

SUMMARY
Faithful DNA replication requires that cells fine-tune their histone pool in coordination with cell-cycle progression. Replication-dependent histone biosynthesis is initiated at a low level upon cell-cycle commitment, followed by a burst at the G1/S transition, but it remains unclear how exactly the cell regulates this burst in histone biosynthesis as DNA replication begins. Here, we use single-cell time-lapse imaging to elucidate the mechanisms by which cells modulate histone production during different phases of the cell cycle. We find that CDK2-mediated phosphorylation of NPAT at the restriction point triggers histone transcription, which results in a burst of histone mRNA precisely at the G1/S phase boundary. Excess soluble histone protein further modulates histone abundance by promoting the degradation of histone mRNA for the duration of S phase. Thus, cells regulate their histone production in strict coordination with cell-cycle progression by two distinct mechanisms acting in concert.
Graphical abstract
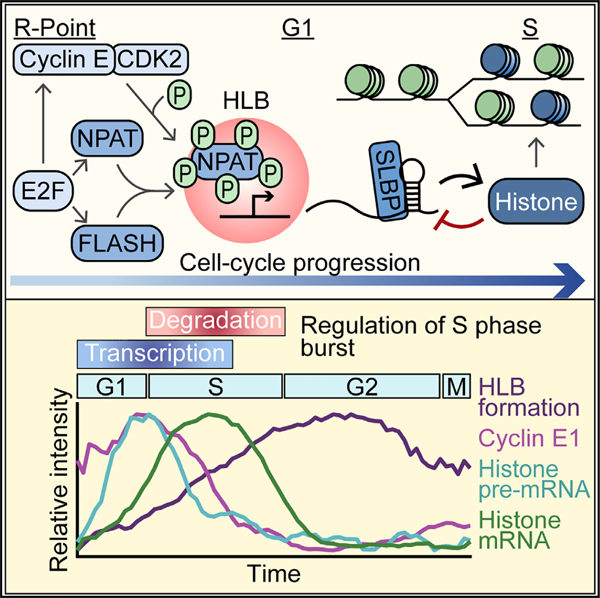
In brief
Histone biosynthesis is coordinated with DNA replication. Synthesis begins at the restriction point and is followed by a burst at the G1/S transition. Armstrong et al. show that CDK2-triggered transcription of histone genes and histone mRNA degradation mediated by unincorporated histones act in tandem to regulate histone levels.
INTRODUCTION
Maintaining genomic stability during DNA replication requires that cells double their histone proteins in coordination with the doubling of the genome beginning at the start of S phase when DNA replication is initiated. Cells must rapidly produce approximately 400 million replication-dependent histone proteins, herein referred to as histone proteins, in as little as 8 h to accommodate genome doubling in S phase.1 Our recent work established a temporal decoupling of the initiation of low levels of histone biosynthesis at cell-cycle commitment and a burst of histone production 5 h later at the G1/S boundary.2 Thus, new questions have arisen as to how cells regulate this transition from low to high levels of histone biosynthesis at the start of S phase.
Metazoans have evolved a tightly regulated system for histone biosynthesis. In humans, histone genes are found in two evolutionarily conserved clusters at chromosomes 1 (HIST2) and 6 (HIST1) around which a nuclear body forms, known as the histone locus body (HLB).1,3,4 HLB formation is dependent on the scaffolding protein and histone transcription factor nuclear protein at the ATM locus (NPAT).3 Several other HLB-specific factors are recruited upon HLB formation, including FLICE-associated huge protein (FLASH), which then recruits U7 small nuclear RNA (snRNA)-associated Sm-like protein (Lsm11) for 3′ end processing of histone mRNA.3,5–8 Once transcribed, histone mRNA is not polyadenylated at the 3′ end but instead ends in a 3′ stem loop that requires binding to the stem loop binding protein (SLBP) for stabilization (Figure 1A).9–11 In the absence of SLBP, histone mRNA will be rapidly degraded by histone-specific exonuclease 3′hExo.9,12–16
Figure 1. HLB factors are recruited in G1 and retained through the cell cycle.
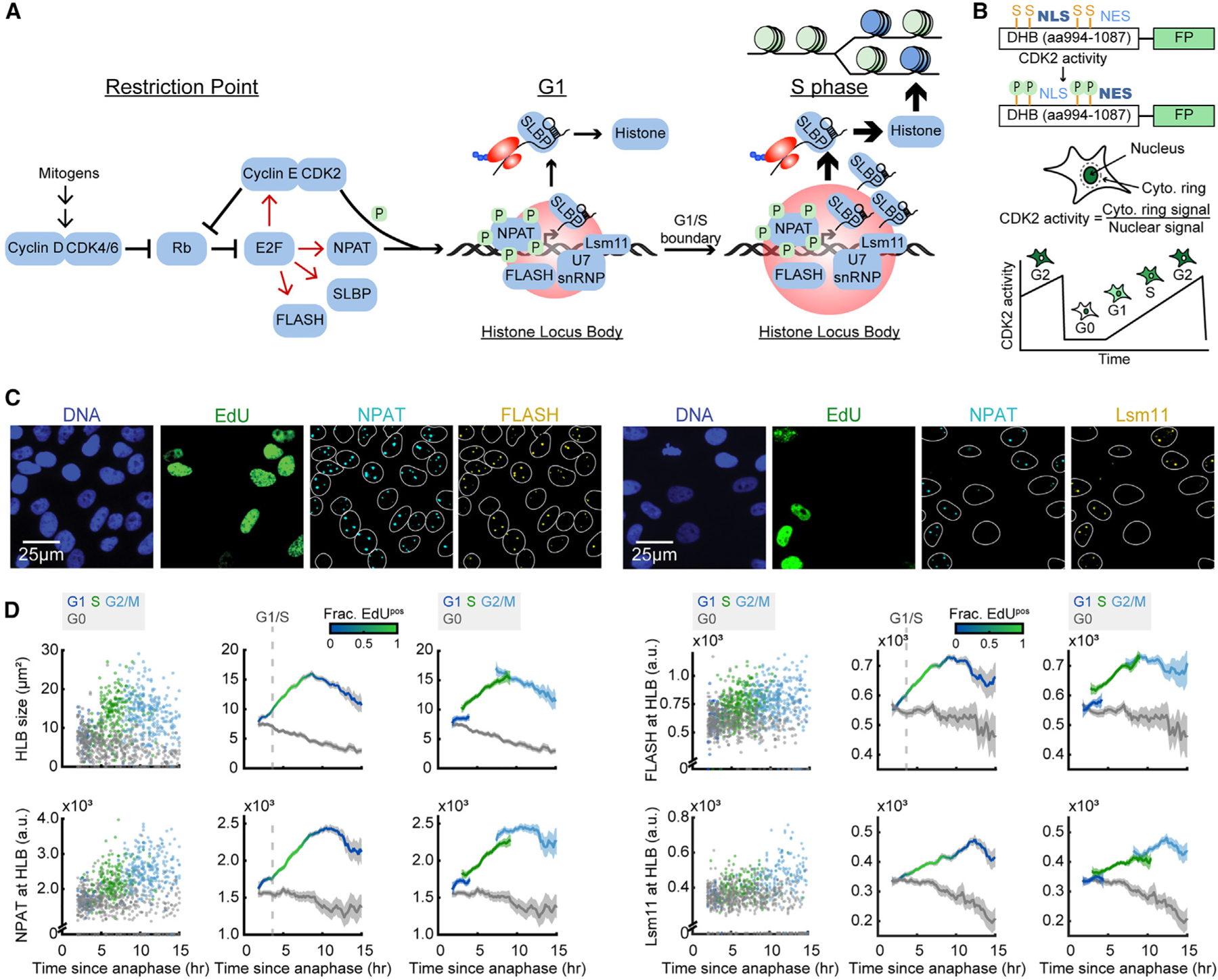
(A) Schematic of histone biosynthesis relative to the restriction point and cell-cycle progression.
(B) Schematic of the CDK2 sensor and live-cell tracking.17
(C) Representative images of cells following time-lapse imaging, stained for DNA content, EdU, NPAT, and either FLASH or Lsm11.
(D) Column 1: raw single-cell data by cell-cycle phase. CDK2 activity was tracked using time-lapse microscopy followed immediately by cell staining of individual cells’ immunofluorescent signal at the HLB as a function of time since anaphase. Cells are color-coded according to whether they are in G0 (CDK2low cells), G1 (CDK2inc, 2N, and EdUneg), S (CDK2inc and EdUpos), or G2/M (CDK2inc, 4N, and EdUneg) during the staining immediately following the last frame of the movie. Column 2: average of each cell’s immunofluorescence signal and 95% confidence intervals as a function of time since anaphase for CDK2inc and CDK2low populations. The G1/S transition is marked as the time when 50% of cells are EdUpos (dashed line). Column 3: CDK2inc population from column 2 segmented into G1, S, and G2/M (see also Figure S1).
Information on biological and technical replicates can be found in Table S2.
Several key factors in histone biosynthesis, including cyclin E, NPAT, FLASH, and SLBP, are transcriptional targets of cell-cycle master transcription factor E2F (Figure 1A).18–20 In quiescent cells, E2F is bound and inhibited by the retinoblastoma protein Rb. Hyper-phosphorylation of Rb by cell-cycle master kinase cyclin-dependent kinase 2 (CDK2)/cyclin E at the point of cell-cycle commitment known as the restriction point causes the release and activation of E2F (Figure 1A).21–23 Cyclin E/CDK2 is also known to phosphorylate NPAT, and this phosphorylation is required for transcriptional activation of histone genes at the HLB (Figure 1A).19,24–27 We previously showed that in unperturbed asynchronously cycling cells, a majority of cells will be born with elevated CDK2 activity having already crossed the restriction point.17,28 We further demonstrated that in these cells born committed to the cell cycle, histone biosynthesis occurs in two phases, with low levels beginning at the restriction point and lasting throughout G1, followed by a burst of histone mRNA biosynthesis at the G1/S boundary.2 However, it remains unclear if cyclin E/CDK2 triggers the low level of histone transcription at the restriction point, if it triggers the burst at the start of S phase, or if the burst of histone production is triggered by stabilization of histone mRNA.
Once transcribed, histone mRNA is subject to secondary regulation via the degradation of mature histone mRNA through destabilization of the interaction between SLBP and the 3′ stem loop.9,11 It has been previously shown that histone mRNA will be rapidly degraded in response to inhibition of DNA synthesis, in part because of the increase in non-nucleosomal histone protein, herein referred to as soluble histone protein, that has not been incorporated into chromatin.29,30 Soluble histone protein must remain bound to histone chaperones from the time it is made until it is incorporated in the chromatin, or it will be subject to degradation.31–33 One such histone chaperone is nuclear autoantigenic sperm protein (NASP), which protects the available soluble histone H1, H3, and H4 pool from degradation.34–36 It remains unclear if soluble histone protein regulates histone mRNA stability in naturally cycling cells, or if this mechanism occurs only in response to DNA synthesis inhibition. Additionally, it is unknown if soluble histone protein’s ability to promote histone mRNA degradation is exclusive to S phase, or if the burst of histone mRNA at the G1/S boundary is due in part to excess soluble histones being incorporated into the chromatin as DNA replication begins, thereby reducing the pool of soluble histones available to promote histone mRNA degradation.
In this study, we use single-cell time-lapse imaging of asynchronously cycling non-transformed human MCF10A cells to determine the regulation of HLB formation, transcriptional activation of histone genes, and histone mRNA stabilization across the cell cycle. MCF10As were chosen because of their normal cell cycle and ease of imaging and computational tracking. We find that HLB formation is initiated at the restriction point and is equally dependent on both NPAT and FLASH protein. Furthermore, we find that phosphorylation of NPAT by cyclin E/CDK2 at the restriction point triggers the initiation of histone transcription, which leads to the burst of histone mRNA at the G1/S boundary approximately 5 h later. By contrast, phosphorylation of NPAT does not alter HLB formation. Finally, our data demonstrate that as cells enter S phase, regulation of histone mRNA levels transfers from CDK2-mediated transcriptional activation to degradation of histone mRNA via negative feedback from excess soluble histone protein. Therefore, cells reach an equilibrium between overlapping mechanisms to coordinate the burst in histone biosynthesis with the start of S phase, and then fine-tune histone production in response to the rate of DNA replication.
RESULTS
HLB factors are recruited in G1 and retained through the cell cycle
To map the temporal dynamics of HLB formation within the cell cycle, we tracked individual cells using single-cell time-lapse imaging of asynchronously cycling cells, followed by post hoc staining, as has been previously described.2,37,38 We first transduced MCF10A cells with fluorescent histone 2B (H2B) as a nuclear marker, and a fluorescent fragment of DNA helicase B (DHB) as a sensor of CDK2 activity, measured by the cytoplasmic to nuclear ratio of DHB (Figure 1B).17 Time-lapse imaging, in which cells can be distinguished as proliferative (CDK2inc) or quiescent (CDK2low) on the basis of DHB translocation, was followed by paraformaldehyde fixation of the cell and photobleaching of the DHB sensor. Cells were then stained for DNA content, thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) as a marker of DNA replication, NPAT as a marker for the HLB, and either FLASH or Lsm11 protein (Figures 1A–1C). Cells were then aligned to the time of anaphase and classified as either CDK2inc or CDK2low. CDK2inc cells were further segmented by cell-cycle phase, as G1 (2N DNA content, EdUneg), S (EdUpos), or G2/M (4N DNA content, EdUneg) (STAR Methods) (see also Figures S1A–S1D).
