

Abstract
The cannabis derivative marijuana is the most widely used recreational drug in the Western world and is consumed by an estimated 83 million individuals (∼3% of the world population). In recent years, there has been a marked transformation in society regarding the risk perception of cannabis, driven by its legalization and medical use in many states in the United States and worldwide. Compelling research evidence and the Food and Drug Administration cannabis-derived cannabidiol approval for severe childhood epilepsy have confirmed the large therapeutic potential of cannabidiol itself, Δ9-tetrahydrocannabinol and other plant-derived cannabinoids (phytocannabinoids). Of note, our body has a complex endocannabinoid system (ECS)—made of receptors, metabolic enzymes, and transporters—that is also regulated by phytocannabinoids. The first endocannabinoid to be discovered 30 years ago was anandamide (N-arachidonoyl-ethanolamine); since then, distinct elements of the ECS have been the target of drug design programs aimed at curing (or at least slowing down) a number of human diseases, both in the central nervous system and at the periphery. Here a critical review of our knowledge of the goods and bads of the ECS as a therapeutic target is presented to define the benefits of ECS-active phytocannabinoids and ECS-oriented synthetic drugs for human health.
Significance Statement
The endocannabinoid system plays important roles virtually everywhere in our body and is either involved in mediating key processes of central and peripheral diseases or represents a therapeutic target for treatment. Therefore, understanding the structure, function, and pharmacology of the components of this complex system, and in particular of key receptors (like cannabinoid receptors 1 and 2) and metabolic enzymes (like fatty acid amide hydrolase and monoacylglycerol lipase), will advance our understanding of endocannabinoid signaling and activity at molecular, cellular, and system levels, providing new opportunities to treat patients.

I. Introduction
Paleobotanical records date the beginning of human cannabis cultivation in Eurasia to > 8000 years ago, while archaeological evidence anchors its use as a psychotropic substance to approximately 2500 years ago (Russo et al., 2008; Long et al., 2017). Today, cannabis is one of the world’s most widely used recreational drugs, after alcohol and tobacco, and is consumed by an estimated 83 million individuals (∼3% of the world population) (https://www.unodc.org/wdr2017/field/Booklet_1_EXSUM.pdf). Cannabis’ increasingly expanding legal status heightens the need for research into its therapeutic potential for a wide range of pathologic conditions (National Academies of Sciences, Engineering, and Medicine, 2017; Cohen et al., 2019; Friedman et al., 2019; Cristino et al., 2020) but also raises concerns about its possible hazards to health. Indeed, medical and nonmedical cannabis use has been associated with short-term and long-term adverse effects, including schizophrenia, alterations in cognition, and mood disorders (Cohen et al., 2019), as well as an impact on adult neurogenesis (Oddi et al., 2020) and female (Cecconi et al., 2020) and male reproductive health (Maccarrone et al., 2021).
A. Phytocannabinoids
The trichomes, specialized structures in the inflorescences of the female cannabis plant, produce a family of terpenophenolic substances, called phytocannabinoids (pCBs), which contain tricyclic, bicyclic, and monocyclic structures. In most cannabis varietals, the most abundant pCBs are the acidic (i.e., carboxylic) precursors of Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD), which are converted to THC and CBD by drying or heating, but many others have been identified whose pharmacological properties are still awaiting clarification (Gomez-Cañas et al., 2023). Indeed, cannabis contains more than 110 pCBs as well as hundreds of terpenoids, flavonoids, sterols, and other non-pCB substances (El Sohly and Gul, 2014; El Sohly et al., 2017; Solymosi and Köfalvi, 2017). THC and its analogs (including Δ8-tetrahydrocannabinol and the propyl derivative Δ9-tetrahydrocannabivarin), CBD and its analogs (including cannabidivarin), cannabinol and its analogs (including the propyl derivative cannabivarin), and cannabigerol and its analogs are highly abundant. In addition, trace amounts of cannabinodiol, cannabichromene, cannabicyclol, cannabielsoin, and cannabitriol are also detectable (Mechoulam, 2005; El Sohly and Gul, 2014; El Sohly et al., 2017; Morales et al., 2017; Li et al., 2022). The structures of the main pCBs identified so far are shown in Table 1.
TABLE 1.
Major phytocannabinoids (pCBs)
| Name (abbreviation) | Chemical Structure |
|---|---|
| More Abundant pCBs | |
| Δ9-Tetrahydrocannabinol (THC) |
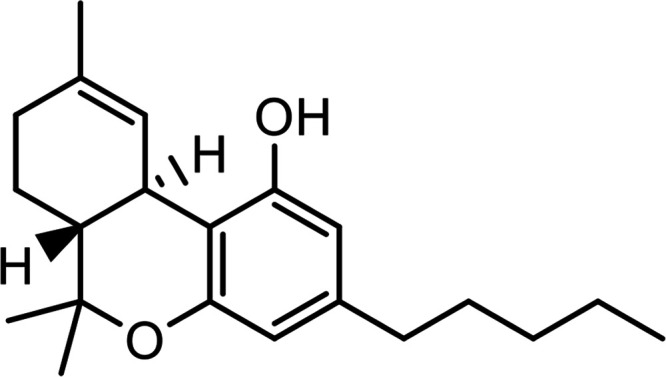
|
| Cannabidiol (CBD) |
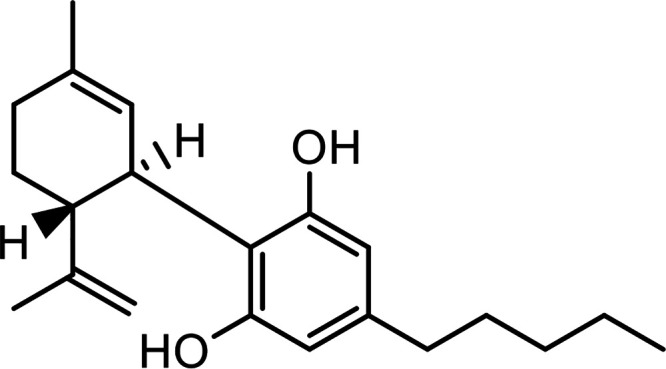
|
| Cannabinol (CBN) |
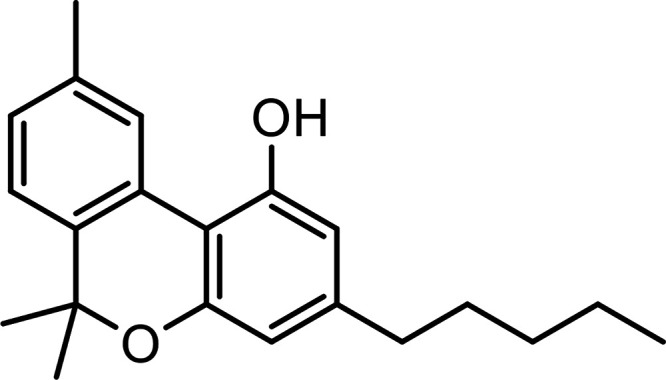
|
| Cannabigerol (CBG) |
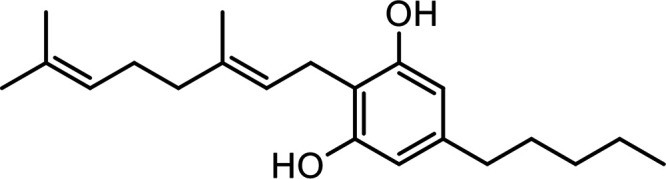
|
| Cannabivarin (CBV) |
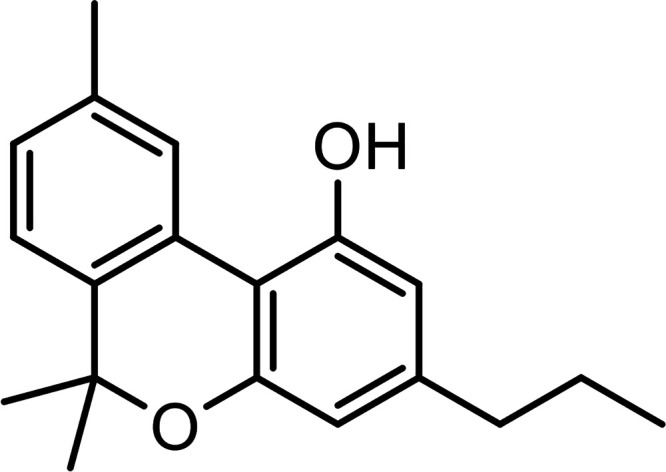
|
| Cannabidivarin (CBDV) |
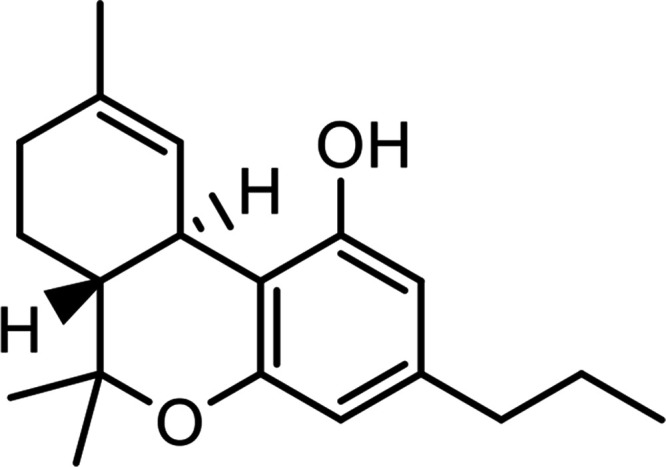
|
| Δ9-Tetrahydrocannabivarin (THCV) |
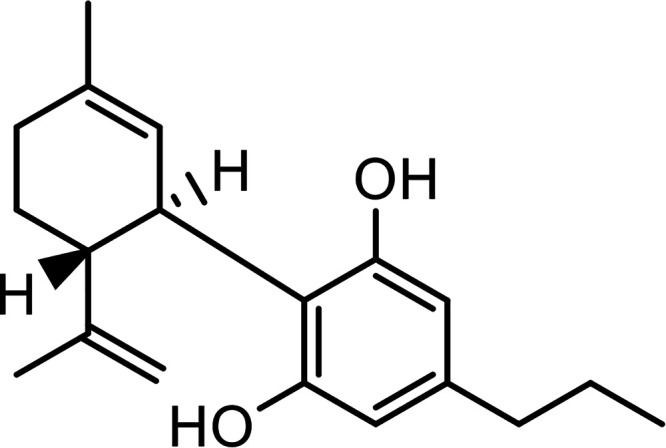
|
| Less Abundant pCBs | |
| Cannabichromene (CBC) |
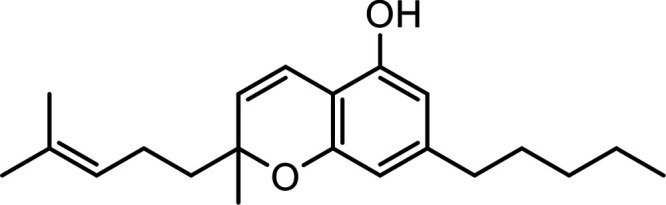
|
| Cannabinodiol (CBND) |
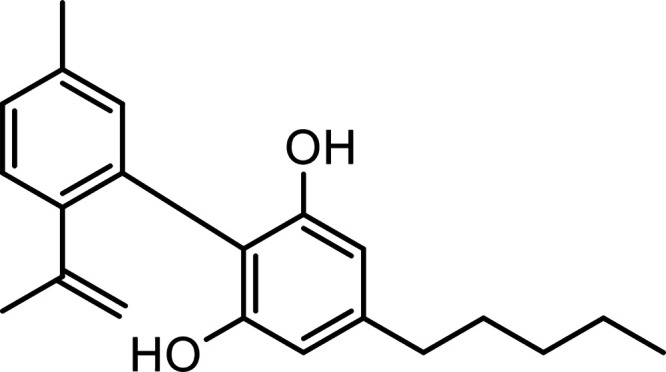
|
| Cannabicyclol (CBL) |
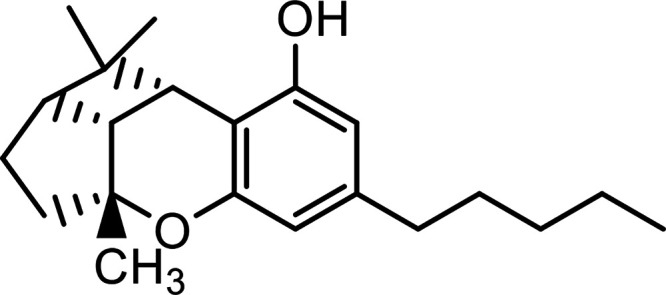
|
| Cannabielsoin (CBE) |
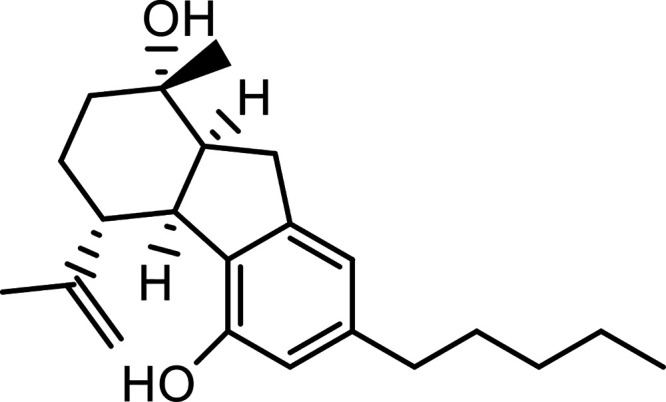
|
| Cannabitriol (CBT) |
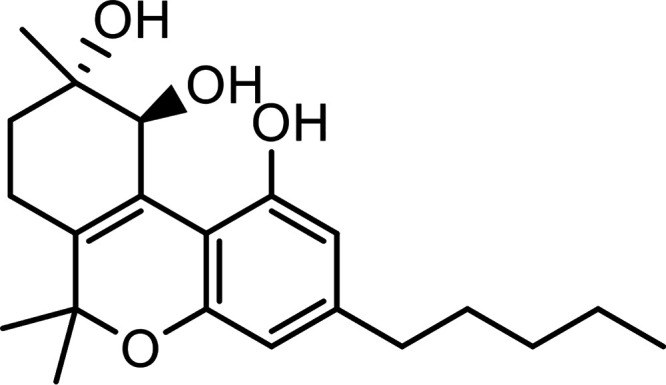
|
To date, the therapeutic potential of THC and CBD, alone or in combination, seems apparent and has been critically discussed in recent reviews (Maccarrone et al., 2017; Friedman et al., 2019; Pacher et al., 2020; Rock et al., 2021; Stella, 2023). Here, the main applications of THC and CBD for human health are summarized in Table 2.
TABLE 2.
Approved and potential indications for THC and CBD
| Cannabinoid | Approved (A) and Potential (B) Indications |
|---|---|
| THC | (A) Chemotherapy-induced nausea and vomiting; appetite stimulant (HIV/AIDS). (B) Spasticity in MS; neuropathic pain in MS; cancer pain unresponsive to opioids; other pain conditions (i.e., postherpetic neuralgia, postoperative pain); intraocular pressure in glaucoma; depression; anxiety/sleep disorder; psychosis; tics of Tourette syndrome; tremor/bladder dysfunction in MS; dyskinesias in HD; levodopa-induced dyskinesias in PD; cervical dystonia; epilepsy; and AD. |
| CBD | (B) Childhood epilepsy; tuberous sclerosis complex seizure; Lennox-Gastaut syndrome; Dravet syndrome and infantile spasms. |
| THC/CBD | (A) Spasticity in MS. (B) Paraplegia and spasticity in amyotrophic lateral sclerosis; cancer pain unresponsive to opioids; other pain conditions (i.e., postherpetic neuralgia, postoperative pain); intraocular pressure in glaucoma; depression; anxiety/sleep disorder; psychosis; tics of Tourette syndrome; tremor/bladder dysfunction due to MS; dyskinesias in HD; levodopa-induced dyskinesias in PD; cervical dystonia; epilepsy; and AD. |
AD, Alzheimer’s disease; CBD, cannabidiol; HD, Huntington’s disease; MS, multiple sclerosis; PD, Parkinson’s disease; THC, Δ9-tetrahydrocannabinol.
By contrast, our understanding of the pharmacological properties of less prevalent pCBs has only scratched the surface, and very little information is available on their effect in the human body (Russo, 2018; Franco et al., 2020; Maccarrone, 2020; Rock et al., 2021; Mechoulam, 2023; Li et al., 2022). For instance, cannabidiolic acid and cannabichromene are used in creams, foods, and beverages (Straiker et al., 2021), and the methyl ester of cannabidiolic acid has been shown to suppress nausea and anxiety (Pertwee et al., 2018), to reduce depression-like effects (Hen-Shoval et al., 2018), and to have a potent antihyperalgesic effect (Zhu et al., 2020). Further research has shown that cannabinol exhibits neuroprotective effects in an experimental model of glaucoma (Somvanshi et al., 2022); cannabigerol reduces inflammation, pain, and obesity (Kogan et al., 2021); and both pCBs hold anticancer potential (Li et al., 2022). Humans and other mammals do not produce pCBs but can effectively remove them via the cytochrome P450 and glucuronidation pathways in the liver and other organs (Huestis, 2007; Watanabe et al., 2007; Schafroth and Carreira, 2017; Solymosi and Köfalvi, 2017).
Overall, it is apparent that the term “phytocannabinoid” serves to cluster different plant-derived lipophilic compounds (Pertwee, 2014; Ligresti et al., 2016). It is also worth noting that different cannabis varietals can have distinct chemical profiles (referred to as “chemovars”) and can thus display both qualitative and quantitative differences in their constituents. Because differences in genetics, cultivation technique, harvest, and extraction can affect the ultimate product consumed by humans, it is reasonable to conclude that there is no “one cannabis” and that caution must be taken in generalizing its effects (Hanuš et al., 2016; Procaccia et al., 2022). This variability may also confound our understanding of cannabis’ pharmacological properties, and, indeed, remaining uncertainties represent a serious obstacle to its clinical applications (Friedman et al., 2019). Unsurprisingly, despite its use for millennia, cannabis remains surrounded by controversies, debates, and misconceptions related to its medical potential, legalization, and long-term health consequences.
Taken together, the complexity of cannabis extracts seems apparent. However, such a complexity is mirrored, and possibly even exceeded, by that of the ensemble of receptors, enzymes, and transporters of endocannabinoid (eCB) substances that together form the “eCB system” (ECS), recently discussed in comprehensive reviews (Iannotti et al., 2016; Maccarrone, 2017; Baggelaar et al., 2018; Cristino et al., 2020; Kilaru and Chapman, 2020; Simard et al., 2022; Piomelli and Mabou Tagne, 2022). Notably, the main components of the ECS support and control the manifold actions of the eCBs both in the central nervous system (CNS) (Maccarrone et al., 2014; Iannotti et al., 2016; Cristino et al., 2020) and the periphery (Maccarrone et al., 2015). Here it should be stressed that little is still known about the effects that pCBs have on the ECS. Emerging evidence indicates that, even at low concentrations, THC can alter eCB signaling, especially when administered during critical periods such as adolescence (Lee et al., 2022). Additionally, 24-hour treatment with cannabigerol, cannabichromene, Δ9-tetrahydrocannabivarin, and cannabigerolic acid has been shown to modulate the function of distinct ECS elements in human HaCaT keratinocytes, where they all increase binding of [3H]CP55940 to cannabinoid receptors 1 and 2 (CB1R and CB2R), stimulation of transient receptor potential vanilloid 1 (TRPV1) channels, as well as catalytic activity of fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) catabolic enzymes (Di Meo et al., 2022). These data extend previous studies on the effects of cannabinoid-enriched cannabis extracts on transient receptor potential (TRP) channels (De Petrocellis et al., 2011) and of cannabidiol- and cannabigerol-type pCBs on CB1R and CB2R (Navarro et al., 2020), suggesting that these minor pCBs could have an impact when present in various cannabis formulations (Di Marzo and Piscitelli, 2015; Turner et al., 2017).
B. Cannabinoid Receptors, Endocannabinoids, and Their Congeners
The discovery of THC in the 1940s (Adams et al., 1948) and its complete structural elucidation 20 years later (Gaoni and Mechoulam, 1964) allowed researchers to synthesize radiolabeled synthetic analogs that were instrumental to the identification and localization of specific cannabinoid binding sites in the brain (Devane et al., 1988; Herkenham, et al., 1990). In particular, a radiolabeled THC congener, the nonclassical bicyclic cannabinoid CP55940, allowed researchers to perform initial binding assays and structure-activity relationship studies of the receptor (Devane et al., 1988; Howlett et al., 1988). This was followed by development of radiolabeled 5′-(1,1-dimethylheptyl)-7-hydroxyhexahydrocannabinol (Devane et al., 1992a). The pharmacological characterization eventually led to the molecular cloning of the CB1R from rat (Matsuda et al., 1990) and human (Gerard et al., 1990, 1991) orphan G protein-coupled receptor (GPCR) clones. CB1R activation in mice led to a standard set of cannabimimetic responses, the so-called “tetrad test,” which sequentially assesses antinociception, catalepsy, hypomotility, and hypothermia (Smith et al., 1994). Shortly afterward a second molecular target of THC was found and named CB2R (Munro et al., 1993), predominantly localized to the immune system (Lynn and Herkenham, 1994), where it leads to immune suppressive responses (Howlett et al., 2002; Klein and Cabral, 2006; Cabral and Griffin-Thomas, 2008; Cabral et al., 2008). For a comprehensive review of both cannabinoid receptors see the report of the International Union of Pharmacology Cannabinoid Receptor Nomenclature Committee (Howlett et al., 2002).
The identification of CB1R, the most abundant GPCR in the mammalian brain, and of CB2R prompted intense research into the endogenous ligands for these receptors (Di Marzo and Fontana, 1995). Such ligands were identified as anandamide [N-arachidonoylethanolamine (AEA)] (Devane et al., 1992b) and 2-arachidonoylglycerol (2-AG) (Mechoulam et al., 1995; Sugiura et al., 1995). The first endogenous ligand of CB1R and CB2R was named anandamide after the Sanskrit word “Ananda,” which means bliss, and on its chemical nature as an amide. Indeed, AEA and 2-AG are an amide and an ester of the ω-6 arachidonic acid (AA), respectively (Table 3), and remain the best-studied eCBs.
TABLE 3.
Major endocannabinoids and congeners
| Name (abbreviation) | Chemical Structure | |
|---|---|---|
| Major ω-6 eCBs | ||
|
N-Arachidonoylethanolamine (Anandamide, AEA) |
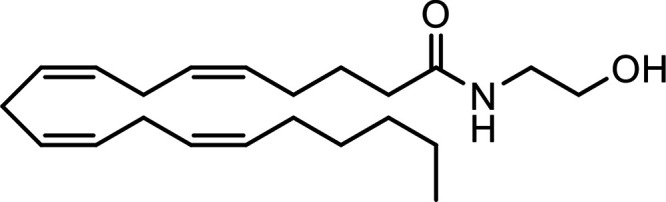
|
|
| 2-Arachidonoylglycerol (2-AG) |
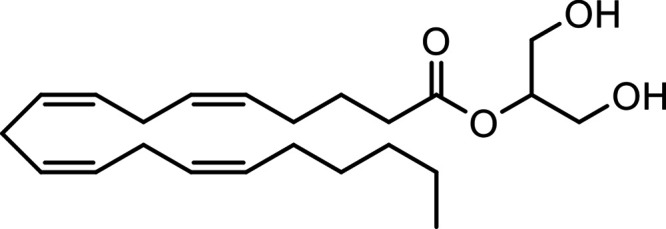
|
|
| 2-Arachidonoylglycerol (Noladin) Ether (2-AGE) |
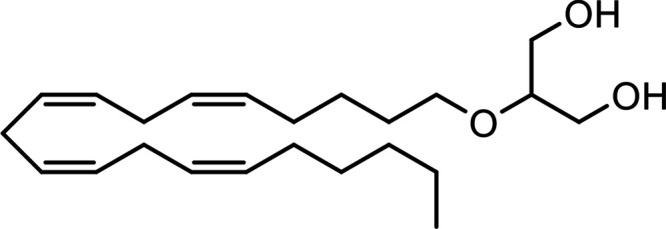
|
|
| Virodhamine (O-Arachidonoylethanolamine, O-AEA) |
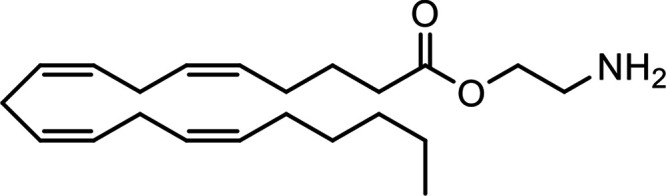
|
|
| Major ω-3 eCBs | ||
|
N-Eicosapentaenoylethanolamine (EPEA) |
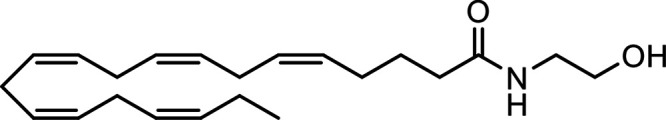
|
|
|
N-Docosahexaenoylethanolamine (DHEA) |
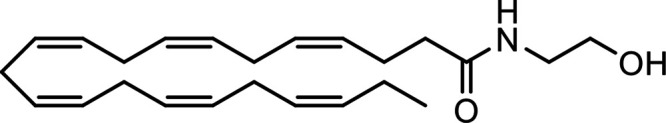
|
|
| Major eCB-like Compounds | ||
|
N-Palmitoylethanolamine (PEA) |
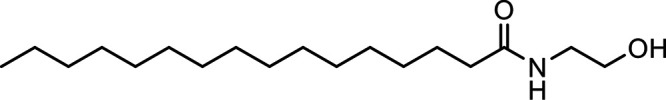
|
|
|
N-Oleoylethanolamine (OEA) |
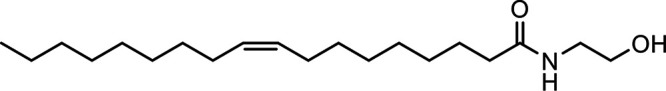
|
|
|
N-Stearoylethanolamine (SEA) |
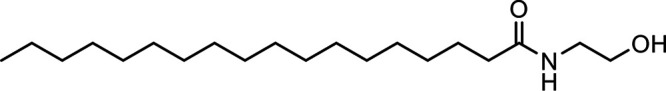
|
|
|
N-Linoleoylethanolamine (LEA) |
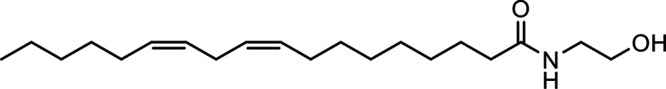
|
|
| 2-Oleoylglycerol (2-OG) |
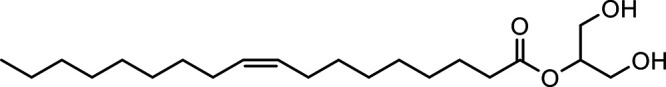
|
|
| Major eCB-Amino Acids | ||
|
N-Arachidonoyl dopamine (NADA) |
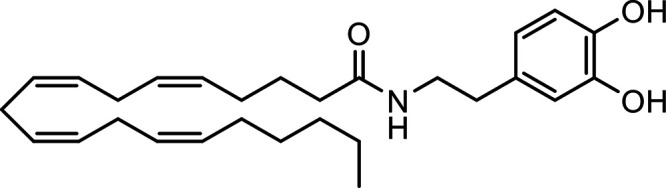
|
|
|
N-Arachidonoyl glycine (NAGly) |
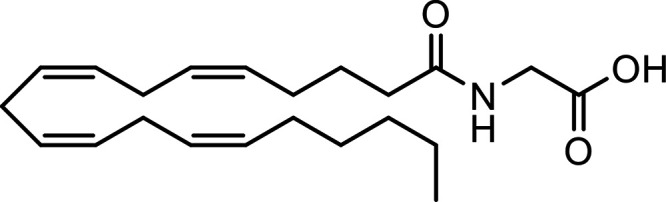
|
|
|
N-Arachidonoyl serine (ARA-S) |
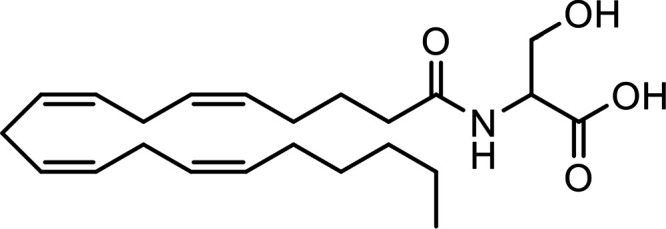
|
|
|
N-Oleoyl glycine (OlGly) |
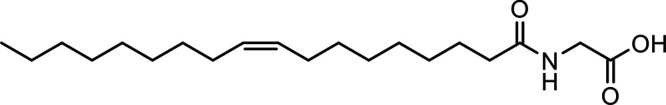
|
|
|
N-Oleoyl alanine (OlAla) |
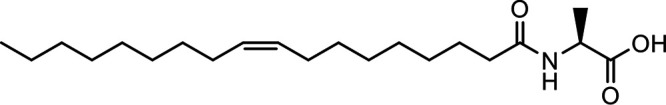
|
|
Other potential members of the eCB family have been discovered, including: (1) ω-6 fatty acid-derived eCBs like AEA, 2-AG, 2-arachidonoylglycerol (noladin) ether, and the “inverted anandamide” virodhamine, reported to have various biologic activities (Maccarrone, 2017; Baggelaar et al., 2018; Cristino et al., 2020), and (2) ω-3 fatty acid-derived eCBs like N-eicosapentaenoylethanolamine and N-docosahexaenoylethanolamine, endowed with promising anticancer activity (Brown et al., 2010, 2020). In addition, various eCB-like fatty acid ethanolamides, including N-palmitoylethanolamine and N-oleoylethanolamine, have been described, which serve important functions in the control of energy metabolism (Rodríguez de Fonseca et al., 2001; Schwartz et al., 2008; Misto et al., 2019), pain (Calignano et al., 1998; Fotio et al., 2021b), and inflammation (Solorzano et al., 2009) by engaging the nuclear receptor peroxisome proliferator-activated receptor (PPAR) α (Fu et al., 2003; Lo Verme et al., 2005). N-stearoylethanolamine also has anti-inflammatory activity but via activation of PPARγ (Kosiakova et al., 2022). Finally, eCB-like amino acids (also known as lipoamino acids) have been isolated, such as N-arachidonoylglycine, N-arachidonoyldopamine, N-arachidonoylserine, N-oleoylglycine, and N-oleoylalanine (Ayoub et al., 2020), which may have a number of distinct biologic activities and hold therapeutic potential against vasodilation and osteoporosis (Table 3).
Athough THC and AEA have completely different structures, with THC being a terpene-resorcinol derivative (Table 1) and AEA being an AA amide linkage with ethanolamine (Table 3), their biologic activities were found to be closely related (Fride and Mechoulam, 1993; Vogel et al., 1993). Also of note is the observation based on phylogenetic analyses that eCBs appear to be much older than pCBs. Cannabis (aged ca. 76 − 107 million years) is much younger than organisms like black truffles (Tuber melanosporum, aged ca. 156 million years) (Pacioni et al., 2015), hydra (De Petrocellis et al., 1999), and tetraymena (Anagnostopoulos et al., 2010) where eCBs can be detected.
C. Diverse Phytocannabinoids and Endocannabinoid Targets and Signaling Pathways
The number of receptors activated by pCBs and eCBs in the same cell, both on the plasma membrane and in the nucleus, appears striking and is schematically depicted in Fig. 1.
Fig. 1.
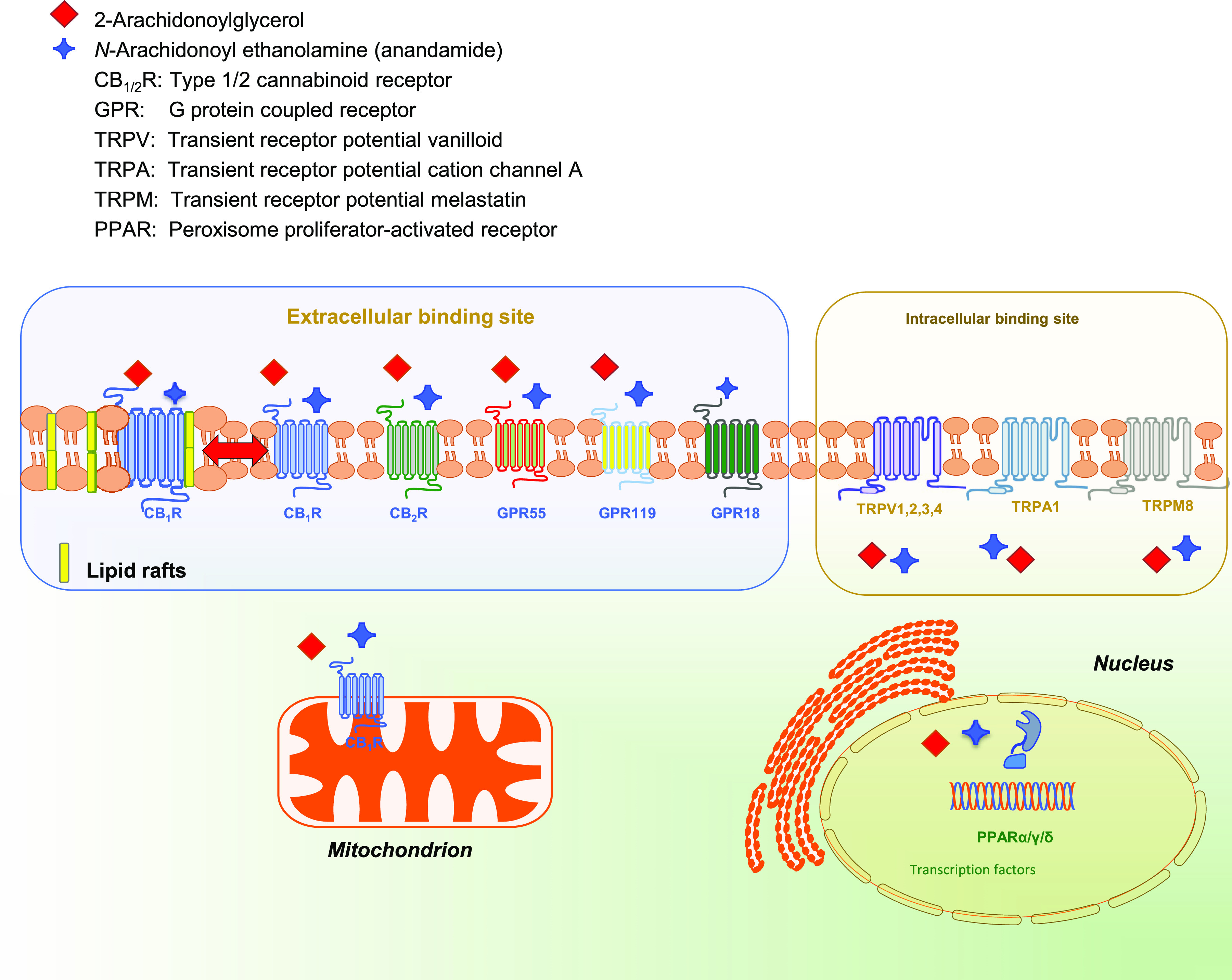
Endocannabinoid binding receptors. The two major endocannabinoids anandamide and 2-arachidonoylglycerol bind to and activate metabotropic and ionotropic membrane receptors (with either an intracellular or an extracellular binding site) and nuclear receptors.
Indeed, pCB- and eCB-binding receptors include (1) seven-transmembrane GPCRs CB1 and CB2 (Howlett et al., 2002), as well the recently deorphanized GPCRs GPR55, GPR119, and GPR18 that can also bind cannabinoid-like ligands (Godlewski et al., 2009; Pertwee et al., 2010; Zhao and Abood, 2013; Shore and Reggio, 2015; Morales and Reggio, 2017; Alhouayek et al., 2018; Morales et al., 2020; Im, 2021); (2) receptors that are located on the plasma membrane and have intracellular binding sites, such as ionotropic TRP vanilloid 1, 2, 3, 4 channels, TRP cation channel A1, and melastatin 8, which are all six-transmembrane spanning receptors; and (3) nuclear PPARs α, γ, and δ, which are transcription factors able to regulate gene expression (Maccarrone, 2020; Gomez-Cañas et al., 2023). Of note, CB1R has been shown to move in and out of distinct microdomains of the plasma membrane known as lipid rafts, which might contribute to the control of their G protein-dependent signaling (Oddi et al., 2017; Saumell-Esnaola et al., 2021). In addition, CB1R appears to localize also in the outer membrane of mitochondria, where it modulates energy metabolism of neuronal and nonneuronal cells (Pagano Zottola et al., 2022). GPCRs, TRPs, and PPARs trigger different transduction pathways, summarized in Fig. 2.
Fig. 2.
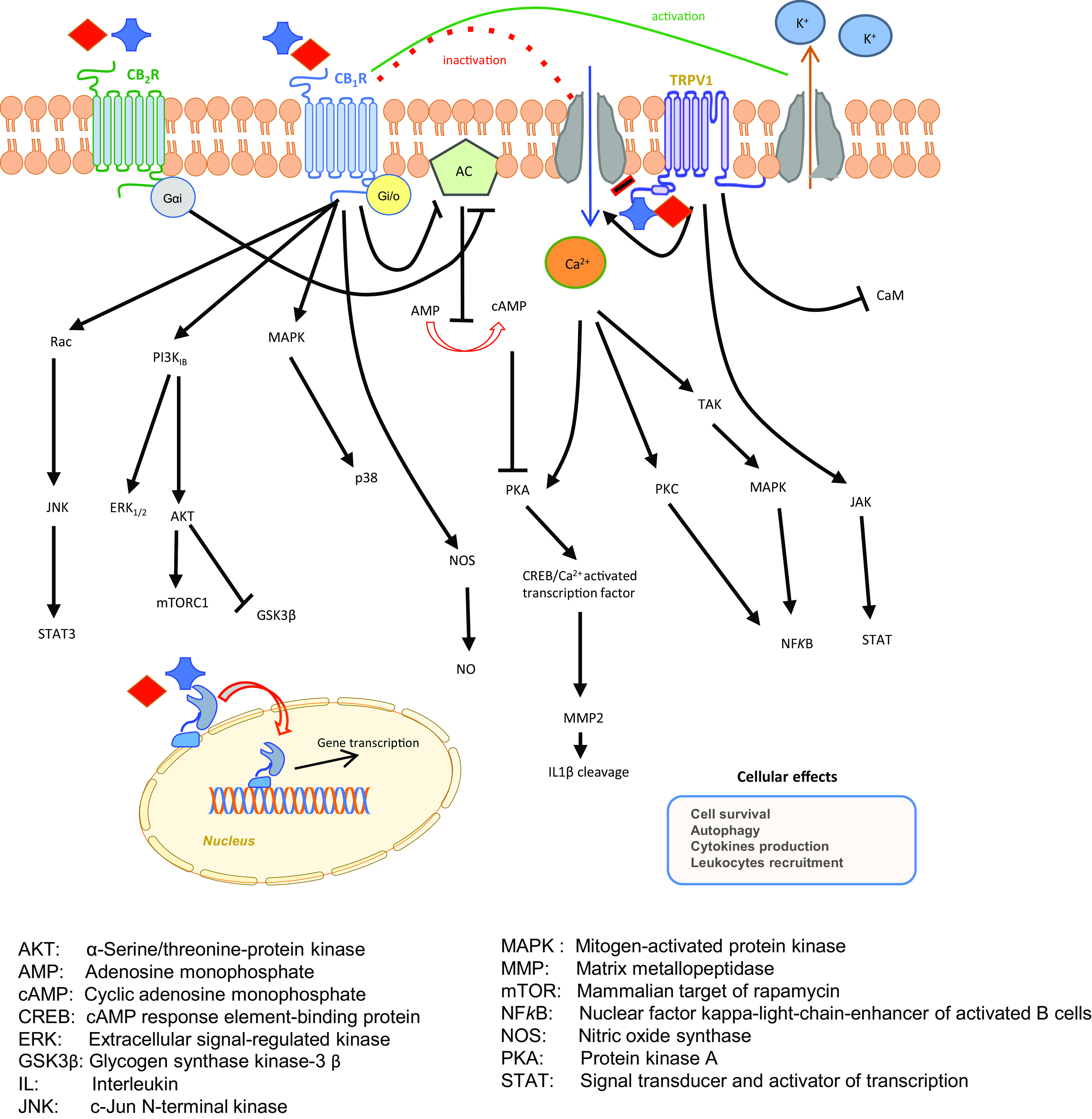
Endocannabinoid signaling pathways. Receptor binding by anandamide and 2-arachidonoylglycerol triggers various signal transduction pathways, which activate G proteins, ion channels, as well as gene transcription.
Therapeutic benefits have been documented by targeting the pCB/eCB-binding receptors and signal transduction thereof, both in CNS and peripheral pathologies as detailed in the following sections. It is now widely appreciated that GPCRs instigate intracellular signaling by two transducer families, heterotrimeric G proteins and GPCR kinases/arrestin. These transducers interact with agonist-bound GPCRs to trigger alternative signaling cascades, so that biased agonists that favor either heterotrimeric G protein or GPCR kinases/arrestin signaling are of profound pharmacological interest (Chen and Tesmer, 2022). In this context, recent advances in understanding biased signaling and off-target activity of CB2R (Soethoudt et al., 2017), also in living cells (Sarott et al., 2020), and molecular mechanism of allosteric modulation of CB1R (Yang et al., 2022) suggest that biased signaling driven by eCBs might be better appreciated in the near future and usher in a new generation of drugs with greatly reduced side effects.
D. Metabolic Routes
Metabolism of AEA and 2-AG has been intensely investigated since their discovery in the mid-1990s, whereas little information is as yet available on the metabolic routes of the additional eCBs and congeners. AEA and 2-AG are metabolized by a complex array of distinct biosynthetic and catalytic enzymes and are transported through the plasma membrane, intracellularly and extracellularly, by distinct and poorly understood mechanisms that engage putative protein carriers.
In general, it is of paramount importance that all biologic activities of eCBs, either receptor dependent or independent, are subjected to a stringent “metabolic control,” which means that they depend on the cellular concentration of eCBs, which in turn depends on a balance between synthesis and degradation by multiple regulated enzymes (Friedman et al., 2019; Cristino et al., 2020; Maccarrone, 2020).
1. Metabolism of N-Arachidonyl Ethanolamine
AEA can be produced by membrane phospholipid precursors via multiple pathways, as schematically depicted in Fig. 3. Among these, N-acyltransferase (NAT), either Ca2+-dependent or independent (iNAT), and N-acylphosphatidylethanolamine-specific phospholipase D (NAPE-PLD) catalyze the most classic route for the release of AEA from phosphatidylethanolamine and phosphatidylcholine precursors. In addition, soluble phospholipase A2, α/β hydrolase domain protein 4, phospholipase C, lyso-phospholipase D, protein tyrosine phosphatase non-receptor type 22, SH2 domain-containing polyinositol-5-phosphatase 1, and various glycerophosphodiesterase family members catalyze parallel routes for the biosynthesis of AEA.
Fig. 3.
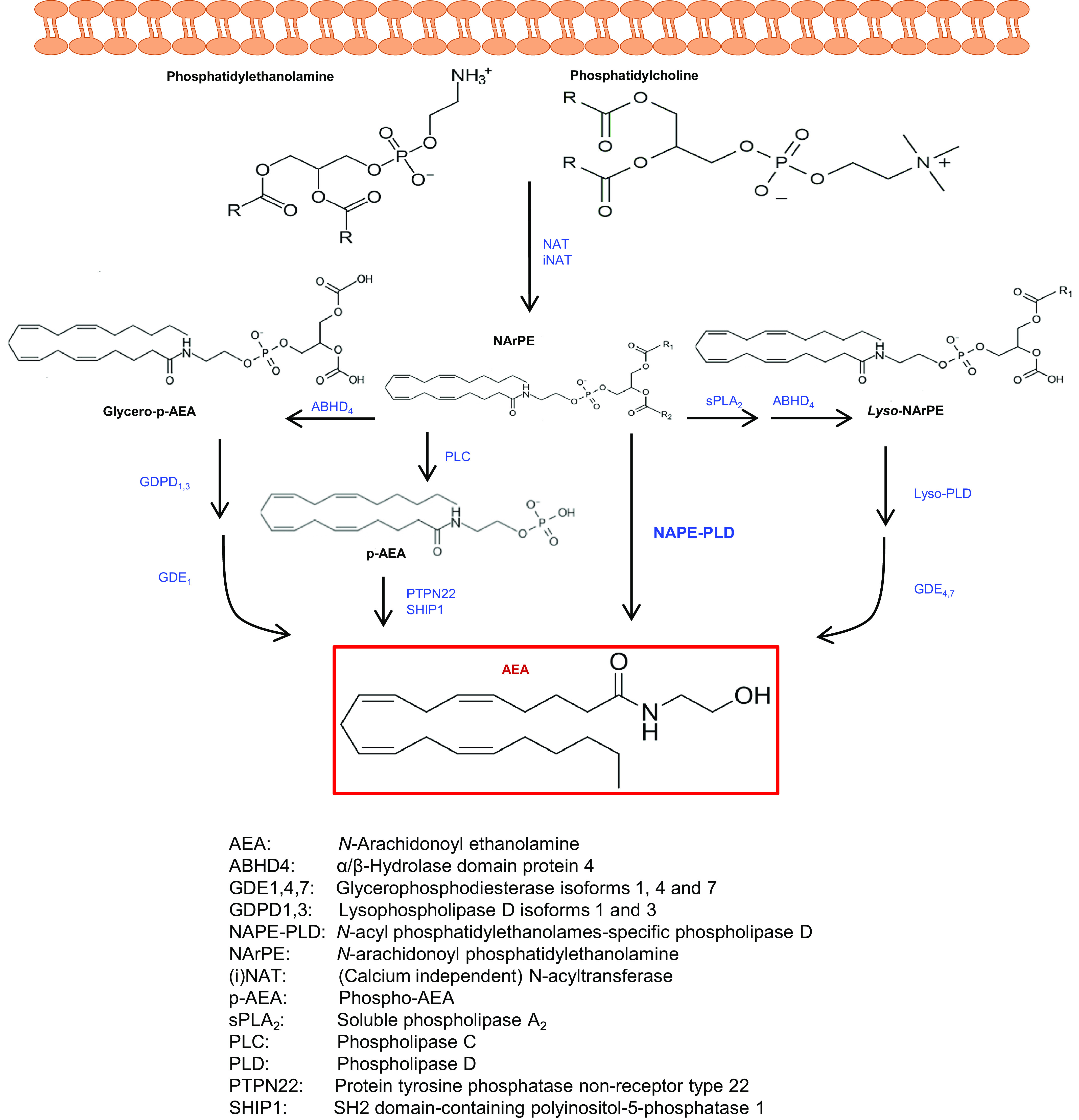
Biosynthetic pathways of anandamide. AEA can be synthesized from membrane phospholipid precursors via different routes. The Ca2+-dependent hydrolysis of NArPE by NAPE-PLD is considered the most relevant among these biosynthetic pathways.
Multiple pathways also exist for the degradation of AEA, which can be cleaved into ethanolamine and AA, thus terminating its biologic activity. This hydrolysis is primarily catalyzed by fatty acid amide hydrolase-1 (FAAH-1) but also by the less widespread FAAH-2 or by the lysosomal enzyme N-acylethanolamine acid amidase (NAAA) (Piomelli et al., 2020), as shown in Fig. 4.
Fig. 4.
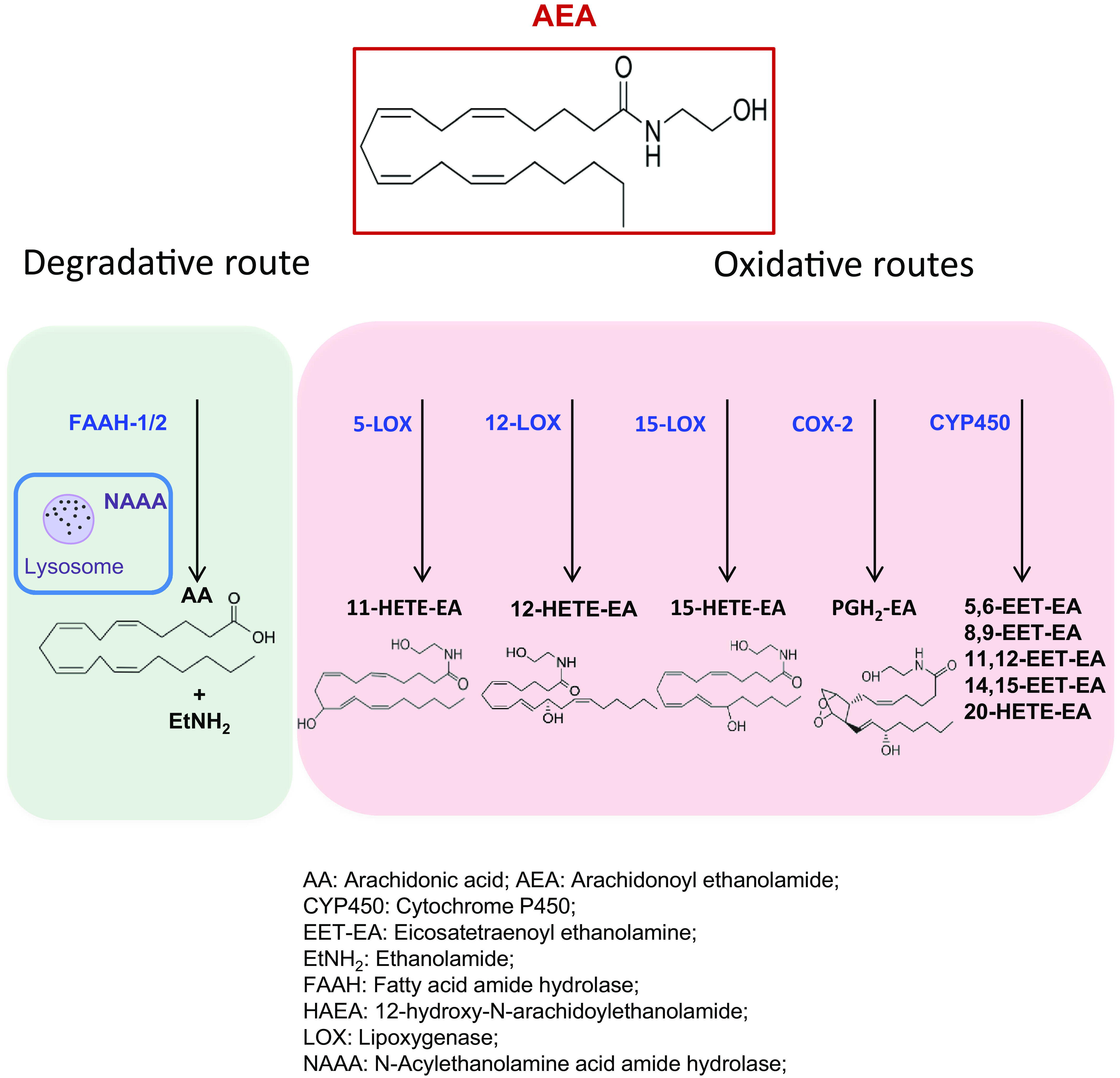
Catabolic pathways of anandamide. AEA can be cleaved to arachidonic acid and ethanolamine by different hydrolytic routes. FAAH-1 is considered the most relevant among these catabolic pathways. Alternatively to hydrolytic routes, AEA can be oxidized by LOXs, COX-2, or cytochrome P450 to generate various eicosanoid-like PG-ethanolamides or hydroxy-AEAs.
As an alternative to degradation, AEA can be biotransformed by oxygenation (i.e., addition of molecular O2) of the AA moiety catalyzed by lipoxygenase 5, 12, 15 isozymes (5-, 12-, 15-LOX), cyclooxygenase-2 (COX-2) or cytochrome P450 (CYP450), as summarized in Fig. 4 and recently reviewed (Rouzer and Marnett, 2011; Fezza et al., 2014; Simard et al., 2022). Remarkably, COX-2-generated prostamides and the other oxidative derivatives of AEA are endowed with biologic activities on their own (Van der Stelt et al., 2002; Simard et al., 2022). To date, their pathophysiological roles remain rather elusive, but apparently they include neuroprotection of the brain (Veldhuis et al., 2003).
2. Metabolism of 2-Arachidonoylglycerol
Much like AEA, membrane phospholipid precursors like phosphatidylinositol and phosphatidic acid are cleaved via phospholipase A1 or phosphohydrolase, respectively, to release 2-arachidonoylglycerol-3-phosphate or diacylglycerol, respectively (Fig. 5). Then, a Ca2+-dependent phospholipase C (PLC) or Ca2+- and glutathione-dependent DAG lipases (DAGL) α and β release 2-AG. The latter DAGLα/β-dependent pathway is the classic biosynthetic route for 2-AG (Bisogno et al., 2003), and glutathione seems to be a key regulator in the brain (Maccarrone et al., 2008).
Fig. 5.
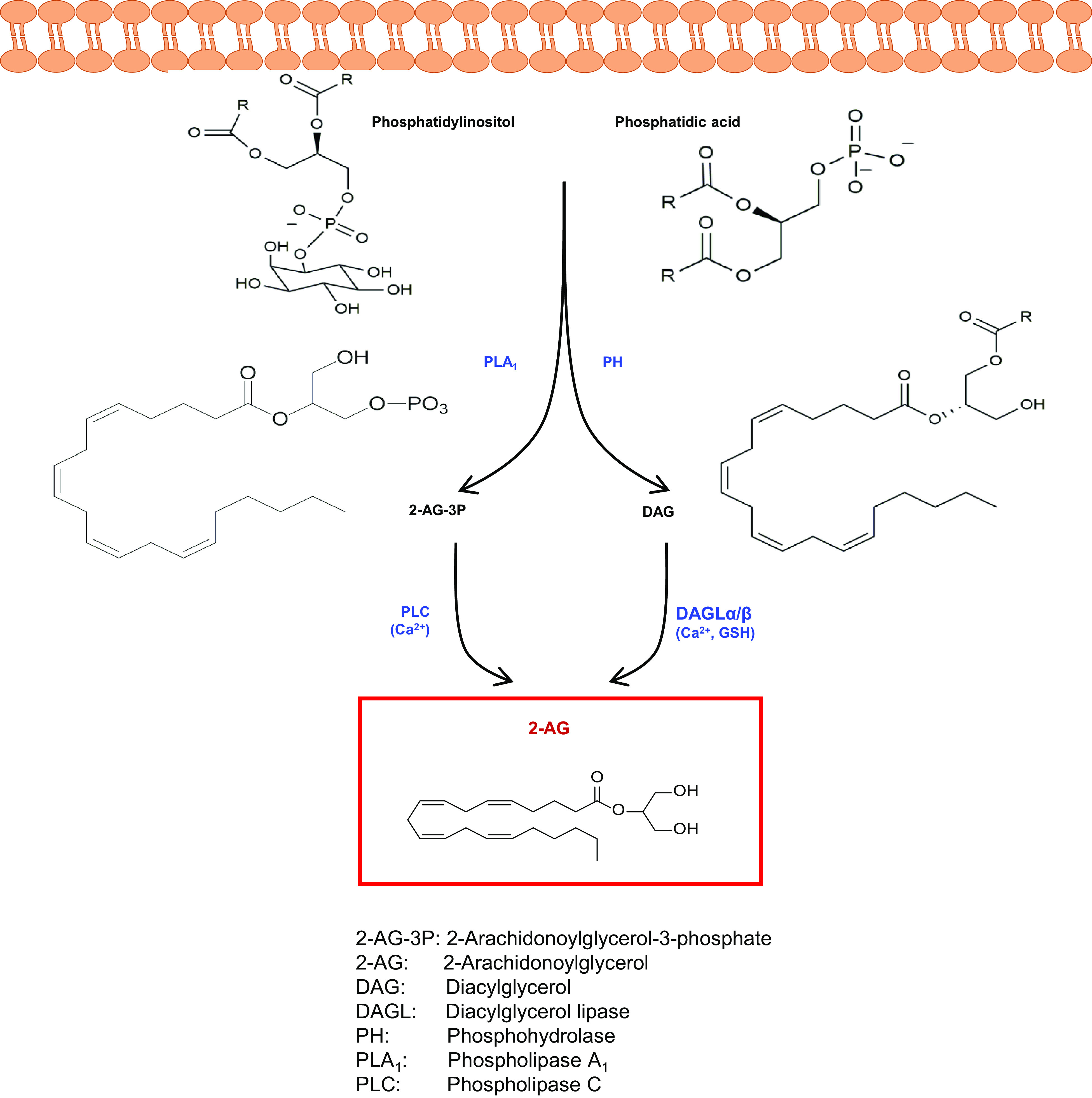
Biosynthetic pathways of 2-arachidonoylglycerol. 2-AG can be synthesized from membrane phospholipid precursors via different routes. The Ca2+- and glutathione-dependent hydrolysis of DAG by DAGLα/β is considered the most relevant among these biosynthetic pathways.
Alternative pathways have been discovered for the degradation of 2-AG, which is primarily cleaved to glycerol and AA by MAGL, as shown in Fig. 6. In addition, α/β hydrolase domain proteins 2, 6, and 12, carboxylesterases 1 and 2, and palmitoyl-protein thioesterase 1 can degrade 2-AG to AA and glycerol (Baggelaar et al., 2018; Maccarrone, 2020), as shown in Fig. 6. Much like AEA, 2-AG can be oxygenated by COX-2, 12- and 15-LOX (Rouzer and Marnett, 2011; Fezza et al., 2014; Simard et al., 2022), leading to oxidative derivatives like prostaglandin- or thromboxane-glyceryl esters with their own biologic activities (Baggelaar et al., 2018; Simard et al., 2022).
Fig. 6.

