

Abstract
Background and aims:
Acute-on-chronic liver failure (ACLF) is an acute liver and multisystem failure in patients with previously stable cirrhosis. A common cause of ACLF is sepsis secondary to bacterial infection. Sepsis-associated ACLF involves a loss of differentiated liver function in the absence of direct liver injury, and its mechanism is unknown. We aimed to study the mechanism of sepsis associated ACLF using a novel mouse model.
Approach and Results:
Sepsis-associated ACLF was induced by cecal ligation and puncture procedure (CLP) in mice treated with thioacetamide (TAA). The combination of TAA and CLP resulted in a significant decrease in liver synthetic function and high mortality. These changes were associated with reduced metabolic gene expression and increased C/EBPβ transcriptional activity. We found that C/EBPβ binding to its target gene promoters was increased. In humans C/EBPβ chromatin binding was similarly increased in ACLF group compared to control cirrhosis. Hepatocyte specific Cebpb knockout mice had reduced mortality and increased gene expression of hepatocyte differentiation markers in TAA/CLP mice, suggesting that C/EBPβ promotes liver failure in these mice. C/EBPβ activation was associated with endothelial dysfunction, characterized by reduced Angiopoietin-1/Angiopoietin-2 ratio and increased endothelial production of HGF. Angiopoietin-1 supplementation or Hgf knockdown reduced hepatocyte C/EBPβ accumulation, restored liver function, and reduced mortality, suggesting that endothelial dysfunction induced by sepsis drives acute-on-chronic liver failure via HGF-C/EBPβ pathway.
Conclusion:
The transcription factor C/EBPβ is activated in both mouse and human ACLF and is a potential therapeutic target to prevent liver failure in patients with sepsis and cirrhosis.
Keywords: ACLF, HNF4α, angiopoietin, endothelial cells, cirrhosis
Graphical Abstract
Introduction
Acute-on-chronic liver failure (ACLF) is a distinct clinical syndrome characterized by acute loss of liver function and multiorgan system failure in patients with chronic liver disease (1, 2). A wide variety intrahepatic and extrahepatic insults can lead to the development of ACLF. Bacterial infection, alcoholism, and reactivated viral hepatitis are among the most common precipitating events (3). Bacterial infection in particular has been associated with increased mortality in ACLF (4). Overall, sepsis-associated ACLF has up to 70% 30-day mortality (4).
Treatment options for ACLF are limited due to a lack of understanding of the molecular mechanisms that underly the loss of liver function. This is mostly due to the absence of a suitable animal model. Past rodent models have generally used an acute insult that causes massive liver necrosis (5, 6) or an acute injury to the liver accompanying bacterial infection (7). A mouse model in which liver failure is induced by sepsis alone on the background of cirrhosis has not been reported.
In this study we introduce a mouse model of sepsis associated ACLF in which liver fibrosis is induced by thioacetamide (TAA) treatment and sepsis is induced by cecal ligation and puncture (CLP). We identified the transcription factor CCAAT enhancer binding protein beta (C/EBPβ) as an important mediator of ACLF induced by sepsis. C/EBPβ has been linked to a variety of physiologic and pathologic functions. It has a role in ovulation as well as stress-induced hematopoiesis (8, 9). C/EBPβ has also been analyzed for its potential association with several cancers, including acute myeloid leukemias, hepatocellular carcinoma, hepatoblastoma, and melanoma (10–13). There is evidence hepatocyte function might become compromised in liver disease states due to altered C/EBPβ activity. Chronic ethanol consumption in rats, for example, has been associated with a shift in C/EBPβ DNA binding and a reduced regenerative response following partial hepatectomy (14).
In the liver C/EBPβ is a key hepatocyte transcription factor known to have roles in liver function and regeneration (15–18). It belongs to the basic region leucine zipper-family and binds DNA as either a homo- or heterodimer with its partner C/EBPα (15). While C/EBPα is highly expressed under normal conditions (19, 20) and regulates metabolic genes, C/EBPβ is rapidly (within 3 hours) upregulated after liver injury and is required for a full regenerative response (19, 21). This is evidenced by the phenotype of knockout mice (C/EBPβ−/−) that cannot undergo regeneration following partial hepatectomy (21). During liver regeneration, C/EBPβ chromatin binding dynamically changes to provide optimal induction of acute response genes followed by proliferation and finally return of metabolic gene activity and hepatocyte re-differentiation (15). Competing C/EBPβ and C/EBPα DNA binding likely contributes to the balance between hepatocyte proliferation following liver injury and normal metabolic function during rest (15).
Here, we found hepatocyte C/EBPβ to be activated in sepsis associated ACLF. C/EBPβ activation is mediated by endothelial cell dysfunction and an increase in endothelial cell production of HGF. C/EBPβ is activated both in TAA/CLP mice and in human ACLF samples. Activated C/EBPβ promoted sustained suppression of metabolic and hepatocyte differentiation genes. Hepatocyte specific Cebpb knockout increased gene expression of hepatocyte differentiation markers and prevented mortality following TAA/CLP treatment.
Results
Sepsis promotes liver failure phenotype in mice with fibrosis
To study the molecular mechanism of sepsis related ACLF, we developed a mouse model using a combination of thioacetamide (TAA) treatment to induce liver fibrosis and cecal ligation and puncture (CLP) procedure to induce sepsis (Figure 1A). TAA treated mice had lower body weights and higher liver/body weight ratios compared to control mice. However, CLP surgery produced similar weight loss of about 6% at 24 hours in both groups (Figure 1B).
Figure 1. TAA and CLP combination results in rapid and sustained loss of liver function.
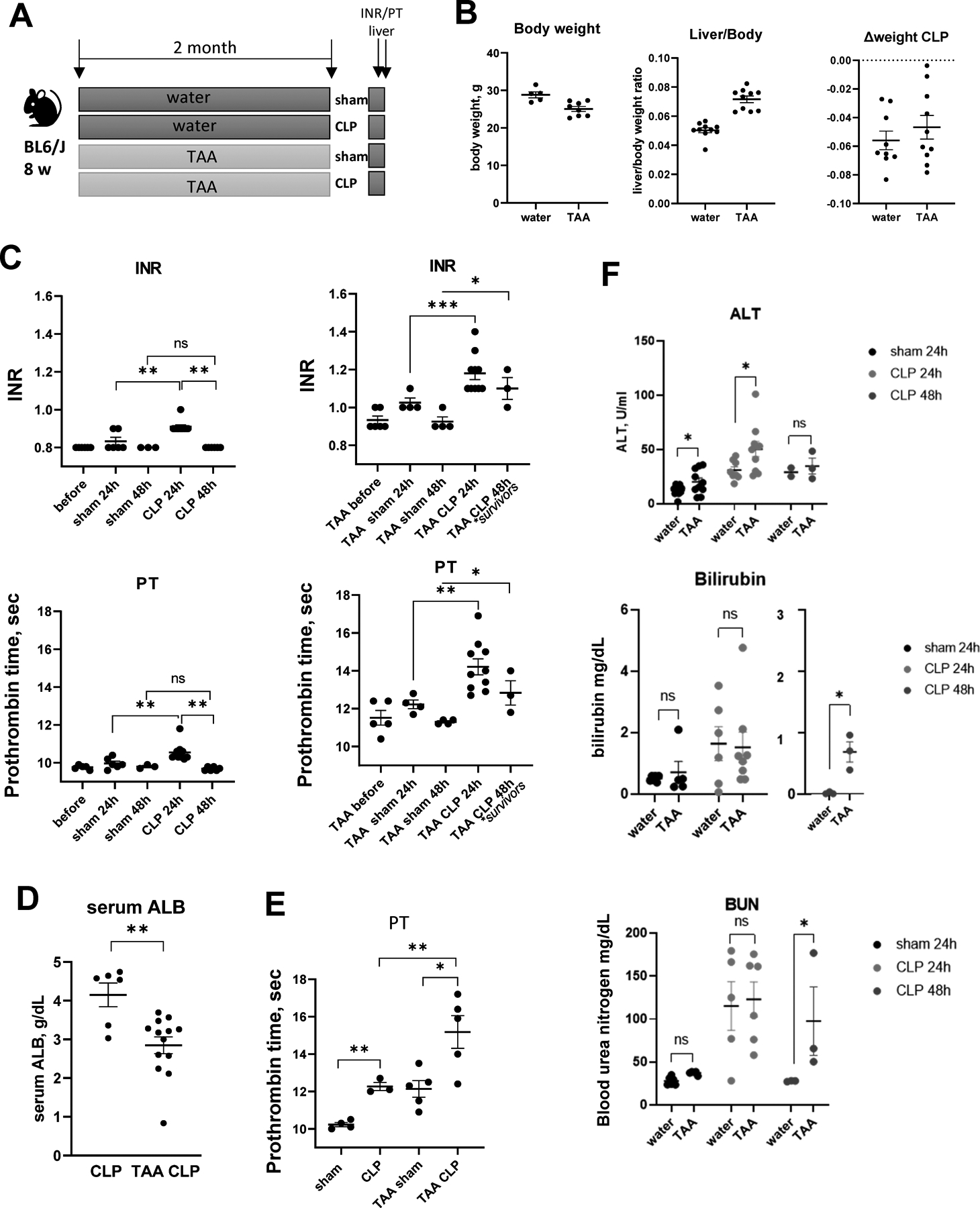
Mice (male and female) were treated with TAA (200mg/L in the drinking water) and subjected to cecal ligation and puncture (CLP) or sham surgery. A. Treatment groups. B. Liver/body weight ratios and weight loss after CLP surgery. C-E. INR, prothrombin time (PT) (C) in male mice and serum albumin (D) and prothrombin time (PT) in female mice (E) as indicated. F. Serum ALT (top), Bilirubin (middle) and blood urea (BUN, bottom) in sham and CLP mice as indicated. *, P<0.05, **, P<0.01.