HLB growth (as determined by the sum of the number of pixels of all the HLBs per cell, ‘‘HLB size,’’ or as the mean NPAT concentration within those HLBs, ‘‘NPAT at HLB’’) is initiated as early as 2 h after anaphase in CDK2inc cells and progresses continuously through G1 and S phase before plateauing in G2 (Figures 1C and 1D). We then quantified the mean concentration of FLASH or Lsm11 protein at the HLB (as defined by the total HLB area) to map the recruitment of these factors relative to cell-cycle phase (Figures 1C and 1D). We found that FLASH is recruited in tandem with NPAT to the HLB, while Lsm11 recruitment remains lower throughout G1, before rising steadily in S phase and plateauing in G2/M (Figure 1D). We found similar HLB formation dynamics for CDK2inc and CDK2low RPE-hTERT cells (Figures S2A and S2B). This indicates that there may be a hierarchical recruitment of processing factors, with delayed recruitment of the U7 snRNP to the HLB as a means to regulate the timing of histone mRNA production, as has been previously suggested in Drosophila.7,39 These data also indicate that although HLB formation is critical, the HLB continues to exist well past when it is needed in S phase, indicating that transcriptional activation of histone genes is also being regulated by an alternative mechanism.
NPAT and FLASH are required for HLB formation
To better quantify recruitment of Lsm11 and FLASH to the HLB, we performed high-resolution three-dimensional (3D) imaging on cells stained for DNA, NPAT, and either FLASH or Lsm11 (Figures 2A and 2B). Individual HLB puncta were segmented as 3D objects on the basis of the NPAT signal, with subsequent FLASH and Lsm11 quantification and segmentation (STAR Methods). For individual HLBs, we quantified FLASH puncta volume relative to NPAT puncta volume and found a strong linear correlation between the size of NPAT puncta and FLASH puncta within the HLB, with FLASH puncta being consistently slightly smaller than NPAT puncta (Figure 2C). This may be due to the FLASH antibody recognizing the C terminus of the protein, which has been shown to localize more strongly to the center of the HLB compared with the N terminus of the protein.40 Accurate volumetric measurements of Lsm11 HLB puncta were not possible, because of the puncta being diffraction limited (STAR Methods).
Figure 2. NPAT and FLASH are required for HLB formation.
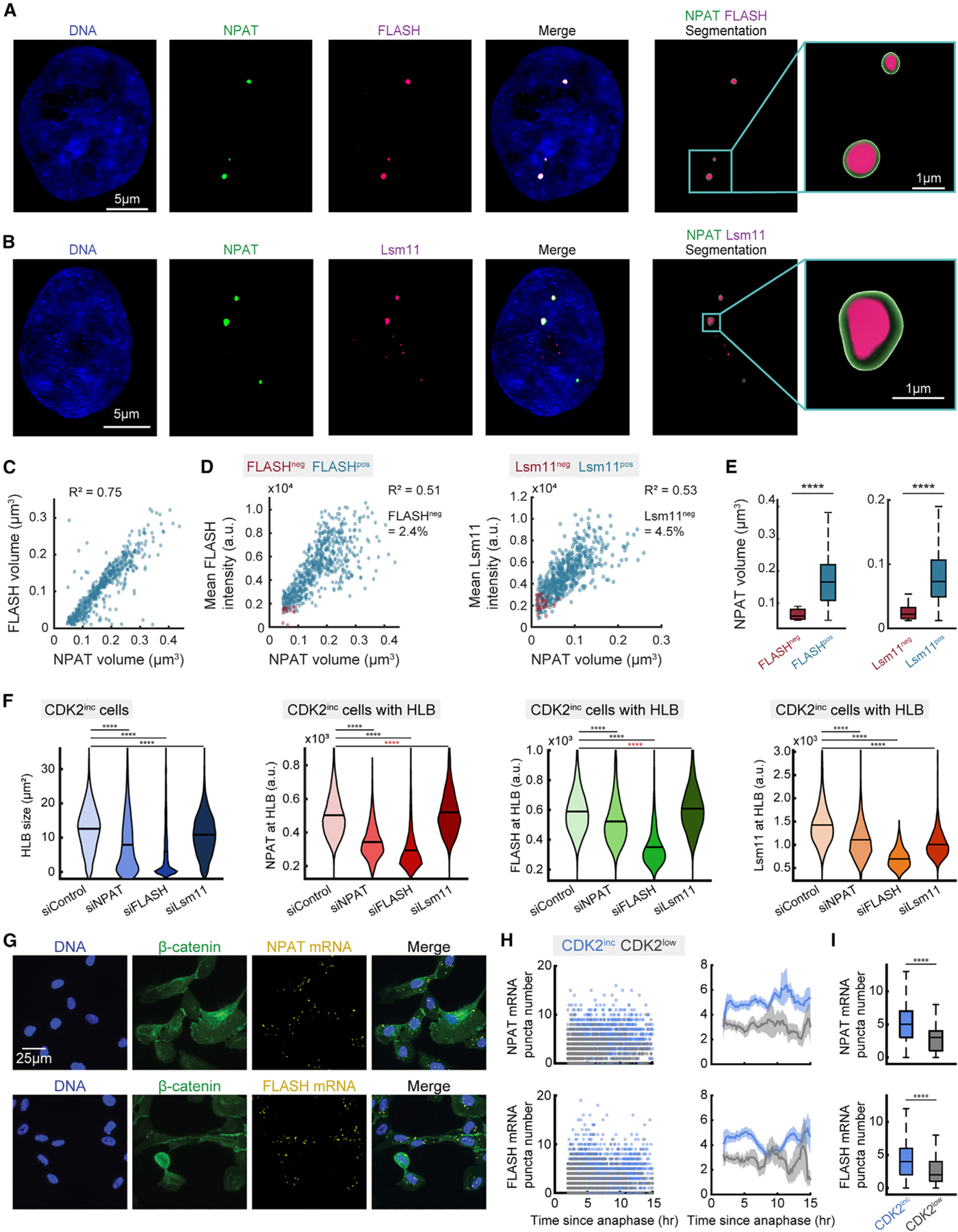
(A and B) Representative images of cells stained for DNA, NPAT, and either FLASH (A) or Lsm11 (B).
(C) Scatter of individual HLBs with FLASH volume vs. NPAT volume.
(D) Mean protein intensity for FLASH (column 1) or Lsm11 (column 2) vs. NPAT volume of HLBs, designated as positive or negative for either FLASH or Lsm11 on the basis of the detection limit for FLASH or Lsm11 puncta.
(E) Distribution of NPAT volume for populations either positive or negative for FLASH (column 1) or Lsm11 (column 2) puncta detection.
(F) Violin plots of average HLB size for all CDK2inc cells (left-most plot) and mean intensity of NPAT, FLASH, or Lsm11 at HLB for CDK2inc cells with HLB following 48 h treatment with siControl, siNPAT, siFLASH, or siLsm11.
(G) Representative images of cells following time-lapse imaging and staining for DNA, β-catenin for cytoplasmic segmentation, and either NPAT or FLASH mRNA.
(H) Column 1: raw single-cell RNA FISH data for CDK2inc and CDK2low populations. Column 2: NPAT or FLASH mRNA FISH puncta number and 95% confidence interval as a function of time since anaphase.
(I) NPAT or FLASH mRNA FISH puncta number for CDK2inc vs. CDK2low cells. p values are indicated as ****p ≤ 0.0001 from two-sample t test, with red asterisks indicating an increase in mean value.
Information on biological and technical replicates can be found in Table S2.
As the Lsm11 puncta were too small to accurately quantify their volume, we measured FLASH and Lsm11 recruitment to the HLB as mean protein intensity for FLASH or Lsm11 within the HLB, relative to NPAT puncta volume (Figure 2C). To confirm that this was an accurate means of measuring HLB recruitment, we also quantified the mean NPAT intensity relative to NPAT volume, showing a strong correlation (Figure S1H). We further categorized HLB puncta as those with (FLASHpos/Lsm11pos) or without (FLASHneg/Lsm11neg) detection for FLASH or Lsm11 protein (Figure 2D) (STAR Methods). Localization of both FLASH and Lsm11 protein to the HLB correlates linearly with HLB growth, with only a small percentage of HLBs lacking either FLASH (2.4%) or Lsm11 (4.5%) protein (Figures 2D and 2E). Lack of Lsm11 or FLASH occurred in the smallest HLBs, indicating that HLB factors are recruited after HLB formation has been initiated (Figure 2E). It is unclear if FLASH and Lsm11 localize to the HLB once the condensate has reached a certain size, or if recruitment occurs later in the cell cycle after HLB growth has already been initiated.
To determine the dependency of HLB formation on each of these essential HLB proteins, we performed a 48 h small interfering RNA (siRNA) knockdown of NPAT, FLASH, or Lsm11, with live-cell imaging in the final 18 h before fixation and HLB staining (Figure 2F; see also Figures S1E and S1F). We found that knockdown of either NPAT or FLASH significantly reduced HLB formation, with each protein being unable to localize to the HLB without the other. By contrast, knockdown of Lsm11 caused only a small reduction in HLB size and no reduction in protein concentrations of NPAT or FLASH (Figure 2F).
In cells that entered a CDK2low quiescent state following mitosis, the HLB reforms initially and then dissipates over time (Figure S1G). Protein concentrations within the HLB in CDK2low cells stayed fairly consistent over time for NPAT, FLASH, and Lsm11, indicating that the HLB condensate retains HLB factors until it has fully dissipated (Figure S1G). Together, these data suggest that FLASH has an equally important role in seeding the HLB as NPAT and that the HLB will form in cells that have both NPAT and FLASH protein available even if that cell is newly quiescent, lacks CDK2 activity, and is not producing histone mRNA, as has been previously shown in Drosophila.8,41
Since NPAT and FLASH are both E2F target genes, we theorized that their expression would begin to rise immediately in CDK2inc cells, as hyper-phosphorylation of Rb and the release of E2F is driven by CDK2 activity (Figure 1A). Following live-cell imaging, we stained for either NPAT or FLASH mRNA by RNA fluorescence in situ hybridization (FISH) along with a cytoplasmic marker and quantified the number of FISH puncta per cell (Figure 2G). We then aligned the single cells relative to their time of anaphase in CDK2inc and CDK2low cells (Figures 2H and 2I). We found that both NPAT and FLASH expression are significantly different between CDK2inc and CDK2low cells as early as 2 h following mitosis and that this difference in mRNA levels between CDK2inc and CDK2low cells is largely sustained throughout the cell cycle (Figures 2H and 2I). Thus, we conclude that HLB formation is fundamentally driven by E2F release at the restriction point and the consequent rise in NPAT and FLASH protein. Following the crossing of the restriction point, HLB formation is dependent on NPAT and FLASH concentration.
Transcriptional activation of histone genes at the HLB is triggered by CDK2-mediated phosphorylation of NPAT
To look more directly at transcriptional activation at the HLB, we designed a probe set against nascent histone mRNA to quantify transcriptional activation of histone genes at the HLB via RNA FISH (STAR Methods; see also Table S1). As histone genes are intron-less, we designed probes against the sequence downstream of the 3′ cleavage site and prior to transcriptional termination for 50 of the histone genes found at the HIST1 and HIST2 clusters (Figure 3A; see also Table S1).4 We also developed a custom antibody against phospho-NPAT (pNPAT) T1270 on the basis of a previously published epitope.25 Cells were stained for DNA, EdU, NPAT, and either pNPAT T1270 or nascent histone RNA FISH following time-lapse imaging (Figure 3B). We normalized pNPAT T1270 and nascent histone mRNA signals to the nuclear background in cells that have HLBs, as both are HLB-specific signals (Figure 3C). Both pNPAT T1270 and nascent histone mRNA levels begin rising upon cell-cycle commitment, peak at the G1/S boundary, and fall to baseline levels at the end of S phase (Figure 3C). Unexpectedly, there is then a second rise in pNPAT T1270 at the HLB that does not correlate with histone gene transcription, beginning at G2 and ending at mitosis in MCF10A cells, which was consistent in RPE-hTERT cells (Figures S2C and S2D).
Figure 3. Transcriptional activation of histone genes at the HLB is triggered by CDK2-mediated phosphorylation of NPAT.