Catabolic pathways of 2-arachidonoylglycerol. 2-AG can be cleaved into arachidonic acid and glycerol by different hydrolytic routes. MAGL is considered the most relevant among these catabolic pathways. Alternatively to hydrolytic routes, 2-AG can be oxidized by LOXs or COX-2 to generate various eicosanoid-like PG-glyceryl esters or hydroxy-2-AGs.
E. Trafficking of Endocannabinoids
The stringent metabolic control of eCB tone is further modulated by distinct transporters that facilitate the movement of eCBs across the plasma membrane (possibly via a purported and as yet elusive eCB membrane transporter), as well as intracellularly and extracellulary. Moreover, not only can eCBs be released from membrane precursors when the cell receives a stimulus “on demand,” but they can be stored in cytosolic organelles like adiposomes (Maccarrone, 2020). The mechanisms underlying the membrane transport of eCBs have been extensively investigated, leading to two prevailing models whereby eCBs are transported either by passive diffusion (Fasia et al., 2003) or by facilitated diffusion through a membrane carrier (Di Marzo et al., 1994; Beltramo et al., 1997). The mechanism(s) of transmembrane transport of eCBs remain(s) a highly debated issue in the field and has/have been the subject of comprehensive critical reviews (Fowler, 2013; Nicolussi and Gertsch, 2015; Kaczocha and Haj-Dahmane, 2022). In addition to passive or facilitated diffusion, eCBs can leave a cell as part of microvesicles that undergo exocytosis, and indeed such a mode of extracellular transport has been demonstrated in the synaptic cleft for both AEA (Gabrielli et al., 2015) and 2-AG (Nakamura et al., 2019). The different modalities of transmembrane transport of eCBs are schematically depicted in Fig. 7A.
Fig. 7.

Transport of endocannabinoids. (A) Anandamide and 2-arachidonoylglycerol can cross the plasma membrane via different mechanisms, which include passive diffusion, exocytosis of microvesicles and a putative membrane transporter. (B) Intracellular trafficking of anandamide and 2-arachidonoylglycerol is driven by various carriers that include structurally unrelated proteins like albumin, RBP2, HSP70, FABPs, SCP2, and FLAT.
The eCBs are lipids, and as such they cannot travel the aqueous cytosol without a suitable carrier (Maccarrone et al., 2010). Unsurprisingly, cytosolic AEA-binding proteins have been demonstrated over the last few years and include structurally unrelated proteins like heat shock protein 70 and albumin (Oddi et al., 2009), fatty acid binding proteins (FABPs) 1, 5, and 7 (Kaczocha et al., 2009; Elmes et al., 2019), FAAH-like anandamide transporter (Fu et al., 2011), sterol carrier protein 2 (Hillard et al., 2017), and retinol-binding protein 2 (Plau et al., 2022). These eCB transporters are schematically depicted in Fig. 7B.
While the pathophysiological relevance of intracellular and extracellular trafficking of eCBs remains elusive (Jacobson et al., 2019; Fauzan et al., 2022), it appears that carriers of these lipids should be actively investigated, because they might be major players in driving eCB signaling. Indeed, these carriers can ferry the right eCB to the right target, at the right time and in the right concentration, thus holding potential as primary action points for the development of effective eCB-oriented therapeutics. Of note, these novel therapeutics should be devoid of unwanted side effects often associated with drugs that target receptors or metabolic enzymes of eCBs (Ciaramellano et al., 2023).
On a final note, to date, 3D structures of only 23 major components of the ECS have been resolved, whereas many other elements still await clarification of their structural features (Maccarrone, 2020). Among the latter, key receptors (e.g., GPR55, GPR119, and TRPV4), enzymes (e.g., NAT, DAGLα/β, GDE1,4,7, ABHD2, 4, 6, 12), and the putative eCB membrane transporter can be listed. It is apparent that such an information gap is particularly troubling for drug discovery programs and must be urgently filled.
In the following sections, the main properties and therapeutic potential of some of the main ECS components are detailed, whereas the other elements suffer from a lack of information.
II. Cannabinoid Receptor Physiology and Pharmacology
The eCBs and THC are dual effectors at both CB1R and CB2R, which share a ligand binding domain sequence identity of 44% (Matsuda et al., 1990; Munro et al., 1993; Howlett et al., 2002; Mackie, 2005). The absolute stereochemistry of THC was deciphered in 1967 (Fig. 1) (Mechoulam and Gaoni, 1967), and this was followed by the development of many analogs by academic chemists (Razdan, 1986). THC is a dual CB1R and CB2R partial agonist exhibiting multiple therapeutically interesting physiologic properties involving both receptor types, which include anti-inflammatory, immunosuppressive, and analgesic effects. THC was the first cannabinoid agonist approved as a medication by the FDA under the generic name dronabinol (Marinol), although its use was restricted due to CNS-mediated psychotropic side effects.
Many additional nonselective cannabinoid agonists have provided insights into pharmacotherapuetic potential (reviewed by Robson, 2001; Pertwee 2008b, 2012). With a goal to develop cannabinoid, nonopioid analgesics, Pfizer produced a series of A-C-bicyclic and A-C-D-tricyclic analogs of THC’s CNS-active metabolite 11-OH-THC, and these are referred to as “non-classical cannabinoids” because of their origin and similarity to the A-B-C-tricyclic structure of THC (Johnson et al., 1981; Howlett et al., 1990; Melvin et al., 1993, 1995). Of these, levonantradol was taken to clinical trials for postoperative pain, but the project was discontinued due to prominent sedative and euphoric/dysphoric properties (Jain et al., 1981). The primary outcome of the Pfizer effort was the development of the CB1R/CB2R nonselective full agonist CP55940, outperforming THC with regard to CB1R/CB2R binding affinity and analgesic activity (Devane et al., 1988; Howlett et al., 1988; Showalter et al., 1996) (Fig. 8). CP55940 is a research tool that has been invaluable in identifying cannabinoid receptor cellular and systems physiology (Devane et al., 1988). Tritiated CP55940 was critically involved in the deorphanization of both CB1R (Matsuda et al., 1990) and CB2R (Munro et al., 1993) and has been broadly applied to quantitate the structure-activity relationships of most novel ligands developed for the investigation of cannabinoid receptors.
Fig. 8.

Chemical structure, CB2R binding affinity and selectivity of relevant nonclassic cannabinoids. aConsensus human CB2R binding affinity values from a multicentric collaborative profiling effort between multiple independent academic laboratories and industry (Soethoudt et al., 2017). bCB2R selectivity (10∧(pKi CB2R-pKi CB1R).
Sterling-Winthrop discovered that structural modifications of the nonsteroidal anti-inflammatory agent pravadoline resulted in greater antinociceptive activity with diminished potential to block prostaglandin production (Bell et al., 1991). Although the Sterling-Winthrop project was terminated in the preclinical stages, the introduction of the CB1R/CB2R nonselective full agonist WIN55212-2 has contributed greatly to investigations of cannabinoid receptor physiology and pharmacology (Fig. 9). WIN55212-2 in its tritiated form is a standard CB1R/CB2R radioligand (D’Ambra et al., 1992; Eissenstat et al., 1995) and with its derivatives is referred to as “aminoalkylindoles” because their structure is built on indole or indene platforms.
Fig. 9.

Chemical structure, CB2R binding affinity and selectivity of representative aminoalkylindole CB2R ligands. aConsensus human CB2R binding affinity values from a multicentric collaborative profiling effort between multiple independent academic laboratories and industry (Soethoudt et al., 2017). bCB2R selectivity (10∧(pKi CB2R-pKi CB1R).
Selective activation of either CB1R or CB2R by THC or the other nonselective agonists seems to be controlled by differential expression (induction or desensitization/downregulation) of the receptors on a wide variety of cells that control differentiated functions (reviewed by Howlett and Abood, 2017).
Research work using ligand-assisted protein structure methodology has characterized the sites of action at CB1R and CB2R (Janero et al., 2017). However, a more detailed CB1R structure was reported in 2016 in its inactive conformation by using the long-acting CB1R antagonist AM6538 (Hua et al., 2016), shown in Fig. 10, and the antagonist/inverse agonist taranabant (Shao et al., 2016). This allowed the docking of several CB1R antagonist analogs and the study of their interactions with the receptor. This work was followed by studies on the structures of the agonist-bound CB1R (Hua et al., 2017; Krishna Kumar et al., 2019; Hua et al., 2020), which demonstrated that activation of CB1R induces dramatic conformational changes of both extracellular and intracellular domains of the receptor, accompanied by a serious contraction of the binding pocket. This more expansive conformation of CB1R in its inactive state explains how several antagonists can be accommodated in the receptor structure.
Fig. 10.

(A) X-ray structure of CB1R (blue) bound to the antagonist AM6538. (B) Cryo-EM structure of CB1R (green) in complex with G proteins (α subunit in yellow, β subunit in blue, γ subunit in purple) and the classic cannabinoid agonist AM841. (C) Chemical structures of AM6538 and AM841.
The high-resolution crystal structure of antagonist-bound CB2R was determined in 2019, which first discloses the binding mode of antagonist AM10257 (Li et al., 2019). The latter locates at the orthosteric ligand-binding pocket and mainly forms hydrophobic and aromatic interactions with residues from extracellular loop 2, as well as the cytoplasmic parts of transmembrane helices 2, 3, 5, and 6 of CB2R (Fig. 11A). However, the antagonist AM10257 adopts a constrained binding pose in CB2R, which is quite different from the extended binding conformation of antagonists in CB1R (Hua et al., 2016). Of note, the adamantyl moiety of AM10257, adapting a vertical conformation, would clash with the residue Phe102N-term of CB1R when two structures are superimposed (Fig. 11, B–D). That is the reason why the N-terminus of CB2R forms a short helix over the orthosteric pocket, instead of the V-shaped loop that directly interacts with the antagonist in CB1R (Hua et al., 2016; Shao et al., 2016). In addition, the extracellular part of transmembrane helices 1 and 2 in CB2R is more compact compared with the conformations of the same helices in CB1R, resulting in a much smaller antagonist-binding pocket than that of CB1R (Hua et al., 2016; Shao et al., 2016). The structural analysis provides the basis for the high degree of antagonist selectivity between CB1R and CB2R.
Fig. 11.

Comparison of ligand binding modes in CB1R and CB2R. (A) The binding pocket of AM10257 in CB2R crystal structure (PDB code 5ZTY). AM10257 and the key residues are shown in sticks as the following color code: CB2R, brown; AM10257, light coral. (B–D) Binding pose comparison of AM6538 in CB1R (PDB code 5TGZ), and AM10257 in CB2R, using color code as follows: CB1R, slate blue; AM6538, dodger blue; CB2R, brown; AM10257, light coral. (E–F) The binding pocket of AM12033 in CB2R (PDB code 6KPF) and WIN55,212-2 in CB2R (PDB code 6TP0). Ligands and the key residues are shown in sticks as the following color code: AM12033, brown; CB2R (6KPF), dark green; WIN55,212-2, royal blue; CB2R (6TP0), dark salmon. (G) The conformational comparison of “toggle switch” residues Trp2586.48 between AM12033- and WIN55,212-2-bound CB2R. (H–J) Binding pose comparison of THC-like agonist in CB1R (PDB code 6KPG) and CB2R (PDB code 6KPF). THC-like agonists are shown as sticks (H) and surface (I–J), the key residues are shown in sticks as the following color code: CB2R, dark green; AM12033, brown; CB1R, maroon; AM841, dark khaki. (K-M) Binding pose comparison of agonist FUB in CB1R (PDB code 6N4B) and agonist WIN55,212-2 in CB2R (PDB code 6TP0). FUB and WIN55,212-2 are shown as sticks (K) and surface (L–M), the key residues are shown in sticks as the following color code: CB2R, dark salmon; WIN55,212-2, royal blue; CB1R, dark cyan; FUB, orange.
In spite of the high selectivity of antagonists or inverse agonists, most agonists can bind both CB receptors with comparable affinity (Pertwee et al., 2010). The recently determined structures of agonist-bound CB2R provide valuable information at the molecular level for subtype-selective agonist design (Hua et al., 2020; Xing et al., 2020) and subtype-selective receptor activation (Li et al., 2023). Both the synthetic THC-like agonist AM12033 and aminoalkylindole agonist WIN55212-2 form mainly hydrophobic and aromatic interactions with CB2R, including residues from transmembrane helices 2–3 and 5–7 and the extracellular loop 2 with similar binding mode in the orthosteric ligand-binding pocket (Fig. 11, E–F). Although the core of WIN55212-2 forms π-π interactions with F1173.36 and W2586.48 of CB2R, the rotamers of F1173.36 and W2586.48 are very similar in these two structures (Fig. 11G). The superposition of agonist-bound CB1R and CB2R structures shows that the agonist binding pocket volume, as well as the key residues that form interactions with ligands, are almost identical (Fig. 11, H–M). This accurate molecular information of the CB receptors’ orthosteric binding pockets obtained so far should aid the design of selective agonists for safer therapeutics.
The CB2R activation mechanism was revealed by the comparison of active and inactive structures. Though the antagonists and agonists of CB2R share similar binding pockets, including the key interaction residues with the receptors, the interaction of CB2R ligands with the “toggle residue” W2586.48 is related to their efficacies. Compared with antagonist AM10257, agonist AM12033 lacks the moiety that extends deeper into the binding cavity to constrain W2586.48 rotation, which can trigger receptor activation (Fig. 12A). Subsequently, the classic rearrangements of N7.49 P7.50 × x Y7.53 and D3.49 R3.50 Y3.51 motifs were observed that contribute to the conformational change of the intracellular part of CB2R, eventually forming the G-protein binding cavity (Fig. 12, B–C). However, in contrast to agonist-bound CB1R, only the intracellular part of CB2R exhibits obvious conformational changes while the extracellular part including the N-terminus of CB2R undergoes minor changes during its activation (Fig. 12, D–F). The balloon-like plasticity of CB1R during its activation indicates its higher ability to respond to a diverse array of ligands than CB2R, which may explain the low selectivity compared with CB1R for most classic THC-like agonists of CB2R.
Fig. 12.

Conformational changes during CB2R activation. (A–C) The conformational change of key residues between inactive- and active-CB2R. “Toggle switch residue” (A), D3.49R3.50Y3.51 motif (B), and N7.49P7.50xxY7.53 motif (C). (D–F) The overall structure (D), the extracellular region (E), and intracellular region (F) comparison of inactive- (brown) and active-state (dark green) CB2R structures.
A. Therapeutic Potential of Cannabinoid Receptor 1
The epigenetic regulation of CB1R expression and signal transduction pathways following Gi/o or β-arrestin activation is related to differentiated cell functions, as reviewed recently (Kendall and Yudowski, 2016; Ligresti et al., 2016; Howlett and Abood, 2017; Lutz, 2020; Schurman et al., 2020). The CB1R is highly abundant in the CNS and many peripheral tissues and organs (Howlett et al., 2002; Pacher et al., 2006). For instance, it is critically involved in the regulation of mood and appetite, pain perception, learning, and memory, as well as motor control (Marsicano and Lutz, 2006; Kano et al., 2009; De Laurentiis et al., 2014). The CB1R has been recognized as a target for pharmacotherapeutic development based on a wealth of preclinical data (for reviews, see Mackie, 2008; Pertwee, 2008b, 2012; Tsang and Giudice, 2016; Lu and Anderson, 2017; Amin and Ali, 2019; Schurman et al., 2020; Wilkerson et al., 2021). However, bringing CB1R agonists and antagonists to market has been fraught with the challenges of selectivity resulting from the abundance of CB1R throughout all areas of the brain, including expression by neuronal and nonneuronal cells. This broad distribution increases the probability of unwanted side effects accompanying the therapeutic benefits.
1. CB1R Agonists and Positive Allosteric Modulators
The only FDA-approved CB1R agonists are THC itself (synthesized as dronabinol) and its dimethylheptyl analog nabilone (LY-109514), specifically to treat cancer chemotherapy-induced nausea and vomiting, and these medicines remain within the US Pharmacopeia (Clarivate, 2022d; https://adisinsight.springer.com/drugs/800025856). The European Medicines Agency (EMA) approved the mixture of THC and CBD extracted and purified from cannabis (nabiximols) for the treatment of spasticity and pain in multiple sclerosis (MS). Dronabinol, nabilone, and nabiximols exhibit agonist activity at both CB1R and CB2R, though therapeutic responses and untoward side effects can be attributed to one or both CB receptors, as determined by pharmacological characterization in in vivo or in vitro models. Nevertheless, targeting CB1R for unmet therapeutic needs has evolved based on preclinical investigations, and these opportunities will be considered in this section.
Dronabinol was developed to counteract nausea and vomiting in cancer chemotherapy and was later approved to promote appetite stimulation and metabolic maintenance to counteract cachexia in AIDS patients (Plasse et al., 1991). Dronabinol is synthetically produced THC formulated in a sesame oil capsule and marketed as Marinol (https://adisinsight.springer.com/drugs/800007811). Dronabinol is also available in a liquid formulation solubilized in ethanol and propylene glycol and marketed as SYNDROS. The pharmacokinetics, dosage recommendations, and drug interactions are available at Prescribers Digital Reference (https://www.pdr.net/drug-summary/Marinol-dronabinol-2726). The warnings reported include bradycardia and seizures in vulnerable populations. Mild to moderate adverse reactions include emotional lability in 8% to 24% of users; impaired cognition, dysphoria or euphoria, depression, hypotension, drowsiness, paranoia, dizziness, or nausea in 3% to 10% of users; and conjunctivitis, hallucinations, confusion, amnesia, ataxia, tinnitus, nightmares, or diarrhea in 0.3% to 1% of users (https://www.pdr.net/drug-summary/Marinol-dronabinol-2726).
Nabilone is synthesized as a 9-ketocannabinoid with a dimethylheptyl side chain (Fig. 13) and is enzymatically reduced in the liver to the hydroxylated S(axial) isomer believed to be the active form (Archer et al., 1977; Rubin et al., 1977; Billings et al., 1980). Nabilone was approved as an antiemetic for cancer chemotherapy but also exhibits anxiolytic properties (Lemberger and Rowe, 1975; Ward and Holmes, 1985). Nabilone is used off-label for treatment of the symptoms of Huntington’s chorea (https://www.pdr.net/drug-summary/Cesamet-nabilone-692). The warnings and adverse reactions are similar to those reported for dronabinol: seizures in vulnerable populations, early euphoria or dysphoria, delayed depression, ataxia, hypotension, drowsiness, vertigo, dizziness, asthenia, or headache.
Fig. 13.

Structures of the clinically tested cannabinoid agonists (A) and the selective CB1R antagonists (B).
Nabiximols is a mixture of THC and CBD (1:1) in ethanol and propylene glycol solvent as an oromucosal spray formulation marketed as Sativex (see the EMA compendium for information: https://www.medicines.org.uk/emc/product/602/smpc#gref). The spray is intended to be applied at the onset of muscle contractions to reduce spasticity and pain in MS patients. Each application provides some fraction of the dosage to be absorbed via the mucosal membranes, and the remainder is swallowed and absorbed from the gastrointestinal tract. Sativex was granted orphan designation by the EMA for the treatment of glioma patients while clinical trials were being conducted; however, this status was later withdrawn (see EMA notices: EMA, 2022). The EMA reports pharmacokinetic data and recommends dosing schedules for use in MS patients (https://www.medicines.org.uk/emc/product/602/smpc#gref). The report includes warnings/precautions for use in patients with histories of seizures or cardiovascular disease. Adverse reactions found in clinical trials include appetite changes, dizziness, disorientation, mood swings, depression, amnesia/memory impairment, somnolence or blurred vision in 1% to 10% of users, and pharyngitis, syncope, anxiety, illusions, paranoia, hallucinations, or delusional beliefs in 0.1% to 1% of users. Adverse effects at the site of application include oral discomfort/pain, altered taste, mouth ulceration, and accompanying pain.
Prior to the recognition of CB receptors, clinical trials provided positive indications for CBD for seizure control and movement disorders (Cunha et al., 1980; Carlini and Cunha, 1981; Consroe et al., 1986, 1991). CBD entered the market for the treatment of Dravet syndrome, infantile severe myoclonic epilepsy, Lennox-Gastaut syndrome, and tuberous sclerosis (Clarivate, 2022b). In addition, CBD and THC combinations have been approved for MS-associated spasticity and pain management (Clarivate, 2022g; https://citeline.informa.com/drugs/details/175074). CBD in the nabiximols formulation may or may not exert its cellular actions via processes involving CB1R. CBD exerts both negative and positive interactions with THC over a range of biologic and behavioral responses in animal models and humans (Pertwee, 2008a; McPartland et al., 2015). In a cloned neuronal cell model, CBD competed with the CB1R agonist [3H]CP55940 in binding to the receptor at concentrations nearly three orders of magnitude greater than did THC (Devane et al., 1988); however, CBD failed to inhibit cAMP production via the CB1R-coupled Gi protein as does THC (Howlett, 1984; Mukhopadhyay et al., 2002). Similar findings of low potency binding to CB1R and inability to stimulate CB1R cellular signaling were reported in multiple studies using other models as compiled in a meta-analysis from a pool of > 200 research publications (McPartland et al., 2015). Two influences of CBD on CB1R pharmacology are most compelling: (1) CBD could exert a noncompetitive antagonism at CB1 receptor (Petitet et al., 1998; Thomas et al., 2007; Laprairie et al., 2015) and (2) CBD could indirectly modulate CB1R activity by FABP competition (Elmes et al., 2015) and FAAH inhibition (Bisogno et al., 2001; De Petrocellis et al., 2011) or activation (Massi et al., 2008), thereby changing eCB tone. Non-CB1R mechanisms proposed for CBD’s neurologic actions minimally include the facilitation of serotonin signaling, activation of TRPV1 or PPARγ receptors, neuroprotection via antioxidant activity, and attenuation of proinflammatory processes (Campos et al., 2012; Ibeas Bih et al., 2015; Campos et al., 2017). Other molecular targets for CBD include additional GPCRs (e.g., GPR55, GPR18, μ and δ opioid receptors) and TRP channels A1, V2, M8 (McPartland et al., 2015; Ligresti et al., 2016).
Because dronabinol, nabilone, and nabiximols are currently approved medicines by regulatory agencies, it is acceptable to repurpose these preparations for treatment or amelioration of other disease symptoms based upon preclinical evidence that justifies their use. Table 4 lists the double-blind clinical trials that have been registered with ClinicalTrials.gov and are completed or ongoing at the date of publication of this review.
TABLE 4.
Diseases/symptoms for treatment with CB1R agonists and antagonists registered with ClinicalTrials.gov
| Generic Name Brand Name Class/Efficacy |
Completed Clinical Trials | Ongoing Clinical Trials |
|---|---|---|
| Dronabinol | Chronic pain (with opioid treatment) | Osteoarthritis pain |
| Marinol | Fibromyalgia, back pain | Diabetic neuropathy |
| Phytocannabinoid | Migraine pain | Knee arthroplasty |
| Synthetically produced | Neuropathic, low back pain | Arthroscopic surgery |
| Δ9-tetrahydrocannabinol (THC) |
Cervical dystonia | Sleep and pain in MS |
| CB1R/CB2R partial agonist | Chest pain | Postsurgical pain-lumbar fusion |
| Neuropathic pain in MS | Postsurgical pain-knee replacement | |
| Cramps in ALS | Pain in opioid-maintained pts | |
| Irritable bowel syndrome | Alzheimer’s agitation | |
| Complex regional pain syndromes | Bipolar disorder | |
| Cannabis dependence | Sleep | |
| Cannabis use disorder | Post-traumatic stress disorder | |
| Marijuana withdrawal | Trauma intrusive memories | |
| Trichotillomania related behaviors | Glaucoma hemodynamics | |
| Post-traumatic stress disorder | ||
| Obstructive sleep apnea | ||
| PostSurgical N/V | ||
| Anti-retroviral therapy N/V | ||
| Brain neoplasms N/V | ||
| Schizophrenia | ||
| Dronabinol derivatives | ||
| BX-1 oral solution | Spasticity | Chemo N/V, pain in pancreatic CA |
| Syndros (dronabinol) | Bone pain metastatic breast CA | |
| Namisol THC | Postsurgical abdominal pain | |
| Namisol THC | Pancreatitis abdominal pain | |
| Namisol THC | Dementia–Alzheimer’s | |
| Namisol THC | Dementia w/ neuropsych symptoms | |
| THC olive oil | Post-traumatic stress disorder | |
| THC olive oil | Fibromyalgia–pain | |
| SCI-110 THC + PEA | Tourette syndrome | Tourette syndrome |
| THX-110 THC + PEA | Tourette syndrome | |
| dronabinol + naltrexone | Opioid dependence | |
| Nabilone | ||
| Cesamet | Phantom limb pain | Spinal neuropathic pain |
| Synthetic THC analog | Fibromyalgia | Pain and insomnia |
| CB1R/CB2R agonist | Failed back surgery pain | End-stage renal disease |
| Inflammatory bowel pain | Obesity | |
| Diabetic neuropathies | Developmental cognitive disability | |
| Spinal injury muscle | Obsessive-compulsive disorder | |
| Spinal cord injury | Alzheimer’s disease agitation | |
| Postsurgical N/V | ||
| Cancer anorexia/cachexia | ||
| Parkinson’s disease | ||
| Parkinson’s nonmotor symptoms | ||
| Alzheimer’s disease | ||
| PP-01 Nabilone+Gabapentin | Cannabis withdrawal | |
| Nabiximols | ||
| Sativex | Chemotherapy neuropathic pain | Diabetic neuropathy |
| Phytocannabinoid | Advanced malignancy pain | MS spasticity and pain |
| Purified Plant Extract | Pain | |
| THC:CBD (1:1) | Tourette syndrome | |
| THC: CB1R/CB2R agonist | Attention deficit hyperactivity disorder | |
| CBD: CB1 NAM | Cannabis dependence | |
| (Negative Allosteric Modulator) | ||
| Mixed THC:CBD | ||
| THC:CBD 1:1 | Endometriosis pain | |
| THC:CBD 1:1, 1:2 | Chronic pain | |
| THC:CBD1:10 | Crohn’s disease | |
| THC:CBD 1: 50 | Childhood epilepsy | |
| NanaBis Oro-MucSpray | Cancer pain | |
| NanaBis Oro-MucSpray | Chronic widespread pain | |
| THC or THC:CBD 1:10 | Chronic spine back and neck pain | |
| LGP1-20 THC:CBD (1:20) | Adolescent migraines | |
| FibroCann Solution | Fibromyalgia | |
| Pure Green SL Tablets | Osteoarthritis pain | |
| MPL-001 THC:CBD 1:25 | Postsurgical osteoarthritis pain | |
| TN-TC11G THC:CBD1:1 | Glioblastoma (w/standard of care) | |
| TIL-T150 THC:CBD 1:5;1:25 | Depression, insomnia | |
| Pure Femme SLTab 1:30 + PEA + terpenes |
Menstrual symptoms | |
| THC or CBD | HIV cognition | |
| THC + CBD + CBG | Chronic migraine | |
| Pro-drug paracetamol (or acetaminophen) | ||
| Biometabolite is AM404 | Pruritis | |
| CB1R/CB2R agonist | Presurgical analgesia | |
| Pain in tonsillectomies | ||
| SR141716 | ||
| Rimonabant | • Carotid atherosclerosis | Recovery spinal cord injury |
| Acomplia, Zimulti | Cannabis dependence | |
| CB1R antagonist/inverse agonist | Diabetes w/ metformin | |
| Obesity, weight loss | ||
| • Metabolic syndrome | ||
| Reduce alcohol consumption | ||
| Fatty liver-NASH in T2D | ||
| Smoking cessation | ||
| MK-0364 | ||
| Taranabant | Obesity | |
| CB1R antagonist/inverse agonist | Smoking cessation | |
| Fatty liver-NASH in T2D | ||
| CP-945598 | ||
| Otenabant | Nonalcoholic steato-hepatitis | |
| CB1R antagonist/inverse agonist | Obesity | |
| SLV319 | ||
| Ibipinabant | Obesity | |
| CB1R antagonist/inverse agonist | ||
| SR147778 | ||
| Surinabant | Obesity | |
| CB1R antagonit/inverse agonist | Smoking cessation | |
| ANEB-001 | ||
| CB1R antagonist/inverse agonist | Acute cannabis intoxication | |
| GFB-024 | ||
| Peripherally acting CB1R inverse agonist monoclonal Ab | Diabetic nephropathies | |
| Nimacimab | ||
| Peripherally acting CB1R antagonist/inverse agonist monoclonal Ab | Diabetic gastroparesis | |
Appropriate preclinical data justify these putative uses and warrant evaluation of both efficacy of these cannabinoid agonists for these purposes and relative safety given the risk:benefits assessment and the circumstances of patient treatment. Review articles are cited that summarize research evidence in animal models, address implications and challenges, and provide original references.
Nausea and vomiting that accompany surgical procedures, retroviral therapy, and neoplasms are unmet needs that build upon the original usage approved by regulatory agencies for patients undergoing cancer chemotherapy (Abrams and Guzman, 2015). Preclinical studies using animal models of nausea and vomiting (“retching” or “gaping”) have demonstrated effective attenuation with CB1R agonists, although the exact neurologic mechanism has not been established (Parker et al., 2011; Sticht et al., 2015). In contrast, in the current population of recreational cannabis users, a novel cannabis-induced hyperemesis syndrome has been attributed to ingestion of very high doses of THC. The mechanism is poorly understood, but it has been suggested that prolonged exposure to high doses of THC might downregulate CB1R or otherwise perturb the endogenous regulation of vomiting centers in the brain stem and/or elicit stress mechanisms at the hypothalamic-pituitary axis (Galli et al., 2011; DeVuono et al., 2020). Thus, there may be a “bell-shaped” dose-response curve suggesting multiple mechanisms for the anti- versus pro-nausea/vomiting endpoints.
The appetite stimulation response was the impetus for regulatory approval of CB1R agonists as “orphan” drugs for the treatment of cachexia in cancer (Plasse et al., 1991). However, it is the appetite suppression by CB1R antagonism that prompted clinical trials for weight loss in morbidly obese individuals and resulted in an explosion of basic science research linking the CB1R to metabolic processes associated with energy storage (Piazza et al., 2017; DiPatrizio, 2021; Miralpeix et al., 2021). Studies of CB1R-mediated inhibition of gut mobility (Pertwee et al., 1992; Pertwee, 2001) led to the consideration of agonist treatments for irritable bowel syndrome and other gastrointestinal pathologies (Lee et al., 2016; Sharkey and Wiley, 2016). Conversely, detrimental influences of CB1R stimulation on pancreatic β-cell function, diabetic insulin resistance, and hepatic steatosis (Gruden et al., 2016; Nagappan et al., 2019), as well as on female (Cecconi et al., 2020) and male (Maccarrone et al., 2021) reproductive functions, must be considered in the safety profile for CB1R agonist medicines.
Control of chronic and episodic pain continues to be an unmet therapeutic need. The development of CB1R agonists as antinociceptive agents by Pfizer Central Research (Johnson et al., 1981; Howlett et al., 1990; Melvin et al., 1993, 1995) was meant to fulfill this need, but the effort was discontinued due to untoward side effects in patients during clinical trials (Jain et al., 1981). A resurgence of interest in cannabinoid analgesics as adjunctive or second/third-line treatments has reassessed the benefits versus risks ratio for pain conditions associated with cancer, neuropathy, fibromyalgia, and spasticity (Tsang and Giudice, 2016; Woodhams et al., 2017). Recent clinical trials suggest that cannabinoid-mediated analgesia in humans could be attributed to a moderate reduction in affective response but not a reduced perception of the experimental pain (Lötsch et al., 2018).
CB1R agonist efficacy in symptomatic relief in MS and amyotrophic lateral sclerosis is related to the reduced spasticity and tremors, as investigated in an animal model of chronic relapsing experimental allergic encephalomyelitis, as well as reports from patients (Pryce and Baker, 2015; Pertwee, 2002). In addition to relieving the spastic pain, cannabinoid agonists at CB1R and CB2R slow the progression of the disease as a result of neuroprotective mechanisms and oligodendrocyte development to promote myelination (Pryce and Baker, 2015; Ilyasov et al., 2018; Khan et al., 2022). Similarly, agonist stimulation of both CB receptors reduces symptomology and disease progression in other neurodegenerative diseases such as Parkinson’s disease, Huntington’s chorea, Alzheimer’s disease, and stroke (Fernández-Ruiz et al., 2015a,b).
Numerous “cannabinoid products” that are not approved by regulatory agencies are being tested for their potential therapeutic value. It is difficult to discern the composition and concentration of active agents in these herbal preparations, which are variously described as cannabis, cannabis oil, smoked cannabis (cigarettes), inhaled cannabis, vaporized cannabis, cannabis extract, or “CBD-rich”/“THC-rich” marijuana or extracts. These preparations are not further discussed here, because of the lack of quantitative analyses of the materials being used by the patients. Of note, these herbal studies are registered in ClinicalTrials.gov as assessments (blinded or unblinded) for symptomatic improvements in neuropsychiatric and neurologic disorders, including attention deficit and hyperactivity disorder, dementia, anxiety, depression, post-traumatic stress disorder, autism spectrum disorder, obsessive-compulsive disorder, refractory epilepsy, MS, amyotrophic lateral sclerosis, Tourettes’ syndrome, pain (migraine, neuropathic, fibromyalgia, pre- and post-surgical, back, and cancer), agitation associated with aging dementia, irritable bowel disease, chronic obstructive pulmonary disease, and retinitis pigmentosa with degeneration. The rationale for using plant products is that the effects of multiple chemical entities (including “cannabinoids,” terpenes, and flavonoids) may synergize, a concept referred to as an “entourage effect.” The idea of combining medicines—referred to as polypharmacology—that provide different but complementary pharmacological responses, such as anti-inflammatory plus analgesic agents, is not new and is often a preferred treatment strategy (Brodie et al., 2015; Ligresti et al., 2016). However, the challenges of determining the active synergistic agents, appropriate dosing schedule, specificity of therapeutic use, and safety profile remain to be overcome when herbals are used as medicinal products.
In an effort to address selectivity for the CB1R, modifications have been made to pCB, aminoalkylindole, and eCB ligands. For example, AEA analogs arachidonylcyclopropylamide (ACPA) and arachidonyl-2-chloroethylamide (ACEA) exhibit 1-2 nM affinity for the CB1R but 1-3 μM affinity for the CB2R, and both inhibit cAMP CB1R selectivity of the ACPA (Hillard et al., 1999). This selectivity led to the CB1R selective (CB1R/CB2R Ki ratio = 0.1) dual CB1R/CB2R agonist CMX-020, which is currently being explored in phase 2 clinical trials for the treatment of osteoarthritis (https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=371547&isReview=true), pain including sciatica, and diabetic neuropathy (Clarivate, 2022c).
Another mechanism for achieving selectivity is found in the recent development of allosteric modulators to modify the CB1R response. Exploiting allosteric modulation is a broadly used approach for targeting GPCRs (Wold et al., 2019). It allows addressing target selectivity issues and associated off-target side effects of orthosteric ligands by binding to a topographically distinct site. Allosteric ligands modify the conformation of the receptor protein, which allows for modulating the affinity of orthosteric ligands. Allosteric ligands can either augment (positive allosteric modulation) or diminish (negative allosteric modulation) the effect of endogenous ligands. Importantly, this provides the opportunity for tissue-specific modulation of ECS signaling, for example, via a local eCB increase as a consequence of an inflammatory stimulus. In contrast to the high evolutionary conservation of orthosteric binding domains, allosteric sites exhibit a greater sequence difference, allowing for the generation of ligands with high subtype selectivity (Kenakin and Miller, 2010). In addition, an interaction with cholesterol was also observed with CB1R, suggesting its endogenous allosteric modulating role (Hua et al., 2020). This observation extended previous in vitro (Bari et al., 2005) and ex vivo (Maccarrone et al., 2009) functional data showing that membrane cholesterol controls CB1R dimerization and binding activity.
Positive allosteric modulation of CB1R is likely to play an increasingly important role for drug discovery (Saleh et al., 2018; Garai et al., 2021). For instance, ZCZ011 increased the potency and reduced tolerance development in the anti-nociceptive activity of CB1 agonists (Ignatowska-Jankowska et al., 2015); GAT211 synergized with FAAH- or MAGL-inhibitor-mediated eCB accumulation to attenuate inflammatory and neuropathic pain (Slivicki et al., 2018, 2020). Preclinical studies of CB1R allosteric modulators have been reviewed recently (Khurana et al., 2017; Hryhorowicz et al., 2019; Manning et al., 2021).
Another promising approach to selectivity is the development of “biased agonists.” The binding mechanism for a biased agonist would be expected to alter the conformation of the CB1R to prefer either an interaction with the Gi/o family or alternatively allow phosphorylation of the receptor via G-protein receptor kinases to facilitate interaction with β-arrestins 1 or 2 (Priestley et al., 2017; Al-Zoubi et al., 2019). The selectivity would be for the signal transduction pathway involved in the beneficial effects while diminishing the signal for unwanted side effects. Preclinical studies that explore possible CB1R-biased agonists have been reviewed recently (Laprairie et al., 2016; Ibsen et al., 2017; Leo and Abood, 2021; Manning et al., 2021).
2. Cannabinoid Receptor 1 Antagonists
Sanofi discovered the first CB1R selective antagonist in the early 2000s (SR141716), and the compound was initially earmarked for use as a medication for loss of weight (rimonabant, marketed as Acomplia or Zimulti). It was reasoned that since the activation of CB1R increased food intake with weight gain, the use of its antagonist as a drug would result in weight loss. The potential success of such a medication prompted other drug companies to produce their own compounds that were structurally different but pharmacologically identical. The first CB1R antagonist to enter clinical trials for several of these indications was rimonabant (Sanofi), followed by taranabant (Merck), otenabant (Pfizer), ibipinabant (Solvay), and surinabant (Sanofi) as shown in Fig. 13. Additional indications that have been explored clinically include hepatic fibrosis and nonalcoholic fatty liver disease, renal diseases, as well as alcohol dependence and smoking cessation (Cinar et al., 2020). Recently, a CB1R antagonist, ANEB-001 (Anebulo), has been under clinical development as an antidote for acute cannabis intoxication.
Based on results showing weight loss and improved cardiometabolic markers in overweight and obese patients (Despres et al., 2005), rimonabant was accepted by the EMA in 2006 as an adjunct to diet and exercise for the treatment of obesity and related metabolic risks. However, approval by the FDA failed because of its unexpected neuropsychiatric side effects, namely depression and suicidal ideation (Christensen et al., 2007). Some additional side effects of CB1R antagonists are related to the gastrointestinal tract and include nausea, vomiting, and frequent bowel movements (Addy et al., 2008; Limebeer et al., 2010). When the use of rimonabant was withdrawn by Sanofi in 2008, the development of CB1R antagonists was discontinued by other pharmaceutical companies. Notwithstanding the failure of rimonabant, its availability allowed research toward understanding the mechanism of action of CB1R antagonists and the potential use of such compounds for other indications. Ligands like SR141716 and AM251 (Rinaldi-Carmona et al., 1995; Lan et al., 1999) were used to establish the role of CB1R in physiology (Varga et al., 1995; Petitet et al., 1996; Gatley et al., 1997; Liu et al., 2000; Di Marzo et al., 2001; Wang et al., 2003).
The apparent therapeutic value of CB1R blockade led to much of the research in developing selective CB1R antagonists and their preclinical and clinical testing for a variety of disorders related to metabolism, the cardiovascular system, and addiction (Pacher et al., 2008; Cinar et al., 2020). Given the clinical efficacy shown by CB1R blockade for several conditions with unmet medical needs, additional approaches have been explored to retain efficacy and circumvent the unwanted neuropsychiatric side effects. Among these, CB1R antagonist/inverse agonists that cannot enter the CNS and CB1R neutral antagonists have shown promising results in preclinical models.
The discovery of functional CB1Rs in the periphery and the realization that they mediate many processes of the cardiovascular system, metabolism, and fibrotic conditions (Liu et al., 2000; Di Marzo et al., 2001; Jourdan et al., 2014; Bowles et al., 2015) have led to the hypothesis that peripherally selective CB1R antagonist/inverse agonists may retain the therapeutic effects of CB1R blockade without the unwanted CNS effects. Small-molecule CB1R antagonist/inverse agonists with minimal brain exposure have shown efficacy in animal models of obesity and metabolic syndrome, alcoholic and nonalcoholic liver steatosis, liver fibrosis, and renal diseases, as recently reviewed by Kunos’ group (Cinar et al., 2020). The primary methods used to determine brain permeability are pharmacokinetic studies (Zhang et al., 2018; Iyer et al., 2022), while for the specific engagement of brain CB1R positron emission tomography tracers are used (Tam et al., 2012; Chang et al., 2019), as well as antagonism of the tetrad effects induced by CB1R agonists (Fulp et al., 2013; Amato et al., 2018). Although many peripherally restricted ligands have minimal brain permeability after acute administration, it remains to be ascertained whether chronic administration would lead to an increase in brain permeability that can affect the profile of unwanted CNS side effects. Furthermore, only a few peripheral CB1R antagonists/inverse agonists have been evaluated in detail for their unwanted effects, with the most extensively studied being JD5037 (Kale et al., 2019). This compound exhibited only minor side effects such as repetitive grooming at doses much higher than the therapeutic doses, which is translated into a safer therapeutic window compared with the brain-permeant CB1R antagonist/inverse agonists (Kale et al., 2019).
In a different approach to achieving peripheral restriction, monoclonal antibodies that act as CB1R antagonists/inverse agonists have been developed and entered clinical evaluation. The two candidates that have been in clinical development for renal diseases and diabetic complications are Nimacimab (Bird Rock Bio) and GFB-024 (Goldfinch Bio), both listed in Table 4. However, there are no publicly available data regarding the efficacy and safety of this innovative approach.
CB1R is a constitutively active receptor that even in the absence of ligands exists in equilibrium between active and inactive states; this condition is translated into increased basal activity (Pertwee, 2005; Fong, 2014) and may be important for cellular homeostasis. While inverse agonists reduce the basal activity of receptors, neutral antagonists do not significantly affect it (Bond and Ijzerman, 2006; Sink et al., 2008). Additionally, the ECS as a whole exhibits an endogenously active tone controlled by the cellular production of eCBs (Howlett et al., 2011). Therefore, CB1R neutral antagonists can compete with the endogenous cannabinoid ligands without affecting the basal activity of the receptor. For this reason, it was hypothesized that CB1R neutral antagonists could produce the therapeutic phenotypes of CB1R antagonism without the unwanted CNS and gastrointestinal side effects. In this regard, the most extensively studied CB1R neutral antagonist, AM4113, exhibited therapeutic efficacy with a better tolerability profile. In animal models of obesity, AM4113 was shown to reduce food intake and weight gain, as well as to suppress food-reinforced operant responding and feeding (Chambers et al., 2007; Sink et al., 2008; Gueye et al., 2016). Ιn addiction-related models, AM4113 was effective in suppressing alcohol consumption, reducing drug-seeking behavior of nicotine and THC, as well as inhibiting the self-administration of heroin (Gueye et al., 2016; Schindler et al., 2016b; Balla et al., 2018; He et al., 2019). Moreover, AM4113 did not induce anxiety-like behaviors in elevated plus maze and electrical brain-stimulation reward paradigm, unlike the CB1R antagonist/inverse agonist AM251 (Sink et al., 2010; Gueye et al., 2016; He et al., 2019). Additionally, in contrast to CB1R inverse agonists AM4113 did not produce gastrointestinal side effects such as nausea, potentiation of vomiting, and increase in whole gut transit (Chambers et al., 2007; Sink et al., 2008; Storr et al., 2010). Other CB1R neutral antagonists, such as the peripherally restricted AM6545 and NESS06SM, have been shown to suppress food intake and improve cardiometabolic risk factors (Cluny et al., 2010; Randall et al., 2010; Tam et al., 2010; Mastinu et al., 2013). AM6545 also exhibited efficacy in animal models of experimental diabetic nephropathy, alone and in combination with the CB2R agonist AM1241 (Barutta et al., 2017; 2018).
On a final note, a novel and attractive dual-targeting approach is represented by the combination of CB1R antagonists and CB2R agonists, as evidenced by the synergy shown by coadministration of AM6545 and AM1241 for treating diabetic nephropathy (Barutta et al., 2017). Indeed, there is early evidence that CB1R and CB2R promote opposing functions in fibrotic and inflammatory conditions of peripheral organs (Gruden et al., 2016), as well as in some preclinical models of addiction (Delis et al., 2017; Gobira et al., 2019) that could be leveraged for a therapeutic benefit by dual-acting CB1R antagonists/CB2R agonists.
B. Therapeutic Potential of Cannabinoid Receptor 2
The CB2R is a class A (rhodopsin-like) GPCR (Fig. 14). It is an essential element of the ECS, and indeed CB2R-mediated signaling plays an important role in many human health and disease conditions (Pacher and Mechoulam, 2011; Gasperi et al., 2023). Therefore, CB2R holds tremendous therapeutic potential for treating major pathologies affecting humans.
Fig. 14.