Next, we used prothrombin time (PT) and INR measurements to assess the reduction in liver function induced by sepsis (Figure 1C). Sham surgery in control mice did not affect INR/PT; in contrast, sepsis induction by CLP resulted in a mild reduction in liver function that returned to baseline levels at 48h post-surgery (Figure 1C). These data agree with previously published studies (22). Mice treated with TAA/sham similarly showed a small increase in INR/PT that was not significant (Figure 1C). In contrast, TAA/CLP combination produced much greater loss of liver function accompanied by high mortality (Figure S1A); moreover, the increase in INR/PT persisted at 48h (in surviving mice) (Figure 1C). In addition, we observed a significant decrease in serum albumin levels in TAA/CLP mice (Figure 1D). Similar INR/PT elevation was present in female mice (Figure 1E, S1B).
Sepsis induction also resulted in mildly elevated serum ALT in both TAA and control mice (Figure 1F). ALT was significantly higher in TAA mice at 24 hours after CLP. However, no difference was observed at 48 h, suggesting that loss of liver function was not due to hepatocyte death. This was further supported by an absence of pathological changes in H&E staining (Figure S2). Additionally, CLP mice showed elevated bilirubin and blood urea levels (Figure 1F). We found no difference in these parameters at 24 hours between TAA and control treatment groups. After 48 hours control mice levels returned to baseline levels, but in TAA treated mice these parameters remained elevated. This suggests that in the presence of liver fibrosis, sepsis results in a more dramatic and prolonged loss of liver function in the absence of hepatocyte death that is similar to human sepsis induced ACLF (4).
Loss of HNF4α transcriptional activity is not rescued with gene overexpression
HNF4α is a master regulator of hepatocyte differentiation (23, 24). HNF4α loss is a driver of liver failure in alcoholic hepatitis, another form of ACLF that is characterized by loss of liver function in the absence of hepatocyte death (23). Thus, we evaluated the status of HNF4α in our model. We observed that CLP alone reduced HNF4α protein expression (Figure 2A). Both TAA and CLP reduced HNF4α activity in the liver as measured by HNF4α target gene expression (Figure 2B). TAA/CLP combination did not further decrease HNF4α activity, suggesting that the TAA and CLP have no additive effect.
Figure 2. HNF4α is inactivated in TAA/CLP mice.
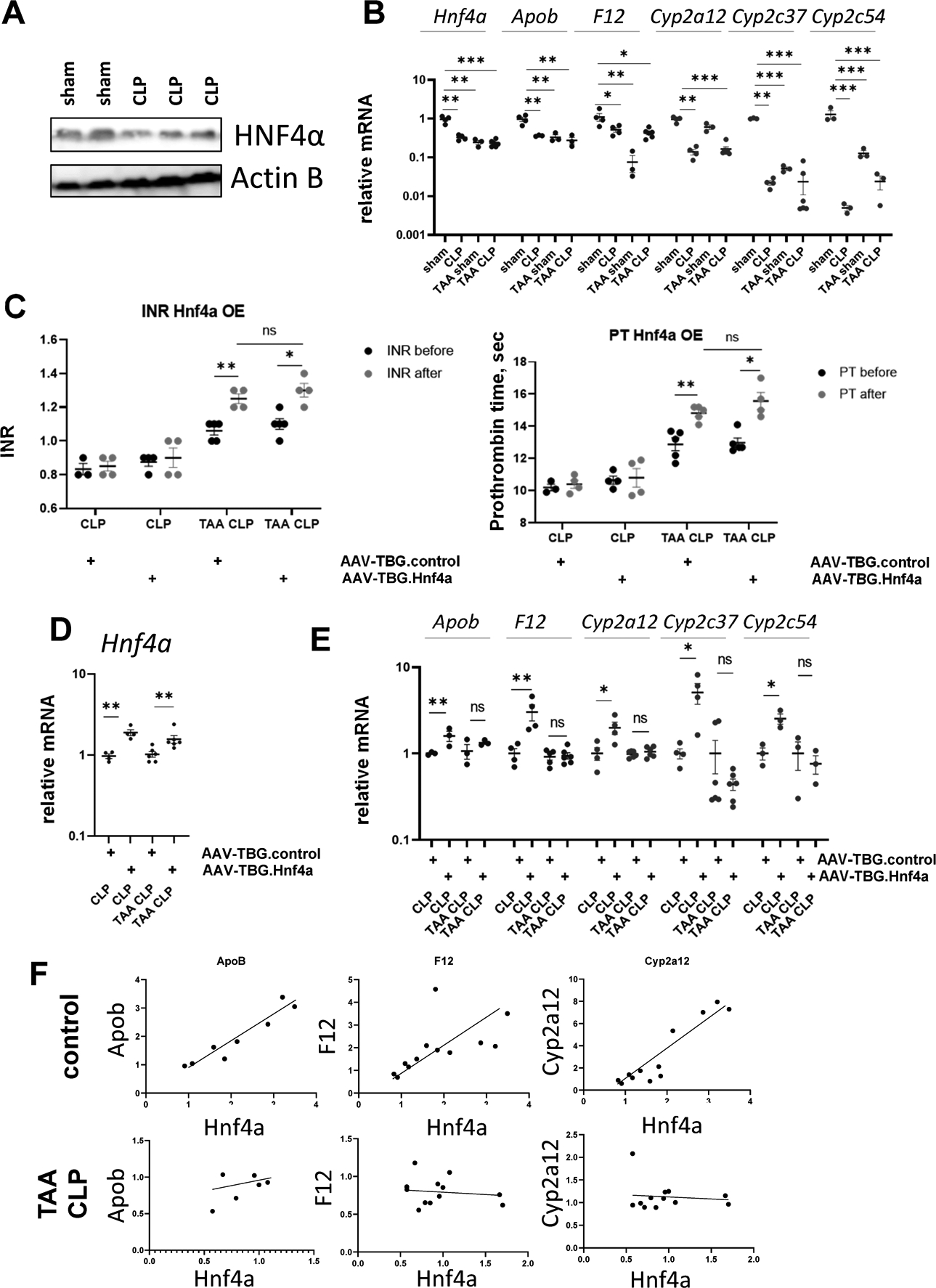
A. HNF4α protein levels in whole liver lysates from mice that were subjected to CLP or sham surgeries. B. HNF4α target gene expression in four groups of mice. C-E. Mice (male and female) were treated with TAA (200mg/L in the drinking water) or received water only and subjected to cecal ligation and puncture (CLP). One week before surgeries mice were treated with AAV.TBG.control or AAV.TBG.Hnf4a vectors at 10^11 gc/mouse. C. INR and PT in these mice one day before or one day after CLP. D-E. Relative gene expression in these mice. F. Correlation between gene expression of Hnf4a and HNF4α target genes in control (water/sham+water/CLP) groups and TAA/CLP group. *, P<0.05, **, P<0.01, ***, P<0.001.
To explore the role of Hnf4a downregulation, we overexpressed Hnf4a in hepatocytes using AAV.TBG.Hnf4a vector at 1011 genome copies per mouse 7 days before CLP surgeries. We used AAV.TBG.control vector as a control (Figure 2C–E). Hnf4a overexpression did not affect baseline INR/PT before the CLP and did not affect INR/PT elevation in TAA/CLP treated mice (Figure 2C). We confirmed that Hnf4a expression in the liver was induced about 2-fold in both treatment groups (Figure 2D). However, Hnf4a overexpression in CLP only mice tended to restore HNF4α target gene expression (Figure 2E), while in TAA/CLP mice Hnf4a overexpression did not affect its target gene expression. This suggests that HNF4α activity is impaired independent of its expression. Another indication of impaired HNF4α activity is that while in control mice we observed strong positive correlation between Hnf4a gene expression and HNF4α target gene expression (Apob, F12 and Cyp2a12), in TAA/CLP mice no significant correlation was present (Figure 2F). This HNF4α dysfunction could be due altered HNF4α posttranslational modifications or TAA/CLP-induced changes in HNF4α co-factors.
C/EBPβ transcriptional activity is increased in TAA/CLP mice
To identify potential mechanism of TAA/CLP mediated loss of liver function, we performed whole liver RNA-seq analysis of four groups of mice at 24 hours in triplicates (Figure 3). We found that TAA/CLP mouse livers had transcriptional signature distinct from either TAA only or CLP only mouse livers (Figure 3A). Compared to CLP only mice, TAA/CLP mice showed elevated expression of Afp, Mmp7, and Krt20, genes associated with loss of hepatocyte differentiation and hepatocellular carcinoma (Figure 3B, S3). Compared to TAA only mice, TAA/CLP mice had elevated levels of Fgf23, Tnfrsf9, and Csf3, genes associated with inflammation and acute response, as well as angiotensinogen Agt (Figure 3B, S3).
Figure 3. TAA and CLP combination results in unique transcriptional changes.
