. Res. Commun. 386, 237–241 [DOI] [PubMed] [Google Scholar]
- 37.Gong Y-N et al. (2017) ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rühl S et al. (2018) ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 [DOI] [PubMed] [Google Scholar]
- 39.Ritter AT et al. (2022) ESCRT-mediated membrane repair protects tumor-derived cells against T cell attack. Science 376, 377–382 [DOI] [PubMed] [Google Scholar]
- 40.Tam C et al. (2010) Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 189, 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holopainen JM et al. (2000) Vectorial budding of vesicles by asymmetrical enzymatic formation of ceramide in giant liposomes. Biophys. J. 78, 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zha X et al. (1998) Sphingomyelinase treatment induces ATP-independent endocytosis. J. Cell Biol. 140, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuida K et al. (1996) Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384, 368–372 [DOI] [PubMed] [Google Scholar]
- 44.Lakhani SA et al. (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311, 847–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamkanfi M et al. (2008) Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteomics 7, 2350–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edelmann B et al. (2011) Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J. 30, 379–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cassidy SKB et al. (2012) Membrane damage during Listeria monocytogenes infection triggers a caspase-7 dependent cytoprotective response. PLoS Pathog. 8, e1002628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonçalves AV et al. (2019) Gasdermin-D and Caspase-7 are the key Caspase-1/8 substrates downstream of the NAIP5/NLRC4 inflammasome required for restriction of Legionella pneumophila. PLoS Pathog. 15, e1007886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Westman J et al. (2022) Calcium-dependent ESCRT recruitment and lysosome exocytosis maintain epithelial integrity during Candida albicans invasion. Cell Rep. 38, 110187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schöneberg J et al. (2018) ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science 362, 1423–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen YA and Scheller RH (2001) SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2, 98–106 [DOI] [PubMed] [Google Scholar]
- 52.Hoffmann EK et al. (2009) Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89, 193–277 [DOI] [PubMed] [Google Scholar]
- 53.Weinberg JM et al. (2016) The role of glycine in regulated cell death. Cell. Mol. Life Sci. 73, 2285–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borges JP et al. (2022) Glycine inhibits NINJ1 membrane clustering to suppress plasma membrane rupture in cell death. Elife 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen KW et al. (2014) The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 8, 570–582 [DOI] [PubMed] [Google Scholar]
- 56.Kovacs SB et al. (2020) Neutrophil Caspase-11 Is Essential to Defend against a Cytosol-Invasive Bacterium. Cell Rep. 32, 107967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karmakar M et al. (2020) N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat. Commun. 11, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oh C et al. (2022) Neutrophil inflammasomes sense the subcellular delivery route of translocated bacterial effectors and toxins. Cell Rep. 41, 111688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brinkmann V et al. (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 [DOI] [PubMed] [Google Scholar]
- 60.Poli V and Zanoni I (2022) Neutrophil intrinsic and extrinsic regulation of NETosis in health and disease. Trends Microbiol. 0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Papayannopoulos V (2017) Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147 [DOI] [PubMed] [Google Scholar]
- 62.Mondal S and Thompson PR (2021) Chemical biology of protein citrullination by the protein A arginine deiminases. Curr. Opin. Chem. Biol. 63, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen KW et al. (2018) Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Science Immunology 3 [DOI] [PubMed] [Google Scholar]
- 64.Sollberger G et al. (2018) Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 3 [DOI] [PubMed] [Google Scholar]
- 65.Thiam HR et al. (2020) NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proceedings of the National Academy of Sciences 117, 7326–7337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Evavold CL et al. (2018) The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jorgensen I et al. (2016) Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 213, 2113–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zanoni I et al. (2017) By Capturing Inflammatory Lipids Released from Dying Cells, the Receptor CD14 Induces Inflammasome-Dependent Phagocyte Hyperactivation. Immunity 47, 697–709.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gudipaty SA and Rosenblatt J (2017) Epithelial cell extrusion: Pathways and pathologies. Semin. Cell Dev. Biol. 67, 132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macdonald TT and Monteleone G (2005) Immunity, inflammation, and allergy in the gut. Science 307, 1920–1925 [DOI] [PubMed] [Google Scholar]
- 71.Marchiando AM et al. (2011) The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology 140, 1208–1218.e1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosenblatt J et al. (2001) An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr. Biol. 11, 1847–1857 [DOI] [PubMed] [Google Scholar]
- 73.Gu Y et al. (2011) Epithelial cell extrusion requires the sphingosine-1-phosphate receptor 2 pathway. J. Cell Biol. 193, 667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sellin ME et al. (2014) Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 16, 237–248 [DOI] [PubMed] [Google Scholar]
- 75.Rauch I et al. (2017) NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and −8. Immunity 46, 649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Law RHP et al. (2010) The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 468, 447–451 [DOI] [PubMed] [Google Scholar]
- 77.Maltez VI et al. (2015) Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity 43, 987–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Voskoboinik I et al. (2015) Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400 [DOI] [PubMed] [Google Scholar]
- 79.Halle S et al. (2016) In Vivo Killing Capacity of Cytotoxic T Cells Is Limited and Involves Dynamic Interactions and T Cell Cooperativity. Immunity 44, 233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Keefe D et al. (2005) Perforin Triggers a Plasma Membrane-Repair Response that Facilitates CTL Induction of Apoptosis. Immunity 23, 249–262 [DOI] [PubMed] [Google Scholar]
- 81.Thiery J et al. (2010) Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood 115, 1582–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thiery J et al. (2011) Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol. 12, 770–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lopez JA et al. (2013) Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 121, 2659–2668 [DOI] [PubMed] [Google Scholar]