Structures of the CB2R in different states. (A) Crystal structure of antagonist AM10257-bound CB2R (PDB code 5ZTY). (B) Crystal structure of agonist AM12033-bound CB2R (PDB code 6KPC). (C) Cryo-EM structure of AM12033-bound CB2R-Gi complex (PDB code 6KPF). (D) Cryo-EM structure of WIN55,212-2-bound CB2R-Gi complex (PDB code 6TP0), using color code as follows: CB2R-AM10257, brown; CB2R-AM12033 (PDB code 6KPC), sky blue; CB2R-AM12033 (PDB code 6KPF), green; CB2R-WIN55,212-2, dark salmon; Gαi in CB2R-AM12033, purple; Gβ in CB2R-AM12033, teal; Gγ in CB2R-AM12033, orchid; scFv16 in CB2R-AM12033, cornflower blue; Gαi in CB2R-WIN55,212-2, medium purple; Gβ in CB2R-WIN55,212-2, turquoise; Gγ in CB2R-WIN55,212-2, plum; scFv16 in CB2R-WIN55,212-2, light blue.
A plethora of preclinical evidence demonstrating the anti-inflammatory and tissue-protective effects of CB2R activation has been generated, triggering the design, synthesis, and evaluation of multiple CB2R ligands. Based on their chemical structure, they can be characterized as pCBs, eCBs, and congeners or synthetic ligands (Han et al., 2013; Guba et al., 2020; Brennecke et al., 2021). While the majority of these molecules are CB2R activators, multiple antagonists/inverse agonists and a few allosteric ligands have also been discovered. Of these, more than 20 CB2R-selective agonists have been advanced to clinical trials. Recently, several 3D structures of CB2R in complex with ligands have been reported (Li et al., 2019; Hua et al., 2020; Xing et al., 2020). Furthermore, a wide variety of labeled chemical probes was generated and applied in mechanistic studies (Basagni et al., 2020; Haider et al., 2020; Sarott et al., 2020; Gazzi et al., 2022; Guberman et al., 2022) and has contributed the understanding of the structural basis of selective CB2R activation (Li et al., 2023). Together this knowledge will facilitate the design of novel, further improved ligands. Here efforts were made to recognize the full range of studies that have contributed to progress CB2R research since the discovery of the receptor. Due to space limitations, the content of this section highlights only foundational studies and key aspects.
The CB2R is primarily expressed in immune cells, including macrophages, T and B cells, monocytes and polymorphonuclear neutrophils, as well as tissues like spleen (Bouaboula et al., 1993; Galiègue et al., 1995; Atwood and Mackie, 2010; http://www.immgen.org/), bone (Ofek et al., 2006), and the gastrointestinal tract (Atwood et al., 2012). CB2R is expressed both on the cell surface and intracellularly (Kleyer et al., 2012; Brailoiu et al., 2014; Castaneda et al., 2017) and is highly inducible, for instance, in microglia upon neuroinflammation (Cabral et al., 2008). The CB2R is a Gi/o coupled GPCR, and its activation leads to an inhibition of cAMP production. In addition, the CB2R recruits β-arrestins, controls the activation and phosphorylation of different mitogen-activated protein kinase family members (ERK1/2, p38 MAPK, JNK), and interacts with PLC as well as G-protein-coupled inwardly rectifying K+-channels (Bouaboula et al., 1993; Felder et al., 1995; Howlett et al., 2002; Cabral et al., 2008; Atwood and Mackie, 2010). Surface and intracellular CB2R might be able to activate distinct signaling responses (Brailoiu et al., 2014). In addition, agonists binding to the orthosteric site exhibit different transduction profiles that might translate into distinct pharmacodynamics read-outs (Oyagawa et al., 2018; Yuan et al., 2021). Downstream effects of CB2R activation encompass the differentiation of B and T lymphocytes (Ziring et al., 2006), the suppression of T cell receptor signaling (Börner et al., 2009), the induction of natural killer cell migration (Kishimoto et al., 2005), and the modulation of cytokine release (Cencioni et al., 2010; Correa et al., 2011). CB2R interactions at the molecular level and its resulting downstream effects translate toward modulation of disease pathogenesis. CB2R ligands have demonstrated a huge therapeutic potential in a large variety of disease models (e.g., in liver; Mallat and Lotersztajn, 2008; Pacher and Gao, 2008), kidney (Mukhopadhyay et al., 2010a,b, 2016; Zoja et al., 2016), lung (Pacher et al., 2006), and heart disorders (Pacher et al., 2008); skin pathologies (Bíró et al., 2009; Maccarrone et al., 2015), neurodegenerative diseases (Centonze et al., 2007; Fernández-Ruiz et al., 2007); and pain (Guindon and Hohmann, 2008; Anand et al., 2009). Generally, the reported effects are a consequence of CB2R-mediated immunosuppressive and anti-inflammatory effects leading to a dampening of tissue injury. In hypoactivated immune states, CB2R activation might, however, enhance tissue damage (Pacher and Mechoulam, 2011). Under these pathologic conditions, CB2R inverse agonists/antagonists might provide therapeutic options.
1. Cannabinoid Receptor 2 Agonists
Due to the huge therapeutic potential of CB2R, multiple ligands have been developed. In 1996, a first patent for a CB2R-selective antagonist was filed (Rinaldi et al., 1996). Since then, more than 1150 CB2R patent applications have been registered. CB2R targeting molecules covered by these papers and patents encompass agonists, modulators, neutral antagonists, inverse agonists, and allosteric ligands. While the majority of these ligands are classic small molecules, including many labeled chemical probes, some are of a peptidic nature. Multiple comprehensive and excellent reviews on this subject have been published (Thakur et al., 2009; Riether, 2012, Han et al., 2013, 2014; Morales et al., 2016; Aghazadeh Tabrizi et al., 2016; Cooper et al., 2017; Guba et al., 2020; Brennecke et al., 2021). Focus within this section has been placed on representative molecules that describe the development of a “CB2R ligand space” with a strong emphasis on those that made it into clinical development, all of them being activators of CB2R. CB2R agonists that are launched or under active development and registered with ClinicalTrials.Gov are listed in Table 5.
TABLE 5.
Diseases/symptoms for treatment with CB2R agonists and antagonists registered with ClinicalTrials.gova
| Generic Name Brand Name Class/Efficacy |
Completed Clinical Trials | Ongoing Clinical Trials |
|---|---|---|
| Dronabinolb | ||
| Dronabinol derivativesb | ||
| Nabiloneb | ||
| Nabiximolsb | ||
| Mixed THC:CBDb | ||
| Cannabidiol | ||
| Epidiolex | Sturge-Weber syndrome | Obsessive-compulsive disorder |
| CB1R/CB2R ligand others | Opioid-use disorder | Tuberous sclerosis complex |
| Prostate cancer | Typical absence seizures | |
| Cannabis use disorder | Autism | |
| Opioid withdrawal | Fibromyalgia | |
| Musculoskeletal pain | Aromatase inhibitor-associated arthralgias | |
| Alcohol use disorder | Back pain | |
| • Post-traumatic stress disorder | Depressive symptoms | |
| • Inflammatory bowel disease | Electrical status epilepticus of slow-wave sleep | |
| • Knee osteoarthritis | Dental pain | |
| Parkinson’s disease | Behavioral problems in children and adolescents with intellectual disability | |
| Opiate addiction | Knee arthritis | |
| Epilepsy | Chemotherapy-induced peripheral neuropathy | |
| Seizures | Bipolar disorder | |
| COVID-19 | Hypertension | |
| Burn-out | Anxiety and fear | |
| Chronic periodontitis | Chronic pain | |
| Urinary stone | Early psychosis | |
| Schizophrenia | Post-traumatic stress disorder | |
| Blepharospasm | Anorexia nervosa | |
| Cocaine craving/dependence | Gastroparesis and functional dyspepsia | |
| Generalized anxiety disorder | Anxiety in advanced breast cancer | |
| Lennox-Gastaut syndrome | Traumatic brain injury | |
| Dravet syndrome | Tobacco cessation | |
| Psychotic disorders | Social anxiety disorder | |
| Infantile spasms | Rheumatoid arthritis | |
| Fragile X syndrome | Diabetes | |
| Tuberous sclerosis complex | Chronic pain | |
| Psoriatic arthritis | Endometriosis pain | |
| Hand osteoarthritis | Social anxiety disorder | |
| Cancer | • Radiculopathy | |
| Diabetic neuropathies | Sleep disturbance | |
| Ulcerative colitis | Insomnia | |
| Fatty liver | Prevention aGVHD | |
| Prader-Willi syndrome | ||
| Musculoskeletal pain | ||
| Lenabasum | ||
| CB2R/CB1R agonist | Cystic fibrosis | |
| Dermatomyositis | ||
| Systemic lupus erythematosus | ||
| Olorinab | ||
| CB2R agonist | Crohn’s disease | |
| Abdominal pain | ||
| RG7774 | ||
| CB2R agonist | Diabetic retinopathy | |
| CNTX-6016 | ||
| CB2R agonist | Chronic pain | Painful diabetic neuropathy |
| Nociceptive pain | ||
| Pain | ||
| EHP-101 | ||
| CB2R agonist PPARγ agonist | Diffuse cutaneous systemic sclerosis | |
aStudies with the status “not yet recruiting, recruiting,” “enrolling by invitation,” “active, not recruiting,” and “completed” were included in this table.
bSee Table 4 for respective CB1R data.
a. Endocannabinoids and related fatty acid derivatives
Polyunsaturated C20 fatty acids such as AA are the basic building blocks of eCBs and related fatty acid derivatives, which include amides such as AEA, esters like 2-AG, and ethers like noladin ether (Hanus et al., 2001) (Figs. 1 and 2). 2-AG was first isolated from canine gut and rat brain (Mechoulam et al., 1995; Sugiura et al., 1995) and is considered as the most relevant signaling component of the ECS. Like AEA, it can be generated by several pathways and enzymes (Fezza et al., 2014; Baggelaar et al., 2018; Tsuboi et al., 2018). These key eCBs are synthesized and released on demand following CB1/2R activation (De Petrocellis et al., 2004; Lambert and Fowler, 2005; Di Marzo, 2018; Cristino et al., 2020). Besides CB1/2R (Fig. 15), they interact also with further molecular targets, e.g., the vanilloid TRPV1 ligand-gated ion channel (De Petrocellis et al., 2000).
Fig. 15.

Chemical structure and CB2R binding affinity of THC, N-arachidonoylethanolamine, and 2-arachidonoyl glycerol. aConsensus human CB2R binding affinity values from a multicentric collaborative profiling effort between multiple independent academic laboratories and industry (Soethoudt et al., 2017). bCB2R selectivity (10∧(pKi CB2R-pKi CB1R).
In the meantime, further eCBs and a multitude of eCB-like mediators have been isolated. Generally, the eCBs are relatively short-acting ligands, especially due to their hydrolysis through FAAH and MAGL. Therefore, synthetic efforts were undertaken to improve the hydrolytic stability of eCBs, e.g., by modifying the amide residue of AEA, which provided ligands such as ACPA (Fig. 16) (Hillard et al., 1999).
Fig. 16.

Chemical structure and CB2R binding affinity of noladin ether and synthetic eCB analogs. aBinding to spleen cannabinoid receptor. bWith phenylmethanesulfonyl fluoride.
b. Plant-derived cannabinoids
THC and its thermodynamically more stable and similarly potent regioisomer Δ8-THC served as a starting point for generating further classic cannabinoids (Razdan, 1986; Mechoulam et al., 1998). Dual CB1R/CB2R agonist Lenabasum, also known as Anabasum, Resunab, ajulemic acid, JBT-101, or CT-3 (Tepper et al., 2014), demonstrated efficacy in reducing chronic neuropathic pain in a phase 2 clinical trial (Karst et al., 2003) (Fig. 17). Currently the ligand is being evaluated in phase 3 for the treatment of dermatomyositis and scleroderma (https://adisinsight.springer.com/drugs/800007180). Although Lenabasum activates CB1R in addition to CB2R, it is not psychoactive (Zurier et al., 1998). Presumably, this is the consequence of its low brain penetrance.
Fig. 17.

Chemical structure, CB2R binding affinity and selectivity of representative classic cannabinoids. aConsensus human CB2R binding affinity values from a multicentric collaborative profiling effort between multiple independent academic laboratories and industry (Soethoudt et al., 2017). bCB2R selectivity (10∧(pKi CB2R-pKi CB1R).
Preferential CB2R activation can be achieved by omitting the phenolic C-1 hydroxyl of THC (Reggio et al., 1990; Gareau et al., 1996; Huffman et al., 1996), a strategy that was successfully applied for the generation of JWH133. The ligand is one of the first CB2R-selective agonists (10^(pKi CB2R-pKi CB1R) > 153), and thus it has been exploited to interrogate CB2R pharmacology (Pertwee, 1999; Soethoudt et al., 2017).The reference compound CP55940 is a potent dual CB1R/CB2R agonist outperforming THC with regard to CB1/2R binding affinity and analgesic activity (Showalter et al., 1996). Its tritiated congener has been broadly applied for the discovery and profiling of many CB1/2R ligands (Devane et al., 1988).
In contrast, nonpsychotropic (-)CBD exhibits moderate affinity for CB2R (Showalter et al., 1996). CBD has been suggested to function as an inverse agonist of CB2R (Thomas et al., 2007) but interacts with multiple other targets as well (Ibeas Bih et al., 2015). Second-generation CBD derivative EHP-101 (VCI-004.8) is a dual CB2R and PPARγ agonist and activator of protein phosphatase 2A, which is currently investigated in phase 2a clinical trials (Del Río et al., 2016; EMA, 2022). Indications in focus are systemic and multiple sclerosis, for which preclinical proof of concept, e.g., in fibrosis models (García-Martín et al., 2018) and in neuroinflammation (Navarrete et al., 2018), has been demonstrated.
Cannabinoid fumaric acid ester PRS-211375 (Cannabinor) is a selective CB2R agonist (CB2R EC50 cAMP = 17.4 nM; 98% efficacy) (Gratzke et al., 2010). It showed efficacy in various rodent in vivo disease models including pain readouts in a chronic constriction injury model (Clarivate, 2022j). Analgesic effects were translated into the clinic. In patients undergoing third molar dental extraction, nociceptive pain was reduced at 12 mg (i.v.) in a phase 2a study (Clarivate, 2022j). Interestingly, no effect was observed at higher doses. This bell-shaped curve behavior is characteristic of the pharmacodynamics studies with other cannabinoid-derived CB1/2R ligands (Martellotta et al., 1998; Linares et al., 2019). Converting the phenolic C-1 hydroxyl group of CBD-dimethylheptyl into a methoxy moiety can enhance selectivity for CB2R as exemplified for HU-308. This potent, selective, and bioavailable CB2R agonist (Soethoudt et al., 2017) has demonstrated anti-inflammatory and tissue-protective effects in multiple rodent disease models such as formalin-induced inflammation (Hanus et al., 1999) and hepatic ischemia/reperfusion injury studies (Rajesh et al., 2007). Attenuated leukostasis, chemotaxis, and oxidative stress associated with reperfusion damage suppressed the acute inflammatory response (Pacher and Haskó, 2008). Structurally close analog HU-910 exhibited high binding and functional selectivity for CB2R over CB1R (Soethoudt et al., 2017). In addition, it is highly selective against a representative set of further off-targets and displays favorable pharmacokinetic properties. Therefore, HU-910 was recommended as a preferred CB2R agonist for studying the role of the receptor in biologic and disease processes (Soethoudt et al., 2017). HU-910 in vivo efficacy studies opened the door for exploring the potential of CB2R activation for the treatment of type 2 diabetic nephropathy (Zoja et al., 2016) and eye diseases such as uveitis (Porter et al., 2019). Importantly, HU-910 exhibits a different signaling preference in the five CB2R signal transduction pathways in human and mouse. In contrast to being an unbiased agonist for the human CB2R, HU-910 exhibited a preference toward G-protein activation as compared with cAMP signaling and β-arrestin recruitment in mice (Soethoudt et al., 2017). Such interspecies differences in signaling preference might influence the translation of preclinical models to the clinic.
The vast majority of synthetic cannabinoids exhibit high lipophilicity, low aqueous solubility, and tight plasma protein binding, which translates into poor pharmacokinetic properties, such as high in vivo clearance and low oral bioavailability (McGilveray, 2005; Huestis, 2007). To overcome these issues, molecules were developed to exhibit favorable physicochemical properties and improved oral bioavailability. In the following paragraphs, key representatives from the most important scaffolds were selected to illustrate the progress made on synthetic CB2R ligands.
Aminoalkylindoles were among the earliest discovered CB2R scaffolds. In particular, dual CB1R and CB2R agonist WIN55212-2 (Eissenstat et al., 1990; Bell et al., 1991) was very important for identifying and deciphering the role of cannabinoid receptors (Fig. 9). It displays antihyperalgesic activity in multiple rodent pain models (D’Ambra et al., 1992; Fox et al., 2001; Johanek and Simone, 2004).
Initial aminoalkylindoles were structurally simplified. Furthermore, CB2R selectivity was improved to lead to CB2R agonists such as A-796260 (Fig. 9) (Frost et al., 2008), which achieved efficacy in various rodent pain models upon intraperitoneal-injection administration (Yao et al., 2008). Importantly, these antihyperalgesic effects could be blocked by pretreatment with a CB2R antagonist.
Bicyclic (het)aryl scaffolds were investigated for CB2R selectivity or minimal CB1R efficacy. Several high throughput screening campaigns were conducted in the search for potent, selective, and orally bioavailable CB2R agonists, e.g., providing benzimidazole (Pagé et al., 2008) and triazolopyrimidine (Nettekoven et al., 2016) derived starting points. Subsequent lead optimization efforts provided development candidates such as dual CB1R/CB2R agonist ART-27.13 (AZD-1940) (Pagé et al., 2010) (Fig. 18). This molecule is currently assessed as oral treatment of cachexia in phase 2 and cancer-related anorexia in phase 1 trials (Clarivate, 2022a; https://artelobio.com/pipeline/). However, due to CNS-related side effects, phase 2 studies for the treatment of nociceptive and neuropathic pain were terminated (Kalliomäki et al., 2013; https://www.astrazenecaclinicaltrials.com/study/D3120C00006/).
Fig. 18.

Chemical structure, CB2R binding affinity or functional activity, and selectivity of clinically evaluated bicyclic (het)aryl derived CB2R ligands.
Phase 2 clinical trials for the oral treatment of osteoarthritic knee pain were conducted with CB2R agonist LY-2828360 (Fig. 17), but they were terminated despite an acceptable side-effect profile (Hollinshead et al., 2013; https://clinicaltrials.gov/ct2/show/NCT01319929; Clarivate, 2022f). The imidazopyrimidine is brain penetrant and exhibits an excellent selectivity over CB1R (ratio CB1R/CB2R EC50 for GTPγS binding was > 5′000). Recently reported triazolopyrimidine-derived CB2R agonist (at 1 nM) RG7774 (Fig. 17) is under active development in phase 2 as an innovative oral treatment of diabetic retinopathy exhibiting very high selectivity over CB1R (ratio CB1R/CB2R EC50 for cAMP > 6’940) (Grether, 2022). Further bicyclic (het)aryl derived ligands reached advanced preclinical stages and were successfully explored in various disease models with an inflammatory pathology. PF-0355009 6 (Kikuchi et al., 2008) and RQ-00202730 (Iwata et al., 2015) were tested in 2,4,6-trinitrobenzene sulfonic acid-induced colonic pain rat models, and RO6871304 was tested in rodent models of kidney ischemia–reperfusion, renal fibrosis, and endotoxin-induced uveitis (Nettekoven et al., 2016; Porter et al., 2019).
Multiple organizations developed bicyclic aliphatic (het)aryl arrays with at least one aliphatic ring. Five-five, five-six, and five-seven systems were elaborated, and four of these CB2R agonists made it into clinical trials. Tedalinab was investigated for the oral treatment of neuropathic pain and osteoarthritis (Clarivate, 2022n) (Fig. 19). The CB2R-selective pyrazole carboxamide exhibits similar binding affinities for human and rat CB2R (human CB2R Ki = rat CB2R Ki ≈ 12 nM) and bioavailabilities > 50% across species. Despite favorable safety and tolerability data in single ascending dose (doses up to 1200 mg) and multiple ascending dose studies (doses up to 300 mg once daily for 14 days), development was halted for unknown reasons.
Fig. 19.
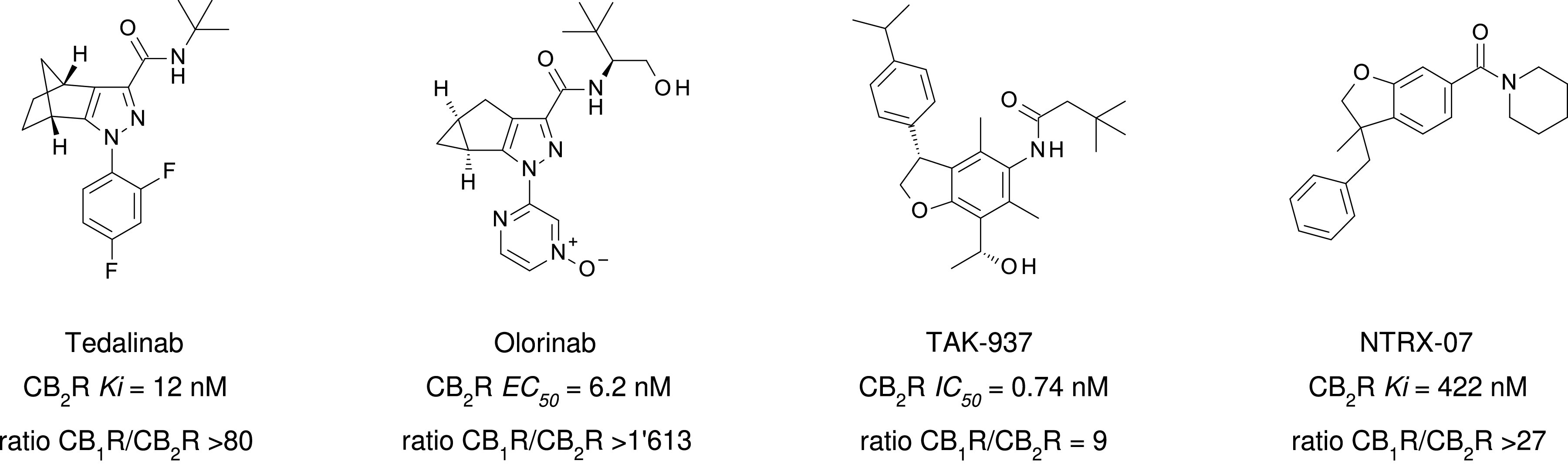
Chemical structure, CB2R binding affinity or functional activity, and selectivity of clinically evaluated bicyclic aliphatic (het)aryl arrays.
Lead optimization toward olorinab was guided by a β-arrestin efficacy assay (Han et al., 2017). This highly potent CB2R full agonist is a peripherally acting molecule that is devoid of psychotropic effects (https://adisinsight.springer.com/drugs/800039670; https://clinicaltrials.gov/ct2/show/NCT04043455). Olorinab (Fig. 19) was clinically assessed as an oral treatment of pain related to irritable bowel syndrome in phase 2. While the drug was well tolerated, it did not meet the primary efficacy endpoint of statistically significant improvement in the overall average abdominal pain score (Pharma Intelligence, 2022). The ligand exhibits a short human half-life and was therefore administered three times a day (Clarivate, 2022i). Dual CB1R/CB2R agonist TAK-937 (Fig. 19) was developed as an injectable for the treatment of stroke after observing cerebroprotective effects in rat and nonhuman primate in vivo efficacy studies (Suzuki et al., 2012; Clarivate, 2022m). Yet, due to a narrow safety margin, development was halted. CB2R-selective agonist dihydro-benzofuran NTRX-07 (Fig. 19) is being explored in phase 1 as an oral drug for the treatment of memory loss in Alzheimer's disease, cognitive disorder, and neuropathic pain (Clarivate, 2022h; https://www.neurotherapia.com/research). Follow-up studies targeting MS and amyotrophic lateral sclerosis are foreseen. NTRX-07 preserves CB2R potency across species and showed efficacy in multiple rodent efficacy studies (Naguib et al., 2008). The (S)-enantiomer is the active stereoisomer (Diaz et al., 2009).
CB2R modulators containing aromatic and aliphatic five-, six-, and seven-membered central cores have been described by multiple organizations. Pyrimidine-based agonist GW-842166X displays high CB2R selectivity over CB1R, and favorable pharmacokinetic properties, translating into potent analgesic effects (ED50 = 0.1 mg/kg) in the Complete Freund’s adjuvant rat model of inflammatory pain without initiating tetrad-like effects such as catalepsy or hypothermia (Giblin et al., 2007) (Fig. 20). The ligand reached phase 2 clinical trials for pain associated with osteoarthritis (https://clinicaltrials.gov/ct2/show/NCT00479427) and dental pain (https://clinicaltrials.gov/ct2/show/NCT00444769).
Fig. 20.
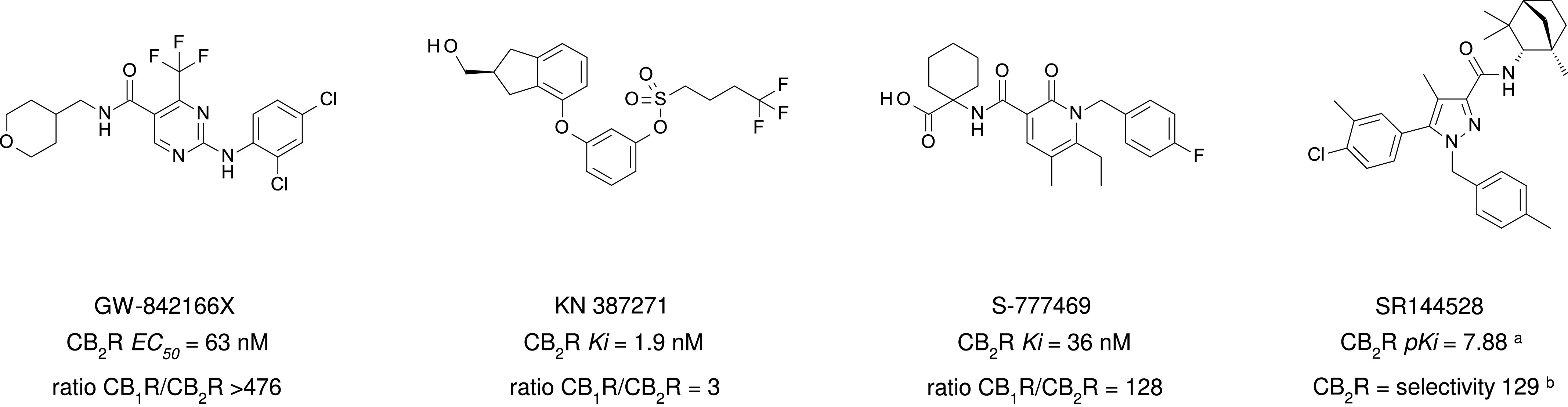
Chemical structure, CB2R binding affinity or functional activity, and selectivity of clinically evaluated CB2R agonists and CB2R inverse agonists SR144528 containing five- and six-membered central cores. aConsensus human CB2R binding affinity values from a multicentric collaborative profiling effort between multiple independent academic laboratories and industry (Soethoudt et al., 2017). bCB2R selectivity (10∧(pKi CB2R-pKi CB1R).
Disubstituted phenyl derivative KN 387271 (Fig. 20) is a dual CB1R/CB2R agonist. Neuroprotective effects in rat models of cerebral ischemia and traumatic brain injury (Mauler et al., 2002; Mauler et al., 2003) enabled phase 1 stroke and phase 2 traumatic brain injury studies in humans (Clarivate, 2022e). The selective orally bioavailable CB2R agonist S-777469 exhibited efficacy in rodent models of scratching and skin inflammation (Odan et al., 2012; Haruna et al., 2015, 2017). However, these effects did not translate into therapeutic benefits in phase 2a trials with patients suffering from atopic dermatitis and pruritus (Clarivate, 2022l; https://www.shionogi.com/content/dam/shionogi/global/investors/pdf/e_p090803.pdf). Many additional CB2R modulators with different central cores such as pyrazoles (Ohta et al., 2007), thiazoles (Yao et al., 2009), diazepanes (Zindell et al., 2011), piperidines (Bartolozzi et al., 2015), pyrrolidones (Riether et al., 2015), imidazoleidine-2,4-diones (Mukhopadhyay et al., 2016), pyridines (Porter et al., 2019), and 4-oxo-1,4-dihydropyridines (El Bakali et al., 2014) have been evaluated in detail. In some cases, minor structural changes triggering a switch from agonism to inverse agonism have been reported (Sellitto et al., 2010; Porter et al., 2019). Insights on how to design CB2R agonists with favorable kinetic profiles were disclosed in a structure kinetics relationship study on a biaryl imidazoleidine-2,4-dione-based scaffold (Soethoudt et al., 2018b). An adamantyl-derived series was investigated for functional activity on the Q63R variant of CB2R (Nettekoven et al., 2013), which is associated with the risk of schizophrenia (Ishiguro et al., 2010) and an increased risk of celiac disease and liver damage in obese children (Rossi et al., 2011).
2. Cannabinoid Receptor 2 Antagonists and Allosteric Ligands
Selective CB2R antagonist/inverse agonist SR144528 (human CB2R selectivity ratio = 129; mouse CB2R selectivity ratio 10^(pKi CB2R-pKi CB1R) = 6’026) (Rinaldi-Carmona et al., 1998; Portier et al., 1999; Soethoudt et al., 2017) is an important pharmacological tool for antagonizing effects triggered by CB2R agonists in vitro and in vivo (Nackley et al., 2003). Interestingly, the ligand shows a bias in suppressing different signal transduction pathways. It effectively blocks the modulation of cAMP signaling but is less potent with regard to antagonizing CB2R-mediated signal transduction pathways (Soethoudt et al., 2017).
Few ligands targeting postulated CB2R allosteric sites (Feng et al., 2014; Pandey et al., 2020) are known. An allosteric CB2R interaction has been suggested for CBD (Martinez-Pinilla et al., 2017). Conversely, it was also experimentally shown that CBD acts as an orthosteric partial agonist (Tham et al., 2019), although it does not follow a simple one-site competition model. An overlap of allosteric and orthosteric binding pockets might provide a suitable explanation for these findings. In contrast, 1,1’-dimethyl heptyl CBD was shown to act as a pathway-specific CB2R allosteric modulator (Fig. 21). While positively modulating the cAMP response, it negatively modulated β-arrestin1 recruitment by CP55940 and SR144528. Interaction with a high-affinity allosteric binding site has been postulated by 5XRA- and 5TGZ-based in silico docking studies.
Fig. 21.
Chemical structure of validated CB2R allosteric modulators.
Endogenously occurring RVD-hemopressin peptide pepcan-12 (Fig. 21) exhibits positive allosteric modulation of CB2R (Petrucci et al., 2017). It was shown to increase binding of orthosteric ligands and to potentiate 2-AG- and CP55940-induced CB2R signaling. Synthetic ligand C2 shows positive allosteric modulation of CB2R in vitro (Gado et al., 2019). Importantly, these effects translated into dose-dependent efficacy in a mouse model of neuropathic pain upon oral administration. Neither an X-ray crystal nor a cryo-electron microscopy structure of a CB2R allosteric modulator in complex with the receptor has been reported. Therefore, the design of novel ligands is mostly aided by in silico predictions including molecular dynamics simulations that can lead to the identification and ranking of multiple putative allosteric binding sites (Yuan et al., 2022). Furthermore, molecular dynamics simulations suggest that cholesterol exerts an allosteric effect on the intracellular CB2R regions that interact with the G-protein complex, thus altering the recruitment of G-protein (Yeliseev et al., 2021). Therefore, cholesterol levels might influence the screening for novel allo- and orthosteric CB2R ligands, which should be taken into account in designing selective drugs directed toward CB2R.
3. Cannabinoid Receptor 2 Chemical Probes for Research and Diagnostics
A labeled chemical probe is a small molecule that is a ligand for a respective target and carries a reporter unit, e.g., a radio, fluorescent, or biotin label that allows characterization of ligand-target interactions. Optionally a linker connects the target recognition element and reporter unit (Prevet and Collins, 2019). Labeled probes are of utmost importance for all research and discovery phases (Guberman et al., 2022). Due to a major debate regarding the specificity of CB2R antibodies (Cécyre et al., 2014; Marchalant et al., 2014; Zhang et al., 2019), labeled chemical CB2R probes are highly important tools for determining CB2R protein expression. While radioligands are generally used for studying binding affinity (Cascio et al., 2016) or drug-target binding kinetics (Martella et al., 2017) of unlabeled ligands, positron emission tomography tracers focus on determining receptor expression in tissues and noninvasively measuring the distribution and receptor occupancy of drug candidates in patients (Honer et al., 2014). Nonselective [3H]CP55940 and [3H] WIN55212-2 are the most relevant probes for measuring equilibrium binding affinities of novel CB2R ligands applying radioligand competition-binding assays. Selective CB2R inverse agonist [35S]SCH225336 (Lavey et al., 2005; Gonsiorek et al., 2006) was successfully applied for quantifying CB2R expression in various cell lines and hemopoietic cells making use of the superior specific activity of its 35S reporter unit, as compared with tritiated cannabinoids (> 1’400 versus ∼20 Ci/mmol) (Fig. 22).
Fig. 22.
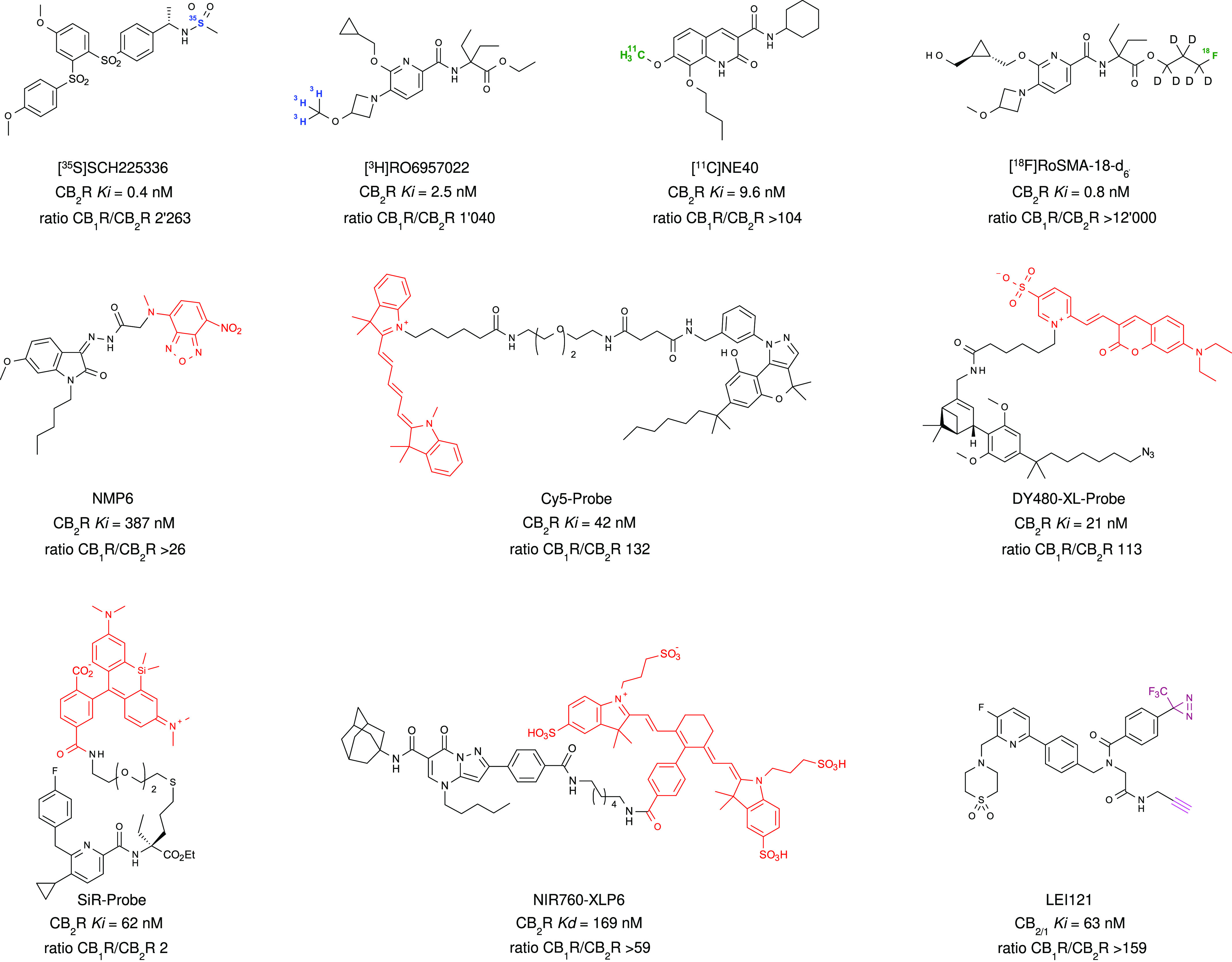
Chemical structure, CB2R binding affinity, and selectivity of CB2R radioligands, PET tracers, fluorescent and pAfBPP probes.
Tritiated pyridine [3H]RO6957022 exhibits high binding selectivity targeting CB2R (Martella et al., 2017). The CB2R inverse agonist was used for studying drug-target binding kinetics. Its 11C-labeled analog [11C]RSR-056 carrying the carbon-11 reporter unit at the methoxy group is a CB2R-specific brain-penetrant positron emission tomography tracer that displayed a higher brain radioactivity in mice with lipopolysaccharide-induced neuroinflammation than in the control group (Slavik et al., 2015). 2-Oxoquinoline-derived [11C]NE40 is the first tracer that has been used for CB2R in vivo positron emission tomography in humans (Ahmad et al., 2013). In agreement with the known expression of CB2R, major uptake was observed in lymphoid tissue. Despite a rapid brain uptake and washout, no CB2R upregulation was detected in the brains of Alzheimer’s disease patients (Ahmad et al., 2016). [18F]RoSMA-18-d6 exhibits subnanomolar affinity for CB2R across species and a remarkable selectivity factor of > 12’000 over CB1R (Haider et al., 2020). It showed specific and reversible target binding in vitro and in vivo and was successfully used for detecting CB2R upregulation on post-mortem human amyotrophic lateral sclerosis spinal cord tissues.
Fluorescently labeled CB2R ligands are highly versatile tools for studying receptor-ligand interactions and cellular trafficking, e.g., applying techniques such as flow cytometry, confocal fluorescence microscopy, and time-resolved fluorescence resonance energy transfer. N-Alkyl isatin acylhydrazone NMP6 was among the first fluorescently labeled ligands that showed selectivity for CB2R over CB1R (Petrov et al., 2011). In flow cytometry and confocal microscopy studies, specific binding to endogenously expressed CB2R in CD4+ T cells and B-lymphocytes was demonstrated. Cy5-labeled (Cy5-) probe is a CB2R inverse agonist with an extended linker moiety showing low levels of nonspecific fluorescence in live-cell experiments (Singh et al., 2019). Combination of favorable structural elements of the two cannabinoid ligands HU-308 and AM841 provided a privileged chimera motif that was functionalized with a range of fluorophores while retaining excellent affinity and selectivity for CB2R (Sarott et al., 2020; Westphal et al., 2020). Coumarin fluorophore-labeled DY480-XL probe allowed for setting up a novel assay based on fluorescence resonance energy transfer, able to characterize equilibrium and kinetic binding constants and visualize in real-time CB2R in endogenously expressing murine splenocytes and human macrophages. The reverse-design approach, in which small molecules previously optimized in medicinal chemistry programs form the basis for the generation of high-quality probes (Guberman et al., 2022), was applied for the generation of cell-permeable agonist-based SiR probe that was used for real-time in vivo tracing of CB2R in zebrafish larvae (Gazzi et al., 2022). Near-infrared fluorophores are best suited for in vivo imaging in higher species due to their deeper light penetration of biologic tissues (Hong et al., 2017). Pyrazolopyrimidine derivative NIR760-XLP6 displays high selectivity over CB1R and improved specific binding as compared with predecessors such as NIR760-mbc94 and therefore holds promise for visualizing CB2R in in vivo imaging studies (Ling et al., 2015). Alternatively to fluorescent probes, biotinylated CB2R ligands have been applied for visualization of the receptor after conjugation with streptavidin-AlexaFluor488 (Martin-Couce et al., 2012).
While reversible noncovalent interaction with CB2R can easily be disrupted under experimental conditions, resulting in the washout of the probe from the binding site, a covalent attachment can surmount these issues (Weichert and Gmeiner, 2015; Yang et al., 2019). The water-stable isothiocyanate group, which reacts preferentially with the nucleophilic amino acid side chains of cysteines, was exploited to covalently attach cannabinoids to CB2R (Szymanski et al., 2011; Mallipeddi et al., 2017). Furthermore, CB2R-selective photoaffinity probes carrying benzophenone (Dixon et al., 2012) or azide (Szymanski et al., 2018) groups as photoreactive moiety have been reported. Two-step photoaffinity-based protein profiling probe LEI121 elegantly combines the covalently modifying photoaffinity technique with a click chemistry approach, allowing for target engagement studies in live human cells by covalent SDS-PAGE visualization, flow cytometry, and mass spectrometry-based proteomics (Soethoudt et al., 2018a).
C. Summary of Clinical Status of Cannabinoid Receptor 1 and Cannabinoid Receptor 2 Agonists
In summary, three phytocannabinoid preparations (dronabinol, nabiximols, and CBD) are currently available for treatment of diseases via stimulation of CB1R, CB2R, both, or neither (Table 6). Although the need for selective full agonist stimulation of CB1R is limited due to side effects, selective CB2R agonists are in phase 2 clinical trials. We are at the stage of defining which human diseases can best be treated with these CB2R agonists. Mixed CB1R/CB2R-directed agonist preparations and numerous selective CB2R ligands are either on the market or under clinical development (reported in Table 6). Overall, more than 20 new molecular entities that activate CB2R have been investigated in humans for a wide range of indications. Structurally, they cover a huge chemical space including fatty acid derivatives, classic and nonclassic cannabinoids, as well as multiple diverse synthetic ligands, thus resulting also in the coverage of a broad range of physicochemical properties.
TABLE 6.
CB2R agonist that are launched or under active clinical development
| Drug | Chemical Class | Mode of Action | CB2R/CB1R in vitro Pharmacology | Indication(s) | Highest Phase of Development |
|---|---|---|---|---|---|
| Dronabinol (THC, Syndros, Marinol) | Classic cannabinoid | CB2R/CB1R agonist | pKi=8.16/8.48a | Appetite loss, CINV, anorexia, cancer pain | Launched |
| Nabilone (Cesamet) | Classic cannabinoid | CB2R/CB1R agonist | Ki=1.84/2.19 nM | CINV | Launched |
| Lenabasum (Ajulemic acid) |
Classic cannabinoid | CB2R/CB1R agonist | Ki=51/628 nM | CF, SLE, RA, systemic sclerosis, dermatomyositis | Phase 3 (systemic sclerosis since 2017; dermatomyositis since 2018) |
| Olorinab (ADP-371) | Tricyclic 3,5,5-fused pyrazole 3-carboxamide | CB2R agonist | EC50=6.2/>104 nMb | IBS-related pain, IBS with predominant constipation or diarrhea | Phase 2b (since 2017) |
| CMX-020 | Arachidonic acid analog | CB2R/CB1R agonist, TRPV1 agonist | Ki=150/21 nM | Pain, OA, DnP | Phase 2 (since 2015) |
| RG7774 | Triazolopyrimidine | CB2R agonist | EC50=1/>104 nMc | DR | Phase 2 (since 2020) |
| CNTX-6016 | Piperidine based ligand | CB2R agonist | — | Pain Np, DnP | Phase 2 (since 2020) |
| ART-27.13 (AZD-1940) |
Benzimidazole | CB2R/CB1R agonist | Ki=0.9/12 nM | Pain, cachexia, CINV | Phase 2 (since 2021) |
| EHP-101 (VCE-004.8) | Cannabidiol derivative | CB2R agonist, PPARγ agonist | Ki=170/>4x104 nM | MS, ScD | Phase 2 (since 2020) |
| NTRX-07 (MDA-7) | 2,3-Dihydro-1-benzofuran | CB2R agonist | Ki=422/>104 nM | AD, Np pain, cognitive disorder | Phase 1 (since 2019) |
AD, Alzheimer’s disease; CF, cystic fibrosis; CINV, chemotherapy induced nausea and vomiting; DnP, diabetic neuropathy; DR, diabetic retinopathy; IBS, irritable bowel syndrome; LGS, Lennox Gastaut syndrome; MS, multiple sclerosis; Np, neuropathic; OA, osteoarthritis; RA, rheumatoid arthritis; ScD, scleroderma; SLE, systemic lupus erythematosus.
aConsensus human CB2R binding affinity values from a multicentric collaborative profiling effort between multiple independent academic laboratories and industry (Soethoudt et al., 2017).
bFunctional activity in β-Arrestin-2 assay on human cannabinoid receptors (Han et al., 2017).
cFunctional activity in cAMP assay on human cannabinoid receptors (Grether, 2022).
Dronabinol, nabilone, and CBD, exerting their action through both CB1R and CB2R activation, have been introduced to the market. Oral THC is used for the treatment of anorexia, cachexia, and chemotherapy-induced emesis (Clarivate, 2022d). Buccal THC has been launched for cancer pain (https://adisinsight.springer.com/drugs/800027102). Other routes of administration, e.g., inhalable and sublingual formulations, are under exploration. Nabilone was launched for treating patients who suffer from chemotherapy-induced nausea and vomiting (https://adisinsight.springer.com/drugs/800025856). Clinical trials for the treatment of Parkinson’s disease and pain are in advanced stages. CBD, a nonclassic cannabinoid for which the main mode of action is still a matter of debate, is marketed for the treatment of infantile severe myoclonic epilepsy, Dravet and Lennox-Gastaut syndrome, and tuberous sclerosis (Clarivate, 2022b). As reported in Table 2, combinations of CBD and THC have been approved for treating MS-associated spasticity and pain management, while glioblastoma trials and studies targeting further indications are ongoing (Clarivate, 2022g; https://citeline.informa.com/drugs/details/175074). Nonpsychoactive dual CB1R/CB2R agonist Lenabasum (Zurier et al., 1998) is in phase 3 trials for the treatment of systemic sclerosis and dermatomyositis (https://adisinsight.springer.com/drugs/800007180; Corbus Pharmaceuticals, 2022). Most advanced selective CB2R agonists are the synthetic cannabinoids olorinab (https://adisinsight.springer.com/drugs/800039670) and RG7774 (Grether, 2022). Clinical focus of olorinab is on pain related to irritable bowel syndrome, as such or with predominant constipation or diarrhea. RG7774 aims to provide an oral treatment for patients suffering from diabetic retinopathy (Clarivate, 2022k). AA analog CMX-020 is studied in phase 2 trials for the treatment of pain, osteoarthritis, and diabetic neuropathy using both oral and intravenous formulations (https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=371547&isReview=true). Pain, in particular neuropathic pain, is also the focus of the selective synthetic CB2R agonists CNTX-6016, whose structure has not been yet disclosed (https://centrexion.com/science/pipeline/; https://clinicaltrials.gov/ct2/show/NCT04857957), and NTRX-07 (https://www.neurotherapia.com/research; Clarivate, 2022h). CNTX-6016 is in phase 2 and NTRX-07 in phase 1 trials. Dual CB1R/CB2R agonist ART-27.13 is in phase 2 trying to provide treatment options for cachexia and cancer-related anorexia (https://artelobio.com/pipeline/). CBD derivative EHP-101, which activates both PPARγ and CB2R, is aimed at MS and scleroderma patient populations in phase 2 clinical trials (EMA, 2022). Ten additional new chemical entities were investigated in phase 1 and 2 clinical trials for different pain indications (neuropathic, dental, pain associated with osteoarthritis of the knee), postherpetic neuralgia, pruritis, atopic dermatitis, stroke, traumatic brain injury, coronary artery bypass graft, and ocular hypertension (Brennecke et al., 2021). Dual CB1R/CB2R agonist TAK-937 was terminated due to a narrow safety margin (Clarivate, 2022m), S-777469 due to the lack of a pharmacological effect (Clarivate, 2022l; https://www.shionogi.com/content/dam/shionogi/global/investors/pdf/e_p090803.pdf), while for KN 387271 (Clarivate, 2022e) and PRS-211375 13 (Clarivate, 2022j) business reasons were reported.
It is clear from this summary that there are many therapeutic opportunities for both CB1R and CB2R agonists (Pacher and Kunos, 2013), yet the untoward effects of the CB1R at the CNS have limited the clinical progression of CB1R agonists that penetrate the blood-brain barrier and preclude their use in the nonhospitalized population. Tissue and cell-type selectivity for therapeutic responses is a challenge, as many cannabinoid and aminoalkylinodle agonists have been relegated to research rather than clinical use. Development of dual-target compounds that act by inhibiting CB1R-mediated side effects while simultaneously activating CB2R-mediated beneficial responses is also ongoing. Current development of peripherally restricted agonists and antagonists that fail to cross the blood-brain barrier will open avenues for treatment of diseases in organs outside of the brain. Research on “biased agonists” that promote cannabinoid receptor conformations that favor G-protein versus β-arrestin signaling is an approach that offers treatment opportunities if one pathway dominates in treatment while the alternative pathway is responsible for side effects. Researchers are screening for allosteric modulators based on the notion that their effects would be limited to only those receptors simultaneously engaged with an eCB agonist in the disease process. Thus, a positive allosteric modulator could potentiate responses if eCBs are understimulating the receptors. In contrast, a negative allosteric modulator would impart noncompetitive antagonism in a situation of excessive eCB tone. Research findings not discussed in the present review have recognized the presence of CB1R and CB2R receptor heterodimers with a wide range of GPCRs, as well as receptor complexes with other associated proteins. As these studies gain maturity, the understanding of the impact of such receptor combinations within the same cell can open avenues for novel therapeutic compounds. Although the present use of pCB and small molecule agonists is meeting unmet needs of many diseases, particularly those involving inflammation, the future for cannabinoid receptor pharmacotherapeutics must advance to agonists, antagonists, and modulators that exhibit greater selectivity to improve treatments and eliminate unwanted side effects.
III. Therapeutic Potential of Metabolic Enzymes of AEA
A. Enzymes of AEA Production
AEA is produced upon demand from the membrane phospholipid precursor N-arachidonoyl- phosphatidylethanolamine via two enzyme-mediated reactions (Fig. 3).
1. NAT and iNAT
The first step is the formation of NArPE, which occurs through N-acylation of phosphatidylethanolamine, mediated by Ca2+-dependent or independent N-acyl transferase (NAT and iNAT). It should be noted that the acyl donor is another phospholipid molecule, such as phosphatidylcholine, rather than acyl-CoA. The presence of N-arachidonoyl-phosphatidylethanolamine in mammalian tissues and the N-acyl-transferase activity responsible for its production were first reported in the late 1990s (Cadas, et al., 1997) and later molecularly identified as cytosolic phospholipase A2ε (cPLA2ε) (Ogura et al., 2016). Members of the phospholipase A and acyltransferase (PLAAT) family (Jin et al., 2007; Uyama et al., 2012) were identified as Ca2+-dependent and -independent NAT, respectively. cPLA2ε belongs to the cPLA2 family with a serine residue as catalytic nucleophile. Since for N-acylation of phosphatidylethanolamine cPLA2ε selectively abstracts an acyl chain from the sn-1 position of the glycerol backbone of glycerophospholipid, which is abundant in saturated and mono-unsaturated fatty acids rather than poly-unsaturated fatty acids like AA, N-arachidonoyl-phosphatidylethanolamine and AEA account for a small percentage of the N-acyl-phosphatidylethanolamine (NAPE) and fatty acid ethanolamides present in cells. The analysis of cPLA2ε-deficient mice revealed the central role of this enzyme in the accumulation of NAPEs and N-acylethanolamines in an imiquimod-induced psoriasis model (Liang et al., 2022), as well as in an ex vivo model of brain ischemia (Rahman et al., 2022). The NAPE-forming activity of cPLA2ε in skin was suggested to be protective against skin inflammation such as psoriasis by producing anti-inflammatory N-acylethanolamines. On the other hand, PLAAT enzymes compose a small protein family with a cysteine residue as catalytic nucleophile (Uyama et al., 2017). Among the five members (1–5) in humans, PLAAT1, 2, and 5 exhibit relatively high NAT activity over the coexisting PLA1/A2 activity (Uyama et al., 2012). Since without any cellular stimulus the NAT activity is easily detected in the cells where recombinant PLAAT is expressed, the role of PLAATs is presumed to maintain the basal levels of NAPEs and N-acylethanolamines in unstimulated cells. However, their contribution to the formation of NAPEs in vivo remains unclarified.
2. N-Acyl Phosphatidylethanolamine-Specific Phospholipase D
NAPE-phospholipase D (PLD) catalyzes the second step of AEA formation (Fig. 3). The enzyme releases AEA and other fatty acid ethanolamides from their corresponding NAPEs in a PLD-type hydrolytic reaction (Okamoto et al., 2004). However, NAPE-PLD is a member of the metallo-β-lactamase superfamily and shows no sequence similarity to classic PLDs converting phosphatidylcholine to phosphatidic acid. Multiple aspartic acid and histidine residues, highly conserved among the family members, are essential for catalytic activity, and metal analysis suggested the presence of Zn2+ coordinated by these amino acid residues (Wang et al., 2006). The crystal structure of human NAPE-PLD clarified the formation of homodimers adapted to associate with phospholipids and the presence of a binuclear Zn2+ center at the active site (Magotti et al., 2015). Purified recombinant NAPE-PLD selectively hydrolyzes NAPE among various phospholipids (Wang et al., 2006). However, the enzyme does not distinguish N-acyl species in NAPE, explaining why the composition of naturally occurring fatty acid ethanolamides is similar to the N-acyl composition of NAPEs. Recently, the role of NAPE-PLD in energy metabolism has received much attention. A common NAPE-PLD haplotype was reported to be protective against obesity (Wangensteen et al., 2011). Conditional knockout of adipocyte, intestinal, or hepatic NAPE-PLD showed the tendency to induce obesity (Geurts et al., 2015; Everard et al., 2019; Lefort et al., 2020). Moreover, LEI-401, the first brain-active NAPE-PLD inhibitor, was instrumental in demonstrating the distinctive role of NAPE-PLD in AEA biosynthesis in the brain (Mock et al., 2020). LEI-401 activated the hypothalamus-pituitary-adrenal axis and impaired fear extinction, thereby emulating the effect of a CB1R antagonist and suggesting the presence of an endogenous AEA tone controlling emotional behavior (Mock et al., 2020).
3. Alternative Pathways
The analysis of NAPE-PLD-deficient mice revealed the existence of alternative pathways for fatty acid ethanolamide biosynthesis in brain (Leung et al., 2006; Tsuboi et al., 2011) and peripheral tissues (Inoue et al., 2017). Among the proposed multistep pathways (Fig. 3), the route via lyso-NAPE and glycerophospho-N-acylethanolamines appears to be the most important, whereby either α/β-hydrolase domain protein 4 (Simon and Cravatt, 2006) or cPLA2γ (Guo et al., 2021) generates glycerophospho-N-acylethanolamines from NAPE via lyso-NAPE in two consecutive esterase reactions. The resultant compound is further hydrolyzed to generate N-acylethanolamines by glycerophosphodiesterase 1 (Simon and Cravatt, 2008) and 4 (Tsuboi et al., 2015; Rahman et al., 2016). The glycerophosphodiesterase family is composed of seven proteins (1–7) in mammals (Yanaka, 2007), and isoforms 4 and 7 also show lyso-PLD activity directly producing N-acylethanolamines from lyso-NAPE (Tsuboi et al., 2015; Rahman et al., 2016). It is not fully elucidated how much these alternative pathways contribute to the generation of AEA and other N-acylethanolamines in the tissues of wild-type mice. The physiologic significance in human tissues also remains unclarified.
B. Enzymes of N-Arachidonyl Ethanolamine Degradation
The major pathway of AEA degradation is hydrolysis to AA and ethanolamine, which is mediated by FAAH (Desarnaud et al., 1995; Hillard et al., 1995; Cravatt et al., 1996), two isoforms of which have been described: FAAH-1 and FAAH-2 (Wei et al., 2006). It should be noted that FAAH-2, sharing 20% sequence identity with FAAH-1, is expressed in humans but not in rodents (Wei et al., 2006), making its complete understanding difficult. Different from FAAH-1, which is found in the endoplasmic reticulum and the nucleus, FAAH-2 may be localized to lipid droplets (Kaczocha et al., 2010). NAAA and acid ceramidase also hydrolyze AEA, albeit with low activity (Ghidini et al., 2021; Tsuboi et al., 2021). In addition to hydrolytic degradation, AEA can be oxygenated by lipoxygenases (5-, 12-, 15-LOX), COX-2, or CYP450 (Fig. 4), all of which have been fully characterized as eicosanoid-generating oxygenase enzymes (Rouzer and Marnett, 2011; Fezza et al., 2014; Simard et al., 2022). The physiologic significance of these AEA oxygenation pathways remains unclear.
1. Fatty Acid Amide Hydrolase
FAAH-1, which is often referred to simply as FAAH, is widely distributed in mammalian tissues with high expression in liver, brain, and small intestine of rats (Katayama et al., 1997). The analysis of FAAH-1-deficient mice revealed increased endogenous AEA levels and hence the central role of FAAH-1 in AEA degradation (Cravatt et al., 2001). FAAH deletion reduced pain sensation, and when AEA was administered, FAAH-1-deficient mice exhibited intense hypomotility, antinociception, catalepsy, and hypothermia in a CB1R-dependent manner. Although FAAH-1 is highly active with AEA, the enzyme shows broad substrate specificity, hydrolyzing other fatty acid ethanolamides, N-acyl taurines, and primary fatty acid amides such as oleamide. FAAH-1 can also catalyze the reverse reaction in which AEA is formed from AA and ethanolamine. However, the equilibrium constant demonstrated the predominance of the hydrolytic action of AEA (Katayama et al., 1999). FAAH-1 is an integral membrane protein functioning as a serine hydrolase and belongs to the amidase signature family characterized by the Ser-Ser-Lys catalytic triad (McKinney and Cravatt, 2005). Rat FAAH was crystallized as a homodimer. In common with bacterial enzymes of the same family, the structure exhibits a core fold comprised of a twisted β-sheet consisting of 11 mixed strands surrounded by a number of α-helices (Bracey et al., 2002). Remarkably, the FAAH dimer is stabilized by the lipid bilayer and shows a higher enzymatic activity within membranes containing cholesterol (Dainese et al., 2014) according to allosteric kinetics (Dainese et al., 2020). Additionally, colocalization of cholesterol, AEA, and FAAH in mouse neuroblastoma cells suggests a mechanism by which cholesterol increases the substrate accessibility of FAAH (Dainese et al., 2014); yet, the pathophysiological implications of these findings remain to be understood. C385A polymorphism of the FAAH-1 gene (rs324420) results in the formation of P129T mutant, which is associated with the reduction of FAAH activity and cellular expression as well as increased risk for substance use disorders (Sipe et al., 2002). This polymorphism also affects susceptibility to various diseases (Hosseinzadeh Anvar and Ahmadalipour, 2023).
2. N-Acylethanolamine Acid Amide Hydrolase
NAAA is a lysosomal hydrolase (Tsuboi et al., 2007a; Ueda et al., 2010) that shows 33% amino acid identity with acid ceramidase, which hydrolyzes ceramide to sphingosine and fatty acid. Similar to other members of the N-terminal nucleophile hydrolase family (Linhorst and Lübke, 2022), NAAA is synthesized as a catalytically inactive precursor and then matured to heterodimer, consisting of α and β subunits, by post-translational autoproteolytic cleavage (Zhao et al., 2007). This reaction proceeds in vitro only at acidic pH, suggesting that the maturation occurs only after its migration to endosomes/lysosomes from the endoplasmic reticulum via the Golgi apparatus. The resultant N-terminal cysteine residue of the β subunit (Cys-126 in human NAAA, Cys-131 in rodents) functions as the catalytic nucleophile. Importantly, this cysteine residue is also indispensable for the autoproteolytic cleavage. The crystal structures of NAAA elucidated that autoproteolysis exposes the buried active site to enable catalysis (Gorelik et al., 2018). NAAA hydrolyzes various fatty acid ethanolamides in vitro, but its highest reactivity is for N-palmitoylethanolamine (Ghidini et al., 2021). The fact that NAAA is highly expressed in macrophages (Tsuboi et al., 2007b) and other immune cells (Ribeiro et al., 2015) suggests that this enzyme may regulate fatty acid ethanolamide levels at the site of inflammation. In fact, in dermatitis induced by treatment of mice with 2,4-dinitrofluorobenzene, NAAA-deficient mice showed elevated N-palmitoylethanolamine, but not N-oleoylethanolamine, levels in ear tissue relative to wild-type controls and exhibited a strong reduction in the inflammatory reaction (Sasso et al., 2018). Furthermore, NAAA deficiency in mice increased N-palmitoylethanolamine and AEA levels in bone marrow and macrophages and AEA levels in lungs (Xie et al., 2022).
C. Fatty Acid Amide Hydrolase Inhibitors
The first potent, selective, and systemically active FAAH inhibitor was the N-biphenylcarbamate derivative URB597, shown in Fig. 23 (Kathuria et al., 2003; Tarzia et al., 2003). This agent acts by forming a carbamoyl adduct with FAAH’s catalytic serine (Mileni et al., 2010) and exhibits robust anxiolytic-like and antidepressant-like properties, which depend on indirect CB1R activation by accumulated anandamide (Kathuria et al., 2003; Gobbi et al., 2005; Bortolato et al., 2007).
Fig. 23.