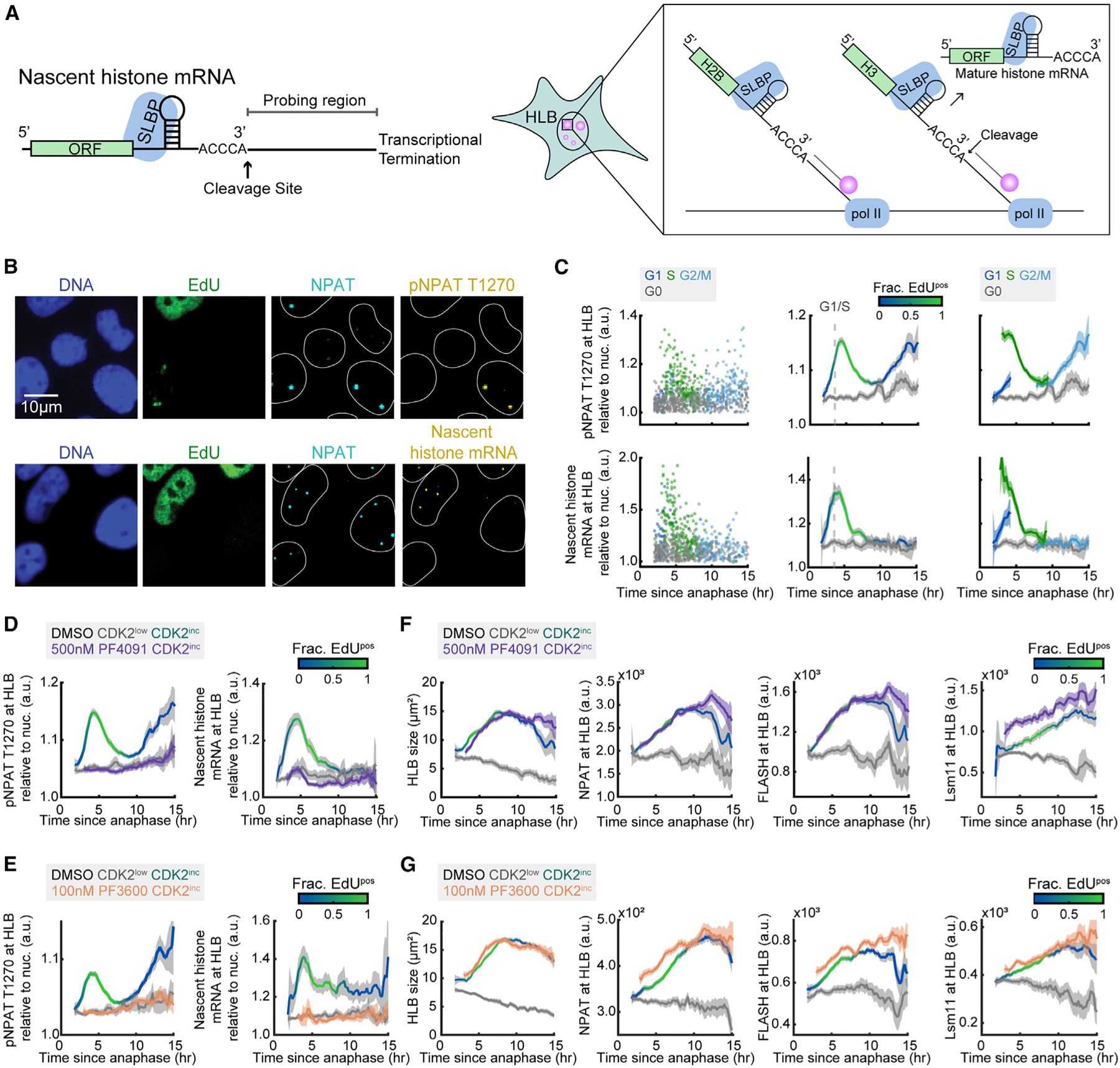
(A) Schematic of nascent histone mRNA FISH probe design (see Table S1).
(B) Representative images following time-lapse imaging of cells stained for DNA, EdU, NPAT, and either pNPAT T1270 or nascent histone mRNA FISH.
(C) Column 1: raw single-cell data by cell-cycle phase. Column 2: mean pNPAT T1270 or nascent histone mRNA at the HLB normalized to nuclear signal and 95% confidence intervals as a function of time since anaphase for CDK2inc and CDK2low populations. The G1/S transition is marked as the time when 50% of cells are EdUpos (dashed line). Column 3: CDK2inc population from column 2 segmented in to G1, S, and G2/M.
(D and E) Mean pNPAT T1270 and nascent histone mRNA FISH at the HLB normalized to nuclear signal for CDK2inc and CDK2low populations for cells treated with DMSO, and CDK2inc population for cells treated with 500 nM PF-07104091 (D) or 100 nM PF-06873600 (E) for the last 1 h of live-cell imaging.
(F and G) Mean HLB size and protein signal at the HLB with 95% confidence interval for NPAT, FLASH, and Lsm11 for CDK2inc and CDK2low populations for cells treated with DMSO, and CDK2inc population for cells treated with 500 nM PF-07104091 (F) or 100 nM PF-06873600 (G) for the last 1 h of live-cell imaging.
Information on biological and technical replicates can be found in Table S2.
To further probe the relationship between pNPAT and nascent histone mRNA, we used two selective small-molecule inhibitors of CDK2 (CDK2i), PF-07104091 (PF4091) and PF-06873600 (PF3600).42–46 Cells were treated with either 500 nM PF4091 or 100 nM PF3600 during the last hour of live-cell imaging before fixing at staining for DNA, EdU, NPAT, and either pNPAT T1270 or nascent histone RNA FISH (Figures 3D–3F; see also Figures S3A, S3B, S4A, and S4B). CDK2 inhibition was verified by the rapid nuclear translocation of the DHB sensor upon drug treatment (Figures S3G, S3H, S4G, and S4H). We found that inhibition of CDK2 led to an immediate and complete loss of both NPAT phosphorylation and nascent histone transcription (Figures 3D and 3E). To determine if this was due to the loss of HLB formation, we treated cells with either 500 nM PF4091 or 100 nM PF3600 during the last hour of live-cell imaging and stained for DNA, EdU, NPAT, and either FLASH or Lsm11 (Figures 3F and 3G; see also Figures S3C, S3D, S4C, and S4D). HLB formation was unchanged with CDK2i treatment, indicating that formation of the HLB and initiation of histone transcription are decouplable once the cells have crossed the restriction point. These effects were consistent at lower and higher doses of CDK2i (100 nM and 2 μM) (Figures S3A–S3F and S4A–S4F). Thus, we find that histone transcription is positively regulated by CDK2 phosphorylation of NPAT from the restriction point to the S/G2 transition, peaking at the G1/S boundary.
Phosphorylation of NPAT at the G1/S boundary is mediated by cyclin E1/CDK2
To gain further insight into the regulation of transcriptional activation of histone genes at the HLB via phosphorylation of NPAT by CDK2, we tracked localization of cyclin E to the HLB, since NPAT has previously been shown to be a cyclin E/CDK2-specific substrate.24,26,47 We began by determining which forms of cyclin E were accumulating in the HLB, by staining for both cyclin E1 and cyclin E2 using antibodies we validated by siRNA knockdown (Figures S5A and S5B), along with DNA, EdU, and NPAT (Figure 4A). We found that the pan-nuclear signal of both cyclin E1 and E2 peaked approximately at the G1/S phase boundary as has been previously reported.37 By contrast, cyclin E1 but not E2 continues to rise in CDK2low cells (Figures 4B and 4C), since cyclin E1 is not being actively degraded in the CDK2low state (Figures 4B and 4C).37,38 We then determined whether cyclin E1 or E2 was specifically accumulating at the HLB by taking the average signal at the HLB as marked by NPAT and normalizing it to the nuclear signal for each cyclin (Figures 4B and 4C). We find that both cyclin E1 and E2 have a pool of protein that localizes specifically to the HLB, with the highest levels in late G1 and early S (Figures 4B and 4C). This enables CDK2 to phosphorylate NPAT locally and in a cell-cycle-dependent manner. This HLB localization however, is much stronger in MCF10A than RPE-hTERT cells and is not necessarily required for NPAT phosphorylation when cyclin E is abundant in the nucleus (Figures S2E and S2F). The strong correlation in the timing of cyclin E, pNPAT T1270, and nascent histone mRNA suggests that activation and cessation of transcription at the HLB is predominantly regulated by cyclin E/CDK2 activity.
Figure 4. Phosphorylation of NPAT at the G1/S boundary is mediated by cyclin E1/CDK2.
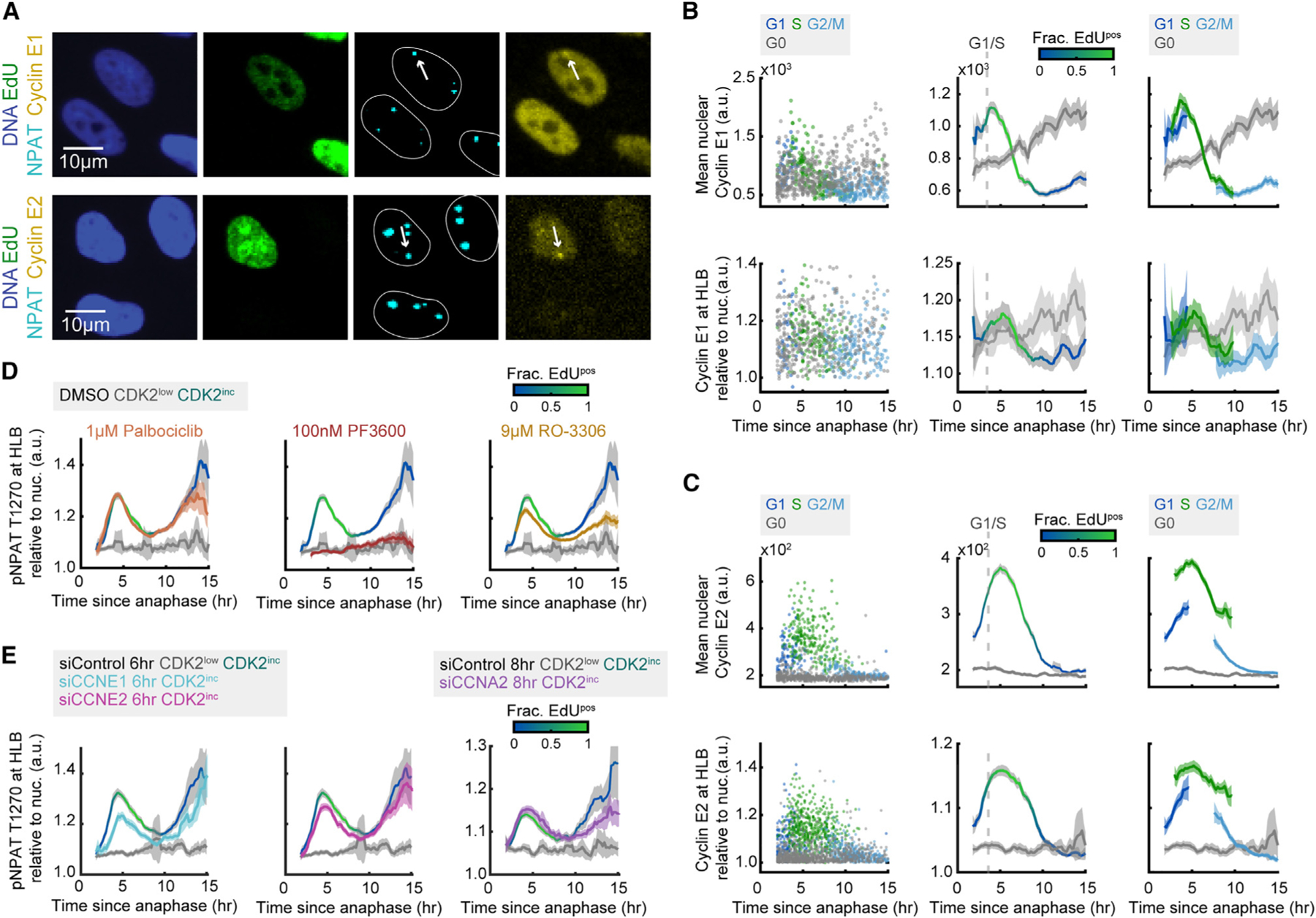
(A) Representative images following time-lapse imaging of cells stained for DNA, EdU, NPAT, and either cyclin E1 or cyclin E2.
(B and C) Column 1: raw single-cell data by cell-cycle phase for nuclear cyclin E1 or E2 (top) or cyclin E1 or E2 at the HLB (bottom). Column 2: mean nuclear signal for cyclin E1 or cyclin E2 and mean HLB signal for cyclin E1 or cyclin E2 normalized to nuclear signal and 95% confidence intervals as a function of time since anaphase for CDK2inc and CDK2low populations. The G1/S transition is marked as the time when 50% of cells are EdUpos (dashed line). Column 3: CDK2inc population from column 2 segmented in to G1, S, and G2/M.
(D) Mean HLB signal for pNPAT T1270 normalized to nuclear signal for CDK2inc and CDK2low populations for cells treated with DMSO, and CDK2inc population for cells treated with 1 μM palbociclib (CDK4/6i), 100 nM PF-06873600 (CDK2i), or 9 μM RO-3306 (CDK1i) for the last 1 h of live-cell imaging.
(E) Mean HLB signal for pNPAT T1270 normalized to nuclear signal for CDK2inc and CDK2low populations for cells treated with siControl, siCCNE1, or siCCNE2 for 6 h (left and middle), or cells treated with siControl or siCCNA2 for 8 h (right).
Information on biological and technical replicates can be found in Table S2.
We next set out to verify the specificity of NPAT as a cyclin E/CDK2 substrate, as our previous data suggests (Figures 3D and 3E). Cells were treated with 1 μM palbociclib (CDK4/6i), 100 nM PF3600 (CDK2i), or 9 μM RO-3306 (CDK1i) for the final 1 h of live-cell imaging, followed by staining for DNA, EdU, NPAT, and pNPAT T1270 (Figure 4D). Although we see no loss of pNPAT with CDK4/6i treatment as expected, we do see a partial loss in signal with CDK1 inhibition, especially in the G2 rise of NPAT phosphorylation (Figure 4D). This reduction, however, is substantially less than the complete loss of NPAT phosphorylation seen with CDK2 inhibition in both the G1/S and G2 peaks and may be due to the partial off-target inhibition of CDK2 by RO-3306 at this concentration. These data indicate that the rise in pNPAT T1270 at the G1/S boundary and in G2 is due predominantly to phosphorylation by CDK2, though CDK1 activity may also be contributing to the rise of pNPAT in G2.
