

SUMMARY
During mouse embryogenesis, expression of the long non-coding RNA (lncRNA) Airn leads to gene repression and recruitment of Polycomb repressive complexes (PRCs) to varying extents over a 15-Mb domain. The mechanisms remain unclear. Using high-resolution approaches, we show in mouse trophoblast stem cells that Airn expression induces long-range changes to chromatin architecture that coincide with PRC-directed modifications and center around CpG island promoters that contact the Airn locus even in the absence of Airn expression. Intensity of contact between the Airn lncRNA and chromatin correlated with underlying intensity of PRC recruitment and PRC-directed modifications. Deletion of CpG islands that contact the Airn locus altered long-distance repression and PRC activity in a manner that correlated with changes in chromatin architecture. Our data imply that the extent to which Airn expression recruits PRCs to chromatin is controlled by DNA regulatory elements that modulate proximity of the Airn lncRNA product to its target DNA.
In brief
Braceros et al. map interactions between DNA regulatory elements, Polycomb repressive complexes (PRCs), and the lncRNA Airn. They find that the presence of Airn lncRNA and PRCs correlates in lock-step on chromatin and that specific DNA regulatory elements modulate intensity of repression by Airn in different ways.
Graphical abstract

INTRODUCTION
Genomic imprinting is a process known to occur for some ~150 mammalian genes, resulting in their preferential expression from one parentally inherited allele over the other. Dysregulated imprinting can lead to developmental disorders, cancer, and changes in metabolism. Studies of genomic imprinting have also yielded important insights into fundamental mechanisms of gene regulation, including the recognition that long non-coding RNAs (lncRNAs) control the expression of genes that are essential for proper human development.1–4
Airn (Antisense of Igf2r Non-Protein Coding RNA) is a gene that in mice is imprinted and produces a lncRNA specifically from the paternal allele of chromosome (chr) 17. The Airn locus produces lncRNA transcripts that are upward of ~90 kb in length but have heterogeneous 3′ ends.5 In addition, Airn transcripts are lowly abundant, short-lived, retained near their site of transcription, predominantly unspliced, and not obviously conserved outside of rodents.6–12 Nevertheless, Airn expression results in transcriptional repression over a domain that spans ~15 Mb in extraembryonic tissues of the mouse, the largest autosomal region known to be under the control of a repressive lncRNA.13,14 Repression by Airn occurs predominantly, if not exclusively, in cis, on the same chromosome from which the lncRNA is transcribed.13,14
The mechanism by which Airn mediates repression over its 15-Mb target domain is not clear. Airn’s lack of conservation, lack of splicing, and the instability and variable length of its RNA product have raised questions about whether it is the Airn lncRNA itself or merely the act of its transcription that mediates repression.15,16 Accumulating data are supportive of a role for the Airn lncRNA product in mediating long-range repression,14,17,18 yet it remains unclear what properties of the RNA may enable it to do so. Moreover, the intensity of repression across the Airn target domain is non-uniform,14 implying that features of the genome influence repression by Airn in ways that are not yet clear.
For its full repressive effect, Airn requires several histone-modifying enzymes, including the Polycomb repressive complexes (PRCs), which are known to be recruited to chromatin by the expression of a handful of other lncRNAs, including Xist.19–21 There are two major PRCs, PRC1 and PRC2, each of which can be classified into different sub-complexes that contain core and auxiliary components.22 PRC1 monoubiquitinates histone H2A at lysine 119 (H2AK119ub) and is composed of canonical and variant complexes termed cPRC1 and vPRC1, respectively. PRC2 tri-methylates histone H3 at lysine 27 (H3K27me3) and is composed of sub-complexes called PRC2.1 and PRC2.2. The different auxiliary factors that distinguish PRC sub-complexes modulate their enzymatic activities, interaction partners, effects on three-dimensional (3D) genome organization, and ultimately, effects on gene expression.22 For example, specific forms of vPRC1 have been shown to interface the most closely with the lncRNA Xist.23,24
We previously found that in mouse trophoblast stem cells (TSCs), expression of Airn represses genes and induces the deposition of H2AK119ub and H3K27me3 in a highly non-uniform fashion across a 15-Mb target domain.14 Intensity of gene repression and PRC-directed modifications could be modulated by altering levels of Airn transcription from its endogenous promoter, supporting a role for the Airn lncRNA product in PRC recruitment and indicating that chromatin within the Airn target domain is highly sensitive to levels of Airn. Within the domain, the regions most highly decorated in PRC-deposited modifications centered around a subset of CpG island (CGI) promoters bound by the catalytic components of PRC1 and PRC2, even on the maternal allele, which does not express Airn. These and other data led us to hypothesize that the non-uniform repression across the Airn target domain was mediated by DNA regulatory elements that preferentially contact the Airn locus through 3D space and focus Airn’s repressive activity over certain genomic regions.
Herein, we set out to test that hypothesis and examine in greater detail the extent to which repression by Airn is influenced by chromatin architecture and underlying features of the genome. In TSCs and mouse embryonic stem cells (ESCs), we found that variation in repression across the domain could be partly explained by 3D DNA contacts that exist in the absence of Airn expression, which appear to bring certain genomic regions in closer proximity to the Airn locus over others. Regions within the domain that associated the most robustly with the Airn lncRNA product associated the most robustly with PRC1 and PRC2. Seemingly similar DNA regulatory elements located in different regions within the target domain had different effects on Airn-dependent repression, possibly by modulating local PRC recruitment or frequency of contact between Airn and target DNA. Our data support the notion that the Airn lncRNA product recruits several forms of PRC1 and PRC2 to chromatin and demonstrates that DNA regulatory elements can control the regional intensity with which it does so.
RESULTS
Airn expression induces large-scale changes to chromatin architecture
Because Airn is monoallelically expressed and cis acting, its target domain exists in different states on each allele; the paternal allele being repressed by Airn, and the maternal allele existing in the non-repressed state. For this reason, we and others have studied Airn in F1-hybrid cell lines or animals derived from different strains of inbred mice.13,14,17 In F1-hybrids, physical events associated with maternal and paternal alleles can be distinguished by single-nucleotide polymorphisms (SNPs) in sequencing reads.25
To determine how Airn expression alters chromatin architecture, we performed in situ Hi-C26 in three F1-hybrid TSC lines: one line derived from a cross between a CAST/EiJ mother and C57BL/6J father (C/B TSCs), one derived from the reciprocal cross, a C57BL/6J mother and CAST/EiJ father (B/C TSCs), and a third TSC line in which we previously used CRISPR to insert a triple-polyadenylation cassette ~3 kb downstream of the Airn transcription start site in C/B TSCs, generating a mutant with an expected null phenotype (C/B Airn truncation TSCs8,14). Hi-C libraries were generated in biological triplicate from each TSC line and sequenced to an aggregate depth per genotype of at least 1.6 billion paired-end 150-nt reads (Table S1).
We constructed allele-specific contact maps to examine how Airn expression alters chromatin architecture across its target domain.27,28 In C/B wild-type TSCs, on the paternal B6 allele, which expresses Airn, relative to the maternal CAST allele, which does not, we observed a reduction in short-range DNA contacts concomitant with an increase in long-range contacts, which were largely contained within the 15-Mb target domain previously shown to be repressed by Airn in TSCs (Figures 1Ai and 1Bi14). The greatest increases in contact occurred within a 4.5-Mb interval that extended from the Airn locus and terminated at the genes Prr18, T, and Pde10a (Figures 1Ai and 1Bi). In Airn truncation TSCs, the increased contacts were not detectable, demonstrating their dependence on Airn expression (Figures 1Aii and 1Bii). Moreover, the overall trends that were observed in C/B wild-type TSCs were also observed in the reciprocal F1-hybrid cell line, B/C wild-type TSCs, in which the paternal, Airn-expressing allele is of CAST origin, confirming that differences in chromatin architecture between paternal and maternal alleles are due to parent-of-origin and not strain-specific effects (Figures 1Aiii and 1Biii). The regions that underwent the strongest Airn-dependent changes in contact frequency were the ones that most clearly shifted from the “A” to the “B” chromosome compartment specifically on the alleles that expressed Airn (Figure 1Ci–iii; Figure S1,26,29). Thus, in TSCs, Airn expression induces large-scale changes to chromatin architecture that are largely contained within a 15-Mb genomic interval previously shown to be subject to Airn-dependent repression.13,14
Figure 1. Airn expression induces large-scale changes to chromatin architecture.

(A) Hi-C contact heatmaps of allelic observed counts in (i) C/B wild-type, (ii) C/B Airn truncation, and (iii) B/C wild-type TSCs; n = 2 or 3. Allelic heatmaps are partitioned and at 50-kb resolution.
(B) Subtraction contact heatmaps of log2-transformed (PAT – MAT) observed counts (i–iii) as in (A).
(C) Eigenvectors at 50-kb resolution for “A” and “B” chromosome compartmentalization.26
In heatmaps: dotted lines, 15-Mb Airn target domain; purple circle, Airn gene; green circles, other loci of interest. Datasets used are listed in Table S4. KR bal., Knight-Ruiz balanced. See STAR Methods for detailed description of analyses. See also Figure S1.
Airn-dependent changes in chromatin architecture coincide with the presence of PRC-deposited modifications
To understand how DNA contacts with the Airn locus correlate with Airn-dependent repression, we created a series of “viewpoint” plots, in which contacts between Airn and surrounding regions were extracted and visualized in two dimensions. Consistent with the heatmaps of Figure 1, we observed a dramatic increase in Airn-dependent contacts with the Airn locus on the paternal allele toward the centromeric end of chr17, including a pronounced shoulder of increased contacts surrounding the genes Prr18, T, and Pde10a (Figure 2A). We also observed augmented contacts between Airn and the gene Qk on both alleles in all TSC lines profiled (Figure 2A). In C/B wild-type TSCs, the intensity of allele-specific contacts with Airn as measured by Hi-C correlated remarkably well with allele-specific distances to the Airn locus that we had previously measured by DNA fluorescence in situ hybridization (FISH), corroborating both forms of measurement (Figure 2B; Spearman’s r of −0.82 and −0.95 and p = 0.007 and 0.001 on maternal and paternal alleles, respectively; FISH probe locations from Schertzer et al.14 shown in Figure 2Ai).
Figure 2. Airn-dependent changes in chromatin architecture coincide with the presence of PRC-deposited modifications.
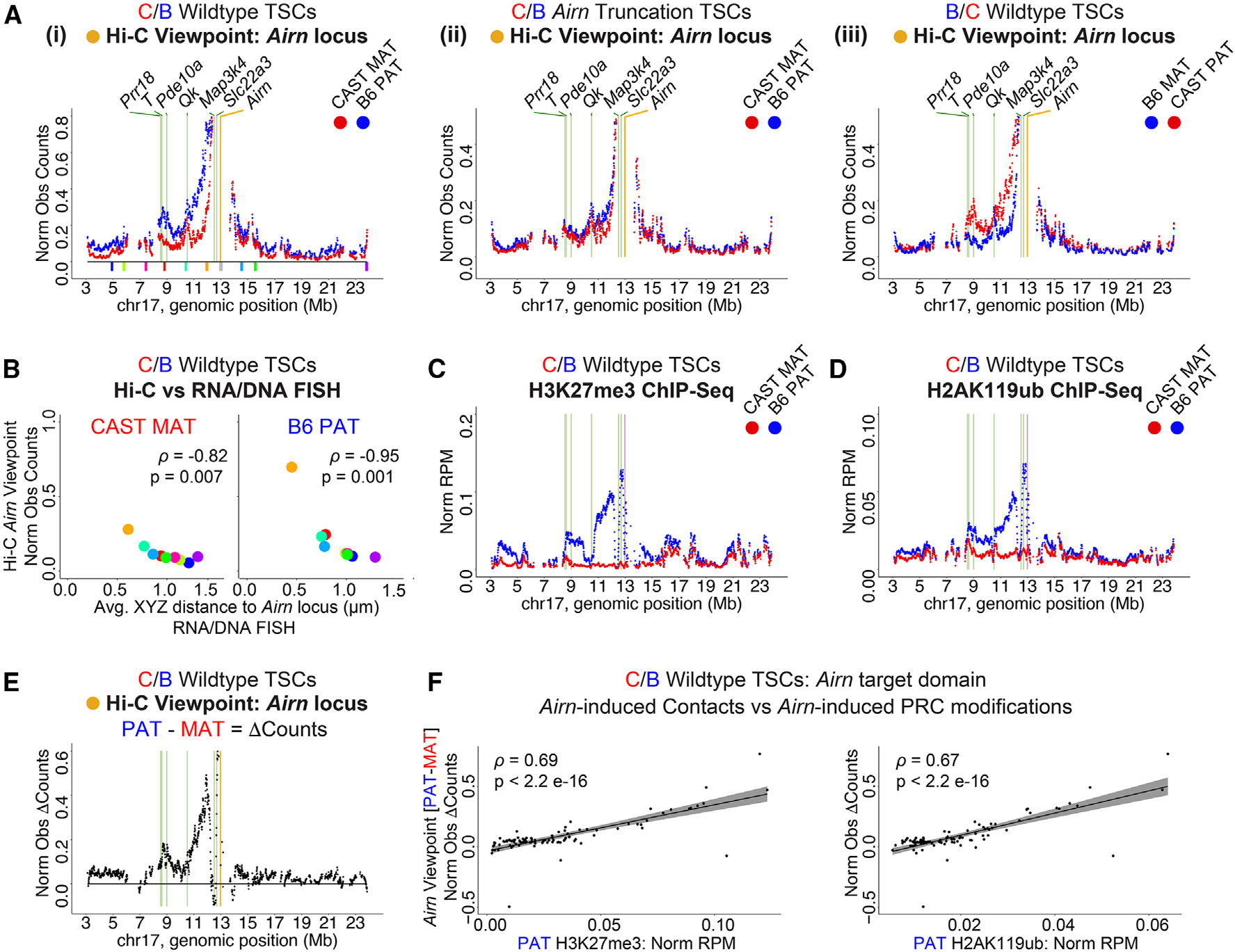
(A) Tiling density plots of allelic Hi-C Airn viewpoint observed contact counts in (i) C/B wild-type, (ii) C/B Airn truncation, and (iii) B/C wild-type TSCs. Colored blocks in (i), and FISH probes analyzed in (B).14
(B) Allelic Hi-C Airn viewpoint contacts from (Ai) vs. average distance to Airn measured by RNA/DNA FISH in C/B wild-type TSCs from Schertzer et al.14 Spearman’s r and p values are shown.