Chemical structures of representative inhibitors of FAAH.
Importantly, unlike direct-acting CB1R agonists such as THC, URB597 is not rewarding to nonhuman primates, suggesting a lack of abuse potential (Justinova et al., 2008). An exploration of its scaffold unexpectedly led to the identification of the first peripherally restricted FAAH inhibitor, URB937 (Fig. 23), which strongly attenuates pain-related responses in animal models (Clapper et al., 2010). The promising pharmacological profile of URB597 prompted efforts by both academe and industry to create more advanced inhibitors. Reviews of this considerable body of work are available (Tuo et al., 2017; Fazio et al., 2020; Piomelli and Mabou Tagne, 2022), but one especially significant chemical class, the piperidine/piperazine-ureas, should be mentioned here. High-throughput screening of a chemical library led scientists at Johnson & Johnson to discover JNJ-1661010 (Fig. 23), which inhibits human FAAH with nanomolar potency (IC50 = 33 nM) and through a covalent mechanism (Keith et al., 2008). Further optimization identified the compound JNJ-42165279, a slowly reversible FAAH inhibitor that was selected for clinical testing. Concomitant work at Pfizer produced several nanomolar piperidine/piperazine-urea covalent FAAH inhibitors (Ahn et al., 2007) and eventually led to PF-04457845 (Fig. 23), which was also moved to clinical development. There are several possible therapeutic indications for which FAAH inhibitors have been or are currently being tested, including anxiety disorders, substance use disorders, and pain.
Building on the observation that URB597 exerts profound anxiolytic-like and antidepressant-like effects in mice and rats, animal and human experiments have shown that AEA signaling at CB1R modulates the emotional response to stress via regulation of prefrontal cortical-amygdala circuits (Patel et al., 2017). For example, subjects carrying the loss-of-function faah gene polymorphism C385A (rs324420) display enhanced fronto-amygdalar connectivity and cued fear extinction (Dincheva et al., 2015). This conclusion was later confirmed by several other human experimental medicine studies. For instance, Paulus and coworkers found that JNJ-42165279 (100 mg) dampens amygdala activity during an emotion face-processing task, an effect that is associated positively with plasma AEA concentrations (Paulus et al., 2021). A lower dose of the drug (25 mg) was tested in a multicenter, placebo-controlled phase 2 trial in patients with social anxiety disorder. The study reported statistically detectable signs of efficacy, but the dosage was considered insufficient to fully inhibit FAAH (Schmidt et al., 2021). Additional clinical testing in anxiety and allied conditions is clearly warranted.
The impact of FAAH inhibitors on tobacco and cannabis use disorders exemplifies well the promise offered by these agents but also their complex actions. URB597 was shown to reduce nicotine reward and to prevent reinstatement of nicotine use in animal models (Justinova et al., 2015), an effect that was associated with reduced burst firing of dopamine neurons in the midbrain and dopamine release in the terminal field of such neurons (Melis et al., 2004). Unexpectedly, the effects of URB597 on nicotine reward were prevented by PPARα rather than CB1R blockade, leading to the suggestion that they were mediated by PPARα agonists, such as N-oleoylethanolamine and N-palmitoylethanolamine, rather than by AEA acting at CB1R. With regard to cannabis, a phase 2 clinical trial demonstrated that PF-04457845 is effective in reducing cannabis use and alleviating cannabis withdrawal symptoms in men (D’Souza et al., 2019).
There is strong preclinical evidence indicating that eCBs are critical regulators of pain sensation (for review, see Finn et al., 2021). The analgesic phenotype of individuals carrying loss-of-function FAAH mutations (C385A, faah-out) supports this conclusion (Habib et al., 2019), but the results of clinical trials have been disappointing (Huggins et al., 2012; Wagenlehner et al., 2017). Possible explanations for this discrepancy include species-specific differences, selection of inadequate clinical pain conditions, inconsistencies between preclinical and clinical study design, and lack of predictive validity of current animal models. Other pathologies where FAAH inhibitors might be clinically useful include chronic cough (Wortley et al., 2017) and urinary tract dysfunction (Wagenlehner et al., 2017). Overall, several FAAH inhibitors have been patented for their potential therapeutic use, as summarized in Table 7 (Fazio et al., 2020).
TABLE 7.
Potential therapeutic use of patented FAAH inhibitorsa
| Compound | Potential Therapeutic Use |
|---|---|
Oxazole Derivatives
|
Treatment of different types of pain: postoperative pain, chronic pain, cancer pain, cancer chemotherapy, neuralgia, nociception pain, inflammatory pain |
Urea Derivatives
|
Treatment of depression, analgesia, and cannabis use disorders |
Urea/Carbamate
|
Treatment of pain, inflammation, neuropathy, neurodegenerative diseases, anxiety, motor function disorder, infertility, eating disorders, THC dependence, metabolic disorders, movement disorders, chemotherapy-induced nausea and vomiting, and cancer |
ARN2508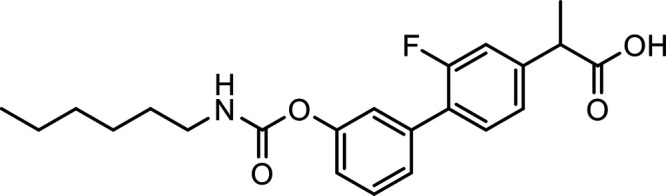
|
Treatment of intestinal inflammation where a pure FAAH inhibitor was weakly active and the pure COX inhibitor flurbiprofen aggravated inflammation Simultaneous blockade of FAAH and COX-1/COX-2 results in a combination of profound anti-inflammatory and tissue protective actions |
Oxazolyl-ketones [replacement of the phenyl hexyl group of OL-135 with a piperidine ring]
|
Treatment of anxiety, pain, sleep disorders, eating disorders, inflammation, or movement disorders (e.g., in multiple sclerosis) |
JNJ-42119779
|
Effective in the spinal nerve ligation (Chung) model of neuropathic pain |
JNJ-40413269
|
Effective in the rat spinal nerve ligation (Chung) model of neuropathic pain |
2,3,4-Tetrahydro-2,6-naphthyridines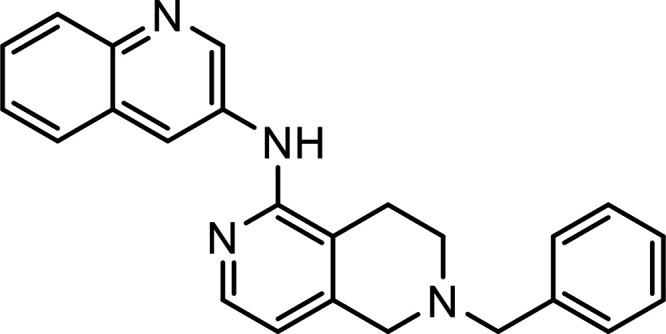
|
Treatment of pain, anxiety, depression, inflammation, cognitive disorders, weight and eating disorders, Parkinson’s disease, Alzheimer’s disease, spasticity, addiction, glaucoma |
aFor further details see Fazio et al. (2020).
Several compounds (URB597, PF-04457845, SSR411298, APD8477, V158866, BIA 10-2474, and JNJ-42165279) have also been tested in clinical trials (Table 8). Of note, the FAAH inhibitor BIA 10-2474 led to adverse neurologic side effects and the death of one healthy volunteer in a phase 1 clinical trial (Kerbrat et al., 2016). Since the other FAAH inhibitors tested in clinical trials did not elicit any adverse neurologic effects and BIA 10-2474 was shown to have multiple off-targets, inhibition of FAAH is considered to be safe.
TABLE 8.
Diseases/symptoms for treatment with FAAH inhibitors registered with ClinicalTrials.gova
| Generic Name Brand Name Class/Efficacy |
Completed Clinical Trials | Ongoing Clinical Trials |
|---|---|---|
| FAAH inhibitors | ||
| PF-04457845 | Tourette syndrome | Cannabis use disorder |
| Cannabis withdrawal | ||
| Fear conditioning | ||
| Acute pain | ||
| • Chronic pain | ||
| • Knee osteoarthritis | ||
| URB597 | Schizophrenia | |
| SSR411298 | Major depressive disorder Cancer pain |
|
| APD8477 | Peripheral neuropathic pain | |
| V158866 | Neuropathic pain | |
| JNJ-42165279 | Major depressive disorder Social anxiety disorder Autism |
aStudies with the status “not yet recruiting, recruiting,” “enrolling by invitation,” “active, not recruiting,” and “completed” were included in this table.
D. N-Acylethanolamine Acid Amide Hydrolase
The search for potent, selective, and systemically active NAAA inhibitors started in 2009 with the identification of the β-lactone derivative N-[(3S)-2-oxo-3-oxetanyl]-3-phenylpropanamide [(S)-OOPP] shown in Fig. 24, which inhibits rat NAAA with submicromolar potency (IC50 = 420 nM on rat NAAA) via a noncompetitive and partially reversible mechanism (Solorzano et al., 2009).
Fig. 24.
Chemical structures of representative inhibitors of NAAA.
Due to the opening of its β-lactone ring, (S)-OOPP undergoes rapid hydrolytic deactivation, which makes it unsuitable for systemic administration. The compound has, however, two interesting properties (Solorzano et al., 2009). First, it is selective for NAAA over other functionally (FAAH) or structurally (acid ceramidase) related lipid amidases. Second, its inhibitory effect is stereospecific, allowing researchers to leverage the enantiomer (R)-OOPP (IC50 = 6 μM) as a negative control in pharmacological experiments. These experiments showed that incubation with S-OOPP increases N-palmitoylethanolamine levels in RAW264.7 macrophages stimulated with bacterial endotoxin, whereas (R)-OOPP does not (Solorzano et al., 2009). Moreover, subdermal application of (S)-OOPP, but not (R)-OOPP, blocked carrageenan-induced neutrophil infiltration and plasma extravasation in mice, two effects that are prevented by genetic PPARα ablation and are mimicked by administration of PPARα agonists. These findings identified NAAA as a druggable target for the treatment of inflammation and encouraged efforts to discover inhibitors with greater potency and stability. The first notable outcome of this search was another β-lactone derivative, ARN077 (also known as URB913), in which the amide group of (S)-OOPP is replaced by a carbamate moiety and a syn-methyl group is introduced at the β position of the lactone ring (Fig. 24).
Compared with (S)-OOPP, ARN077 exhibits better chemical stability and greater NAAA inhibitory potency (IC50 = 50 nM on rat NAAA) (Ponzano et al., 2013). ARN077 was found to be selective for NAAA when assessed in a broad panel of potential off-targets. Importantly, topical application of ARN077 on the mouse or rat skin attenuated inflammation and pain-related responses (Sasso et al., 2013, 2018). Despite these significant steps forward, the low chemical and enzymatic stability of the β-lactone ring remained a challenge to the systemic use of ARN077 and other chemically related inhibitors. Efforts were thus undertaken to overcome this problem, which led to the discovery of several new classes of NAAA inhibitors, including β-lactam derivatives (e.g., ARN726) (Ribeiro et al., 2015), isothiocyanate derivatives (e.g., AM9023) (Alhouayek et al., 2015), azetidine-nitrile derivatives (Malamas et al., 2020), and benzothiazole derivatives (e.g., ARN19702) (Migliore et al., 2016), shown in Fig. 24. The discovery, inhibitory properties, and mechanism of action of these agents were recently reviewed (Piomelli et al., 2020). Thus far, three main therapeutic indications have emerged for NAAA inhibitors: inflammation, pain, and neuroinflammation/neurodegeneration.
A chemically diverse set of NAAA inhibitors exhibit notable anti-inflammatory properties in animal models. For example, topical application of the β-lactone ARN077 was shown to suppress skin inflammation elicited by exposure to UV B-radiation in rats or phorbol ester in mice (Sasso et al., 2013). The compound also attenuated itch and skin inflammation in sensitized mice challenged with 2,4-dinitrofluorobenzene (Sasso et al., 2018). Confirming that ARN077 acts by protecting N-palmitoylethanolamine from NAAA-mediated hydrolysis, the effects of ARN077 were accompanied by restoration of normal N-palmitoylethanolamine content in inflamed skin tissue and were dependent on PPARα activation (Sasso et al., 2013, 2018). The striking effects produced by ARN077 on critical mediators of the allergic response (e.g., interleukin 4 and immunoglobulin E) (Sasso et al., 2018) and the efficacy demonstrated by N-palmitoylethanolamine as an adjuvant treatment of eczema (Eberlein et al., 2008) encourage further evaluation of NAAA as a target for the treatment of the atopic diathesis, a disease cluster that includes atopic dermatitis, bronchial asthma, hay fever, and allergic rhinitis. Other inflammatory diseases in which NAAA inhibitor might find clinical use, as suggested by animal model studies, include osteoarthritis (Bonezzi et al., 2016; Zhou et al., 2019b) and colitis (Alhouayek et al., 2015; Xiu et al., 2020).
In addition to inflammation, NAAA inhibitors may also be effective in the treatment of pain and neuroinflammation/neurodegeneration. For example, the systemically active NAAA inhibitor ARN19702 exhibited a broad antinociceptive profile in mouse models of acute and chronic pain (Fotio et al., 2021a) and alleviated symptoms of neuroinflammation in mouse models of multiple sclerosis (Migliore et al., 2016) and Parkinson’s disease (Palese et al., 2022). Similarly, the topically active β-lactone derivative ARN077 alleviated hypersensitivity in mouse and rat models of neuropathic pain (Sasso et al., 2013), while the oxazolidinone imide derivative F96 (Fig. 24) attenuated acetic acid-induced writhing and tactile allodynia evoked by sciatic nerve injury in mice (Yang et al., 2015). No NAAA-targeting compound has yet reached clinical trials.
IV. Therapeutic Potential of Metabolic Enzymes of 2-AG
A. Metabolism of 2-Arachidonoylglycerol
The endocannabinoid 2-AG can be produced via two distinct biologic pathways. The metabolic pathway uses sn-2 arachidonoyl-containing triglycerides, which are hydrolyzed by hormone-sensitive lipase, carboxyl esterases, or other lipases toward sn-2 arachidonoyl DAGs (Baggelaar et al., 2018). The signaling pathway utilizes phosphatidylinositol-4,5-bisphosphate, which is converted by PLC β in the CNS or PLCγ2 in immune cells. The PLC enzymes are activated by Ca2+ ions and integrate Gq protein-coupled receptor activation and extracellular Ca2+ influx via ionotropic receptors and voltage-gated Ca2+-channels, thereby also producing DAGs. The diglycerides activate protein kinase C and are the central precursors for the production of 2-AG in both the metabolic and signaling pathways. The sn-1 acyl group from DAGs is predominantly hydrolyzed by two isoenzymes, diacylglycerol lipase-α and -β (DAGLα and DAGLβ, also termed diacylglyceride lipases), which produce 2-AG and other sn-2 acylglycerides. The DAGLs were discovered by Doherty’s group in 2003 (Bisogno et al., 2003), and the generation of genetically modified animals lacking daglα and daglß, the genes encoding the DAGL proteins, demonstrated that these enzymes are essential for 2-AG production in the brain (Gao et al., 2010; Tanimura et al., 2010). Of note, the DAGLs also terminate protein kinase C signaling by hydrolyzing DAGs; thus, these enzymes are an important hub to connect lipid and kinase signaling.
Termination of 2-AG signaling at CB1R or CB2R occurs through hydrolysis of the ester bond, thereby generating AA and glycerol. MAGL (also termed monoglyceride lipase) is the main enzyme responsible for the inactivation of 2-AG in the brain (Dinh et al., 2002), whereas α/β-hydrolase domain protein 6 and 12 may play a role in 2-AG hydrolysis in specific cell types (Marrs et al., 2010; Blankman et al., 2007). In various tissues, including the brain, 2-AG is responsible for the main supply of AA, which is the central precursor for proinflammatory signaling lipids, such as the prostaglandins (Nomura et al., 2010). Thus, MAGL is a central node that connects endocannabinoid and eicosanoid signaling. Modulators of 2-AG metabolism are listed in Table 9, and in the next sections their therapeutic potential is described. For an extensive review on chemical probes of the endocannabinoid system, see also Punt et al. (2023).
TABLE 9.
Modulators of 2-AG metabolism
| Name | Target | Phase | Structure | Reference |
|---|---|---|---|---|
| LEI-105 | DAGL | Preclinical |
|
Baggelaar et al., 2015 |
| DO34 | DAGL | Preclinical |
|
Ogasawara et al., 2016 |
| DH376 | DAGL | Preclinical |
|
Ogasawara et al., 2016 |
| DO53 | Negative control compound | Preclinical |
|
Ogasawara et al., 2016 |
| KT109 | DAGL | Preclinical |
|
Hsu et al., 2012 |
| JZL184 | MAGL | Preclinical |
|
Long et al., 2009 |
| MJN110 | MAGL | Preclinical |
|
Niphakis et al., 2013 |
| ABX-1431 (Lu-AG06466) | MAGL | Phase 2 |
|
B. Therapeutic Potential of Diacylglycerol Lipase-α
DAGLα belongs to the family of serine hydrolases, and is responsible for the production of 2-AG in the CNS (Bisogno et al., 2003), where it is primarily found in the dendrites and soma of neurons and to a lower extent in astrocytes, but not in microglial cells. DAGLα is expressed in various brain regions, such as cortex, hippocampus, cerebellum, and striatum, and its activity is highest in the cerebellum (Baggelaar et al., 2017). DAGLα is a 120 kDa integral plasma membrane protein with multiple domains (Fig. 25) and has four transmembrane helices followed by a lipase domain, which contains the catalytic triad Ser, His, Asp (Bisogno et al., 2003).
Fig. 25.

(A) Structured part of the AlphaFold model for human DAGLα, residues 1-681; red: transmembrane domain, blue: catalytic domain, green: regulatory loop. (B) Unstructured tail region from the AlphaFold model, residues 682–1042 highlighting potential phosphorylation sites, as discussed in the text, and Homer binding domain. (C) Schematic representation with highlighted regions and relevant serines shown.
DAGLα produces 2-AG on demand as a retrograde messenger upon depolarization of the post-synaptic neuron or by stimulation of Gq/11-coupled metabotropic receptors, with or without activation of ionotropic receptors at both excitatory and inhibitory synapses (Gao et al., 2010; Tanimura et al., 2010). Animals with constitutive genetic disruption of DAGLα show a variety of neurologic phenotypes, including impaired synaptic transmission, disturbed memory and learning, compromised adult neurogenesis (Gao et al., 2010), hypophagia (Powell et al., 2015), enhanced anxiety and fear responses (Shonesy et al., 2014; Jenniches et al., 2016), and susceptibility to spontaneous seizures (Powell et al., 2015). Multiple selective pharmacological tools have been developed to modulate DAGLα (as well as DAGLβ) activity in an acute and temporary manner (Baggelaar et al., 2018; Punt et al., 2023). LEI-105, DO34, and DH376 are currently widely used DAGL inhibitors to study the involvement of these enzymes in physiologic processes (Baggelaar et al., 2015; Ogasawara et al., 2016). For example, the same inhibitors were instrumental, in conjunction with genetic models, to unequivocally demonstrate that 2-AG production is “on demand,” i.e., when and where needed upon stimuli during short-term synaptic plasticity, such as depolarization-induced suppression of inhibition or excitation (DSE) in hippocampal and cerebellar slices (Baggelaar et al., 2015; Ogasawara et al., 2016). DAGL inhibitors also contributed to our understanding of the role of 2-AG in cocaine seeking (McReynolds et al., 2018), alcohol addiction, food intake (Deng et al., 2017), neuroinflammation (Ogasawara et al., 2016), anxiety and stress (Bluett et al., 2017), learning and memory (Schurman et al., 2019), pain sensation (Wilkerson et al., 2017), and voluntary movement (Farrell et al., 2021). It should be noted that DO34 and DH376, but not LEI-105, also inhibited other serine hydrolases suABHD6. Thus, it is advisable to include DO53 as a negative control in the experimental design when using DO34 or DH376 (Deng et al., 2017).
DAGLα is very well conserved throughout evolution. Human DAGLα has 97% homology with its mouse ortholog, whereas it has only 79% homology to DAGLβ. DAGLα has a long unstructured C-terminal tail, which contains many phosphorylation sites that regulate its activity and subcellular localization through protein-protein interactions. It has been shown that CaMKII phosphorylates Ser782 and Ser808, thereby reducing the enzyme activity (Shonesy et al., 2013). On the other hand, protein kinase A, which is activated by cAMP, has been shown to phosphorylate multiple sites in the C-terminus of DAGLα, including Ser798, thereby activating the enzyme (Shonesy et al., 2020). It has been suggested that the opposing actions of protein kinase A and CaMKII on DAGLα activity may be important in setting the level of tonic 2-AG signaling. Of note, cAMP-induced phosphorylation of Ser738 of DAGLα has been shown to enhance the interaction of DAGLα with ankyrin-G, a scaffolding protein in dendritic spines (Yoon et al., 2021). This led to increased spine size and decreased DAGLα surface diffusion. Repeated strong excitatory dendritic spine stimulation resulted in a feedback signal that promoted the growth of an inhibitory γ-aminobutyric acid bouton onto the same dendrite in a DAGL-dependent manner. The C-terminus also contains the consensus motif PPxxF, needed to bind the coiled-coil domain of Homer proteins, which are adapter proteins that localize DAGLα close to the post-synaptic density in the vicinity of metabotropic glutamate receptor 5 (Jung et al., 2007). Interestingly, the surface localization of DAGLα was shown to be a dynamic process controlled by protein kinase C. DAGLα colocalized with β-tubulin and cycled between the plasma membrane and endosomal compartments via EEA1- and Rab5-positive early endosomes in a clathrin-independent pathway (Zhou et al., 2016). This process could be disrupted by protein kinase C inhibitors but not by protein kinase A inhibitors. In a mouse model of Fragile X syndrome, which is the most commonly known genetic cause of autism, aberrant subcellular localization of DAGLα was found to cause a disruption in glutamatergic signaling, thereby impairing long-term depression (Jung et al., 2012). Recently, the first clinical evidence was presented that a daglα variant, which led to a disrupted cellular localization of the protein, was connected to a human genetic disorder. Nine children from eight families with heterozygous de novo truncating variants in the last exon of DAGLα exhibited developmental delay, ataxia, and complex oculomotor abnormalities (Bainbridge et al., 2022). Altogether, these observations demonstrate that the post-translational regulation of DAGLα activity and its subcellular localization enable a tight spatiotemporal control on 2-AG-dependent synaptic transmission. Disturbances in the subcellular localization of DAGLα and its activity result in abnormal neurotransmission and neurologic disorders. Unfortunately, pharmacological inhibition of DAGLα in the CNS is unlikely to be of therapeutic value due to on-target toxicity.
C. Therapeutic Potential of Diacylglycerol Lipase-β
DAGLβ is the main enzyme responsible for the production of 2-AG in immune cells, including microglia that are the brain resident macrophages. DAGLβ is a 70 kDa multidomain, integral membrane serine hydrolase that lacks the unstructured C-terminal tail observed in DAGLα. This suggests that the activity and subcellular localization of DAGLβ is differently regulated. DAGLβ has a similar substrate preference as DAGLα, but it is also capable of hydrolyzing polyunsaturated fatty acid-specific triacylglycerides (Shin et al., 2020). DAGLβ knockout mice show 50% reduction in 2-AG levels in the brain, whereas in the liver, a > 90% reduction was observed (Gao et al., 2010). DAGLβ is not involved in the regulation of depolarization-induced suppression of inhibition or DSE in hippocampal or cerebellar slices (Gao et al., 2010), and in the developing brain, it is detected in the axonal growth cone of neurons (Bisogno et al., 2003). DAGLβ is transported to the cone via the adaptor protein complex AP-4 (Davies et al., 2022). A patient deficient in AP-4 was shown to accumulate DAGLβ in the trans-Golgi network of cells, and AP-4 knockout mice had reduced eCB levels in the brain (Davies et al., 2022). Recently, a specific subset of nigral dopaminergic neurons in the adult brain was found to express DAGLβ. This expression was implicated in the inhibition of γ-aminobutyric acid release from dorsal striatal spiny projection neurons and is supposed to be involved in locomotor skill learning across sessions (Liu et al., 2022). Multiple homozygous loss-of-function mutations in DAGLβ were linked to sporadic, early-onset autosomal recessive Parkinsonism in Chinese families (Liu et al., 2022). PLCγ2, for which activating mutations are associated with autoinflammatory disorders and Alzheimer’s disease, has recently been shown to serve as the principal enzyme providing the DAG pool for DAGLβ-MAGL axis in human innate immune cells and microglia (Jing et al., 2021). Mouse microglia lacking PLCγ2 displayed a suppressed endocannabinoid-eicosanoid cross-talk and an impaired in vivo inflammatory response to lipopolysaccharide that led to reduced CD68-expression but not to release of proinflammatory cytokines. These findings extend the previous observations that genetic and pharmacological inhibition of DAGLβ exerts anti-inflammatory properties in mouse macrophages and microglia (Hsu et al., 2012; Viader et al., 2016). Overall, it was suggested that selective inhibitors of DAGLβ (and MAGL) may be therapeutically of interest for immune pathologies caused by activation of PLCγ2.
Currently, no selective DAGLβ inhibitors are available. KT-109 was originally reported as a selective DAGLβ inhibitor (Hsu et al., 2012), which displayed analgesic efficacy in an inflammatory and neuropathic pain model (Wilkerson et al., 2016; Shin et al., 2018), as well as in a sickle cell disease model (Khasabova et al., 2023). However, it should be noted that KT109 also inhibits DAGLα to the same extent as DAGLβ (Deng et al., 2017); thus care should be taken in the interpretation of the effects of this compound. As noted earlier, nonselective dual DAGL inhibitors, such as DO34 and DH376, have anti-neuroinflammatory properties (Ogasawara et al., 2016; Viader et al., 2016). They reduced production of proinflammatory cytokines and prostaglandins in microglia and impaired lipopolysaccharide-induced hypothermia in mice. In summary, selective compounds are still required to test the therapeutic potential of DAGLβ inhibition in neuroinflammatory diseases and inflammatory pain.
D. Therapeutic Potential of Monoacylglycerol Lipase
MAGL is a membrane-associated serine hydrolase, which was cloned in 1997, and consists of two tissue-specific splice-variants with a molecular weight of 33 kDa and 36 kDa. It has the typical catalytic triade Ser122, Asp239, and His269 and uses monoacylglycerols with different chain length and saturation, including 2-AG, as a substrate (Dinh et al., 2002). Oxidation of two noncatalytic cysteines (C201 and C208) reduces its enzymatic activity (Dotsey et al., 2015). MAGL is abundantly expressed in various tissues (e.g., brain, lung, liver, spleen, kidney, heart, and intestines) and is active in different brain regions including hippocampus, cerebellum, cortex, and striatum (Baggelaar et al., 2017). MAGL is found in neurons and astrocytes, and to a lesser extent in microglia (Viader et al., 2016), and notably is localized at the presynaptic site along with the CB1 receptor and opposed to DAGLα. This lipase terminates the retrograde eCB signaling mediated by 2-AG, and indeed mice lacking the mgll gene that encodes for MAGL show robust elevations of 2-AG in the brain and less pronounced elevations in liver, spleen, and thymus (Long et al., 2009). This observation suggests that other esterases may participate in the hydrolysis of 2-AG at the periphery. MAGL knockout mice also have significantly reduced AA levels in their brain, which indicates that the DAGL-MAGL axis is responsible for the pool of free AA in the brain (Nomura et al., 2010). Furthermore, MAGL knockout animals have a desensitized CB1R, and show impaired eCB-dependent synaptic plasticity and physical dependence (Schlosburg et al., 2010).
Several in vivo active MAGL inhibitors, including JZL184 and MJN110, have been described in the literature (Long et al., 2009; Niphakis et al., 2013). Together with the MAGL knockout animals, these inhibitors have been instrumental in studies of the therapeutic potential of MAGL inhibition in a broad range of diseases, spanning from cancer (Nomura et al., 2011), Parkinson’s disease (Nomura et al., 2010), Alzheimer’s disease (Chen et al., 2012), and MS (Hernández-Torres et al., 2014) to inflammatory and neuropathic pain (Hohmann et al., 2005; Kinsey et al., 2009), acute liver injury (Cao et al., 2013), and anxiety and depression (Bluett et al., 2017; Zhang et al., 2015a). For recent reviews, see Gil-Ordóñez et al. (2018), Deng and Li (2020), and Van Egmond et al. (2021). Of note, chronic, high dosing of MAGL inhibitors caused desensitization and downregulation of CB1R, and behavioral tolerance to CB1R agonists. A therapeutic window for anti-nociceptive efficacy without CB1R desensitization was observed upon acute and chronic low dosing. In this respect, MAGL inhibition may have therapeutic potential for treating inflammatory and neuropathic pain, as well as neurodegenerative diseases accompanied by neuroinflammation like MS, Alzheimer’s, and Parkinson’s diseases. Several pharmaceutical companies, including Johnson & Johnson, Lundbeck, Takeda Pharmaceuticals, Pfizer, and Hoffman-LaRoche, have filed patents describing a diverse range of chemotypes of MAGL inhibitors (Bononi et al., 2021). Among these compounds, the covalent, irreversible MAGL inhibitor Lu-AG06466, developed by Lundbeck (formerly ABX-1431 from Abide Therapeutics), is the most advanced experimental drug. It has been reported that Lu-AG06466 exerts adverse effects in the CNS and appeared ineffective in a phase 2 clinical trial for Tourette syndrome (Müller-Vahl et al., 2021, 2022), yet this compound is currently being investigated in phase 2 trials for other indications, such as post-traumatic stress disorder and spasticity in multiple sclerosis. Clinical trials for Lu-AG06466 listed in Clinical Trials.gov at the date of this review are shown in Table 10.
TABLE 10.
Clinical trials for MAGL inhibitor Lu-AG06466 listed in ClinicalTrials.gov
| Identifier | Status | Condition | Title |
|---|---|---|---|
| NCT04597450 | Ongoing | PTSD | Lu-AG06466 in Participants With Post Traumatic Stress Disorder |
| NCT04990219 | Ongoing | Multiple sclerosis | A Study of Lu-AG06466 for the Treatment of Spasticity in Participants With Multiple Sclerosis |
| NCT05028673 | Completed | Healthy | A Study to Evaluate a New Tablet Formulation of Lu-AG06466 in Healthy Participants |
| NCT04713254 | Completed | Healthy | Drug Drug Interaction Study With Lu-AG06466 in Young Healthy Men |
| NCT04405323 | Completed | Healthy | Study That Evaluates the Effect of CYP3A4 Inhibition on Lu-AG06466 in Healthy Men and Women |
| NCT04419636 | Completed | Healthy | Binding of Lu-AG06466 in the Brain in Healthy Men |
| NCT05081518 | Terminated | Focal epilepsy | A Study of Lu-AG06466 in Participants With Treatment Resistant Focal Epilepsy |
| NCT05201092 | Completed | Healthy | A Study Investigating Lu-AG06466 in Healthy Men |
| NCT05219838 | Completed | Healthy | Binding and Effects of Lu-AG06466 in the Brain of Healthy Men |
| NCT04974359 | Terminated | Fibromyalgia | A Study to Evaluate Lu-AG06466 in Participants With Fibromyalgia |
| NCT05177029 | Terminated | Healthy | Safety and Tolerability Study of Lu-AG06466 in Healthy Young Japanese and Caucasian Participants |
Overall, it is hypothesized that reversible MAGL inhibitors may avoid some of the adverse effects observed with covalent, irreversible inhibitors (Van Egmond et al., 2021). However, another strategy to avoid CNS-mediated side effects could be to generate peripherally restricted MAGL inhibitors for the potential treatment of cancer, tissue ischemic-reperfusion injury, and/or antinociception.
V. Therapeutic Potential of Transmembrane, Intracellular, and Extracellular Transporters
While translational efforts toward the development of ECS modulators have been primarily dedicated to eCB degradation inhibitors, in particular FAAH and MAGL blockers (Blankman and Cravatt, 2013; Fowler, 2021; van Egmond et al., 2021), translating research on inhibitors of eCB cellular uptake or cellular trafficking remains slow. The development of such transport inhibitors has been convoluted by the fact that extra- and intracellular eCB-binding proteins are promiscuous, as well as by the lack of a concrete target responsible for plasma membrane transport, whose identity remains elusive. Nevertheless, RT126, a FABP inhibitor that competes with eCB binding for FABP4 and FABP5, and SYT510, a selective eCB reuptake inhibitor (SERI) which increases extracellular eCB levels in the brain by targeting the putative eCB membrane transporter, are under development by the pharmaceutical industry (https://ir.artelobio.com/news-events/press-releases/detail/90/artelo-biosciences-reports-positive-pre-clinical-results, https://www.synendos.com). Unlike active cellular transport mechanisms that are energy-driven, the lipophilic eCBs seem to traffic between membranes and across aqueous barriers through interactions with binding proteins and pass the plasma membrane by energy-independent mechanisms (Fig. 7). Among these, facilitated diffusion is influenced by both the interaction of eCBs with extra- and intracellular binding proteins and their metabolic enzymes. The measurement of facilitated diffusion and plasma membrane lipid transport is challenging and demands special phenotypic assays not easily accessible for routine screening (Oddi et al., 2010; Fowler, 2013; Rau et al., 2016; Reynoso-Moreno et al., 2023). A major challenge has been to differentiate FAAH and AEA uptake inhibitors as these processes are intrinsically coupled (Fowler et al., 2004; Vandevoorde and Fowler, 2005; Hillard et al., 2007). Therefore, only recently selective and potent eCB cellular uptake inhibitors have been developed (Chicca et al., 2017).
In 2009, the identification of intracellular carrier proteins, primarily FABPs, and lipid droplets as potential sequestration domains for AEA provided a new perspective in AEA transport research (Oddi et al., 2008, 2009; Kaczocha et al., 2009). FABPs facilitate the spatial organization of eCBs into domains and enable the trafficking between plasma and intracellular membranes. In this section, an update on previous reviews on the topic (Fowler, 2013; Nicolussi and Gertsch, 2015; Reynoso-Moreno and Gertsch, 2021) is provided, focusing on the molecular pharmacology and possible implications for therapeutic intervention of using the diverse eCB transport inhibitors shown in Fig. 26. Since such inhibitors show CB1R/CB2R-dependent indirect cannabimimetic effects like analgesia, anti-inflammatory, and anxiolytic effects, they constitute a new class of pharmacological inhibitors that indirectly activate the ECS, showing a differential effect on the system compared with FAAH and MAGL inhibitors.
Fig. 26.