To further test the specificity of NPAT to cyclin E/CDK2, we partially knocked down cyclin E1 or cyclin E2 via siRNA for the last 6 h of live-cell imaging, and cyclin A2 for the last 8 h of imaging, followed by staining for DNA, EdU, NPAT, and pNPAT T1270 (Figure 4E; see also Figures S5C and S5D). We found that the reduction of cyclin E1 had a greater effect on the pNPAT peak at G1/S than cyclin E2, despite having a less complete knockdown (Figures S5C and S5D), suggesting that phosphorylation of NPAT and corresponding transcriptional activation of histone genes at the G1/S boundary are regulated primarily by cyclin E1/CDK2, with a smaller contribution from cyclin E2 (Figure 4E). Additionally, we found that partial knockdown of cyclin A2 caused a reduction in the G2 wave of NPAT phosphorylation (Figure 4E; see also Figures S5C and S5D). These data suggest that there may be cyclin A2/CDK2-regulated phosphorylation of NPAT late in the cell cycle independent of histone gene transcription.
Histone mRNA degrades rapidly and proportionally upon inhibition of DNA synthesis
Having shown that transcription of histone genes is primarily triggered by cyclin E1/CDK2 activity upon cell-cycle commitment and that the rate of mature histone mRNA production peaks at the G1/S phase boundary, we next sought to understand if there was a secondary regulation mechanism suppressing excess histone biosynthesis in a cell-cycle-dependent manner. We began by establishing the expression dynamics of the histone mRNA stabilizing protein, SLBP, and histone H4.2 mRNA relative to the cell cycle (Figures 5A and 5B; see also Figures S5E and S5F). The results were consistent with our previously published findings that SLBP protein levels are high throughout G1 and S phase and fall at the S/G2 boundary, with elevated SLBP mRNA in CDK2inc vs. CDK2low cells (Figure 5C; see also Figures S6A–S6C).2 Histone mRNA is sustained at low to moderate levels throughout G1 before a rapid accumulation of histone mRNA at the G1/S phase boundary, coincident with EdU incorporation, as we and others showed previously (Figure 5D).2,48–51 Since histone mRNA levels fall prior to SLBP degradation (Figures 5C and 5D), these data indicate that histone mRNA stabilization must depend not only on SLBP abundance but also on an alternative mechanism more closely linking histone production with DNA synthesis in S phase.
Figure 5. Histone mRNA degrades rapidly and proportionally upon inhibition of DNA synthesis.
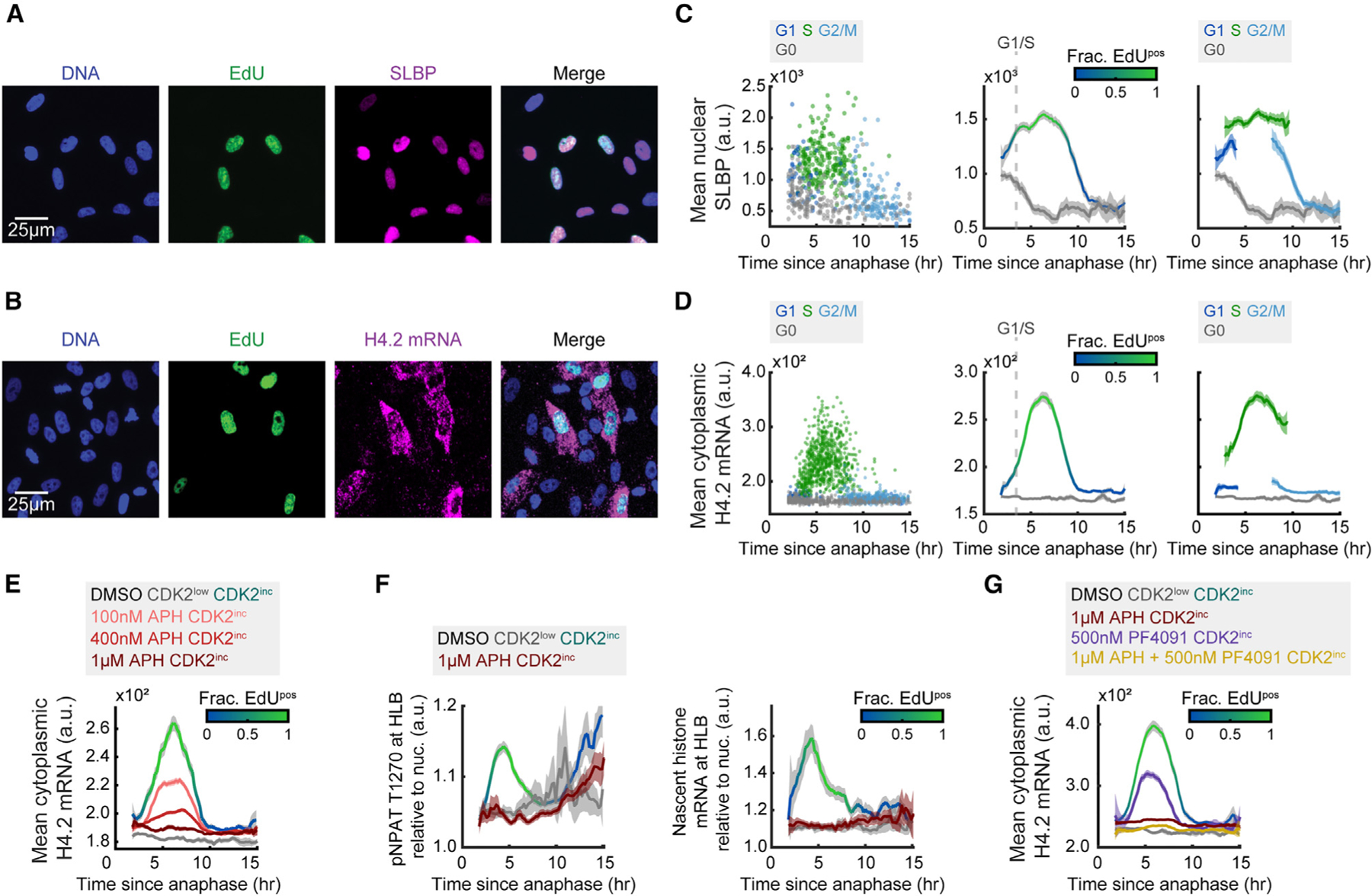
(A and B) Representative images following time-lapse imaging of cells stained for DNA, EdU, and SLBP (A) or H4.2 mRNA (B).
(C and D) Column 1: raw single-cell data by cell-cycle phase. Column 2: mean nuclear SLBP signal (C) or cytoplasmic histone H4.2 mRNA (D) and 95% confidence intervals as a function of time since anaphase for CDK2inc and CDK2low populations. The G1/S transition is marked as the time when 50% of cells are EdUpos (dashed line). Column 3: CDK2inc population from column 2 segmented in to G1, S, and G2/M.
(E) Mean cytoplasmic histone H4.2 mRNA signal for CDK2inc and CDK2low populations for cells treated with DMSO, and CDK2inc population for cells treated with 100 nM, 400 nM, or 1 μM APH for the last 1 h of live-cell imaging.
(F) Mean HLB signal for pNPAT T1270 and nascent histone mRNA FISH normalized to nuclear signal for CDK2inc and CDK2low populations for cells treated with DMSO, and CDK2inc population for cells treated with 1 μM APH for the last 1 h of live-cell imaging.
(G) Mean cytoplasmic histone H4.2 mRNA signal and 95% confidence interval for CDK2inc and CDK2low populations for cells treated with DMSO, and CDK2inc population for cells treated with 1 μM APH, 500 nM PF-07104091 (PF4091), or a combination of 1 μM APH and 500 nM PF-07104091 (PF4091) for the last 1 h of live-cell imaging.
Information on biological and technical replicates can be found in Table S2.
To test this hypothesis, we inhibited DNA synthesis by treating cells with aphidicolin (APH) at a range of doses to inhibit DNA synthesis partially or completely (100 nM, 400 nM, and 1 μM) for 1 h before the end of live-cell imaging. We then fixed and stained the cells for DNA, EdU, and cytoplasmic histone H4.2 mRNA (Figure 5E; see also Figures S6D–S6F). Histone H4.2 mRNA levels fell proportionally to the degree of DNA synthesis inhibition, regardless of the timing within S phase (Figure 5E), suggesting that cells have a mechanism by which they can immediately sense and respond to changes in the rate of DNA replication. To address if this change in cytoplasmic histone H4.2 mRNA was due to a loss of transcription or increased degradation of the mature mRNA, we treated cells with 1 μM APH for 1 h before staining for pNPAT T1270 or nascent histone mRNA FISH (Figure 5F; see also Figures S6G and S6H). We found that inhibition of DNA synthesis prevents NPAT phosphorylation as well as new histone transcription (Figure 5F; see also Figures S6G and S6H). However, when we treated cells with 500 nM PF4091, which fully inhibits nascent histone transcription (Figure 3D), cytoplasmic histone H4.2 mRNA is only partially reduced, suggesting that the dramatic loss of mature histone H4.2 mRNA with 1 μM APH treatment is due at least in part to the degradation of the mature histone mRNA (Figure 5G; see also Figures S6I–S6J).
The partial reduction in cytoplasmic histone H4.2 mRNA upon 500 nM PF4091 treatment is likely due to a decrease in DNA synthesis, as CDK2 activity is also needed for DNA replication to proceed (Figures S6I and S6J). We also noted that when we treated cells with a combination of 1 μM APH and 500 nM PF4091, cytoplasmic histone H4.2 mRNA levels fell even further than with 1 μM APH treatment alone, despite DNA synthesis being fully inhibited in both conditions (Figure 5G; see also Figures S6I and S6J). This suggests that CDK2 activity might not only activate transcription of histone genes but may potentially also play a smaller role in protecting cytoplasmic histone mRNA from degradation. By contrast, inhibition of DNA synthesis has no effect on HLB formation (Figures S7A–S7C) or SLBP levels (Figures S7D and S7E), demonstrating that the reduction of cytoplasmic histone mRNA upon APH treatment is due specifically to loss of transcription and an increase in mature histone mRNA degradation. Together, these data indicate that cells respond to changes in DNA synthesis rate in S phase both by preventing further histone gene expression and by actively degrading the cytoplasmic histone mRNA that is already present.
Soluble histone protein promotes the degradation of histone mRNA in S phase
Previous studies have suggested that soluble histone protein may promote the degradation of histone mRNA, but it is not known if this negative feedback regulates the burst of histone mRNA at the G1/S boundary (Figure 6A).30 To answer this question, we used siRNA to deplete cells of Eri1 or NASP. Knockdown of Eri1, the gene for histone mRNA-specific exonuclease 3′hExo, should prevent histone mRNA degradation, and result in an increase in histone mRNA. Partial knockdown of histone chaperone NASP provides a means to modulate the soluble histone pool of histones H1, H3, and H4.34,35 NASP is a histone chaperone that is not required for nucleosome remodeling within the chromatin but instead specifically shields the soluble histone pool from degradation, though it is unknown if NASP can act on histone mRNA levels in additional ways, such as by regulating histone transcription (Figure 6A).34,52 Knockdown of NASP should decrease the amount of soluble histone protein and thereby increase the amount of histone mRNA.
Figure 6. Soluble histone protein promotes the degradation of histone mRNA in S phase.
