

Graphical abstract
Keywords: In-silico Analysis, Pharmacokinetics, Toxicology, Phytochemicals, Drug-Likeness, ADME/Tox Profiling
Highlights
-
•
Herbal syrups have sparked interest among traditional medicine practitioners and contemporary healthcare providers.
-
•
Limited knowledge exists on the pharmacokinetic and toxicological properties of phytochemicals in these herbal mixtures.
-
•
This study employs novel computational analysis methodologies, leveraging in-silico techniques and prediction tools.
-
•
Valuable insights into the pharmacokinetic and toxicological profiles of herbal concoctions are provided, advancing environmental and alternative medicine.
-
•
Future studies should explore molecular dynamics, docking, and system-wide analyses to better understand interactions between herbal syrups and biological systems.
Abstract
Herbal medications have an extensive history of use in treating various diseases, attributed to their perceived efficacy and safety. Traditional medicine practitioners and contemporary healthcare providers have shown particular interest in herbal syrups, especially for respiratory illnesses associated with the SARS-CoV-2 virus. However, the current understanding of the pharmacokinetic and toxicological properties of phytochemicals in these herbal mixtures is limited. This study presents a comprehensive computational analysis utilizing novel approach methodologies (NAMs) to investigate the pharmacokinetic and toxicological profiles of phytochemicals in herbal syrup, leveraging in-silico techniques and prediction tools such as PubChem, SwissADME, and Molsoft's database. Although molecular dynamics, docking, and broader system-wide analyses were not considered, future studies hold potential for further investigation in these areas. By combining drug-likeness with molecular simulation, researchers identify diverse phytochemicals suitable for complex medication development examining their pharmacokinetic-toxicological profiles in phytopharmaceutical syrup. The study focuses on herbal solutions for respiratory infections, with the goal of adding to the pool of all-natural treatments for such ailments. This research has the potential to revolutionize environmental and alternative medicine by leveraging in-silico models and innovative analytical techniques to identify novel phytochemicals with enhanced therapeutic benefits and explore network-based and systems biology approaches for a deeper understanding of their interactions with biological systems. Overall, our study offers valuable insights into the computational analysis of the pharmacokinetic and toxicological profiles of herbal concoction. This paves the way for advancements in environmental and alternative medicine. However, we acknowledge the need for future studies to address the aforementioned topics that were not adequately covered in this research.
Introduction:
Several tribes all around the world have utilized herbal treatments for ages. Due to its alleged efficacy and safety, there has been a resurgence in interest in herbal medicines recently (Komolafe et al., 2021). Several of these medications are created using phytochemicals, which are organic substances found in plants. These phytochemicals have the potential to be turned into medicinal medicines since they have a variety of pharmacological characteristics (Kothandan et al., 2021). Unfortunately, creating medications from phytochemicals is sometimes a time-consuming, expensive, and ineffective procedure. An effective and organized method for handling this procedure is provided by in-silico analysis (Nasim et al., 2022). A heightened interest in the possibility of herbal remedies, particularly herbal mixtures, as potential therapies or preventative measures against the disease has been sparked by the current COVID-19 epidemic (Barghash et al., 2021). Numerous studies have looked into the antiviral qualities of specific herbs and their active ingredients, emphasizing their promise as an alternative treatment to conventional medication (Prajapati et al., 2022). Studies have focused on the potential of natural remedies with components like ginger, turmeric, and garlic to strengthen the immune system and reduce respiratory illness symptoms. However, thorough scientific analysis is necessary to determine the effectiveness and safety of herbal remedies as therapies for COVID-19 or any other illness (Aware & Rohane, 2021). A useful instrument for this assessment is in-silico analysis, which enables scientists to forecast the pharmacokinetics, drug-likeness, and toxicological profiles of the mixture's phytochemical constituents (Xiong et al., 2021). Before performing additional preclinical and clinical research, this can help spot possible safety issues or medication interactions. In-silico analysis has been used recently in research to investigate the possibility of herbal concoctions as COVID-19 treatments (Gowrishankar et al., 2021). For instance, research published in the Journal of Biomolecular Structure and Dynamics examined the interactions of the active components of the plant remedy Kabasura Kudineer with the SARS-CoV-2 spike protein using molecular docking and molecular dynamics models. The findings indicated that the plant mixture's active ingredients might prevent the virus from entering host cells and lessen its infectiousness (Jayaraj et al., 2022). The possibility of herbal remedies as COVID-19 treatments or preventative steps emphasizes the need for more study in this field (Pranskuniene et al., 2022). In-silico analysis can be a useful instrument for assessing the effectiveness and safety of these treatments, paving the way for further preclinical and clinical studies (Singh et al., 2021c).
To anticipate the physicochemical characteristics, pharmacokinetics, drug-likeness, and therapeutic chemistry of phytochemicals, in-silico research uses computational models (Rajalakshmi et al., 2021). To show the role of phytochemicals on metabolic profile of liver inhibition of CYP family proteins was investigated as an additional factor to shed light on the toxicological characteristics of the phytochemicals, thus expanding the scope of the in-silico models. This approach can be used in conjunction with ethnopharmacology to find compounds that might have therapeutic effects (Prakash et al., 2021). One of in-silico analysis's major benefits is rapid assessment of phytonutrients. This may result in the finding of novel compounds with therapeutic promise (Umadevi et al., 2022). In-silico analysis can also be used to find prospective drug targets, forecast the effectiveness of brand-new medications, and enhance the pharmacokinetic characteristics of already-approved medications.
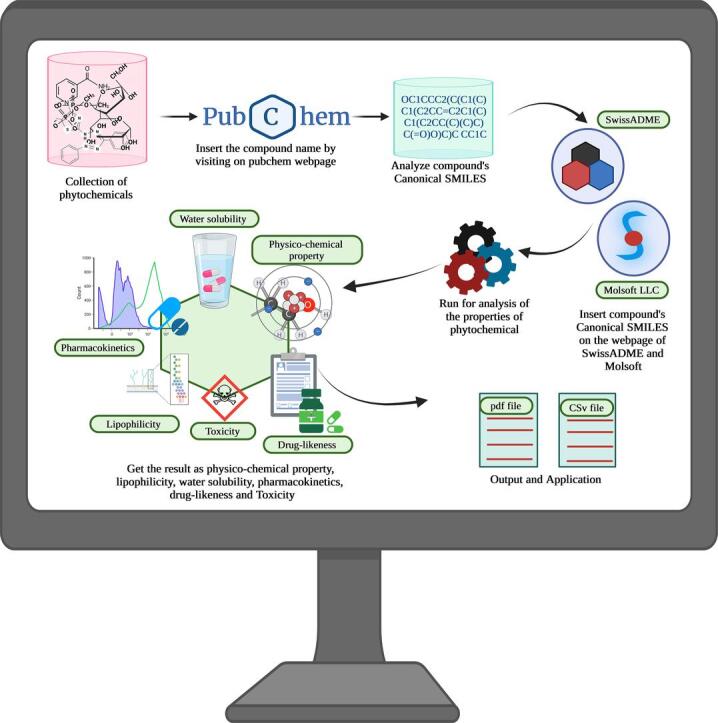
In this research, we use in-silico drug-likeness and ADMET analysis to assess the pharmacokinetic and toxicological characteristics of phytochemicals in a Phytopharmaceutical syrup (Ali et al., 2023). We will research the physicochemical characteristics and medicinal chemistry of the compounds in the syrup using a variety of prediction tools, including PubChem, SwissADME, and Molsoft's database. Additionally, we will apply Lipinski's Rule of Five (RO5) to forecast these phytochemicals' likelihood of becoming drugs. In addition to these conventional methods, we'll use cutting-edge techniques like network pharmacology and machine learning (Inamdar & Suryawanshi, 2023). A comparatively new discipline called network pharmacology seeks to comprehend the intricate relationships that exist between medicines and the biological systems they target (Liu et al., 2015). We can find possible drug targets and forecast the effectiveness of novel medications by looking at these interactions. Another effective method for forecasting the pharmacokinetic and toxicological characteristics of medicines is machine learning (Lima et al., 2016). We can accurately anticipate the characteristics of novel drugs by training machine-learning algorithms on huge datasets of known drugs. Our research will shed light on how successful herbal medications can be created using in-silico analysis. We can unearth phytochemicals with potential medicinal qualities and turn them into secure and efficient medicines by merging conventional ethnopharmacology with contemporary computational methods (Zhang et al., 2020). The exploration of natural sources for novel medicines presents an opportunity to evaluate and minimize the environmental impact associated with drug development, which is crucial for environmental research and sustainability efforts.