(C and D) Tiling density plots of allelic (C) H3K27me3 ChIP-seq and (D) H2AK119ub ChIP-seq signal in C/B wild-type TSCs. Data are from Schertzer et al.14 n=4 and 2.
(E) Tiling density plot of allelic Airn viewpoint (PAT – MAT) observed counts in C/B wild-type TSCs from (Ai).
(F) Scatterplots of Airn viewpoint (PAT – MAT) observed counts vs. (left) H3K27me3 and (right) H2AK119ub. Spearman’s r and p values are shown.
In tiling plots: yellow/purple bar, Airn viewpoint/gene; green bars, other loci of interest. Datasets used are listed in Table S4. Norm Obs Counts, counts normalized for SNP density; Norm RPM, reads per million total reads normalized for SNP density. See STAR Methods for detailed description of analyses. See also Figure S2.
The magnitude of Airn-dependent contacts (i.e., paternal contacts subtracted from maternal contacts) also correlated well with the underlying intensity of Airn-dependent, PRC-directed chromatin modifications (Figures 2C–2F; Spearman’s r between Airn-dependent contacts and H3K27me3 and H2AK119ub in C/B TSCs, 0.69 and 0.67, respectively, p < 2e–16 for both comparisons). Moreover, we used chromatin immunoprecipitation sequencing (ChIP-seq) to profile seven individual components of PRC1 and PRC2 (RING1B, RYBP, CBX7, KDM2B, EZH2, MTF2, and JARID2). Within the target domain, six out of seven PRC components exhibited a signature of Airn-responsiveness, defined as a broad shoulder of paternal enrichment on the centromeric side of the Airn locus (Figure S2A). The same six PRC components appeared responsive to Xist (Figure S3). The sole exception was the PRC1.1 component KDM2B (Figures S2A and S3). Because the PRCs and their modifications to chromatin can induce DNA compaction,7,22,26 and Airn expression correlates with the presence of PRCs within its target domain, our data are consistent with the notion that the large-scale changes in genome architecture induced by expression of Airn depend, at least in part, on the PRCs and their modifications to chromatin.
Airn-dependent repression centers around regions that form pre-existing contacts with the Airn locus and harbor CGIs bound by vPRC1
DNA contacts detected by Hi-C can occur by chance with a frequency that increases with decreasing distance from the locus in question.26 To quantify the relative intensity of contacts that occur with the Airn locus after correcting for distance-dependent effects, we created a series of observed-over-expected (O/E) plots with Airn as the viewpoint, in which detected contacts were normalized by those expected from a distance-dependent decay model.26
These O/E viewpoint plots revealed three local maxima of contact with Airn that fell within the 4.5-Mb genomic interval most intensely repressed by Airn, extending from the Airn locus and terminating at Prr18, T, and Pde10a (Figures 2, 3A, and S4). Specifically, maxima were detected surrounding the genes Prr18/T/Pde10a, the gene Qk, and the gene Slc22a3 (Figures 3A and S4). Although intensity of O/E contact with Prr18/T/Pde10a increased dramatically upon expression of Airn, intensity of O/E contact with Qk and Slc22a3 changed to lesser extents or not at all (Figures 3A and S4). All three maxima were present in Airn truncation TSCs, highlighting contact with the Airn locus even in the absence of Airn expression (Figures 3Aii and S4). Each gene within these maxima is driven by CGI promoters that we found in previous work to either bind high levels of RING1B and EZH2 (in the cases of Prr18/T/Pde10a and Qk) or be present in the region of the Airn target domain that accumulates the highest levels of Airn-dependent, PRC-directed chromatin modifications (in the case of Slc22a3) (Figure 2).14
Figure 3. Airn-dependent repression centers around regions that form pre-existing contacts with the Airn locus and harbor CGIs bound by vPRC1.

(A) Tiling density plots of allelic Hi-C Airn viewpoint observed-over-expected (O/E) contact counts in (i) C/B wild-type, (ii) C/B Airn truncation, and (iii) B/C wild-type TSCs. y axes as in Figure 2. Yellow bar, viewpoint; green bars, other loci of interest.
(B) Genome browser graphics of regions harboring peaks of O/E contact. ChIP-seq tracks, non-allelic read density from C/B wild-type TSCs; n = 2 or 3. Red or blue asterisks, significant enrichment on maternal or paternal alleles, respectively (p < 0.05, permutation). Pink rectangles, DNA regions deleted in Figures 5, 6, and 7.
Datasets used are listed in Table S4. See STAR Methods for detailed description of analyses. See also Figure S4 and Table S3.
We were intrigued that points of contact with Airn centered around PRC-bound CGIs. CGIs often mark high-density sites of PRC binding, and PRCs and the modifications that they deposit on chromatin can mediate long-range 3D contacts independently of CTCF and Cohesin.30–39 Moreover, the genes within the points of contact–Pde10a, Qk, and Slc22a3–are all repressed by Airn in TSCs14 (Table S2). Also, the Airn gene body itself harbors two CGIs that bind RING1B/PRC1,14 and the Airn lncRNA has previously been found to associate with the Slc22a3 CGI.18 Lastly, although the reasons remain unclear, we previously found that deletion of the Slc22a3 CGI resulted in a dramatic loss of Airn-induced accumulation of H3K27me3, most notably in the 4.5-Mb interval beginning at Airn and terminating at Prr18, T, and Pde10a.14 These data raise the possibility that features associated with the CGIs in regions that form augmented DNA contacts with the Airn locus play roles in modulating the local intensity of Airn-dependent repression.
With that possibility in mind, we used ChIP-seq to examine what factors and chromatin modifications were enriched over CGIs contained within points of 3D contact with Airn. These included CGIs at Pde10a, Qk, Slc22a3, and Airn itself, as well as the CGI promoter of the gene Map3k4. Like Qk, Map3k4 is repressed by Airn and forms a detectable contact with the Airn locus by Hi-C (Figures 3A and S4B) but partially escapes silencing and sits within a region that resists the local accumulation of Airn-induced, PRC-deposited chromatin modifications (Figures 2C and 2D; Table S2). In total, we examined ChIP-seq data for the same seven PRC components whose allelic tiling density profiles are shown in Figures S2 and S3, four chromatin modifications (H3K27me3, H2K119ub, H3K4me2, and H3K27ac), and two architectural factors (SMC1A/Cohesin and CTCF; data from this study, Schertzer et al.,14 and Calabrese et al.40). A summary of the allele-specific enrichment of each factor is found in Table S3, and the total, non-allelic genome browser density tracks are shown in Figure 3B. The asterisks above each CGI indicate whether the factor was detected on the maternal allele, paternal allele, or both (Figure 3B; Table S3).
Although there was not one singular pattern of enrichment, notable similarities emerged. Each CGI except for the one found at the promoter of Airn showed some level of peak-like enrichment for vPRC1 on the maternal allele (Figure 3B). Likewise, all of the CGIs examined except for the one at the Airn promoter showed peak-like enrichment of at least one of the two chromatin modifications associated with transcriptional activation (H3K4me2 or H3K27ac), also on the maternal allele. SMC1A/Cohesin was also detected on the maternal allele of these same sets of CGIs, although its intensity of enrichment was low relative to intergenic peaks (Figure 3B). Thus, the regions within the target domain that form 3D contacts with the Airn locus on the maternal allele all harbor CGI promoters that are enriched in vPRC1, Cohesin, and signatures of transcriptional activity.
In contrast, CGIs in the region that underwent the strongest Airn-dependent changes in chromatin architecture, surrounding the genes Prr18, T, and Pde10a, were associated with sharp peaks of cPRC1 and PRC2, as well as vPRC1 (Figures 3B and S2A). Of those CGIs, the CGI at Pde10a was associated with the highest levels of both PRC1 and PRC2 (Table S3). Likewise, the intergenic regions that underwent the strongest Airn-dependent changes in chromatin architecture were similarly enriched in cPRC1 and PRC2, as well as vPRC1 (Figure S2A). Thus, although the presence of vPRC1, Cohesin, and transcription can identify regions that contact the Airn locus in the absence of Airn expression, the presence of cPRC1 and PRC2 at both CGIs and intergenic regions correlate more strongly with the presence of Airn-dependent, PRC-deposited modifications and long-range changes to architecture.
Presence of Airn lncRNA on chromatin correlates with the presence of PRC1 and PRC2 and centers around pre-existing contacts with the Airn locus
We next sought to determine whether the Airn lncRNA itself was preferentially associated with specific DNA regions. To address this question, we used CHART-seq, an approach to identify genomic regions located proximal to lncRNAs.41 We performed CHART-seq for Airn (Figure S5A) in C/B wild-type TSCs, in Airn truncation TSCs, and in a C/B TSC line from Schertzer et al.,14 in which we used CRISPR-Cas9 to over-express Airn from its endogenous promoter (Airn highly expressing [H-E] TSCs). qRT-PCR confirmed expected levels of Airn expression in each TSC line (Figure S5B).
In C/B wild-type TSCs, Airn CHART-seq revealed enrichment of DNA on the paternal, but not maternal, allele across the Airn target domain, beginning near the centromere and ending ~3 Mb downstream of the Airn locus, the same region where the last Airn-induced PRC-dependent modifications are visible (Figures 4Ai, 4Bi, S5C, and S5D). In H-E TSCs, the enrichment increased with over-expression of Airn (Figures 4Aii, S5C, and S5D). Conversely, in Airn truncation cells, DNA enrichment from the paternal allele was lost (Figures 4Aiii, S5C, and S5D). These data indicate that DNA recovered by CHART is sensitive to overall levels of Airn expression. Moreover, we observed a strong correlation between paternal Airn CHART-seq and H3K27me3, RING1B, and EZH2 ChIP-seq signal throughout the target domain (lowest Spearman’s r across C/B wild-type and H-E TSC comparisons is 0.53, p < 2e−9 for all comparisons; Figures 4A–4D). Calibrated and non-calibrated ChIP-seq for H3K27me3 and EZH2 showed the same patterns of enrichment (Figures 4B and 4D vs. S2B and S2C). We were especially struck by the lower CHART-seq signal in H-E TSCs that began just upstream of Map3k4 and Qk, genes that are repressed by Airn but located at inflection points where the intensity of PRCs and PRC-directed modifications drops precipitously (Figures 4A–4D). Likewise, particularly in H-E TSCs, local maxima of Airn lncRNA association coincide with genomic regions that form local maxima of Hi-C O/E contacts with the Airn locus even in the absence of Airn expression (Figures 4A and 3Aii).
Figure 4. Presence of Airn lncRNA on chromatin correlates with presence of PRC1 and PRC2 and centers around pre-existing contacts with the Airn locus.

(A–D) Tiling density plots of allelic (A) Airn CHART-seq, (B) H3K27me3 ChIP-seq, (C) RING1B ChIP-seq, and (D) EZH2 ChIP-seq signal in C/B (i) wild-type, (ii) Airn highly expressing (H-E), and (iii) Airn truncation TSCs; n = 1 or 2. y axes are as in Figure 2. H3K27me3 and RING1B ChIP-seq data are from Schertzer et al.14
(E and F) Tiling density plots of (E) Airn CHART-seq and (F) H3K27me3 ChIP-seq signal in untreated (Airn OFF) or dox-treated (Airn ON) H-E ESCs; n = 1.
(G) Tiling density plot of Hi-C Airn viewpoint O/E counts in ESCs.42 Yellow/purple bar, Airn viewpoint/gene; green bars, other loci of interest.
Datasets used are listed in Table S4. See STAR Methods for detailed description of analyses. See also Figures S2 and S5.
We also performed Airn CHART-seq in mouse ESCs, in which we used CRISPR-Cas9 to force expression of Airn from its endogenous promoter (H-E ESCs). In H-E ESCs, Airn was expressed at a level approximately equal to wild-type TSCs (Figure S5B). As a negative control, we performed CHART-seq in uninduced ESCs, which do not express meaningful levels of Airn. We also performed ChIP-seq to determine the extent to which DNA retrieved by CHART correlated with intensity of H3K27me3. We observed associations between the Airn lncRNA and DNA in H-E ESCs across a domain that was remarkably similar in size and contour to the domain associated with Airn in TSCs (Figure 4F vs. 4A). Also, as in TSCs, Airn associations were significantly correlated with underlying H3K27me3 (Figure 4F vs. 4G; Spearman’s r = 0.64, p = 7.9e–14), and Map3k4 and Qk resisted Airn and PRC-directed modifications in H-E ESCs (Figures 4E and 4F). Furthermore, we observed that local maxima of Hi-C O/E contacts with the Airn locus in ESCs coincided or were proximal to local maxima of Airn lncRNA association and H3K27me3, most notably at Prr18/T/Pde10a, Slc22a3, and on the telomeric side of Airn, in a region surrounding the genes Dact2 and Wdr27 (Hi-C;42 Figure 4G vs. 4E and 4F). The only exception to this pattern was at Qk, which despite forming DNA contacts with the Airn locus, resisted contacting the Airn lncRNA and accumulating PRC-directed modifications in both TSCs and ESCs.