Model of eCB membrane transport and trafficking showing druggable targets. Molecular pharmacology and possible implications for therapeutic intervention of using diverse eCB transport inhibitors are shown. See text for details and abbreviations.
A. Endocannabinoid Trafficking and Transport
Although 2-AG is the major eCB in tissues such as the brain and is generally more soluble in water than AEA (1400 ng/ml versus 250 ng/ml, respectively; see Tetko et al., 2005), most research on eCB transporters has been carried out on AEA. Intriguingly, almost 30 years after the identification of AEA in porcine brain (Devane et al., 1992b), the mechanisms of eCB membrane transport (i.e., release into the extracellular space and cellular reuptake) remain only partially understood. However, different hypothetical models have been proposed and reviewed for AEA uptake and trafficking (Felder et al., 2006; Yates and Barker, 2009; Nicolussi and Gertsch, 2015), as shown in Fig. 7. The currently the best substantiated model of facilitated diffusion is discussed here in the context of the emerging specific pharmacological modulators.
In the 1990s, the first reports on the cellular uptake of AEA designated a temperature- and time-dependent transport, which was linked to the enzymatic hydrolysis by FAAH in C6 glioma cells, N18TG2 neuroblastoma cells, and primary neuronal cells (Deutsch and Chin, 1993; Di Marzo et al., 1994). This cellular uptake process of AEA was rapid (t1/2 = 2.5 minutes), saturable, and, importantly, did not compete with closely related N-acylethanolamines such as N-stearoylethanolamine, N-linoleoylethanolamine, or N-palmitoylethanolamine, shown in Table 3 (Di Marzo et al., 1994). Since all N-acylethanolamines compete for FAAH hydrolysis, being ideal substrates for this enzyme, the fact that they showed no competition for cellular AEA uptake clearly suggested a mechanism independent of AEA metabolism (Chicca et al., 2012).
The early investigations in the 1990s suggested a carrier-mediated uptake process for AEA that was not dependent on either ATP or coupled to ion (Na+, Cl-, H+) gradients (Beltramo et al., 1997; Hillard et al., 1997; Hillard and Jarrahian, 2000). The transport process of AEA displayed high-affinity Michaelis-Menten constants in astrocytes (Km = 0.3 µM), cortical neurons (Km = 1.2 µM), and human neuroblastoma CHP100 cells (Km = 0.2 µM) (Beltramo et al., 1997; Maccarrone et al., 1998), with values comparable to those obtained with the transporters of serotonin (Km = 0.3–0.5 µM), dopamine (Km = 0.9–1.2 µM), and noradrenaline (Km = 0.4 µM) (Masson et al., 1999; Piomelli, 2003). Among more than 25 cell lines, and considering different assay protocols and confounding factors such as sticking of lipids to plastic and vials, the range of the apparent Km values for AEA uptake diverges dramatically, from 0.1 µM to 45 µM (Felder et al., 2006; Oddi et al., 2010). Although different routes of AEA catabolism exist (Fig. 4), which in principle can influence AEA cellular uptake, their contribution seems insignificant compared with that of FAAH. In an experiment on [3H]AEA uptake competition in U937 cells, different eCBs congeners (AEA, 2-AGE, O-AEA, and NADA, shown in Table 3) competed with [3H]AEA uptake, suggesting that a common cellular membrane uptake mechanism seemingly competes for one target related to cellular eCB uptake (Chicca et al., 2012).
Albumin and Hsp70 have been identified as cytosolic AEA-binding proteins in mouse skin keratinocytes using proteomics and functional assays (Oddi et al., 2009). Another candidate in the list of intracellular carrier proteins for AEA is the reported FAAH-like AEA transporter (FLAT) (Fu et al., 2011). The latter was proposed to be a partially cytosolic, catalytically silent variant of the AEA-degrading enzyme FAAH. The role and existence of FLAT as an AEA transporter were subsequently questioned, as no expression in either mouse brain, spinal cord, or dorsal root ganglia could be detected by independent groups (Leung et al., 2013; Fowler, 2014). Furthermore, a certain enzymatic activity could still be detected in artificial FLAT-transfected HeLa cells (Leung et al., 2013). On the other hand, an inhibitor of FLAT (ARN272) showed promising indirect cannabimimetic effects in a mouse model of nausea and vomiting (O’Brien et al., 2013).
In a docking study, the sterol carrier protein 2 (SCP2) was shown to be yet another potential eCB carrier protein (Liedhegner et al., 2014). Although an increase of AEA accumulation could be detected in SCP2-transfected HEK-293 cells, competition experiments with AM404 and 2-AG did not show a significant difference in their IC50 values between SCP2-expressing and wild-type cells. It was concluded that SCP2 is a low affinity binding protein for AEA and that it might facilitate AEA cellular uptake to a minor degree. A fluorescent probe displacement assay was developed to screen for SCP2 inhibitors, which might help to elucidate the role of SCP2 in eCB transport. Using this assay, the binding affinities of AEA (Ki = 0.68 ± 0.05 μM) and 2-AG (Ki = 0.37 ± 0.02 μM) to SCP2 were calculated (Hillard et al., 2017). The binding affinities of a library of previously reported SCP2 inhibitors were tested along with a new series of analogs, where SCPI-1 was the most potent probe with a Ki = 1.0 ± 0.1 µM (Hillard et al., 2017). SCP-2/SCP-x gene ablation in FABP1 null (LKO) mice antagonized the impact of LKO and high-fat diet on brain AA and, subsequently, on eCB levels, suggesting that both FABP1 and SCP-2 directly or indirectly participate in regulating the ECS (Martin et al., 2019). In principle, any protein with hydrophobic surfaces/cavities may serve as an acceptor for lipids like AEA and other eCBs. This is confirmed by the recent crystal structure of cellular retinol-binding protein in complex with 2-AG (Lee et al., 2020). Currently, the pharmacological competition of eCBs at extracellular binding proteins like albumin, Hsp70, SCP2, or extracellular FABPs by synthetic ligands has not been studied in sufficient detail to allow conclusions regarding their druggability; i.e., it remains unclear whether competing for extracellular AEA protein binding would exert robust cannabimimetic effects, as well as diverse CB1R/CB2R-dependent pharmacological effects. Therefore, the current focus is on plasma membrane-associated and intracellular processes. Notably, the involvement of FABPs in the transport of eCBs was suggested already in the context of intracellular PPAR activation (Sun et al., 2008). Consequently, FABP5 and FABP7 were shown to mediate AEA intracellular transport from the plasma membrane to FAAH in COS-7-FAAH-eGFP and N18TG2 neuroblastoma cells (Kaczocha et al., 2009).
As shown in Fig. 1, membrane-derived AEA and 2-AG can initiate cellular signaling at both extracellularly accessible (e.g., CB and other GPCRs) and intracellularly accessible (e.g., TRPVs, TRPs, and PPARs) sites (Ross, 2003; Watanabe et al., 2003; Goodfellow and Glass, 2009; Sigel et al., 2011; Baur et al., 2013). Because any eCB agonist needs to be removed from the orthosteric binding site of its receptor targets, the evolution of a membrane protein that facilitates reuptake and CB1R/CB2R clearance would make sense. The interference with the movement of eCBs through competitive inhibition at binding sites or the putative eCB membrane transporter, therefore, has great potential to modulate pathophysiological processes through the ECS, with a range of possible therapeutic applications like FAAH and MAGL inhibitors.
B. Evolution of Pharmacological Inhibitors of N-Arachidonyl Ethanolamine Transport
In the 1990s, the first AEA uptake inhibitors were synthesized. Based on the observed substratespecificity, initially mainly structural analogs of AEA were synthesized and tested for [3H]AEA uptake inhibition in rat brain neurons and astrocytes (Khanolkar et al., 1996; Beltramo et al., 1997). The first inhibitor of AEA cellular uptake was the N-(4-hydroxyphenyl)-arachidonamide AM404, which exhibited an IC50 value ∼1 µM in neurons and an IC50 value ∼5 µM in astrocytes (see Table 11) (Beltramo et al., 1997).
TABLE 11.
Potent inhibitors of AEA cellular uptake relative to inhibition of AEA degradation (by FAAH) or intracellular transport (by FABP5)
| Compound | AEA Cellular Uptake Inhibition | ||||
|---|---|---|---|---|---|
| IC50 (µM) | Cell Type | AEA Cellular Uptake Kinetics | FAAH IC50 value (µM) |
FABP5 Ki value (µM) |
|
 Thiazolidinone-type second-generation selective Thiazolidinone-type second-generation selectiveendocannabinoid reuptake inhibitors (SERIs) Patent ES2845636T3 |
0.08-0.50 | U937 | ND | >10 | >10 |
 WOBE437 WOBE437Chicca et al., 2017 |
0.01 0.137 0.055 ∼1 [50% inh.] |
U937 HMC-a Neuro2A Rat cortical neurons |
Km = 0.25 µM, Vmax = 67.4 fmol/min/cell in U937 cells | >10 | >50 |
 RX-055 RX-055Chicca et al., 2017 |
0.014 ∼1 [35% inh.] |
U937 Neuro2A |
ND | 4.0 | ND |
 Guineensine GuineensineNicolussi et al., 2014b |
1.32 0.29 0.62 |
U937 U937 HMC-1a |
ND | 46.8 | >100 |
 Macamide 7 Macamide 7Hajdu et al., 2014 |
0.67 | U937 | ND | 4.1 | ND |
 Farinosone-C derivative (BSL-34) Farinosone-C derivative (BSL-34)Burch et al., 2013 |
0.23 | U937 | ND | >10 | ND |
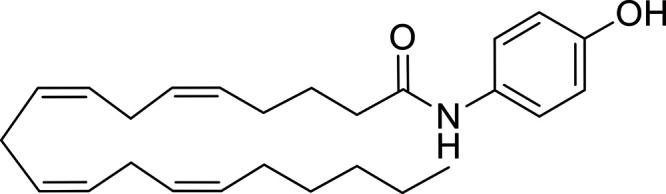 AM404 AM404Beltramo et al., 1997; Maccarrone et al., 1998; Piomelli et al., 1999; De Petrocellis et al., 2000; Rakhshan et al., 2000; Jarrahian et al., 2000; Deutsch et al., 2001; Porter et al., 2002; Glaser et al., 2003; López-Rodríguez et al., 2003a; Fowler et al., 2004; Vandevoorde and Fowler, 2005; Dickason-Chesterfield et al., 2006; Hillard et al., 2007; Nicolussi et al., 2014a |
1.0 5.0 2.2 10.2 10.9 8.1 10.2 4 Ki ≈ 14 14.9 20 1.8 4.9 >100 44.4 |
Rat cortical neurons Rat cortical astrocytes Astrocytoma C6 glioma RBL-2H3 RBL-2H3 RBL-2H3 RBL-2H3 RBL-2H3 RBL-2H3 RBL-2H3 U937 Cerebellar granular neurons HMC- a HeLaa |
Km = 1.2 µM, Vmax = 90.9 pmol/min/mg in neurons Km = 0.32 ± 0.1 µM, Vmax = 171 pmol/min/mg in astrocytes Km = 0.6 ± 0.1 µM, Vmax = 14.7 ± 1.5 pmol/min/mg in astrocytoma Km = 0.7 ± 0.1 µM, Vmax = 0.39 fmol/min/cell in C9 glioma cells Km = 11.4 ± 2.3 µM, Vmax = 17.5 ± 2.1 × 10−17 mol/min/cell in RBL-2H3 cells Km = 0.13 ± 0.01 µM, Vmax = 140 ± 15 pmol/min/mg in U937 cells |
5.9 3.6 0.5 0.8 >30 Ki = 0.60 6 2.1 |
0.39 |
 AM1172 AM1172Fegley et al., 2004; Dickason-Chesterfield et al., 2006; Kaczocha et al. 2006; Hillard et al., 2007 |
2.5 2.1 24.0 86.6 68.9 |
Astrocytoma Rat cortical neurons Cerebellar granular neurons RBL-2H3 HeLa a |
ND | >5 Ki ≈3.2 >100 >50 |
ND |
|
VDM11 De Petrocellis et al., 2000; Fowler et al., 2004; Vandevoorde and Fowler, 2005; Dickason-Chesterfield et al., 2006; Kaczocha et al., 2006; Hillard et al., 2007; Kaczocha et al., 2012; Nicolussi et al., 2014a |
10.2 6.1 11.2 9.9 1.1 5.5 >100 23.8 |
C6 glioma C6 glioma RBL-2H3 RBL-2H3 U937 Cerebellar granular neurons HMC-1a HeLa a |
ND | 2.0 1.2 0.4 Ki ≈ 0.44 11.174 5111 1.6 - 2.9 |
1.75 |
 OMDM-1/2 OMDM-1/2Ortar et al., 2003; Fowler et al., 2004; Dickason-Chesterfield et al., 2006; Kaczocha et al., 2006; Hillard et al., 2007; Chicca et al., 2012; Kaczocha et al., 2012; Nicolussi et al., 2014a |
Ki ≈ 3 16.6 3.2 9.1 3.93 5.2 3.2 3.1 >100 4.9 |
RBL-2H3 C6 glioma RBL-2H3 RBL-2H3 U937 U93764 U937 HMC-1a HeLaa Cerebellar granular neurons |
ND | >50 54 23.4 >100 Ki ≈ 9.7 >100 >50 |
3.85 >100 |
 UCM707 UCM707López-Rodríguez et al., 2003a; Fegley et al., 2004; Fowler et al., 2004; Dickason-Chesterfield et al., 2006; Kaczocha et al., 2006; Hillard et al., 2007; Chicca et al., 2012; Nicolussi et al., 2014a |
0.8 1.34 1.8 41 25 20.1 3.6 56.4 30.3 4 3 |
U937 U937 U937 C6 glioma RBL-2H3 RBL-2H3 HMC-1a HeLaa Cerebellar granular neurons Cortical neurons Cortical neurons FAAH−/− |
Km = 1.1 µM, Vmax = 151 pmol/min/mg in WT cortical neurons Km = 1.3 µM, Vmax = 157 pmol/min/mg in FAAH−/− cortical neurons |
30 8.32 >100 Ki ≈ 0.37 20.5 50 |
25.8 |
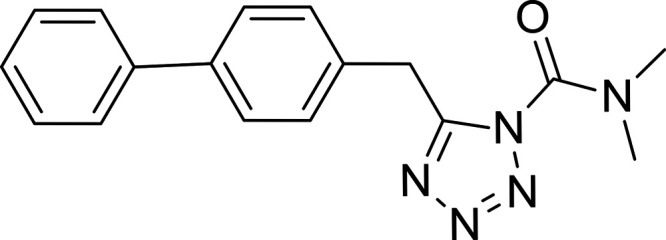 LY2183240 LY2183240Moore et al., 2005; Alexander and Cravatt, 2006; Dickason-Chesterfield et al., 2006; Ortar et al., 2008; Nicolussi, 2014; Nicolussi et al., 2014a |
0.000270 0.015 0.00095 1.93 29.7 |
RBL-2H3 RBL-2H3 U937 HMC-1a HeLaa |
Km = 4.69 ± 0.46 µM, Vmax = 0.02 fmol/min/cell in RBL-2H3 cells | 0.014 0.0021 0.0124 0.001 |
ND |
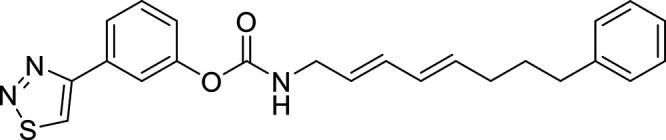 WOBE492 WOBE492Nicolussi et al., 2014 |
0.000005 0.12 |
U937 HMC-1 |
ND | 0.000014 | ND |
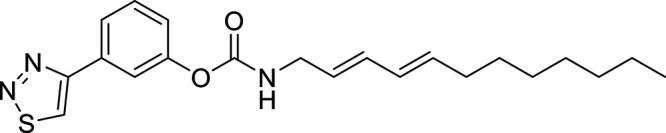 WOBE498 WOBE498Nicolussi et al., 2014 |
0.00005 0.25 |
U937 HMC-1 |
ND | 0.000015 | ND |
ND, not determined.
aCells lacking FAAH-activity.
This probe, which was later discovered to be a bioactive AA conjugated metabolite of paracetamol (also known as acetaminophen) (Högestätt et al., 2005), was initially reported to be selective toward uptake inhibition over FAAH inhibition (IC50 > 30 µM). AM404 was later suggested to be a competitive inhibitor of AEA uptake and was found to be transported as a pseudo-substrate of the postulated AEA transporter (Beltramo and Piomelli, 1999). Yet, independent groups showed that AM404 inhibits FAAH with IC50 values close to those obtained for AEA uptake inhibition (Table 11), thus questioning the selectivity of this compound. In addition, AM404 may also interact with other targets of the ECS. The reversed amide analog AM1172 apparently solved the problem of selectivity by being an equally potent inhibitor of AEA uptake as AM404 but resistant to hydrolysis by FAAH (Fegley et al., 2004). Yet, it was subsequently reported that AM1172 also inhibits FAAH (Hillard et al., 2007).
The quest for better AEA cellular uptake inhibitors continued with the aim to increase their potency and generate structure-activity relationship studies for the postulated transporter target (Jarrahian et al., 2000; Di Marzo et al., 2004). Since the inhibition of FAAH also leads to the inhibition of [3H]AEA uptake, the aspect of selectivity over FAAH became a crucial differentiation criterion (Day et al., 2001; Deutsch et al., 2001). Besides the well-studied AEA uptake inhibitor VDM11 (De Petrocellis et al., 2000), more selective inhibitors such as the oleic acid derivatives OMDM-1 and OMDM-2 (Ortar et al., 2003) or UCM707 (López-Rodríguez et al., 2001, 2003a) were synthesized (Table 11). Despite their initially published selectivity over FAAH, some of these inhibitors were later found to inhibit FAAH with similar or even identical IC50 values obtained for AEA uptake inhibition (Fowler et al., 2004; Vandevoorde and Fowler, 2005; Hillard et al., 2007). Unfortunately, almost nothing is known about the pharmacokinetics and tissue distribution of these compounds, and pharmacological effects are difficult to attribute to either FAAH inhibition or AEA transport inhibition. UCM707 was investigated in neuronal preparations of FAAH−/− mice and still showed an IC50 = 3 ± 1 µM for AEA accumulation (Ortega-Gutiérrez et al., 2004). This finding agreed with its selectivity for AEA uptake inhibition over FAAH (López-Rodríguez et al., 2003b). A direct comparison of the data obtained with neuronal cells of FAAH+/+ mice demonstrated that AEA cellular uptake is a facilitated process in which a specific “UCM707-binding protein” was proposed to participate with a relative contribution of at least 30% (Ortega-Gutiérrez et al., 2004). FABP5 as an intracellular eCB carrier protein (Kaczocha et al., 2012; Sanson et al., 2014) was therefore a possible candidate. However, the affinity of UCM707 to FAPB5 was measured (Table 11) and resulted in a Ki = 25.8 µM (19.5–44.7 µM) (Nicolussi, 2014). This low affinity interaction of UCM707 with FABP5 clearly does not match the determined IC50 value for AEA cellular uptake. Moreover, given that UCM707 still works in FAAH-lacking cells and synergizes with FAAH inhibitors for AEA uptake inhibition and inhibits AEA efflux (Chicca et al., 2012), the possibility that UCM707 targets a membrane transport mechanism is still valid. Unsurprisingly, the highly potent FAAH inhibitors LY2183240 and URB597 (Table 11) resulted in pronounced AEA cellular uptake inhibition in different cell types (Mor et al., 2004; Moore et al., 2005; Dickason-Chesterfield et al., 2006) and were essentially representative of all FAAH inhibitors. The unexpected and paradoxical inhibition of passive diffusion by small organic molecules, as the primary evidence of the carrier-mediated model, was readily refuted because inhibitors like AM404 did not inhibit AEA cellular uptake at short incubation times (< 40 seconds) and inhibited FAAH (Glaser et al., 2003). Ligresti and colleagues convincingly showed saturable AEA uptake within 90 seconds not only in RBL-2H3 and C6 glioma cell lines but also in mouse brain synaptosomes from FAAH−/− mice (Ligresti et al., 2004). In the study by Glaser and colleagues (Glaser et al., 2003), where simple diffusion of AEA was measured, very high nonphysiologic AEA concentrations (1–100 µM) were used, which may easily mask the transport kinetics seen with concentrations of 50 to 500 nM (as a note, at ≥ 1 µM AEA simple diffusion kinetics can be measured). Yet, such high AEA concentrations are not found in tissues, and much less is needed for receptor activation. Eli Lilly developed the highly potent tetrazole inhibitor called LY2183240 with an astonishing IC50 = 270 ± 29 pM for AEA cellular uptake in RBL-2H3 cells (Moore et al., 2005; Ortar et al., 2008) (Table 11). Using the modified radiolabeled probe [125I]-LY2318912, a high-affinity membrane binding site involved in the transport of AEA could be identified, curiously also in FAAH-lacking HeLa cells (Kd = 7.06 ± 1.69 nM, Bmax = 32.2 ± 2.98 fmol/mg). In human FAAH-transfected HeLa cells, neither the binding affinity (Kd) nor the Bmax value changed significantly, indicating that one binding site is independent of FAAH (Moore et al., 2005). Having raised hopes for the molecular identification of the postulated AEA transporter, shortly afterward, LY2183240 was shown to be an ultrapotent, irreversible, and nonspecific inhibitor of FAAH, MAGL, and other serine hydrolases (Alexander and Cravatt, 2006).
Additional indirect evidence for the existence of a transporter-mediated (facilitated) AEA uptake mechanism was provided by the demonstration of AEA uptake in synaptosomes from human, mouse, and rat brain (Battista et al., 2002) and in neuronal preparations of FAAH knockout mice (Fegley et al., 2004; Ligresti et al., 2004; Ortega-Gutiérrez et al., 2004). Known AEA uptake inhibitors like UCM707 still reduced the accumulation of AEA, but the uptake efficacy was much lower in cells lacking FAAH compared with those from wild-type mice (Fegley et al., 2004; Ligresti et al., 2004; Ortega-Gutiérrez et al., 2004). However, FAAH activity alone did not seem to be causative of all AEA uptake phenomena (Ligresti et al., 2004). In agreement with the view that FAAH is not the only player in AEA transport, cells lacking FAAH like HMC-1 cells (Maccarrone et al., 2000; Nicolussi et al., 2014b) show robust AEA uptake kinetics, although with a lower Vmax than in FAAH-expressing cells. Moreover, an energy-independent and saturable export of [3H]AEA was demonstrated in human endothelial cells (Maccarrone et al., 2002). Obviously, hydrolysis by FAAH has no impact on AEA efflux. Additionally, it was demonstrated that the transport inhibitor VDM11 inhibited the release of de novo–generated AEA in HEK-293 cells (Ligresti et al., 2004). Taken together, these studies pointed toward a bidirectional membrane transport mechanism for AEA shown by independent groups (Hillard et al., 1997; Maccarrone et al., 2002; Ligresti et al., 2004; Chicca et al., 2012). In this context, the release of AEA and 2-AG was assessed in an electrophysiological study measuring striatal long-term depression in acute brain slice preparation, where postsynaptic blockage of eCB membrane transport using VDM11 achieved a disruption of eCB release (Ronesi et al., 2004). In another study, OMDM-2 and AM404 increased activity-dependent AEA and 2-AG levels in the hypothalamus and inhibited the synaptically driven spiking activity in postsynaptic neurons upon enhanced retrograde signaling (Di et al., 2005). In urethane-anesthetized rats, VDM11 inhibited the micturition reflex at least in part through CB1R (Honda et al., 2016), suggesting a possible therapeutic role of AEA transport inhibitors in disturbances of the storage function of the bladder or disturbances of the emptying function. The only pharmacological study that uses AEA release inhibition as an explanation for the effect was the comparison of OMDM-2 versus the FAAH inhibitor URB597 on social withdrawal in rodents (Seillier and Giuffrida, 2018). Systemic administration of OMDM-2 reduced social interaction, but, in contrast to URB597-induced social deficit, this effect was not reversed by the TRPV1 antagonist capsazepine. Conversely, the CB1R antagonist AM251, which did not affect URB597-induced social withdrawal, exacerbated OMDM-2 effect (Seillier and Giuffrida, 2018). The infusion of OMDM-2 and VMD11 in both cases reduced the extracellular levels of dopamine collected from nucleus accumbens and suggested a role for AEA transport in sleep modulation (Murillo-Rodriguez et al., 2013). Interestingly, AM404 but not VDM11 reduced the acute freezing response in male mice in a strong auditory-cued fear memory via CB1R- and TRPV1-mediated mechanisms (Llorente-Berzal et al., 2015). Finally, in nonhuman primates, AM404 reinforced anandamide or cocaine self-administration behavior and induced reinstatement of drug-seeking behavior in abstinent monkeys by a CB1R-dependent mechanism (Schindler et al., 2016a).
C. Preclinical Development of Selective Endocannabinoid Reuptake Inhibitors
The N-isobutylamide guineensine from Piper species (Table 11) was identified as a nanomolar and strongly selective inhibitor of AEA cellular uptake over FAAH inhibition and other ECS targets (Nicolussi et al., 2014b). Guineensine did not show a relevant inhibition of FAAH activity (IC50 > 50 µM) or FABP5 binding (Ki > 100 µM) and dose-dependently induced cannabimimetic effects in BALB/c mice shown by strong catalepsy, hypothermia, reduced locomotion, and analgesia in the hot plate test. The catalepsy and analgesia were blocked by the CB1R antagonist rimonabant (SR141716A) (Reynoso-Moreno et al., 2017). The pharmacological evidence of indirect cannabimimetic effects strongly suggests that guineensine also targets eCB cellular reuptake in vivo (Reynoso-Moreno et al., 2017). An efficient total synthesis of guineensine was published that may facilitate the provision of this rare natural product for research (Bartholomäus et al., 2019). Another compound in the list of plant-derived natural AEA uptake inhibitors is the N-benzyl-(9Z,12Z)-octadecadieneamide (macamide 7, shown in Table 11), which exhibited a nanomolar IC50 value for AEA uptake inhibition but also inhibits FAAH at low micromolar concentrations (Hajdu et al., 2014). Furthermore, an analog of the natural product farinosone-C (BSL-34, Table 11) was found to be a more selective inhibitor of AEA uptake (IC50 = 232 nM) over FAAH inhibition (IC50 > 10 µM), with close structural similarity to OMDM-2 (Burch et al., 2013).
Building on previous work on N-alkyl-2,4-dodecadienamides from Echinacea purpurea, which have been shown to interact with the ECS (Raduner et al., 2007; Chicca et al., 2009), a series of derivatives and analogs were synthesized. Diverse N-alkylcarbamates were also synthesized and tested in U937 cells for their ability to inhibit AEA hydrolysis and uptake, showing ultrapotent FAAH inhibition that led to hyperpotent AEA uptake inhibition (Nicolussi et al., 2014a). Interestingly, some of these N-alkylcarbamates (e.g., WOBE492 and WOBE498) showed a FAAH-independent AEA uptake inhibition in HMC-1 cells with IC50 values below 300 nM (Nicolussi et al., 2014a). This study led to the identification of (2E,4E)-N-[2-(3,4-dimethoxyphenyl)ethyl] dodeca-2,4-dienamide (WOBE437, shown in Table 11) as a highly potent and selective eCB uptake inhibitor, which was extensively profiled (Chicca et al., 2017). For instance, WOBE437 inhibits AEA and 2-AG uptake in U937 cells with IC50 values of 10 ± 8 nM and 283 ± 121 nM, respectively (Chicca et al., 2017). Furthermore, WOBE437 was tested in Neuro2a mouse neuroblastoma cells, primary rat cortical neurons, and FAAH-deficient HMC-1 cells, showing differential but significant inhibition of AEA uptake in all the cell lines. WOBE437 did not inhibit FAAH, MAGL, α/β-hydrolase domain proteins 6 and 12, or COX-2, nor did it show a significant interaction with CB1R/CB2R, FABP5, or any of 45 relevant CNS-related receptors/transporters/ion channels/enzymes tested (Chicca et al., 2017). Moreover, in C57BL6/J male mice WOBE437 was found to be orally bioavailable (Reynoso-Moreno et al., 2018), and in a clinically relevant mouse model of MS-like experimental autoimmune encephalomyelitis, it significantly reduced disease severity and accelerated recovery through CB1R/CB2R-dependent mechanisms (Reynoso-Moreno et al., 2021). A structure-activity relationship study on the WOBE437 scaffold for cellular AEA uptake inhibition was recently published (Mäder et al., 2021). However, using a clickable analog of the WOBE437-derived photoaffinity probe RX-055 (Table 11), saccharopine dehydrogenase-like oxidoreductase, vesicle amine transport 1, and ferrochelatase were identified as low affinity (10 µM) off-targets of WOBE437 in Neuro-2a cells (Gagestein et al., 2022), calling for attention on the therapeutic exploitation of this inhibitor at higher doses.
Currently, a new class of SERIs, the thiazolidinones (Table 11), are being developed for the treatment of psychiatric or neurologic disorders and inflammation at Synendos Therapeutics, though their target protein has not yet been published.
D. Preclinical Development of Fatty Acid Binding Protein Inhibitors
In liver, it has been shown that FABP1 not only acts as an eCB and pCB binding protein but also regulates hepatic eCB levels (Huang et al., 2018). Studies using FABP1 knockout mice revealed a markedly diminished impact of a high-fat diet on brain eCB levels, especially in male mice, suggesting the involvement of FABP1 in the biosynthesis of these lipids (Martin et al., 2017). FABPs seem to be generally involved in modulating AEA trafficking as the overexpression of FABP5 and FABP7 in COS-7-FAAH-eGFP cells increased AEA uptake and hydrolysis by 32% and 35%, respectively (Kaczocha et al., 2009). N18TG2 cells showed an increase of 36% upon FABP5 and 42% upon FABP7 overexpression. In the same cells, a reduction of AEA uptake and hydrolysis could be monitored after preincubation with the FABP4/5 inhibitor BMS309403 (Table 12). While BMS309403 exhibited a Ki = 350 ± 3 nM for FABP5 binding, a concentration of 100 µM of this probe was needed to reach ∼50% inhibition of cellular AEA uptake (Sulsky et al., 2007; Furuhashi and Hotamisligil, 2008; Kaczocha et al., 2009). FABP5 was suggested as the main target of the AEA uptake inhibitors OMDM-2, VDM11, and AM404, because these blockers showed binding affinities to FABP5 comparable to the published Ki values for AEA-FABP5 binding (Kaczocha et al., 2012; Nicolussi et al., 2014a).
TABLE 12.
Additional and less potent inhibitors of AEA cellular uptake relative to inhibition of AEA degradation (by FAAH) or intracellular transport (by FABP5)
| Compound | AEA Cellular Uptake Inhibition | |||
|---|---|---|---|---|
| IC50 (µM) | Cell Type | FAAH IC50 Value (µM) |
FABP5 Ki Value (µM) |
|
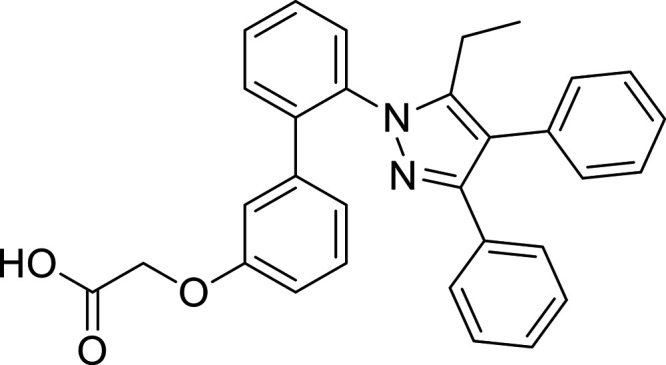 BMS309403 BMS309403Sulsky et al., 2007; Kaczocha et al., 2009; Berger et al., 2012; Kaczocha et al., 2012; Nicolussi et al., 2014a |
∼100 [57% inh.] >100 [48% inh.] 24.7 >100 [30% inh.] |
N18TG2 COS7-FAAH-eGFP U937 HMC-1* |
>100 >100 |
0.35 0.75 0.89 |
 SBFI-26 (RT26) SBFI-26 (RT26)Berger et al., 2012: Zhou et al., 2019a |
∼20 [40% inh.] |
HeLaa | >50 | 0.9 |
aCells lacking FAAH-activity.
Surprisingly, AA also showed a strong affinity to FABP5 (Kaczocha et al., 2012). It is generally accepted that AA does not affect AEA cellular uptake up to a concentration of 100 µM (Beltramo et al., 1997; Hillard et al., 1997; Piomelli et al., 1999), a finding that would challenge the role of FABP5 in AEA transport. The development of potent and specific FABP5 inhibitors with the aim to modulate AEA cellular transport is ongoing (Berger et al., 2012; Zhou et al., 2019a), and a first in vivo evaluation of SBFI-26 (one of these compounds shown in Table 12) was reported (Kaczocha et al., 2014). SBFI-26 is an α-truxillic acid 1-naphthyl monoester, originally identified using a computational docking protocol and synthesized as a mixture of both the (S) and (R) enantiomers (Berger et al., 2012). SBFI-26 produced antinociceptive and anti-inflammatory effects in mice and inhibited the activities of FABP5 and FABP7 with Ki values of 0.9 µM and 0.4 µM, respectively (Berger et al., 2012; Kaczocha et al., 2014). In FABP5, SBFI-26 was unexpectedly found to bind at the substrate entry portal region in addition to binding at the canonical ligand-binding pocket (Hsu et al., 2017). However, it is noted that the high concentrations needed in vitro for AEA cellular uptake inhibition experiments do not match the reported FABP5 affinity of SBFI-26. In rodents, SBFI-16 showed peripheral and supraspinal analgesic effects (Peng et al., 2017) and abrogated pulmonary artery remodeling in pulmonary hypertension secondary to left heart disease and improved cardiac function (Lei et al., 2022). Yet, the involvement of the ECS in these effects was not elucidated. As already pointed out above, the Ki values obtained for AEA binding to FABP5 are not in agreement with the Km values in many cells that show AEA transport. Recently, it was demonstrated that FABP5 both promotes the hydrolysis of AEA to AA and thus reduces brain eCB levels and directly shuttles AA to the nucleus, where it delivers it to PPARβ/δ, enabling its activation (Yu et al., 2014). Interestingly, in adult neurons, neither FABP5 nor FABP7 seems to be expressed in significant amounts (Liedhegner et al., 2014).
The first evidence for intracellular carriers of 2-AG was provided by two independent groups. The known cytosolic FABP5, which is an AEA carrier and binds numerous highly abundant fatty acids, was also shown to bind 2-AG. Using fluorescence polarization and a labeled fatty acid probe that was displaced from FABP5, a Ki = 8.7 µM was determined (Nicolussi, et al., 2014a). Simultaneously, a crystallographic study of FABP5 as an intracellular carrier protein of eCBs confirmed the binding data (Sanson et al., 2014). Of note, the Kd for 2-AG binding to FABP5 more closely matches the Km for 2-AG transport than in the case of AEA.
It was recently reported that FABP5 could act as a synaptic (i.e., extracellular) transporter of 2-AG and control the retrograde signaling by this eCB (Haj-Dahmane et al., 2018). Using dorsal raphe neurons incubated with SBFI-26 (a FABP5 and FABP7 inhibitor) or from FABP5−/− mice, it was shown that FABP5 inhibition or absence prevented DSE, which under normal conditions occurs after depolarization of postsynaptic neurons and phasic 2-AG release, followed by presynaptic CB1R activation, reduction of glutamate release, and a reduction in excitatory postsynaptic currents. Furthermore, FABP5 inhibition or absence prevented the increase seen in excitatory postsynaptic currents after incubation with AM251 (a CB1R antagonist/inverse agonist), showing that by acting as a carrier FABP5 modulates the effect of phasic and tonic levels of 2-AG in the control of retrograde signaling. Additionally, in a coculture of primary hippocampal astrocytes and neurons, it was shown that FABP5 is secreted by astrocytes to the extracellular media in a time-depended manner, supporting its role as an extracellular synaptic transporter of 2-AG. In FABP5−/− neurons, no changes were observed in the protein expression of CB1R, DAGLα (the neuronal isoform) or MAGL, concluding that there are no changes in either CB1R activation or 2-AG metabolism. Although nonsignificant changes in 2-AG levels were observed after incubation with SBFI-26, the opposite occurred in FABP5−/− neurons, which showed a significant increase. Furthermore, dorsal raphe neurons incubated with SBFI-26 showed an increase in AEA levels, which agrees with previous reports (Kaczocha et al., 2009; 2014); however, there were no changes in AEA levels measured in FABP5−/− neurons compared with wild-types. These contradictions raise a question regarding other possible changes in the metabolic pathways of 2-AG and AEA, respectively, that might not be observed at the protein level. As shown recently, deletion of FABP5 impaired tonic 2-AG and AEA signaling at striatal γ-aminobutyric acid synapses of medium spiny neurons and blunted phasic 2-AG mediated short-term synaptic plasticity without altering CB1R expression or function (Fauzan et al., 2022). Based on the expression of FABP5 in TRPV1-positive nociceptors, a conditional knockout strategy was employed that showed that deletion of FABP5 specifically in nociceptors augments AEA levels, resulting in antinociceptive effects mediated by CB1R (Bogdan et al., 2022). Given that the concentration of free fatty acids including AA may be much higher in the synaptic cleft and in neuronal membranes, it is intriguing that FABP5 binds 2-AG in a physiologic environment, which is found at significantly lower concentrations and competes for the same binding site as AA in this protein, especially because there are not multiple lipid binding sites in FABP5. Overall, it cannot be ruled out that the effects observed with FABP KO mice may also be related to AA metabolism.
E. Translational Implications of Endocannabinoid Transport Inhibitors
The different models of eCB cellular uptake and trafficking offer different druggable sites, that are schematically depicted in Fig. 26. The identification of intracellular carrier proteins for AEA has clearly provided a missing link to explain how eCBs are able to cross the cytosol, which constitutes a hydrophilic barrier for these lipophilic compounds (Hillard and Jarrahian, 2003; Fegley et al., 2004; Glaser et al., 2005; Hillard et al., 2007; Kaczocha et al., 2009; Oddi et al., 2009; Fowler, 2012, 2013).
To date, different AEA transport inhibitors and detailed pharmacological assessment of their in vivo effects have led to a better understanding of the druggability of such processes within the ECS. However, only a few inhibitors used in pharmacological experiments have been studied for their bioavailability, tissue distribution, and overall pharmacokinetics; thus in vivo effects of such inhibitors are difficult to interpret. As yet, only FABP5 inhibitors AT26 and SYT510 have shown efficacy in models of pain, anxiety, and inflammation through mechanisms involving the ECS and are drug candidates in a late preclinical stage (Table 13).
TABLE 13.
Drug candidates that target eCB transport in late preclinical stage
| Name | Company | Indication(s) | Target |
|---|---|---|---|
| RT26 | Artelo Biosciences | Prostate cancer Chemotherapy-Induced Peripheral Neuropathy |
FABP5 inhibitor |
| SYT510 | Synendos Therapeutics |
Neuropsychiatric Disorders |
SERI, undisclosed target |
SERI, selective eCB reuptake inhibitor.
The new generation of selective inhibitors for AEA uptake (WOBE437, RX-055, guineensine) also blocks 2-AG uptake but does not interact with any of the known metabolic enzymes or AEA binding proteins, suggesting an additional common target that is competitive with eCB membrane transport. This observation has inspired the development of thiazolidinones like SYT510 that act as SERIs for the treatment of neuropsychiatric disorders—in a manner that is comparable to MAGL inhibitors—and are in early clinical development (Table 13). Based on the pharmacological profiles of these SERIs, it can be expected that they are more specific and do not interfere with metabolic classes. Moreover, unlike FAAH, MAGL, and FABP5 inhibitors, SERIs rather selectively inhibit the uptake of both AEA and 2-AG, without modulating other lipids (Chicca et al., 2017). Remarkably, their mild modulation of the eCB tone may be beneficial when it comes to issues related to the desensitization of CB1R (Reynoso-Moreno et al., 2021).
VI. Therapeutic Potential of Additional Targets Within the “Endocannabinoidome”
A. Definition of the Endocannabinoidome
Two realizations during the past 20 years have brought to the attention of the scientific community that the ECS should be considered as part of a much wider signaling system, now referred to as the endocannabinoidome (eCBome) (Balvers et al., 2009; Piscitelli et al., 2011; Di Marzo, 2018; Cristino et al., 2020): (1) the discovery that several endogenous congeners of AEA and 2-AG, i.e., the N-acyl-ethanolamines and 2-monoacylglycerols, respectively, and other eCB analogs like long chain fatty acid derivatives are present in tissues and biologic fluids, although they seldom share with eCBs the capability of modulating the activity of CB1R and CB2R (while often being biosynthesized and/or degraded by the same enzymes) (Di Marzo, 2018), and (2) the finding that most plant cannabinoids other than THC, such as cannabidiol, cannabigerol, cannabidivarine, and cannabichromene, to name a few, also do not share with THC, AEA, and 2-AG their activity at cannabinoid receptors, although they often interact, among others, with several receptors of the aforementioned eCB-like molecules (Di Marzo, 2018). The main components of the eCBome are summarized in Table 14.
TABLE 14.
Lipid signals from the eCBomes, their targets and metabolic enzymes
| Family | Most Studied Subfamily (If Applicable) | Most Studied And/Or Most Tissue Abundant Members | Best Established Molecular Target(s) | Best Established Anabolic Enzyme(s) | Best Established Catabolic Enzyme(s) | Main Potential Therapeutic Applications (Based On The Best Established Pharmacological Actions) | References | |
|---|---|---|---|---|---|---|---|---|
| eCBome | NAEs (AEA and its congeners) |
N-Arachidonoyl-ethanolamine (anandamide) | CB1R (↑), CB2R (↑), TRPV1(↑), TRPV2(↓), TRPM8(↓), Cav3.3(↓) |
NAPE-PLD, ABHD4+GDE1 | FAAH-1 | Chronic and inflammatory pain; anxiety; depression; (neuro)inflammatory disorders; cancer. Obesity, NASH and type 2 diabetes |
Mechoulam, 2023 | |
| N-Docosa-hexaenoyl-ethanolamine | GPR110(↑),CB2R(↑) | FAAH-1 | Inflammation; neurodegenerative disorders; cancer. | Watson et al., 2019 | ||||
| N-Oleoyl-ethanolamine | PPARα(↑), TRPV1(↑), GPR119(↑) | FAAH-1, FAAH-2 | Obesity, type 2 diabetes, steatosis and related disorders | Bowen et al., 2017 | ||||
| N-Palmitoyl-ethanolamine | PPARα(↑), GPR55(↑) | NAAA, FAAH-1 |
Chronic and inflammatory pain; eczema, neuroinflammatory disorders; migraine; COVID-19 | Petrosino and Di Marzo, 2017 | ||||
| MAGs (2-AG and its congeners) |
2-Arachidonoyl-glycerol | CB1R(↑), CB2R (↑), TRPV1(↑), TRPM8(↓) |
DAGLα, DAGLβ | MAGL, ABHD6, ABHD12 |
Chronic and inflammatory pain; anxiety; depression; (neuro)inflammatory disorders, obesity and type 2 diabetes |
Baggelaar et al., 2018 | ||
| 2-Oleoyl- and 2-Linoleoyl-glycerol | GPR119(↑), TRPV1(↑) | Type 2 diabetes | Hansen and Vana, 2019 | |||||
| 1- or 2-Palmitoyl-glycerol | PPARα(↑) | Obesity, type 2 diabetes, steatosis and related disorders | Depommier et al., 2021 | |||||
| Primary amides | Oleamide | TRPV2(↓), CB1R(?)(↑) | GLYATL3+PAM | FAAH-1 | Sleep disorders | Leggett et al., 2004; Schiano Moriello et al., 2018 | ||
| N-Acyl-amino acids (lipo-aminoacids) | N-Acyl-glycines |
N-Oleoyl-glycine N-Palmitoyl-glycine N-Arachidonoyl-glycine |
PPARα(↑), FAAH-1(↓), Cav3.3(↓), GPR132(↑), GPR118 (?)(↑) |
GLYATL3 | PAM FAAH-1 |
Brain trauma and its consequences, neuroprotection, nicotine and opiate addiction, pain | Huang et al., 2001; Foster et al., 2019; Donvito et al., 2019; Piscitelli et al., 2020 | |
| N-Acyl-alanines | N-Oleoyl-alanine | PPARα(↑), FAAH-1(↓) | ? | ? | Nicotine and opiate addiction | Ayoub et al., 2020 | ||
| N-Acyl-serines | N-Oleoyl-serine | Cav3.3(↓) | ? | Osteoporosis | Milman et al., 2006 | |||
| N-Acyl-taurines | N-Oleoyl- and N-Arachidonoyl-taurine | TRPV1(↑), TRPV4(↑), Cav3.3(↓) |
FAAH-1 | Skin wound healing, type 2 diabetes and dyslipidemia | Grevengoed et al., 2019; Sasso et al., 2016. | |||
| N-Acyl-neurotransmit-ters | N-Acyl-dopamines | N-Arachidonoyl-dopamine | TRPV1(↑), CB1R(↑), FAAH(↓), Cav3.3(↓) | ? | COMT, Cyp epoxygenases |
Pain, cancer, nausea, neuroinflammatory and neurodegenerative disorders | De Petrocellis and Di Marzo, 2014 | |
| N-Acyl-serotonins | N-Arachidonoyl-serotonin | TRPV1(↓), FAAH(↓), Cav3.3(↓) | Cyp epoxygenases | Chronic and inflammatory pain, anxiety, depression, (neuro)inflammatory disorders, epilepsy, IBS | ||||
| OxyeCBome | COX-2 derivatives | Prostamides | Prostamide F2α | FP/Alt4-FP heteromer (↑) | COX-2 +PGFS | ? | Glaucoma, obesity, alopecia pain |
Woodward et al., 2013 |
| Prostaglandin-glycerol esters | Prostaglandin E2-glycerol ester | P2Y6(↑) | COX-2 +PGES | MAGL | Hippocampal excitotoxity | Brüser et al., 2017 | ||
| 15-LOX derivatives | Linoleoylethanolamide derivatives | 13-hydroxy-octadecaenoyl-ethanolamide | TRPV1(↑) | 15-LOX | ? | ? | Simard et al., 2022 | |
| Anandamide derivatives | 15-hydroxy-eicosatetraenoyl-ethanolamide | CB1R(↑), CB2R (↑) TRPV1(↑) |
? | |||||
| Cyp epoxygenase derivatives | Epoxyeicosatrienoyl-ethanolamides | 5,6- Epoxy-eicosatrienoyl-ethanolamide | CB2R(↑), TRPV4(?) (↑) |
Cyp epoxygenase | ? | Cardiovascular disorders, hypertension, inflammation | Snider et al., 2009 | |
| Epoxyeicosatrienoyl-neurotransmitters | Epoxyeicosatrienoyl-dopamine and -serotonin | CB1R(↑), CB1R(↑) TRPV1(↑) |
? | Neuroinflammation | Arnold et al., 2021 | |||
| μbeCBome | Microbiota-derived N-Acyl-amides | Glycine and alanine derivatives | Commendamide, N-Myristoyl-alanine |
GPR132(↑) | Bacterial N-acyl-transferase (choA/glsB) | ? | Cancer, intestinal inflammation | Cohen et al., 2017 |
| Diacylated glycine lipids | ? | Bacterial N-acyl-transferase (choA/glsB) + and O-acyl-transferases (choB/glsA) | ? | Intestinal fitness, eubiosis | Lynch et al., 2019 | |||
| Serinol derivatives | N-Oleoyl-serinol | GPR119(↑) | Bacterial N-acyl-transferases | ? | Type 2 diabetes | Cohen et al., 2017 | ||
| N-Acyl-tyramines, N-Acyl-tryptamines, N-Acyl-aminoacids | N-Lauroyl-tryptamine | GPR183(↓) | Bacterial N-acyl-transferases | ? | Immune disorders | Chang et al., 2021 |
The main potential therapeutic applications of the manipulation of their levels and activity is also shown. Upward and downward arrows denote agonism/activation or antagonism/inhibition. Endocannabinoid-like mediators derived from gut microbiota are also included. Notably, several receptors other than CB1R and CB2R have been found to be also modulated by non-euphoric plant cannabinoids such as cannabidiol, i.e., GPR55, GPR118, GPR132, TRPV1, TRPV2, TRPM8, PPARs, and Cav3.3. Potential therapeutic applications obtained from counteracting the action or the formation of the mediators are shown in italics. ABHD, α/β-hydrolase domain; Alt4-FP, splicing variant 4 of the FP receptor; Cav3.3, T-type Ca2+ channels; COMT, catechol-O-methyl transferase; Covid-19, coronavirus disease type 19; COX-2, cyclooxygenase-2; Cyp, cytochrome p450; DAGL, diacylglycerol lipase; eCBome, endocannabinoidome; FAAH, fatty acid amide hydrolase; FP, prostaglandin F receptor; GDE1, glycerodiesterase type 1; GLYATL3, glycine-N-Acyltransferase Like 3; GPR, orphan G-protein couple receptor; IBS, irritable bowel syndrome; MAGL, monoacylglycerol lipase; μbeCBome, microbendocannabinoidome; NAPE-PLD, N-acyl-phosphatidylethanolamines-specific phospholipase D-like enzyme; oxyeCBome, oxyendocannabinoidome; PAM, peptidyl-glycine alpha-amidating monooxygenase; PGES, prostaglandin E synthase; PGFS, prostaglandin F synthase; PPAR, peroxisome proliferator-activated receptor; TRPM8, transient receptor potential melastatin type-8; TRPV1, transient receptor potential vanilloid type-1; TRPV2, transient receptor potential vanilloid type-2.
In particular, beyond N-acyl-ethanolamines and 2-monoacylglycerols, whose existence was known even before the discovery of AEA and 2-AG, several other subfamilies of eCB-like molecules have been recently discovered, including (1) primary amides of long chain fatty acid, of which the sleep-inducing factor oleamide is the prototypical member; (2) amides between long chain fatty acids and several amino acids, such as N-acyl-taurines, -glycines, and –serines (Huang et al., 2001; Milman et al., 2006; Saghatelian et al., 2006); (3) amides of long chain fatty acids with neurotransmitters and other amines, such as N-acyl-dopamines and -serotonines (Huang et al., 2002; Verhoeckx et al., 2011), and (4) oxidation products (usually, but not necessarily, produced by the action of “arachidonate cascade” enzymes COX-2 and LOXs) of the di- and poly-unsaturated members of the aforementioned families (namely N-acyl-ethanolamines and 2-monoacylglycerols), a subfamily of lipids that we can refer to as the “oxyendocannabinoidome” (oxyeCBome) (reviewed in Simard et al., 2022). Therefore, considering that several long chain fatty acids exist, from the C16-containing and completely saturated palmitic acid, to the C22-containing hexa-unsaturated docosahexeanoic acid, it can be reckoned that, in principle, hundreds of such eCBome mediators exist. However, the actual occurrence of only a few dozens of them has been ascertained so far, through the use of bidimensional liquid chromatography mass spectrometry approaches (Piscitelli et al., 2011; Leishman et al., 2016; Kantae et al., 2017; Lacroix et al., 2019).
Importantly, each different member of these subfamilies, depending on its fatty acid and amine moieties, can modulate the activity of one or more different receptors that in a few cases, have been suggested to be also targeted by AEA or 2-AG (Table 14), although often at concentrations higher than those required to activate CB1R and CB2R (Di Marzo, 2018; Gómez-Cañas et al., 2023). In fact, at least three classes of receptors have been suggested to act as targets for eCBome mediators (Morales and Reggio, 2017; Muller et al., 2019; Lago-Fernandez et al., 2021): (1) GPCRs, like CB1R and CB2R and beyond, and including some orphan GPCRs like GPR18, GPR55, GPR110, GPR119, and GPR130 or GPCRs that are known to be activated or inhibited by other mediators, such as some serotonin receptors; (2) ligand-activated ion channels, such as (i) TRP channels of the V1-4, A1, and M8 types and T-type Ca2+ (Cav3) channels, which have been suggested to be modulated also by other lipid mediators, and (ii) amino acid neurotransmitter-activated targets, such as γ-aminobutyric acid or glycine receptors; and (3) nuclear PPARs. However, for some eCBome mediators, such as the primary amides, the molecular targets are still unknown, although for oleamide evidence of it being a weak agonist of CB1R (Leggett et al., 2004) and a TRPV2 antagonist (Schiano Moriello et al., 2018) exist. Additionally, some eCB-like molecules have also been shown to produce some of their pharmacological actions by interacting with eCB metabolic enzymes. While stimulation of DAGLs by N-palmitoylethanolamine, which possibly explains why this mediator can enhance 2-AG levels in vitro and in vivo, has been only recently shown (Petrosino et al., 2019), inhibition of FAAH leading to increased levels of AEA and other endogenous substrates for these enzymes (e.g., N-acyl-taurines and, under certain circumstances, 2-AG) has been suggested as the basis of some of the pharmacological effects of other NAEs and unsaturated N-acyl-serotonins, -glycines, and -alanines (Jonsson et al., 2001; Petrosino and Di Marzo, 2017; Bashashati et al., 2017; Ayoub et al., 2020). Interestingly, the capability of inhibiting FAAH is shared also by the noneuphoric cannabinoid CBD (Watanabe et al., 1996; Bisogno et al., 2001).
As mentioned, several non-THC cannabinoids, as well as AEA and 2-AG, have also been suggested to influence the activity of the aforementioned eCBome receptors, including (1) orphan GPCRs, particularly for CBD, which antagonizes GPR55 and seems to modulate some opioid and serotonin receptor subtypes (reviewed by de Almeida and Devi, 2020); (2) TRPV1-4 or TRPA1 channels, which are activated by several noneuphoric cannabinoids—with TRPV1 now being widely considered also as an alternative physiopathological target for AEA, unsaturated NAEs, and 2-AG—and TRPM8, which is antagonized by all tested cannabinoids (with the only exception of cannabichromene) as well as by both AEA and 2-AG (Zygmunt et al., 1999; De Petrocellis et al., 2007; De Petrocellis et al., 2011; De Petrocellis et al., 2012; Muller et al., 2019); 3) Cav3.3 channels and glycine receptors, which can be variedly inhibited by THC, cannabidiol, and several types of eCBome mediators, as well as by AEA and some N-acylethanolamines, whereas some subunits of the γ-aminobutyric acid receptor are instead activated by 2-AG and CBD (Sigel et al., 2011; Baur et al., 2013; Chemin et al., 2014; Bakas et al., 2017; Mirlohi et al., 2022); and (4) PPARs, which can be activated by CBD and cannabigerol, particularly in their acidic forms normally found in cannabis flowers, as well as, particularly in the case of PPARα, by some N-acylethanolamines, 2-palmitoyl-glycerol, and N-acyl-glycines (O’Sullivan, 2016; Donvito et al., 2019; D’Aniello et al., 2019; Tutunchi et al., 2020; Depommier et al., 2021; Lago-Fernandez et al., 2021). These eCBome receptors are schematically depicted in Fig. 27.
Fig. 27.