Materials and methods
Common herbal syrup ingredients and their phytochemicals coding:
A number of the frequently used herbs in the preparations of herbal syrup consist of Tulsi (Ocimum sanctum), Haldi (Curcuma longa), Giloy (Tinospora cordiofolia), Black pepper (Piper nigrum), Ginger (Zingiber officinale), Clove (Syzygium aromaticum), Cardamom (Elettaria cardamomum), lemon (Citrus limon) and Ashwagandha (Withania somnifera). A catalog of 108 phytochemicals is present in herbs that are used in the preparation of herbal syrup. Phytochemicals discovered in Ocimum sanctum (Oleanolic acid, Ursolic acid, Rosmarinic acid, Eugenol, Carvacrol, Linalool, Beta-caryophyllene, Estragole, Eugenic acid, Apigenin, Cirsimaritin, Isothymusin, Isothymonin, Vicenin, Orientin and Cirsilineol), Curcuma longa (Curcumin, Demethoxycurcumin, Bisdemethoxycurcumin, Ar-turmerone, Alpha-turmerone, Beta-turmerone, Atlantone, Cyclocurcumin, Calebin A, Trans-Ferulic acid, Vanillin and Vanillic acid), Tinospora cordiofolia (Magnoflorine, Berberine, Choline, Jatrorrhizine, Beta-Sitosterol, Tinosporide, Tinosporaside, Cordifolioside A, Tinocordioside, Cordioside, Tinocordifolioside and Tinocordifolin), Piper nigrum (Piperine, Piperamide, Piperamine, Pipericide, Sarmentosine, Sarmentine, Brachyamide B, Dihydropipericide, N-Formylpiperidine, Guineensine, Pentadienoylpiperidine, Tricholein, Trichostachine, Piperettine, Piperolein B, Retrofractamide A, Chavicine, Isochavicine, Isopiperine, Nerolidol, Beta-caryophyllene and Piperic acid), Zingiber officinale (6-gingerol, 6- shogaol, 6-paradol, Zingiberene, Bisabolene, 1-dehydrogingerdione, Gingerdione, 10-gingerdione, 4-gingerdiol, 6-gingerdiol, 10- gingerdiol, Citral and Eucalyptol), Syzygium aromaticum (Beta-caryophyllene, Vanillin, Eugenol, Acetyl eugenol, Crategolic acid, Eugenin, Methyl salicylate, Kaempferol, Rhamnetin, Eugenitin, Oleanolic acid, Stigmasterol, Campesterol, Gallic acid and Flavonol glucosides), Elettaria cardamomum (Protocatechualdehyde, Protocatechuic acid, Alpha- terpinyl acetate, 1,8-cineole, Linalool, Linalyl acetate, Limonene, 4-terpineol and Geraniol), Citrus limon (Eriodictyol, Quercetin, Hesperetin, Phloroglucinol, Umbelliferone, Vitamin C) and Withania somnifera (Withaferin A, Somniferine, Choline, Anaferine, Withanolide A, Withanolide B, Withanone and Withanolide)(Maurya & Sharma, 2022).
In-silico analysis of the pharmacokinetic and toxicological profiles of phytochemicals
SwissADME is a freely accessible web tool designed to assess the pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of small molecules (Daina et al., 2017). The tool is based on in-silico models and utilizes a variety of descriptors, including Molecular Weight, LogP, Number of Hydrogen Bond Donors and Acceptors, and Topological Polar Surface Area (TPSA).
The Supporting Figure S1 illustrates the step-by-step process, from inputting the compound to interpreting the results through SwissADME, the user uploads a small molecule structure in SMILES, SDF or MOL format, or draws the structure using a built-in sketcher. The tool then calculates various physicochemical properties, including LogP, TPSA, Molecular Weight, Number of Rotatable Bonds, and Aqueous Solubility (Singh et al., 2022b). These properties are used to evaluate the drug-likeness of the molecule, based on the rule-of-five (RO5) criteria proposed by Lipinski et al. The RO5 criteria state that a molecule with a molecular weight of less than 500, logP of less than 5, less than 5 hydrogen bond donors, and less than 10 hydrogen bond acceptors is more likely to have good oral bioavailability and be orally active.
SwissADME also provides predictions for several pharmacokinetic properties, including human intestinal Absorption, Blood-Brain Barrier permeation, P-glycoprotein (P-gp) substrate potential, and Cytochrome P450 (CYP) inhibition. These predictions are based on in-silico models trained on experimentally validated data and are used to evaluate the potential efficacy and safety of the molecule. In addition, SwissADME provides information on the medicinal chemistry friendliness of the molecule, such as the presence of reactive groups, drug-like scaffolds, and common substructures found in bioactive compounds.
The chemical structure and canonical SMILES of the molecule described alongside the bioavailability radar
The two-dimensional chemical structure with canonical SMILES was extracted for each phytochemical molecule from Chemspider or Pubchem databases. The bioavailability radar provides an initial assessment of the drug-likeness of the molecules of interest. It takes into account six physicochemical properties, including Lipophilicity (LIPO), Size (SIZE), Polarity (POLAR), Insolubility (INSOLU), Insaturation (INSATU), and Flexibility (FLEX) (Maharjan et al., 2022). Lipophilicity is measured by XLOGP3, which ranges from −0.7 to + 5.0. Size is determined by molecular weight (MW), which falls between 150 and 500 g/mol. Polarity is represented by topological polar surface area (TPSA), which ranges from 20 to 130 Å2. Solubility is indicated by log S, which should not exceed 6. Insaturation is determined by the fraction of carbons in the sp3 hybridization, which should be no less than 0.25. Flexibility is assessed by the number of rotatable bonds, which should not exceed 9.
Brain or intestinal estimated permeation method (BOILED-Egg) for BBB and gastrointestinal absorption prediction
The BOILED-Egg method was used to predict the permeability of small molecules across both the blood–brain barrier and the gastrointestinal tract (Daina & Zoete, 2016). The method involved estimating the partition coefficients of the molecules between an octanol phase and both brain and intestinal tissue, and then using these values to calculate predicted permeability coefficients. The method was validated using a set of known compounds with experimental permeability data and was shown to be accurate in predicting brain penetration and gastrointestinal absorption of small molecules. Once the partition coefficients were estimated, the predicted permeability coefficients were calculated using equations based on Fick's law of diffusion (Suenderhauf et al., 2012). Specifically, the predicted permeability across the Blood-Brain Barrier (BBB) was calculated using the following equation:
log(Pe_brain) = log(P_octanol) − 0.67log(P_intestine) − 0.0062.
where, Pe_brain is the predicted permeability coefficient across the BBB, P_octanol is the octanol–water partition coefficient, and P_intestine is the partition coefficient between intestinal tissue and octanol.
Similarly, the predicted permeability across the Gastrointestinal tract (GIT) was calculated using the following equation:
log (Pe_GIT) = log(P_octanol) + 0.43log(P_intestine) − 0.34.
where, Pe_GIT is the predicted permeability coefficient across the GIT.
To validate the BOILED-Egg method, a set of known compounds with experimental permeability data were used. The predicted permeability coefficients were compared to the experimental data, and the method was found to be accurate in predicting both brain penetration (Singh et al., 2022a) and gastrointestinal absorption of small molecules.
Statistical analysis
The Student’s two-tailed t-test was used for statistical significance for comparison between groups with normal distributions. For the statistical analysis, SPSS (v25) was used as mentioned in our previous report (Singh et al., 2023b). The primary analysis focused on calculating p-values to determine the significance of the correlations between pairs of chemical properties viz included Fraction Csp3, Molar Refractivity, TPSA(Å2), MolPSA (A2), MolVol (A3), Druglikeness score, Consensus Log Po/w, Log S (ESOL), Log S (Ali), and Log S (SILICOS-IT). A p-value (usually less than 0.05) indicated a statistically significant correlation.
Results and discussion
Physicochemical property analysis of phytochemicals presents in herbal concoction
Phytochemicals, derived from plants, have various therapeutic benefits (Dillard & Bruce German, n.d.). When extracting these substances from herbal concoctions, it is important to consider their physicochemical characteristics. In our study, we aimed to identify potential drug candidates from phytochemical pools by employing a drug-likeness scoring system. This system evaluates the molecular properties of a compound to determine its resemblance to known drugs, helping predict its drug-like characteristics. By applying this scoring criteria, we narrowed down the list of phytochemicals to those with positive scores, focusing on compounds with desirable properties for further drug development. Selecting compounds with positive drug-likeness scores is a widely accepted practice in drug discovery, as it increases the likelihood of identifying potential drug candidates with favourable pharmacokinetics, safety, and efficacy. Our goal is to increase the probability of identifying potential drug candidates for further investigation and potential development into therapeutics.
Following is a discussion of the significance of some of these characteristics, including Fraction Csp3, Molar Refractivity, TPSA, MolPSA, MolVol and pKa value.
The percentage of sp3-hybridized carbon atoms in a molecule is expressed as fraction Csp3. Since compounds with a greater Fraction Csp3 number are more likely to be metabolized and eliminated from the body than those with a lower value, this characteristic is crucial. The pharmacokinetics of phytochemicals with a greater Fraction Csp3 number may be impacted as a result of their shorter half-life (Dehelean et al., 2020).
The percentage of carbon atoms in a molecule that are sp3 bonded is known as the fraction Csp3, and it is a crucial factor in figuring out a compound's lipophilicity. A compound's capacity to penetrate cell membranes, which is crucial for its bioavailability, depends on how lipophilic it is (Mahmud et al., 2022). Our research revealed that the phytochemicals' Fraction Csp3 values varied from 0 to 0.93, with Beta-sitosterol having the greatest value. Beta-sitosterol, Cordioside, Cordifolioside A, and Magnoflorine showed the greatest Fraction Csp3 values. Fraction Csp3 showed highly significant correlations with Molar Refractivity (p = 1.7613E-37), MolVol (A3) (p = 4.2254E-26), and Druglikeness score (p = 9.35371E-33), while exhibiting marginally significant associations with TPSA(Å2) (p = 0.059100432) and MolPSA (A2) (p = 0.055785406).Fig. 1.
Fig. 1.

In-silico drug-likeness and ADMET analysis to assess the pharmacokinetic and toxicological characteristics of phytochemicals.
As shown in Fig. 2, the Fraction of carbon atoms with sp3 hybridization (Csp3), Molar Refractivity, Topological Polar Surface Area (TPSA), Molecular Polar Surface Area (MolPSA), Molecular Volume (MolVol), pKa value and Drug-likeness score analysis of various phytochemicals in supporting Table S6 including Oleanolic acid, Ursolic acid, Rosmarinic acid, Apigenin, Cirsimaritin, Isothymonin, Vicenin, Orientin, Cirsilineol, Cyclocurcumin, Magnoflorine, Berberine, Choline, Jatrorrhizine, Beta-sitosterol, Cordifolioside A, Cordioside, Tinocordifolioside, Piperamide, Piperamine, Pipericide, Dihydropipericide, Guineensine, Retrofractamide A, Piperolein B, 4-gingerdiol, Crategolic acid, Kaempferol, and Rhamnetin.