Altogether, our data support the notion that a major function of the Airn lncRNA product is to recruit the PRCs to chromatin over a 15-Mb domain, and that proximity to the Airn lncRNA product dictates the intensity with which the recruitment occurs. Moreover, pre-existing DNA contacts, those that occur with the Airn locus in the absence of Airn expression, center within regions of chromatin that become decorated in PRC-directed modifications upon Airn expression. Lastly, Map3k4 and Qk resist associations with the Airn lncRNA despite forming DNA contacts with the Airn locus.
DNA regulatory element deletions alter levels of PRC-directed modifications and gene repression throughout the Airn target domain
Our data indicate that regional proximity to the Airn lncRNA product correlates with local intensity of gene repression and PRC recruitment. To study the extent to which DNA regulatory elements might control proximity to Airn, we focused on the region surrounding the genes Prr18/T/Pde10a, which harbors several CGIs that bind PRCs and undergoes increased frequency of 3D contact with the Airn locus upon Airn expression (Figures 2 and 3). We used CRISPR-Cas9 to individually delete the CGI promoters of T and Pde10a, as well as a 190-kb cluster of intergenic CTCF and SMC1A/Cohesin peaks located between T and Pde10a (pink rectangles in Figures 3B and S6A–S6C). We derived heterozygous clonal TSC lines harboring deletions for each element on their paternal alleles (ΔT, four lines; ΔPde10a, two lines; ΔCluster, two lines; Figures S5A–S5C). As controls, we derived four clonal non-targeting (NTG) TSC lines that harbor the same doxycycline-inducible Cas9 transgene and underwent the same process of electroporation, clonal selection, and induction as above, but that express a NTG sgRNA that does not match the mouse genome (Figures S6A–S6D; Table S5). In addition, we revived the two TSC lines in which we previously deleted the Slc22a3 CGI on the paternal allele (ΔSlc22a3 TSCs from Schertzer etal.14; Figure S6D). In that study, we found that Slc22a3 CGI deletion caused a ~4.5-Mb reduction in the intensity of H3K27me3, beginning essentially at the Slc22a3 gene and extending through the distal cluster of PRC-bound CGIs at Prr18/T/Pde10a.14 qRT-PCR and RNA sequencing (RNA-seq) showed that Airn expression levels varied by no more than 2-fold across our panel of lines (Figure S6E).
We performed H3K27me3 and H2AK119ub ChIP-seq, as well as RNA-seq, to examine how the deletions affected PRC activity and gene repression. Relative to NTG controls, ChIP-seq in ΔSlc22a3 TSCs revealed a dramatic loss of H3K27me3 and H2AK119ub throughout the Airn target domain (Figure 5A vs. S6F and S6G), consistent with and extending results from Schertzer et al.14 However, whereas deletion of the T CGI had little to no effect, deletion of the Pde10a CGI unexpectedly caused a dramatic increase in the levels of H3K27me3 and H2AK119ub throughout the target domain, opposite to that observed in ΔSlc22a3 TSCs (Figures 5B, 5C, and S6G). Deletion of the cluster of CTCF and SMC1A/Cohesin peaks similarly increased H3K27me3 and H2AK119ub (Figure 5D). Cross-genotype comparisons within the target domain and across the remainder of chr17 are shown in Figure S6G.
Figure 5. DNA regulatory element deletions alter levels of PRC-directed modifications and gene repression throughout the Airn target domain.

(A–D) Tiling density plots of allelic (left) H3K27me3 and (right) H2AK119ub ChIP-seq signal in C/B (A) ΔSlc22a3, (B) ΔT, (C) ΔPde10a, and (D) ΔCluster TSCs; n = 1 per clonal line. y axes are as in Figure 2. Data from NTG clones were averaged; n = 4.
(E) Paternal expression of the 27 Airn target genes across genotypes relative to NTG; n = 1, 2, or 4. Asterisks, significant changes relative to NTG (p < 0.05, Welch two-sample t test).
In tiling plots: purple bar, Airn gene; green bars, other loci of interest; pink bars, DNA regions deleted. Datasets used are listed in Table S4. See STAR Methods for detailed description of analyses. See also Figure S6 and Table S2.
RNA-seq from deletion clones showed changes in gene expression consistent with changes in PRC-deposited modifications (Figure 5E vs. 5A–5D). In our previous study, we identified 27 genes within the target domain that were subject to repression by Airn.14 In ΔSlc22a3 TSCs, the relative paternal expression of these 27 genes increased significantly compared with their baseline in NTG, up to an average level that was slightly less than that observed in Airn truncation TSCs, which are effectively null mutants14 (NTG vs. ΔSlc22a3 [A12 and A13 clones], p = 0.038 and 0.025, respectively, Welch two-sample t test; Figure 5E). Conversely, in ΔPde10a and ΔCluster TSCs, paternal expression of these genes decreased relative to NTG TSCs, albeit with variability between clones (NTG vs. ΔPde10a and NTG vs. ΔCluster; Figure 5E). The decreased expression in ΔPde10a and ΔCluster TSCs was not as strong as that observed in Airn H-E TSCs (Figure 5E) but was nevertheless consistent with the increase in PRC-deposited modifications (Figures 5C and 5D).14 Thus, seemingly similar DNA regulatory elements play critical but different roles in dictating the regional intensity of gene repression and PRC recruitment induced by Airn within its 15-Mb target domain.
Changes in DNA contacts with Airn mirror changes in PRC activity caused by regulatory element deletion
To gain insight into the effects caused by regulatory element deletion, we used in situ Hi-C to examine DNA contacts in NTG, ΔSlc22a3, and ΔPde10a TSCs (Table S1). Consistent with changes in gene repression and PRC-deposited modifications, deletion of the Slc22a3 and Pde10a CGIs alternately diminished and increased Airn-dependent contacts across the entire target domain, coincident with corresponding changes in the intensities of compartmentalization (Figures 6A, 6B, and S7).
Figure 6. Changes in DNA contacts with Airn mirror changes in PRC activity caused by regulatory element deletion.

(A) Hi-C subtraction contact heatmaps of log2-transformed (PAT – MAT) observed counts in C/B (i) NTG, (ii) ΔSlc22a3, and (iii) ΔPde10a TSCs; n = 2.
(B) Eigenvectors at 50-kb resolution for “A” and “B” chromosome compartmentalization (i–iii) as in (A).
(C) Tiling density plots of allelic Airn viewpoint observed contact counts (i–iii) as in (A). y axes as in Figure 2.
(D) Tiling density plots of allelic Airn viewpoint (PAT – MAT) observed counts (i–iii) as in (A).
In heatmaps: dotted lines, 15-Mb Airn target domain; purple circles, Airn gene; green circles, other loci of interest. In tiling plots: yellow bar, Airn locus viewpoint; green bars, other loci of interest; pink bar, DNA region deleted. Datasets used are listed in Table S4. See STAR Methods for detailed description of analyses. See also Figure S7.
Examining contacts from the Airn viewpoint provided additional insights (Figures 6C and 6D). Deletion of the Slc22a3 CGI was coincident with reduced levels of Airn-dependent contacts throughout the domain, with a possible exception at Qk (Figures 6Cii and 6Dii). Proportionally, the greatest decreases in Airn-dependent contacts surrounded Prr18/T/Pde10a (Figures 6Cii and 6Dii). In contrast, in ΔPde10a cells, Airn-dependent contacts increased uniformly except at Prr18/T/Pde10a (Figures 6Ciii and 6Diii). Thus, deletion of the Slc22a3 CGI reduced the interaction between Airn and DNA throughout its target domain, whereas deletion of the Pde10a CGI increased the interaction between Airn and all other regions in the domain save Prr18/T/Pde10a.
Airn expression is coincident with dissolution of DNA loops encasing Slc22a3 and a local increase in PRC-directed modifications
We next examined our data to determine how deletion of the Slc22a3 CGI might restrict repression by Airn. In our major Hi-C datasets (described in Figure 1), we noted that the Slc22a3 and Airn genes are located within the same contact domain (a contiguous region exhibiting high levels of inter-locus interactions), where they sit within nested DNA loops anchored by CTCF and Cohesin (Figures 7A and 7B). By Hi-C, the loops that surround Slc22a3 were reduced by Airn expression, to the extent that they are no longer detectable by the significant interaction peak (SIP) algorithm on Airn-expressing alleles (Figure 7A).43 Likewise, we observed a relative reduction in SMC1A and CTCF binding at those same loop anchors, again on Airn-expressing alleles, consistent with the reduced loop intensity (Figure 7B). We also searched for these DNA loops in NTG, ΔSlc22a3, and ΔPde10a Hi-C data, but lower sequencing depth (~600 million read pairs per genotype) precluded a high-confidence analysis. Nonetheless, visual inspection of loops identified in our high-depth datasets was consistent with the notion that the Slc22a3 CGI is required for Airn-dependent dissolution of the nested loops encasing Slc22a3 (Figure 7C, note relative increase in intensity at loop anchor regions in the ΔSlc22a3 genotype, denoted by the two gray arrows). We also observed that deletion of the Slc22a3 CGI led to a local drop in H3K27me3 and had a lesser yet significant effect on H2AK119ub (Figure 7D). Thus, in contrast with the Pde10a CGI, which may effectively restrict surrounding regions from contacting Airn through mechanisms that remain to be determined, it is conceivable that prior to Airn expression, nested loops that encase Slc22a3 and Airn may reduce the latter’s ability to interact with distal DNA. Upon Airn expression, recruitment of PRCs locally, promoted by the Slc22a3 CGI, could antagonize Cohesin37–39 and disrupt loops that then enable Airn to contact distal regions of chromatin more efficiently.
Figure 7. Airn expression is coincident with dissolution of DNA loops encasing Slc22a3 and a local increase in PRC-directed modifications.

(A) Allelic Hi-C contact heatmaps of the Airn contact domain at 5-kb resolution in (i) C/B wild-type, (ii) C/B Airn truncation, and (iii) B/C wild-type TSCs. Black arrows, SIP-called DNA loops.
(B) SMC1A/Cohesin and CTCF ChIP-seq. Percent parental biases at loops (i and ii) as in (A).
(C) Allelic contact heatmaps of Airn contact domain at 5-kb resolution in C/B (i) NTG, (ii) ΔSlc22a3, and (iii) ΔPde10a TSCs. Gray arrows, DNA loops of interest from (A). In heatmaps: circles, 5′ end of genes/CGIs; rectangles, gene bodies; purple, Airn gene; green, Slc22a3 gene.
(D) Boxplots of average allelic (left) H3K27me3 and (right) H2AK119 ChIP-seq signal over Airn contact domain in NTG, ΔSlc22a3, and ΔPde10a TSCs; n = 2. Asterisks, significant changes relative to NTG (p < 0.05, Welch two-sample t test). Error bars, data points outside the interquartile range.
(E) Model: DNA regulatory elements modulate frequency of contact with and repression by Airn. (i) On maternal allele, pre-existing contacts with Airn locus render certain regions more susceptible torepression than others. (ii) On paternal allele, PRCs and PRC-deposited modifications to chromatin increase contacts with Airn locus, which in turn increase intensity of long-distance repression. (iii) Loss of Slc22a3 CGI attenuates repression by reducing local PRC recruitment and contact with distal regions. (iv) Loss of Pde10a CGI increases frequency with which surrounding regions contact Airn.
Datasets used are listed in Table S4. See STAR Methods for detailed description of analyses.
DISCUSSION
We report a series of intriguing relationships between 3D DNA contacts, DNA regulatory elements, and PRC recruitment within the largest autosomal region known to be repressed by a mammalian lncRNA. Our results support the view that Airn is a potent cis-acting lncRNA that functions to maintain gene repression and recruit the PRCs within a 15-Mb domain on mouse chr17. We show that extent of repression maintained by Airn can be modulated by discrete DNA regulatory elements that control the proximity of Airn to its genomic targets, a paradigm likely relevant to other domains governed by strong locus control regions, including the inactive X chromosome.
Using in situ Hi-C, we observed that expression of Airn is accompanied by changes in 3D contacts and compartmentalization on the centromeric side of the Airn locus. These changes correlated in step with the intensity of Airn-induced H3K27me3 and H2AK119ub and centered around three regions that contact the Airn locus even in the absence of Airn expression. Each of these regions harbors CGI promoters that bind components of vPRC1, exhibits signatures of transcriptional activity, and is located proximal to peaks of Cohesin on both the maternal and the paternal alleles. Extent of association with the Airn lncRNA, as assessed by CHART-seq, also correlated in step with the intensity of PRCs and PRC-directed modifications and centered around pre-existing DNA contacts, in both TSCs and ESCs. Of the seven different PRC subunits we profiled by ChIP-seq, all except KDM2B were responsive to Airn. Two genes, Map3k4 and Qk, resisted accumulating PRC-directed modifications, despite both genes being repressed by Airn and forming contacts with the Airn locus. Moreover, the intensity of DNA contacts between Map3k4, Qk, and the Airn locus was relatively unchanged by Airn expression, and relative to surrounding regions, Map3k4 and Qk resisted association with the Airn lncRNA.