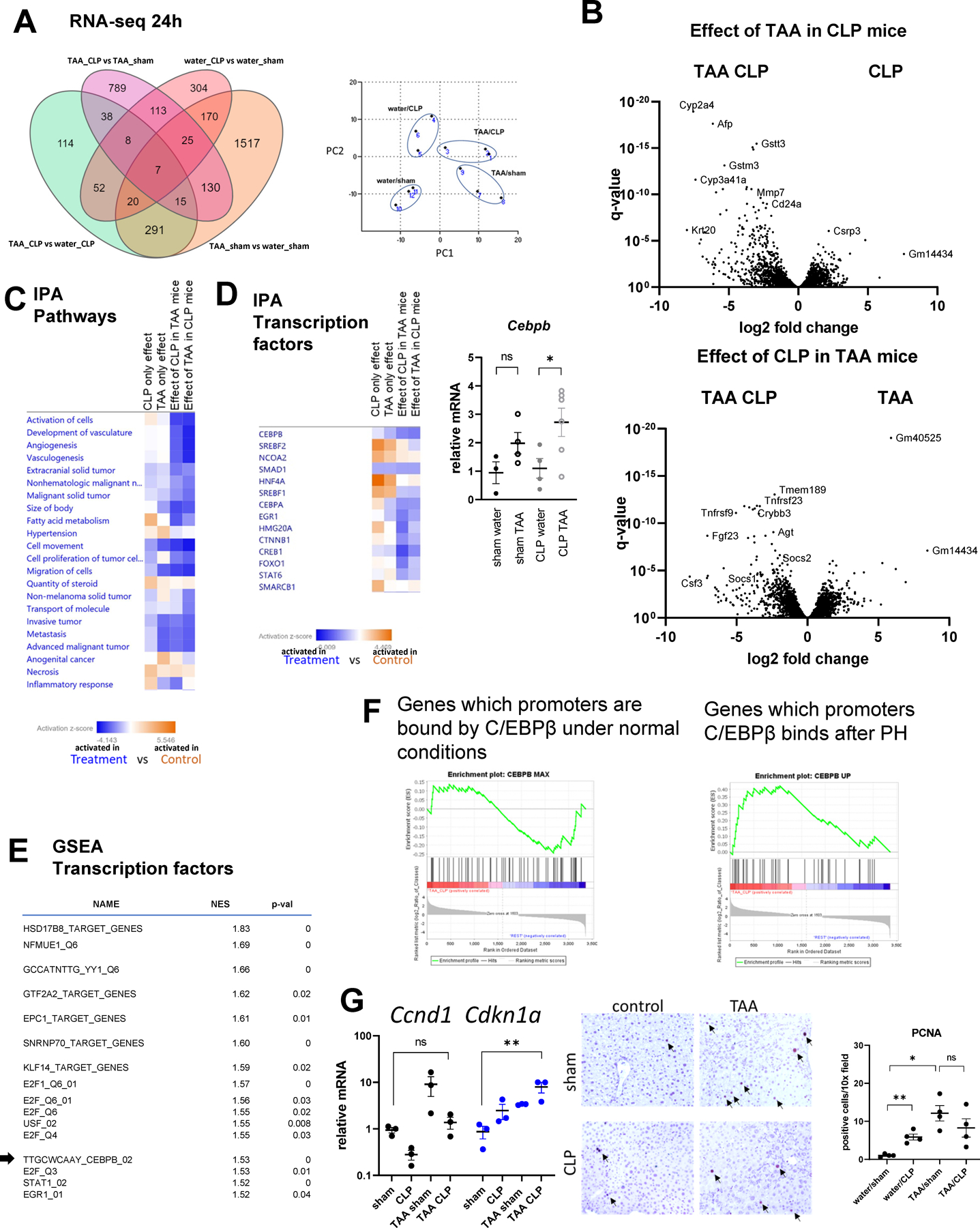
Mice (male and female) were treated with TAA and subjected to cecal ligation and puncture or sham surgery. Whole liver mRNA from 4 groups (3 mice per group) at 24h post-CLP was analyzed using RNA-seq. A. Number of differentially expressed genes in pairwise comparisons. Right. Principal component analysis. B. Volcano plots comparing TAA/CLP combination to single treatments. C-D. Ingenuity pathway analysis for each of the pairwise comparisons. Pathway and transcription regulators that are activated in treatment group (blue) and in control group (orange). Right. Cebpb gene expression in four groups of mice. *, P<0.05. E. Gene set enrichment analysis. Top transcription factors predicted to be activated in TAA/CLP group compared to all other groups. F. C/EBPβ target genes enrichment in genes activated in TAA/CLP group compared to the rest. G. Cell cycle and proliferation associated gene expression in four groups of mice. *, P<0.05, **, P<0.01. Middle. PCNA staining in four groups. Right. Average number of PCNA potive cells per 10x field. *, P<0.05, **, P<0.01.
Next, we performed bioinformatic analysis of pathways (Figure 3C, S4) and transcriptional regulators (Figure 3D, S5) affected in these mice using Ingenuity Pathway Analysis (IPA, Qiagen). TAA/CLP group showed significant activation of pathways related to tumorigenesis when compared to TAA only or CLP only groups (Figure 3C). These data agree with loss of hepatocyte differentiation observed in these mice. Among upstream regulators, C/EBPβ showed the highest activation in TAA/CLP group compared to TAA or CLP groups (Z-score = 3.8 and 2.8, TAA/CLP compared to TAA/sham and water/CLP, respectively, p-value < 10−9) (Figure 3D). Moreover, C/EBPβ was not significantly activated by CLP or TAA alone (Z-score < 2). Cebpb mRNA levels followed this prediction (Figure 3D). Moreover, in agreement with data presented in Figure 2, IPA predicted that CLP and TAA suppress the activity of HNF4α (Z-score = −4.2 and −5.6, TAA/sham and water/CLP respectively compared to water/sham, p-value < 10−15). However, TAA/CLP combination had no additional effect on HNF4α activity (Z-score < 2).
Next, we performed Gene Set Enrichment Analysis (GSEA) to validate our results (Figure 3E). We analyzed genes significantly up or downregulated in TAA/CLP mice compared to three other groups and compared them to transcription factor target gene sets. C/EBPβ targets were enriched among genes activated in TAA/CLP mice (NES = 1.53).
C/EBPβ is a multifunctional protein (15, 17, 18). Its levels in the liver are low under normal conditions, but it is rapidly upregulated when liver regeneration is required (15). Thus, we next explored what set of C/EBPβ targets is activated in TAA/CLP mice. We compared genes up or downregulated in TAA/CLP mice with gene sets previously identified as C/EBPβ targets at various timepoints during liver regeneration after partial hepatectomy (15). Genes upregulated in TAA/CLP mice were enriched in genes induced by C/EBPβ at 3 hours after partial hepatectomy. This time point is when C/EBPβ induces acute response genes while suppressing metabolic genes (15), but no hepatocyte proliferation is yet present. In contrast, genes bound by C/EBPβ under normal conditions were not enriched in TAA/CLP mice (Figure 3F).
Moreover, C/EBPβ activation was not associated with increased hepatocyte proliferation (Figure 3G). CLP treatment in TAA mice resulted in a decrease in proliferation genes such as Ccnd1 and an increase in cell cycle arrest genes such as Cdkn1a. Additionally, there was no increase in PCNA positive cells in TAA/CLP mice compared to TAA alone or CLP alone, suggesting that liver regeneration does not occur.
C/EBPβ levels are increased in TAA/CLP mice
In agreement with above data, C/EBPβ protein levels in hepatocytes were greatly increased in TAA/CLP mice compared to other groups of mice (Figure 4A). C/EBPβ protein levels remained elevated in TAA/CLP mice 48 h post-CLP (Figure 4B).
Figure 4. TAA and CLP combination results in C/EBPβ activation.
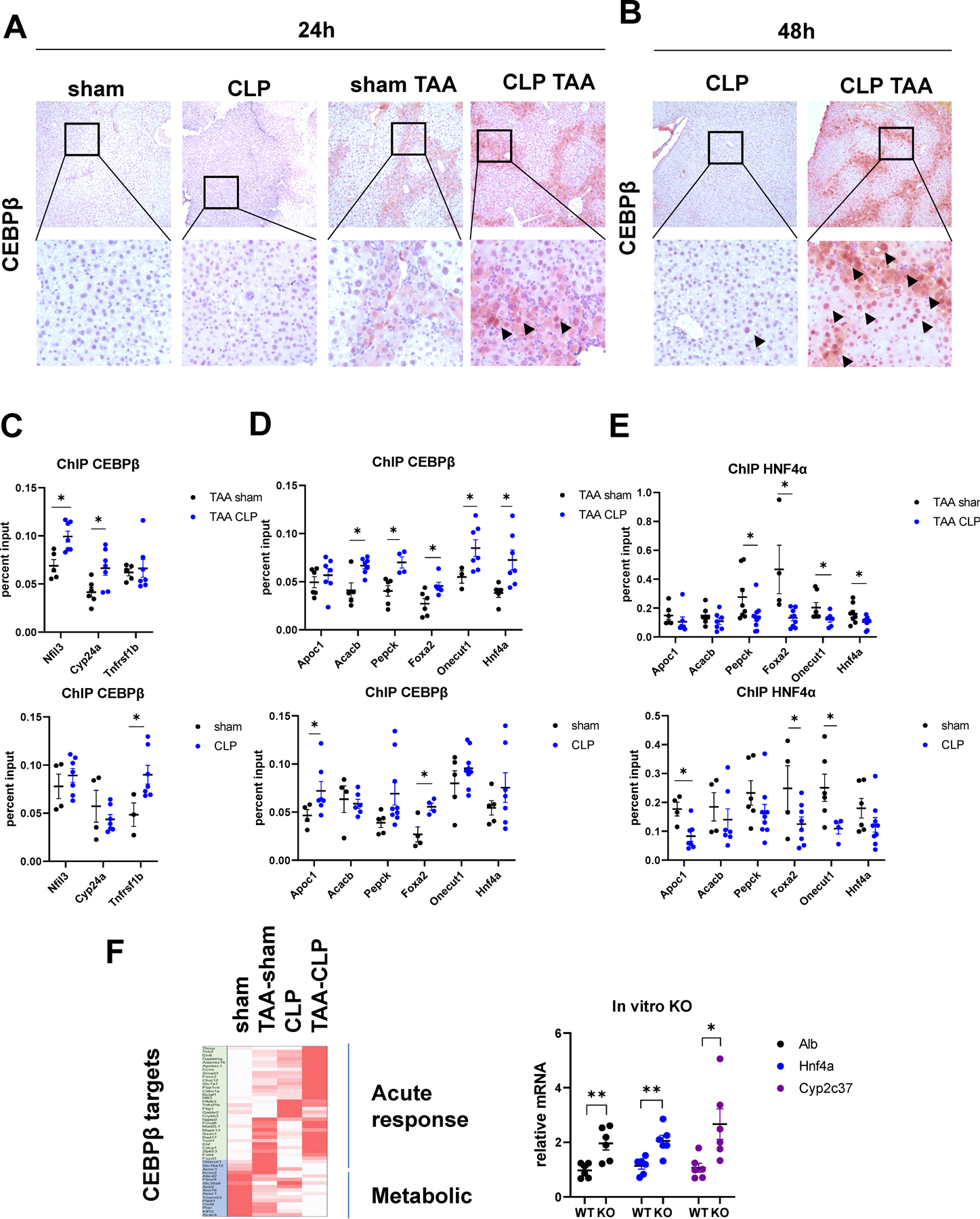
A-B. Mice (male and female) were treated with TAA and subjected to cecal ligation and puncture or sham surgery. Liver section from indicated groups of mice at 24 hours (A) and 48 hours (B) post-surgery were stained using C/EBPβ specific antibodies (total C/EBPβ). C-E Chromatin immunoprecipitation using C/EBPβ specific antibodies (total C/EBPβ) or HNF4α specific antibodies in four groups of mice. Data is presented as percent input. N= 4–8 mice per group. *, P<0.05. F. Relative gene expression changes in TAA, CLP and TAA/CLP mice of top genes bound by C/EBPβ at 3 hours after partial hepatectomy. Right. Relative gene expression in mouse primary hepatocytes isolated from Cebpb floxed mice treated with Ad-Cre vector or control for 48 hours. *, P<0.05, **, P<0.01.