The eCBome receptors as a pharmacological substrate for plant-derived cannabinoids and host or commensal bacteria-derived eCBs and eCB-like molecules. The elements of the ECS as part of the eCBome are shown squared in red. The chemical structures of commendamide, N-miristoyl-alanine, and N-oleoyl-serinol are shown from the top right and down. CBD, cannabidiol; THC, Δ9-tetrahydrocannabinol; THCV, Δ9-tetrahydrocannabidivarin.
In summary, the eCBome and the oxyeCBome potentially include perhaps hundreds of mediators (several combinations of amides between long chain fatty acids and amino acids or bioactive amines and the plethora of oxidation products that can be generated from polyunsaturated eCBome mediators, to name a few) and dozens of receptors. Of the latter, however, many had been previously described as molecular targets for other mediators (neurotransmitters, fatty acids, etc.) and cannot be listed as “specific” eCBome receptors, at least not until their preferential role is ascertained as intermediates of the biologic effects of eCBome mediators, which, however, are often very promiscuous in their modulation of the activity of pharmacologically relevant proteins. While it is of crucial importance to know that AEA and 2-AG are accompanied in tissues by several congeners and metabolites with similar biochemistry and different pharmacology, a discussion of the pharmacological and therapeutic importance of these non-CB1R, non-CB2R receptors is too speculative and goes beyond the scope of this article.
Indeed, the existence of the eCBomes both opens new opportunities and raises new challenges for the development of new therapeutics from the study of the ECS. On the one hand, the recognition that some plant cannabinoids, which are devoid of the typical psychotropic and unwanted effects of THC, owe some of their pharmacological effects—and hence potential therapeutic actions—to modulation of the activity of eCBome receptors beyond CB1R and CB2R, widens their potential applications in medicine (Fig. 27). This same realization is also at the basis of the use of some eCBome mediators, such as N-palmitoylethanolamine and N-oleoylethanolamine, as either synthetic drugs, nutraceuticals, and tissue-targeted nanoparticles (Bowen et al., 2017; Petrosino and Di Marzo, 2017; Wu et al., 2021) or through diets rich in their fatty acid precursors (Sihag and Di Marzo, 2022) as potential new therapeutic approaches in inflammatory and metabolic disorders. On the other hand, the fact that several eCBome mediators that have noncannabinoid receptors as their main targets and share with AEA or 2-AG the capability of being inactivated by FAAH (as in the case of N-acyl-ethanolamines, -glycines and -taurines) or MAGL (as in the case of 2-monoacylglycerols), respectively, may limit the clinical applicability of FAAH and MAGL inhibitors. In fact, such drugs might concomitantly elevate the tissue levels not only of AEA or 2-AG, thus indirectly activating CB1 and CB2 receptors, but also of other eCBome mediators with targets whose functions in disease might also be opposite to those exerted by cannabinoid receptors in disease. A typical example of this potential problem might be represented by the failure, to date, of FAAH inhibitors to counteract inflammatory and chronic pain in clinical trials, when the fact that such molecules elevate the levels not only of AEA but also of N-palmitoylethanolamine and N-oleoylethanolamine, which may consequently act at targets such as TRPV1 and GPR55, may explain the lack of efficacy. Likewise, inhibitors of DAGLs, which have been proposed as a potential treatment of obesity and metabolic disorders through the impairment of 2-AG biosynthesis and subsequent CB1R activation (Bisogno et al., 2013; Janssen and van der Stelt, 2016), might reduce the levels of 2-monoacylglycerols acting at CB2R, GPR119, and TRPV1, which, unlike CB1R, may have beneficial effects in such pathologic conditions (Di Marzo and Silvestri, 2019). In yet other pathologic conditions, however, where both cannabinoid and other eCBome receptors play similar functions (e.g., inflammation, neurodegeneration, and mood control), such an intrinsic lack of functional selectivity of inhibitors of eCB metabolism may provide additional advantages. It is, therefore, clear that the use of such inhibitors requires (1) first the understanding of what eCBome receptors are involved in a given disorder and (2) in case of conflicting effects being predicted for the blockade of a given enzyme, the development of multitarget drugs, capable also of modifying the activity of targets that exert opposing roles in a given disorder (see, for review, Maione et al., 2013). The eCBome mediators and their synthetic analogs that are under clinical testing are listed in Table 15.
TABLE 15.
Endocannabinoidome mediators and their synthetic analogs under clinical testing
| Name | Company (and Commercial Name) when Available | Indication(s) | Main Proposed Target | Clinical Trials Gov Identifier |
|---|---|---|---|---|
| N-Palmitoylethanolamine (PEA) | Epitech Italy (Normast, Pelvilen, Glialia) | Neuropathic pain from spinal cord injury Covid-19 Fibromyalgia Chronic pain Chronic pelvic pain in endometriosis Frontotemporal Dementia Tourette’s syndrome (in combination with dronabinol) |
PPARα GPR55a TRPV1b CB2Rb |
NCT01851499 NCT04568876 NCT04488926 NCT02699281 NCT02372903 NCT04489017 NCT03066193 |
| N-Oleoylethanolamine (OEA) | University of California Davis Davis, California, United States NutriForward LLC (RiduZone, 90% OEA) |
Post-prandial inflammation Overweight and obesity |
PPARα TRPV1 GPR119 |
NCT05017428 NCT04614233 |
| Bimatoprost (Prostamide F2α analog) | Allergan (Lumigan) | Intraocular Pressure, Glaucoma, Ocular Hypertension Eyelash hypotrichosis, alopecia |
FP/Alt4-FP heteromer FPa |
Several completed studies Several completed studies |
Reported studies are mostly completed (only in a few cases recruitment is still ongoing), and results of most of them have not been disclosed yet. An exception is the case of bimatoprost, which has proven to be very effective on eyelash hypotrichosis and promising on various forms of alopecia (Jha et al., 2018). N-Palmitoylethanolamine is often administered as the ultramicronised solid, and/or in combination with other molecules, such as transpolydatin (in Pelvilen, for pelvic pain) or luteolin (Glialia, for some CNS disorders) (Petrosino and Di Marzo, 2017). Abbreviations: see Table 14.
aStill controversial.
bThese targets are activated indirectly via elevation of endogenous ligand levels or activity.
B. Interaction with the Gut Microbiome
An additional possibility that is attracting attention is to explore targeted nutritional strategies for the therapeutic manipulation of the eCBome, based on the concept that the content of different eCBome mediators is strongly affected by the presence of their fatty acid precursors in the diet (Castonguay-Paradis et al., 2020). Indeed, an important opportunity opened by the discovery of the eCBome lies in its capability of interacting, much more than the ECS does, with another fundamental player in mammalian physiology and pathology that, like the eCBome, is also strongly influenced by diet, medications, and other environmental and lifestyle as well as genetic factors: the gut microbiome (Di Marzo and Silvestri, 2019). Trillions of microorganisms, belonging to thousands of species from several phyla (bacteria, archea, viruses, yeasts, eukaryote parasites), populate the mammalian gut and communicate with, and subsequently regulate, the activity of host cells, mostly through the production of a plethora of chemical signals that are capable of interacting with host targets. In particular, some small molecules typically produced by gut bacteria following the digestion (fermentation) of dietary macro- and micro-nutrients, have been well characterized and include, among others, (1) short chain fatty acids (derived from the processing of dietary complex carbohydrates), (2) branched chain fatty acids and amino acids (usually derived from the processing of dietary proteins), (3) tryptophan derivatives, and (4) secondary bile acids (Gold and Zhu J, 2022). These molecules usually act at receptors located in host cells (e.g., GPCRs and PPARs, in the case of branched chain fatty acids, and the aryl hydrocarbon receptor, in the case of some tryptophan derivatives), as recently reviewed (Ikeda et al., 2022; Wang et al., 2022). However, only recently it has become evident that some gut bacteria and yeasts can produce eCB-like molecules, such as N-acylated glycines, dopamines, tyramines, phenylethylamines, and tryptamines, as well as oxyeCBome mediators capable of binding to the same receptors as the host eCBome mediators (De Petrocellis et al., 2009; Cohen et al., 2017; Chang et al., 2021). This emerging “microbendocannabinoidome,” also summarized in Table 14, enlarges the span of microbe-host communication and expands it to the eCBome, as depicted in Fig. 28. It also adds to previous evidence suggesting that, reciprocally, the ECS modulates the gut microbiome.
Fig. 28.