Together, our data support the notion that spatial proximity to the Airn lncRNA product induces gene repression and, in most cases, also induces the accumulation of multiple forms of PRC1, PRC2, and PRC-directed chromatin modifications. Airn is a short-lived RNA that does not diffuse away from its site of transcription.12,14 Thus, consequently, the regions that are most sensitive to repression by Airn are predisposed to contacting the Airn locus even in the absence of Airn expression. In turn, considering our findings along with prior data showing that the PRCs and their modifications to chromatin can induce DNA compaction,7,22,26 it seems likely that Airn-recruited PRCs and the modifications they deposit on chromatin are responsible for the major changes in chromatin architecture induced by Airn expression. Such changes would presumably potentiate Airn-dependent repression, stabilizing the process by positive feedback.
Three notable regions within the Airn target domain, encompassing the genes Slc22a3, Qk, and Prr18/T/Pde10a, exhibited augmented contacts with the Airn locus even in the absence of Airn expression. Of these regions, only Slc22a3 formed a detectable DNA loop anchored at the Airn locus.43 However, each of the regions harbored CGI promoters that themselves were associated with vPRC1, signatures of transcriptional activity, and nearby peaks of Cohesin. Prior works would suggest that any or all of these features could facilitate interactions between the regions and Airn DNA.26,31,44 Upon expression of Airn, the region encompassing Prr18/T/Pde10a exhibited peak-like increases in contact with the Airn locus, whereas the intensity of detectable contacts with Qk and Slc22a3 remained relatively unchanged. Deletion of specific DNA regulatory elements within the Prr18/T/Pde10a and Slc22a3 regions affected the ability of Airn to repress genes, induce PRC-directed modifications, and induce changes to chromatin architecture over megabases. Thus, DNA elements shape long-range contacts within the Airn target domain in ways that extend beyond single loop-based models of regulation.44
Considered together, our data support the view that the extent of repression across the Airn target domain is governed by an equilibratory network of DNA regulatory elements that through direct or indirect means controls spatial proximity to the Airn lncRNA product (Figure 7E). Shifting the equilibrium in either direction has consequences on gene expression, chromatin modifications, and chromatin architecture. Indeed, we identified one CGI that appears to promote certain long-range contacts while restricting others (Pde10a) and another CGI that may promote long-range contacts by serving as a local Polycomb response element (Slc22a3; Figure 7E).45 To the latter point, a prior study demonstrated that Airn DNA preferentially interacts with the Slc22a3 locus when the Airn lncRNA is not expressed,17 consistent with a model whereby Airn-induced, PRC-directed chromatin modifications at Slc22a3 help to disengage the Airn locus from local DNA interactions and promote distal ones. Meanwhile, the CGI promoters of Map3k4 and Qk gave the appearance of serving as boundary elements that attenuate local spread of repression by Airn. Our data suggest that variation in the genetic or epigenetic content of a DNA regulatory element has the potential to control gene expression by altering spatial equilibria between genes and locus control regions, be those repressors or enhancers.46–51 By extension, unrecognized alterations to spatial equilibria that modulate contact with locus control regions may contribute to the challenge of assigning target genes to disease-associated SNPs.52
Repression of the Airn-target gene Igf2r is due to the act of Airn transcription and does not depend on the Airn lncRNA product.15 The mechanisms responsible for long-range repression by Airn remain unclear. Our observation that Airn lncRNA association and PRC-directed chromatin modifications correlate in lock-step over a 15-Mb domain in both TSCs and ESCs, together with data from Schertzer et al.,14 Andergassen et al.,17 and Nagano et al.18 support the idea that Airn is a potent cis-acting repressive lncRNA that recruits the PRCs and possibly other repressive enzymes to chromatin. Future studies are needed to definitively prove this notion and demonstrate the mechanism by which the Airn lncRNA might mediate its longrange repressive effects.
Limitations of the study
Considering that Airn acts largely, if not exclusively, in cis, F1-hybrid TSCs harbor an internal control in all genomic profiling experiments, in the form of the allele on which Airn is not expressed. Throughout our study, we interpret local differences in signal between Airn-expressing and non-expressing alleles to reflect Airn-dependent effects. Within individual TSC lines, these differences can be interpreted as absolute. However, when comparing signals between different TSC lines, particularly on Airn-expressing alleles, our Hi-C, CHART, and almost all ChIPs were performed under conditions that enable us to comment on relative, but not absolute, differences. Also, although the model in Figure 7 provides a parsimonious explanation of our data, our observations regarding 3D DNA contacts are correlative and we have not proven causation. It is also unclear to what extent Airn expression may induce changes in chromatin conformation independent of the PRCs. Lastly, the mechanisms by which Airn might recruit PRCs to chromatin are unclear. It is also unclear what molecular constituents occupy the interface between the Airn lncRNA and its genomic targets. If studies of Xist are any guide,53,54 it is likely that many hundreds of molecules of protein surround each molecule of Airn.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mauro Calabrese (jmcalabr@med.unc.edu).
Materials availability
Plasmids and cell lines generated in this study are available upon request.
Data and code availability
All raw and processed sequencing data generated in this study have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All new and published datasets analyzed are summarized in Table S4. Oligonucleotides used are detailed in Table S5.
All original code has been deposited at Zenodo and Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| H3K27me3 | Cell Signaling | Cat#9733; RRID: AB_2616029 |
| H2AK119ub | Cell Signaling | Cat#8240; RRID: AB_10891618 |
| RING1B | Cell Signaling | Cat#5694; RRID: AB_10705604 |
| CBX7 | Abcam | Cat#ab21873; RRID: AB_726005 |
| DEDAF (RYBP) | Millipore | Cat#AB3637; RRID: AB_11213333 |
| JHDM1B (KDM2B) | Millipore | Cat#09–864; RRID: AB_10806072 |
| EZH2 | Cell Signaling | Cat#5246; RRID: AB_10694683 |
| MTF2 | Proteintech | Cat#16208–1-AP; RRID: AB_2147370 |
| JARID2 | Cell Signaling | Cat#13594; RRID: AB_2798269 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Heparin Sodium Salt | Sigma-Aldrich | Cat#H3149 |
| FGF4 Recombinant Human Protein | Gibco | Cat#PHG0154 |
| RPMI 1640 | Gibco | Cat#11875093 |
| DMEM, high glucose, sodium pyruvate | Gibco | Cat#11995073 |
| Fetal Bovine Serum, qualified | Gibco | Cat#26140079 |
| Penicillin-Streptomycin | Gibco | Cat#15140122 |
| L-glutamine | Gibco | Cat#25030081 |
| MEM Non-essential Amino Acids | Gibco | Cat#11140050 |
| Sodium Pyruvate | Gibco | Cat#11360070 |
| β-mercaptoethanol | Sigma-Aldrich | Cat#63689 |
| LIF | Lif-1Cα/COS-conditioned ESC media | N/A |
| Trypsin-EDTA, phenol red | Gibco | Cat#2520072 |
| LipoFectamine 3000 | Invitrogen | Cat#L3000015 |
| 10X Phosphate-Buffered Saline (PBS) | Corning | Cat#46–013-CM |
| Pierce 16% Formaldehyde | Thermo Scientific | Cat#28906 |
| 37% Formaldehyde | Fisher Scientific | Cat#BP531–500 |
| TRIzol Reagent | Invitrogen | Cat#15596026 |
| Chloroform | Fisher Scientific | Cat#BP1145–1 |
| 25:24:1 Phenol:Chloroform:Isoamyl Alcohol | Sigma-Aldrich | Cat#6805 |
| Protein A/G PLUS Agarose Beads | Santa Cruz Biotechnology | Cat#sc-2003 |
| AMPure XP Beads | Beckman Coulter | Cat#A63880 |
| Dynabeads M-280 Streptavidin | Invitrogen | Cat#11205D |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | Cat#65002 |
| Ribonuclease A (RNase A) from bovine pancreas | Sigma-Aldrich | Cat#R4642 |
| Proteinase K Solution | Bioline | Cat#BIO-37084 |
| Proteinase K (Fungal) | Invitrogen | Cat#25530015 |
| Linear Acrylamide | Invitrogen | Cat#AM9520 |
| Choice Taq | Denville | Cat#CB4050–2 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0530S |
| Biotin-16-dUTP | Roche | Cat#11093070910 |
| Terminal Deoxynucleotidyl Transferase | Thermo Scientific | Cat#EP0161 |
| Klenow Fragment (3'->5' exo) | New England Biolabs | Cat#M0212S |
|
| ||
| dATP Solution | Invitrogen | Cat#10–216-018 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Neon Transfection System 100μL Kit | Invitrogen | Cat#MPK10025 |
| QIAquick Nucleotide Removal Kit | Qiagen | Cat#28304 |
| Zymo Research RNA Clean & Concentrator | Zymo Research Kit | Cat#50–125-1669 |
| Arima-HiC + Kit | Arima Genomics | Cat#A510008 |
| KAPA RNA HyperPrep with RiboErase | Roche/KAPA Biosystems (HMR) | Cat#KK8561 |
| NEBNext End Repair Module | New England Biolabs | Cat#E6050S |
| Quick Ligation Kit | New England Biolabs | Cat#M2200S |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat#M0541S |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#1725125 |
| PureLink HiPure Plasmid Midiprep Kit | Invitrogen | Cat#K2100004 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed sequencing data | This study; see Table S4 | GEO: GSE217262 |
| Mouse reference genome NCBI build 37, NCBI37/mm9 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/mouse |
| Variant sequence data | Sanger Institute | https://www.sanger.ac.uk/data/mouse-genomes-project/ |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: C/B and B/C trophoblast stem cells | This study | N/A |
| Mouse: E14 embryonic stem cells | This study | N/A |
| Mouse: DR4 embryonic fibroblast cells | ATCC | Cat#SCRC-1045 |
|
| ||
| Oligonucleotides | ||
|
| ||
| CRISPR-Cas9 sgRNAs, see Table S5 | This study | N/A |
| Genotyping PCR primers, see Table S5 | This study | N/A |
| qPCR primers, see Table S5 | This study | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| PB_tre_dCas9_VP160 vector | Schertzer et al.55 | Cat#126031;https://doi.org/10.1261/rna.068932.118 |
| PB_tre_Cas9 vector | Schertzer et al.55 | Cat#126029;https://doi.org/10.1261/rna.068932.118 |
| PB_rtTA_BsmBI vector Schertzer et al.55 | Schertzer et al.55 | Cat#126028;https://doi.org/10.1261/rna.068932.118 |
| pUC19-piggyBac transposase | Schertzer et al.55 | https://doi.org/10.1261/rna.068932.118 |
|
| ||
| Software and algorithms | ||
|
| ||
| Custom Scripts | This study | https://github.com/aki-kb/calabrese_lab/tree/main/braceros_airn_2023;https://doi.org/10.5281/zenodo.7988247 |
| Juicer/Juicebox (v1.5.6) | Durand et al.27,28 | https://github.com/aidenlab/juicer;https://github.com/aidenlab/Juicebox |
| Juicer Tools (v1.9.9) | Durand et al.27 | https://github.com/aidenlab/juicer |
| HiCExplorer (v3.7.2) | Wolff et al.,56 Ramirez et al.57 | https://hicexplorer.readthedocs.io/en/latest |
| Bowtie2 (v2.4.5) | Langmead and Salzberg58 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR (v2.7.9a) | Dobin et al.58 | https://github.com/alexdobin/STAR |
|
| ||
| Samtools (v1.16) | Li et al.59 | https://github.com/samtools/samtools |
| Bedtools (v2.30) | Quinlan and Hall60 | https://bedtools.readthedocs.io/en/latest |
| Significant Peak Interaction (SIP; v1.5) | Rowley et al.43 | https://github.com/PouletAxel/SIP |
| MACS2 (v2.1.2) | Zhang et al.61 | https://github.com/taoliu/MACS |
| Subread: featureCounts (v1.6.3) | Liao et al.62 | http://subread.sourceforge.net |
| RStudio (v4.2.0) | N/A | https://posit.co/products/open-source/rstudio |
| ggplot2, ggpubr (v3.3.5) | Wickham63 | https://ggplot2.tidyverse.org |
| GraphPad Prism (v9.4.1) | N/A | https://www.graphpad.com |
|
| ||
| Other | ||
|
| ||
| Vibra-Cell™ VCX 130 Sonicator & 6mm Probe | Sonics | Cat#H-1002–2 |
| Bioruptor® Plus sonication device | Diagenode | Cat#B01020001 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
TSC culture
The mouse C/B TSC and B/C TSC lines used in this work correspond to the CAST/EiJ maternal/C57BL/6J paternal (C/B) and C57BL/6J maternal/CAST/EiJ paternal (B/C) TSCs used in40,64 and are referred to as CB.1 and BC.1 TSCs in.64 TSCs were cultured as in.65 Briefly, TSCs were cultured on gelatin-coated, pre-plated irradiated mouse embryonic fibroblast (irMEF) feeder cells in TSC media (RPMI [Gibco, cat #: 11875093], 20% qualified FBS [Gibco, cat #: 26140079], 0.1mM penicillin-streptomycin [Gibco, cat #: 15140122], 1mM sodium pyruvate [Gibco, cat #: 11360070], 2mM L-glutamine [Gibco, cat #: 25030081], 100μM β-mercaptoethanol [Sigma-Aldrich, cat #: 63689]) supplemented with 25 ng/mL FGF4 (Gibco, cat #: PHG0154) and 1 μg/mL Heparin (Sigma-Aldrich, cat #: H3149) just before use, at 37°C in a humidified incubator at 5% CO2. At passage, TSCs were trypsinized with 0.125% Trypsin-EDTA in PBS solution (Gibco, cat #: 25200–072) for ~4 min at room temperature and gently dislodged from the plate with a sterile, cotton-plugged Pasteur pipette. To deplete irMEFs from TSCs prior to all harvests, TSCs were pre-plated for 45 min at 37°C, transferred to a fresh culture plate, and then cultured for three days in 70% irMEF-conditioned TSC media supplemented with growth factors as above.