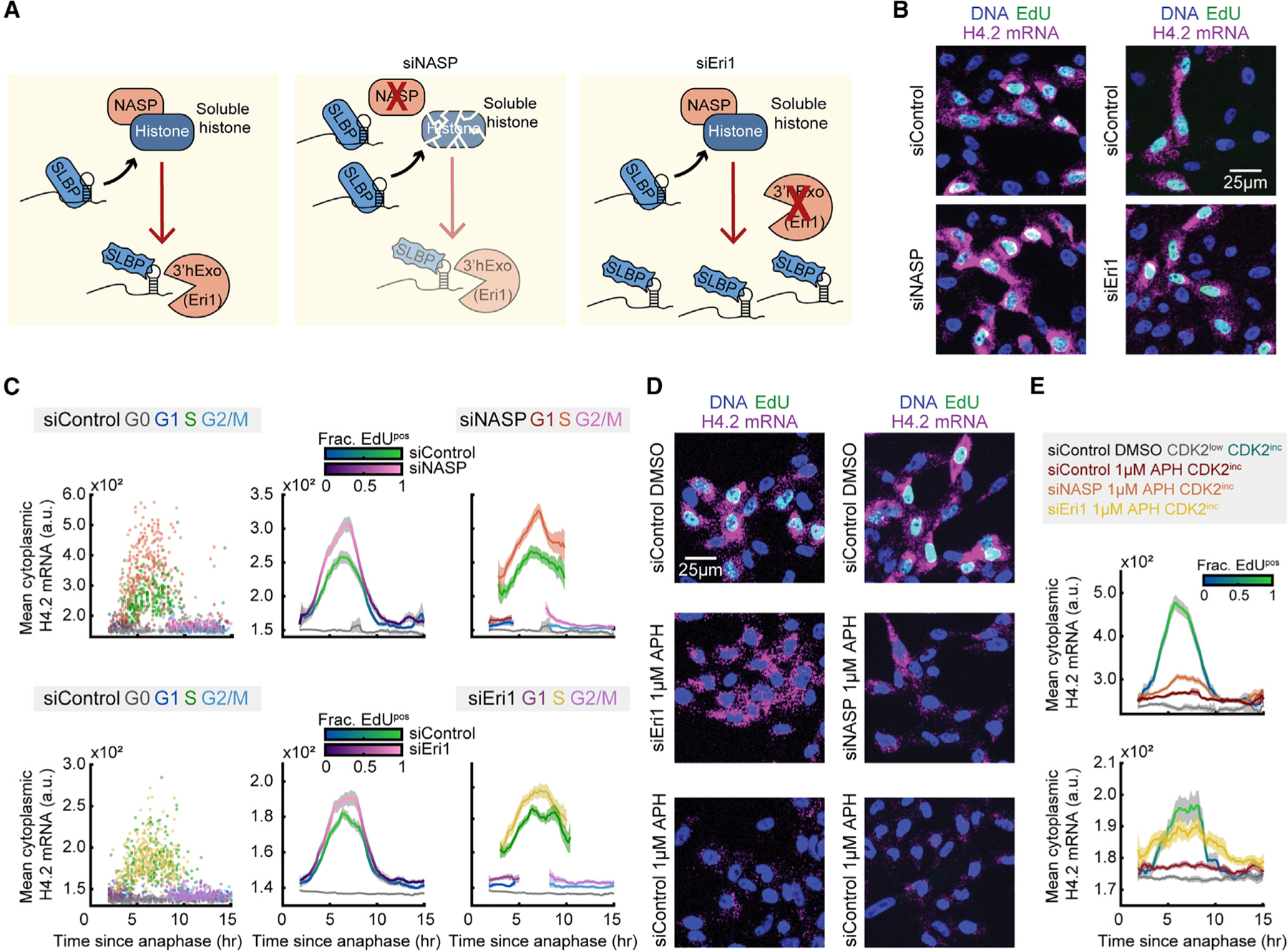
(A) Schematic of histone protein and histone mRNA stabilization with siRNA knockdown of NASP and Eri1. Soluble histone protein disrupts the interaction between SLBP and histone mRNA (squiggly SLBP), and Eri1 (3′hExo) is required for histone mRNA degradation.
(B) Representative images of cells treated with 20 nM siControl or siNASP for 24 h before fixation, or cells treated with 25 nM siControl or siEri1 for 48 h before fixation.
(C) Column 1: raw single-cell data. Column 2: mean cytoplasmic histone H4.2 mRNA signal and 95% confidence intervals as a function of time since anaphase for CDK2inc and CDK2low populations for cells treated with siControl vs. siNASP or siControl vs. siEri1. Column 3: CDK2inc population from column 2 segmented in to G1, S, and G2/M populations.
(D) Representative images of cells treated with 20 nM siControl for 24 h with 1 h of DMSO, 20 nM siControl for 24 h with 1 h of 1 μM APH, or 20 nM siNASP for 24 h with 1 h of 1 μM APH before fixation; and cells treated with 25 nM siControl for 48 h with 1 h of DMSO, 25 nM siControl for 48 h with 1 h of 1 μM APH, or 25 nM siEri1 for 48 h with 1 h of 1 μM APH before fixation.
(E) Mean histone H4.2 mRNA signal and 95% confidence interval for cells in (D), showing partial rescue of cytoplasmic histone H4.2 mRNA after APH treatment with siNASP or siEri1 knockdown.
Information on biological and technical replicates can be found in Table S2.
To directly test if soluble histone protein can promote the degradation of histone mRNA, we partially knocked down NASP via siRNA for 24 h, filming cells for the last 18 h of the knockdown period, followed by fixing and staining for DNA, EdU, and histone H4.2 mRNA (Figure 6B). Similarly, we knocked down Eri1 via siRNA for 48 h prior to fixation, with the last 18 h of the siRNA knockdown being filmed, followed by staining for DNA, EdU, and histone H4.2 mRNA (Figure 6B). The siRNA knockdown of both NASP and Eri1 were validated by immunofluorescent staining of NASP and 3′hExo (Figures S5E and S5F). We found that reduction of the soluble histone pool via partial knockdown of NASP led to higher levels of histone H4.2 mRNA in S phase specifically. Additionally, prevention of histone mRNA degradation via knockdown of Eri1 led to an increase in histone mRNA levels in S phase and a slight increase in histone mRNA levels in G1 (Figure 6C). We conclude that soluble histone protein promotes histone mRNA degradation specifically in S phase, while an alternative mechanism promotes histone mRNA degradation in G1 to a lesser degree.
To further test whether the decrease of histone mRNA upon inhibition of DNA synthesis is caused by an increase in soluble histone protein, we paired the siRNA knockdown of NASP and Eri1 with a 1 h treatment of 1 μM APH prior to fixing and staining for DNA, EdU, and histone H4.2 mRNA (Figure 6D). In the presence of APH, S phase histone H4.2 mRNA levels were increased by the partial knockdown of NASP and a consequent decrease in the soluble histone pool (Figures 6D and 6E). Additionally, histone H4.2 mRNA levels in the presence of APH were largely rescued via knockdown of Eri1, indicating that the loss in histone mRNA upon inhibition of DNA synthesis is due predominantly to Eri-mediated degradation (Figures 6D and 6E). Rescue of histone mRNA following the knockdown of NASP or Eri1 suggests that upon inhibition of DNA synthesis, soluble histone protein directly acts to promote the degradation of histone mRNA. Thus, as cells enter S phase, the incorporation rate of soluble histone into the chromatin reduces the degradation of histone mRNA, thereby balancing histone production with the rate of DNA synthesis.
DISCUSSION
In this study, we investigated the temporal dynamics of the mechanisms that regulate histone biosynthesis at the G1/S boundary. It was long thought that the crossing of the restriction point and the start of S phase happened close in time, and thus that histone biosynthesis was tied solely with DNA replication.24,53 Our recent studies, however, have illustrated that crossing of the restriction point occurs about 5 h before the start of S phase, and thus these cell-cycle milestones are decoupled temporally.17 Therefore, the mechanistic basis of the burst of histone biosynthesis at the G1/S boundary was not properly understood. Here, we demonstrate that histone biosynthesis undergoes positive and negative regulation from the restriction point to the start of S phase via three cell-cycle-dependent mechanisms: (1) formation of the HLB driven by E2F-mediated expression of NPAT and FLASH, (2) initiation of histone transcription upon phosphorylation of NPAT by cyclin E/CDK2, and (3) the rate of histone incorporation into chromatin during DNA replication dictating soluble histone protein levels and contributing to the rate of histone mRNA degradation in S phase. As cells transition from G1 to S phase, they reach an equilibrium between cyclin E/CDK2-mediated transcriptional activation of histone genes and soluble histone protein-mediated degradation of histone mRNA, with both processes responding rapidly to inhibition of DNA replication. Therefore, the burst of mature histone mRNA observed at the start of S phase is caused by the initiation of nascent histone transcription by cyclin E/CDK2’s phosphorylation of NPAT several hours earlier at the restriction point. The peak of nascent histone pre-mRNA at the G1/S boundary correlates temporally with the fastest rate of increase of mature histone mRNA, since the actively transcribed histone pre-mRNA must be processed before accumulating as mature histone mRNA, whereas the nascent pre-mRNA is transitory (Figure 7A).
Figure 7. Temporal dynamics and cell-cycle regulation of histone biosynthesis.
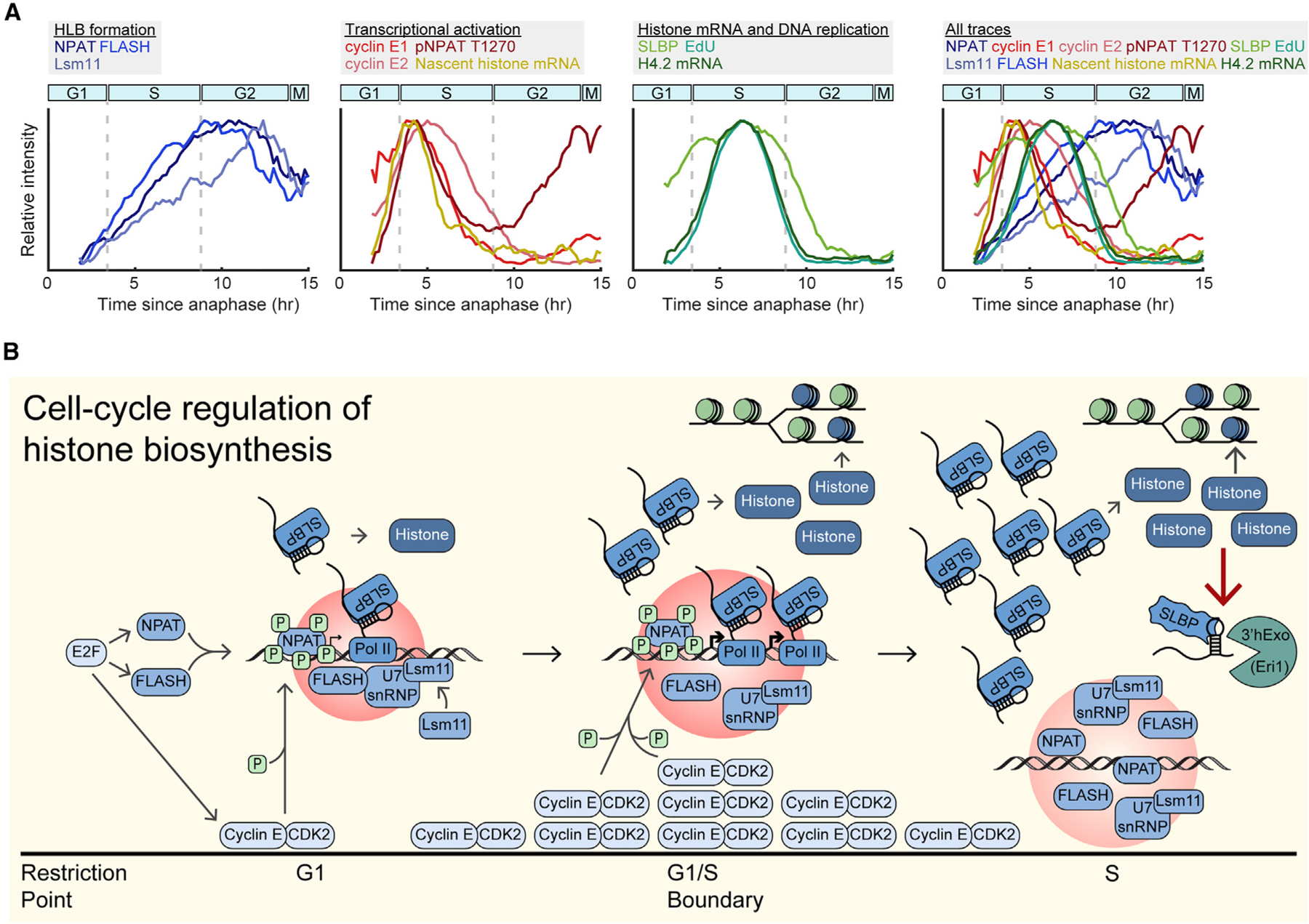
(A) Overlay of normalized mean signals presented in this study, categorized by HLB formation, transcriptional activation, histone mRNA and DNA replication, and all traces. Cyclin E signals are mean nuclear intensity.
(B) Schematic of cell-cycle regulation of histone biosynthesis relative to cell-cycle progression.
Decoupling of HLB formation and function
Formation of transcriptional condensates, such as super enhancers, myc condensates, and HLBs, are essential for the high levels of transcription and processing of the genes they envelope. However, the correlation between formation and activation of these transcriptional condensates has remained unclear. Here, we demonstrate that on short timescales, the phosphorylation of the HLB seeding and scaffolding protein NPAT activates transcription at the HLBs but does not affect HLB formation. This is in contrast to what has been seen in Drosophila embryos where CDK2 inhibition results in a rapid decrease in nuclear Mxc and consequent reduction of HLB size.27 We further show that the cessation of transcription upon CDK2 inhibition does not occur through a dissociation of the HLB, but through loss of NPAT phosphorylation. Additionally, NPAT appears to undergo a later wave of cyclin A2/CDK2-mediated phosphorylation in G2 whose functional role is unknown. This late phosphorylation of NPAT late may play a role in HLB dissociation in mitosis, or in preparing the cell for histone biosynthesis in its subsequent cell cycle. Together, these data suggest that once cells have crossed the restriction point, HLB formation and transcription activation are decouplable in human cells, a mechanism which ensures prolonged stability of the HLB while tightly regulating the timing of histone gene transcription.