Fig. 2.

(A) Molar Refractivity, Topological polar surface area (TPSA), molecular polar surface area (MolPSA) and molecular volume (MolVol) analysis of selected phytochemicals; (B) Fraction (Csp3) and Drug-likeness score analysis of selected phytochemicals.
A molecule's capacity to polarize light is gauged by its molar refractivity. It has to do with a molecule's size and polarizability because these factors influence a molecule's solubility, absorption, and porosity (A. Kumar et al., 2021). According to our study, Vicenin had the greatest molar refractivity value of all the phytochemicals, ranging from 29.69 to 139.23. Vicenin and Crategolic acid, two substances with high molar refractivity values. Molar Refractivity displayed highly significant relationships with TPSA(Å2) (p = 4.83509E-17), MolPSA (A2) (p = 2.5582E-17), and MolVol (A3) (p = 9.38202E-33), but no significant correlation with Druglikeness score (p = 0.393342662).
Important metrics that characterize a molecule's polarity are the TPSA (total polar surface area) and MolPSA (molecular polar surface area). A compound's solubility, absorption, and protein binding are all influenced by the TPSA (Tripathi et al., 2023) and MolPSA readings of that compound (Belal, 2018). According to our study, Vicenin had the greatest TPSA value of all the phytochemicals, ranging from 6.48 to 271.2 Å2. Vicenin, Orientin, and Cordioside, which have the greatest TPSA values. TPSA(Å2) exhibited highly significant correlations with MolPSA (A2) (p = 6.58262E-05), MolVol (A3) (p = 3.82268E-24), and Druglikeness score (p = 1.73845E-37).
Similar to this, the MolPSA values of the phytochemicals varied from 5.7 to 214.06 A2, with Tinocordifolioside having the greatest value. Tinocordifolioside, Orientin, and Cordifolioside A are among the compounds with the greatest MolPSA values that may have excellent protein binding affinity, which is crucial for their biological activity (Rudrapal et al., 2022). A molecule's space-occupied volume, or MolVol, is what determines a molecule's solubility, porosity, and absorption. According to our research, the MolVol values of the phytochemicals varied from 260.14 to 598.51 A3, with crategolic acid having the greatest value. The substances with the greatest MolVol values, including Cratagolic acid, Oleanolic acid, and Cordioside, may have excellent solubility and be easily absorbed by the body (Vandecruys et al., 2007). Furthermore, significant associations were found between MolPSA (A2) and MolVol (A3) (p = 4.68128E-24), MolPSA (A2) and Druglikeness score (p = 4.78768E-17), and MolVol (A3) and Druglikeness score (p = 2.52657E-17).
The acidity of a particle is determined by its pKa value. The molecule's solubility, stability, and ionization state in biological fluids can all be impacted by this crucial characteristic. A more acidic substance is indicated by a lower pKa number, and vice versa (Settimo et al., 2014). Furthermore, some of the phytochemicals have low pKa values, which show that they are acidic and might not be viable under specific pH circumstances. For instance, the pKa values of Magnoflorine and Berberine are below 5.0, which indicates that they might not be viable in the acidic environment of the gut. The absorption of these compounds may be impacted by this characteristic (Tsume et al., 2012).
One can see from the given chart that some of the phytochemicals have high TPSA and MolPSA values, which indicate a high hydrogen bonding capacity and a significant polar surface area. These molecules might not effectively penetrate cellular membranes and have reduced bioavailability. Higher Molar Refractivity Numbers, on the other hand, tend to be more polar and can lead to greater solubility and dispersion in the body (Ngbolua et al., 2022). A measure known as the “Drug-likeness index” calculates how closely a molecule resembles a given drug. It is founded on a collection of guidelines that outline the physicochemical characteristics of substances that are most likely to be ingested and have pharmacological effects (Singh et al., 2020). Higher Drug-likeness ratings indicate molecules with a better likelihood of being effective drug candidates. The broad spread of Drug-likeness scores in the table is another intriguing finding (Leeson & Springthorpe, 2007). For instance, compared to other phytochemicals like Isothymonin and Magnoflorine, Piperamide and Piperamine have greater drug-likeness ratings.
Solubility and lipophilicity analysis of phytochemicals: Critically ignored parameters for phytochemicals
Based on their Consensus Log Po/w, Log S (ESOL), Log S (Ali), and Log S (SILICOS-IT) values, the aforementioned findings from in-silico study of different phytochemicals, including alkaloids, flavonoids, terpenoids, and other compounds, are provided. Understanding these compounds' physicochemical characteristics is crucial for forecasting their bioavailability, toxicity, and possible therapeutic effects. The research offers significant insights into these properties (Adeosun et al., 2022).
As shown Table 1 and in Fig. 3 in-silico analysis of lipophilicity vs. water solubility of various phytochemicals. The figure shows the Consensus Log Po/w and Log S values obtained using different software modules, namely ESOL, Ali, and SILICOS-IT, for a range of phytochemicals. The data in supporting Table S7 reveals the lipophilicity (Log Po/w) fluctuation for different chemicals and correlation of aqueous solubility (Log S) of the molecules, which are important parameters in drug design and development.Table 2.Table 3.
Table 1.
A Statistical Analysis of Correlation Coefficients between Log P and Log S Values.
| Value | Consensus Log Po/w | Log S (ESOL) | Log S (Ali) | Log S (SILICOS-IT) |
|---|---|---|---|---|
| Mean | 2.66 | −4.18 | −4.83 | −3.68 |
| Standard Deviation | 2.36 | 1.87 | 2.27 | 2.46 |
| Covariance with Consensus Log Po/w | −4.08 | −4.89 | −4.19 | |
| Correlation Coefficient with Consensus Log Po/w | −0.95 | −0.94 | −0.74 |
Fig. 3.

Correlation Coefficients between Log P and Log S Values predicted by ESOL, Ali and SILICOS-IT models.
Table 2.
Various rulesets for drug-likeness along with key results Lipinski's rule of five, the Ghose filter, the Veber, Egan, and Muegge rules.
| Ruleset | Lipinski | Veber | Ghose | Muegge | Egan |
|---|---|---|---|---|---|
| Rule 1 | Molecular weight <= 500 Da | Total number of rotatable bonds <= 10 | LogP (octanol–water partition coefficient) between −0.4 and + 5.6 | Number of hydrogen bond donors <= 5 | LogP (octanol–water partition coefficient) between −0.4 and + 5.6 |
| Rule 2 | LogP (octanol–water partition coefficient) <= 5 | Number of hydrogen bond donors <= 5 | Molecular weight <= 480 Da | Molecular weight <= 450 Da | Molecular weight <= 500 Da |
| Rule 3 | Hydrogen bond donors <= 5 | Number of hydrogen bond acceptors <= 10 | Number of hydrogen bond acceptors <= 10 | Molecular refractivity between 40 and 130 | Number of hydrogen bond acceptors <= 10 |
| Rule 4 | Hydrogen bond acceptors <= 10 | Total polar surface area <= 140 Å2 | LogP (octanol–water partition coefficient) between −0.4 and + 5.6 | N/A | Molecular refractivity between 40 and 130 |
| Rule 5 | Number of rotatable bonds <= 10 | N/A | Number of rotatable bonds <= 10 | N/A | N/A |
Table 3.
Various rulesets for medicinal chemistry along with key results PAINs, Brenk and Lead-likeness rules.
| Ruleset | Key Results | Criteria |
|---|---|---|
| PAINS | Identifies frequent hitters and promiscuous compounds that can interfere with assay results or exhibit undesirable properties. | - Consists of substructural patterns associated with problematic behavior. - Alerts against compounds with potential for false positives or misleading data. - Does not guarantee drug-likeness. |
| Brenk | Filters out compounds that violate certain chemical rules associated with undesirable properties. | - Based on chemical fragments and structural motifs. - Identifies compounds with potential for toxicity, reactivity, or instability. - Does not ensure drug-like properties. |
| Leadlikeness | Screens compounds based on structural features commonly found in lead compounds. | - Identifies compounds with desirable properties for further development. - Considers molecular weight, lipophilicity, and structural complexity. - Aims to prioritize compounds with lead-like characteristics. |
The partition coefficient between octanol and water, or Log Po/w, is a metric that is frequently used to estimate a compound's lipophilicity. A compound's affinity for lipid membranes and hydrophobicity are both indicated by high Log Po/w values, whereas its affinity for aqueous phase and hydrophilia is indicated by low Log Po/w values (Kumer et al., 2022). The tested compounds' Log Po/w values in the current research varied from −1.98 to 7.24. The greatest Log Po/w value was displayed by Beta-sitosterol (7.24), demonstrating its high lipophilicity and promise for use in lipid-based drug delivery systems. High Log Po/w values for Oleanolic acid and Ursolic acid were also observed (6.07 and 5.93, respectively), suggesting the possibility for effective cellular uptake (Singh et al., 2021b). The Consensus Log Po/w demonstrated highly significant correlations with Log S (ESOL) (p = 5.06575E-25), Log S (Ali) (p = 3.39957E-25), and Log S (SILICOS-IT) (p = 4.03081E-20).