ESC culture
Mouse E14 ESCs were cultured on gelatin-coated plates in ESC media (DMEM high glucose and sodium pyruvate [Gibco, cat #: 11995073], 15% qualified FBS, 0.1mM MEM non-essential AA [Gibco, cat #: 11140050], 0.1mM penicillin-streptomycin, 2mM L-glutamine, 100μM β-mercaptoethanol, 1:500 LIF) at 37°C in a humidified incubator with 5% CO2. At passage, ESCs were trypsinized with 0.125% Trypsin-EDTA in PBS solution for ~5 min at room temperature and dislodged from the plate at single-cell suspension. ESCs were passaged every other day and provided fresh media daily.
Generation of regulatory element deletions
Per regulatory element deletion, four unique sgRNAs were designed using CRISPOR,66 with two sgRNAs flanking the target site (Figures S2A–S2D, Table S5). As a negative control, an sgRNA using a non-targeting (NTG) sequence from (Invitrogen, cat #: A35526) was designed. Each sgRNA was cloned into the BsmbI site of the piggyBac-cargo rtTA vector (PB_rtTA_BsmBI) from55 and transformed in DH5-alpha competent bacterial cells. Starter transformant cultures for each sequence-verified sgRNA were pooled together in equal volume amounts prior to liquid culture expansion and plasmid purification using the PureLink HiPure Plasmid Midiprep kit (Invitrogen, cat #: K2100004). The pooled sgRNAs were then co-electroporated with doxycycline-inducible Cas9-cargo (PB_tre_Cas9) and pUC19-piggyBac transposase vectors from55 at an 8:2:1 plasmid ratio of 2.5μg total DNA into 1 million C/B TSCs on irradiated drug-resistant MEFs (irDR4-MEFs; ATCC, cat #: SCRC-1045) in a single well of a 6-well plate. The electroporations were performed using a Neon Instrument (electroporation settings: 950V, 30ms, 2 pulses). Two days after electroporation, TSCs were selected with 150 μg/mL hygromycin B (Corning, cat #: MT30240CR)) and 200 μg/mL G418 (Gibco, cat #: 10131035) in irMEF-conditioned TSC media with growth factors for 11 days, followed by four days of 1 μg/mL doxycycline treatment (Sigma-Aldrich, cat #: D9891) to induce Cas9 expression. 2,000 doxycycline-induced TSCs were then plated onto a pre-plated irMEFs 100-mm dish for clonal selection and expansion. Prior to harvesting for genotyping assays, clonal TSC lines were passaged once off of irMEFs as above.
For genotyping assays, PCR was used to detect the presence or absence of target deletion DNA. “Wildtype” primers were designed to amplify either the flanking end or internal region of the deletion site. “Deletion” primers were designed to externally flank both ends of the deletion sites that would efficiently amplify a sizable PCR product if the deletion occurred (Table S5). Sanger sequencing (Eton) was then used to detect the presence of informative B6/CAST SNPs in the PCR products for allelic identification.
Generation of Airn-overexpressing ESCs
750,000 ESCs were seeded in a single gelatin-coated well of a six-well plate, and the next day transfected with 2.5μg of an 8:2:1 plasmid ratio of piggyBac-cargo rtTA-Airn sgRNA, doxycycline-inducible piggyBac-cargo dCas9-VP160 (PB_tre_dCas9_VP160), and pUC19-piggyBac transposase from55 using Lipofectamine 3000 (Invitrogen, cat #: L3000015) according to manufacturer instructions. The next day, transfected ESCs were selected with ESC media containing 150 μg/mL hygromycin B and 200 μg/mL G418 for 10 days, followed by 4 days of doxycycline treatment to induce Airn expression via dCas9-VP160 prior to harvests.
METHOD DETAILS
In situ Hi-C
Prior to crosslinking for Hi-C, TSCs were passaged once off of irMEFs as described above. TSCs were then trypsinized and washed once with PBS. 5–10 million cells were crosslinked in resuspension with 10mL of 1% formaldehyde (Thermo Scientific, cat #: 28906) in PBS solution for 10 min at room temperature, quenched with 200mM glycine for 5 min at room temperature, and then washed twice with ice-cold PBS. Cells were then divided into 5 aliquots (1–2 million cells/aliquot), where one aliquot was used for each Hi-C experiment. Importantly, for the removal of all PBS washes and crosslinking solution, cells were spun at 160 × g for 5 min.
Hi-C libraries from C/B wild-type, B/C wild-type, and C/B Airn truncation TSCs were generated and sequenced as in the detailed protocol from,26 including DNA fragmentation with MboI and MseI restriction enzymes. Hi-C libraries from regulatory element deletion TSCs were generated using the Arima-HiC + kit (Arima Genomics, cat #: A510008) according to the manufacturer instructions. Paired-end, 150-bp sequencing was performed using Illumina NovaSeq 6000 System.
(Calibrated) ChIP-seq
Prior to crosslinking for ChIP, TSCs were passaged once off of irMEFs as above. For all ChIP experiments, except those for PRC components, adhered cells were crosslinked with 0.6% formaldehyde (Fisher Scientific, cat #: BP531–500) in RPMI media with 10% FBS for 10 min at room temperature, then quenched with 125mM glycine for 5 min at room temperature. Crosslinked cells were then washed twice with ice-cold PBS and scraped with ice-cold PBS with 0.05% Tween (Fisher Scientific, cat #: EW-88065–31) and PIC (Sigma Aldrich, cat #: P8340). The cells were then spun at 1,200 × g at 4°C to remove PBS, followed by resuspension in ice-cold PBS with PIC and divided into 10-million cell aliquots. For PRC component ChIPs, adhered C/B TSCs were crosslinked in PBS with 2mM DSG (disuccinimidyl glutarate; Thermo Scientific, cat #: 20593) for 45 min at room temperature and then in PBS with 1% formaldehyde (Thermo Scientific, cat #: 28906) for 15 min at room temperature. Crosslinking was quenched with 200mM glycine for 5 min at room temperature. Cells were then washed, scraped, and aliquoted as above. All ChIPs were performed using 10 million cells, 10μL of antibody, and 30μL of Protein A/G agarose beads (Santa Cruz, cat #: sc-2003). Input chromatin was isolated accordingly to each antibody (see below) and sonicated to 100–500bp fragments using a Vibra-Cell VX130 (Sonics) with the following parameters: 8–10 cycles of 30% intensity for 30 s with 1 min of rest on ice between cycles. Antibody-conjugated beads were prepared by incubating antibody with beads in 300μL Blocking Buffer (PBS, 0.5% BSA [Invitrogen, cat #: AM2616]) overnight at 4°C with rotation.
For H3K27me3 and H2AK119ub ChIPs, crosslinked TSCs were resuspended in 1mL Lysis Buffer 1 (50mM HEPES pH 7.5, 140mM NaCl, 1mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, PIC) and incubated with rotation for 10 min at 4°C. Cells were then resuspended in 1mL Lysis Buffer 2 (10mM Tris-HCl pH 8.0, 200mM NaCl, 1mM EDTA, 0.5 mM EGTA, PIC) for 10 min at room temperature. All buffer removal steps were performed with 5-min 1,200 × g spins at 4°C. The extracted nuclei pellet was then resuspended and sonicated in 500μL Lysis Buffer 3 (10mM Tris-HCl pH 8.0, 100mM NaCl, 1mM EDTA, 0.5mM EGTA, 0.1% sodium-deoxycholate, 0.5% N- lauroylsarcosine, PIC). Soluble chromatin was obtained with a 30-min max speed spin at 4°C, mixed with 1% Triton X-100, and then incubated with pre-conjugated antibody beads overnight at 4°C with rotation. The ChIP beads were then washed five times in 1mL RIPA Buffer (50mM HEPES pH 7.5, 500mM LiCl, 1mM EDTA, 1% NP-40, 0.7% sodium-deoxycholate, PIC) and once with 1mL TE, each for 5 min at 4°C with rotation and spun at 2,000 × g for 2 min for buffer removal.
For PRC component ChIPs, crosslinked C/B TSCs were resuspended and sonicated in 500μL Low Salt Pol II ChIP Buffer (50mM Tris-HCl pH 7.5, 140mM NaCl, 1mM EDTA, 1mM EGTA, 0.1% sodium-deoxycholate, 0.1% SDS, PIC). Soluble chromatin was obtained with a 30-min max speed spin at 4°C, mixed with 1% Triton X-100, and then incubated with pre-conjugated antibody beads overnight at 4°C with rotation. The ChIP beads were then washed three times with 1mL Low Salt Pol II ChIP Buffer with 1% Triton X-100 and PIC, once with 1mL High Salt Pol II ChIP Buffer (50mM Tris-HCl pH 7.5, 500mM NaCl, 1mM EDTA, 1mM EGTA, 0.1% sodium-deoxycholate, 0.1% SDS, PIC), once with 1mL LiCl Wash Buffer (20mM Tris-HCl pH 8.0, 250mM LiCl, 1mM EDTA, 0.5% Nadeoxycholate, 0.5% NP-40, PIC), and once with 1 mL TE, each for 5 min at 4°C with rotation and spun at 2,000 × g for 2 min for buffer removal.
For all ChIP DNA elution steps, washed beads were resuspended in Elution buffer (50mM Tris-HCl pH 8.0, 10mM EDTA, 1% SDS) and placed on a 65°C heat block for 17 min with frequent vortexing. ChIP DNA was then reverse crosslinked in 0.5% SDS and 100mM NaCl overnight at 65°C, followed by a 1-h RNaseA (3μL; Thermo Scientific, cat #: EN0531) treatment at 37°C and a 2.5-h Proteinase K (10μL; Invitrogen, cat #: 25530015) treatment at 56°C. DNA was then extracted with 1 volume of phenol:chloroform:isoamyl alcohol (Sigma-Aldrich, cat #: P3803) and precipitated with 2 volumes 100% ethanol, 1/10 volume 3M sodium-acetate pH 5.4, and 1/1000 volume linear acrylamide (Invitrogen, cat #: AM9520) overnight at −20°C. Precipitated DNA was then extracted with a 30-min max speed spin at 4°C, washed once with ice-cold 80% ethanol, and resuspended in TE.
Calibrated ChIPs were performed to validate Airn-induced changes across genotypes. At the IP step, 5% sonicated chromatin from HEK293T cells was added to a standardized protein amount of input TSC chromatin across samples. Bradford protein assays (Bio-Rad, cat #: 5000006) with BSA protein standard were performed to determine protein quantity as a proxy for input chromatin amount. HEK293T chromatin was sonicated as above to achieve 100–500bp fragments. All other steps were performed as normal.
ChIP-seq libraries were prepared with NEBNext End Repair Module (NEB, cat #: E6050S), A-tailing by Klenow Fragment (3’→5′ exo-; NEB, cat #: M0212S), and TruSeq 6-bp index adaptor ligation by Quick ligase (NEB, cat #: M2200S), and NEBNext High-Fidelity 2X PCR Master Mix (NEB, cat #: M0541S). All DNA size-selection purification steps were performed with AMPure XP beads (Beckman Coulter, cat #: A63880). Single-end, 75-bp sequencing was performed using an Illumina NextSeq 500/550 High Output v2.5 kit (Illumina, cat #: 20024906) on a NextSeq 500 System.
CHART-seq
CHART was performed in duplicate as in the detailed protocol from.67 TSCs were passaged once off of irMEFs as above. Airn highly-expressing TSCs from14 and ESCs were induced with 1 μg/mL doxycycline 4 days prior to crosslinking. Adhered TSCs and ESCs were crosslinked with 1% formaldehyde (Fisher Scientific, cat #: BP531–500) in PBS solution for 10 min at room temperature. On ice, cells were washed twice with ice-cold PBS and twice with ice-cold PBS +0.05% Tween before being scraped, spun at 1,000 × g for 5 min at 4°C, and divided into 25-million cell aliquots.
Per aliquot, nuclei was extracted with two rounds of douncing in 4mL sucrose buffer (300mM sucrose, 10mM HEPES pH 7.5, 100mM KOAc, 1% Triton X-100, 0.1mM EGTA, 0.5mM spermidine, 0.15mM spermine, cOmplete EDTA-free protease inhibitor cocktail [Millipore, cat #: 11873580001], 1mM DTT, 80U SUPERase-in [Invitrogen, cat #: AM2696]), mixing 1:1 with glycerol buffer (25% glycerol, 10mM HEPES pH 7.5, 100mM KOAc, 1mM EDTA, 0.1mM EGTA, 0.5mM spermidine, 0.15mM spermine, cOmplete EDTA-free protease inhibitor cocktail, 1mM DTT, 80U SUPERase-in), and centrifugation through 4mL glycerol buffer at 1,000 × g for 15 min at 4°C. The nuclei pellet was then washed twice with ice-cold PBS +0.05% Tween, then further crosslinked with 3% formaldehyde (Fisher Scientific, cat #: BP531–500) in PBS +0.05% Tween for 30 min at room temperature. Formaldehyde was then washed out twice with ice-cold PBS +0.05% Tween, then resuspended in freshly prepared 250μL Sonication Buffer (50mM HEPES pH 7.5, 75mM NaCl, 0.5% N-lauroylsarcosine solution, 0.1% sodium-deoxycholate, 0.1mM EGTA, cOmplete EDTA-free protease inhibitor cocktail, 1mMDTT, 300U SUPERase-in). Chromatin was sonicated to 2–10kb fragments using a Bioruptor Plus sonication device (Diagenode; sonication parameters: 30 s on, 30 s off cycles on high setting), then spun at max speed for 30 min at 4°C to retrieve soluble chromatin.