Self-regulation of histone protein
Once histone mRNA has been transcribed and processed, it is no longer under the regulation of HLB activation. Yet high levels of histone production in the event of a slowing of DNA synthesis would be toxic to the cell because of the accumulation of excess soluble histone and the aberrant interactions they would form with negatively charged molecules.33 We find that cells address this via a negative-feedback mechanism in which excess soluble histone protein promotes the degradation of histone mRNA. This likely occurs through destabilization of the SLBP interaction with the 3′ stem loop, which normally blocks degradation of histone mRNA by 3′hExo,9 although the precise mechanism of this destabilization is unknown. Activation of ATR upon inhibition of DNA synthesis has been found to destabilize the SLBP-3′ stem loop interaction via Upf1, as well as inhibit CDK2, likely causing the loss of histone transcription.9,16,29,54 Additionally, it was surprising to find that soluble histone protein suppresses histone mRNA even when DNA synthesis is unperturbed. These data indicate that this is a mechanism in naturally cycling cells to keep histone biosynthesis in step with DNA synthesis (Figure 7A). Together, our work maps the timing by which various mechanisms regulate histone biosynthesis as cells transition from the point of cell-cycle commitment to the end of DNA replication (Figure 7B), thereby precisely detailing one of the most critical processes for faithful replication of the genome.
Limitations of the study
Although we were able to use single-cell live-cell imaging linked with immunofluorescent and RNA FISH staining to measure our proteins and RNAs of interest at each time point relative to each cell’s last mitosis, a limitation of this study is the absence of live-cell sensors for our histone biosynthesis factors. Development of a live-cell sensor for NPAT, as exists for its Drosophila homolog Mxc, would allow tracking of HLB growth and formation in live single cells.27,55 Observing HLB growth in individual cells would provide deeper understanding of the heterogeneity of HLB formation that may exist within the population. Quantifying HLB formation in tandem with live-cell sensors for other HLB factors, SLBP, or expression of histone genes, would serve to validate the temporal dynamics presented herein and determine if variation in histone biosynthesis correlates with changes in DNA replication stress or cell-cycle fate.
Additionally, because of the challenging and laborious nature of these experiments, the results presented in this study were performed primarily in MCF10A, and partially in RPE-hTERT cells. To test the universality of these dynamics, histone biosynthesis would ideally be mapped in several cell types, including normal primary cells and cancer cells. This would provide a comparison between healthy and aberrant histone biosynthesis dynamics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sabrina Spencer (sabrina.spencer@colorado.edu).
Materials availability
The custom pNPAT antibody is available upon request.
Data and code availability
Representative original images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. All raw movies and images are available upon request.
All code has been deposited at Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-NPAT clone 27 | Santa Cruz, BD Biosciences | sc-136007, 611344 |
| Rabbit anti-SLBP | Abcam | ab221166 |
| Rabbit anti-CASP8AP2 (FLASH) | Sigma Aldrich | HPA053573 |
| Rabbit anti-Lsm11 | Sigma Aldrich | HPA039587 |
| Rabbit anti-Eri1 | Thermo Fisher | 14592-1-AP |
| Rabbit polyclonal anti-NASP | Sigma Aldrich | HPA030520 |
| Rabbit monoclonal anti-Cyclin E1 EP435E | abcam | ab33911 |
| Rabbit monoclonal anti-Cyclin E2 EP454Y | abcam | ab40890 |
| Mouse anti-Cyclin A2 | Santa Cruz | sc-271682 |
| Rabbit polyclonal anti-phospho-NPAT T1270 | Thermo Fisher-custom antibody | N/A |
| Alexa Fluor 488 anti-mouse secondary | Thermo Fisher | A-11029 |
| Alexa Fluor 546 anti-mouse secondary | Thermo Fisher | A-11030 |
| Alexa Fluor 647 anti-mouse secondary | Thermo Fisher | A-21236 |
| Alexa Flour 546 anti-rabbit secondary | Thermo Fisher | A-11035 |
| Alexa Fluor 647 anti-rabbit secondary | Thermo Fisher | A-21245 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| PF-07104091 | ChemieTek | CT-PF0710 |
| PF-06873600 | SelleckChem | S8816 |
| Palbociclib (PD-0332991) | SelleckChem | S1116 |
| RO-3306 | SelleckChem | S7747 |
| Aphidicolin | Cell Signaling | 32774 |
| ddUTP-ATTO550 | Jena Bioscience | NU-1619-550 |
| Terminal Deoxynucleotidyl Transferase | Thermo Fisher | EP0161 |
| DharmaFECT 1 | Dharmacon | T-2001-02 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ViewRNA™ ISH Cell Assay Kit | Thermo Fisher | QVC0001 |
| Click-iT™ EdU Cell Proliferation Kit for Imaging | Fisher Scientific | C10340 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MCF10A | ATCC | CRL-10317 |
| RPE-hTERT | ATCC | CRL-4000 |
|
| ||
| Software and algorithms | ||
|
| ||
| MATLAB R2020a | Mathworks | N/A |
| Adobe Illustrator 2022 | Adobe | N/A |
| ImageJ | Fiji | N/A |
| Tracking code | Meyer Lab | https://github.com/scappell/Cell_tracking. |
| Imaris Analysis | Oxford Instruments | N/A |
|
| ||
| Deposited data | ||
|
| ||
| Imaging Data | Mendeley Data | https://doi.org/10.17632/d2b55pmjmk.1 |
|
| ||
| Oligonucleotides | ||
|
| ||
| siNPAT | Horizon Discovery | J-019599-10-0002 |
| siFLASH | Horizon Discovery | J-012413-05-0002 |
| siLsm11 | Horizon Discovery | J-018455-17-0002 |
| siCCNE1 | Horizon Discovery | M-003213-02-0005 |
| siCCNE2 | Horizon Discovery | M-003214-02-0005 |
| siCCNA2 | Horizon Discovery | M-003205-02-0005 |
| siSLBP | Horizon Discovery | J-012286-05-0002 |
| siNASP | Horizon Discovery | J-011740-13-0002 |
| siEri1 | Horizon Discovery | J-021497-09-0002 |
| siControl | Horizon Discovery | D-001810-02-05 |
| HIST2H4A | Thermo Fisher | VA6-3174283-VC |
| NPAT | Thermo Fisher | VA6-3172751-VC |
| CASP8AP2 (FLASH) | Thermo Fisher | VA6-3175253-VC |
| SLBP | Thermo Fisher | VA6-3174137-VCP |
| See Table S1 for oligonucleotides used for nascent histone RNA FISH | Integrated DNA Technologies | N/A |
|
| ||
| Other | ||
|
| ||
| Stellaris® RNA FISH Hybridization Buffer | Biosearch Technologies | SMF-HB1-10 |
| Stellaris® RNA FISH Wash Buffer A | Biosearch Technologies | SMF-WA1-60 |
| Stellaris® RNA FISH Wash Buffer B | Biosearch Technologies | SMF-WB1-20 |
| DMEM/F-12 phenol red free | Thermo Fisher | 11039047 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
MCF10A were obtained from ATCC (CRL-10317) and were cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) supplemented with 5% horse serum, 100 ng/mL cholera toxin, 20 ng/mL EGF, 10 μg/mL insulin, 0.5 μg/mL hydrocortisone, and 100 μg/mL of both penicillin and streptomycin. All cells were cultured at 37°C with 5% CO2. RPE-hTERT cells were obtained from ATCC (CRL-4000) and were cultured in DMEM/F12 supplemented with 10% Fetal Bovine Serum (FBS), 1× Glutamax, and 100 μg/mL of both penicillin and streptomycin. Live-cell imaging of MCF10A and RPE-hTERT cells was done using a phenol red free version of the growth media (Thermo Fisher, 11039047).
METHOD DETAILS
Antibodies
Primary antibodies were used at the following dilutions: NPAT (sc-136007 1:400, 611344 1:4000), SLBP (ab221166, 1:1000), FLASH (HPA053573, 1:4000), Lsm11 (HPA039587, 1:1000), Eri1 (14592–1-AP, 1:250), NASP (HPA030520, 1:2000), Cyclin E1 (ab33911, 1:4000), Cyclin E2 (ab40890, 1:500), Cyclin A2 (sc-271682, 1:500), pNPAT T1270 (1:2000). Secondary antibodies were used at a dilution of 1:500, except for FLASH, which was stained with a secondary dilution of 1:1000.
Custom antibody development
The antibody against phospho-NPAT T1270 is a polyclonal rabbit antibody produced by the Thermo Fisher custom antibody services. The construct was designed as previously described, with the sequence Asp-Leu-Pro-Val-Pro-Arg-phosphoThr-Pro-Gly-Ser-Gly-Ala-Gly-Cys, generated after coupling to keyhole limpet hemocyanin.25
Immunofluorescent staining
Cells were fixed with 4% paraformaldehyde for 15 min, washed 3 times with PBS, then permeabilized in 0.1% Triton X-100 for 20 min. Standard protocols were then used for immunofluorescent staining: cells were first blocked in 3% BSA for 1 h at 37°C, primary antibodies were incubated in 3% BSA overnight at 4°C, cells were washed three times in PBS, secondary antibodies were then incubated at room temperature for 2 h, cells were washed three times in PBS before being incubated with Hoechst at 1:10,000 at room temperature for 20 min. Imaging was done on a Nikon Ti-E with a 103 0.45 NA objective with the appropriate filter applied. Exposure times were set to 400ms for DAPI, 300ms for Cy3, and 400ms for Cy5.
RNA FISH staining
Cells were fixed with 4% paraformaldehyde for 15 min. Histone H4.2 (Thermo Fisher, VA6–3174283-VC), NPAT (Thermo Fisher, VA6–3172751-VC), FLASH (Thermo Fisher, VA6–3175253-VC), and SLBP (Thermo Fisher, VA6–3174137-VCP) mRNA were visualized according to the manufacturer’s protocol (ViewRNA ISH Cell Assay Kit, ThermoFisher QVC0001), with cells being permeabilized for 30 min and mRNA probes hybridized for 4 h at 40°C. Exposure times were set to 600ms for Cy5.
Nascent histone RNA FISH design and staining
Probe design
We designed probes targeted against the sequence downstream of the 3′ cleavage site for 50 histone genes found at clusters HIST1 and HIST2 (see Table S1 for sequence and position relative to the cleavage site), to measure nascent histone mRNA expression at the HLB. Probes were designed with the Stellaris RNA FISH Probe Designer (Biosearch Technologies, Inc., Petaluma, CA) available online at www.biosearchtech.com/stellarisdesigner (Version 4.2). Probes were set to have an oligo length of 20 nt, a masking level of 5, and a minimum spacing length of 2nt. Starting positions were chosen to be within 25nt of the cleavage site for HIST1, and 150nt of the cleavage site for HIST2.
Probe set labeling
Probes for nascent histone RNA FISH were ordered from IDT. Each oligo was resuspended at a concentration of 250μM in nuclease free H2O, before being pooled in an oligo master mix. Probes were then labeled with ATTO 550 using ddUTP-ATTO550 (Jena Bioscience, NU-1619–550) with terminal deoxynucleotidyl transferase (Thermo Fisher, EP0161).56 The oligo pool was incubated with the ddUTP fluorophore for 18 h at 37°C, before being heat inactivated at 70°C for 10 min. Upon completion of the enzymatic reaction, labeled oligos were mixed with phenol:chloroform:isoamyl alcohol and extracted from the aqueous layer, before undergoing ethanol precipitation for 16 h at 4°C. Labeled oligos were then resuspended in nuclease-free H2O at a final concentration of 12.5μM.
Staining
Cells were fixed with 4% paraformaldehyde for 15 min, before being permeabilized with 0.1% Triton X-100 for 30 min. Nascent histone RNA was then visualized in accordance with the manufacture’s protocol (https://biosearch-cdn.azureedge.net/assetsv6/protocol_stellaris-adherent-cells-96-well-glass-bottom-plates.pdf) (Biosearch Technologies, Inc., Petaluma, CA; Wash Buffer A, SMF-WA1–60; Wash Buffer B, SMF-WB1–20; Hybridization Buffer, SMF-HB1–10). Labeled nascent histone mRNA probes were hybridized for 16 h at 37°C at a dilution of 1:50. Exposure times were set to 800ms for Cy3. Following nascent histone RNA FISH staining and imaging, cells were stained for NPAT protein as described above.
siRNA transfection
siRNA transfections were performed using the DharmaFECT 1 (Dharmacon, #T-2001–02) reagent as described by the manufacturer. For live-cell imaging, the transfection mix was added to cells with 20nM final concentration for siNASP and 25nM final concentration for siEri1, siNPAT, siFLASH, and siLsm11, and removed after 6 h. Imaging was started immediately and proceeded for an additional 18 h for siNASP, or imaging began 24 h after transfection mixture was removed and proceeded for an additional 18 h for siEri1, siNPAT, siFLASH, and siLsm11. For the validation of antibodies for Cyclin E1, Cyclin E2, and SLBP, transfection mixture was added to cells with 25nM final concentration of siRNA and was removed after 6 h. Cells were fixed and stained as described above after 24 h for siCCNE1 and siCCNE2, and after 48hr for siSLBP. Oligonucleotides used in this study include: control (Horizon, D-001810–02-05), NPAT (Horizon, J-019599–10-0002), SLBP (Horizon, J-012286–05-0002), FLASH (Horizon, J-012413–05-0002), Lsm11 (Horizon, J-018455–17-0002), Eri1 (Horizon, J-021497–09-0002), NASP (Horizon, J-011740–13-0002), CCNE1 (Horizon, M-003213–02-0005), CCNE2 (Horizon, M-003214–02-0005), CCNA2 (Horizon, M-003205–02-0005).