Next, we confirmed that C/EBPβ binding to its target genes was increased in TAA/CLP mice. We performed chromatin immunoprecipitation assays to detect C/EBPβ binding to acute response genes, which are induced by C/EBPβ, and metabolic genes known to be suppressed by C/EBPβ binding (Figure 4C, D). We found that for both acute response genes (Figure 4C) and metabolic genes (Figure 4D), C/EBPβ binding was significantly increased by CLP in control and TAA treated animals. However, the magnitude of the effect varied between the groups; for example, metabolic genes in TAA/CLP group showed greater increases in C/EBPβ binding compared to control/TAA group.
In addition, we measured HNF4α binding to these genes. In both groups HNF4α binding was significantly decreased by CLP (Figure 4E). Figure 4F shows relative mRNA expression in four groups of mice with top acute response/proliferation and metabolic genes that are regulated by C/EBPβ at 3 hours after partial hepatectomy from a published dataset (15). We observed that CLP induces C/EBPβ-dependent acute response genes expression. Interestingly, induced genes were different in control and TAA treated mice (Figure 4F). These data are in agreement with chromatin binding data (Figure 4C). Both TAA and CLP treatment result in reduced expression of metabolic genes, and TAA/CLP combination further decreased metabolic gene expression, likely due to combined decrease in HNF4α binding (Figure 4E) and an increase in C/EBPβ binding (Figure 4D).
We confirmed that C/EBPβ in hepatocytes can suppress metabolic/differentiation gene expression using primary mouse hepatocytes isolated from Cebpb floxed mice. Hepatocytes were treated with Ad-Cre vector to induce KO or control vector (WT) for 48 h. Cebpb knockout resulted in an upregulation of genes associated with hepatocyte differentiation (Alb, Hnf4a, Cyp2c37), suggesting that C/EBPβ activation can promote liver failure.
C/EBPβ transcriptional activity is increased in human ACLF
To confirm that our findings are relevant in human ACLF, we analyzed gene expression changes in ACLF patients from a published dataset (GSE139602) using GSEA (Figure 5A). C/EBPβ targets were enriched among those that are upregulated in ACLF group compared to healthy controls (NES = 1.27), suggesting that C/EBPβ is activated in ACLF (Figure 5A). Next, we analyzed C/EBPβ promoter binding to acute response and metabolic genes promoters in a cohort of ACLF patients from KUMC Liver Bank (Figure 5B). We compared ACLF patients to control cirrhosis of matching etiology and two samples of acute liver failure livers that were used as controls. C/EBPβ binding to acute response/proliferation genes and metabolic genes was increased in ACLF group compared to control cirrhosis (Figure 5B). Increase in binding was specific to ACLF for a subset of genes, including 20-fold increase in binding to Foxa2, a key transcription factor of hepatocyte identity. We did not observe major changes in HNF4α chromatin binding in these samples (Figure 5C), suggesting that HNF4α may not play a role in ACLF development in these patients.
Figure 5. C/EBPβ promoter binding is increased in human ACLF compared to control cirrhosis.
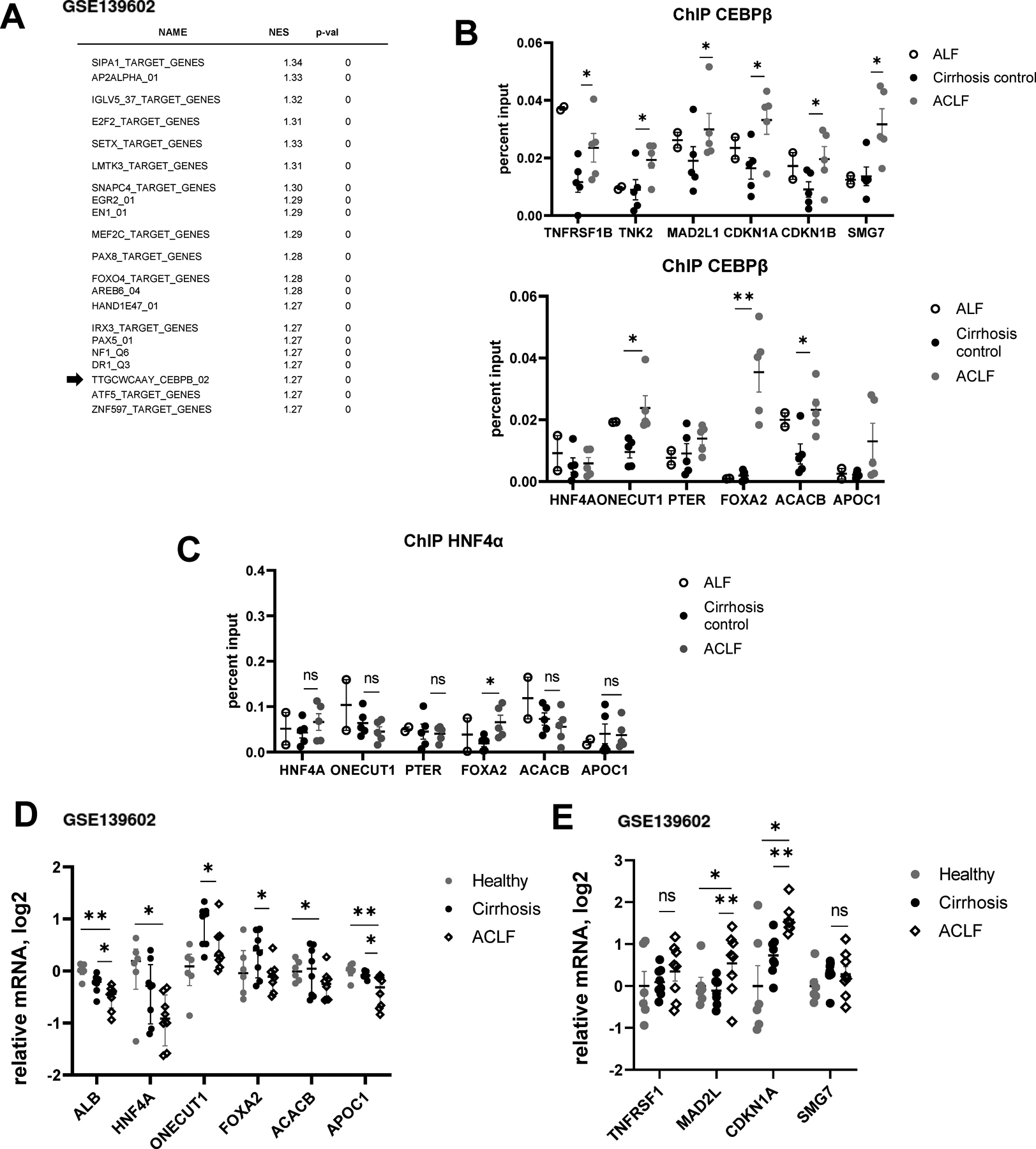
A. Gene set enrichment analysis. Top transcription factors predicted to be activated in ACLF group compared to healthy controls using published dataset GSE139602. B-C. Chromatin immunoprecipitation using C/EBPβ specific antibodies (total C/EBPβ) or HNF4α specific antibodies. Data are presented as percent input. N= 5 (ACLF and cirrhosis) N=2 (ALF). *, P<0.05, **, P<0.01. D-E. C/EBPβ target gene expression in ACLF, cirrhosis and healthy controls (GSE139602). *, P<0.05, **, P<0.01.
Finally, we confirmed that chromatin binding translated into gene expression changes. To validate our findings, we assessed gene expression in ACLF samples from a published study (GSE139602). We observed that ACLF samples have lowest expression of ALB and HNF4A in agreement with liver failure phenotype. In addition, we found a significant reduction in metabolic/differentiation genes (Figure 5D) and an increase in acute response/proliferation genes (Figure 5E). These data agree with C/EBPβ chromatin binding changes (Figure 5B).
Cebpb knockout mice are protected from liver failure phenotype
To investigate the role of C/EBPβ in vivo, we induced hepatocyte specific Cebpb knockout using AAV/TBG,cre vector (or AAV.TBG.control) in mice treated with TAA one week before CLP surgery (Figure 6A). We observed that knockout mice have reduced C/EBPβ protein levels in the liver at 24 h post CLP, suggesting that majority of C/EBPβ in TAA/CLP mice is expressed in hepatocytes. In addition, we confirmed that Cebpb knockout did not affect the levels of liver fibrosis in these mice (Figure 6A).
Figure 6. Hepatocyte specific Cebpb knockout protects from mortality and loss of liver function in TAA/CLP mice.