For Airn CHART, we designed 22-nucleotide complementary oligos that tile across the first 75 kb of the Airn RNA sequence using the ChIRP Probe Designer (LGC Biosearch Technologies) under parameters of high masking for specificity and ≥500-nt spacings (Table S5). The resulting 51 oligo probes were then mixed to a 100μM pool for in-house oligo biotin labeling.68 Briefly, 20μM oligo probe mix was labeled with 300μM biotin-16-dUTP (Roche, cat #: 11093070910) and 30U terminal deoxynucleotidyl transferase (Thermo Scientific, cat #: EP0161), then labeled for 15 min at 37°C and inactivated for 20 min at 75°C. Biotinylated oligo probes were then purified using the QIAquick Nucleotide Removal kit (Qiagen, cat #: 28304), eluting to ~20μM.
Per Airn CHART reaction, 12.5 million cells worth of chromatin was mixed with 0.5 volume PAB (8M Urea, 100mM HEPES pH 7.5, 200mM NaCl, 2% SDS) and 1.5 volumes of freshly prepared Hybridization Buffer (1.5M NaCl, 1.12M Urea, 10X Denhardt’s solution [Invitrogen, cat #: 750018], 10mM EDTA), then pre-cleared with 50μL Dynabeads M-280 Streptavidin beads (Invitrogen, cat #: 11205D) for 1 h at room temperature with rotation. The pre-cleared chromatin was then isolated from the beads by spinning at 1,000 × g for 30 s. 1% of pre-cleared chromatin was saved as “Input” sample, and the remaining pre-cleared chromatin was incubated with 750pmol biotinylated oligo probes overnight at room temperature. The next day, the sample was spun at max speed for 20 min at 20°C, and the supernatant was incubated with 200μL worth of Dynabeads MyOne Streptavidin C1 beads (Invitrogen, cat #: 65001) resuspended in 125μL 2:1 diluted PAB overnight at room temperature with rotation. CHART beads were then placed on the magnet and washed 4 times with 400μL CHART Wash Buffer (250mM NaCl, 10mM HEPES pH 7.5, 2mM EDTA, 2mM EGTA, 0.2% SDS, 0.1% N-lauroylsarcosine) and once with RNase H Elution Buffer (75mM NaCl, 50mM HEPES pH 7.5, 0.02% sodium-deoxycholate, 0.1% N-lauroylsarcosine, 10mM DTT, 3mM MgCl2, 200U SUPERase-in). Beads were resuspended in 100μL RNase H Elution Buffer and treated with 2μL RNase H (NEB, cat #: M0297) for 10 min at room temperature. To stop the RNase H reaction, 1/4 volume of naXLR (166.7mM Tris-HCl pH 7.2, 1.67% SDS, 83.3mM EDTA, 600μg Proteinase K [Bioline, cat #: BIO-37084]) was added, and the CHART eluate was then subject to proteinase K digestion and reverse-crosslinking for 1 h at 55°C, followed by 1h at 65°C. The sample was then split for DNA (90%) and RNA (10%) analysis.
The Airn CHART-enriched RNA sample was treated with 1mL TRIZol and 200μL chloroform, and the RNA was DNase-treated and purified with Zymo Research RNA Clean & Concentrator kit (Zymo Research, cat #: 50–125-1669). To determine the extent of Airn RNA enrichment, 50% of both Input and CHART RNA samples were reverse transcribed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, cat #: 4368814). qPCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad, cat #: 1725125) and primers targeting 45 kb downstream of the Airn TSS and Gapdh (Table S5).
The Airn CHART-enriched DNA sample was extracted with 1 volume of phenol:chloroform:isoamyl and purified with ethanol precipitation and TE resuspension as above for ChIP DNA. qPCR was performed as above with the same primers to check Airn DNA enrichment.
CHART-seq libraries were prepared as above for ChIP-seq libraries. However, prior to library prep, CHART-enriched DNA samples were further sonicated to 100–500bp fragments using the Bioruptor Plus with 10 cycles of 30 s on, 30 s off on high setting, then purified with one round of 1:1 AMPure XP beads size selection purification. Single-end, 75-bp sequencing was performed using an Il-lumina NextSeq 500/550 High Output v2.5 kit on a NextSeq 500 System.
RNA isolation, RT-qPCR, RNA-seq
TSCs were passaged once off of irMEFs as above onto a single well of a 6-well plate. ESCs were grown on a single well of a 6-well plate. Both were grown to ≥75% confluency prior to RNA harvest using 1mL TRIzol, followed by the addition of 200μL chloroform, which were vortexed and subsequently spun at max speed for 5 min at 4°C for phase separation. The aqueous layer was collected and combined with 1 volume of 100% isopropanol and 5μL linear acrylamide. Precipitation was achieved at −80°C for 1 h, followed by a max speed spin for 30 min at 4°C and one wash of the RNA pellet with ice-cold 80% ethanol. The pellet was then resuspended in 100μL H2O and quantified via Qubit (Invitrogen, cat #: Q32855).
For RT-qPCR assays in Figures 5A and S4E, 1μg of RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit, and qPCR was performed using iTaq Universal SYBR Green Supermix and custom primers (Table S5).
RNA-seq libraries were prepared from 1μg of total RNA using KAPA RNA HyperPrep Kit with Ribose Erase (Kapa Biosystems, cat #: KR1351) according to the manufacturer instructions. Single-end, 75-bp sequencing was performed using an Illumina NextSeq 500/550 High Output v2.5 kit on a NextSeq 500 System.
Sequence alignment and processing
All mouse reference NCBI build 37/mm9 genome annotations were obtained from the UCSC genome browser.69 Variant sequence data was obtained from the Sanger Institute (http://www.sanger.ac.uk/resources/mouse/genomes/.25; The CAST/EiJ (CAST) pseudogenome creation was performed as in.40,64 Hi-C reads were aligned using BWA as a part of the Juicer pipeline (Durand et al. 2016a). ChIP- and CHART-seq reads were aligned using bowtie2 with default parameters.70 RNA-seq reads were aligned using STAR with default parameters.58
For Hi-C analyses in this study, read pairs that had a mapping quality greater than or equal to 10 were used for allelic TSC analysis and read pairs that had greater than or equal to 30 were used for ESC analysis. For all ChIP-, CHART- and RNA-seq analyses in this study, reads that had a mapping quality greater than or equal to 30 were extracted with samtools,59 and allele-specific read retention (i.e., reads that overlap at least one B6 or CAST SNP) was performed as in40,64 using a custom perl script (intersect_reads_snps16.pl: see github).
Chromosome tiling density plots
For all chromosome-scale tiling density plots in Figures 2–6 and S2–S6, reads were summed in 10kb bins across each chromosome. MAPQ≥30 aligned reads were then divided by the total number of reads in the dataset and divided by a million (i.e., RPM). For allelic TSC ChIP- and CHART-seq data, binned counts were divided by the number of B6/CAST SNPs detected in the bin genomic coordinates (i.e., SNP-norm RPM). Finally, bins were averaged every 9 bins in 1bin increments. For allelic TSC Hi-C viewpoint data, we excluded bins whose aggregate SNP-overlapping read count across merged Hi-C datasets fell within the bottom quintile relative to bins in the rest of the genome. The allelic data in this group of bins were too sparse to be interpreted with confidence. For the same reason, for allelic CHART- and ChIP-seq data, only bins with greater or equal to 25 SNPs were plotted. Bins of replicate datasets were averaged.
For total, non-allelic ESC Airn CHART-seq and H3K27me3 ChIP-seq data, reads were RPM converted, binned, and averaged as above. For all ESC data in Figure 4, the same genomic bins as the allelic TSC ChIP- and CHART-seq data were plotted.
All plots were generated using ggplot263 in RStudio.
Tiling density correlations
To derive Spearman’s r and p values for tiling density correlations in Figures 2 and 4, reads were processed as described in “Chromosome tiling density plots” and every 10th bin along the genomic region of interest was correlated.
To determine significant changes in H3K27me3 and H2AK119ub density across genotypes in Figures 5 and S6, all binned SNP-norm RPM values over the genomic region of analysis were subjected to a Welch two-sample t test in RStudio.
Genome browser density tracks
Wiggle density files were created using a custom perl script (bigbowtie_to_wig3_mm9.pl; see github) and loaded into a UCSC Genome Browser session to generate the graphics in Figures 3 and 7. All density tracks were auto-scaled to data view and set to a maximum windowing function with 3-pixel smoothing.
Hi-C analysis
Juicer processing, quality control, and allele-specific read retention
Hi-C analyses were carried out using a combination of Juicer (default parameters) and HiCExplorer.27,57,71 For exact Juicer commands used, see github.
For quality control, Hi-C statistics of each dataset were generated by Juicer (Table S1) and were referenced to the standard guidelines in.26 In addition, long-range DNA interactions (25kb-10Mb) were correlated between Hi-C replicate datasets at 10kb and 25kb resolutions using HiCExplorer’s hicCorrelate56 with the following parameters: –method = pearson –range 25000:10000000.
For allele-specific retention of Hi-C contacts, read pairs in which at least one read end overlaps a B6/CAST SNP were extracted from the Juicer output merged_nodups.txt file using a modified Juicer diploid script (juicer_diploid_v6.sh; see github).
2D contact heatmaps
Allelic 2D contact heatmaps in Figures 1, 6, 7, S1 and S7 were generated with Juicebox.71 Contact matrices of observed counts were viewed in KR (Knight-Ruiz) balance mode at 5kb and 50kb resolutions. Subtraction heatmaps, where maternal contacts were subtracted from paternal contacts, were viewed under the same conditions.
Loop calling
DNA loops were detected using the Significant Interaction Peak (SIP) caller43 with the following parameters: -factor 4 -g 2.0 -t 2000 -fdr 0.05. The output finalLoops.txt file (i.e., a merged list of unique DNA loop anchors detected across 5k, 10kb, and 25kb resolutions) was used to determine loops over the 2D contact heatmaps in Juicebox for Figure 7.
“A” and “B” compartmentalization
“A” and “B” chromosome compartments for each allele were delineated by eigenvector analysis using the Juicer eigenvector tool with the following parameters: -p KR 17 BP 50000. Extracted eigenvector values were then visualized and plotted with Juicebox in Figures 1 and 6.
Allelic viewpoints
Allelic viewpoints of locus-specific contact matrices were extracted at 10 or 25kb resolution for observed and observed-over-expected (O/E) counts using the Juicer dump tool with the following parameters: observed/oe NONE chr:start:end chr:start:end BP 10000/25000. If applicable, all viewpoint loci of interest were extended to 100kb lengths from their centers to improve contact matrix coverage. Extracted counts were then processed and plotted as described in the Chromosome-scale tiling density plots section.
Correlation with FISH
Allelic observed contacts from Airn viewpoint data in C/B wild-type TSCs were summed over the genomic coordinates for each of the 9 FISH probes across the Airn target domain analyzed in.14 For each allele, the summed Hi-C counts at the probe locations were then correlated by Spearman’s test (using in RStudio) with the corresponding average distance to the Airn probe as measured by RNA/DNA co-FISH in C/B TSCs.14 Scatterplots in Figure 2 were generated with ggplot2 in RStudio.63
ChIP-seq analysis
Calibrated ChIP-seq spike-in normalization
Reads from calibrated ChIP-seq samples were also aligned to human GRCh37/hg19 genome build. A normalization factor was calculated for each sample using the formula 1/h, where h is the number of hg19-aligned reads in millions, as described previously.72,73 Raw B6 and CAST reads were then scaled by multiplying the corresponding normalization factor. For tiling density plots, binned spike-in normalized reads were divided by the total number of B6/CAST SNPs.
Peak calling
ChIP-seq peaks were called from non-allelic reads against an H3 ChIP-seq dataset (from40) using the MACS2 peak calling algorithm61 with the following parameters: -g mm –broad –broad-cutoff 0.01.
Allelic enrichment at CpG islands and other features
A statistical permutation test was performed to determine how significantly enriched PRC components, CTCF, SMC1A/Cohesin, and epigenetic marks are at loci of interest relative to the rest of the genome (see Table S3). All datasets analyzed were generated from C/B TSCs. H3K27ac, H3K4me2, CTCF, and SMC1A data were generated in previous studies, as a part of.14,40 If applicable, all genomic features of interest were standardized to 1.5kb lengths (i.e., the largest CGI of interest) relative to their center positions. Using bedtools’ ‘shuffle’,60 we created a list of 80,000 1.5kb regions randomly selected from within ‘gene’ coordinates from gencode.v-M1.annotation.gtf74 with 100kb extended start and end sites while excluding any regions that fell within 2.5kb of a region annotated by MACS as an H3K27me3 or PRC subunit peak. Shuffled coordinates were filtered to retain regions encompassing at least one B6/CAST SNP, leaving 67,262 shuffled regions. B6- and CAST-overlapping ChIP-seq reads were then counted over the features of interest and shuffled coordinates using a custom script (ase_analyzer10_adj2.pl; see github), then divided by the number of B6/CAST SNPs detected in the genomic coordinates (SNP-norm counts). The features were then ranked by SNP-norm counts for each allele in each dataset (1 = highest allelic signal), and a percentile rank was used to determine an empirical p value for allele-specific enrichment of the ChIP target at the loci of interest.