Reciprocal modulation of the ECS and the gut microbiome. The emerging microbendocannabinoidome (μbeCBome), also summarized in Table 14, enlarges the span of microbe-host communication and expands it to the eCBome.
This evidence came from experiments carried out in animal models of obesity, gut dysbiosis (i.e., perturbation of gut microbiota composition and function), and ensuing metabolic endotoxaemia using CB1R antagonists and TRPV1 agonists (namely capsaicin) (Cluny et al., 2015; Shen et al., 2017; Mehrpouya-Bahrami et al., 2017), as well as in mice where eCB metabolic enzymes were knocked out (Geurts et al., 2015; Dione et al., 2020). These interventions, together with the expected alterations of eCB and eCBome signaling, were found to lead to concomitant and interrelated modulation of metabolic and inflammatory parameters and gut microbiota composition, with increases of the relative abundance of beneficial gut bacteria species, such as Akkermansia muciniphila. More recently, some therapeutic effects of pCBs were likewise found to be accompanied by corresponding beneficial actions on the gut microbiome. Clearly, it is difficult to understand solely from these in vivo studies whether the effects observed on the gut microbiome were the direct consequence of the pharmacological and genetic manipulation of the eCBome or only an indirect and host-mediated effect of the latter.
In fact, there is now also accumulating ex vivo and in vitro evidence that host-derived eCB-like mediators can directly affect the composition and function of the gut microbiome. In particular, both N-acylethanolamines and 2-monoacylglycerols were found to affect the function (e.g., proliferation, biofilm formation, and virulence) of gut bacteria, clearly through mechanisms and at experimental concentrations that are quite different from those underlying their effects on host cells (Ellermann et al., 2020; Dione et al., 2020; Fornelos et al., 2020; Sionov and Steinberg, 2022). On the other hand, it was also shown that the manipulation of the gut microbiome, either in germ-free mice or following prolonged treatment of mice with antibiotics, directly affects the expression of eCBome receptors, metabolic enzymes, and mediators in the gut and other, more distal host tissues like the brain (Muccioli et al., 2010; Aguilera et al., 2015; Manca et al., 2020a, b). The mechanisms through which this influence is exerted are not yet known but are likely to be due to the action on host cells of the aforementioned microbiome-derived metabolites (i.e., short chain fatty acids) and may include epigenetic regulation of genes encoding for eCBome proteins, since these changes can be reversed upon reinstatement of the gut microbiome.
Also, probiotics are known to affect the intestinal ECS and to potentially owe to this interaction part of their pharmacological actions (Rousseaux et al., 2007; Ringel-Kulka et al., 2014; Rossi et al., 2020; Cuozzo et al., 2021). Yet, it remains unclear whether these effects are due to a direct action of probiotic bacterial species on host cells or to their capability of modulating the gut microbiome. Interestingly A. muciniphila, a proposed gut microbiota-derived probiotic whose administration produces beneficial actions in both animal models of obesity and dysmetabolism and authentic obese subjects with metabolic syndrome (Everard et al., 2013; Plovier et al., 2017; Depommier et al., 2019), was found to increase the intestinal levels of pharmacologically active 2-monoacylglycerols (i.e., 2-AG, 2-palmitoyl-glycerol, and 2-oleoyl-glycerol) in mice with diet-induced obesity (Everard et al., 2013), as well as the circulating levels of 2-palmitoyl-glycerol—a PPARα agonist with potential metabolic beneficial activity—in obese individuals (Depommier et al., 2021). Probiotics can also reverse the gut dysbiosis-induced alterations of N-acyl-serotonin and N-acylethanolamine concentrations in gut (Guida et al., 2018) or adipose tissue (Geurts et al., 2015), respectively, with a corresponding amelioration of dysmetabolism and mood disturbances, respectively; again it is not clear whether these effects were exerted directly on eCBome mediator biosynthesis or degradation. Clearly, in vitro studies, using, for example, cocultures of commensal bacteria or probiotic species (or their culture media) with mammalian intestinal cells or organoids, are again needed to understand whether these in vivo effects are the consequence of direct bacterial interactions with host cells.
VII. Conclusion
Plant-derived cannabinoids and endocannabinoids represent two different but equally complex systems, so that the terms “(phyto)cannabinoids” and “endocannabinoids” are actually used to identify rather heterogeneous groups of lipophilic substances. It is striking how some of these molecules happened to share 3D structures, allowing exogenous pCBs to play so many biologic activities in our body, because they mimic eCBs. The additional layer of complexity brought about by these structural similarities makes extremely challenging the use of pCBs and ECS-oriented drugs as potential therapeutics to combat human diseases and requires deeper knowledge of the structural and functional details of their potential targets in the cell. Undoubtedly, a better understanding of these fine molecular clues will allow us to turn pCBs and ECS-oriented drugs from threats to a treasure trove for human health.
Among the various components of the ECS, CB1R, CB2R, and FAAH have been the most largely exploited to develop therapeutic drugs for human diseases.
Shortly after the discovery of CB1R, many therapeutic opportunities identified it for its agonists and antagonists; yet, improvements in medicinal cannabinoids are continually meeting novel challenges (Pacher and Kunos, 2013). Selectivity for specific tissue responses is necessary to promote beneficial therapeutic responses while minimizing side effects. Developing agonists that are highly selective for CB1R but devoid of activity at other receptors (e.g., CB2R, GPR55) continues to remain a challenge. Organ-system selectivity is a second goal for minimizing CNS actions of CB1R agonists and antagonists and is being met by the development of peripherally restricted ligands that have limited access to brain CB1R (Amato et al., 2019). A third approach to selectivity involves tuning the functional outcome of ligands such as “biased agonists” that would modify the active CB1R conformation to direct signaling preferentially through either G protein pathways or β-arrestin pathways and, further, to select for individual G protein subtypes (Gs or G12/13 versus Gi/o) and for β-arrestin 1 versus β-arrestin 2. A fourth mechanism for selectivity is to develop allosteric modulators whose effects would be limited to only those receptors concurrently being stimulated by an endogenous agonist. A positive allosteric modulator would augment the response to eCBs and could potentiate ongoing stimulatory signals, whereas a negative allosteric modulator would be expected to provide noncompetitive inhibition to those receptors receiving an endocannabinoid signal. Additionally, future goals for antagonists would be the development of biased antagonists, negative allosteric modulators, as well as neutral antagonists that do not affect the basal activity of CB1R. A still unexplored approach with therapeutic potential that is closer to the concept of polypharmacology involves specifically inhibiting CB1R and activating CB2R. Thus, the future for CB1R pharmacotherapeutics can be predicted to move from phytocannabinoid preparations to agonists and antagonists that exhibit greater selectivity through one of these strategies.
Also, CB2R is a key element of the ECS. It is highly expressed in immune cells, and its activation limits inflammation and associated tissue injury under multiple pathologic conditions. Efficacy in preclinical models of pain, neurodegenerative, cardiovascular, gastrointestinal, liver, kidney, and lung diseases has been demonstrated. Due to this enormous therapeutic potential, a multitude of CB2R ligands has been developed that can be categorized as eCBs and related fatty acid derivatives, pCBs, or synthetic CB2R ligands. The majority of these ligands include agonists, modulators, neutral antagonists, inverse agonists, allosteric ligands, as well as labeled chemical probes. Altogether, a large, structurally diverse chemical space is covered. Generally, early CB2R modulators are dual CB1R/CB2R agonists that are mostly not quite “drug-like.” In contrast, recent ligands often combine high potency for CB2R with favorable overall ADME profiles including low lipophilicity, aqueous solubility, and favorable plasma protein binding, which translate into excellent pharmacokinetic profiles and consequently improved developability. To overcome CB1R-driven psychotropic effects, two strategies were followed: limiting exposure toward the periphery or enhancing the selectivity over CB1R through excellent structure activity relationship work in the lead optimization phase, thus enabling clinical studies with more than 20 new molecular entities. First, trials focused on diseases of the CNS and pain. Most recent ligands and clinical studies focus on peripheral indications with a strong inflammatory/immunomodulatory and/or fibrotic background. Three phytocannabinoids (THC, nabilone, and cannabidiol) have been launched. Most advanced selective CB2R agonists are in phase 2 clinical trials. While no CB2R-related toxicity issues have been reported from clinical studies, the demonstration of target engagement and the identification of best-suited human disease condition(s) for the therapeutic use of CB2R modulators still poses challenges for the development of CB2R-based therapies. The generation of translational animal models and a better understanding of CB2R and the ECS in general will help unlock the receptor’s full therapeutic potential. Recently discovered high-quality labeled chemical probes have enabled a better understanding of CB2R expression, mechanism of action, and translatability of results toward the human situation. The in-depth understanding of signaling bias as well as CB2R receptor homo- and heterodimers might translate into different functional properties and ultimately tailor-made CB2R therapeutics. Deeper insights into drug-target binding kinetics, their impact on receptor function, and, in particular, the recently reported structures of antagonist- and agonist-bound CB2R and knowledge on allosterism will facilitate rational drug design. Together with the huge chemical space available to generate tailor-made CB2R modulators, this will hopefully guide us to the discovery of potent, effective, and safe medicines for indications with a dire or even unmet need.
Finally, since the discovery of FAAH and MAGL many compounds have been developed starting from inhibitors of other known serine hydrolases to study enzyme activity, its regulation, and relevance in the pathophysiological process. Over the years, different approaches have been used to identify and synthesize new classes of single or dual inhibitors, paying more and more attention to the analysis of their selectivity, potency, and mechanism of action. The development of selective FAAH and MAGL inhibitors remains one of the major issues in drug discovery, as also demonstrated by the large number of research papers and review articles devoted to this topic. The main interest in this field is to develop a therapeutic alternative to the use of cannabinoid receptor agonists, able to prevent or minimize serious psychotropic side effects due to direct receptor activation. The possibility to increase eCB tone by reducing degradation, and to apply new multitarget strategies that include additional receptors and enzymes, have boosted FAAH and MAGL inhibition studies to generate therapeutics against peripheral and CNS-related pathologies. However, more details seem to be necessary on 3D structure, catalytic mechanism, and regulation of both hydrolases to design effective inhibitors devoid of off-target activity. Novel in silico approaches like computer-aided drug discovery may be useful to reach this goal. In this context, it has to be recalled the tragic use of BIA10-2474, a purported FAAH inhibitor that killed one volunteer and led four others to hospitalization in phase I clinical trials because of serious adverse neurologic events (Kerbrat et al., 2016). This BIA 10-2474 disaster clearly reminds us that accurate preclinical characterization of the biochemical profile of any new chemical entity must be performed, before claiming that a new selective drug has been discovered, and especially before allowing its use in clinical trials.
Overall, the same potentialities and limitations revealed by accumulated evidence in so many studies on CB1R, CB2R, FAAH, and MAGL are likely to apply also to the more recently characterized elements of the ECS and hopefully will be instructive to avoid making the same mistakes. In particular, a better appreciation of the 3D structure of the desired targets, also via cryo-electron microscopy (Hua et al., 2020), and the application of powerful in silico tools like CADD, virtual screening (Stasiulewicz et al., 2022), and machine learning (Atz et al., 2023) hold promise to shorten the path from the bench to the patient’s bed in drug discovery programs oriented toward the ECS. This knowledge of structural details will also help to decipher complex interactions between pCBs/eCBs and other bioactive lipids (e.g., eicosanoids and specialized pro-resolving mediators) that are receiving increasing attention for their therapeutic potential to treat human diseases (Maccarrone, 2023).
Acknowledgments
M.M. wishes to express gratitude to all colleagues who contributed over the last 30 years to his studies on the ECS and its implications for human health and disease. M.M. also thanks Cinzia Rapino (University of Teramo, Teramo, Italy) and Marina Fava (Campus Bio-Medico University of Rome, Rome, Italy) for their help in preparing Tables 1 to 3 and Figures 1 to 7, respectively. A.M. thanks Christos Iliopoulos Tsoutsouvas (Northeastern University, Boston, MA, USA) for his support in collecting information on CB1R and preparing Table 4 and Figures 8 and 9. D.P. thanks Yannick Fotio (University of California, Irvine, CA, USA) for his support in preparing Figures 23 and 24. M.vd S. thanks Na Zhu and Anthe Janssen (Leiden University, Leiden, Netherlands) for preparing the schemes in Tables 1, 3, 7, and 9 and for Figure 25.
Abbreviations
- AA
arachidonic acid
- 2-AG
2-arachidonoylglycerol
- AEA
N-arachidonyl ethanolamine (anandamide)
- CADD
computer-aided drug discovery
- CBD
cannabidiol
- CB1R
cannabinoid receptor 1
- CB2R
cannabinoid receptor 2
- CNS
central nervous system
- COVID-19
coronavirus disease of 2019
- COX
cyclooxygenase
- cPLA
cytosolic phospholipase A
- CYP450
cytochrome P450
- DAGL
diacylglycerol lipase
- DSE
depolarization-induced suppression of excitation
- eCB
endocannabinoid
- eCBome
endocannabinoidome
- ECS
endocannabinoid system
- EMA
European Medicines Agency
- FAAH
fatty acid amide hydrolase
- FABP
fatty acid binding protein
- FDA
Food and Drug Administration
- GPCR
G protein-coupled receptor
- LOX
lipoxygenase
- MAGL
monoacylglycerol lipase
- MS
multiple sclerosis
- NAAA
N-acylethanolamine acid amide hydrolase
- NAPE-PLD
N-acyl phosphatidylethanolamine-specific phospholipase D
- NAT
N-acyltransferase
- NOS
nitric oxide synthase
- PAM
peptidyl-glycine alpha-amidating monooxygenase
- PGE2-G
prostaglandin E2 glyceryl ester
- PGH2-EA
prostaglandin H2 ethanolamide
- PLC
phospholipase C
- PPAR
peroxisome proliferator-activated receptor
- SAR
structure-activity relationship
- SERI
selective eCB reuptake inhibitor
- (S)-OOPP
N-[(3S)-2-oxo-3-oxetanyl]-3-phenylpropanamide
- THC
Δ9-tetrahydrocannabinol
- TRP(V)
transient receptor potential (vanilloid).
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Maccarrone, Di Marzo, Gertsch, Grether, Howlett, Hua, Makriyannis, Piomelli, Ueda, Van der Stelt.
Note Added in Proof: Two mistakes were found in Figure 4 and Figure 6 published in the Fast Forward version published May 10, 2023. Figures 4 and 6 have now been corrected.
Footnotes
All authors have contributed equally to this study.
For this investigation M.M. was partly supported by the Italian Ministry of Health through the competitive Ricerca Finalizzata 2018 [Grant RF-2018-12365391]. A.C.H. was supported by National Institutes of Health National Institute on Drug Abuse [Grant R01-DA042157]. A.M. was supported by the National Institute on Drug Abuse [Grant P01-DA009158] and by the National Institute on Alcohol Abuse and Alcoholism [Grant U01-AA028963].
No author has an actual or perceived conflict of interest with the contents of this article.
References
- Abrams DI, Guzman M (2015) Cannabis in cancer care. Clin Pharmacol ther 97:575–586. [DOI] [PubMed] [Google Scholar]
- Adams R, MacKenzie S Jr, Loewe S (1948) Tetrahydrocannabinol homologs with double branched alkyl groups in the 3-position. J Am Chem Soc 70:664–668. [DOI] [PubMed] [Google Scholar]
- Addy C, Wright H, Van Laere K, Gantz I, Erondu N, Musser BJ, Lu K, Yuan J, Sanabria-Bohórquez SM, Stoch A, et al. (2008) The acyclic CB1R inverse agonist taranabant mediates weight loss by increasing energy expenditure and decreasing caloric intake. Cell Metab 7:68–78. [DOI] [PubMed] [Google Scholar]
- Aghazadeh Tabrizi M, Baraldi PG, Borea PA, Varani K (2016) Medicinal chemistry, pharmacology, and potential therapeutic benefits of cannabinoid CB2 receptor agonists. Chem Rev 116:519–560. [DOI] [PubMed] [Google Scholar]
- Aguilera M, Cerdà-Cuéllar M, Martínez V (2015) Antibiotic-induced dysbiosis alters host-bacterial interactions and leads to colonic sensory and motor changes in mice. Gut Microbes 6:10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad R, Koole M, Evens N, Serdons K, Verbruggen A, Bormans G, Van Laere K (2013) Whole-body biodistribution and radiation dosimetry of the cannabinoid type 2 receptor ligand [11C]-NE40 in healthy subjects. Mol Imaging Biol 15:384–390. [DOI] [PubMed] [Google Scholar]
- Ahmad R, Postnov A, Bormans G, Versijpt J, Vandenbulcke M, Van Laere K (2016) Decreased in vivo availability of the cannabinoid type 2 receptor in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 43:2219–2227. [DOI] [PubMed] [Google Scholar]
- Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, et al. (2007) Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry 46:13019–13030. [DOI] [PubMed] [Google Scholar]
- Alexander JP, Cravatt BF (2006) The putative endocannabinoid transport blocker LY2183240 is a potent inhibitor of FAAH and several other brain serine hydrolases. J Am Chem Soc 128:9699–9704. [DOI] [PubMed] [Google Scholar]
- Alhouayek M, Bottemanne P, Subramanian KV, Lambert DM, Makriyannis A, Cani PD, Muccioli GG (2015) N-Acylethanolamine-hydrolyzing acid amidase inhibition increases colon N-palmitoylethanolamine levels and counteracts murine colitis. FASEB J 29:650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M, Masquelier J, Muccioli GG (2018) Lysophosphatidylinositols, from cell membrane constituents to GPR55 ligands. Trends Pharmacol Sci 39:586–604. [DOI] [PubMed] [Google Scholar]
- Al-Zoubi R, Morales P, Reggio PH (2019) Structural insights into CB1 receptor biased signaling. Int J Mol Sci 20:1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato G, Khan NS, Maitra R (2019) A patent update on cannabinoid receptor 1 antagonists (2015-2018). Expert Opin Ther Pat 29:261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato GS, Manke A, Harris DL, Wiethe RW, Vasukuttan V, Snyder RW, Lefever TW, Cortes R, Zhang Y, Wang S, et al. (2018) Blocking alcoholic steatosis in mice with a peripherally restricted purine antagonist of the type 1 cannabinoid receptor. J Med Chem 61:4370–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin MR, Ali DW (2019) Pharmacology of medical cannabis. Adv Exp Med Biol 1162:151–165. [DOI] [PubMed] [Google Scholar]
- Anagnostopoulos D, Rakiec C, Wood J, Pandarinathan L, Zvonok N, Makriyannis A, Siafaka-Kapadai A (2010) Identification of endocannabinoids and related N-acylethanolamines in tetrahymena. A new class of compounds for Tetrahymena. Protist 161:452–465. [DOI] [PubMed] [Google Scholar]
- Anand P, Whiteside G, Fowler CJ, Hohmann AG (2009) Targeting CB2 receptors and the endocannabinoid system for the treatment of pain. Brain Res Brain Res Rev 60:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer RA, Blanchard WB, Day WA, Johnson DW, Lavagnino ER, Ryan CW, Baldwin JE (1977) Cannabinoids. 3. Synthetic approaches to 9-ketocannabinoids. Total synthesis of nabilone. J Org Chem 42:2277–2284. [DOI] [PubMed] [Google Scholar]
- Arnold WR, Carnevale LN, Xie Z, Baylon JL, Tajkhorshid E, Hu H, Das A (2021) Anti-inflammatory dopamine- and serotonin-based endocannabinoid epoxides reciprocally regulate cannabinoid receptors and the TRPV1 channel. Nat Commun 12: 926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Mackie K (2010) CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol 160:467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Wager-Miller J, Haskins C, Straiker A, Mackie K (2012) Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB(2) ligands. Mol Pharmacol 81:250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atz K, Guba W, Grether U, Schneider G (2023) Machine learning and computational chemistry for the endocannabinoid system. Methods Mol Biol 2576:477–493. [DOI] [PubMed] [Google Scholar]
- Ayoub SM, Smoum R, Farag M, Atwal H, Collins SA, Rock EM, Limebeer CL, Piscitelli F, Iannotti FA, Lichtman AH, et al. (2020) Oleoyl alanine (HU595): a stable monomethylated oleoyl glycine interferes with acute naloxone precipitated morphine withdrawal in male rats. Psychopharmacology (Berl) 237:2753–2765. [DOI] [PubMed] [Google Scholar]
- Baggelaar MP, Chameau PJ, Kantae V, Hummel J, Hsu KL, Janssen F, van der Wel T, Soethoudt M, Deng H, den Dulk H, et al. (2015) Highly selective, reversible inhibitor identified by comparative chemoproteomics modulates diacylglycerol lipase activity in neurons. J Am Chem Soc 137:8851–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggelaar MP, Maccarrone M, van der Stelt M (2018) 2-Arachidonoylglycerol: a signaling lipid with manifold actions in the brain. Prog Lipid Res 71:1–17. [DOI] [PubMed] [Google Scholar]
- Baggelaar MP, van Esbroeck AC, van Rooden EJ, Florea BI, Overkleeft HS, Marsicano G, Chaouloff F, van der Stelt M (2017) Chemical proteomics maps brain region specific activity of endocannabinoid hydrolases. ACS Chem Biol 12:852–861. [DOI] [PubMed] [Google Scholar]
- Bainbridge MN, Mazumder A, Ogasawara D, Abou Jamra R, Bernard G, Bertini E, Burglen L, Cope H, Crawford A, Derksen A, et al. ; Rady Children’s Institute for Genomic Medicine; Undiagnosed Disease Network (2022) Endocannabinoid dysfunction in neurological disease: neuro-ocular DAGLA-related syndrome. Brain 145:3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakas T, van Nieuwenhuijzen PS, Devenish SO, McGregor IS, Arnold JC, Chebib M (2017) The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABAA receptors. Pharmacol Res 119:358–370. [DOI] [PubMed] [Google Scholar]
- Balla A, Dong B, Shilpa BM, Vemuri K, Makriyannis A, Pandey SC, Sershen H, Suckow RF, Vinod KY (2018) Cannabinoid-1 receptor neutral antagonist reduces binge-like alcohol consumption and alcohol-induced accumbal dopaminergic signaling. Neuropharmacology 131:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balvers MG, Verhoeckx KC, Witkamp RF (2009) Development and validation of a quantitative method for the determination of 12 endocannabinoids and related compounds in human plasma using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 877:1583–1590. [DOI] [PubMed] [Google Scholar]
- Bari M, Battista N, Fezza F, Finazzi-Agrò A, Maccarrone M (2005) Lipid rafts control signaling of type-1 cannabinoid receptors in neuronal cells. Implications for anandamide-induced apoptosis. J Biol Chem 280:12212–12220. [DOI] [PubMed] [Google Scholar]
- Bartholomäus R, Nicolussi S, Baumann A, Rau M, Simão AC, Gertsch J, Altmann KH (2019) Total synthesis of the endocannabinoid uptake inhibitor guineensine and SAR studies. ChemMedChem 14:1590–1596. [DOI] [PubMed] [Google Scholar]
- Bartolozzi A, Cirillo PF, Berry AK, Hickey ER, Thomson DS, Wu L, Zindell R, Albrecht C, Ceci A, Gemkow MJ, et al. (2015) Selective CB2 receptor agonists. Part 3: the optimization of a piperidine-based series that demonstrated efficacy in an in vivo neuropathic pain model. Bioorg Med Chem Lett 25:587–592. [DOI] [PubMed] [Google Scholar]
- Barutta F, Bellini S, Mastrocola R, Gambino R, Piscitelli F, di Marzo V, Corbetta B, Vemuri VK, Makriyannis A, Annaratone L, et al. (2018) Reversal of albuminuria by combined AM6545 and perindopril therapy in experimental diabetic nephropathy. Br J Pharmacol 175:4371–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutta F, Grimaldi S, Gambino R, Vemuri K, Makriyannis A, Annaratone L, di Marzo V, Bruno G, Gruden G (2017) Dual therapy targeting the endocannabinoid system prevents experimental diabetic nephropathy. Nephrol Dial Transplant 32:1655–1665. [DOI] [PubMed] [Google Scholar]
- Basagni F, Rosini M, Decker M (2020) Functionalized cannabinoid subtype 2 receptor ligands: fluorescent, PET, photochromic and covalent molecular probes. ChemMedChem 15:1374–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashashati M, Fichna J, Piscitelli F, Capasso R, Izzo AA, Sibaev A, Timmermans JP, Cenac N, Vergnolle N, Di Marzo V, et al. (2017) Targeting fatty acid amide hydrolase and transient receptor potential vanilloid-1 simultaneously to modulate colonic motility and visceral sensation in the mouse: a pharmacological intervention with N-arachidonoyl-serotonin (AA-5-HT). Neurogastroenterol Motil 29:12. [DOI] [PubMed] [Google Scholar]
- Battista N, Bari M, Finazzi-Agrò A, Maccarrone M (2002) Anandamide uptake by synaptosomes from human, mouse and rat brain: inhibition by glutamine and glutamate. Lipids Health Dis 1: PMC139962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur R, Gertsch J, Sigel E (2013) Do N-arachidonyl-glycine (NA-glycine) and 2-arachidonoyl glycerol (2-AG) share mode of action and the binding site on the β2 subunit of GABAA receptors? PeerJ 1:e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MR, D’Ambra TE, Kumar V, Eissenstat MA, Herrmann JL Jr, Wetzel JR, Rosi D, Philion RE, Daum SJ, Hlasta DJ, et al. (1991) Antinociceptive (aminoalkyl)indoles. J Med Chem 34:1099–1110. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Piomelli D (1999) Anandamide transport inhibition by the vanilloid agonist olvanil. Eur J Pharmacol 364:75–78. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D (1997) Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 277:1094–1097. [DOI] [PubMed] [Google Scholar]
- Berger WT, Ralph BP, Kaczocha M, Sun J, Balius TE, Rizzo RC, Haj-Dahmane S, Ojima I, Deutsch DG (2012) Targeting fatty acid binding protein (FABP) anandamide transporters - a novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS One 7:e50968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings RE, Whitaker GW, McMahon RE (1980) The stereoselective enzymic reduction of the synthetic 9-ketocannabinoid, nabilone, in vivo, in isolated liver cells and in liver homogenate. Xenobiotica 10:33–36. [DOI] [PubMed] [Google Scholar]
- Bíró T, Tóth BI, Haskó G, Paus R, Pacher P (2009) The endocannabinoid system of the skin in health and disease: novel perspectives and therapeutic opportunities. Trends Pharmacol Sci 30:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Hanus L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I, Moriello AS, Davis JB, Mechoulam R, Di Marzo V (2001) Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol 134:845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, et al. (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163:463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Mahadevan A, Coccurello R, Chang JW, Allarà M, Chen Y, Giacovazzo G, Lichtman A, Cravatt B, Moles A, et al. (2013) A novel fluorophosphonate inhibitor of the biosynthesis of the endocannabinoid 2-arachidonoylglycerol with potential anti-obesity effects. Br J Pharmacol 169:784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Cravatt BF (2013) Chemical probes of endocannabinoid metabolism. Pharmacol Rev 65:849–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14:1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluett RJ, Báldi R, Haymer A, Gaulden AD, Hartley ND, Parrish WP, Baechle J, Marcus DJ, Mardam-Bey R, Shonesy BC, et al. (2017) Endocannabinoid signalling modulates susceptibility to traumatic stress exposure. Nat Commun 8:14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan DM, Studholme K, DiBua A, Gordon C, Kanjiya MP, Yu M, Puopolo M, Kaczocha M (2022) FABP5 deletion in nociceptors augments endocannabinoid signaling and suppresses TRPV1 sensitization and inflammatory pain. Sci Rep 12:9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond RA, Ijzerman AP (2006) Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol Sci 27:92–96. [DOI] [PubMed] [Google Scholar]
- Bonezzi FT, Sasso O, Pontis S, Realini N, Romeo E, Ponzano S, Nuzzi A, Fiasella A, Bertozzi F, Piomelli D (2016) An important role for N-acylethanolamine acid amidase in the complete Freund’s adjuvant rat model of arthritis. J Pharmacol Exp Ther 356:656–663. [DOI] [PubMed] [Google Scholar]
- Bononi G, Poli G, Rizzolio F, Tuccinardi T, Macchia M, Minutolo F, Granchi C (2021) An updated patent review of monoacylglycerol lipase (MAGL) inhibitors (2018-present). Expert Opin Ther Pat 31:153–168. [DOI] [PubMed] [Google Scholar]
- Börner C, Smida M, Höllt V, Schraven B, Kraus J (2009) Cannabinoid receptor type 1- and 2-mediated increase in cyclic AMP inhibits T cell receptor-triggered signaling. J Biol Chem 284:35450–35460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2007) Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry 62:1103–1110. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Rinaldi M, Carayon P, Carillon C, Delpech B, Shire D, Le Fur G, Casellas P (1993) Cannabinoid-receptor expression in human leukocytes. Eur J Biochem 214:173–180. [DOI] [PubMed] [Google Scholar]
- Bowen KJ, Kris-Etherton PM, Shearer GC, West SG, Reddivari L, Jones PJH (2017) Oleic acid-derived oleoylethanolamide: a nutritional science perspective. Prog Lipid Res 67:1–15. [DOI] [PubMed] [Google Scholar]
- Bowles NP, Karatsoreos IN, Li X, Vemuri VK, Wood JA, Li Z, Tamashiro KL, Schwartz GJ, Makriyannis AM, Kunos G, et al. (2015) A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolic syndrome. Proc Natl Acad Sci USA 112:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF (2002) Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 298:1793–1796. [DOI] [PubMed] [Google Scholar]
- Brailoiu GC, Deliu E, Marcu J, Hoffman NE, Console-Bram L, Zhao P, Madesh M, Abood ME, Brailoiu E (2014) Differential activation of intracellular versus plasmalemmal CB2 cannabinoid receptors. Biochemistry 53:4990–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke B, Gazzi T, Atz K, Fingerle J, Kuner P, Schindler T, Weck G, Nazaré M, Grether U (2021) Cannabinoid receptor type 2 ligands: an analysis of granted patents since 2010. Pharm Pat Anal 10:111–163. [DOI] [PubMed] [Google Scholar]
- Brodie JS, Di Marzo V, Guy GW (2015) Polypharmacology shakes hands with complex aetiopathology. Trends Pharmacol Sci 36:802–821. [DOI] [PubMed] [Google Scholar]
- Brown I, Cascio MG, Wahle KW, Smoum R, Mechoulam R, Ross RA, Pertwee RG, Heys SD (2010) Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines. Carcinogenesis 31:1584–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown I, Lee J, Sneddon AA, Cascio MG, Pertwee RG, Wahle KWJ, Rotondo D, Heys SD (2020) Anticancer effects of n-3 EPA and DHA and their endocannabinoid derivatives on breast cancer cell growth and invasion. Prostaglandins Leukot Essent Fatty Acids 156:102024. [DOI] [PubMed] [Google Scholar]
- Brüser A, Zimmermann A, Crews BC, Sliwoski G, Meiler J, König GM, Kostenis E, Lede V, Marnett LJ, Schöneberg T (2017) Prostaglandin E2 glyceryl ester is an endogenous agonist of the nucleotide receptor P2Y6. Sci Rep 7:2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch P, Chicca A, Gertsch J, Gademann K (2013) Functionally optimized neuritogenic farinosone C analogs: SAR r-study and investigations on their mode of action. ACS Med Chem Lett 5:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral GA, Griffin-Thomas L (2008) Cannabinoids as therapeutic agents for ablating neuroinflammatory disease. Endocr Metab Immune Disord Drug Targets 8:159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F (2008) CB2 receptors in the brain: role in central immune function. Br J Pharmacol 153:240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadas H, di Tomaso E, Piomelli D (1997) Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J Neurosci 17:1226–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D (1998) Control of pain initiation by endogenous cannabinoids. Nature 394:277–281. [DOI] [PubMed] [Google Scholar]
- Campos AC, Fogaça MV, Scarante FF, Joca SRL, Sales AJ, Gomes FV, Sonego AB, Rodrigues NS, Galve-Roperh I, Guimarães FS (2017) Plastic and neuroprotective mechanisms involved in the therapeutic effects of cannabidiol in psychiatric disorders. Front Pharmacol 8:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS (2012) Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Philos Trans R Soc Lond B Biol Sci 367:3364–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Mulvihill MM, Mukhopadhyay P, Xu H, Erdélyi K, Hao E, Holovac E, Haskó G, Cravatt BF, Nomura DK, et al. (2013) Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology 144:808–817.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlini EA, Cunha JM (1981) Hypnotic and antiepileptic effects of cannabidiol. J Clin Pharmacol 21(Suppl 1):417S–427S. [DOI] [PubMed] [Google Scholar]
- Cascio MG, Marini P, Pertwee RG (2016) The displacement binding assay using human cannabinoid CB2 receptor-transfected cells. Methods Mol Biol 1412:57–63. [DOI] [PubMed] [Google Scholar]
- Castaneda JT, Harui A, Roth MD (2017) Regulation of cell surface CB2 receptor during human B cell activation and differentiation. J Neuroimmune Pharmacol 12:544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay-Paradis S, Lacroix S, Rochefort G, Parent L, Perron J, Martin C, Lamarche B, Raymond F, Flamand N, Di Marzo V, et al. (2020) Dietary fatty acid intake and gut microbiota determine circulating endocannabinoidome signaling beyond the effect of body fat. Sci Rep 10:15975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi S, Rapino C, Di Nisio V, Rossi G, Maccarrone M (2020) The (endo)cannabinoid signaling in female reproduction: what are the latest advances? Prog Lipid Res 77:101019. [DOI] [PubMed] [Google Scholar]
- Cécyre B, Thomas S, Ptito M, Casanova C, Bouchard JF (2014) Evaluation of the specificity of antibodies raised against cannabinoid receptor type 2 in the mouse retina. Naunyn Schmiedebergs Arch Pharmacol 387:175–184. [DOI] [PubMed] [Google Scholar]
- Cencioni MT, Chiurchiù V, Catanzaro G, Borsellino G, Bernardi G, Battistini L, Maccarrone M (2010) Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLoS One 5:e8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Finazzi-Agrò A, Bernardi G, Maccarrone M (2007) The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol Sci 28:180–187. [DOI] [PubMed] [Google Scholar]
- Chambers AP, Vemuri VK, Peng Y, Wood JT, Olszewska T, Pittman QJ, Makriyannis A, Sharkey KA (2007) A neutral CB1 receptor antagonist reduces weight gain in rat. Am J Physiol Regul Integr Comp Physiol 293:R2185–R2193. [DOI] [PubMed] [Google Scholar]
- Chang C-P, Huang H-L, Huang J-K, Hung M-S, Wu C-H, Song J-S, Lee C-J, Yu C-S, Shia K-S (2019) Fluorine-18 isotope labeling for positron emission tomography imaging. Direct evidence for DBPR211 as a peripherally restricted CB1 inverse agonist. Bioorg Med Chem 27:216–223. [DOI] [PubMed] [Google Scholar]
- Chang FY, Siuti P, Laurent S, Williams T, Glassey E, Sailer AW, Gordon DB, Hemmerle H, Voigt CA (2021) Gut-inhabiting Clostridia build human GPCR ligands by conjugating neurotransmitters with diet- and human-derived fatty acids. Nat Microbiol 6:792–805. [DOI] [PubMed] [Google Scholar]
- Chemin J, Cazade M, Lory P (2014) Modulation of T-type calcium channels by bioactive lipids. Pflugers Arch 466:689–700. [DOI] [PubMed] [Google Scholar]
- Chen Q, Tesmer JJG (2022) G protein-coupled receptor interactions with arrestins and GPCR kinases: the unresolved issue of signal bias. J Biol Chem 298:102279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP, Teng Z, Chen C (2012) Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep 2:1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicca A, Marazzi J, Nicolussi S, Gertsch J (2012) Evidence for bidirectional endocannabinoid transport across cell membranes. J Biol Chem 287:34660–34682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicca A, Nicolussi S, Bartholomäus R, Blunder M, Aparisi Rey A, Petrucci V, Reynoso-Moreno IDC, Viveros-Paredes JM, Dalghi Gens M, Lutz B, et al. (2017) Chemical probes to potently and selectively inhibit endocannabinoid cellular reuptake. Proc Natl Acad Sci USA 114:E5006–E5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicca A, Raduner S, Pellati F, Strompen T, Altmann KH, Schoop R, Gertsch J (2009) Synergistic immunomopharmacological effects of N-alkylamides in Echinacea purpurea herbal extracts. Int Immunopharmacol 9:850–858. [DOI] [PubMed] [Google Scholar]
- Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A (2007) Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet 370:1706–1713. [DOI] [PubMed] [Google Scholar]
- Ciaramellano F, Fanti F, Scipioni L, Maccarrone M, Oddi S (2023) Endocannabinoid metabolism and transport as drug targets. Methods Mol Biol 2576:201–211. [DOI] [PubMed] [Google Scholar]
- Cinar R, Iyer MR, Kunos G (2020) The therapeutic potential of second and third generation CB1R antagonists. Pharmacol Ther 208:107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, et al. (2010) Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci 13:1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarivate (2022a) Drug report on ART-27.13. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/54930 (accessed October 2022).
- Clarivate (2022b) Drug report on cannabidiol. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/57457 (accessed October 2022).
- Clarivate (2022c) Drug report on CMX-020. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/89531 (accessed October 2022).
- Clarivate (2022d) Drug report on dronabinol. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/12866 (accessed October 2022).
- Clarivate (2022e) Drug report on KN 387271. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/22439 (accessed October 2022).
- Clarivate (2022f) Drug report on LY-2828360. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/70612 (accessed October 2022).
- Clarivate (2022g) Drug report on nabiximols. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/94779 (accessed October 2022).
- Clarivate (2022h) Drug report on NTRX-07. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/104592 (accessed October 2022).
- Clarivate (2022i) Drug report on olorinab. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/70435 (accessed October 2022).
- Clarivate (2022j) Drug report on PRS-211375. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/27212 (accessed October 2022).
- Clarivate (2022k) Drug report on RG7774. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/121857 (accessed October 2022).
- Clarivate (2022l) Drug report on S-777469. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/56044 (accessed October 2022).
- Clarivate (2022m) Drug report on TAK-937. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/63642 (accessed October 2022).
- Clarivate (2022n) Drug report on tedalinab. Cortellis data base, https://www.cortellis.com/intelligence/report/ci/nextgendrugall/55827 (accessed October 2022).
- Cluny NL, Keenan CM, Reimer RA, Le Foll B, Sharkey KA (2015) Prevention of diet-induced obesity effects on body weight and gut microbiota in mice treated chronically with Δ9-tetrahydrocannabinol. PLoS One 10:e0144270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluny NL, Vemuri VK, Chambers AP, Limebeer CL, Bedard H, Wood JT, Lutz B, Zimmer A, Parker LA, Makriyannis A, et al. (2010) A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol 161:629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen K, Weizman A, Weinstein A (2019) Positive and negative effects of cannabis and cannabinoids on health. Clin Pharmacol Ther 105:1139–1147. [DOI] [PubMed] [Google Scholar]
- Cohen LJ, Esterhazy D, Kim SH, Lemetre C, Aguilar RR, Gordon EA, Pickard AJ, Cross JR, Emiliano AB, Han SM, et al. (2017) Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 549:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consroe P, Laguna J, Allender J, Snider S, Stern L, Sandyk R, Kennedy K, Schram K (1991) Controlled clinical trial of cannabidiol in Huntington’s disease. Pharmacol Biochem Behav 40:701–708. [DOI] [PubMed] [Google Scholar]
- Consroe P, Sandyk R, Snider SR (1986) Open label evaluation of cannabidiol in dystonic movement disorders. Int J Neurosci 30:277–282. [DOI] [PubMed] [Google Scholar]
- Cooper A, Singh S, Hook S, Tyndall JDA, Vernall AJ (2017) Chemical tools for studying lipid-binding class A G protein-coupled receptors. Pharmacol Rev 69:316–353. [DOI] [PubMed] [Google Scholar]
- Corbus Pharmaceuticals (2022) Drug report on lenabasum. Adis Insight data base, https://adisinsight.springer.com/drugs/800007180.
- Correa F, Hernangómez-Herrero M, Mestre L, Loría F, Docagne F, Guaza C (2011) The endocannabinoid anandamide downregulates IL-23 and IL-12 subunits in a viral model of multiple sclerosis: evidence for a cross-talk between IL-12p70/IL-23 axis and IL-10 in microglial cells. Brain Behav Immun 25:736–749. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH (2001) Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA 98:9371–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87. [DOI] [PubMed] [Google Scholar]
- Cristino L, Bisogno T, Di Marzo V (2020) Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol 16:9–29. [DOI] [PubMed] [Google Scholar]
- Cunha JM, Carlini EA, Pereira AE, Ramos OL, Pimentel C, Gagliardi R, Sanvito WL, Lander N, Mechoulam R (1980) Chronic administration of cannabidiol to healthy volunteers and epileptic patients. Pharmacology 21:175–185. [DOI] [PubMed] [Google Scholar]
- Cuozzo M, Castelli V, Avagliano C, Cimini A, d’Angelo M, Cristiano C, Russo R (2021) Effects of chronic oral probiotic treatment in paclitaxel-induced neuropathic pain. Biomedicines 9:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dainese E, De Fabritiis G, Sabatucci A, Oddi S, Angelucci CB, Di Pancrazio C, Giorgino T, Stanley N, Del Carlo M, Cravatt BF, et al. (2014) Membrane lipids are key modulators of the endocannabinoid-hydrolase FAAH. Biochem J 457:463–472. [DOI] [PubMed] [Google Scholar]
- Dainese E, Oddi S, Simonetti M, Sabatucci A, Angelucci CB, Ballone A, Dufrusine B, Fezza F, De Fabritiis G, Maccarrone M (2020) The endocannabinoid hydrolase FAAH is an allosteric enzyme. Sci Rep 10:2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambra TE, Estep KG, Bell MR, Eissenstat MA, Josef KA, Ward SJ, Haycock DA, Baizman ER, Casiano FM, Beglin NC, et al. (1992) Conformationally restrained analogues of pravadoline: nanomolar potent, enantioselective, (aminoalkyl)indole agonists of the cannabinoid receptor. J Med Chem 35:124–135. [DOI] [PubMed] [Google Scholar]
- D’Aniello E, Fellous T, Iannotti FA, Gentile A, Allarà M, Balestrieri F, Gray R, Amodeo P, Vitale RM, Di Marzo V (2019) Identification and characterization of phytocannabinoids as novel dual PPARα/γ agonists by a computational and in vitro experimental approach. Biochim Biophys Acta, Gen Subj 1863:586–597. [DOI] [PubMed] [Google Scholar]
- Davies AK, Alecu JE, Ziegler M, Vasilopoulou CG, Merciai F, Jumo H, Afshar-Saber W, Sahin M, Ebrahimi-Fakhari D, Borner GHH (2022) AP-4-mediated axonal transport controls endocannabinoid production in neurons. Nat Commun 13:1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TA, Rakhshan F, Deutsch DG, Barker EL (2001) Role of fatty acid amide hydrolase in the transport of the endogenous cannabinoid anandamide. Mol Pharmacol 59:1369–1375. [DOI] [PubMed] [Google Scholar]
- de Almeida DL, Devi LA (2020) Diversity of molecular targets and signaling pathways for CBD. Pharmacol Res Perspect 8:e00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Laurentiis A, Araujo HA, Rettori V (2014) Role of the endocannabinoid system in the neuroendocrine responses to inflammation. Curr Pharm Des 20:4697–4706. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V (2000) Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett 483:52–56. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Cascio MG, Di Marzo V (2004) The endocannabinoid system: a general view and latest additions. Br J Pharmacol 141:765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Deva R, Mainieri F, Schaefer M, Bisogno T, Ciccoli R, Ligresti A, Hill K, Nigam S, Appendino G, et al. (2009) Chemical synthesis, pharmacological characterization, and possible formation in unicellular fungi of 3-hydroxy-anandamide. J Lipid Res 50:658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Di Marzo V (2014) N-Acyldopamines and N-acylserotonins: from synthetic pharmacological tools to endogenous multitarget mediators, in The Endocannabinoidome: The World of Endocannabinoids and Related Mediators (Di Marzo V, Wang J, eds) pp 67–84, Elsevier, Boston. [Google Scholar]
- De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, Stott CG, Di Marzo V (2011) Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol 163:1479–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Marini P, Matias I, Moriello AS, Starowicz K, Cristino L, Nigam S, Di Marzo V (2007) Mechanisms for the coupling of cannabinoid receptors to intracellular calcium mobilization in rat insulinoma beta-cells. Exp Cell Res 313:2993–3004. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Melck D, Bisogno T, Milone A, Di Marzo V (1999) Finding of the endocannabinoid signalling system in Hydra, a very primitive organism: possible role in the feeding response. Neuroscience 92:377–387. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Orlando P, Moriello AS, Aviello G, Stott C, Izzo AA, Di Marzo V (2012) Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol (Oxf) 204:255–266. [DOI] [PubMed] [Google Scholar]
- del Río C, Navarrete C, Collado JA, Bellido ML, Gómez-Cañas M, Pazos MR, Fernández-Ruiz J, Pollastro F, Appendino G, Calzado MA, et al. (2016) The cannabinoid quinol VCE-004.8 alleviates bleomycin-induced scleroderma and exerts potent antifibrotic effects through peroxisome proliferator-activated receptor-γ and CB2 pathways. Sci Rep 6:21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delis F, Polissidis A, Poulia N, Justinova Z, Nomikos GG, Goldberg SR, Antoniou K (2017) Attenuation of cocaine-induced conditioned place preference and motor activity via cannabinoid CB2 receptor agonism and CB1 receptor antagonism in rats. Int J Neuropsychopharmacol 20:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Li W (2020) Monoacylglycerol lipase inhibitors: modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm Sin B 10:582–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Kooijman S, van den Nieuwendijk AM, Ogasawara D, van der Wel T, van Dalen F, Baggelaar MP, Janssen FJ, van den Berg RJ, den Dulk H, et al. (2017) Triazole ureas act as diacylglycerol lipase inhibitors and prevent fasting-induced refeeding. J Med Chem 60:428–440. [DOI] [PubMed] [Google Scholar]
- Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, Falony G, Raes J, Maiter D, Delzenne NM, et al. (2019) Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med 25:1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depommier C, Vitale RM, Iannotti FA, Silvestri C, Flamand N, Druart C, Everard A, Pelicaen R, Maiter D, Thissen JP, et al. (2021) Beneficial effects of Akkermansia muciniphila are not associated with major changes in the circulating endocannabinoidome but linked to higher mono-palmitoyl-glycerol levels as new PPARα agonists. Cells 10:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desarnaud F, Cadas H, Piomelli D (1995) Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem 270:6030–6035. [DOI] [PubMed] [Google Scholar]
- Després JP, Golay A, Sjöström L; Rimonabant in Obesity-Lipids Study Group (2005) Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med 353:2121–2134. [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Chin SA (1993) Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 46:791–796. [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Glaser ST, Howell JM, Kunz JS, Puffenbarger RA, Hillard CJ, Abumrad N (2001) The cellular uptake of anandamide is coupled to its breakdown by fatty-acid amide hydrolase. J Biol Chem 276:6967–6973. [DOI] [PubMed] [Google Scholar]
- Devane WA, Breuer A, Sheskin T, Järbe TUC, Eisen MS, Mechoulam R (1992a) A novel probe for the cannabinoid receptor. J Med Chem 35:2065–2069. [DOI] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA 3rd, Johnson MR, Melvin LS, Howlett AC (1988) Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol 34:605–613. [PubMed] [Google Scholar]
- Devane WA, Hanuš L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992b) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949. [DOI] [PubMed] [Google Scholar]
- DeVuono MV, Hrelja KM, Petrie GN, Limebeer CL, Rock EM, Hill MN, Parker LA (2020) Nausea-induced conditioned gaping reactions in rats produced by high-dose synthetic cannabinoid, JWH-018. Cannabis Cannabinoid Res 5:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V (2018) New approaches and challenges to targeting the endocannabinoid system. Nat Rev Drug Discov 17:623–639. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A (1995) Anandamide, an endogenous cannabinomimetic eicosanoid: “killing two birds with one stone.” Prostaglandins Leukot Essent Fatty Acids 53:1–11. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Piscitelli F (2015) The endocannabinoid system and its modulation by phytocannabinoids. Neurotherapeutics 12:692–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Silvestri C (2019) Lifestyle and metabolic syndrome: contribution of the endocannabinoidome. Nutrients 11:1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372:686–691. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Goparaju SK, Wang L, Liu J, Bátkai S, Járai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, et al. (2001) Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 410:822–825. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Ligresti A, Morera E, Nalli M, Ortar G (2004) The anandamide membrane transporter. Structure-activity relationships of anandamide and oleoylethanolamine analogs with phenyl rings in the polar head group region. Bioorg Med Chem 12:5161–5169. [DOI] [PubMed] [Google Scholar]
- Di Meo C, Tortolani D, Standoli S, Angelucci CB, Fanti F, Leuti A, Sergi M, Kadhim S, Hsu E, Rapino C, et al. (2022) Effects of rare phytocannabinoids on the endocannabinoid system of human keratinocytes. Int J Mol Sci 23:5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, Bazan NG, Tasker JG (2005) Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol 569:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz P, Phatak SS, Xu J, Fronczek FR, Astruc-Diaz F, Thompson CM, Cavasotto CN, Naguib M (2009) 2,3-Dihydro-1-benzofuran derivatives as a series of potent selective cannabinoid receptor 2 agonists: design, synthesis, and binding mode prediction through ligand-steered modeling. ChemMedChem 4:1615–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickason-Chesterfield AK, Kidd SR, Moore SA, Schaus JM, Liu B, Nomikos GG, Felder CC (2006) Pharmacological characterization of endocannabinoid transport and fatty acid amide hydrolase inhibitors. Cell Mol Neurobiol 26:407–423. [DOI] [PubMed] [Google Scholar]
- Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC, Ra S, Gray JM, Yang R, DeGruccio AM, et al. (2015) FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat Commun 6:6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA 99:10819–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dione N, Lacroix S, Taschler U, Deschênes T, Abolghasemi A, Leblanc N, Di Marzo V, Silvestri C (2020) Mgll knockout mouse resistance to diet-induced dysmetabolism is associated with altered gut microbiota. Cells 9:2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPatrizio NV (2021) Endocannabinoids and the gut-brain control of food intake and obesity. Nutrients 13:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon DD, Tius MA, Thakur GA, Zhou H, Bowman AL, Shukla VG, Peng Y, Makriyannis A (2012) C3-heteroaroyl cannabinoids as photolabeling ligands for the CB2 cannabinoid receptor. Bioorg Med Chem Lett 22:5322–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donvito G, Piscitelli F, Muldoon P, Jackson A, Vitale RM, D’Aniello E, Giordano C, Ignatowska-Jankowska BM, Mustafa MA, Guida F, et al. (2019) N-oleoyl-glycine reduces nicotine reward and withdrawal in mice. Neuropharmacology 148:320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotsey EY, Jung KM, Basit A, Wei D, Daglian J, Vacondio F, Armirotti A, Mor M, Piomelli D (2015) Peroxide-dependent MGL sulfenylation regulates 2-AG-mediated endocannabinoid signaling in brain neurons. Chem Biol 22:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza DC, Cortes-Briones J, Creatura G, Bluez G, Thurnauer H, Deaso E, Bielen K, Surti T, Radhakrishnan R, Gupta A, et al. (2019) Efficacy and safety of a fatty acid amide hydrolase inhibitor (PF-04457845) in the treatment of cannabis withdrawal and dependence in men: a double-blind, placebo-controlled, parallel group, phase 2a single-site randomised controlled trial. Lancet Psychiatry 6:35–45. [DOI] [PubMed] [Google Scholar]
- Eberlein B, Eicke C, Reinhardt HW, Ring J (2008) Adjuvant treatment of atopic eczema: assessment of an emollient containing N-palmitoylethanolamine (ATOPA study). J Eur Acad Dermatol Venereol 22:73–82. [DOI] [PubMed] [Google Scholar]
- Eissenstat MA, Bell MR, D’Ambra TE, Alexander EJ, Daum SJ, Ackerman JH, Gruett MD, Kumar V, Estep KG, Olefirowicz EM, et al. (1995) Aminoalkylindoles: structure-activity relationships of novel cannabinoid mimetics. J Med Chem 38:3094–3105. [DOI] [PubMed] [Google Scholar]
- Eissenstat MA, Bell MR, D’Ambra TE, Estep KG, Haycock DA, Olefirowicz EM, Ward SJ (1990) Aminoalkylindoles (AAIs): structurally novel cannabinoid-mimetics. NIDA Res Monogr 105:427–428. [PubMed] [Google Scholar]
- El Bakali J, Muccioli GG, Body-Malapel M, Djouina M, Klupsch F, Ghinet A, Barczyk A, Renault N, Chavatte P, Desreumaux P, et al. (2014) Conformational restriction leading to a selective CB2 cannabinoid receptor agonist orally active against colitis. ACS Med Chem Lett 6:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Sohly MA, Gul W (2014) Constituents of Cannabis sativa, in Handbook of Cannabis (Pertwee RG, ed) pp 3–22, Oxford University Press, Oxford, U.K. [Google Scholar]
- El Sohly MA, Radwan MM, Gul W, Chandra S, Galal A (2017) Phytochemistry of Cannabis sativa L. Prog Chem Org Nat Prod 103:1–36. [DOI] [PubMed] [Google Scholar]
- Ellermann M, Pacheco AR, Jimenez AG, Russell RM, Cuesta S, Kumar A, Zhu W, Vale G, Martin SA, Raj P, et al. (2020) Endocannabinoids inhibit the induction of virulence in enteric pathogens. Cell 183:650–665.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmes MW, Kaczocha M, Berger WT, Leung K, Ralph BP, Wang L, Sweeney JM, Miyauchi JT, Tsirka SE, Ojima I, et al. (2015) Fatty acid-binding proteins (FABPs) are intracellular carriers for Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J Biol Chem 290:8711–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmes MW, Prentis LE, McGoldrick LL, Giuliano CJ, Sweeney JM, Joseph OM, Che J, Carbonetti GS, Studholme K, Deutsch DG, et al. (2019) FABP1 controls hepatic transport and biotransformation of Δ9-THC. Sci Rep 9:7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Medicines Agency (2022) Sativex (https://www.ema.europa.eu/en/medicines?search_api_views_fulltext=Sativex). Emerald Health Pharmaceuticals, https://emeraldpharma.com/clinical-trials/ (accessed October 2022).
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, et al. (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110:9066–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Plovier H, Rastelli M, Van Hul M, de Wouters d’Oplinter A, Geurts L, Druart C, Robine S, Delzenne NM, Muccioli GG, et al. (2019) Intestinal epithelial N-acylphosphatidylethanolamine phospholipase D links dietary fat to metabolic adaptations in obesity and steatosis. Nat Commun 10:457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JS, Colangeli R, Dong A, George AG, Addo-Osafo K, Kingsley PJ, Morena M, Wolff MD, Dudok B, He K, et al. (2021) In vivo endocannabinoid dynamics at the timescale of physiological and pathological neural activity. Neuron 109:2398–2403.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasia L, Karava V, Siafaka-Kapadai A (2003) Uptake and metabolism of [3H]anandamide by rabbit platelets. Lack of transporter? Eur J Biochem 270:3498–3506. [DOI] [PubMed] [Google Scholar]
- Fauzan M, Oubraim S, Yu M, Glaser ST, Kaczocha M, Haj-Dahmane S (2022) Fatty acid-binding protein 5 modulates brain endocannabinoid tone and retrograde signaling in the striatum. Front Cell Neurosci 16:936939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio D, Criscuolo E, Piccoli A, Barboni B, Fezza F, Maccarrone M (2020) Advances in the discovery of fatty acid amide hydrolase inhibitors: what does the future hold? Expert Opin Drug Discov 15:765–778. [DOI] [PubMed] [Google Scholar]
- Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, Piomelli D (2004) Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci USA 101:8756–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, Dickason-Chesterfield AK, Moore SA (2006) Cannabinoid biology. Mol Interv 6:149–161. [DOI] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL (1995) Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48:443–450. [PubMed] [Google Scholar]
- Feng Z, Alqarni MH, Yang P, Tong Q, Chowdhury A, Wang L, Xie XQ (2014) Modeling, molecular dynamics simulation, and mutation validation for structure of cannabinoid receptor 2 based on known crystal structures of GPCRs. J Chem Inf Model 54:2483–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Moro MA, Martínez-Orgado J (2015a) Cannabinoids in neurodegenerative disorders and stroke/brain trauma: from preclinical models to clinical applications. Neurotherapeutics 12:793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Romero J, Ramos JA (2015b) Endocannabinoids and neurodegenerative disorders: Parkinson’s disease, Huntington’s chorea, Alzheimer’s disease, and others. Handb Exp Pharmacol 231:233–259. [DOI] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Romero J, Velasco G, Tolón RM, Ramos JA, Guzmán M (2007) Cannabinoid CB2 receptor: a new target for controlling neural cell survival? Trends Pharmacol Sci 28:39–45. [DOI] [PubMed] [Google Scholar]
- Fezza F, Bari M, Florio R, Talamonti E, Feole M, Maccarrone M (2014) Endocannabinoids, related compounds and their metabolic routes. Molecules 19:17078–17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn DP, Haroutounian S, Hohmann AG, Krane E, Soliman N, Rice ASC (2021) Cannabinoids, the endocannabinoid system, and pain: a review of preclinical studies. Pain 162(Suppl 1):S5–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong TM (2014) Constitutive activity in cannabinoid receptors. Adv Pharmacol 70:121–133. [DOI] [PubMed] [Google Scholar]
- Fornelos N, Franzosa EA, Bishai J, Annand JW, Oka A, Lloyd-Price J, Arthur TD, Garner A, Avila-Pacheco J, Haiser HJ, et al. (2020) Growth effects of N-acylethanolamines on gut bacteria reflect altered bacterial abundances in inflammatory bowel disease. Nat Microbiol 5:486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JR, Ueno S, Chen MX, Harvey J, Dowell SJ, Irving AJ, Brown AJ (2019) N-Palmitoylglycine and other N-acylamides activate the lipid receptor G2A/GPR132. Pharmacol Res Perspect 7:e00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotio Y, Jung KM, Palese F, Obenaus A, Tagne AM, Lin L, Rashid TI, Pacheco R, Jullienne A, Ramirez J, et al. (2021b) NAAA-regulated lipid signaling governs the transition from acute to chronic pain. Sci Adv 7:eabi8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotio Y, Sasso O, Ciccocioppo R, Piomelli D (2021a) Antinociceptive profile of ARN19702,(2-ethylsulfonylphenyl)-[(2S)-4-(6-fluoro-1, 3-benzothiazol-2-yl)-2-methylpiperazin-1-yl] methanone, a novel orally active N-acylethanolamine acid amidase inhibitor, in animal models. J Pharmacol Exp Ther 378:70–76. [DOI] [PubMed] [Google Scholar]
- Fowler CJ (2012) Anandamide uptake explained? Trends Pharmacol Sci 33:181–185. [DOI] [PubMed] [Google Scholar]
- Fowler CJ (2013) Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J 280:1895–1904. [DOI] [PubMed] [Google Scholar]
- Fowler CJ (2014) Has FLAT fallen flat? Trends Pharmacol Sci 35:51–52. [DOI] [PubMed] [Google Scholar]
- Fowler CJ (2021) The endocannabinoid system—current implications for drug development. J Intern Med 290:2–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Tiger G, Ligresti A, López-Rodríguez ML, Di Marzo V, López-Rodríguez ML, Di Marzo V (2004) Selective inhibition of anandamide cellular uptake versus enzymatic hydrolysis—a difficult issue to handle. Eur J Pharmacol 492:1–11. [DOI] [PubMed] [Google Scholar]
- Fox A, Kesingland A, Gentry C, McNair K, Patel S, Urban L, James I (2001) The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain 92:91–100. [DOI] [PubMed] [Google Scholar]
- Franco R, Rivas-Santisteban R, Reyes-Resina I, Casanovas M, Pérez-Olives C, Ferreiro-Vera C, Navarro G, Sánchez de Medina V, Nadal X (2020) Pharmacological potential of varinic-, minor-, and acidic phytocannabinoids. Pharmacol Res 158:104801. [DOI] [PubMed] [Google Scholar]
- Fride E, Mechoulam R (1993) Pharmacological activity of the cannabinoid receptor agonist, anandamide, a brain constituent. Eur J Pharmacol 231:313–314. [DOI] [PubMed] [Google Scholar]
- Friedman D, French JA, Maccarrone M (2019) Safety, efficacy, and mechanisms of action of cannabinoids in neurological disorders. Lancet Neurol 18:504–512. [DOI] [PubMed] [Google Scholar]
- Frost JM, Dart MJ, Tietje KR, Garrison TR, Grayson GK, Daza AV, El-Kouhen OF, Miller LN, Li L, Yao BB, et al. (2008) Indol-3-yl-tetramethylcyclopropyl ketones: effects of indole ring substitution on CB2 cannabinoid receptor activity. J Med Chem 51:1904–1912. [DOI] [PubMed] [Google Scholar]
- Fu J, Bottegoni G, Sasso O, Bertorelli R, Rocchia W, Masetti M, Guijarro A, Lodola A, Armirotti A, Garau G, et al. (2011) A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat Neurosci 15:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, et al. (2003) Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 425:90–93. [DOI] [PubMed] [Google Scholar]
- Fulp A, Bortoff K, Zhang Y, Snyder R, Fennell T, Marusich JA, Wiley JL, Seltzman H, Maitra R (2013) Peripherally selective diphenyl purine antagonist of the CB1 receptor. J Med Chem 56:8066–8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Hotamisligil GS (2008) Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov 7:489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielli M, Battista N, Riganti L, Prada I, Antonucci F, Cantone L, Matteoli M, Maccarrone M, Verderio C (2015) Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep 16:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gado F, Di Cesare Mannelli L, Lucarini E, Bertini S, Cappelli E, Digiacomo M, Stevenson LA, Macchia M, Tuccinardi T, Ghelardini C, et al. (2019) Identification of the first synthetic allosteric modulator of the CB2 receptors and evidence of its efficacy for neuropathic pain relief. J Med Chem 62:276–287. [DOI] [PubMed] [Google Scholar]
- Gagestein B, Stevens AF, Fazio D, Florea BI, van der Wel T, Bakker AT, Fezza F, Dulk HD, Overkleeft HS, Maccarrone M, et al. (2022) Chemical proteomics reveals off-targets of the anandamide reuptake inhibitor WOBE437. ACS Chem Biol 17:1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P (1995) Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 232:54–61. [DOI] [PubMed] [Google Scholar]
- Galli JA, Sawaya RA, Friedenberg FK (2011) Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev 4:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, et al. (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30:2017–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R (1964) Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc 86:1646–1647. [Google Scholar]
- Garai S, Schaffer PC, Laprairie RB, Janero DR, Pertwee RG, Straiker A, Thakur GA (2021) Design, synthesis, and pharmacological profiling of cannabinoid 1 receptor allosteric modulators: preclinical efficacy of C2-group GAT211 congeners for reducing intraocular pressure. Bioorg Med Chem 50:116421. [DOI] [PubMed] [Google Scholar]
- García-Martín A, Garrido-Rodríguez M, Navarrete C, Del Río C, Bellido ML, Appendino G, Calzado MA, Muñoz E (2018) EHP-101, an oral formulation of the cannabidiol aminoquinone VCE-004.8, alleviates bleomycin-induced skin and lung fibrosis. Biochem Pharmacol 157:304–313. [DOI] [PubMed] [Google Scholar]
- Gareau Y, Dufresne C, Gallant M, Rochette C, Sawyer N, Slipetz DM, Tremblay N, Weech PK, Metters KM, Labelle M (1996) Structure activity relationships of tetrahydrocannabinol analogs on human cannabinoid receptors. Bioorg Med Chem Lett 6:189–194. [Google Scholar]
- Gasperi V, Guzzo T, Topai A, Gambacorta N, Ciriaco F, Nicolotti O, Maccarrone M (2023) Recent advances on type-2 cannabinoid (CB2) receptor agonists and their therapeutic potential. Curr Med Chem 30:1420–1457 [DOI] [PubMed] [Google Scholar]
- Gatley SJ, Lan R, Pyatt B, Gifford AN, Volkow ND, Makriyannis A (1997) Binding of the non-classical cannabinoid CP 55,940, and the diarylpyrazole AM251 to rodent brain cannabinoid receptors. Life Sci 61:191–197. [DOI] [PubMed] [Google Scholar]
- Gazzi T, Brennecke B, Atz K, Korn C, Sykes D, Forn-Cuni G, Pfaff P, Sarott RC, Westphal MV, Mostinski Y, et al. (2022) Detection of cannabinoid receptor type 2 in native cells and zebrafish with a highly potent, cell-permeable fluorescent probe. Chem Sci (Camb) 13:5539–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard C, Mollereau C, Vassart G, Parmentier M (1990) Nucleotide sequence of a human cannabinoid receptor cDNA. Nucleic Acids Res 18:7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard CM, Mollereau C, Vassart G, Parmentier M (1991) Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem J 279:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts L, Everard A, Van Hul M, Essaghir A, Duparc T, Matamoros S, Plovier H, Castel J, Denis RG, Bergiers M, et al. (2015) Adipose tissue NAPE-PLD controls fat mass development by altering the browning process and gut microbiota. Nat Commun 6:6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghidini A, Scalvini L, Palese F, Lodola A, Mor M, Piomelli D (2021) Different roles for the acyl chain and the amine leaving group in the substrate selectivity of N-acylethanolamine acid amidase. J Enzyme Inhib Med Chem 36:1411–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giblin GM, O’Shaughnessy CT, Naylor A, Mitchell WL, Eatherton AJ, Slingsby BP, Rawlings DA, Goldsmith P, Brown AJ, Haslam CP, et al. (2007) Discovery of 2-[(2,4-dichlorophenyl)amino]-N-[(tetrahydro- 2H-pyran-4-yl)methyl]-4-(trifluoromethyl)- 5-pyrimidinecarboxamide, a selective CB2 receptor agonist for the treatment of inflammatory pain. J Med Chem 50:2597–2600. [DOI] [PubMed] [Google Scholar]
- Gil-Ordóñez A, Martín-Fontecha M, Ortega-Gutiérrez S, López-Rodríguez ML (2018) Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem Pharmacol 157:18–32. [DOI] [PubMed] [Google Scholar]
- Glaser ST, Abumrad NA, Fatade F, Kaczocha M, Studholme KM, Deutsch DG (2003) Evidence against the presence of an anandamide transporter. Proc Natl Acad Sci USA 100:4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser ST, Kaczocha M, Deutsch DG (2005) Anandamide transport: a critical review. Life Sci 77:1584–1604. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, et al. (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA 102:18620–18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobira PH, Oliveira AC, Gomes JS, da Silveira VT, Asth L, Bastos JR, Batista EM, Issy AC, Okine BN, de Oliveira AC, et al. (2019) Opposing roles of CB1 and CB2 cannabinoid receptors in the stimulant and rewarding effects of cocaine. Br J Pharmacol 176:1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski G, Offertaler L, Wagner JA, Kunos G (2009) Receptors for acylethanolamides-GPR55 and GPR119. Prostagl Other Lipid Med 89:105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold A, Zhu J (2022) Not just a gut feeling: a deep exploration of functional bacterial metabolites that can modulate host health. Gut Microbes 14:2125734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Cañas M, Rodríguez-Cueto C, Satta V, Hernández-Fisac I, Navarro E, Fernández-Ruiz J (2023) Endocannabinoid-binding receptors as drug targets. Methods Mol Biol 2576:67–94. [DOI] [PubMed] [Google Scholar]
- Gonsiorek W, Hesk D, Chen SC, Kinsley D, Fine JS, Jackson JV, Bober LA, Deno G, Bian H, Fossetta J, et al. (2006) Characterization of peripheral human cannabinoid receptor (hCB2) expression and pharmacology using a novel radioligand, [35S]Sch225336. J Biol Chem 281:28143–28151. [DOI] [PubMed] [Google Scholar]
- Goodfellow CE, Glass M (2009) Anandamide receptor signal transduction. Vitam Horm 81:79–110. [DOI] [PubMed] [Google Scholar]
- Gorelik A, Gebai A, Illes K, Piomelli D, Nagar B (2018) Molecular mechanism of activation of the immunoregulatory amidase NAAA. Proc Natl Acad Sci USA 115:E10032–E10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratzke C, Streng T, Stief CG, Downs TR, Alroy I, Rosenbaum JS, Andersson KE, Hedlund P (2010) Effects of cannabinor, a novel selective cannabinoid 2 receptor agonist, on bladder function in normal rats. Eur Urol 57:1093–1100. [DOI] [PubMed] [Google Scholar]
- Grether U(2022) First disclosure of cannabinoid receptor type 2 agonist RG7774—an innovative oral treatment for diabetic retinopathy. XXVII EFMC International Symposium on Medicinal Chemistry, Nice, France. [Google Scholar]
- Grevengoed TJ, Trammell SAJ, McKinney MK, Petersen N, Cardone RL, Svenningsen JS, Ogasawara D, Nexøe-Larsen CC, Knop FK, Schwartz TW, et al. (2019) N-acyl taurines are endogenous lipid messengers that improve glucose homeostasis. Proc Natl Acad Sci USA 116:24770–24778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruden G, Barutta F, Kunos G, Pacher P (2016) Role of the endocannabinoid system in diabetes and diabetic complications. Br J Pharmacol 173:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guba W, Nazaré M, Grether U (2020) Natural compounds and synthetic drugs to target type-2 cannabinoid (CB2) receptor, in New Tools to Interrogate Endocannabinoid Signaling. From Natural Compounds to Synthetic Drugs (Maccarrone M, ed) pp 89–167, Royal Society of Chemistry, Cambridge, U.K. [Google Scholar]
- Guberman M, Kosar M, Omran A, Carreira EM, Nazare M, Grether U (2022) Reverse design toward optimized labeled chemical probes—examples from the endocannabinoid system. Chimia (Aarau) 76:425–434. [DOI] [PubMed] [Google Scholar]
- Gueye AB, Pryslawsky Y, Trigo JM, Poulia N, Delis F, Antoniou K, Loureiro M, Laviolette SR, Vemuri K, Makriyannis A, et al. (2016) The CB1 neutral antagonist AM4113 retains the therapeutic efficacy of the inverse agonist rimonabant for nicotine dependence and weight loss with better psychiatric tolerability. Int J Neuropsychopharmacol 19:pyw068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guida F, Turco F, Iannotta M, De Gregorio D, Palumbo I, Sarnelli G, Furiano A, Napolitano F, Boccella S, Luongo L, et al. (2018) Antibiotic-induced microbiota perturbation causes gut endocannabinoidome changes, hippocampal neuroglial reorganization and depression in mice. Brain Behav Immun 67:230–245. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG (2008) Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol 153:319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Uyama T, Rahman SMK, Sikder MM, Hussain Z, Tsuboi K, Miyake M, Ueda N (2021) Involvement of the γ isoform of cPLA2 in the biosynthesis of bioactive N-acylethanolamines. Molecules 26:5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib AM, Okorokov AL, Hill MN, Bras JT, Lee MC, Li S, Gossage SJ, van Drimmelen M, Morena M, Houlden H, et al. (2019) Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br J Anaesth 123:e249–e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider A, Gobbi L, Kretz J, Ullmer C, Brink A, Honer M, Woltering TJ, Muri D, Iding H, Bürkler M, et al. (2020) Identification and preclinical development of a 2,5,6-trisubstituted fluorinated pyridine derivative as a radioligand for the positron emission tomography imaging of cannabinoid type 2 receptors. J Med Chem 63:10287–10306. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Shen R-YY, Elmes MW, Studholme K, Kanjiya MP, Bogdan D, Thanos PK, Miyauchi JT, Tsirka SE, Deutsch DG, et al. (2018) Fatty-acid-binding protein 5 controls retrograde endocannabinoid signaling at central glutamate synapses. Proc Natl Acad Sci USA 115:3482–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdu Z, Nicolussi S, Rau M, Lorántfy L, Forgo P, Hohmann J, Csupor D, Gertsch J (2014) Identification of endocannabinoid system-modulating N-alkylamides from Heliopsis helianthoides var. scabra and Lepidium meyenii. J Nat Prod 77:1663–1669. [DOI] [PubMed] [Google Scholar]