Live-cell imaging
Live-cell movies with HLB imaging
Cells were plated on a 96-well plate (Cellvis Cat. No. P96–1.5H-N) coated with collagen at a 1:50 dilution in water (Advanced BioMatrix, #5005). Cells were plated at densities of 3,600 cells per well 24 h prior to the start of imaging for all movies except for those with 48 h siRNA knockdowns, which were plated at 1,600 cells per well 48 h prior to the start of imaging. All live cell-imaging was done on a Nikon Ti-E microscope with a 10× 0.45 NA objective, except for movies followed by staining for nascent histone mRNA, which were done with a 20× 0.45 NA objective, with the appropriate filter applied, at a frequency of 5 frames per hour. For the duration of the movie, cells were maintained in a humidified incubation chamber at 37°C, with 5% CO2. Exposure times were set to 70ms per frame for CFP, corresponding to H2B-mTurquoise2, and 100ms for mCherry, corresponding to DHB-mCherry for MCF10A cells, and 70ms per frame for CFP, corresponding to H2B-mTurquoise2, and 100ms for YFP, corresponding to DHB-mVenus for RPE-hTERT cells. Prior to the last frame of imaging, all cells were pulsed with 10μM of EdU for 12 min when applicable, before being immediately fixed with 4% paraformaldehyde. Following permeabilization with 0.1% Triton X-100 for 20 min, cells were photobleached with 3% H2O2 for 30 min until the DHB-mCherry signal was no longer present for MCF10A cells. Cells were then processed for EdU visualization as described by the manufacturer’s protocol (Invitrogen, C10340) with Alexa Fluor 488. Subsequent immunofluorescent or nascent histone mRNA FISH staining was done as described above. Exposure times were set to 400ms for DAPI, 600ms for YFP, 300ms for Cy3, and 400ms for Cy5 unless otherwise specified.
All other live-cell movies
Cells were plated as described above 24 h prior to the start of imaging at a density of 3,600 cells per well for all movies expect for those with 48 h siRNA knockdowns, which were plated at 1,600 cells per well 48 h prior to the start of imaging. Live cell-imaging was done as described above, with exposure times set to 70ms per frame for CFP, corresponding to H2B-mTurquoise2, and 100ms for YFP, corresponding to DHB-mVenus. Prior to the last frame of imaging, cell were pulsed with 10μM of EdU for 12 min when applicable, before being immediately fixed with 4% paraformaldehyde, and processed for EdU visualization as described by the manufacturer’s protocol (Invitrogen, C10340) with Alexa Fluor 555. Cells were then processed for RNA FISH or immunofluorescent staining, as described above. Exposure times were set to 400ms for DAPI, 200ms for Cy3, and 400ms for Cy5, unless otherwise specified.
3D imaging
Following immunofluorescent staining, cells were imaged using a Nikon A1R Laser Scanning Confocal microscope equipped with an Andor iXon 897 Ultra EMCCD and NIS Elements software (v. 5.30). Cells with detectable HLB puncta were selected using a 10× NA 0.45 air objective and their XY coordinates were saved. Individual Z-stacks of selected cells were then captured sequentially using a Plan Apo λ 100×1.45 NA oil objective with 0.1 μm step size and 1.0 AU pinhole size per channel. Chromatic aberration between the TRITC and Cy5 channels was quantified using 0.5 μm fluorescent microspheres on a FocalCheck test slide (Invitrogen, F36909). The Cy5 channel was computationally shifted by +0.06 μm in the X plane for all nuclear Z-stacks to compensate for the measured aberration.
QUANTIFICATION AND STATISTICAL ANALYSIS
Tracking of live-cell imaging
Cell tracking was achieved by generating a nuclear mask based on segmentation of the H2B-mTurquoise fluorescence signal for each frame of the live-cell imaging, with each cell being identified as an individual object, and regional nearest neighbor calculations being used to track single cells from one frame to the next.17,37,57 The nuclear mask was then applied to the DHB fluorescence channel and the nuclear DHB signal was measured as the mean signal of the pixels in each nucleus. Cytoplasmic DHB signal was determined by dilating the nuclear mask by 2 pixels and quantifying the mean pixel intensity of the cytoplasmic ring. CDK2 activity was calculated as the ratio of cytoplasmic to nuclear DHB signal. Following fixed-cell staining, Hoechst signal was used to generate the nuclear mask. The tracking code available at https://github.com/scappell/Cell_tracking. Cyclin E1, Cyclin E2, and SLBP protein signals, as well as EdU incorporation, were quantified as the median pixel value in the nuclear mask, histone H4.2 mRNA FISH signal was quantified as the median pixel value of a two-pixel-wide cytoplasmic ring around the nuclear mask. Protein or mRNA signals were matched back to the last frame of the live-cell movie using nearest neighbor screening after jitter correction. HLB puncta were identified from NPAT images, as previously described.2,57 The MATLAB function regionprops was used to label all puncta and to retrieve the xy coordinates of all pixels belonging to all the puncta of a given nucleus, which was then converted to total HLB size in μm2 based on the objective magnification. Protein signal at the HLB was measured as the total protein intensity normalized to the total HLB size per cell. Pre-imaging of the nascent histone mRNA FISH was linked to post-imaging of NPAT and EdU in the same cells by matching of nuclear centroids. Average protein or RNA FISH signal at the HLB was determined as the sum of the signal of all puncta pixels per cell normalized to the HLB size, with the HLB size being defined as the total number of puncta pixels per cell. For Cyclin E1, Cyclin E2, pNPAT T1270, and nascent histone mRNA FISH, the average signal at the HLB in cells with HLB puncta was normalized to the median nuclear intensity.
Classification of cell populations
Following alignment to anaphase, cells were classified as either CDK2inc or CDK2low based on the level of CDK2 activity (cytoplasmic/nuclear ratio of the DHB sensor) 2 h after anaphase. Cells with CDK2 activity above 0.5 were classified as CDK2inc, while cells that stayed under 0.5 for the remainder of imaging were classified as CDK2low, followed by removing any EdUpos or 4N DNA content cells from the population. CDK2inc cells were further classified by cell-cycle phase, with G1 cells as 2N DNA content, EdUneg; S cells as EdUpos; G2/M cells as 4N DNA content, EdUneg (see also Figures S1A–S1D). CDK2inc vs. CDK2low classification was extended to 3 h after anaphase for cells treated with PF-07104091 (CDK2i), and the duration of time required for cells to have a CDK2 activity above 0.5 to be classified as CDK2inc was shorted from 4 h to 0.2 h, since CDK2i treatment caused the DHB sensor to translocate back to the nucleus from the cytoplasm (see Figures S4A and S4B). All n and p values can be found in Table S2.
HLB quantification from 3D imaging
Laser scanning confocal deconvolution was applied to each z stack using the Richardson-Lucy deconvolution algorithm for 20 iterations using Imaris software (v. 9.9.0). Nuclei were segmented for 3D object creation using a local contrast threshold and filtered with a minimum diameter gate of 15 μm. NPAT and FLASH puncta within the nucleus were segmented using a local contrast threshold and filtered for volumes above the diffraction limit of the imaging system. Lsm11 foci were detected using the Imaris Spots function as they were too small for accurate volume measurement. The distances between centroids of all segmented NPAT and FLASH or NPAT and Lsm11 puncta were calculated and colocalization events were called when the calculated distance was at or below the resolution limit of the imaging system.
Supplementary Material
Highlights.
Histone locus body (HLB) formation is driven by E2F-mediated expression of NPAT and FLASH
Histone transcription is triggered by cyclin E-CDK2’s phosphorylation of NPAT
HLB formation and activation of histone transcription are decouplable
DNA replication regulates histone mRNA degradation via soluble histone levels
ACKNOWLEDGMENTS
We thank Francesca Mattiroli for insights on the project and feedback on the manuscript, as well as the members of the laboratory of S.L.S. for general help and feedback on the manuscript. This work was supported by NIH training grant T32 (GM065103–16) (to C.A.), a Pew-Stewart Scholar for Cancer Research Award, an American Cancer Society Research Scholar Grant (RSG–18–008–01), and an NIH Director’s New Innovator Award (1DP2CA238330–01) (to S.L.S.). The 3D imaging work, imaging data analysis, and visualization were performed at the BioFrontiers Institute Advanced Light Microscopy Core (RRID: SCR_018302). Laser scanning confocal microscopy was performed on a Nikon A1R microscope supported by NIST-CU Cooperative Agreement award number 70NANB15H226. The Analysis Workstation and the software package Imaris were supported by NIH 1S10RR026680–01A1.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112768.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Duronio RJ, and Marzluff WF (2017). Coordinating cell cycle-regulated histone gene expression through assembly and function of the Histone Locus Body. RNA Biol 14, 726–738. 10.1080/15476286.2016.1265198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong C, and Spencer SL (2021). Replication-dependent histone biosynthesis is coupled to cell-cycle commitment. Proc. Natl. Acad. Sci. USA 118, e2100178118. 10.1073/pnas.2100178118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tatomer DC, Terzo E, Curry KP, Salzler H, Sabath I, Zapotoczny G, McKay DJ, Dominski Z, Marzluff WF, and Duronio RJ (2016). Concentrating pre-mRNA processing factors in the histone locus body facilitates efficient histone mRNA biogenesis. J. Cell Biol 213, 557–570. 10.1083/jcb.201504043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marzluff WF, Gongidi P, Woods KR, Jin J, and Maltais LJ (2002). The human and mouse replication-dependent histone genes. Genomics 80, 487–498. [PubMed] [Google Scholar]
- 5.Yang XC, Burch BD, Yan Y, Marzluff WF, and Dominski Z (2009). FLASH, a proapoptotic protein involved in activation of caspase-8, is essential for 3’ end processing of histone pre-mRNAs. Mol. Cell 36, 267–278. 10.1016/j.molcel.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang XC, Sabath I, Dębski J, Kaus-Drobek M, Dadlez M, Marzluff WF, and Dominski Z (2013). A complex containing the CPSF73 endonuclease and other polyadenylation factors associates with U7 snRNP and is recruited to histone pre-mRNA for 3’-end processing. Mol. Cell Biol 33, 28–37. 10.1128/MCB.00653-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skrajna A, Yang XC, Bucholc K, Zhang J, Hall TMT, Dadlez M, Marzluff WF, and Dominski Z (2017). U7 snRNP is recruited to histone pre-mRNA in a FLASH-dependent manner by two separate regions of the stem-loop binding protein. RNA 23, 938–951. 10.1261/rna.060806.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White AE, Burch BD, Yang XC, Gasdaska PY, Dominski Z, Marzluff WF, and Duronio RJ (2011). Drosophila histone locus bodies form by hierarchical recruitment of components. J. Cell Biol 193, 677–694. 10.1083/jcb.201012077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marzluff WF, and Koreski KP (2017). Birth and Death of Histone mRNAs. Trends Genet 33, 745–759. 10.1016/j.tig.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whitfield ML, Zheng LX, Baldwin A, Ohta T, Hurt MM, and Marzluff WF (2000). Stem-loop binding protein, the protein that binds the 3’ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol. Cell Biol 20, 4188–4198. 10.1128/MCB.20.12.4188-4198.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sullivan KD, Mullen TE, Marzluff WF, and Wagner EJ (2009). Knockdown of SLBP results in nuclear retention of histone mRNA. RNA 15, 459–472. 10.1261/rna.1205409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullen TE, and Marzluff WF (2008). Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5’ to 3’ and 3’ to 5. Genes Dev 22, 50–65. 10.1101/gad.1622708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koseoglu MM, Graves LM, and Marzluff WF (2008). Phosphorylation of threonine 61 by cyclin a/Cdk1 triggers degradation of stem-loop binding protein at the end of S phase. Mol. Cell Biol 28, 4469–4479. 10.1128/MCB.01416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng L, Dominski Z, Yang XC, Elms P, Raska CS, Borchers CH, and Marzluff WF (2003). Phosphorylation of stem-loop binding protein (SLBP) on two threonines triggers degradation of SLBP, the sole cell cycle-regulated factor required for regulation of histone mRNA processing, at the end of S phase. Mol. Cell Biol 23, 1590–1601. 10.1128/MCB.23.5.1590-1601.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendiratta S, Gatto A, and Almouzni G (2019). Histone supply: Multitiered regulation ensures chromatin dynamics throughout the cell cycle. J. Cell Biol 218, 39–54. 10.1083/jcb.201807179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meaux SA, Holmquist CE, and Marzluff WF (2018). Role of oligouridylation in normal metabolism and regulated degradation of mammalian histone mRNAs. Philos. Trans. R. Soc. Lond. B Biol. Sci 373, 20180170. 10.1098/rstb.2018.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer M, Grossmann P, Padi M, and DeCaprio JA (2016). Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res 44, 6070–6086. 10.1093/nar/gkw523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao G, Bracken AP, Burkard K, Pasini D, Classon M, Attwooll C, Sagara M, Imai T, Helin K, and Zhao J (2003). NPAT expression is regulated by E2F and is essential for cell cycle progression. Mol. Cell Biol 23, 2821–2833. 10.1128/MCB.23.8.2821-2833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sokolova M, Turunen M, Mortusewicz O, Kivioja T, Herr P, Vähärautio A, Björklund M, Taipale M, Helleday T, and Taipale J (2017). Genome-wide screen of cell-cycle regulators in normal and tumor cells identifies a differential response to nucleosome depletion. Cell Cycle 16, 189–199. 10.1080/15384101.2016.1261765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zarkowska T, and Mittnacht S (1997). Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem 272, 12738–12746. 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- 22.Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, and Weinberg RA (1992). Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70, 993–1006. 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 23.Pardee AB (1974). A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 71, 1286–1290. 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao J, Dynlacht B, Imai T, Hori T, and Harlow E (1998). Expression of NPAT, a novel substrate of cyclin E-CDK2, promotes S-phase entry. Genes Dev 12, 456–461. 10.1101/gad.12.4.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma T, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD, Wang J, Qin J, Chow LT, and Harper JW (2000). Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev 14, 2298–2313. 10.1101/gad.829500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, and Harlow E (2000). NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev 14, 2283–2297. [PMC free article] [PubMed] [Google Scholar]