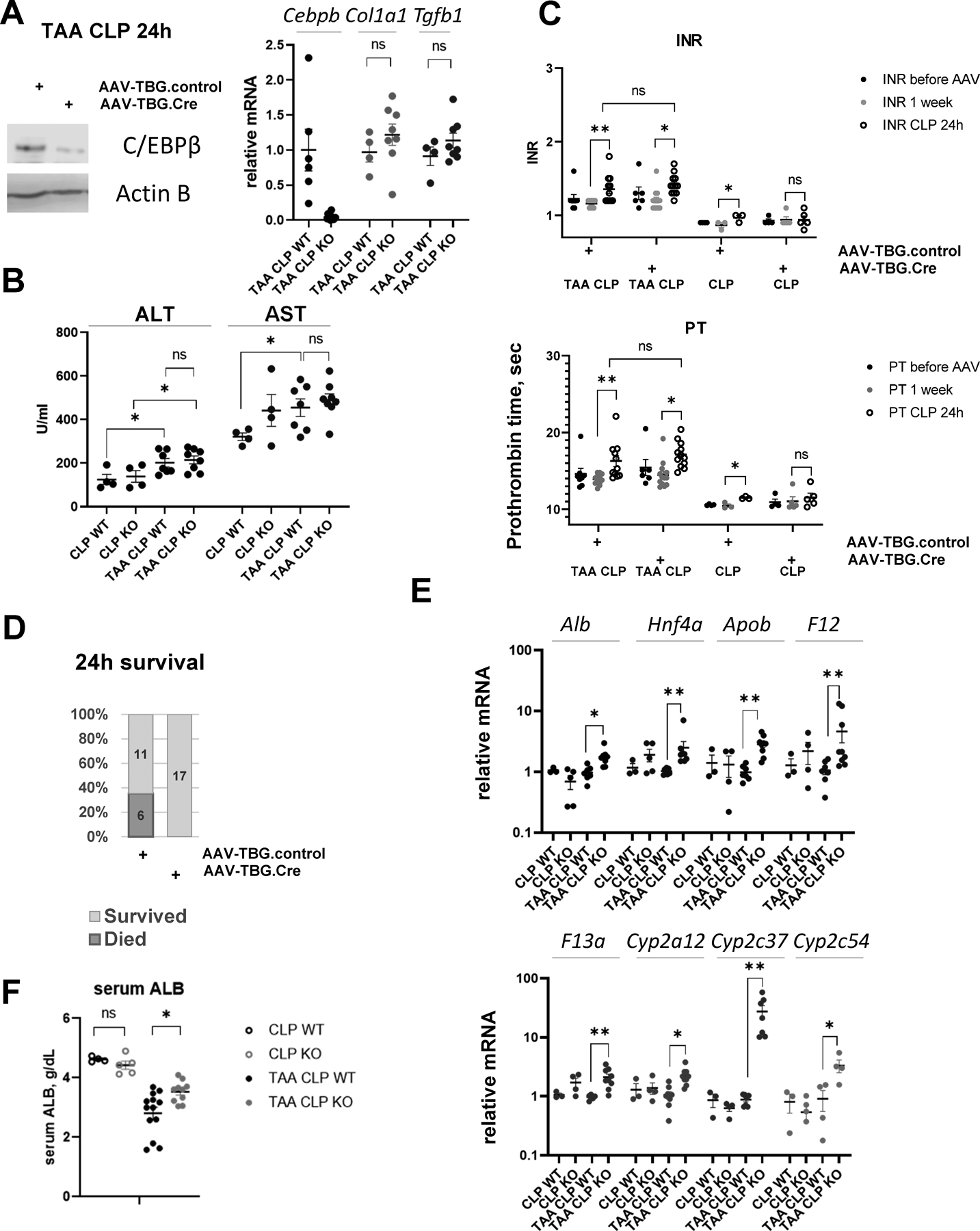
Cebpb floxed mice received AAV-TBG.CRE (KO) or AAV.TBG.control (WT) at 10^11 gc/mouse 1 week before surgeries. A. C/EBPβ protein levels in hepatocyte specific knockout mice. Right. Relative gene expression at one day after CLP. B. Serum ALT and AST in wild type (WT) and knockout mice (KO). C. INR and PT in mice before AAV injection, one day before CLP and one day after CLP. *, P<0.05, **, P<0.01. D. Survival in TAA/CLP mice. N=17 per group. E-F. Whole liver mRNA in these mice *, P<0.05, **, P<0.01.
Cebpb knockout did not affect ALT/AST (Figure 6B) and INR/PT levels after CLP in TAA treated mice (Figure 6C). However, we found that hepatocyte specific Cebpb knockout mice are protected from TAA/CLP induced mortality (Figure 6D); have increased gene expression of hepatocyte differentiation markers including Alb, Hnf4a, and HNF4α target genes (Figure 6E); and have significantly higher levels of serum albumin compared to control wild type mice (Figure 6F). In agreement with ChIP data, Cebpb knockout did not affect hepatocyte differentiation in CLP controls (Figure 6E, F).
HGF expression is induced in TAA/CLP mice
C/EBPβ can be activated by a variety of extracellular and intracellular signals. It integrates signals such as cytokines, growth factors, and hormones to activate transcriptional program fit for a specific condition. To assess the possible mechanisms of C/EBPβ activation in TAA/CLP mice, we performed cytokine arrays to detect 111 cytokines and circulating molecules in mice from control CLP and TAA/CLP groups (Figure 7A).
Figure 7. TAA and CLP combination causes endothelial cell dysfunction and HGF upregulation.
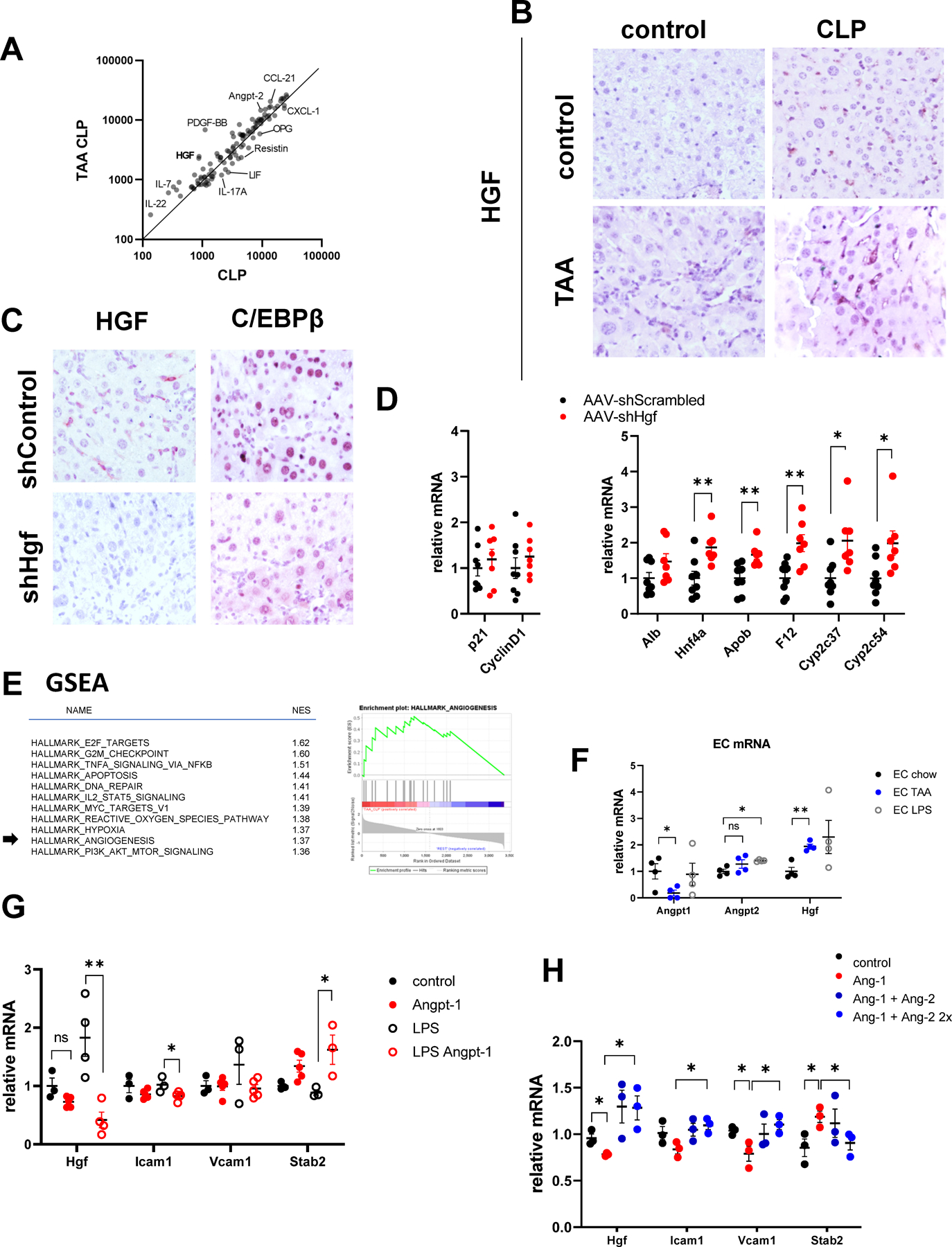
A. Serum samples from TAA/CLP and CLP mice (pooled from N=3 mice each) were used for cytokine array. Relative abundance of cytokines and circulating molecules in two groups. B. Liver section from indicated groups of mice at 24 hours post-CLP were stained using HGFα specific antibodies. C-D. Mice were treated with AAV.shScrambled or AAV.shHgf at 10^11 gc/mouse one week before CLP. C/EBPβ expression and relative gene expression in the livers of TAA/CLP mice 24 hours after CLP. E. Left. Gene set enrichment analysis. Top pathways predicted to be activated in TAA/CLP group compared to all other groups. Right. Angiogenesis genes enrichment in genes activated in TAA/CLP group compared to the rest. F. CD146 positive liver endothelial cells were isolated from control and TAA treated mice or treated with 100ng/ml of LPS. Relative gene expression. N=4 mice per group. G. LSECs were treated with recombinant Angiopoietin-1 (10 ng/ml) in the presence or absence of LPS for 48 hours. N=3–5. *, P<0.05, **, P<0.01. Unpaired t-test. LPS vs LPS Ang-1:Hgf P=0.006, Icam1 P=0.032, Stab2 P=0.045. H. LSECs were treated with recombinant Angiopoietin-1 (10 ng/ml) and Angiopoietin-2 (10 and 20 ng/ml). N=3 independent experiments. *, P<0.05 using paired ttest. Ang-1 vs control: Hgf P=0.040, Vcam1 P=0.048, Stab2 P=0.0096. Ang2+Ang1 vs Ang-1 Hgf P=0.049, Icam P=0.0002, Vcam1 P=0.009, Stab2 P=0.0079.
We found that circulating HGF levels were 3-fold higher in the TAA/CLP group compared to CLP alone (Figure 7A). We confirmed that HGF levels were significantly elevated in the livers of TAA/CLP mice compared to all other groups (Figure 7B). HGF is known to activate C/EBPβ in hepatocytes (16).