Measures of aqueous solubility, or Log S (ESOL), Log S (Ali), and Log S (SILICOS-IT), are crucial for forecasting the absorption, diffusion, metabolism, and excretion (ADME) of medications (Daina et al., 2017). While a substance with low solubility is more likely to be excreted and ineffective, one with high solubility is more likely to be ingested and dispersed throughout the body (Gupta et al., 2013). The Log S (ESOL), Log S (Ali), and Log S (SILICOS-IT) values in the current research varied from −9.67 to 5.69, −8.53 to 0.44, and −6.12 to 1.49, respectively, for the investigated substances. The greatest Log S (SILICOS-IT) value was obtained for Magnoflorine (5.69), suggesting both its high aqueous solubility and potential for oral delivery. However, Log S (ESOL) showed only a marginally significant correlation with Log S (Ali) (p = 0.080057533) and a significant correlation with Log S (SILICOS-IT) (p = 0.01418434). There was no significant correlation between Log S (ESOL) and Log S (SILICOS-IT) (p = 0.144665119).
Numerous conclusions about the physicochemical characteristics of these substances can be derived from the provided chart of phytochemicals and their corresponding agreement Log Po/w values (Singh et al., 2020). First, it is clear that the compounds in the table's Log Po/w numbers vary from −1.98 to 7.24. A compound's lipophilicity is quantified by its Log Po/w number; a larger value typically denotes a more lipid-soluble compound. As a result, substances with larger Log Po/w values, like Beta-sitosterol (7.24) and Campesterol (6.92), are probably more likely to have an attraction for lipid-based membranes and biomolecules. Conversely, it is anticipated that substances with lower Log Po/w ratios, like Choline (-1.86) and Vitamin C (-1.42), will be more water-soluble.
The fact that some substances, like Oleanolic acid and Choline, appear numerous times in the table is another intriguing finding. With a Log Po/w number of 6.07, Oleanolic acid is one of the table's most lipophilic substances. It is a triterpenoid that is present in a variety of plant species and has been linked to several biological functions, including hepatoprotective, anti-inflammatory, and antioxidant benefits. The water-soluble nutrient Choline, on the other hand, has a much lower Log Po/w number of −1.86 and is essential for a variety of cellular processes, including the creation of cell membranes and the production of neurotransmitters (Misra et al., n.d.).
The biochemical activity of several other substances listed in the table is also well-known, and these properties include anti-cancer, anti-inflammatory, and anti-microbial effects (Singh et al., 2018a). For instance, it has been reported that the pentacyclic triterpenoid Ursolic acid (5.93), which is found in a variety of plant species, has anti-cancer properties through several mechanisms, including the suppression of cell proliferation, the induction of apoptosis, and the modification of signaling pathways (Majid et al., 2023). Like this, Withanolides are a class of steroidal lactones discovered in the Ayurveda plant Withania somnifera. These include Withanolide A (3.39), Withanolide B (4.14), and Withanone (3.36) (also known as Ashwagandha). They have a variety of biological functions that are understood to exist, such as anti-cancer, anti-inflammatory, and immunomodulatory impacts (Article et al., n.d.). The Log Po/w values of the phytochemicals in the chart can be analyzed to gain useful information about their physicochemical characteristics and possible biological activities. The information in the table can help direct additional experimental research targeted at examining the pharmacological potential of these compounds.
Exploring the mechanisms of GI Absorption, BBB Permeation, Cytochrome P450 Inhibition, and lipophilicity (Log Kp) analysis
The normal distribution of phytochemicals exhibits predictions with YES (Red) and NO (blue) with mean (µ) and standard deviation (σ).
As shown in Fig. 4(A) schematic showing how different in-silico and QSAR models help to identify the key Physchem/ADMET data in supporting Table S8 essential to understand PK/PD of herbal concoction components understand (Singh et al., 2018b). The data in 4(B) Histogram plot showing the comparative analysis of pharmacokinetic and toxicological profiles of phytochemicals in the herbal concoction in supporting Table S8. Each phytochemical's drug-likeliness, solubility, lipophilicity, permeability, and toxicity characteristics are compared on the graph. The length of each spoke represents the results for that measure, and the enclosed region displays the general profile of each phytochemical. The caption lists the matching color codes, and the phytochemicals are identified as numbers 1–42 on the chart's exterior perimeter. Affirmative for permeability receives a score of 1, while NO receives a value of 0. Comparable high BBB permeability receives a score of 0.99, while low atom-additive permeability receives a score of 0.001.
Fig. 4.

(A) Schematic representation of factors used to analyze ADMET property; (B-C) plot showing the frequency range with normal distribution of the logKp and GI absorption.
Along with effectiveness and toxicity, poor pharmacokinetics and bioavailability are frequently to blame for failed drug research (Mohamed et al., n.d.). A novel predictive model known as the Brain or Intestinal Estimate permeation technique (BOILED-Egg) has been suggested in order to correctly anticipate significant pharmacokinetic characteristics such as gastrointestinal absorption and brain access (Olasupo et al., 2020). It is worth noting that while lipophilicity and reduced polarity can enhance BBB penetration for certain compounds, they are not the sole determinants of drug candidacy or bioavailability. Several other factors, including solubility, metabolism, efflux transporters, molecular weight, and other physicochemical properties, play critical roles in determining a compound's overall bioavailability and suitability as a drug candidate. Thus, a comprehensive evaluation encompassing these factors is necessary for a more robust assessment of drug candidacy and bioavailability in drug discovery. This model is helpful for molecular design at different phases of the drug discovery process because it is theoretically straightforward, highly accurate, and generates understandable graphical outputs (Elmezayen et al., 2020). The BOILED-Egg has many uses, including screening chemical libraries and assessing drug prospects for development. The majority of P-glycoprotein (PGP) substrate molecules that can penetrate both the Blood-Brain Barrier (BBB) and Intestinal Barrier have certain properties that make them excellent substrates for PGP (Hsiao et al., 2021). We implemented the BOILED-Egg model as shown in Fig. 5.
Fig. 5.

Boiled-EGG model for analyzing GI absorption, BBB permeant P-gp substrate.
PGP is an efflux pump that blocks the entrance of possibly hazardous compounds into the brain and systemic blood and is found in intestinal epithelial cells and the BBB (Cordoncardo et al., 1989). PGP can identify and expel PGP + molecules, or molecules that are PGP substrates, from the cells, lowering their content in the brain or general circulation. While PGP is the primary transporter for some PGP + molecules, there are other transporters that can help PGP + molecules enter the brain and circulate throughout the body (Kim et al., 2016). These molecules frequently possess specific physicochemical traits, such as low molecular weight, high water solubility, and mild lipophilicity, which enable them to travel through the intestinal and BBB barriers via passive diffusion or other transport mechanisms (Di et al., 2020).
Because of this, compounds that are PGP + and have adequate physicochemical characteristics for traversing biological barriers, such as the Intestinal Barrier, can also penetrate the BBB (Vyas et al., 2006). In the aforementioned dataset, the BOILED-Egg model is a helpful method for locating outliers that may indicate mistakes or odd values. In this instance, the algorithm has recognized the given list of compounds' molecules 33, 35, 45, 81, and 82 as anomalies. To understand why these anomalies have been labeled as such, it is crucial to look into them further.
In traditional medicine, phytochemicals—naturally occurring substances found in plants are frequently used due to their therapeutic benefits. These substances have attracted a lot of attention in recent years due to their numerous biochemical actions, including their anti-inflammatory, antioxidant, and anticancer effects. The characteristics of these substances, such as their absorption through the GI tract, BBB permeability, P-gp substrate, and suppression of different Cytochrome P450 enzymes, are important in deciding how effective they are as medications (Y. S. Kumar et al., 2010). The SwissADME program was used in this research to evaluate the properties of different phytochemicals found in herbal concoctions (Sravika et al., 2021).
As shown in Fig. 5, the Boiled-Egg model implementation for a list of 107 molecules in supporting Table S3 and S9. Outliers 33, 35, 45, 81, and 82 are indicated by red dots, while the remaining 102 molecules are represented by blue dots. Outlier molecules may have characteristics or attributes that make them less likely to be absorbed or pass through the GIT (gastrointestinal tract) or BBB (blood–brain barrier) in the case of these particular biological barriers. Such anomalies could have an impact on drug design or optimization because they might create obstacles or possibilities for enhancing the bioavailability, effectiveness, or safety of therapeutic candidates. The model suggests that most molecules cross both the Blood-Brain Barrier and the Intestinal Barrier, although there are some exceptions. Results are illustrated in Fig. 5, along with a summary of the phytochemicals examined' characteristics in supporting Table S3. We also plot individual groups of phytochemicals obtained from 9 plants in supporting Figure S2. A-I for clarity.
However, upon closer examination of supporting Table S8, it becomes evident that most phytochemicals have poor gastrointestinal (GI) absorption, except for Apigenin, Cirsimaritin, Isothymonin, Magnoflorine, Berberine, Jatrorrhizine, Piperamide, Piperamine, Piperolein B, 4-gingeridol, and Hesperetin, which demonstrate significant GI absorption. This suggests that these compounds may have higher bioavailability than others, potentially increasing their therapeutic efficacy.
Additionally, among the substances studied, Magnoflorine, Berberine, Jatrorrhizine, Retrofractamide A, and Hesperetin exhibit BBB permeability. This indicates their ability to pass through the Blood-Brain Barrier and potentially impact the central nervous system (Singh et al., 2021a). This characteristic is particularly desirable for medications used to address neurological conditions. Furthermore, Vicenin, Jatrorrhizine, Magnoflorine, Berberine, Piperamide, Piperamine, Retrofractamide A, and Dihydropipericide demonstrate the property of being P-gp substrates. This implies that these substances may be subject to efflux transport by the P-glycoprotein efflux transporter, which can limit their therapeutic efficacy.