RNA-seq analysis
For non-allelic expression analysis in Table S2, featureCounts62 was used to count reads over ‘gene’ entries in gencode.vM1.annotation.gtf.74 Counts were then divided by total reads in the dataset and divided by a million (RPM).
For allelic expression analysis in Table S2 and Figures 5 and S6, a custom perl script (ase_analyzer10.pl; see github) was used to count B6- and CAST SNP-overlapping reads over ‘gene’ entries in gencode.vM1.annotation.gtf.74 Read counts were divided by the total number of reads in the dataset, divided by a million, and divided by the number of SNPs detected within the ‘gene’ coordinates (SNP-norm RPM). To determine the relative paternal expression of Airn in Figure S5E, paternal SNP-norm RPM values over Airn for each genotype were divided by was divided by the averaged NTG value from all four NTG clone data. To determine the relative paternal bias of Airn target gene expression for each genotype in Figure 5, paternal SNP-norm RPM values for the 27 Airn target genes (Table S2;14) were divided by the sum of the paternal and maternal values. The paternal biases for each genotype were then divided by the averaged NTG value from all four NTG clone data, and a Welch t test was used to determine the p value of the relative change in expression relative to NTG in RStudio. Boxplots of these data were generated using GraphPad Prism v9.
QUANTIFICATION AND STATISTICAL ANALYSIS
In Figure 2B, we used Spearman’s Rank Correlation to determine the relationship between KR-balanced Hi-C counts with the Airn viewpoint (normalized for SNP density) versus our previous measurements of spatial distance to the Airn locus made by DNA FISH in.14 In Figure 2F, we used Spearman’s Rank Correlation to determine the relationship between KR-balanced Hi-C counts with the Airn viewpoint (normalized for SNP density) and the density of H3K27me3 and H2AK119ub on the paternal allele. In Figure 3, we used an empirical sampling of ChIP-seq read density in intergenic regions to determine the likelihood that each analyzed histone modification and chromatin-associated factor was enriched within a given region above what we would have expected by chance (p < 0.05). In Figures 4B–4D, we used Spearman’s Rank Correlation to determine the relationship the density of Airn CHART-seq signal on the paternal allele and H3K27me3, RING1B, and EZH2 ChIP-seq signal, respectively, also on the paternal allele. In Figure 4F, we used Spearman’s Rank Correlation to determine the relationship the density of Airn CHART signal and H3K27me3 in ESCs (non-allelic). In Figure 5E, we used a Welch two-sample t test to determine the likelihood (p < 0.05) that the relative paternal bias of 27 Airn target genes from14 changed in each individual genotype relative to NTG control. In Figure 7D, we also used a Welch t test to determine the likelihood that the allelic H3K27me3 and H2AK119ub density was altered in the ΔSlc22a3 and ΔPde10a genotypes relative to the NTG control. Throughout our study, the term “n” refers to a biological replicate. Statistical analyses were performed in R. Software packages used are listed in the key resources table. Additional details can be found in the figure legends, the body of the manuscript, and in the method details section above.
Supplementary Material
Highlights.
Airn-induced architecture changes coincide with PRC-directed chromatin modifications
Changes center around regions that form prior contacts with Airn locus
Airn lncRNA, PRCs, and PRC-directed modifications are linked by proximity
Specific DNA regulatory elements can modulate repression by Airn in different ways
ACKNOWLEDGMENTS
This work was supported by NIH grants R01GM136819 (to J.M.C.), UM1HG009375 (to E.L.A.), R35GM128645 (to D.H.P.), and R35GM124764 (to J.M.D.); NICHD grant F31HD103370 (to A.K.B.); and NIGMS grants T32GM119999 (to A.K.B.), T32CA217824 (to J.B.T.), and T32GM067553 (to E.S.D.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112803.
REFERENCES
- 1.Monk D, Mackay DJG, Eggermann T, Maher ER, and Riccio A (2019). Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 20, 235–248. 10.1038/s41576-018-0092-0. [DOI] [PubMed] [Google Scholar]
- 2.Tucci V, Isles AR, Kelsey G, and Ferguson-Smith AC; Erice Imprinting Group (2019). Genomic Imprinting and Physiological Processes in Mammals. Cell 176, 952–965. 10.1016/j.cell.2019.01.043. [DOI] [PubMed] [Google Scholar]
- 3.MacDonald WA, and Mann MRW (2020). Long noncoding RNA functionality in imprinted domain regulation. PLoS Genet. 16, e1008930. 10.1371/journal.pgen.1008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llères D, Imaizumi Y, and Feil R (2021). Exploring chromatin structural roles of non-coding RNAs at imprinted domains. Biochem. Soc. Trans. 49, 1867–1879. 10.1042/BST20210758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang R, Jaritz M, Guenzl P, Vlatkovic I, Sommer A, Tamir IM, Marks H, Klampfl T, Kralovics R, Stunnenberg HG, et al. (2011). An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS One 6, e27288. 10.1371/journal.pone.0027288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyle R, Watanabe D, te Vruchte D, Lerchner W, Smrzka OW, Wutz A, Schageman J, Hahner L, Davies C, and Barlow DP (2000). The imprinted antisense RNA at the Igf2r locus overlaps but does not imprint Mas1. Nat. Genet. 25, 19–21. 10.1038/75546. [DOI] [PubMed] [Google Scholar]
- 7.Terranova R, Yokobayashi S, Stadler MB, Otte AP, van Lohuizen M, Orkin SH, and Peters AHFM (2008). Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev. Cell 15, 668–679. 10.1016/j.devcel.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 8.Sleutels F, Zwart R, and Barlow DP (2002). The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415, 810–813. 10.1038/415810a.415810a [pii]. [DOI] [PubMed] [Google Scholar]
- 9.Killian JK, Nolan CM, Wylie AA, Li T, Vu TH, Hoffman AR, and Jirtle RL (2001). Divergent evolution in M6P/IGF2R imprinting from the Jurassic to the Quaternary. Hum. Mol. Genet. 10, 1721–1728. 10.1093/hmg/10.17.1721. [DOI] [PubMed] [Google Scholar]
- 10.Yotova IY, Vlatkovic IM, Pauler FM, Warczok KE, Ambros PF, Oshimura M, Theussl HC, Gessler M, Wagner EF, and Barlow DP (2008). Identification of the human homolog of the imprinted mouse Air non-coding RNA. Genomics 92, 464–473. 10.1016/j.ygeno.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki S, Shaw G, and Renfree MB (2018). Identification of a novel antisense noncoding RNA, ALID, transcribed from the putative imprinting control region of marsupial IGF2R. Epigenet. Chromatin 11, 55. 10.1186/s13072-018-0227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seidl CIM, Stricker SH, and Barlow DP (2006). The imprinted Air ncRNA is an atypical RNAPII transcript that evades splicing and escapes nuclear export. EMBO J. 25, 3565–3575. 10.1038/sj.emboj.7601245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andergassen D, Dotter CP, Wenzel D, Sigl V, Bammer PC, Muckenhuber M, Mayer D, Kulinski TM, Theussl HC, Penninger JM, et al. (2017). Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. Elife 6, e25125. 10.7554/eLife.25125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schertzer MD, Braceros KCA, Starmer J, Cherney RE, Lee DM, Salazar G, Justice M, Bischoff SR, Cowley DO, Ariel P, et al. (2019). lncRNA-Induced Spread of Polycomb Controlled by Genome Architecture, RNA Abundance, and CpG Island DNA. Mol. Cell. 75, 523–537.e10. 10.1016/j.molcel.2019.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Latos PA, Pauler FM, Koerner MV, Sxenergin HB, Hudson QJ, Stocsits RR, Allhoff W, Stricker SH, Klement RM, Warczok KE, et al. (2012). Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338, 1469–1472, 338/6113/1469 [pii]. 10.1126/science.1228110. [DOI] [PubMed] [Google Scholar]
- 16.Pauler FM, Barlow DP, and Hudson QJ (2012). Mechanisms of long range silencing by imprinted macro non-coding RNAs. Curr. Opin. Genet. Dev. 22, 283–289. 10.1016/j.gde.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andergassen D, Muckenhuber M, Bammer PC, Kulinski TM, Theussl HC, Shimizu T, Penninger JM, Pauler FM, and Hudson QJ (2019). The Airn lncRNA does not require any DNA elements within its locus to silence distant imprinted genes. PLoS Genet. 15, e1008268. 10.1371/journal.pgen.1008268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, and Fraser P (2008). The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720, 1163802 [pii]. 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 19.Grosswendt S, Kretzmer H, Smith ZD, Kumar AS, Hetzel S, Wittler L, Klages S, Timmermann B, Mukherji S, and Meissner A (2020). Epigenetic regulator function through mouse gastrulation. Nature 584, 102–108. 10.1038/s41586-020-2552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andergassen D, Smith ZD, Kretzmer H, Rinn JL, and Meissner A (2021). Diverse epigenetic mechanisms maintain parental imprints within the embryonic and extraembryonic lineages. Dev. Cell 56, 2995–3005.e4. 10.1016/j.devcel.2021.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trotman JB, Braceros KCA, Cherney RE, Murvin MM, and Calabrese JM (2021). The control of polycomb repressive complexes by long noncoding RNAs. Wiley Interdiscip Rev RNA 12, e1657. 10.1002/wrna.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blackledge NP, and Klose RJ (2021). The molecular principles of gene regulation by Polycomb repressive complexes. Nat. Rev. Mol. Cell Biol. 22, 815–833. 10.1038/s41580-021-00398-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almeida M, Pintacuda G, Masui O, Koseki Y, Gdula M, Cerase A, Brown D, Mould A, Innocent C, Nakayama M, et al. (2017). PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 356, 1081–1084. 10.1126/science.aal2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bousard A, Raposo AC, Żylicz JJ, Picard C, Pires VB, Qi Y, Gil C, Syx L, Chang HY, Heard E, and da Rocha ST (2019). The role of Xist-mediated Polycomb recruitment in the initiation of X-chromosome inactivation. EMBO Rep. 20, e48019. 10.15252/embr.201948019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294, nature10413 [pii]. 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101. 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98. 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293, 326/5950/289 [pii]. 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blackledge NP, Fursova NA, Kelley JR, Huseyin MK, Feldmann A, and Klose RJ (2020). PRC1 Catalytic Activity Is Central to Polycomb System Function. Mol. Cell. 77, 857–874.e9. 10.1016/j.molcel.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyle S, Flyamer IM, Williamson I, Sengupta D, Bickmore WA, and Illingworth RS (2020). A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev. 34, 931–949. 10.1101/gad.336487.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oksuz O, Narendra V, Lee CH, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PP, Garcia BA, et al. (2018). Capturing the Onset of PRC2-Mediated Repressive Domain Formation. Mol. Cell. 70, 1149–1162.e5. 10.1016/j.molcel.2018.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isono K, Endo TA, Ku M, Yamada D, Suzuki R, Sharif J, Ishikura T, Toyoda T, Bernstein BE, and Koseki H (2013). SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell 26, 565–577. 10.1016/j.devcel.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 34.Eeftens JM, Kapoor M, Michieletto D, and Brangwynne CP (2021). Polycomb condensates can promote epigenetic marks but are not required for sustained chromatin compaction. Nat. Commun. 12, 5888. 10.1038/s41467-021-26147-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraft K, Yost KE, Murphy SE, Magg A, Long Y, Corces MR, Granja JM, Wittler L, Mundlos S, Cech TR, et al. (2022). Polycomb-mediated genome architecture enables long-range spreading of H3K27 methylation. Proc. Natl. Acad. Sci. USA 119, e2201883119. 10.1073/pnas.2201883119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai Y, Zhang Y, Loh YP, Tng JQ, Lim MC, Cao Z, Raju A, Lieberman Aiden E, Li S, Manikandan L, et al. (2021). H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 12, 719. 10.1038/s41467-021-20940-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cuadrado A, Giménez-Llorente D, Kojic A, Rodríguez-Corsino M, Cuartero Y, Martín-Serrano G, Gómez-López G, Marti-Renom MA, and Losada A (2019). Specific Contributions of Cohesin-SA1 and Cohesin-SA2 to TADs and Polycomb Domains in Embryonic Stem Cells. Cell Rep. 27, 3500–3510.e4. 10.1016/j.celrep.2019.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kriz AJ, Colognori D, Sunwoo H, Nabet B, and Lee JT (2021). Balancing cohesin eviction and retention prevents aberrant chromosomal interactions, Polycomb-mediated repression, and X-inactivation. Mol. Cell. 81, 1970–1987.e9. 10.1016/j.molcel.2021.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rhodes JDP, Feldmann A, Hernández-Rodríguez B, Dáz N, Brown JM, Fursova NA, Blackledge NP, Prathapan P, Dobrinic P, Huseyin MK, et al. (2020). Cohesin Disrupts Polycomb-Dependent Chromosome Interactions in Embryonic Stem Cells. Cell Rep. 30, 820–835.e10. 