To test the role of HGF in C/EBPβ activation we used AAV-shHgf (or AAV-shScrambled control) vector to knockdown Hgf in the liver. Hgf knockdown reduced C/EBPβ accumulation in hepatocytes (Figure 7C) and promoted expression of genes associated with hepatocyte differentiation such as Alb, Hnf4a, and HNF4α target genes Apob, F12, Cyp2c37, and Cyp2c54. In contrast, HGF depletion in TAA/CLP mice did not affect proliferation genes (Figure 7D). Taken together, these data suggest that HGF is upstream of C/EBPβ upregulation in TAA/CLP mice and that HGF-C/EBPβ signaling is involved in hepatocyte de-differentiation in ACLF.
Endothelial dysfunction drives HGF-C/EBPβ signaling in hepatocytes
HGF can be produced by several cell types in the liver; the most common source in various models is endothelial cells. Both IPA analysis (Figure 3C) and GSEA analysis (Figure 7E) suggested that pathways related to endothelial cell function, such as angiogenesis, are dysregulated in TAA/CLP mice compared to other groups. Thus, we decided to study the role of endothelial cells in HGF production. We isolated CD146 positive liver sinusoidal endothelial cells from control and TAA treated mice and assessed Hgf expression. We found that LSECs from TAA mice had significantly higher levels of Hgf compared to control (Figure 7F).
Angiopoietins are proteins with important roles in vascular development and angiogenesis (25, 26). They are implicated in liver regeneration and can regulate endothelial HGF production.
We observed that elevated Hfg expression was accompanied by increased Angiopoietin-2/ Angiopoietin-1 ratio in LSECs (Figure 7F). In addition, Angiopoietin-2 circulating levels were increased in TAA/CLP mice compared to CLP controls (Figure 7A), suggesting that angiopoietin imbalance might be involved in HGF upregulation in TAA/CLP mice.
Angiopoietin-1 supplementation prevents HGF elevation and C/EBPβ activation
To test this hypothesis, we treated LSECs with recombinant Angiopoietin-1 (Figure 7G) for 48h. We observed that Angiopoietin-1 treatment increased markers of LSEC differentiation such as Stab2 and reduced Icam1 and Vcam1 expression in agreement with its anti-inflammatory properties. Angiopoietin-1 treatment also reduced Hgf expression (Figure 7G). The effect was more noticeable in the presence of LPS. In contrast Angiopoietin-2 treatment prevented Angiopoietin-1-mediated effects in LSECs. We observed that Angiopoietin-2 treatment significantly increased Hgf, Icam1, Vcam1 and reduced Stab2 (Figure 7H).
We tested whether Angiopoietin-1 supplementation affects hepatocyte differentiation. First, we studied the effect of Angiopoietin-1 treatment in vitro in LSEC-Hepatocyte co-culture system. Hepatocytes were treated with Angiopoietin-1 in the presence or absence of endothelial cells (Figure 8A). We observed that Angiopoietin-1 treatment significantly upregulated hepatocyte expression of Alb and Hnf4a when endothelial cells were present but had no effect on gene expression in the absence of LSECs. In contrast Angiopoietin-2 treatment prevented Angiopoietin-1-mediated increase in Alb and promoted Cebpb expression in hepatocytes when endothelial cells were present, suggesting that LSEC-hepatocyte crosstalk is mediating angiopoietin effects on hepatocyte differentiation (Figure 8B).
Figure 8. Angiopoietin-1 supplementation prevents C/EBPβ activation in hepatocytes.
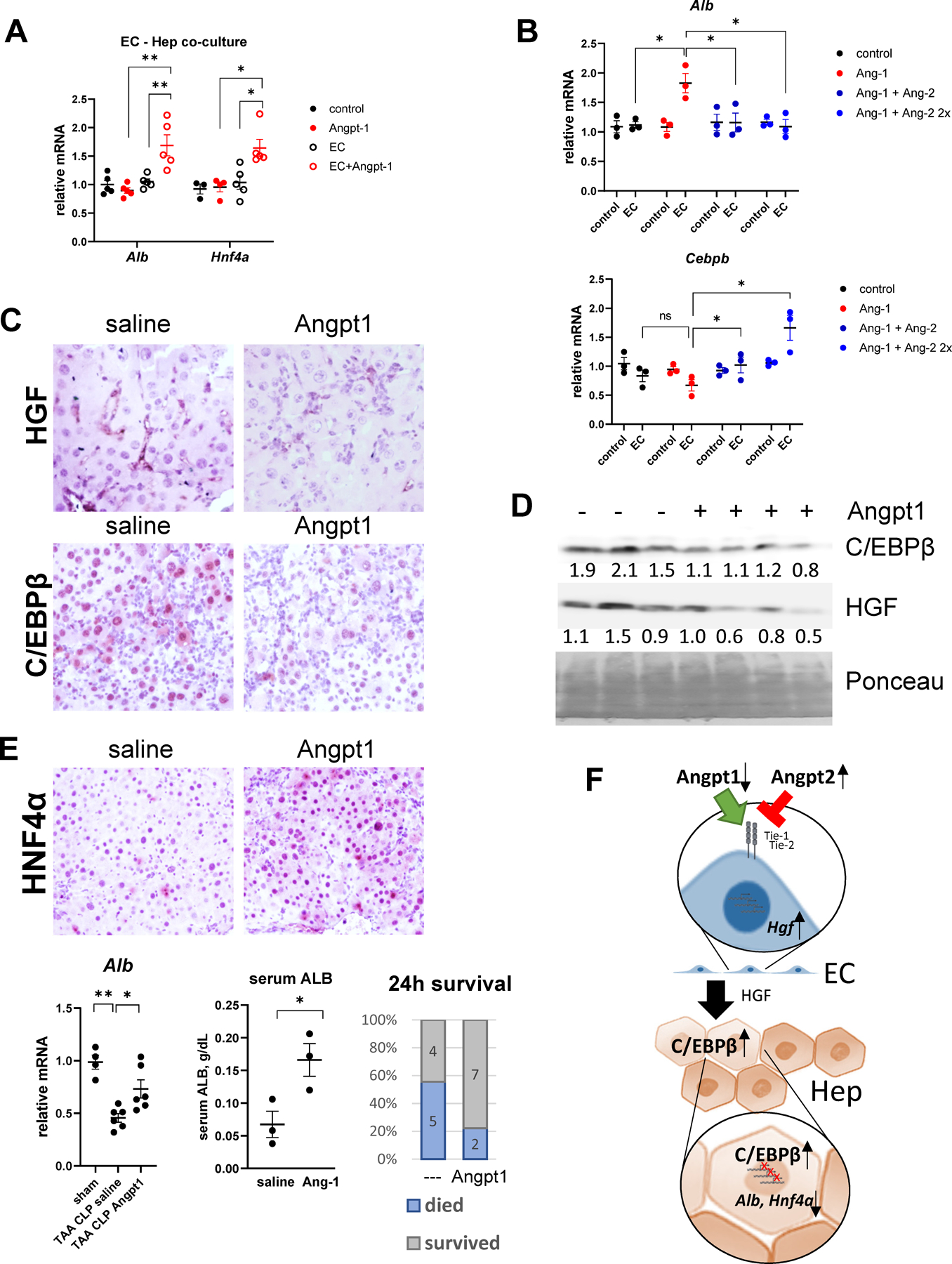
A. Primary mouse hepatocytes alone or in co-culture with LSECs were treated with recombinant Angiopoietin-1 (10 ng/ml). Relative gene expression. N=3–4 per group. *, P<0.05, **, P<0.01. B. Primary mouse hepatocytes alone or in co-culture with LSECs were treated with recombinant Angiopoietin-1 (10 ng/ml) and Angiopoietin-2 (10 and 20 ng/ml). Relative gene expression. N=3 independent experiments. *, P<0.05 using paired ttest. C-E. TAA/CLP mice received 5 μg of recombinant Angiopoietin-1 or saline control at 2 hours after CLP. C. Liver section from indicated groups of mice at 24 hours post-CLP were stained using HGFα or C/EBPβ specific antibodies (total C/EBPβ). D. Western blot analysis of C/EBPβ and HGF protein levels in the livers. D. Whole liver mRNA gene expression and mortality in TAA/CLP mice. E. Top. Liver section from indicated groups of mice at 24 hours post-CLP were stained using HNF4α specific antibodies. Bottom. Albumin liver mRNA and serum levels and mortality at 24h. F. Proposed model of ACLF pathogenesis.
Moreover, we observed that HGF protein and mRNA levels and ANG-2 protein levels were elevated in human ACLF compared to control cirrhosis, suggesting this mechanism is relevant in human ACLF (Figure S6, S7).
Next, we tested the effect of Angiopoietin-1 treatment in vivo. Angiopoietin-1 is a very short half-life protein; thus, we supplemented with recombinant Angiopoietin-1 two hours after CLP, when we expect C/EBPβ upregulation to occur, and studied the effect of the treatment 24h post-CLP. We observed that Angiopoietin-1 prevented upregulation of HGF in the liver (Figure 8C) and reduced levels of nuclear C/EBPβ in hepatocytes (Figure 8C). Similar results were observed in whole liver protein extracts (Figure 8D).
In agreement with in vitro data, Angiopoietin-1 treatment partially restored hepatocytes differentiation markers (Alb, Cyp2c37) in TAA/CLP mice in vivo, increased HNF4α protein expression and serum albumin levels, and reduced mortality in TAA/CLP mice (Figure 8E).
Discussion
While its diagnostic criteria vary among international consortia, acute-on-chronic liver failure (ACLF) is widely considered to be a severe form of acute decompensated cirrhosis associated with a systemic inflammatory response, multiorgan system failure, and high 30-day mortality (1, 2). Regardless of etiology, the molecular pathogenesis of ACLF involves loss of differentiated liver function associated with systemic inflammatory response. However, the complex pathogenesis of ACLF remains largely unknown, and a better understanding is necessary before targeted therapies can be developed. Given the rapid disease course of ACLF, further research into its pathogenesis is heavily reliant on the development of accurate animal models. Several murine models have been described in the literature. A common attempt to replicate ACLF in mice has been the induction of chronic liver injury with CCl4 or TAA followed by an acute insult by high dose of CCl4, APAP or D-GalN and bacterial infection (27, 28).