In terms of drug metabolism, it is crucial for drugs to inhibit different Cytochrome P450 enzymes, as this affects their pharmacokinetics and metabolism. The analysed phytochemicals inhibit various Cytochrome P450 enzymes. Apigenin, Cirsimaritin, Isothymonin, Cirsilineol, Cyclocurcumin, Piperamide, Piperamine, Pipericide, Dihydropipericide, Guineensine, Retrofractamide A, 4-gingerdiol, Kaempferol, Rhamnetin, Protocatechuic acid, Eriodictyol, and Quercetin demonstrate inhibition of CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Notably, Apigenin, Cirsimaritin, Isothymonin, Piperamide, Piperamine, Pipericide, Dihydropipericide, Retrofractamide A, 4-gingerdiol, Kaempferol, Rhamnetin, Protocatechuic acid, Eriodictyol, and Quercetin specifically prevent CYP1A2, an important enzyme involved in drug metabolism.
The lipophilicity of the compounds is indicated by the Log Kp number, with larger values indicating increased lipophilicity. Piperamide, Piperamine, Retrofractamide A, and Guineensine demonstrate the highest lipophilicity among the examined phytochemicals, with Log Kp values of −3.8, −5.08, −4.36, and −3.8 cm/s, respectively (Singh et al., 2020). Conversely, compounds such as Vicenin, Cordifolioside A, Cordioside, and others exhibit low Log Kp values. Finally, it is imperative to note that some of the phytochemicals in the herbal mixture under study may inhibit key enzymes involved in drug metabolism, such as CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 (Zhou et al., 2003). Consequently, combining the mixture with other medications processed by these enzymes may result in alterations in drug concentrations and potential side effects.
A number of intriguing bioactive substances with possible pharmacological activities were discovered after SwissADME software was used to evaluate the phytochemical properties in the examined plant concoction (Nasrin et al., 2021). These include substances with high GI absorption, BBB permeability, and enzymatic suppression of key drug metabolism enzymes. However, due to the possibility of some of the chemicals interacting with vital drug-metabolizing enzymes, care should be exercised when administering this product in conjunction with other medicines (Venkatachalam et al., 2016). To completely comprehend the possible therapeutic benefits and safety profile of the plant concoction, additional research is required to examine the pharmacological actions of the mixture in vitro and in vivo as well (Kaur & Bhatti, 2021).
Comparative analysis of LogPo/w (Consensus Log P) vs. Log Kp
LogPo/w and LogKp are distinct parameters used to evaluate a compound's lipophilicity, hydrophobicity, and membrane permeability. LogPo/w is the logarithm of the partition coefficient between octanol and water, reflecting a compound's affinity for lipid-rich environments and solubility in water. A higher LogPo/w value indicates higher lipophilicity, while a lower value suggests greater hydrophilicity.
On the other hand, LogKp is the logarithm of the apparent permeability coefficient across biological membranes. A higher LogKp value signifies better membrane permeability, while a lower value indicates reduced ability to cross cell membranes.
From the Supporting Figure S3 both LogPo/w and LogKp can provide insights into a compound's lipophilicity and membrane permeability, but they are different parameters measured using different experimental setups.
LogPo/w primarily reflects the compound's hydrophobic/hydrophilic balance, while LogKp provides information about the compound's ability to pass through cell membranes.
For most of the phytochemicals listed, there seems to be a general trend: compounds with higher LogPo/w values also tend to have higher LogKp values, and vice versa. This indicates that more lipophilic compounds are generally more permeable across biological membranes.
However, there are some exceptions to this trend. For example, “Vicenin” has a relatively low LogPo/w value (-1.98), suggesting hydrophilicity, but its LogKp value (-11.53) is extremely low, indicating low permeability across membranes. Similarly, “Withanone” has a relatively high LogPo/w value (3.36), indicating lipophilicity, but its LogKp value (-7.01) is relatively low compared to other lipophilic compounds.
Overall, comparing LogPo/w and LogKp values can provide complementary information about a compound's lipophilicity and membrane permeability, but it is essential to interpret each measure in the context of its specific meaning and experimental conditions. It is also crucial to consider other factors that can influence a compound's behavior in biological systems, such as molecular size, charge, and specific interactions with cellular components.
Different rule-based modules for bioavailability and Drug-likeness analysis
Drug-likeness analysis is a crucial part of the drug discovery process (Terstappen2001, n.d.). It involves assessing a molecule's potential as a drug candidate using a variety of criteria, including the Lipinski's rule of five, the Ghose filter, the Veber, Egan, and Muegge rules, the Bioavailability Score, PAINS, Brenk, and Lead likeness (Associate Professor, 2019). Using in-silico modules, we will assess the Drug-likeness of 36 phytochemicals in this study.
A popular method for assessing a molecule's Drug-likeness is Lipinski's rule of five. It says that a molecule has an increased probability of being directly bioavailable if it has no more than five hydrogen bond donors, ten hydrogen bond acceptors, a molecular weight under 500, and a LogP number under 5 (Chen et al., 2020). 32 of the 36 phytochemicals meet Lipinski's rule of five, suggesting a high possibility for oral bioavailability. Nevertheless, four substances—Rosmarinic acid, Magnoflorine, Quercetin, and Hesperetin—violate the LogP portion of the Lipinski's law. This suggests that the potential for dietary bioavailability of these substances may be constrained.
Another popular method for determining how druglike a substance is the Ghose filter. If a molecule has a molecular weight of between 160 and 480 Daltons, a LogP number between −0.4 and 5.6, and between 20 and 70 rotatable bonds, it is more likely to be directly bioavailable, according to the study (Schneider et al., 2008). 30 of the 36 phytochemicals meet the requirements of the Ghose filter, suggesting a high possibility for oral bioavailability. But six substances—Quercetin, Rhamnetin, Apigenin, Cirsimaritin, Isothymonin, and Withaferin A—are particularly noteworthy violate one or more of the Ghose filter's criteria. This indicates that these compounds may have limited oral bioavailability potential.
As shown in Fig. 6(A) and according to supporting Table S10, Veber's rule says that a molecule has one Polar Surface area (PSA) larger than 140 Å2 along with no more than ten rotatable bonds if it is likely to be orally bioavailable. 31 of the 36 phytochemicals meet Veber's criterion, suggesting a high possibility for oral bioavailability. Rosmarinic acid, Magnoflorine, Quercetin, Hesperetin, and Withanolide A are the five substances that defy Veber's law by having multiple PSA values above 140 Å2. This suggests that the potential for dietary bioavailability of these substances may be constrained.
Fig. 6.

(A) Lipinski's rule of five, the Ghose filter, the Veber, Egan, and Muegge rule analysis of selected phytochemicals; (B) Bioavailability Score and Synthetic accessibility analysis of selected phytochemicals.
Egan's rule states that for a molecule to be easily absorbed orally, it should have a molecular weight ranging from 200 to 600 Daltons, a LogP value between −0.4 and 5.0, less than or equal to six hydrogen bond donors, less than or equal to five hydrogen bond acceptors, and no more than 12 hydrogen bond acceptors (Brandão et al., 2021). In the analyzed molecules, 28 of the 36 phytochemicals meet Egan's criterion, suggesting a high possibility for oral bioavailability. Quercetin, Rhamnetin, Apigenin, Cirsimaritin, Isothymonin, Withaferin A, Choline, and Somniferine are eight substances, however, that fail to meet one or more of the requirements of Egan's formula. This suggests that the potential for dietary bioavailability of these substances may be constrained.
The basis for Muegge's formula is how a molecule's molecular weight and hydrogen bond sources are distributed. If a molecule has a molecular weight of between 250 and 400 Daltons and no more than three hydrogen bond producers, it is more likely to be easily bioavailable, according to the study (Şahin & Dege, 2021). The percentage of a medication that is given that enters the systemic circulation is determined in part by the bioavailability score. The ratings vary from 0 to 1, with 1 denoting the greatest bioavailability level (Shanmugavelan & Mohamed Musthafa, 2022). The bioavailability number for the majority of the phytochemicals in the chart is 0.55, which denotes modest bioavailability. A better number of 0.85, however, for Oleanolic acid and Ursolic acid indicates adequate bioavailability (Fig. 6(B) above).
In biological tests, PAINS—substructures or molecular pieces that are frequently linked to promiscuous or non-specific protein binding—can result in misleading positive results. Several of the compounds in the chart have PAINS warnings, which may preclude their use as lead molecules in the development of new drugs (Bajorath, 2014). A few compounds, including Apigenin, Cirsimaritin, and Jatrorrhizine, have one PAINS warning while others, including Vicenin, Cordifolioside A, Cordioside, and Vitamin C, have none. Two PAINS warnings are present for Rosmarinic acid and Eriodictyol.
Brenk rules are a collection of criteria that can be used to find substances that are more likely to be physiologically active and have characteristics of drugs. Brenk rules-compliant compounds are more likely to be appropriate as lead molecules in the search for new drug (Yang et al., 2020). All of the substances in the chart, with the exception of a few like Orientin and Cordifolioside A, satisfy the Brenk criteria. A notion called Synthetic Accessibility and Lead-likeness seeks to find substances that resemble recognized drugs or compounds that are comparable to drugs in terms of their structural and physicochemical characteristics (Nadin et al., 2012). More lead-like compounds are better candidates for drug development because they are more apt to have favorable pharmacological characteristics (Keserü & Makara, 2009). With the exception of a few, including Apigenin, Cirsimaritin, Isothymonin, Piperamide, Rhamnetin, and Withaferin A, the majority of the substances in the chart are not lead-like (Fig. 6(B) above and supporting Table S10 for drug-likeness).