10.1016/j.celrep.2019.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D, Starmer J, Mieczkowski P, Crawford GE, and Magnuson T (2012). Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 151, 951–963, S0092–8674(12)01300–1 [pii]. 10.1016/j.cell.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg-Schoenberg M, Bowman SK, Kesner BA, Maier VK, Kingston RE, and Lee JT (2013). High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 504, 465–469. 10.1038/nature12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, and Cavalli G (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572.e24. 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rowley MJ, Poulet A, Nichols MH, Bixler BJ, Sanborn AL, Brouhard EA, Hermetz K, Linsenbaum H, Csankovszki G, Lieberman Aiden E, and Corces VG (2020). Analysis of Hi-C data using SIP effectively identifies loops in organisms from C. elegans to mammals. Genome Res. 30, 447–458. 10.1101/gr.257832.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirny L, and Dekker J (2022). Mechanisms of Chromosome Folding and Nuclear Organization: Their Interplay and Open Questions. Cold Spring Harbor Perspect. Biol. 14, a040147. 10.1101/cshper-spect.a040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Højfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, Mohammad F, Jensen ON, and Helin K (2018). Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 25, 225–232. 10.1038/s41594-018-0036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnold PR, Wells AD, and Li XC (2019). Diversity and Emerging Roles of Enhancer RNA in Regulation of Gene Expression and Cell Fate. Front. Cell Dev. Biol. 7, 377. 10.3389/fcell.2019.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Statello L, Guo CJ, Chen LL, and Huarte M (2021). Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118. 10.1038/s41580-020-00315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galupa R, Picard C, Servant N, Nora EP, Zhan Y, van Bemmel JG, El Marjou F, Johanneau C, Borensztein M, Ancelin K, et al. (2022). Inversion of a topological domain leads to restricted changes in its gene expression and affects interdomain communication. Development 149, dev200568. 10.1242/dev.200568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenwald WW, Li H, Benaglio P, Jakubosky D, Matsui H, Schmitt A, Selvaraj S, D’Antonio M, D’Antonio-Chronowska A, Smith EN, and Frazer KA (2019). Subtle changes in chromatin loop contact propensity are associated with differential gene regulation and expression. Nat. Commun. 10, 1054. 10.1038/s41467-019-08940-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zuin J, Roth G, Zhan Y, Cramard J, Redolfi J, Piskadlo E, Mach P, Kryzhanovska M, Tihanyi G, Kohler H, et al. (2022). Nonlinear control of transcription through enhancer-promoter interactions. Nature 604, 571–577. 10.1038/s41586-022-04570-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gazal S, Weissbrod O, Hormozdiari F, Dey KK, Nasser J, Jagadeesh KA, Weiner DJ, Shi H, Fulco CP, O’Connor LJ, et al. (2022). Combining SNP-to-gene linking strategies to identify disease genes and assess disease omnigenicity. Nat. Genet. 54, 827–836. 10.1038/s41588-022-01087-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markaki Y, Gan Chong J, Wang Y, Jacobson EC, Luong C, Tan SYX, Jachowicz JW, Strehle M, Maestrini D, Banerjee AK, et al. (2021). Xist nucleates local protein gradients to propagate silencing across the X chromosome. Cell 184, 6174–6192.e32. 10.1016/j.cell.2021.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jachowicz JW, Strehle M, Banerjee AK, Blanco MR, Thai J, and Guttman M (2022). Xist spatially amplifies SHARP/SPEN recruitment to balance chromosome-wide silencing and specificity to the X chromosome. Nat. Struct. Mol. Biol. 29, 239–249. 10.1038/s41594-022-00739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schertzer MD, Thulson E, Braceros KCA, Lee DM, Hinkle ER, Murphy RM, Kim SO, Vitucci ECM, and Calabrese JM (2019). A piggyBac-based toolkit for inducible genome editing in mammalian cells. RNA 25, 1047–1058. 10.1261/rna.068932.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolff J, Bhardwaj V, Nothjunge S, Richard G, Renschler G, Gilsbach R, Manke T, Backofen R, Ramírez F, and Grüning BA (2018). Galaxy HiCExplorer: a web server for reproducible Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 46, W11–W16. 10.1093/nar/gky504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, and Manke T (2018). High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189. 10.1038/s41467-017-02525-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137, gb-2008–9-9-r137 [pii]. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 63.Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; ). [Google Scholar]
- 64.Calabrese JM, Starmer J, Schertzer MD, Yee D, and Magnuson T (2015). A survey of imprinted gene expression in mouse trophoblast stem cells. G3 (Bethesda) 5, 751–759. 10.1534/g3.114.016238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinn J, Kunath T, and Rossant J (2006). Mouse trophoblast stem cells. Methods Mol. Med. 121, 125–148. [DOI] [PubMed] [Google Scholar]
- 66.Concordet JP, and Haeussler M (2018). CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245. 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davis CP, and West JA (2015). Purification of specific chromatin regions using oligonucleotides: capture hybridization analysis of RNA targets (CHART). Methods Mol. Biol. 1262, 167–182. 10.1007/978-1-4939-2253-6_10. [DOI] [PubMed] [Google Scholar]
- 68.Flickinger JL, Gebeyehu G, Buchman G, Haces A, and Rashtchian A (1992). Differential incorporation of biotinylated nucleotides by terminal deoxynucleotidyl transferase. Nucleic Acids Res. 20, 2382. 10.1093/nar/20.9.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee BT, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M, Fischer C, Gonzalez JN, Hinrichs AS, Lee CM, et al. (2022). The UCSC Genome Browser database: 2022 update. Nucleic Acids Res. 50, D1115–D1122. 10.1093/nar/gkab959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robinson JT, Turner D, Durand NC, Thorvaldsdóttir H, Mesirov JP, and Aiden EL (2018). Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 6, 256–258.e1. 10.1016/j.cels.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170. 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 73.Justice M, Carico ZM, Stefan HC, and Dowen JM (2020). A WIZ/Cohesin/CTCF Complex Anchors DNA Loops to Define Gene Expression and Cell Identity. Cell Rep. 31, 107503. 10.1016/j.celrep.2020.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Frankish A, Diekhans M, Jungreis I, Lagarde J, Loveland JE, Mudge JM, Sisu C, Wright JC, Armstrong J, Barnes I, et al. (2021). Gencode 2021. Nucleic Acids Res. 49, D916–D923. 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed sequencing data generated in this study have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All new and published datasets analyzed are summarized in Table S4. Oligonucleotides used are detailed in Table S5.
All original code has been deposited at Zenodo and Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| H3K27me3 | Cell Signaling | Cat#9733; RRID: AB_2616029 |
| H2AK119ub | Cell Signaling | Cat#8240; RRID: AB_10891618 |
| RING1B | Cell Signaling | Cat#5694; RRID: AB_10705604 |
| CBX7 | Abcam | Cat#ab21873; RRID: AB_726005 |
| DEDAF (RYBP) | Millipore | Cat#AB3637; RRID: AB_11213333 |
| JHDM1B (KDM2B) | Millipore | Cat#09–864; RRID: AB_10806072 |
| EZH2 | Cell Signaling | Cat#5246; RRID: AB_10694683 |
| MTF2 | Proteintech | Cat#16208–1-AP; RRID: AB_2147370 |
| JARID2 | Cell Signaling | Cat#13594; RRID: AB_2798269 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Heparin Sodium Salt | Sigma-Aldrich | Cat#H3149 |
| FGF4 Recombinant Human Protein | Gibco | Cat#PHG0154 |
| RPMI 1640 | Gibco | Cat#11875093 |
| DMEM, high glucose, sodium pyruvate | Gibco | Cat#11995073 |
| Fetal Bovine Serum, qualified | Gibco | Cat#26140079 |
| Penicillin-Streptomycin | Gibco | Cat#15140122 |
| L-glutamine | Gibco | Cat#25030081 |
| MEM Non-essential Amino Acids | Gibco | Cat#11140050 |
| Sodium Pyruvate | Gibco | Cat#11360070 |
| β-mercaptoethanol | Sigma-Aldrich | Cat#63689 |
| LIF | Lif-1Cα/COS-conditioned ESC media | N/A |
| Trypsin-EDTA, phenol red | Gibco | Cat#2520072 |
| LipoFectamine 3000 | Invitrogen | Cat#L3000015 |
| 10X Phosphate-Buffered Saline (PBS) | Corning | Cat#46–013-CM |
| Pierce 16% Formaldehyde | Thermo Scientific | Cat#28906 |
| 37% Formaldehyde | Fisher Scientific | Cat#BP531–500 |
| TRIzol Reagent | Invitrogen | Cat#15596026 |
| Chloroform | Fisher Scientific | Cat#BP1145–1 |
| 25:24:1 Phenol:Chloroform:Isoamyl Alcohol | Sigma-Aldrich | Cat#6805 |
| Protein A/G PLUS Agarose Beads | Santa Cruz Biotechnology | Cat#sc-2003 |
| AMPure XP Beads | Beckman Coulter | Cat#A63880 |
| Dynabeads M-280 Streptavidin | Invitrogen | Cat#11205D |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | Cat#65002 |
| Ribonuclease A (RNase A) from bovine pancreas | Sigma-Aldrich | Cat#R4642 |
| Proteinase K Solution | Bioline | Cat#BIO-37084 |
| Proteinase K (Fungal) | Invitrogen | Cat#25530015 |
| Linear Acrylamide | Invitrogen | Cat#AM9520 |
| Choice Taq | Denville | Cat#CB4050–2 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0530S |
| Biotin-16-dUTP | Roche | Cat#11093070910 |
| Terminal Deoxynucleotidyl Transferase | Thermo Scientific | Cat#EP0161 |
| Klenow Fragment (3'->5' exo) | New England Biolabs | Cat#M0212S |
|
| ||
| dATP Solution | Invitrogen | Cat#10–216-018 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Neon Transfection System 100μL Kit | Invitrogen | Cat#MPK10025 |
| QIAquick Nucleotide Removal Kit | Qiagen | Cat#28304 |
| Zymo Research RNA Clean & Concentrator | Zymo Research Kit | Cat#50–125-1669 |
| Arima-HiC + Kit | Arima Genomics | Cat#A510008 |
| KAPA RNA HyperPrep with RiboErase | Roche/KAPA Biosystems (HMR) | Cat#KK8561 |
| NEBNext End Repair Module | New England Biolabs | Cat#E6050S |
| Quick Ligation Kit | New England Biolabs | Cat#M2200S |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat#M0541S |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#1725125 |
| PureLink HiPure Plasmid Midiprep Kit | Invitrogen | Cat#K2100004 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed sequencing data | This study; see Table S4 | GEO: GSE217262 |
| Mouse reference genome NCBI build 37, NCBI37/mm9 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/mouse |
| Variant sequence data | Sanger Institute | https://www.sanger.ac.uk/data/mouse-genomes-project/ |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: C/B and B/C trophoblast stem cells | This study | N/A |
| Mouse: E14 embryonic stem cells | This study | N/A |
| Mouse: DR4 embryonic fibroblast cells | ATCC | Cat#SCRC-1045 |
|
| ||
| Oligonucleotides | ||
|
| ||
| CRISPR-Cas9 sgRNAs, see Table S5 | This study | N/A |
| Genotyping PCR primers, see Table S5 | This study | N/A |
| qPCR primers, see Table S5 | This study | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| PB_tre_dCas9_VP160 vector | Schertzer et al.55 | Cat#126031;https://doi.org/10.1261/rna.068932.118 |
| PB_tre_Cas9 vector | Schertzer et al.55 | Cat#126029;https://doi.org/10.1261/rna.068932.118 |
| PB_rtTA_BsmBI vector Schertzer et al.55 | Schertzer et al.55 | Cat#126028;https://doi.org/10.1261/rna.068932.118 |
| pUC19-piggyBac transposase | Schertzer et al.55 | https://doi.org/10.1261/rna.068932.118 |
|
| ||
| Software and algorithms | ||
|
| ||
| Custom Scripts | This study | https://github.com/aki-kb/calabrese_lab/tree/main/braceros_airn_2023;https://doi.org/10.5281/zenodo.7988247 |
| Juicer/Juicebox (v1.5.6) | Durand et al.27,28 | https://github.com/aidenlab/juicer;https://github.com/aidenlab/Juicebox |
| Juicer Tools (v1.9.9) | Durand et al.27 | https://github.com/aidenlab/juicer |
| HiCExplorer (v3.7.2) | Wolff et al.,56 Ramirez et al.57 | https://hicexplorer.readthedocs.io/en/latest |
| Bowtie2 (v2.4.5) | Langmead and Salzberg58 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR (v2.7.9a) | Dobin et al.58 | https://github.com/alexdobin/STAR |
|
| ||
| Samtools (v1.16) | Li et al.59 | https://github.com/samtools/samtools |
| Bedtools (v2.30) | Quinlan and Hall60 | https://bedtools.readthedocs.io/en/latest |
| Significant Peak Interaction (SIP; v1.5) | Rowley et al.43 | https://github.com/PouletAxel/SIP |
| MACS2 (v2.1.2) | Zhang et al.61 | https://github.com/taoliu/MACS |
| Subread: featureCounts (v1.6.3) | Liao et al.62 | http://subread.sourceforge.net |
| RStudio (v4.2.0) | N/A | https://posit.co/products/open-source/rstudio |
| ggplot2, ggpubr (v3.3.5) | Wickham63 | https://ggplot2.tidyverse.org |
| GraphPad Prism (v9.4.1) | N/A | https://www.graphpad.com |
|
| ||
| Other | ||
|
| ||
| Vibra-Cell™ VCX 130 Sonicator & 6mm Probe | Sonics | Cat#H-1002–2 |
| Bioruptor® Plus sonication device | Diagenode | Cat#B01020001 |