In this study we developed a mouse model of sepsis associated ACLF, where liver fibrosis is induced by thioacetamide (TAA) treatment and sepsis is induced by cecal ligation and puncture (CLP). We observed that in TAA treated mice, CLP resulted in significant reduction in liver function and reduction in HNF4α expression and HNF4 α transcriptional activity. In contrast to previously published models, for example model developed by Dr. Gao and colleagues (27) and others (28), our model does not produce massive cell death, and thus the loss of liver function in these mice was due to reduced hepatocyte differentiation. There are, however, some similarities with other models. We observed that loss of hepatocyte function was accompanied by increased BUN suggesting presence of kidney injury. Another similarity is loss of regeneration. However, unlike in other models we observed elevated IL-22/STAT3 (Figure 7A and S5) signaling in TAA/CLP mice, suggesting an alternative mechanism is at play.
Preliminary data indicate that loss of liver function in TAA/CLP mice is linked to endothelial dysfunction induced by downregulation of Angiopoietin-1 and an increase in Angiopoietin-2. Angiopoietins bind TIE-1 and TIE-2 receptors on endothelial cells and have roles in vascular development and angiogenesis (25, 26). Angiopoietin-1 promotes endothelial cell differentiation and inhibits inflammation. Angiopoietin-2 prevents Angiopoietin-1 receptor binding and is pro-inflammatory, promotes angiogenesis, and is implicated in endothelial dysfunction. The Angiopoietin-2/Angiopoietin-1 ratio is elevated in patients with ACLF and sepsis (29) and correlates with mortality in patients with cirrhosis (29, 30). Recent studies demonstrated that Angiopoietin-2/Angiopoietin-1 ratio was elevated in patients with liver cirrhosis and severe SARS-CoV-2 infection. Another study showed that Angiopoietin-2/Angiopoietin-1 was increased in patients with alcoholic hepatitis and was associated with an increase in circulating HGF. Taken together angiopoietin imbalance is a common feature of ACLF pathogenesis; however, the link between these factors and liver failure in ACLF has not been previously studied.
We found that the increase Angiopoietin-2/Angiopoietin-1 ratio promotes endothelial HGF production in the liver. Relationship between angiopoietins and HGF production was previously established in liver regeneration studies. Both Angiopoietin-2 and Angiopoietin-1 protein levels change dynamically to promote hepatocyte and endothelial cell proliferation in timely manner after partial hepatectomy. We found that in TAA/CLP mice, reduced Angiopoietin-1 levels were responsible for an increase in endothelial HGF and downstream activation of C/EBPβ in hepatocytes. This situation is similar to the early events during liver regeneration after partial hepatectomy including changes in angiopoietins, HGF expression, C/EBPβ activation, and hepatocyte de-differentiation (31, 32). However, we observed no increase in hepatocyte proliferation in TAA/CLP mice, suggesting that regeneration program was initiated but not completed. We found that similarities between TAA/CLP mice and early events in liver regeneration were present at the transcriptional level as well. RNA-seq analysis confirmed that in TAA/CLP mice there was activation of C/EBPβ-dependent gene expression program much like at 3 hours after PH. However, unlike after PH, C/EBPβ levels were persistently elevated even after 48h, suggesting that signals responsible for the termination of C/EBPβ activation were absent.
C/EBPβ is a transcription factor that regulates the expression of genes involved in the acute phase response, metabolic functions, and cell cycle progression (15, 17, 18). C/EBPβ is transiently activated during liver regeneration (15, 18) and is necessary for full regenerative response. However, persistent activation may result is permanent suppression of metabolic and hepatocyte differentiation gene expression. By analyzing levels of hepatic C/EBPβ, we found that in TAA/CLP mice and human ACLF patients C/EBPβ binds and suppresses metabolic and hepatocyte differentiation genes such as Hnf4a and Foxa2. Hepatocyte specific Cebpb knockout mice were protected from TAA/CLP induced loss of hepatocyte differentiation markers and had reduced mortality. Moreover, we found that C/EBPβ activation was mediated by endothelial cell dysfunction, since supplementation with Angiopoietin-1 prevented C/EBPβ accumulation, increased liver function markers, and improved mortality.
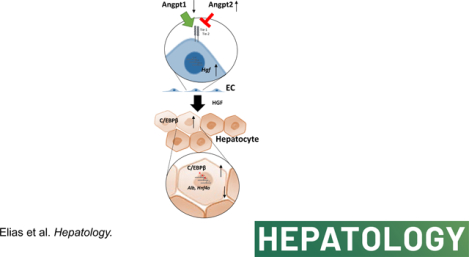
Taken together our data suggest that sepsis in mice and humans with liver fibrosis triggers endothelial dysfunction, which in turn promotes HGF production, activation of C/EBPβ in hepatocytes, and loss of hepatocyte differentiation resulting in liver failure or ACLF (Figure 8F). C/EBPβ inhibition or Angiopoietin-1 supplementation could represent promising therapeutic strategies for ACLF treatment.
Materials and methods
Mice
Cebpb floxed mice (BALB/cJ-Cebpbtm1.1Elgaz ) were obtained from Jackson lab and backcrossed for 7 generations to the C57BL6/J background.
All mice were housed in a temperature-controlled, specific pathogen-free environment with 12-hour light-dark cycles. All animal handling procedures were approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center (Kansas City, KS).
For fibrosis induction mice were treated with 200 mg/L of TAA in the drinking water for 2 months.
Recombinant Angiopoietin-1 was injected IP at 5 μg/mouse.
Vectors
AAV8-U6-m-Hgf-shRNA and AAV8-GFP-U6-scrmb-shRNA; AAV8-TBG-Cre, AAV8-TBG- m-Hnf4a, and AAV8-TBG-EGFP were from Vector BioLabs, Malvern, PA, and were used at 1011 genome copies per mouse.
Cecal ligation and puncture
Cecal ligation and puncture (CLP) was used to produce sepsis and was performed as described previously (33). Mice were anesthetized and an abdominal incision was made. The distal one-third was ligated, punctured with a 25-G needle, a small amount of cecal material was expressed before the cecum was replaced, and the incision was closed with 4–0 surgical suture. Following surgery, all animals received 0.9% saline (1 ml) and Buprenorphine (Sustained release, 0.2 mg/kg, SC). 24h and 48h after the induction of sepsis, mice were euthanized, and livers were collected.
ChIP
Chromatin immunoprecipitation was performed as described previously (34). Whole liver cells were cross-linked with 1% formaldehyde. Cells were lysed and nuclei collected by centrifugation, resuspended in [1% SDS, 5 mmol/L EDTA, 50 mmol/L Tris-HCl (pH 8.0)] and sonicated to generate chromatin to an average length of ~100 to 500 bp. Next, samples in [1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl of pH 8.1, 150 mM NaCl], were immunoprecipitated overnight at 4°C with 4 μg ChIP-grade antibody. 20 μl of magnetic beads (Dynabeads M-280, Invitrogen) were used to purify immunocomplexes. Following purification, DNA were purified with Qiagen DNA purification kit.
Cell isolation
Liver cells were isolated as previously described (35). Mouse livers were digested by retrograde perfusion with liberase via the inferior vena cava. The dissociated cell mixture was placed into a 50 mL conical tube and centrifuged twice at 50 g for 2 min to pellet hepatocytes. The NPC-containing cell supernatant was further used to isolate LSEC with CD146+MicroBeads (MiltenyiBiotec) according to the manufacturer’s instructions.
Transwell co-culture
For co-culture experiments, primary LSECs were placed in cell inserts of 24 well transwell (Corning Incorporated, Acton, MA, 0.4 μm pore size) at a seeding density of 5 × 104/well. Cells were treated as indicated. Freshly isolated hepatocytes were seeded in bottom well at a seeding density 1 × 105/well. The cells were then cultured for 48 hours, and hepatocytes were harvested for RNA isolation.
RNA-Seq
For RNA-Seq analysis total RNA was isolated from liver using Qiagen RNA isolation kit. Three individual mice per condition were used. Library generation and sequencing was performed by BGI genomics services (BGI, Cambridge, MA). 24 samples were sequenced using the BGISEQ platform, on average generating about 4.57G Gb bases per sample. HISAT was used to align the clean reads to the reference genome. Bowtie2 was used to align the clean reads to the reference genes The average mapping ratio with a reference genome (GRCm38.p6) was 96.14%, 16869 genes were identified. Differential gene expression was identified with DESeq2.
Immunohistochemistry
Immunostaining on formalin-fixed sections was performed by deparaffinization and rehydration followed by antigen retrieval by heating in a pressure cooker (121°C) for 5 minutes in 10 mM sodium citrate, pH 6.0 as described previously (36). Peroxidase activity was blocked by incubation in 3% hydrogen peroxide for 10 minutes. Sections were rinsed three times in PBS-T (0.1% Tween-20) and incubated in Protein Block (Dako) at room temperature for 1 hour. Slides were then placed into a humidified chamber and incubated overnight with a primary antibody, diluted 1:300 in Protein Block at 4°C. Antigen was detected using the SignalStain Boost IHC detection reagent (catalogue # 8114; Cell Signaling Technology, Beverly, MA), developed with diaminobenzidene (Dako, Carpinteria, CA), counterstained with hematoxylin (Sigma-Aldrich), and mounted.
RT-PCR
RNA was extracted from livers using the RNeasy Mini Kit (Qiagen). cDNA was generated using the RNA reverse transcription kit (Applied Biosystems, Cat.No 4368814). Quantitative real time RT-PCR was performed in a CFX96 Real time system (Bio-Rad) using specific sense and antisense primers combined with iQ SYBR Green Supermix (Bio-Rad) for 40 amplification cycles: 5 s at 95 °C, 10 s at 57 °C, 30 s at 72 °C. mRNA concentrations were calculated relative to Actb.
WB
Protein extracts (100 μg) were subjected to 10% SDS-PAGE, transferred to nitrocellulose membranes (Amersham Hybond ECL, GE Healthcare), and blocked in 3% BSA/PBS at RT for 1 hour. Primary antibodies were incubated overnight at manufacturer recommended concentrations. Immunoblots were detected with the ECL Plus Western Blotting Detection System (Amersham Biosciences, Piscataway, NJ) with the ODYSSEY Fc, Dual-Mode Imaging system (Li-COR).