Potential drug candidates among phytochemicals based on key results
In this article, preliminary analysis of several phytochemicals based on commonly used drug candidacy criteria. It is important to note that further studies and validation are necessary to confirm their suitability as drug candidates.
To identify potential drug candidates among phytochemicals, we applied a set of established criteria commonly used in drug discovery. These criteria include molecular weight (MW), hydrogen bond donors (HBD), hydrogen bond acceptors (HBAs), lipophilicity (LogP), topological polar surface area (TPSA), Lipinski's Rule of Five, and the absence of toxic alerts or undesirable features identified by filters such as PAINS, Brenk, and Lead likeness. Based on the analysis of phytochemicals compounds such as Oleanolic acid, Ursolic acid, Rosmarinic acid, Apigenin, Cirsimaritin, Isothymonin, Magnoflorine, Berberine, Piperamide, Piperamine, Piperolein B and Kaempferol emerged as potential drug candidates that satisfy most of the drug candidacy criteria.
These phytochemicals exhibit favorable characteristics for drug candidacy, as indicated by their molecular weight below 500 Da, an optimal range for oral absorption and reduced toxicity. The number of hydrogen bond donors and acceptors falls within the desirable range, indicating potential for favorable interactions with biological targets. Furthermore, their lipophilicity (LogP) values suggest a balance between solubility and permeability, essential for efficient drug delivery. The topological polar surface area (TPSA) values below 140 Å2 indicate good permeability across biological membranes (Chandrasekar et al., 2023). Importantly, these phytochemicals do not violate Lipinski's Rule of Five, which helps assess drug-like properties. Compliance with this rule, which limits violations to two of the four criteria, suggests potential for favorable pharmacokinetic properties. Moreover, these compounds have been screened for toxic alerts and undesirable features using established filters, such as PAINS, Brenk, and Lead likeness. The absence of such alerts implies reduced risk of toxicity (Singh et al., 2023a) or undesirable side effects.
The preliminary analysis of these phytochemicals indicates their potential as drug candidates based on commonly used drug discovery criteria. However, it is essential to emphasize that further studies, including in vitro and in vivo experiments, are necessary to confirm their pharmacological properties, safety profiles, efficacy, and potential drug interactions.
Conclusion
In conclusion, the in-silico study of the phytochemicals in the table reveals that a number of the substances have characteristics that make them highly suitable for use as lead molecules in the discovery of new drugs. Indigenous healing remedies have been practiced for years and are listed in the data, although they cannot be regarded as objectively legitimate. For the objective of Assembling, Evaluating, Propagating, Merging, and Exhibiting the comprehensive data on edible herbs used among various tribes in India, SwissADME is an accessible catalogue for it. After scientific verification, such data may be utilized in the formulation of conventional medicinal herbs, phytochemical assessment, or the synthesis of novel drugs. Drug corporations in drug discovery, can use extensive details of phytochemicals and their structures, physiochemical properties, and ADMET analysis as a coherent and consistent asset to investigate the medicinal prospects of curative plants against diverse therapeutic aspirations. SwissADME seeks to deliver a centralized site for scholars, professionals, as well as other research organizations in domains like medicine, drug development, ethno-medicinal research, etc. However, some substances have PAINS warnings, so they should be further examined before being suspected of as lead molecules. The compounds that are Lead-like and adhere to the Brenk rules are superior prospects for drug development because they are more likely to possess favorable pharmacological characteristics (Andricopulo et al., 2009). Overall, the in-silico study offers helpful data for the finding of phytochemicals that may be useful for drug discovery (Siddiqui et al., 2022).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Financial assistance from the BfR (SFP 1322-807) is thankfully acknowledged.
Footnotes
Appendix A
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crtox.2023.100118.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- Adeosun I.J., Baloyi I., Aljoundi A.K., Salifu E.Y., Ibrahim M.A., Cosa S. Molecular modelling of SdiA protein by selected flavonoid and terpenes compounds to attenuate virulence in Klebsiella pneumoniae. J. Biomol. Struct. Dyn. 2022 doi: 10.1080/07391102.2022.2148753. [DOI] [PubMed] [Google Scholar]
- Ali, A., Cottrell, J. J., & Dunshea, F. R. (2023). Antioxidant, Alpha-Glucosidase Inhibition Activities, In Silico Molecular Docking and Pharmacokinetics Study of Phenolic Compounds from Native Australian Fruits and Spices. Antioxidants, 12(2). https://doi.org/10.3390/antiox12020254. [DOI] [PMC free article] [PubMed]
- Andricopulo, A. D., Salum, L. B., & Abraham, D. J. (2009). Structure-Based Drug Design Strategies in Medicinal Chemistry. In Current Topics in Medicinal Chemistry (Vol. 9). [DOI] [PubMed]
- Associate Professor R.C. SwissADME predictions of pharmacokinetics and drug-likeness properties of small molecules present in Ipomoea mauritiana Jacq. ∼ 2063 ∼ Journal of Pharmacognosy and Phytochemistry. 2019;8(5):2063–2073. http://www.swissadme.ch/index.php [Google Scholar]
- Aware D., Rohane S. A Role of Herbal Drug as an Immunity Booster during Covid-19 Pandemic. Asian Journal of Pharmaceutical Research. 2021:206–211. doi: 10.52711/2231-5691.2021.00037. [DOI] [Google Scholar]
- Bajorath J. Activity artifacts in drug discovery and different facets of compound promiscuity. 2014;F1000Research:3. doi: 10.12688/f1000research.5426.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghash R.F., Fawzy I.M., Chandrasekar V., Singh A.V., Katha U., Mandour A.A. In Silico Modeling as a Perspective in Developing Potential Vaccine Candidates and Therapeutics for COVID-19. In silico modeling as a perspective in developing potential vaccine candidates and therapeutics for covid-19. In Coatings. 2021;11(11):1273. [Google Scholar]
- Belal A. Drug likeness, targets, molecular docking and ADMET studies for some indolizine derivatives. Pharmazie. 2018;73(11):635–642. doi: 10.1691/ph.2018.8061. [DOI] [PubMed] [Google Scholar]
- Brandão P., Marques C., Burke A.J., Pineiro M. The application of isatin-based multicomponent-reactions in the quest for new bioactive and druglike molecules. Eur. J. Med. Chem. 2021;211:113102. doi: 10.1016/j.ejmech.2020.113102. [DOI] [PubMed] [Google Scholar]
- Chandrasekar V., Ansari M.Y., Singh A.V., Uddin S., Prabhu K.S., Dash S., Khodor S.A., Terranegra A., Avella M., Dakua S.P. Investigating the Use of Machine Learning Models to Understand the Drugs Permeability Across Placenta. IEEE Access. 2023;11:52726–52739. doi: 10.1109/ACCESS.2023.3272987. [DOI] [Google Scholar]
- Chen X., Li H., Tian L., Li Q., Luo J., Zhang Y. Analysis of the Physicochemical Properties of Acaricides Based on Lipinski’s Rule of Five. J. Comput. Biol. 2020;27(9):1397–1406. doi: 10.1089/cmb.2019.0323. [DOI] [PubMed] [Google Scholar]
- Cordoncardo C., O’brien, J. P., Casals, D., Rittman-Grauert, L., Biedler, J. L., Melamed, M. R., & Bertino, J. R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites (monoclonal antibodies/immunohistochemistry/cancer chemotherapy) In Proc. NatI. Acad. Sci. USA. 1989;86 doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Zoete V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem. 2016;11(11):1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7 doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehelean, C. A., Lazureanu, V., Coricovac, D., Mioc, M., Oancea, R., Marcovici, I., Pinzaru, I., Soica, C., Tsatsakis, A. M., & Cretu, O. (2020). SARS-CoV-2: Repurposed drugs and novel therapeutic approaches—insights into chemical structure—biological activity and toxicological screening. In Journal of Clinical Medicine (Vol. 9, Issue 7, pp. 1–41). MDPI. https://doi.org/10.3390/jcm9072084. [DOI] [PMC free article] [PubMed]
- Di L., Artursson P., Avdeef A., Benet L.Z., Houston J.B., Kansy M., Kerns E.H., Lennernäs H., Smith D.A., Sugano K. The Critical Role of Passive Permeability in Designing Successful Drugs. ChemMedChem. 2020;15(20):1862–1874. doi: 10.1002/cmdc.202000419. [DOI] [PubMed] [Google Scholar]
- Dillard, C. J., & Bruce German, J. (n.d.). Review Phytochemicals: nutraceuticals and human health.
- Elmezayen A.D., Al-Obaidi A., Şahin A.T., Yelekçi K. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2020;39(8):2980–2992. doi: 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S., Muthumanickam S., Kamaladevi A., Karthika C., Jothi R., Boomi P., Maniazhagu D., Pandian S.K. Promising phytochemicals of traditional Indian herbal steam inhalation therapy to combat COVID-19 – An in silico study. Food Chem. Toxicol. 2021;148:111966. doi: 10.1016/j.fct.2020.111966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Kesarla R., Omri A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharmaceutics. 2013;2013:1–16. doi: 10.1155/2013/848043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao, Y., Su, B. H., & Tseng, Y. J. (2021). Current development of integrated web servers for preclinical safety and pharmacokinetics assessments in drug development. In Briefings in Bioinformatics (Vol. 22, Issue 3). Oxford University Press. https://doi.org/10.1093/bib/bbaa160. [DOI] [PubMed]
- Inamdar, A., & Suryawanshi, S. S. (2023). Physicochemical and Pharmacokinetic Properties’ Screening of Selected Cardiovascular Agents: an in-silico Approach. https://doi.org/10.21203/rs.3.rs-2653667/v1.