- 27.Hur W, Kemp JP Jr., Tarzia M, Deneke VE, Marzluff WF, Duronio RJ, and Di Talia S (2020). CDK-Regulated Phase Separation Seeded by Histone Genes Ensures Precise Growth and Function of Histone Locus Bodies. Dev. Cell 54, 379–394.e6. 10.1016/j.devcel.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moser J, Miller I, Carter D, and Spencer SL (2018). Control of the Restriction Point by Rb and p21. Proc. Natl. Acad. Sci. USA 115, E8219–E8227. 10.1073/pnas.1722446115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaygun H, and Marzluff WF (2005). Regulated degradation of replication-dependent histone mRNAs requires both ATR and Upf1. Nat. Struct. Mol. Biol 12, 794–800. 10.1038/nsmb972. [DOI] [PubMed] [Google Scholar]
- 30.Kaygun H, and Marzluff WF (2005). Translation termination is involved in histone mRNA degradation when DNA replication is inhibited. Mol. Cell Biol 25, 6879–6888. 10.1128/MCB.25.16.6879-6888.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jimeno-González S, Payán-Bravo L, Muñoz-Cabello AM, Guijo M, Gutierrez G, Prado F, and Reyes JC (2015). Defective histone supply causes changes in RNA polymerase II elongation rate and cotranscriptional pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 112, 14840–14845. 10.1073/pnas.1506760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ransom M, Dennehey BK, and Tyler JK (2010). Chaperoning histones during DNA replication and repair. Cell 140, 183–195. 10.1016/j.cell.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hogan AK, and Foltz DR (2021). Reduce, Retain, Recycle: Mechanisms for Promoting Histone Protein Degradation versus Stability and Retention. Mol. Cell Biol 41, e0000721. 10.1128/MCB.00007-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cook AJL, Gurard-Levin ZA, Vassias I, and Almouzni G (2011). A specific function for the histone chaperone NASP to fine-tune a reservoir of soluble H3-H4 in the histone supply chain. Mol. Cell 44, 918–927. 10.1016/j.molcel.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 35.Bao H, Carraro M, Flury V, Liu Y, Luo M, Chen L, Groth A, and Huang H (2022). NASP maintains histone H3-H4 homeostasis through two distinct H3 binding modes. Nucleic Acids Res 50, 5349–5368. 10.1093/nar/gkac303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finn RM, Browne K, Hodgson KC, and Ausió J (2008). sNASP, a histone H1-specific eukaryotic chaperone dimer that facilitates chromatin assembly. Biophys. J 95, 1314–1325. 10.1529/biophysj.108.130021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gookin S, Min M, Phadke H, Chung M, Moser J, Miller I, Carter D, and Spencer SL (2017). A map of protein dynamics during cell-cycle progression and cell-cycle exit. PLoS Biol 15, e2003268. 10.1371/journal.pbio.2003268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min M, and Spencer SL (2019). Spontaneously slow-cycling subpopulations of human cells originate from activation of stress-response pathways. PLoS Biol 17, e3000178. 10.1371/journal.pbio.3000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabath I, Skrajna A, Yang XC, Dadlez M, Marzluff WF, and Dominski Z (2013). 3’-End processing of histone pre-mRNAs in Drosophila: U7 snRNP is associated with FLASH and polyadenylation factors. RNA 19, 1726–1744. 10.1261/rna.040360.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kemp JP Jr., Yang XC, Dominski Z, Marzluff WF, and Duronio RJ (2021). Superresolution light microscopy of the Drosophila histone locus body reveals a core-shell organization associated with expression of replication-dependent histone genes. Mol. Biol. Cell 32, 942–955. 10.1091/mbc.E20-10-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JL, Murphy C, Buszczak M, Clatterbuck S, Goodman R, and Gall JG (2006). The Drosophila melanogaster Cajal body. J. Cell Biol 172, 875–884. 10.1083/jcb.200511038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rana S, Mallareddy JR, Singh S, Boghean L, and Natarajan A (2021). Inhibitors, PROTACs and Molecular Glues as Diverse Therapeutic Modalities to Target Cyclin-Dependent Kinase. Cancers 13, 5506. 10.3390/cancers13215506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mezi S, Botticelli A, Pomati G, Cerbelli B, Scagnoli S, Amirhassankhani S, d’Amati G, and Marchetti P (2021). Standard of Care and Promising New Agents for the Treatment of Mesenchymal Triple-Negative Breast Cancer. Cancers 13, 1080. 10.3390/cancers13051080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fassl A, Geng Y, and Sicinski P (2022). CDK4 and CDK6 kinases: From basic science to cancer therapy. Science 375, eabc1495. 10.1126/science.abc1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jhaveri K, Burris Rd HA, Yap TA, Hamilton E, Rugo HS, Goldman JW, Dann S, Liu F, Wong GY, Krupka H, and Shapiro GI (2021). The evolution of cyclin dependent kinase inhibitors in the treatment of cancer. Expert Rev. Anticancer Ther 21, 1105–1124. 10.1080/14737140.2021.1944109. [DOI] [PubMed] [Google Scholar]
- 46.Freeman-Cook KD, Hoffman RL, Behenna DC, Boras B, Carelli J, Diehl W, Ferre RA, He YA, Hui A, Huang B, et al. (2021). Discovery of PF-06873600, a CDK2/4/6 Inhibitor for the Treatment of Cancer. J. Med. Chem 64, 9056–9077. 10.1021/acs.jmedchem.1c00159. [DOI] [PubMed] [Google Scholar]
- 47.Rogers S, Gloss BS, Lee CS, Sergio CM, Dinger ME, Musgrove EA, Burgess A, and Caldon CE (2015). Cyclin E2 is the predominant E-cyclin associated with NPAT in breast cancer cells. Cell Div 10, 1. 10.1186/s13008-015-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris ME, Böhni R, Schneiderman MH, Ramamurthy L, Schümperli D, and Marzluff WF (1991). Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Mol. Cell Biol 11, 2416–2424. 10.1128/mcb.11.5.2416-2424.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heintz N, Sive HL, and Roeder RG (1983). Regulation of human histone gene expression: kinetics of accumulation and changes in the rate of synthesis and in the half-lives of individual histone mRNAs during the HeLa cell cycle. Mol. Cell Biol 3, 539–550. 10.1128/mcb.3.4.539-550.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sittman DB, Graves RA, and Marzluff WF (1983). Histone mRNA concentrations are regulated at the level of transcription and mRNA degradation. Proc. Natl. Acad. Sci. USA 80, 1849–1853. 10.1073/pnas.80.7.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Graves RA, and Marzluff WF (1984). Rapid reversible changes in the rate of histone gene transcription and histone mRNA levels in mouse myeloma cells. Mol. Cell Biol 4, 351–357. 10.1128/mcb.4.2.351-357.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hormazabal J, Saavedra F, Espinoza-Arratia C, Martinez NW, Cruces T, Alfaro IE, and Loyola A (2022). Chaperone mediated autophagy contributes to the newly synthesized histones H3 and H4 quality control. Nucleic Acids Res 50, 1875–1887. 10.1093/nar/gkab1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marzluff WF, Wagner EJ, and Duronio RJ (2008). Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet 9, 843–854. 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daigh LH, Liu C, Chung M, Cimprich KA, and Meyer T (2018). Stochastic Endogenous Replication Stress Causes ATR-Triggered Fluctuations in CDK2 Activity that Dynamically Adjust Global DNA Synthesis Rates. Cell Syst 7, 17–27.e3. 10.1016/j.cels.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terzo EA, Lyons SM, Poulton JS, Temple BRS, Marzluff WF, and Duronio RJ (2015). Distinct self-interaction domains promote Multi Sex Combs accumulation in and formation of the Drosophila histone locus body. Mol. Biol. Cell 26, 1559–1574. 10.1091/mbc.E14-10-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaspar I, Wippich F, and Ephrussi A (2017). Enzymatic production of single-molecule FISH and RNA capture probes. RNA 23, 1582–1591. 10.1261/rna.061184.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arora M, Moser J, Phadke H, Basha AA, and Spencer SL (2017). Endogenous Replication Stress in Mother Cells Leads to Quiescence of Daughter Cells. Cell Rep 19, 1351–1364. 10.1016/j.cel-rep.2017.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Representative original images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. All raw movies and images are available upon request.
All code has been deposited at Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-NPAT clone 27 | Santa Cruz, BD Biosciences | sc-136007, 611344 |
| Rabbit anti-SLBP | Abcam | ab221166 |
| Rabbit anti-CASP8AP2 (FLASH) | Sigma Aldrich | HPA053573 |
| Rabbit anti-Lsm11 | Sigma Aldrich | HPA039587 |
| Rabbit anti-Eri1 | Thermo Fisher | 14592-1-AP |
| Rabbit polyclonal anti-NASP | Sigma Aldrich | HPA030520 |
| Rabbit monoclonal anti-Cyclin E1 EP435E | abcam | ab33911 |
| Rabbit monoclonal anti-Cyclin E2 EP454Y | abcam | ab40890 |
| Mouse anti-Cyclin A2 | Santa Cruz | sc-271682 |
| Rabbit polyclonal anti-phospho-NPAT T1270 | Thermo Fisher-custom antibody | N/A |
| Alexa Fluor 488 anti-mouse secondary | Thermo Fisher | A-11029 |
| Alexa Fluor 546 anti-mouse secondary | Thermo Fisher | A-11030 |
| Alexa Fluor 647 anti-mouse secondary | Thermo Fisher | A-21236 |
| Alexa Flour 546 anti-rabbit secondary | Thermo Fisher | A-11035 |
| Alexa Fluor 647 anti-rabbit secondary | Thermo Fisher | A-21245 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| PF-07104091 | ChemieTek | CT-PF0710 |
| PF-06873600 | SelleckChem | S8816 |
| Palbociclib (PD-0332991) | SelleckChem | S1116 |
| RO-3306 | SelleckChem | S7747 |
| Aphidicolin | Cell Signaling | 32774 |
| ddUTP-ATTO550 | Jena Bioscience | NU-1619-550 |
| Terminal Deoxynucleotidyl Transferase | Thermo Fisher | EP0161 |
| DharmaFECT 1 | Dharmacon | T-2001-02 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ViewRNA™ ISH Cell Assay Kit | Thermo Fisher | QVC0001 |
| Click-iT™ EdU Cell Proliferation Kit for Imaging | Fisher Scientific | C10340 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MCF10A | ATCC | CRL-10317 |
| RPE-hTERT | ATCC | CRL-4000 |
|
| ||
| Software and algorithms | ||
|
| ||
| MATLAB R2020a | Mathworks | N/A |
| Adobe Illustrator 2022 | Adobe | N/A |
| ImageJ | Fiji | N/A |
| Tracking code | Meyer Lab | https://github.com/scappell/Cell_tracking. |
| Imaris Analysis | Oxford Instruments | N/A |
|
| ||
| Deposited data | ||
|
| ||
| Imaging Data | Mendeley Data | https://doi.org/10.17632/d2b55pmjmk.1 |
|
| ||
| Oligonucleotides | ||
|
| ||
| siNPAT | Horizon Discovery | J-019599-10-0002 |
| siFLASH | Horizon Discovery | J-012413-05-0002 |
| siLsm11 | Horizon Discovery | J-018455-17-0002 |
| siCCNE1 | Horizon Discovery | M-003213-02-0005 |
| siCCNE2 | Horizon Discovery | M-003214-02-0005 |
| siCCNA2 | Horizon Discovery | M-003205-02-0005 |
| siSLBP | Horizon Discovery | J-012286-05-0002 |
| siNASP | Horizon Discovery | J-011740-13-0002 |
| siEri1 | Horizon Discovery | J-021497-09-0002 |
| siControl | Horizon Discovery | D-001810-02-05 |
| HIST2H4A | Thermo Fisher | VA6-3174283-VC |
| NPAT | Thermo Fisher | VA6-3172751-VC |
| CASP8AP2 (FLASH) | Thermo Fisher | VA6-3175253-VC |
| SLBP | Thermo Fisher | VA6-3174137-VCP |
| See Table S1 for oligonucleotides used for nascent histone RNA FISH | Integrated DNA Technologies | N/A |
|
| ||
| Other | ||
|
| ||
| Stellaris® RNA FISH Hybridization Buffer | Biosearch Technologies | SMF-HB1-10 |
| Stellaris® RNA FISH Wash Buffer A | Biosearch Technologies | SMF-WA1-60 |
| Stellaris® RNA FISH Wash Buffer B | Biosearch Technologies | SMF-WB1-20 |
| DMEM/F-12 phenol red free | Thermo Fisher | 11039047 |