Cytokine array
Proteome Profiler Mouse Cytokine Array Kit (R&D Systems) was used according to manufacturer’s instructions.
Human samples
De-identified human specimens were obtained from the Liver Center Tissue Bank at the University of Kansas Medical Center. All studies using human tissue samples were approved by the Human Subjects Committee of the University of Kansas Medical Center.
Statistics
Results are expressed as mean ± SD. The student t test, paired t test, Pearson’s correlation, or one-way ANOVA with Bonferroni post hoc test was used for statistical analyses. P value < 0.05 was considered significant.
Supplementary Material
Financial Support:
This study was supported by grants AA027586 and AA012863 from the National Institute on Alcoholism and Alcohol Abuse, VA Merit Award I01BX004694, and Lied Pre-Clinical Pilot Grant from University of Kansas Medical Center.
List of Abbreviations:
- ACLF
acute-on-chronic liver failure
- ALF
acute liver failure
- CLP
cecal ligation and puncture
- C/EBPβ
CCAAT enhancer binding protein beta
- HGF
hepatocyte growth factor
- LSEC
liver sinusoidal endothelial cells
- TAA
thioacetamide
- WD
western diet
- WDA
western diet and alcohol
Footnotes
Conflict of Interest statement: The authors declare that they have no conflicts of interest with the contents of this article.
Data availability statement
Raw sequencing data for all reported datasets is available upon request.
References
- 1.Moreau R, Gao B, Papp M, Bañares R, Kamath PS. Acute-on-chronic liver failure: A distinct clinical syndrome. J Hepatol 2021;75 Suppl 1:S27–s35. [DOI] [PubMed] [Google Scholar]
- 2.Heron M Deaths: Leading Causes for 2019. Natl Vital Stat Rep 2021;70:1–114. [PubMed] [Google Scholar]
- 3.Solé C, Solà E. Update on acute-on-chronic liver failure. Gastroenterol Hepatol 2018;41:43–53. [DOI] [PubMed] [Google Scholar]
- 4.Mucke MM, Rumyantseva T, Mucke VT, Schwarzkopf K, Joshi S, Kempf VAJ, Welsch C, et al. Bacterial infection-triggered acute-on-chronic liver failure is associated with increased mortality. Liver Int 2018;38:645–653. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Wang LK, Wang LW, Han XQ, Yang F, Gong ZJ. Blockade of high-mobility group box-1 ameliorates acute on chronic liver failure in rats. Inflamm Res 2013;62:703–709. [DOI] [PubMed] [Google Scholar]
- 6.Tripathi DM, Vilaseca M, Lafoz E, Garcia-Caldero H, Viegas Haute G, Fernandez-Iglesias A, Rodrigues de Oliveira J, et al. Simvastatin Prevents Progression of Acute on Chronic Liver Failure in Rats With Cirrhosis and Portal Hypertension. Gastroenterology 2018;155:1564–1577. [DOI] [PubMed] [Google Scholar]
- 7.Xiang X, Feng D, Hwang S, Ren T, Wang X, Trojnar E, Matyas C, et al. Interleukin-22 ameliorates acute-on-chronic liver failure by reprogramming impaired regeneration pathways in mice. J Hepatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan HY, Liu Z, Johnson PF, Richards JS. CCAAT/enhancer-binding proteins (C/EBP)-α and -β are essential for ovulation, luteinization, and the expression of key target genes. Mol Endocrinol 2011;25:253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato A, Kamio N, Yokota A, Hayashi Y, Tamura A, Miura Y, Maekawa T, et al. C/EBPβ isoforms sequentially regulate regenerating mouse hematopoietic stem/progenitor cells. Blood Adv 2020;4:3343–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerzoni C, Bardini M, Mariani SA, Ferrari-Amorotti G, Neviani P, Panno ML, Zhang Y, et al. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood 2006;107:4080–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jinesh GG, Napoli M, Ackerman HD, Raulji PM, Montey N, Flores ER, Brohl AS. Regulation of MYO18B mRNA by a network of C19MC miRNA-520G, IFN-γ, CEBPB, p53 and bFGF in hepatocellular carcinoma. Sci Rep 2020;10:12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vidarsdottir L, Fernandes RV, Zachariadis V, Das I, Edsbäcker E, Sigvaldadottir I, Azimi A, et al. Silencing of CEBPB-AS1 modulates CEBPB expression and resensitizes BRAF-inhibitor resistant melanoma cells to vemurafenib. Melanoma Res 2020;30:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu L, Zhang YD, Zhou J, Yao DM, Li X. Identification of target genes of transcription factor CEBPB in acute promyelocytic leukemia cells induced by all-trans retinoic acid. Asian Pac J Trop Med 2013;6:473–480. [DOI] [PubMed] [Google Scholar]
- 14.Kuttippurathu L, Patra B, Cook D, Hoek JB, Vadigepalli R. Pattern analysis uncovers a chronic ethanol-induced disruption of the switch-like dynamics of C/EBP-β and C/EBP-α genome-wide binding during liver regeneration. Physiol Genomics 2017;49:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakobsen JS, Waage J, Rapin N, Bisgaard HC, Larsen FS, Porse BT. Temporal mapping of CEBPA and CEBPB binding during liver regeneration reveals dynamic occupancy and specific regulatory codes for homeostatic and cell cycle gene batteries. Genome Res 2013;23:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang B, Gao C, Ponder KP. C/EBPbeta contributes to hepatocyte growth factor-induced replication of rodent hepatocytes. J Hepatol 2005;43:294–302. [DOI] [PubMed] [Google Scholar]
- 17.Timchenko NA, Harris TE, Wilde M, Bilyeu TA, Burgess-Beusse BL, Finegold MJ, Darlington GJ. CCAAT/enhancer binding protein alpha regulates p21 protein and hepatocyte proliferation in newborn mice. Mol Cell Biol 1997;17:7353–7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trautwein C, Rakemann T, Pietrangelo A, Plumpe J, Montosi G, Manns MP. C/EBP-beta/LAP controls down-regulation of albumin gene transcription during liver regeneration. J Biol Chem 1996;271:22262–22270. [DOI] [PubMed] [Google Scholar]
- 19.Diehl AM, Yang SQ. Regenerative changes in C/EBP alpha and C/EBP beta expression modulate binding to the C/EBP site in the c-fos promoter. Hepatology 1994;19:447–456. [PubMed] [Google Scholar]
- 20.Nerlov C The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol 2007;17:318–324. [DOI] [PubMed] [Google Scholar]
- 21.Greenbaum LE, Li W, Cressman DE, Peng Y, Ciliberto G, Poli V, Taub R. CCAAT enhancer- binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J Clin Invest 1998;102:996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li JL, Li G, Jing XZ, Li YF, Ye QY, Jia HH, Liu SH, et al. Assessment of clinical sepsis-associated biomarkers in a septic mouse model. J Int Med Res 2018;46:2410–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Argemi J, Latasa MU, Atkinson SR, Blokhin IO, Massey V, Gue JP, Cabezas J, et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun 2019;10:3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4alpha in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol 2013;304:G26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefere S, Devisscher L, Geerts A. Angiogenesis in the progression of non-alcoholic fatty liver disease. Acta Gastroenterol Belg 2020;83:301–307. [PubMed] [Google Scholar]
- 26.Bocca C, Novo E, Miglietta A, Parola M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cell Mol Gastroenterol Hepatol 2015;1:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiang X, Feng D, Hwang S, Ren T, Wang X, Trojnar E, Matyas C, et al. Interleukin-22 ameliorates acute-on-chronic liver failure by reprogramming impaired regeneration pathways in mice. J Hepatol 2020;72:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nautiyal N, Maheshwari D, Tripathi DM, Kumar D, Kumari R, Gupta S, Sharma S, et al. Establishment of a murine model of acute-on-chronic liver failure with multi-organ dysfunction. Hepatol Int 2021;15:1389–1401. [DOI] [PubMed] [Google Scholar]
- 29.Kaur S, Hussain S, Kolhe K, Kumar G, Tripathi DM, Tomar A, Kale P, et al. Elevated plasma ICAM1 levels predict 28-day mortality in cirrhotic patients with COVID-19 or bacterial sepsis. JHEP Rep 2021;3:100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allegretti AS, Vela Parada X, Ortiz GA, Long J, Krinsky S, Zhao S, Fuchs BC, et al. Serum Angiopoietin-2 Predicts Mortality and Kidney Outcomes in Decompensated Cirrhosis. Hepatology 2019;69:729–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang R, Huebert RC, Shah VH. Sinusoidal endothelial cells coordinate liver regeneration and angiogenesis via angiopoietin-2: an ode to prometheus. Gastroenterology 2014;147:533–534. [DOI] [PubMed] [Google Scholar]
- 32.Ross MA, Sander CM, Kleeb TB, Watkins SC, Stolz DB. Spatiotemporal expression of angiogenesis growth factor receptors during the revascularization of regenerating rat liver. Hepatology 2001;34:1135–1148. [DOI] [PubMed] [Google Scholar]
- 33.Tikhanovich I, Zhao J, Olson J, Adams A, Taylor R, Bridges B, Marshall L, et al. Protein arginine methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor gamma-dependent macrophage differentiation. J Biol Chem 2017;292:6882–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao J, Adams A, Roberts B, O’Neil M, Vittal A, Schmitt T, Kumer S, et al. Protein arginine methyl transferase 1- and Jumonji C domain-containing protein 6-dependent arginine methylation regulate hepatocyte nuclear factor 4 alpha expression and hepatocyte proliferation in mice. Hepatology 2018;67:1109–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schonfeld M, Villar MT, Artigues A, Weinman SA, Tikhanovich I. Arginine Methylation of Integrin Alpha-4 Prevents Fibrosis Development in Alcohol-Associated Liver Disease. Cell Mol Gastroenterol Hepatol 2022;15:39–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao J, Adams A, Roberts B, O’Neil M, Vittal A, Schmitt T, Kumer S, et al. PRMT1 and JMJD6 dependent arginine methylation regulate HNF4alpha expression and hepatocyte proliferation. Hepatology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data for all reported datasets is available upon request.