- Jayaraj J.M., Jothimani M., Palanisamy C.P., Pentikäinen O.T., Pannipara M., Al-Sehemi A.G., Muthusamy K., Gopinath K., Aruni W. Computational Study on the Inhibitory Effect of Natural Compounds against the SARS-CoV-2 Proteins. Bioinorg. Chem. Appl. 2022;2022:1–19. doi: 10.1155/2022/8635054. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kaur, A., & Bhatti, R. (2021). Understanding the phytochemistry and molecular insights to the pharmacology of Angelica archangelica L. (garden angelica) and its bioactive components. In Phytotherapy Research (Vol. 35, Issue 11, pp. 5961–5979). John Wiley and Sons Ltd. https://doi.org/10.1002/ptr.7206. [DOI] [PubMed]
- Keserü G.M., Makara G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009;8(3):203–212. doi: 10.1038/nrd2796. [DOI] [PubMed] [Google Scholar]
- Kim, M. S., Batrakova, E., Huang, L., Kabanov, A., & O’connor-Semmes, R. (2016). EXOSOME MEDIATED DELIVERY OF PACLITAXEL FOR THE TREATMENT OF MULTI DRUG RESISTANT PULMONARY METASTASES.
- Komolafe K., Komolafe T.R., Fatoki T.H., Akinmoladun A.C., Brai B.I.C., Olaleye M.T., Akindahunsi A.A. Coronavirus Disease 2019 and Herbal Therapy: Pertinent Issues Relating to Toxicity and Standardization of Phytopharmaceuticals. Rev. Bras. Farmacogn. 2021;31(2):142–161. doi: 10.1007/s43450-021-00132-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothandan R., Rajan C.A.S.G., Arjun J., Raj R.R.M., Syed S. Virtual screening of phytochemical compounds as potential inhibitors against SARS-CoV-2 infection. Beni-Suef University Journal of Basic and Applied Sciences. 2021;10(1) doi: 10.1186/s43088-021-00095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Choudhir G., Shukla S.K., Sharma M., Tyagi P., Bhushan A., Rathore M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2021;39(10):3760–3770. doi: 10.1080/07391102.2020.1772112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, Y. S., Adukondalu, D., Sathish, D., Vishnu, Y. V., Ramesh, G., Latha, A. B., Reddy, P. C., Sarangapani, M., & Rao, Y. M. (2010). P-Glycoprotein- and cytochrome P-450-mediated herbal drug interactions. In Drug Metabolism and Drug Interactions (Vol. 25, Issue 1, pp. 3–16). https://doi.org/10.1515/DMDI.2010.006. [DOI] [PubMed]
- Kumer A., Chakma U., Rana M.M., Chandro A., Akash S., Elseehy M.M., Albogami S., El-Shehawi A.M. Investigation of the New Inhibitors by Sulfadiazine and Modified Derivatives of α-D-glucopyranoside for White Spot Syndrome Virus Disease of Shrimp by In Silico: Quantum Calculations, Molecular Docking, ADMET and Molecular Dynamics Study. Molecules. 2022;27(12) doi: 10.3390/molecules27123694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P.D., Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007;6(11):881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Lima A.N., Philot E.A., Trossini G.H.G., Scott L.P.B., Maltarollo V.G., Honorio K.M. Vol. 11(3. Taylor and Francis Ltd.; 2016. Use of machine learning approaches for novel drug discovery; pp. 225–239. (Expert Opinion on Drug Discovery). [DOI] [PubMed] [Google Scholar]
- Liu C., Liu R., Fan H., Xiao X., Chen X., Xu H., Lin Y. Network Pharmacology Bridges Traditional Application and Modern Development of Traditional Chinese Medicine. Chinese Herbal Medicines. 2015;7(1):3–17. doi: 10.1016/s1674-6384(15)60014-4. [DOI] [Google Scholar]
- Maharjan R.S., Singh A.V., Hanif J., Rosenkranz D., Haidar R., Shelar A., Singh S.P., Dey A., Patil R., Zamboni P., Laux P., Luch A. Investigation of the Associations between a Nanomaterial’s Microrheology and Toxicology. ACS Omega. 2022;7(16):13985–13997. doi: 10.1021/acsomega.2c00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud, S., Paul, G. K., Biswas, S., Kazi, T., Mahbub, S., Mita, M. A., Afrose, S., Islam, A., Ahaduzzaman, S., Hasan, R., Shimu, S. S., Promi, M. M., Shehab, M. N., Rahman, E., Sujon, K. M., Alom, W., Modak, A., Zaman, S., Uddin, S., … Saleh, A. (2022). phytochemdb: a platform for virtual screening and computer-aided drug designing. Database, 2022(2022). https://doi.org/10.1093/database/baac002. [DOI] [PMC free article] [PubMed]
- Majid M., Farhan A., Baig M.W., Khan M.T., Kamal Y., Hassan S.S.u., Bungau S., Haq I.-u. Ameliorative Effect of Structurally Divergent Oleanane Triterpenoid, 3-Epifriedelinol from Ipomoea batatas against BPA-Induced Gonadotoxicity by Targeting PARP and NF-κB Signaling in Rats. Molecules. 2023;28(1):290. doi: 10.3390/molecules28010290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurya D.K., Sharma D. Evaluation of traditional ayurvedic Kadha for prevention and management of the novel Coronavirus (SARS-CoV-2) using in silico approach. J. Biomol. Struct. Dyn. 2022;40(9):3949–3964. doi: 10.1080/07391102.2020.1852119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra, A., Shahiwala, A., & Shah, S. P. (n.d.). Drug Delivery To The Central Nervous System: A Review. www.ualberta.ca/∼csps. [PubMed]
- Mohamed, A., Tharik, S., & Meyyanathan, S. N. (n.d.). Swiss ADME predictions for anti cancer drug molecules prior In Vitro In Vivo Correlations (IVIVC). http://xisdxjxsu.asia.
- Nadin A., Hattotuwagama C., Churcher I. Lead-Oriented Synthesis: A New Opportunity for Synthetic Chemistry. Angew. Chem. Int. Ed. 2012;51(5):1114–1122. doi: 10.1002/anie.201105840. [DOI] [PubMed] [Google Scholar]
- Nasim N., Sandeep I.S., Mohanty S. Vol. 65(3. Springer; 2022. Plant-derived natural products for drug discovery: current approaches and prospects; pp. 399–411. (Nucleus (India)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrin S., Watson C.J.W., Perez-Paramo Y.X., Lazarus P. Cannabinoid Metabolites as Inhibitors of Major Hepatic CYP450 Enzymes, with Implications for Cannabis-Drug Interactions. Drug Metab. Dispos. 2021;49(12):1070–1080. doi: 10.1124/DMD.121.000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngbolua J.-P.-K.-T.-N., Kilembe J.T., Matondo A., Ashande C.M., Mukiza J., Nzanzu C.M., Ruphin F.P., Baholy R., Mpiana P.T., Mudogo V. In silico studies on the interaction of four cytotoxic compounds with angiogenesis target protein HIF-1α and human androgen receptor and their ADMET properties. Bulletin of the National Research Centre. 2022;46(1) doi: 10.1186/s42269-022-00793-1. [DOI] [Google Scholar]
- Olasupo S.B., Uzairu A., Shallangwa G., Uba S. QSAR modeling, molecular docking and ADMET/pharmacokinetic studies: a chemometrics approach to search for novel inhibitors of norepinephrine transporter as potent antipsychotic drugs. J. Iran. Chem. Soc. 2020;17(8):1953–1966. doi: 10.1007/s13738-020-01902-5. [DOI] [Google Scholar]
- Prajapati S.K., Malaiya A., Mishra G., Jain D., Kesharwani P., Mody N., Ahmadi A., Paliwal R., Jain A. An exhaustive comprehension of the role of herbal medicines in Pre- and Post-COVID manifestations. J. Ethnopharmacol. 2022;296:115420. doi: 10.1016/j.jep.2022.115420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P., Kumari N., Gayathiri E., Selvam K., Ragunathan M.G., Chandrasekaran M., Al-Dosary M.A., Hatamleh A.A., Nadda A.K., Kumar M. In vitro and in silico toxicological properties of natural antioxidant therapeutic agent azima tetracantha. lam. Antioxidants. 2021;10(8) doi: 10.3390/antiox10081307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pranskuniene Z., Balciunaite R., Simaitiene Z., Bernatoniene J. Herbal Medicine Uses for Respiratory System Disorders and Possible Trends in New Herbal Medicinal Recipes during COVID-19 in Pasvalys District, Lithuania. Int. J. Environ. Res. Public Health. 2022;19(15) doi: 10.3390/ijerph19158905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalakshmi R., Lalitha P., Sharma S.C., Rajiv A., Chithambharan A., Ponnusamy A. In silico studies: Physicochemical properties, drug score, toxicity predictions and molecular docking of organosulphur compounds against Diabetes mellitus. J. Mol. Recognit. 2021;34(11) doi: 10.1002/jmr.2925. [DOI] [PubMed] [Google Scholar]
- Rudrapal M., Gogoi N., Chetia D., Khan J., Banwas S., Alshehri B., Alaidarous M.A., Laddha U.D., Khairnar S.J., Walode S.G. Repurposing of phytomedicine-derived bioactive compounds with promising anti-SARS-CoV-2 potential: Molecular docking, MD simulation and drug-likeness/ADMET studies. Saudi Journal of Biological Sciences. 2022;29(4):2432–2446. doi: 10.1016/j.sjbs.2021.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şahin S., Dege N. A newly synthesized small molecule: the evaluation against Alzheimer’s Disease by in silico drug design and computational structure analysis methods. J. Mol. Struct. 2021;1236:130337. [Google Scholar]
- Schneider N., Jäckels C., Andres C., Hutter M.C. Gradual in silico filtering for druglike substances. J. Chem. Inf. Model. 2008;48(3):613–628. doi: 10.1021/ci700351y. [DOI] [PubMed] [Google Scholar]
- Settimo L., Bellman K., Knegtel R.M.A. Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm. Res. 2014;31(4):1082–1095. doi: 10.1007/s11095-013-1232-z. [DOI] [PubMed] [Google Scholar]
- Shanmugavelan R., Mohamed Musthafa M. Pharmacokinetics and Molecular Docking Study of Siddha Polyherbal Preparation Shailam Against COVID-19 Mutated s Gene. Tropical Journal of Natural Product Research. 2022;6(4):502–513. doi: 10.26538/tjnpr/v6i4.8. [DOI] [Google Scholar]
- Siddiqui S., Upadhyay S., Ahmad R., Barkat M.A., Jamal A., Alothaim A.S., Hassan M.Z., Rahman M.A., Arshad M., Ahamad T., Khan M.F., Shankar H., Ali M., Kaleem S., Ahmad J. Interaction of Bioactive Compounds of Moringa oleifera Leaves with SARS-CoV-2 Proteins to Combat COVID-19 Pathogenesis: a Phytochemical and In Silico Analysis. Appl. Biochem. Biotechnol. 2022;194(12):5918–5944. doi: 10.1007/s12010-022-04040-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.V., Jahnke T., Kishore V., Park B.W., Batuwangala M., Bill J., Sitti M. Cancer cells biomineralize ionic gold into nanoparticles-microplates via secreting defense proteins with specific gold-binding peptides. Acta Biomater. 2018;71:61–71. doi: 10.1016/j.actbio.2018.02.022. [DOI] [PubMed] [Google Scholar]
- Singh A.V., Jahnke T., Wang S., Xiao Y., Alapan Y., Kharratian S., Onbasli M.C., Kozielski K., David H., Richter G., Bill J., Laux P., Luch A., Sitti M. Anisotropic Gold Nanostructures: Optimization via in Silico Modeling for Hyperthermia. ACS Applied Nano Materials. 2018;1(11):6205–6216. doi: 10.1021/acsanm.8b01406. [DOI] [Google Scholar]
- Singh A.V., Ansari M.H.D., Rosenkranz D., Maharjan R.S., Kriegel F.L., Gandhi K., Kanase A., Singh R., Laux P., Luch A. Vol. 9, Issue 17. Wiley-VCH Verlag; 2020. Artificial Intelligence and Machine Learning in Computational Nanotoxicology: Unlocking and Empowering Nanomedicine. (Advanced Healthcare Materials). [DOI] [PubMed] [Google Scholar]
- Singh A.V., Chandrasekar V., Janapareddy P., Mathews D.E., Laux P., Luch A., Yang Y., Garcia-Canibano B., Balakrishnan S., Abinahed J., Al Ansari A., Dakua S.P., Chemical A. Emerging Application of Nanorobotics and Artificial Intelligence to Cross the BBB: Advances in Design, Controlled Maneuvering, and Targeting of the Barriers. In ACS Chemical Neuroscience. Society. 2021;Vol. 12(11:1835–1853. doi: 10.1021/acschemneuro.1c00087. [DOI] [PubMed] [Google Scholar]
- Singh A.V., Maharjan R.S., Kanase A., Siewert K., Rosenkranz D., Singh R., Laux P., Luch A. Machine-Learning-Based Approach to Decode the Influence of Nanomaterial Properties on Their Interaction with Cells. ACS Appl. Mater. Interfaces. 2021;13(1):1943–1955. doi: 10.1021/acsami.0c18470. [DOI] [PubMed] [Google Scholar]
- Singh A.V., Maharjan R.S., Kromer C., Laux P., Luch A., Vats T., Chandrasekar V., Dakua S.P., Park B.-W. Advances in Smoking Related In Vitro Inhalation Toxicology: A Perspective Case of Challenges and Opportunities from Progresses in Lung-on-Chip Technologies. Chem. Res. Toxicol. 2021;34(9):1984–2002. doi: 10.1021/acs.chemrestox.1c00219. [DOI] [PubMed] [Google Scholar]
- Singh A.V., Chandrasekar V., Laux P., Luch A., Dakua S.P., Zamboni P., Shelar A., Yang Y., Pandit V., Tisato V., Gemmati D. Micropatterned Neurovascular Interface to Mimic the Blood-Brain Barrier’s Neurophysiology and Micromechanical Function: A BBB-on-CHIP Model. Cells. 2022;11(18) doi: 10.3390/cells11182801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.V., Kayal A., Malik A., Maharjan R.S., Dietrich P., Thissen A., Siewert K., Curato C., Pande K., Prahlad D., Kulkarni N., Laux P., Luch A. Interfacial Water in the SARS Spike Protein: Investigating the Interaction with Human ACE2 Receptor and In Vitro Uptake in A549 Cells. Langmuir. 2022;38(26):7976–7988. doi: 10.1021/acs.langmuir.2c00671. [DOI] [PubMed] [Google Scholar]
- Singh A.V., Bansod G., Mahajan M., Dietrich P., Singh S.P., Rav K., Thissen A., Bharde A.M., Rothenstein D., Kulkarni S., Bill J. Digital Transformation in Toxicology: Improving Communication and Efficiency in Risk Assessment. ACS Omega. 2023;8(24):21377–21390. doi: 10.1021/acsomega.3c00596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.V., Katz A., Maharjan R.S., Gadicherla A.K., Richter M.H., Heyda J., del Pino P., Laux P., Luch A. Coronavirus-mimicking nanoparticles (CorNPs) in artificial saliva droplets and nanoaerosols: Influence of shape and environmental factors on particokinetics/particle aerodynamics. Sci. Total Environ. 2023;860:160503. doi: 10.1016/j.scitotenv.2022.160503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sravika N., Priya S., Divya N., Jyotsna P.M.S., Anusha P., Kudumula N., Bai S.A. Swiss ADME properties screening of the phytochemical compounds present in Bauhinia acuminata. Journal of Pharmacognosy and Phytochemistry. 2021;10(4):411–419. doi: 10.22271/phyto.2021.v10.i4e.14193. [DOI] [Google Scholar]
- Suenderhauf, C., Hammann, F., & Huwyler, J. (2012). Computational prediction of blood-brain barrier permeability using decision tree induction. Molecules, 17(9), 10429–10445. terstappen2001. (n.d.). [DOI] [PMC free article] [PubMed]
- Tripathi, D., Ray, P., Singh, A. V., Kishore, V., & Singh, S. L. (2023). Durability of Slippery Liquid-Infused Surfaces: Challenges and Advances. Coatings, 13(6), 1095. https://doi.org/10.3390/coatings13061095.
- Tsume Y., Langguth P., Garcia-Arieta A., Amidon G.L. In silico prediction of drug dissolution and absorption with variation in intestinal pH for BCS class II weak acid drugs: Ibuprofen and ketoprofen. Biopharm. Drug Dispos. 2012;33(7):366–377. doi: 10.1002/bdd.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umadevi P., Manivannan S., Fayad A.M., Shelvy S. In silico analysis of phytochemicals as potential inhibitors of proteases involved in SARS-CoV-2 infection. J. Biomol. Struct. Dyn. 2022;40(11):5053–5059. doi: 10.1080/07391102.2020.1866669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandecruys R., Peeters J., Verreck G., Brewster M.E. Use of a screening method to determine excipients which optimize the extent and stability of supersaturated drug solutions and application of this system to solid formulation design. Int. J. Pharm. 2007;342(1–2):168–175. doi: 10.1016/j.ijpharm.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Venkatachalam A., Parashar A., Manoj K.M. Functioning of drug-metabolizing microsomal cytochrome P450s: In silico probing of proteins suggests that the distal heme ‘active site’ pocket plays a relatively ‘passive role’ in some enzyme-substrate interactions. In Silico. Pharmacology. 2016;4(1) doi: 10.1186/s40203-016-0016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas T.K., Shah L., Amiji M.M. Nanoparticulate drug carriers for delivery of HIV/AIDS therapy to viral reservoir sites. Expert Opin. Drug Deliv. 2006;3(5):613–628. doi: 10.1517/17425247.3.5.613. [DOI] [PubMed] [Google Scholar]
- Xiong G., Wu Z., Yi J., Fu L., Yang Z., Hsieh C., Yin M., Zeng X., Wu C., Lu A., Chen X., Hou T., Cao D. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49(W1):W5–W14. doi: 10.1093/nar/gkab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.-Y., He J.-H., Lu A.-P., Hou T.-J., Cao D.-S. Application of Negative Design To Design a More Desirable Virtual Screening Library. J. Med. Chem. 2020;63(9):4411–4429. doi: 10.1021/acs.jmedchem.9b01476. [DOI] [PubMed] [Google Scholar]
- Zhang D.-H., Wu K.-L., Zhang X., Deng S.-Q., Peng B. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. Journal of Integrative Medicine. 2020;18(2):152–158. doi: 10.1016/j.joim.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Gao Y., Jiang W., Huang M., Xu A., Paxton J.W. Interactions of Herbs with Cytochrome P450. Drug Metab. Rev. 2003;35(1):35–98. doi: 10.1081/dmr-120018248. [DOI] [PubMed] [Google Scholar]
Further reading
- Verma S. Therapeutic uses of Withania somnifera (Ashwagandha) with a note on withanolides and its pharmacological actions. Asian J. Pharm. Clin. Res. 2011;4:1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.