- Han S, Chen JJ, Chen JZ (2014) Latest progress in the identification of novel synthetic ligands for the cannabinoid CB2 receptor. Mini Rev Med Chem 14:426–443. [DOI] [PubMed] [Google Scholar]
- Han S, Thatte J, Buzard DJ, Jones RM (2013) Therapeutic utility of cannabinoid receptor type 2 (CB(2)) selective agonists. J Med Chem 56:8224–8256. [DOI] [PubMed] [Google Scholar]
- Han S, Thoresen L, Jung JK, Zhu X, Thatte J, Solomon M, Gaidarov I, Unett DJ, Yoon WH, Barden J, et al. (2017) Discovery of APD371: identification of a highly potent and selective CB2 agonist for the treatment of chronic pain. ACS Med Chem Lett 8:1309–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen HS, Vana V (2019) Non-endocannabinoid N-acylethanolamines and 2-monoacylglycerols in the intestine. Br J Pharmacol 176:1443–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE, Kustanovich I, Mechoulam R (2001) 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci USA 98:3662–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RG, Ross RA, Mechoulam R, Fride E (1999) HU-308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci USA 96:14228–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanuš LO, Meyer SM, Muñoz E, Taglialatela-Scafati O, Appendino G (2016) Phytocannabinoids: a unified critical inventory. Nat Prod Rep 33:1357–1392. [DOI] [PubMed] [Google Scholar]
- Haruna T, Soga M, Morioka Y, Hikita I, Imura K, Furue Y, Yamamoto M, Imura C, Ikeda M, Yamauchi A, et al. (2015) S-777469, a novel cannabinoid type 2 receptor agonist, suppresses itch-associated scratching behavior in rodents through inhibition of itch signal transmission. Pharmacology 95:95–103. [DOI] [PubMed] [Google Scholar]
- Haruna T, Soga M, Morioka Y, Imura K, Furue Y, Yamamoto M, Hayakawa J, Deguchi M, Arimura A, Yasui K (2017) The inhibitory effect of S-777469, a cannabinoid type 2 receptor agonist, on skin inflammation in mice. Pharmacology 99:259–267. [DOI] [PubMed] [Google Scholar]
- He X-H, Jordan CJ, Vemuri K, Bi G-H, Zhan J, Gardner EL, Makriyannis A, Wang Y-L, Xi Z-X (2019) Cannabinoid CB1 receptor neutral antagonist AM4113 inhibits heroin self-administration without depressive side effects in rats. Acta Pharmacol Sin 40:365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hen-Shoval D, Amar S, Shbiro L, Smoum R, Haj CG, Mechoulam R, Zalsman G, Weller A, Shoval G (2018) Acute oral cannabidiolic acid methyl ester reduces depression-like behavior in two genetic animal models of depression. Behav Brain Res 351:1–3. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC (1990) Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA 87:1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Torres G, Cipriano M, Hedén E, Björklund E, Canales Á, Zian D, Feliú A, Mecha M, Guaza C, Fowler CJ, et al. (2014) A reversible and selective inhibitor of monoacylglycerol lipase ameliorates multiple sclerosis. Angew Chem Int Ed Engl 53:13765–13770. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A (2000) The movement of N-arachidonoylethanolamine (anandamide) across cellular membranes. Chem Phys Lipids 108:123–134. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A (2003) Cellular accumulation of anandamide: consensus and controversy. Br J Pharmacol 140:802–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB (1997) Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem 69:631–638. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Huang H, Vogt CD, Rodrigues BE, Neumann TS, Sem DS, Schroeder F, Cunningham CW (2017) Endocannabinoid transport proteins: discovery of tools to study sterol carrier protein-2. Methods Enzymol 593:99–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB (1999) Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J Pharmacol Exp Ther 289:1427–1433. [PubMed] [Google Scholar]
- Hillard CJ, Shi L, Tuniki VR, Falck JR, Campbell WB (2007) Studies of anandamide accumulation inhibitors in cerebellar granule neurons: comparison to inhibition of fatty acid amide hydrolase. J Mol Neurosci 33:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB (1995) Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta 1257:249–256. [DOI] [PubMed] [Google Scholar]
- Högestätt ED, Jönsson BAG, Ermund A, Andersson DA, Björk H, Alexander JP, Cravatt BF, Basbaum AI, Zygmunt PM (2005) Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem 280:31405–31412. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, et al. (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435:1108–1112. [DOI] [PubMed] [Google Scholar]
- Hollinshead SP, Tidwell MW, Palmer J, Guidetti R, Sanderson A, Johnson MP, Chambers MG, Oskins J, Stratford R, Astles PC (2013) Selective cannabinoid receptor type 2 (CB2) agonists: optimization of a series of purines leading to the identification of a clinical candidate for the treatment of osteoarthritic pain. J Med Chem 56:5722–5733. [DOI] [PubMed] [Google Scholar]
- Honda M, Yoshimura N, Kawamoto B, Hikita K, Muraoka K, Shimizu S, Saito M, Chancellor MB, Takenaka A (2016) Anandamide transporter-mediated regulation of the micturition reflex in urethane-anesthetized rats. Int Urol Nephrol 48:1407–1412. [DOI] [PubMed] [Google Scholar]
- Honer M, Gobbi L, Martarello L, Comley RA (2014) Radioligand development for molecular imaging of the central nervous system with positron emission tomography. Drug Discov Today 19:1936–1944. [DOI] [PubMed] [Google Scholar]
- Hong G, Antaris AL, Dai H (2017) Near-infrared fluorophores for biomedical imaging. Nat Biomed Eng 1:0010. [Google Scholar]
- Hosseinzadeh Anvar L, Ahmadalipour A (2023) Fatty acid amide hydrolase C385A polymorphism affects susceptibility to various diseases. Biofactors 49:62–78. [DOI] [PubMed] [Google Scholar]
- Howlett AC (1984) Inhibition of neuroblastoma adenylate cyclase by cannabinoid and nantradol compounds. Life Sci 35:1803–1810. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Abood ME (2017) CB1 and CB2 receptor pharmacology. Adv Pharmacol 80:169–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, et al. (2002) International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 54:161–202. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Johnson MR, Melvin LS (1990) Classical and nonclassical cannabinoids: mechanism of action—brain binding. NIDA Res Monogr 96:100–111. [PubMed] [Google Scholar]
- Howlett AC, Johnson MR, Melvin LS, Milne GM (1988) Nonclassical cannabinoid analgetics inhibit adenylate cyclase: development of a cannabinoid receptor model. Mol Pharmacol 33:297–302. [PubMed] [Google Scholar]
- Howlett AC, Reggio PH, Childers SR, Hampson RE, Ulloa NM, Deutsch DG (2011) Endocannabinoid tone versus constitutive activity of cannabinoid receptors. Br Journal Pharmacol 163:1329–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryhorowicz S, Kaczmarek-Ryś M, Andrzejewska A, Staszak K, Hryhorowicz M, Korcz A, Słomski R (2019) Allosteric modulation of cannabinoid receptor 1—current challenges and future opportunities. Int J Mol Sci 20:5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Tong S, Zhou Y, Elmes MW, Yan S, Kaczocha M, Deutsch DG, Rizzo RC, Ojima I, Li H (2017) The antinociceptive agent SBFI-26 binds to anandamide transporters FABP5 and FABP7 at two different sites. Biochemistry 56:3454–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF (2012) DAGLβ inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol 8:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Li X, Wu L, Iliopoulos-Tsoutsouvas C, Wang Y, Wu M, Shen L, Brust CA, Nikas SP, Song F, et al. (2020) Activation and signaling mechanism revealed by cannabinoid receptor-Gi complex structures. Cell 180:655–665.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, Ho JH, et al. (2017) Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547:468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, Korde A, et al. (2016) Crystal structure of the human cannabinoid receptor CB1. Cell 167:750–762.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, McIntosh AL, Martin GG, Dangott LJ, Kier AB, Schroeder F (2018) Structural and Functional Interaction of Δ9-Tetrahydrocannabinol with Liver Fatty Acid Binding Protein (FABP1). Biochemistry 57:6027–6042. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Petros TJ, Chang SY, Zavitsanos PA, Zipkin RE, Sivakumar R, Coop A, Maeda DY, De Petrocellis L, et al. (2001) Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J Biol Chem 276:42639–42644. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, et al. (2002) An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci USA 99:8400–8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huestis MA (2007) Human cannabinoid pharmacokinetics. Chem Biodivers 4:1770–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman JW, Yu S, Showalter V, Abood ME, Wiley JL, Compton DR, Martin BR, Bramblett RD, Reggio PH (1996) Synthesis and pharmacology of a very potent cannabinoid lacking a phenolic hydroxyl with high affinity for the CB2 receptor. J Med Chem 39:3875–3877. [DOI] [PubMed] [Google Scholar]
- Huggins JP, Smart TS, Langman S, Taylor L, Young T (2012) An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153:1837–1846. [DOI] [PubMed] [Google Scholar]
- Iannotti FA, Di Marzo V, Petrosino S (2016) Endocannabinoids and endocannabinoid-related mediators: targets, metabolism and role in neurological disorders. Prog Lipid Res 62:107–128. [DOI] [PubMed] [Google Scholar]
- Ibeas Bih C, Chen T, Nunn AV, Bazelot M, Dallas M, Whalley BJ (2015) Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics 12:699–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibsen MS, Connor M, Glass M (2017) Cannabinoid CB(1) and CB(2) receptor signaling and bias. Cannabis Cannabinoid Res 2:48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, Damaj IM, Poklis J, Wiley JL, Zanda M, et al. (2015) A cannabinoid CB1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology 40:2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T, Nishida A, Yamano M, Kimura I (2022) Short-chain fatty acid receptors and gut microbiota as therapeutic targets in metabolic, immune, and neurological diseases. Pharmacol Ther 239:108273. [DOI] [PubMed] [Google Scholar]
- Ilyasov AA, Milligan CE, Pharr EP, Howlett AC (2018) The endocannabinoid system and oligodendrocytes in health and disease. Front Neurosci 12:733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im DS(2021) GPR119 and GPR55 as receptors for fatty acid ethanolamides, oleoylethanolamide and palmitoylethanolamide. Int J Mol Sci 22:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Tsuboi K, Okamoto Y, Hidaka M, Uyama T, Tsutsumi T, Tanaka T, Ueda N, Tokumura A (2017) Peripheral tissue levels and molecular species compositions of N-acyl-phosphatidylethanolamine and its metabolites in mice lacking N-acyl-phosphatidylethanolamine-specific phospholipase D. J Biochem 162:449–458. [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Horiuchi Y, Ishikawa M, Koga M, Imai K, Suzuki Y, Morikawa M, Inada T, Watanabe Y, Takahashi M, et al. (2010) Brain cannabinoid CB2 receptor in schizophrenia. Biol Psychiatry 67:974–982. [DOI] [PubMed] [Google Scholar]
- Iwata Y, Ando K, Taniguchi K, Koba N, Sugiura A, Sudo M (2015) Identification of a highly potent and selective CB2 agonist, RQ-00202730, for the treatment of irritable bowel syndrome. Bioorg Med Chem Lett 25:236–240. [DOI] [PubMed] [Google Scholar]
- Iyer MR, Cinar R, Wood CM, Zawatsky CN, Coffey NJ, Kim KA, Liu Z, Katz A, Abdalla J, Hassan SA, et al. (2022) Synthesis, biological evaluation, and molecular modeling studies of 3, 4-diarylpyrazoline series of compounds as potent, nonbrain penetrant antagonists of cannabinoid-1 (CB1R) receptor with reduced lipophilicity. J Med Chem 65:2374–2387. [DOI] [PubMed] [Google Scholar]
- Jacobson MR, Watts JJ, Boileau I, Tong J, Mizrahi R (2019) A systematic review of phytocannabinoid exposure on the endocannabinoid system: implications for psychosis. Eur Neuropsychopharmacol 29:330–348. [DOI] [PubMed] [Google Scholar]
- Jain AK, Ryan JR, McMahon FG, Smith G (1981) Evaluation of intramuscular levonantradol and placebo in acute postoperative pain. J Clin Pharmacol 21(Suppl 1):320S–326S. [DOI] [PubMed] [Google Scholar]
- Janero DR, Korde A, Makriyannis A (2017) Ligand-assisted protein structure (LAPS): an experimental paradigm for characterizing cannabinoid-receptor ligand-binding domains. Methods Enzymol 593:217–235. [DOI] [PubMed] [Google Scholar]
- Janssen FJ, van der Stelt M (2016) Inhibitors of diacylglycerol lipases in neurodegenerative and metabolic disorders. Bioorg Med Chem Lett 26:3831–3837. [DOI] [PubMed] [Google Scholar]
- Jarrahian A, Manna S, Edgemond WS, Campbell WB, Hillard CJ (2000) Structure-activity relationships among N-arachidonylethanolamine (Anandamide) head group analogues for the anandamide transporter. J Neurochem 74:2597–2606. [DOI] [PubMed] [Google Scholar]
- Jenniches I, Ternes S, Albayram O, Otte DM, Bach K, Bindila L, Michel K, Lutz B, Bilkei-Gorzo A, Zimmer A (2016) Anxiety, stress, and fear response in mice with reduced endocannabinoid levels. Biol Psychiatry 79:858–868. [DOI] [PubMed] [Google Scholar]
- Jha AK, Sarkar R, Udayan UK, Roy PK, Jha AK, Chaudhary RKP (2018) Bimatoprost in dermatology. Indian Dermatol Online J 9:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin XH, Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N (2007) Discovery and characterization of a Ca2+-independent phosphatidylethanolamine N-acyltransferase generating the anandamide precursor and its congeners. J Biol Chem 282:3614–3623. [DOI] [PubMed] [Google Scholar]
- Jing H, Reed A, Ulanovskaya OA, Grigoleit JS, Herbst DM, Henry CL, Li H, Barbas S, Germain J, Masuda K, et al. (2021) Phospholipase Cγ2 regulates endocannabinoid and eicosanoid networks in innate immune cells. Proc Natl Acad Sci USA 118:e2112971118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanek LM, Simone DA (2004) Activation of peripheral cannabinoid receptors attenuates cutaneous hyperalgesia produced by a heat injury. Pain 109:432–442. [DOI] [PubMed] [Google Scholar]
- Johnson MR, Melvin LS, Althuis TH, Bindra JS, Harbert CA, Milne GM, Weissman A (1981) Selective and potent analgetics derived from cannabinoids. J Clin Pharmacol 21(Suppl 1):271S–282S. [DOI] [PubMed] [Google Scholar]
- Jonsson KO, Vandevoorde S, Lambert DM, Tiger G, Fowler CJ (2001) Effects of homologues and analogues of palmitoylethanolamide upon the inactivation of the endocannabinoid anandamide. Br J Pharmacol 133:1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan T, Szanda G, Rosenberg AZ, Tam J, Earley BJ, Godlewski G, Cinar R, Liu Z, Liu J, Ju C, et al. (2014) Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy. Proc Natl Acad Sci USA 111:E5420–E5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KM, Astarita G, Zhu C, Wallace M, Mackie K, Piomelli D (2007) A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor-dependent endocannabinoid mobilization. Mol Pharmacol 72:612–621. [DOI] [PubMed] [Google Scholar]
- Jung KM, Sepers M, Henstridge CM, Lassalle O, Neuhofer D, Martin H, Ginger M, Frick A, DiPatrizio NV, Mackie K, et al. (2012) Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat Commun 3:1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, King AR, Redhi GH, Yasar S, Piomelli D, et al. (2008) Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiatry 64:930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justinova Z, Panlilio LV, Moreno-Sanz G, Redhi GH, Auber A, Secci ME, Mascia P, Bandiera T, Armirotti A, Bertorelli R, et al. (2015) Effects of fatty acid amide hydrolase (FAAH) inhibitors in non-human primate models of nicotine reward and relapse. Neuropsychopharmacology 40:2185–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Haj-Dahmane S (2022) Mechanisms of endocannabinoid transport in the brain. Br J Pharmacol 179:4300–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Glaser ST, Deutsch DG (2009) Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci USA 106:6375–6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Glaser ST, Chae J, Brown DA, Deutsch DG (2010) Lipid droplets are novel sites of N-acylethanolamine inactivation by fatty acid amide hydrolase-2. J Biol Chem 285:2796–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Hermann A, Glaser ST, Bojesen IN, Deutsch DG (2006) Anandamide uptake is consistent with rate-limited diffusion and is regulated by the degree of its hydrolysis by fatty acid amide hydrolase. J Biol Chem 281:9066–9075. [DOI] [PubMed] [Google Scholar]
- Kaczocha M, Rebecchi MJ, Ralph BP, Teng Y-HG, Berger WT, Galbavy W, Elmes MW, Glaser ST, Wang L, Rizzo RC, et al. (2014) Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS One 9:e94200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Vivieca S, Sun J, Glaser ST, Deutsch DG (2012) Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J Biol Chem 287:3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale VP, Gibbs S, Taylor JA, Zmarowski A, Novak J, Patton K, Sparrow B, Gorospe J, Anand S, Cinar R, et al. (2019) Preclinical toxicity evaluation of JD5037, a peripherally restricted CB1 receptor inverse agonist, in rats and dogs for treatment of nonalcoholic steatohepatitis. Regul Toxicol Pharmacol 109:104483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliomäki J, Annas P, Huizar K, Clarke C, Zettergren A, Karlsten R, Segerdahl M (2013) Evaluation of the analgesic efficacy and psychoactive effects of AZD1940, a novel peripherally acting cannabinoid agonist, in human capsaicin-induced pain and hyperalgesia. Clin Exp Pharmacol Physiol 40:212–218. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89:309–380. [DOI] [PubMed] [Google Scholar]
- Kantae V, Ogino S, Noga M, Harms AC, van Dongen RM, Onderwater GL, van den Maagdenberg AM, Terwindt GM, van der Stelt M, Ferrari MD, et al. (2017) Quantitative profiling of endocannabinoids and related N-acylethanolamines in human CSF using nano LC-MS/MS. J Lipid Res 58:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst M, Salim K, Burstein S, Conrad I, Hoy L, Schneider U (2003) Analgesic effect of the synthetic cannabinoid CT-3 on chronic neuropathic pain: a randomized controlled trial. JAMA 290:1757–1762. [DOI] [PubMed] [Google Scholar]
- Katayama K, Ueda N, Katoh I, Yamamoto S (1999) Equilibrium in the hydrolysis and synthesis of cannabimimetic anandamide demonstrated by a purified enzyme. Biochim Biophys Acta 1440:205–214. [DOI] [PubMed] [Google Scholar]
- Katayama K, Ueda N, Kurahashi Y, Suzuki H, Yamamoto S, Kato I (1997) Distribution of anandamide amidohydrolase in rat tissues with special reference to small intestine. Biochim Biophys Acta 1347:212–218. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, et al. (2003) Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9:76–81. [DOI] [PubMed] [Google Scholar]
- Keith JM, Apodaca R, Xiao W, Seierstad M, Pattabiraman K, Wu J, Webb M, Karbarz MJ, Brown S, Wilson S, et al. (2008) Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett 18:4838–4843. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62:265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall DA, Yudowski GA (2016) Cannabinoid receptors in the central nervous system: their signaling and roles in disease. Front Cell Neurosci 10:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbrat A, Ferré J-C, Fillatre P, Ronzière T, Vannier S, Carsin-Nicol B, Lavoué S, Vérin M, Gauvrit J-Y, Le Tulzo Y, et al. (2016) Acute neurologic disorder from an inhibitor of fatty acid amide hydrolase. N Engl J Med 375:1717–1725. [DOI] [PubMed] [Google Scholar]
- Khan H, Ghori FK, Ghani U, Javed A, Zahid S (2022) Cannabinoid and endocannabinoid system: a promising therapeutic intervention for multiple sclerosis. Mol Biol Rep 49:5117–5131. [DOI] [PubMed] [Google Scholar]
- Khanolkar AD, Abadji V, Lin S, Hill WA, Taha G, Abouzid K, Meng Z, Fan P, Makriyannis A (1996) Head group analogs of arachidonylethanolamide, the endogenous cannabinoid ligand. J Med Chem 39:4515–4519. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Gable J, Johns M, Khasabov SG, Kalyuzhny AE, Golovko MY, Golovko SA, Kiven S, Gupta K, Seybold VS, et al. (2023) Inhibition of DAGLβ as a therapeutic target for pain in sickle cell disease. Haematologica 108:859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Mackie K, Piomelli D, Kendall DA (2017) Modulation of CB1 cannabinoid receptor by allosteric ligands: pharmacology and therapeutic opportunities. Neuropharmacology 124:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi A, Ohashi K, Sugie Y, Sugimoto H, Omura H (2008) Pharmacological evaluation of a novel cannabinoid 2 (CB2) ligand, PF-03550096, in vitro and in vivo by using a rat model of visceral hypersensitivity. J Pharmacol Sci 106:219–224. [DOI] [PubMed] [Google Scholar]
- Kilaru A, Chapman KD (2020) The endocannabinoid system. Essays Biochem 64:485–499. [DOI] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH (2009) Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther 330:902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto S, Muramatsu M, Gokoh M, Oka S, Waku K, Sugiura T (2005) Endogenous cannabinoid receptor ligand induces the migration of human natural killer cells. J Biochem 137:217–223. [DOI] [PubMed] [Google Scholar]
- Klein TW, Cabral GA (2006) Cannabinoid-induced immune suppression and modulation of antigen-presenting cells. J Neuroimmune Pharmacol 1:50–64. [DOI] [PubMed] [Google Scholar]
- Kleyer J, Nicolussi S, Taylor P, Simonelli D, Furger E, Anderle P, Gertsch J (2012) Cannabinoid receptor trafficking in peripheral cells is dynamically regulated by a binary biochemical switch. Biochem Pharmacol 83:1393–1412. [DOI] [PubMed] [Google Scholar]
- Kogan NM, Lavi Y, Topping LM, Williams RO, McCann FE, Yekhtin Z, Feldmann M, Gallily R, Mechoulam R (2021) Novel CBG derivatives can reduce inflammation, pain and obesity. Molecules 26:5601–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosiakova H, Berdyshev A, Dosenko V, Drevytska T, Herasymenko O, Hula N (2022) The involvement of peroxisome proliferator-activated receptor gamma (PPARγ) in anti-inflammatory activity of N-stearoylethanolamine. Heliyon 8:e11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna Kumar K, Shalev-Benami M, Robertson MJ, Hu H, Banister SD, Hollingsworth SA, Latorraca NR, Kato HE, Hilger D, Maeda S, et al. (2019) Structure of a signaling cannabinoid receptor 1-G protein complex. Cell 176:448–458.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix S, Pechereau F, Leblanc N, Boubertakh B, Houde A, Martin C, Flamand N, Silvestri C, Raymond F, Di Marzo V, et al. (2019) Rapid and concomitant gut microbiota and endocannabinoidome response to diet-induced obesity in mice. mSystems 4:e00407–e00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lago-Fernandez A, Zarzo-Arias S, Jagerovic N, Morales P (2021) Relevance of peroxisome proliferator activated receptors in multitarget paradigm associated with the endocannabinoid system. Int J Mol Sci 22:1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DM, Fowler CJ (2005) The endocannabinoid system: drug targets, lead compounds, and potential therapeutic applications. J Med Chem 48:5059–5087. [DOI] [PubMed] [Google Scholar]
- Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A (1999) Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem 42:769–776. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME, Denovan-Wright EM (2015) Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol 172:4790–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME, Denovan-Wright EM (2016) Biased type 1 cannabinoid receptor signaling influences neuronal viability in a cell culture model of Huntington disease. Mol Pharmacol 89:364–375. [DOI] [PubMed] [Google Scholar]
- Lavey BJ, Kozlowski JA, Hipkin RW, Gonsiorek W, Lundell DJ, Piwinski JJ, Narula S, Lunn CA (2005) Triaryl bis-sulfones as a new class of cannabinoid CB2 receptor inhibitors: identification of a lead and initial SAR studies. Bioorg Med Chem Lett 15:783–786. [DOI] [PubMed] [Google Scholar]
- Lee HL, Jung KM, Fotio Y, Squire E, Palese F, Lin L, Torrens A, Ahmed F, Mabou Tagne A, Ramirez J, et al. (2022) Frequent low-dose Δ9-tetrahydrocannabinol in adolescence disrupts microglia homeostasis and disables responses to microbial infection and social stress in young adulthood. Biol Psychiatry 92:845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SA, Yang KJZ, Brun P-J, Silvaroli JA, Yuen JJ, Shmarakov I, Jiang H, Feranil JB, Li X, Lackey AI, et al. (2020) Retinol-binding protein 2 (RBP2) binds monoacylglycerols and modulates gut endocrine signaling and body weight. Sci Adv 6:eaay8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Jo J, Chung HY, Pothoulakis C, Im E (2016) Endocannabinoids in the gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 311:G655–G666. [DOI] [PubMed] [Google Scholar]
- Lefort C, Roumain M, Van Hul M, Rastelli M, Manco R, Leclercq I, Delzenne NM, Marzo VD, Flamand N, Luquet S, et al. (2020) Hepatic NAPE-PLD is a key regulator of liver lipid metabolism. Cells 9:1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett JD, Aspley S, Beckett SR, D’Antona AM, Kendall DA, Kendall DA (2004) Oleamide is a selective endogenous agonist of rat and human CB1 cannabinoid receptors. Br J Pharmacol 141:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Q, Yu Z, Li H, Cheng J, Wang Y (2022) Fatty acid-binding protein 5 aggravates pulmonary artery fibrosis in pulmonary hypertension secondary to left heart disease via activating wnt/β-catenin pathway. J Adv Res 40:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leishman E, Mackie K, Luquet S, Bradshaw HB (2016) Lipidomics profile of a NAPE-PLD KO mouse provides evidence of a broader role of this enzyme in lipid metabolism in the brain. Biochim Biophys Acta 1861:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberger L, Rowe H (1975) Clinical pharmacology of nabilone, a cannabinol derivative. Clin Pharmacol Ther 18:720–726. [DOI] [PubMed] [Google Scholar]
- Leo LM, Abood ME (2021) CB1 cannabinoid receptor signaling and biased signaling. Molecules 26:5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D, Saghatelian A, Simon GM, Cravatt BF (2006) Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry 45:4720–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K, Elmes MW, Glaser ST, Deutsch DG, Kaczocha M (2013) Role of FAAH-like anandamide transporter in anandamide inactivation. PLoS One 8:e79355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Ilnytskyy Y, Ghasemi Gojani E, Kovalchuk O, Kovalchuk I (2022) Analysis of anti-cancer and anti-inflammatory properties of 25 high-THC cannabis extracts. Molecules 27:6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chang H, Bouma J, de Paus LV, Mukhopadhyay P, Paloczi J, Mustafa M, van der Horst C, Kumar SS, Wu L, et al. (2023) Structural basis of selective cannabinoid CB2 receptor activation. Nat Commun 14:1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Hua T, Vemuri K, Ho JH, Wu Y, Wu L, Popov P, Benchama O, Zvonok N, Locke K, et al. (2019) Crystal structure of the human cannabinoid receptor CB2. Cell 176:459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Takamiya R, Miki Y, Heike K, Taketomi Y, Sugimoto N, Yamaguchi M, Shitara H, Nishito Y, Kobayashi T et al. (2022) Group IVE cytosolic phospholipase A2 limits psoriatic inflammation by mobilizing the anti-inflammatory lipid N-acylethanolamine. FASEB J 36:e22301. [DOI] [PubMed] [Google Scholar]
- Liedhegner ES, Vogt CD, Sem DS, Cunningham CW, Hillard CJ (2014) Sterol carrier protein-2: binding protein for endocannabinoids. Mol Neurobiol 50:149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligresti A, De Petrocellis L, Di Marzo V (2016) From phytocannabinoids to cannabinoid receptors and endocannabinoids: pleiotropic physiological and pathological roles through complex pharmacology. Physiol Rev 96:1593–1659. [DOI] [PubMed] [Google Scholar]
- Ligresti A, Morera E, Van Der Stelt M, Monory K, Lutz B, Ortar G, Di Marzo V (2004) Further evidence for the existence of a specific process for the membrane transport of anandamide. Biochem J 380:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limebeer CL, Vemuri VK, Bedard H, Lang ST, Ossenkopp KP, Makriyannis A, Parker LA (2010) Inverse agonism of cannabinoid CB1 receptors potentiates LiCl-induced nausea in the conditioned gaping model in rats. Br J Pharmacol 161:336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares IM, Zuardi AW, Pereira LC, Queiroz RH, Mechoulam R, Guimarães FS, Crippa JA (2019) Cannabidiol presents an inverted U-shaped dose-response curve in a simulated public speaking test. Br J Psychiatry 41:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling X, Zhang S, Shao P, Li W, Yang L, Ding Y, Xu C, Stella N, Bai M (2015) A novel near-infrared fluorescence imaging probe that preferentially binds to cannabinoid receptors CB2R over CB1R. Biomaterials 57:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhorst A, Lübke T (2022) The human Ntn-hydrolase superfamily: structure, functions and perspectives. Cells 11:1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gao B, Mirshahi F, Sanyal AJ, Khanolkar AD, Makriyannis A, Kunos G (2000) Functional CB1 cannabinoid receptors in human vascular endothelial cells. Biochem J 346:835–840. [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Yang N, Dong J, Tian W, Chang L, Ma J, Guo J, Tan J, Dong A, He K, et al. (2022) Deficiency in endocannabinoid synthase DAGLB contributes to early onset parkinsonism and murine nigral dopaminergic neuron dysfunction. Nat Commun 13:3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente-Berzal A, Terzian AL, di Marzo V, Micale V, Viveros MP, Wotjak CT (2015) 2-AG promotes the expression of conditioned fear via cannabinoid receptor type 1 on GABAergic neurons. Psychopharmacology (Berl) 232:2811–2825. [DOI] [PubMed] [Google Scholar]
- Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D (2005) The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol 67:15–19. [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, et al. (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long T, Wagner M, Demske D, Leipe C, Tarasov PE (2017) Cannabis in Eurasia: origin of human use and Bronze Age trans-continental connections. Veg Hist Archaeobot 26:245–258. [Google Scholar]
- López-Rodríguez ML, Viso A, Ortega-Gutiérrez S, Fowler CJ, Tiger G, de Lago E, Fernández-Ruiz J, Ramos JA (2003a) Design, synthesis and biological evaluation of new endocannabinoid transporter inhibitors. Eur J Med Chem 38:403–412. [DOI] [PubMed] [Google Scholar]
- López-Rodríguez ML, Viso A, Ortega-Gutiérrez S, Fowler CJ, Tiger G, de Lago E, Fernández-Ruiz J, Ramos JA (2003b) Design, synthesis, and biological evaluation of new inhibitors of the endocannabinoid uptake: comparison with effects on fatty acid amidohydrolase. J Med Chem 46:1512–1522. [DOI] [PubMed] [Google Scholar]
- López-Rodríguez ML, Viso A, Ortega-Gutiérrez S, Lastres-Becker I, González S, Fernández-Ruiz J, Ramos JA (2001) Design, synthesis and biological evaluation of novel arachidonic acid derivatives as highly potent and selective endocannabinoid transporter inhibitors. J Med Chem 44:4505–4508. [DOI] [PubMed] [Google Scholar]
- Lötsch J, Weyer-Menkhoff I, Tegeder I (2018) Current evidence of cannabinoid-based analgesia obtained in preclinical and human experimental settings. Eur J Pain 22:471–484. [DOI] [PubMed] [Google Scholar]
- Lu Y, Anderson HD (2017) Cannabinoid signaling in health and disease. Can J Physiol Pharmacol 95:311–327. [DOI] [PubMed] [Google Scholar]
- Lutz B (2020) Neurobiology of cannabinoid receptor signaling. Dialogues Clin Neurosci 22:207–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch A, Tammireddy SR, Doherty MK, Whitfield PD, Clarke DJ (2019) The glycine lipids of Bacteroides thetaiotaomicron are important for fitness during growth in vivo and in vitro. Appl Environ Microbiol 85:e02157–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn AB, Herkenham M (1994) Localization of cannabinoid receptors and nonsaturable high-density cannabinoid binding sites in peripheral tissues of the rat: implications for receptor-mediated immune modulation by cannabinoids. J Pharmacol Exp Ther 268:1612–1623. [PubMed] [Google Scholar]
- Maccarrone M (2017) Metabolism of the endocannabinoid anandamide: open questions after 25 years. Front Mol Neurosci 10:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M (2020) Missing pieces to the endocannabinoid puzzle. Trends Mol Med 26:263–272. [DOI] [PubMed] [Google Scholar]
- Maccarrone M (2023) Deciphering complex interactions in bioactive lipid signaling. Molecules 28:2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Bab I, Bíró T, Cabral GA, Dey SK, Di Marzo V, Konje JC, Kunos G, Mechoulam R, Pacher P, et al. (2015) Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol Sci 36:277–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Bari M, Battista N, Finazzi-Agrò A (2002) Estrogen stimulates arachidonoylethanolamide release from human endothelial cells and platelet activation. Blood 100:4040–4048. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Dainese E, Oddi S (2010) Intracellular trafficking of anandamide: new concepts for signaling. Trends Biochem Sci 35:601–608. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, De Chiara V, Gasperi V, Viscomi MT, Rossi S, Oddi S, Molinari M, Musella A, Finazzi-Agrò A, Centonze D (2009) Lipid rafts regulate 2-arachidonoylglycerol metabolism and physiological activity in the striatum. J Neurochem 109:371–381. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Fiorucci L, Erba F, Bari M, Finazzi-Agrò A, Ascoli F (2000) Human mast cells take up and hydrolyze anandamide under the control of 5-lipoxygenase and do not express cannabinoid receptors. FEBS Lett 468:176–180. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Guzmán M, Mackie K, Doherty P, Harkany T (2014) Programming of neural cells by (endo)cannabinoids: from physiological rules to emerging therapies. Nat Rev Neurosci 15:786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Maldonado R, Casas M, Henze T, Centonze D (2017) Cannabinoids therapeutic use: what is our current understanding following the introduction of THC, THC:CBD oromucosal spray and others? Expert Rev Clin Pharmacol 10:443–455. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Rapino C, Francavilla F, Barbonetti A (2021) Cannabinoid signalling and effects of cannabis on the male reproductive system. Nat Rev Urol 18:19–32. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agrò A, et al. (2008) Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci 11:152–159. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, van der Stelt M, Rossi A, Veldink GA, Vliegenthart JFG, Agrò AF (1998) Anandamide hydrolysis by human cells in culture and brain. J Biol Chem 273:32332–32339. [DOI] [PubMed] [Google Scholar]
- Mackie K (2005) Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol 168:299–325. [DOI] [PubMed] [Google Scholar]
- Mackie K (2008) Signaling via CNS cannabinoid receptors. Mol Cell Endocrinol 286(1-2, Suppl 1):S60–S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäder P, Bartholomäus R, Nicolussi S, Baumann A, Weis M, Chicca A, Rau M, Simão AC, Gertsch J, Altmann KH (2021) Synthesis and biological evaluation of endocannabinoid uptake inhibitors derived from WOBE437. ChemMedChem 16:145–154. [DOI] [PubMed] [Google Scholar]
- Magotti P, Bauer I, Igarashi M, Babagoli M, Marotta R, Piomelli D, Garau G (2015) Structure of human N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: regulation of fatty acid ethanolamide biosynthesis by bile acids. Structure 23:598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione S, Costa B, Di Marzo V (2013) Endocannabinoids: a unique opportunity to develop multitarget analgesics. Pain 154(Suppl 1):S87–S93. [DOI] [PubMed] [Google Scholar]
- Malamas MS, Farah SI, Lamani M, Pelekoudas DN, Perry NT, Rajarshi G, Miyabe CY, Chandrashekhar H, West J, Pavlopoulos S, et al. (2020) Design and synthesis of cyanamides as potent and selective N-acylethanolamine acid amidase inhibitors. Bioorg Med Chem 28:115195. [DOI] [PubMed] [Google Scholar]
- Mallat A, Lotersztajn S (2008) Endocannabinoids and liver disease. I. Endocannabinoids and their receptors in the liver. Am J Physiol Gastrointest Liver Physiol 294:G9–G12. [DOI] [PubMed] [Google Scholar]
- Mallipeddi S, Kreimer S, Zvonok N, Vemuri K, Karger BL, Ivanov AR, Makriyannis A (2017) Binding site characterization of AM1336, a novel covalent inverse agonist at human cannabinoid 2 receptor, using mass spectrometric analysis. J Proteome Res 16:2419–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Shen M, Boubertakh B, Martin C, Flamand N, Silvestri C, Di Marzo V (2020a) Alterations of brain endocannabinoidome signaling in germ-free mice. Biochim Biophys Acta Mol Cell Biol Lipids 1865:158786. [DOI] [PubMed] [Google Scholar]
- Manca C, Boubertakh B, Leblanc N, Deschênes T, Lacroix S, Martin C, Houde A, Veilleux A, Flamand N, Muccioli GG, et al. (2020b) Germ-free mice exhibit profound gut microbiota-dependent alterations of intestinal endocannabinoidome signaling. J Lipid Res 61:70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning JJ, Green HM, Glass M, Finlay DB (2021) Pharmacological selection of cannabinoid receptor effectors: signalling, allosteric modulation and bias. Neuropharmacology 193:108611. [DOI] [PubMed] [Google Scholar]
- Marchalant Y, Brownjohn PW, Bonnet A, Kleffmann T, Ashton JC (2014) Validating antibodies to the cannabinoid CB2 receptor: antibody sensitivity is not evidence of antibody specificity. J Histochem Cytochem 62:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, et al. (2010) The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci 13:951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Lutz B (2006) Neuromodulatory functions of the endocannabinoid system. J Endocrinol Invest 29(3, Suppl):27–46. [PubMed] [Google Scholar]
- Martella A, Sijben H, Rufer AC, Grether U, Fingerle J, Ullmer C, Hartung T, IJzerman AP, van der Stelt M, Heitman LH (2017) A novel selective inverse agonist of the CB2 receptor as a radiolabeled tool compound for kinetic binding studies. Mol Pharmacol 92:389–400. [DOI] [PubMed] [Google Scholar]
- Martellotta MC, Cossu G, Fattore L, Gessa GL, Fratta W (1998) Self-administration of the cannabinoid receptor agonist WIN 55,212-2 in drug-naive mice. Neuroscience 85:327–330. [DOI] [PubMed] [Google Scholar]
- Martin GG, Landrock D, Chung S, Dangott LJ, Seeger DR, Murphy EJ, Golovko MY, Kier AB, Schroeder F (2017) Fabp1 gene ablation inhibits high-fat diet-induced increase in brain endocannabinoids. J Neurochem 140:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GG, Seeger DR, McIntosh AL, Milligan S, Chung S, Landrock D, Dangott LJ, Golovko MY, Murphy EJ, Kier AB, et al. (2019) Sterol carrier protein-2/sterol carrier protein-x/fatty acid binding protein-1 ablation impacts response of brain endocannabinoid to high-fat diet. Lipids 54:583–601. [DOI] [PubMed] [Google Scholar]
- Martín-Couce L, Martín-Fontecha M, Palomares O, Mestre L, Cordomí A, Hernangomez M, Palma S, Pardo L, Guaza C, López-Rodríguez ML, et al. (2012) Chemical probes for the recognition of cannabinoid receptors in native systems. Angew Chem Int Ed Engl 51:6896–6899. [DOI] [PubMed] [Google Scholar]
- Martínez-Pinilla E, Varani K, Reyes-Resina I, Angelats E, Vincenzi F, Ferreiro-Vera C, Oyarzabal J, Canela EI, Lanciego JL, Nadal X, et al. (2017) Binding and signaling studies disclose a potential allosteric site for cannabidiol in cannabinoid CB2 receptors. Front Pharmacol 8:744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi P, Valenti M, Vaccani A, Gasperi V, Perletti G, Marras E, Fezza F, Maccarrone M, Parolaro D (2008) 5-Lipoxygenase and anandamide hydrolase (FAAH) mediate the antitumor activity of cannabidiol, a non-psychoactive cannabinoid. J Neurochem 104:1091–1100. [DOI] [PubMed] [Google Scholar]
- Masson J, Sagné C, Hamon M, El Mestikawy S (1999) Neurotransmitter transporters in the central nervous system. Pharmacol Rev 51:439–464. [PubMed] [Google Scholar]
- Mastinu A, Pira M, Pinna GA, Pisu C, Casu MA, Reali R, Marcello S, Murineddu G, Lazzari P (2013) NESS06SM reduces body weight with an improved profile relative to SR141716A. Pharmacol Res 74:94–108. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564. [DOI] [PubMed] [Google Scholar]
- Mauler F, Hinz V, Augstein KH, Fassbender M, Horváth E (2003) Neuroprotective and brain edema-reducing efficacy of the novel cannabinoid receptor agonist BAY 38-7271. Brain Res 989:99–111. [DOI] [PubMed] [Google Scholar]
- Mauler F, Mittendorf J, Horváth E, De Vry J (2002) Characterization of the diarylether sulfonylester (-)-(R)-3-(2-hydroxymethylindanyl-4-oxy)phenyl-4,4,4-trifluoro-1-sulfonate (BAY 38-7271) as a potent cannabinoid receptor agonist with neuroprotective properties. J Pharmacol Exp Ther 302:359–368. [DOI] [PubMed] [Google Scholar]
- McGilveray IJ (2005) Pharmacokinetics of cannabinoids. Pain Res Manag 10(Suppl A):15A–22A. [DOI] [PubMed] [Google Scholar]
- McKinney MK, Cravatt BF (2005) Structure and function of fatty acid amide hydrolase. Annu Rev Biochem 74:411–432. [DOI] [PubMed] [Google Scholar]
- McPartland JM, Duncan M, Di Marzo V, Pertwee RG (2015) Are cannabidiol and Δ(9) -tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol 172:737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McReynolds JR, Doncheck EM, Li Y, Vranjkovic O, Graf EN, Ogasawara D, Cravatt BF, Baker DA, Liu QS, Hillard CJ, et al. (2018) Stress promotes drug seeking through glucocorticoid-dependent endocannabinoid mobilization in the prelimbic cortex. Biol Psychiatry 84:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R (2005) Plant cannabinoids: a neglected pharmacological treasure trove. Br J Pharmacol 146:913–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R (2023) A delightful trip along the pathway of cannabinoid and endocannabinoid chemistry and pharmacology. Annu Rev Pharmacol Toxicol 63:1–13 10.1146/annurev-pharmtox-051921-083709. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Gaoni Y (1967) The absolute configuration of delta-1-tetrahydrocannabinol, the major active constituent of hashish. Tetrahedron Lett 12:1109–1111. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Hanus L, Fride E (1998) Towards cannabinoid drugs--revisited. Prog Med Chem 35:199–243. [DOI] [PubMed] [Google Scholar]
- Mehrpouya-Bahrami P, Chitrala KN, Ganewatta MS, Tang C, Murphy EA, Enos RT, Velazquez KT, McCellan J, Nagarkatti M, Nagarkatti P (2017) Blockade of CB1 cannabinoid receptor alters gut microbiota and attenuates inflammation and diet-induced obesity. Sci Rep 7:15645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL (2004) Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci 24:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin LS, Milne GM, Johnson MR, Subramaniam B, Wilken GH, Howlett AC (1993) Structure-activity relationships for cannabinoid receptor-binding and analgesic activity: studies of bicyclic cannabinoid analogs. Mol Pharmacol 44:1008–1015. [PubMed] [Google Scholar]
- Melvin LS, Milne GM, Johnson MR, Wilken GH, Howlett AC (1995) Structure-activity relationships defining the ACD-tricyclic cannabinoids: cannabinoid receptor binding and analgesic activity. Drug Des Discov 13:155–166. [PubMed] [Google Scholar]
- Migliore M, Pontis S, Fuentes de Arriba AL, Realini N, Torrente E, Armirotti A, Romeo E, Di Martino S, Russo D, Pizzirani D, et al. (2016) Second‐generation non‐covalent NAAA inhibitors are protective in a model of multiple sclerosis. Angew Chem Int Ed Engl 55:11193–11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC (2010) Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J Mol Biol 400:743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milman G, Maor Y, Abu-Lafi S, Horowitz M, Gallily R, Batkai S, Mo FM, Offertaler L, Pacher P, Kunos G, et al. (2006) N-arachidonoyl L-serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc Natl Acad Sci USA 103:2428–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralpeix C, Reguera AC, Fosch A, Zagmutt S, Casals N, Cota D, Rodríguez-Rodríguez R (2021) Hypothalamic endocannabinoids in obesity: an old story with new challenges. Cell Mol Life Sci 78:7469–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirlohi S, Bladen C, Santiago MJ, Arnold JC, McGregor I, Connor M (2022) Inhibition of human recombinant T-type calcium channels by phytocannabinoids in vitro. Br J Pharmacol 179:4031–4043. [DOI] [PubMed] [Google Scholar]
- Misto A, Provensi G, Vozella V, Passani MB, Piomelli D (2019) Mast cell-derived histamine regulates liver ketogenesis via oleoylethanolamide signaling. Cell Metab 29:91–102.e5. [DOI] [PubMed] [Google Scholar]
- Mock ED, Mustafa M, Gunduz-Cinar O, Cinar R, Petrie GN, Kantae V, Di X, Ogasawara D, Varga ZV, Paloczi J, et al. (2020) Discovery of a NAPE-PLD inhibitor that modulates emotional behavior in mice. Nat Chem Biol 16:667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SA, Nomikos GG, Dickason-Chesterfield AK, Schober DA, Schaus JM, Ying BP, Xu YC, Phebus L, Simmons RM, Li D, et al. (2005) Identification of a high-affinity binding site involved in the transport of endocannabinoids. Proc Natl Acad Sci USA 102:17852–17857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D (2004) Cyclohexylcarbamic acid 3′- or 4′-substituted biphenyl-3-yl esters as fatty acid amide hydrolase inhibitors: synthesis, quantitative structure-activity relationships, and molecular modeling studies. J Med Chem 47:4998–5008. [DOI] [PubMed] [Google Scholar]
- Morales P, Reggio PH (2017) An update on non-CB1, non-CB2 cannabinoid related G-protein-coupled receptors. Cannabis Cannabinoid Res 2:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales P, Hernandez-Folgado L, Goya P, Jagerovic N (2016) Cannabinoid receptor 2 (CB2) agonists and antagonists: a patent update. Expert Opin Ther Pat 26:843–856. [DOI] [PubMed] [Google Scholar]
- Morales P, Hurst DP, Reggio PH (2017) Molecular targets of the phytocannabinoids: a complex picture. Prog Chem Org Nat Prod 103:103–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales P, Lago-Fernandez A, Hurst DP, Sotudeh N, Brailoiu E, Reggio PH, Abood ME, Jagerovic N (2020) Therapeutic exploitation of GPR18: beyond the cannabinoids? J Med Chem 63:14216–14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli GG, Naslain D, Bäckhed F, Reigstad CS, Lambert DM, Delzenne NM, Cani PD (2010) The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol 6:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Baggelaar M, Erdelyi K, Cao Z, Cinar R, Fezza F, Ignatowska-Janlowska B, Wilkerson J, van Gils N, Hansen T, et al. (2016) The novel, orally available and peripherally restricted selective cannabinoid CB2 receptor agonist LEI-101 prevents cisplatin-induced nephrotoxicity. Br J Pharmacol 173:446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Pan H, Rajesh M, Bátkai S, Patel V, Harvey-White J, Mukhopadhyay B, Haskó G, Gao B, Mackie K, et al. (2010b) CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br J Pharmacol 160:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Pan H, Patel V, Mukhopadhyay B, Bátkai S, Gao B, Haskó G, Pacher P (2010a) Cannabinoid-2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic Biol Med 48:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Shim JY, Assi AA, Norford D, Howlett AC (2002) CB(1) cannabinoid receptor-G protein association: a possible mechanism for differential signaling. Chem Phys Lipids 121:91–109. [DOI] [PubMed] [Google Scholar]
- Muller C, Morales P, Reggio PH (2019) Cannabinoid ligands targeting TRP channels. Front Mol Neurosci 11:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Vahl KR, Fremer C, Beals C, Ivkovic J, Loft H, Schindler C (2021) Monoacylglycerol lipase inhibition in Tourette syndrome: a 12-week, randomized, controlled study. Mov Disord 36:2413–2418. [DOI] [PubMed] [Google Scholar]
- Müller-Vahl KR, Fremer C, Beals C, Ivkovic J, Loft H, Schindler C (2022) Endocannabinoid modulation using monoacylglycerol lipase inhibition in Tourette syndrome: a phase 1 randomized, placebo-controlled study. Pharmacopsychiatry 55:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65. [DOI] [PubMed] [Google Scholar]
- Murillo-Rodríguez E, Palomero-Rivero M, Millán-Aldaco D, Di Marzo V (2013) The administration of endocannabinoid uptake inhibitors OMDM-2 or VDM-11 promotes sleep and decreases extracellular levels of dopamine in rats. Physiol Behav 109:88–95. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Makriyannis A, Hohmann AG (2003) Selective activation of cannabinoid CB(2) receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience 119:747–757. [DOI] [PubMed] [Google Scholar]
- Nagappan A, Shin J, Jung MH (2019) Role of cannabinoid receptor type 1 in insulin resistance and its biological implications. Int J Mol Sci 20:2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naguib M, Diaz P, Xu JJ, Astruc-Diaz F, Craig S, Vivas-Mejia P, Brown DL (2008) MDA7: a novel selective agonist for CB2 receptors that prevents allodynia in rat neuropathic pain models. Br J Pharmacol 155:1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Dryanovski DI, Kimura Y, Jackson SN, Woods AS, Yasui Y, Tsai S-Y, Patel S, Covey DP, Su T-P, et al. (2019) Cocaine-induced endocannabinoid signaling mediated by sigma-1 receptors and extracellular vesicle secretion. eLife 8:e47209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Academies of Sciences, Engineering, and Medicine (2017) The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. Washington, DC: National Academies Press. 10.17226/24625 [DOI] [PubMed] [Google Scholar]
- Navarrete C, Carrillo-Salinas F, Palomares B, Mecha M, Jiménez-Jiménez C, Mestre L, Feliú A, Bellido ML, Fiebich BL, Appendino G, et al. (2018) Hypoxia mimetic activity of VCE-004.8, a cannabidiol quinone derivative: implications for multiple sclerosis therapy. J Neuroinflammation 15:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G, Varani K, Lillo A, Vincenzi F, Rivas-Santisteban R, Raïch I, Reyes-Resina I, Ferreiro-Vera C, Borea PA, Sánchez de Medina V, et al. (2020) Pharmacological data of cannabidiol- and cannabigerol-type phytocannabinoids acting on cannabinoid CB1, CB2 and CB1/CB2 heteromer receptors. Pharmacol Res 159:104940. [DOI] [PubMed] [Google Scholar]
- Nettekoven M, Adam JM, Bendels S, Bissantz C, Fingerle J, Grether U, Grüner S, Guba W, Kimbara A, Ottaviani G, et al. (2016) Novel triazolopyrimidine-derived cannabinoid receptor 2 agonists as potential treatment for inflammatory kidney diseases. ChemMedChem 11:179–189. [DOI] [PubMed] [Google Scholar]
- Nettekoven M, Fingerle J, Grether U, Grüner S, Kimbara A, Püllmann B, Rogers-Evans M, Röver S, Schuler F, Schulz-Gasch T, et al. (2013) Highly potent and selective cannabinoid receptor 2 agonists: initial hit optimization of an adamantyl hit series identified from high-through-put screening. Bioorg Med Chem Lett 23:1177–1181. [DOI] [PubMed] [Google Scholar]
- Nicolussi S (2014) Identification and characterization of potent and selective inhibitors of endocannabinoid uptake. GCB Grad Sch Univ Bern, Switz 14:1–173. [Google Scholar]
- Nicolussi S, Gertsch J (2015) Endocannabinoid transport revisited. Vitam Horm 98:441–485. [DOI] [PubMed] [Google Scholar]
- Nicolussi S, Chicca A, Rau M, Rihs S, Soeberdt M, Abels C, Gertsch J (2014a) Correlating FAAH and anandamide cellular uptake inhibition using N-alkylcarbamate inhibitors: from ultrapotent to hyperpotent. Biochem Pharmacol 92:669–689. [DOI] [PubMed] [Google Scholar]
- Nicolussi S, Viveros-Paredes JM, Gachet MS, Rau M, Flores-Soto ME, Blunder M, Gertsch J (2014b) Guineensine is a novel inhibitor of endocannabinoid uptake showing cannabimimetic behavioral effects in BALB/c mice. Pharmacol Res 80:52–65. [DOI] [PubMed] [Google Scholar]
- Niphakis MJ, Cognetta AB 3rd, Chang JW, Buczynski MW, Parsons LH, Byrne F, Burston JJ, Chapman V, Cravatt BF (2013) Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem Neurosci 4:1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF (2010) Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, et al. (2011) Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334:809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien LD, Limebeer CL, Rock EM, Bottegoni G, Piomelli D, Parker LA (2013) Anandamide transport inhibition by ARN272 attenuates nausea-induced behaviour in rats, and vomiting in shrews (Suncus murinus). Br J Pharmacol 170:1130–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan SE (2016) An update on PPAR activation by cannabinoids. Br J Pharmacol 173:1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odan M, Ishizuka N, Hiramatsu Y, Inagaki M, Hashizume H, Fujii Y, Mitsumori S, Morioka Y, Soga M, Deguchi M, et al. (2012) Discovery of S-777469: an orally available CB2 agonist as an antipruritic agent. Bioorg Med Chem Lett 22:2803–2806. [DOI] [PubMed] [Google Scholar]
- Oddi S, Fezza F, Catanzaro G, De Simone C, Pucci M, Piomelli D, Finazzi-Agrò A, Maccarrone M (2010) Pitfalls and solutions in assaying anandamide transport in cells. J Lipid Res 51:2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddi S, Fezza F, Pasquariello N, D’Agostino A, Catanzaro G, De Simone C, Rapino C, Finazzi-Agrò A, Maccarrone M (2009) Molecular identification of albumin and Hsp70 as cytosolic anandamide-binding proteins. Chem Biol 16:624–632. [DOI] [PubMed] [Google Scholar]
- Oddi S, Fezza F, Pasquariello N, De Simone C, Rapino C, Dainese E, Finazzi-Agrò A, Maccarrone M (2008) Evidence for the intracellular accumulation of anandamide in adiposomes. Cell Mol Life Sci 65:840–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddi S, Scipioni L, Maccarrone M (2020) Endocannabinoid system and adult neurogenesis: a focused review. Curr Opin Pharmacol 50:25–32. [DOI] [PubMed] [Google Scholar]
- Oddi S, Stepniewski TM, Totaro A, Selent J, Scipioni L, Dufrusine B, Fezza F, Dainese E, Maccarrone M (2017) Palmitoylation of cysteine 415 of CB1 receptor affects ligand-stimulated internalization and selective interaction with membrane cholesterol and caveolin 1. Biochim Biophys Acta Mol Cell Biol Lipids 1862:523–532. [DOI] [PubMed] [Google Scholar]
- Ofek O, Karsak M, Leclerc N, Fogel M, Frenkel B, Wright K, Tam J, Attar-Namdar M, Kram V, Shohami E, et al. (2006) Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci USA 103:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara D, Deng H, Viader A, Baggelaar MP, Breman A, den Dulk H, van den Nieuwendijk AM, Soethoudt M, van der Wel T, Zhou J, et al. (2016) Rapid and profound rewiring of brain lipid signaling networks by acute diacylglycerol lipase inhibition. Proc Natl Acad Sci USA 113:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Parsons WH, Kamat SS, Cravatt BF (2016) A calcium-dependent acyltransferase that produces N-acyl phosphatidylethanolamines. Nat Chem Biol 12:669–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H, Ishizaka T, Tatsuzuki M, Yoshinaga M, Iida I, Tomishima Y, Toda Y, Saito S (2007) N-Alkylidenearylcarboxamides as new potent and selective CB(2) cannabinoid receptor agonists with good oral bioavailability. Bioorg Med Chem Lett 17:6299–6304. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279:5298–5305. [DOI] [PubMed] [Google Scholar]
- Ortar G, Cascio MG, Moriello AS, Camalli M, Morera E, Nalli M, Di Marzo V (2008) Carbamoyl tetrazoles as inhibitors of endocannabinoid inactivation: a critical revisitation. Eur J Med Chem 43:62–72. [DOI] [PubMed] [Google Scholar]
- Ortar G, Ligresti A, De Petrocellis L, Morera E, Di Marzo V (2003) Novel selective and metabolically stable inhibitors of anandamide cellular uptake. Biochem Pharmacol 65:1473–1481. [DOI] [PubMed] [Google Scholar]
- Ortega-Gutiérrez S, Hawkins EG, Viso A, López-Rodríguez ML, Cravatt BF (2004) Comparison of anandamide transport in FAAH wild-type and knockout neurons: evidence for contributions by both FAAH and the CB1 receptor to anandamide uptake. Biochemistry 43:8184–8190. [DOI] [PubMed] [Google Scholar]
- Oyagawa CRM, de la Harpe SM, Saroz Y, Glass M, Vernall AJ, Grimsey NL (2018) Cannabinoid receptor 2 signalling bias elicited by 2,4,6-trisubstituted 1,3,5-triazines. Front Pharmacol 9:1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Kunos G (2013) Modulating the endocannabinoid system in human health and disease--successes and failures. FEBS J 280:1918–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Gao B (2008) Endocannabinoids and liver disease. III. Endocannabinoid effects on immune cells: implications for inflammatory liver diseases. Am J Physiol Gastrointest Liver Physiol 294:G850–G854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Haskó G (2008) Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br J Pharmacol 153:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Mechoulam R (2011) Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 50:193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, Kunos G (2006) The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 58:389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Kogan NM, Mechoulam R (2020) Beyond THC and Endocannabinoids. Annu Rev Pharmacol Toxicol 60:637–659. [DOI] [PubMed] [Google Scholar]
- Pacher P, Mukhopadhyay P, Mohanraj R, Godlewski G, Bátkai S, Kunos G (2008) Modulation of the endocannabinoid system in cardiovascular disease: therapeutic potential and limitations. Hypertension 52:601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacioni G, Rapino C, Zarivi O, Falconi A, Leonardi M, Battista N, Colafarina S, Sergi M, Bonfigli A, Miranda M, et al. (2015) Truffles contain endocannabinoid metabolic enzymes and anandamide. Phytochemistry 110:104–110. [DOI] [PubMed] [Google Scholar]
- Pagano Zottola AC, Severi I, Cannich A, Ciofi P, Cota D, Marsicano G, Giordano A, Bellocchio L (2022) Expression of functional cannabinoid type-1 (CB1) receptor in mitochondria of white adipocytes. Cells 11:2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagé D, Balaux E, Boisvert L, Liu Z, Milburn C, Tremblay M, Wei Z, Woo S, Luo X, Cheng YX, et al. (2008) Novel benzimidazole derivatives as selective CB2 agonists. Bioorg Med Chem Lett 18:3695–3700. [DOI] [PubMed] [Google Scholar]
- Pagé D, Wei Z, Liu Z, Tremblay M, Desfosses H, Milburn C, Srivastava S, Yang H, Brown W, Walpole C, et al. (2010) 5-Sulfonamide benzimidazoles: a class of cannabinoid receptors agonists with potent in vivo antinociception activity. Lett Drug Des Discov 7:208–213. [Google Scholar]
- Palese F, Pontis S, Realini N, Torrens A, Ahmed F, Assogna F, Pellicano C, Bossù P, Spalletta G, Green K, et al. (2022) Targeting NAAA counters dopamine neuron loss and symptom progression in mouse models of parkinsonism. Pharmacol Res 182:106338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P, Roy KK, Doerksen RJ (2020) Negative allosteric modulators of cannabinoid receptor 2: protein modeling, binding site identification and molecular dynamics simulations in the presence of an orthosteric agonist. J Biomol Struct Dyn 38:32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker LA, Rock EM, Limebeer CL (2011) Regulation of nausea and vomiting by cannabinoids. Br J Pharmacol 163:1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Hill MN, Cheer JF, Wotjak CT, Holmes A (2017) The endocannabinoid system as a target for novel anxiolytic drugs. Neurosci Biobehav Rev 76 (Pt A):56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus MP, Stein MB, Simmons AN, Risbrough VB, Halter R, Chaplan SR (2021) The effects of FAAH inhibition on the neural basis of anxiety-related processing in healthy male subjects: a randomized clinical trial. Neuropsychopharmacology 46:1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Studholme K, Kanjiya MP, Luk J, Bogdan D, Elmes MW, Carbonetti G, Tong S, Gary Teng YH, Rizzo RC, et al. (2017) Fatty-acid-binding protein inhibition produces analgesic effects through peripheral and central mechanisms. Mol Pain 13:1744806917697007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG (1999) Pharmacology of cannabinoid receptor ligands. Curr Med Chem 6:635–664. [PubMed] [Google Scholar]
- Pertwee RG (2001) Cannabinoids and the gastrointestinal tract. Gut 48:859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG (2002) Cannabinoids and multiple sclerosis. Pharmacol Ther 95:165–174. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2005) Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci 76:1307–1324. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2008a) The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol 153:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG (2008b) Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addict Biol 13:147–159. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2012) Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos Trans R Soc Lond B Biol Sci 367:3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG (2014) Handbook of Cannabis. Oxford University press, Oxford, U.K. [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, et al. (2010) International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev 62:588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Rock EM, Guenther K, Limebeer CL, Stevenson LA, Haj C, Smoum R, Parker LA, Mechoulam R (2018) Cannabidiolic acid methyl ester, a stable synthetic analogue of cannabidiolic acid, can produce 5-HT1A receptor-mediated suppression of nausea and anxiety in rats. Br J Pharmacol 175:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Stevenson LA, Elrick DB, Mechoulam R, Corbett AD (1992) Inhibitory effects of certain enantiomeric cannabinoids in the mouse vas deferens and the myenteric plexus preparation of guinea-pig small intestine. Br J Pharmacol 105:980–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitet F, Jeantaud B, Reibaud M, Imperato A, Dubroeucq MC (1998) Complex pharmacology of natural cannabinoids: evidence for partial agonist activity of delta9-tetrahydrocannabinol and antagonist activity of cannabidiol on rat brain cannabinoid receptors. Life Sci 63:PL1–PL6. [DOI] [PubMed] [Google Scholar]
- Petitet F, Marin L, Doble A (1996) Biochemical and pharmacological characterization of cannabinoid binding sites using [3H]SR141716A. Neuroreport 7:789–792. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Di Marzo V (2017) The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br J Pharmacol 174:1349–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Schiano Moriello A, Verde R, Allarà M, Imperatore R, Ligresti A, Mahmoud AM, Peritore AF, Iannotti FA, Di Marzo V (2019) Palmitoylethanolamide counteracts substance P-induced mast cell activation in vitro by stimulating diacylglycerol lipase activity. J Neuroinflammation 16:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov RR, Ferrini ME, Jaffar Z, Thompson CM, Roberts K, Diaz P (2011) Design and evaluation of a novel fluorescent CB2 ligand as probe for receptor visualization in immune cells. Bioorg Med Chem Lett 21:5859–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucci V, Chicca A, Glasmacher S, Paloczi J, Cao Z, Pacher P, Gertsch J (2017) Pepcan-12 (RVD-hemopressin) is a CB2 receptor positive allosteric modulator constitutively secreted by adrenals and in liver upon tissue damage. Sci Rep 7:9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharma Intelligence (2022) Olorinab. Arena cannabinoid pain compound misses in phase IIb IBS trial. Scrip, informa.com (accessed October 2022).
- Piazza PV, Cota D, Marsicano G (2017) The CB1 receptor as the cornerstone of exostasis. Neuron 93:1252–1274. [DOI] [PubMed] [Google Scholar]
- Piomelli D (2003) The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4:873–884. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Mabou Tagne A (2022) Endocannabinoid-based therapies. Annu Rev Pharmacol Toxicol 62:483–507. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Beltramo M, Glasnapp S, Lin SY, Goutopoulos A, Xie X-Q, Makriyannis A (1999) Structural determinants for recognition and translocation by the anandamide transporter. Proc Natl Acad Sci USA 96:5802–5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D, Scalvini L, Fotio Y, Lodola A, Spadoni G, Tarzia G, Mor M (2020) N-acylethanolamine acid amidase (NAAA): structure, function, and inhibition. J Med Chem 63:7475–7490. [DOI] [PubMed] [Google Scholar]
- Piscitelli F, Carta G, Bisogno T, Murru E, Cordeddu L, Berge K, Tandy S, Cohn JS, Griinari M, Banni S, et al. (2011) Effect of dietary krill oil supplementation on the endocannabinoidome of metabolically relevant tissues from high-fat-fed mice. Nutr Metab (Lond) 8:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscitelli F, Guida F, Luongo L, Iannotti FA, Boccella S, Verde R, Lauritano A, Imperatore R, Smoum R, Cristino L, et al. (2020) Protective effects of N-oleoylglycine in a mouse model of mild traumatic brain injury. ACS Chem Neurosci 11:1117–1128. [DOI] [PubMed] [Google Scholar]
- Plasse TF, Gorter RW, Krasnow SH, Lane M, Shepard KV, Wadleigh RG (1991) Recent clinical experience with dronabinol. Pharmacol Biochem Behav 40:695–700. [DOI] [PubMed] [Google Scholar]
- Plau J, Golczak M, Paik J, Calderon RM, Blaner WS (2022) Retinol-binding protein 2 (RBP2): More than just dietary retinoid uptake. Biochim Biophys Acta Mol Cell Biol Lipids 1867:159179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, Chilloux J, Ottman N, Duparc T, Lichtenstein L, et al. (2017) A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med 23:107–113. [DOI] [PubMed] [Google Scholar]
- Ponzano S, Bertozzi F, Mengatto L, Dionisi M, Armirotti A, Romeo E, Berteotti A, Fiorelli C, Tarozzo G, Reggiani A, et al. (2013) Synthesis and structure-activity relationship (SAR) of 2-methyl-4-oxo-3-oxetanylcarbamic acid esters, a class of potent N-acylethanolamine acid amidase (NAAA) inhibitors. J Med Chem 56:6917–6934. [DOI] [PubMed] [Google Scholar]
- Porter AC, Sauer J-M, Knierman MD, Becker GW, Berna MJ, Bao J, Nomikos GG, Carter P, Bymaster FP, Leese AB, et al. (2002) Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther 301:1020–1024. [DOI] [PubMed] [Google Scholar]
- Porter RF, Szczesniak AM, Toguri JT, Gebremeskel S, Johnston B, Lehmann C, Fingerle J, Rothenhäusler B, Perret C, Rogers-Evans M, et al. (2019) Selective cannabinoid 2 receptor agonists as potential therapeutic drugs for the treatment of endotoxin-induced uveitis. Molecules 24:3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portier M, Rinaldi-Carmona M, Pecceu F, Combes T, Poinot-Chazel C, Calandra B, Barth F, le Fur G, Casellas P (1999) SR 144528, an antagonist for the peripheral cannabinoid receptor that behaves as an inverse agonist. J Pharmacol Exp Ther 288:582–589. [PubMed] [Google Scholar]
- Powell DR, Gay JP, Wilganowski N, Doree D, Savelieva KV, Lanthorn TH, Read R, Vogel P, Hansen GM, Brommage R, et al. (2015) Diacylglycerol lipase α knockout mice demonstrate metabolic and behavioral phenotypes similar to those of cannabinoid receptor 1 knockout mice. Front Endocrinol (Lausanne) 6:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevet H, Collins I (2019) Labelled chemical probes for demonstrating direct target engagement in living systems. Future Med Chem 11:1195–1224. [DOI] [PubMed] [Google Scholar]
- Priestley R, Glass M, Kendall D (2017) Functional selectivity at cannabinoid receptors. Adv Pharmacol 80:207–221. [DOI] [PubMed] [Google Scholar]
- Procaccia S, Lewitus GM, Lipson Feder C, Shapira A, Berman P, Meiri D (2022) Cannabis for medical use: versatile plant rather than a single drug. Front Pharmacol 13:894960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce G, Baker D (2015) Endocannabinoids in multiple sclerosis and amyotrophic lateral sclerosis. Handb Exp Pharmacol 231:213–231. [DOI] [PubMed] [Google Scholar]
- Punt JM, van der Vliet D, van der Stelt M (2023) Chemical probes to control and visualize lipid metabolism in the brain. Acc Chem Res 55:3205–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raduner S, Bisson W, Abagyan R, Altmann K-H, Gertsch J (2007) Self-assembling cannabinomimetics: supramolecular structures of N-alkyl amides. J Nat Prod 70:1010–1015. [DOI] [PubMed] [Google Scholar]
- Rahman IAS, Tsuboi K, Hussain Z, Yamashita R, Okamoto Y, Uyama T, Yamazaki N, Tanaka T, Tokumura A, Ueda N (2016) Calcium-dependent generation of N-acylethanolamines and lysophosphatidic acids by glycerophosphodiesterase GDE7. Biochim Biophys Acta 1861(12 Pt A):1881–1892. [DOI] [PubMed] [Google Scholar]
- Rahman SMK, Hussain Z, Morito K, Takahashi N, Sikder MM, Tanaka T, Ohta KI, Ueno M, Takahashi H, Yamamoto T, et al. (2022) Formation of N-acyl-phosphatidylethanolamines by cytosolic phospholipase A2ε in an ex vivo murine model of brain ischemia. Biochim Biophys Acta Mol Cell Biol Lipids 1867:159222. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Pan H, Mukhopadhyay P, Bátkai S, Osei-Hyiaman D, Haskó G, Liaudet L, Gao B, Pacher P (2007) Cannabinoid-2 receptor agonist HU-308 protects against hepatic ischemia/reperfusion injury by attenuating oxidative stress, inflammatory response, and apoptosis. J Leukoc Biol 82:1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhshan F, Day TA, Blakely RD, Barker EL (2000) Carrier-mediated uptake of the endogenous cannabinoid anandamide in RBL-2H3 cells. J Pharmacol Exp Ther 292:960–967. [PubMed] [Google Scholar]
- Randall PA, Vemuri VK, Segovia KN, Torres EF, Hosmer S, Nunes EJ, Santerre JL, Makriyannis A, Salamone JD (2010) The novel cannabinoid CB1 antagonist AM6545 suppresses food intake and food-reinforced behavior. Pharmacol Biochem Behav 97:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau M, Nicolussi S, Chicca A, Gertsch J (2016) Assay of endocannabinoid uptake. Methods Mol Biol 1412:191–203. [DOI] [PubMed] [Google Scholar]
- Razdan RK (1986) Structure-activity relationships in cannabinoids. Pharmacol Rev 38:75–149. [PubMed] [Google Scholar]
- Reggio PH, Seltzman HH, Compton DR, Prescott WR Jr, Martin BR (1990) Investigation of the role of the phenolic hydroxyl in cannabinoid activity. Mol Pharmacol 38:854–862. [PubMed] [Google Scholar]
- Reynoso-Moreno I, Gertsch J (2021) Small-molecule inhibitors of endocannabinoid transport and trafficking. Drug Discov Series 76:414–450. [Google Scholar]
- Reynoso-Moreno I, Chicca A, Flores-Soto ME, Viveros-Paredes JM, Gertsch J (2018) The endocannabinoid reuptake inhibitor WOBE437 is orally bioavailable and exerts indirect polypharmacological effects via different endocannabinoid receptors. Front Mol Neurosci 11:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoso-Moreno I, Najar-Guerrero I, Escareño N, Flores-Soto MEME, Gertsch J, Viveros-Paredes JMJM, Escareño N, Flores-Soto MEME, Gertsch J, Viveros-Paredes JMJM (2017) An endocannabinoid uptake inhibitor from black pepper exerts pronounced anti-inflammatory effects in mice. J Agric Food Chem 65:9435–9442. [DOI] [PubMed] [Google Scholar]
- Reynoso-Moreno I, Rau M, Chicca A, Nicolussi S, Gertsch J (2023) Assay of endocannabinoid uptake. Methods Mol Biol 2576:329–348. [DOI] [PubMed] [Google Scholar]
- Reynoso-Moreno I, Tietz S, Vallini E, Engelhardt B, Gertsch J, Chicca A (2021) Selective endocannabinoid reuptake inhibitor WOBE437 reduces disease progression in a mouse model of multiple sclerosis. ACS Pharmacol Transl Sci 4:765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro A, Pontis S, Mengatto L, Armirotti A, Chiurchiù V, Capurro V, Fiasella A, Nuzzi A, Romeo E, Moreno-Sanz G, et al. (2015) A potent systemically active N-acylethanolamine acid amidase inhibitor that suppresses inflammation and human macrophage activation. ACS Chem Biol 10:1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riether D (2012) Selective cannabinoid receptor 2 modulators: a patent review 2009--present. Expert Opin Ther Pat 22:495–510. [DOI] [PubMed] [Google Scholar]
- Riether D, Zindell R, Wu L, Betageri R, Jenkins JE, Khor S, Berry AK, Hickey ER, Ermann M, Albrecht C, et al. (2015) Selective CB2 receptor agonists. Part 2: structure-activity relationship studies and optimization of proline-based compounds. Bioorg Med Chem Lett 25:581–586. [DOI] [PubMed] [Google Scholar]
- Rinaldi M, Barth F, Casellas P, Congy C, Oustric D, Bell MR, D’Ambra TE, Philion RE (1996) Utilisation de composes agonistes du recepteur cb2 humain pour la preparation de medicaments immunomodulateurs, nouveaux composes agonistes du recepteur cb2 et les compositions pharmaceutiques les contenant. FR Pat, FR2735774A1. [Google Scholar]
- Rinaldi-Carmona M, Barth F, Héaulme M, Alonso R, Shire D, Congy C, Soubrié P, Brelière JC, Le Fur G (1995) Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci 56:1941–1947. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Millan J, Derocq JM, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, et al. (1998) SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther 284:644–650. [PubMed] [Google Scholar]
- Ringel-Kulka T, Goldsmith JR, Carroll IM, Barros SP, Palsson O, Jobin C, Ringel Y (2014) Lactobacillus acidophilus NCFM affects colonic mucosal opioid receptor expression in patients with functional abdominal pain - a randomised clinical study. Aliment Pharmacol Ther 40:200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson P (2001) Therapeutic aspects of cannabis and cannabinoids. Br J Psychiatry 178:107–115. [DOI] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Pertwee RG, Mechoulam R, Parker LA (2021) Therapeutic potential of cannabidiol, cannabidiolic acid, and cannabidiolic acid methyl ester as treatments for nausea and vomiting. Cannabis Cannabinoid Res 6:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez de Fonseca F, Navarro M, Gómez R, Escuredo L, Nava F, Fu J, Murillo-Rodríguez E, Giuffrida A, LoVerme J, Gaetani S, et al. (2001) An anorexic lipid mediator regulated by feeding. Nature 414:209–212. [DOI] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM (2004) Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci 24:1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA (2003) Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol 140:790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F, Bellini G, Nobili B, Maione S, Perrone L, del Giudice EM (2011) Association of the cannabinoid receptor 2 (CB2) Gln63Arg polymorphism with indices of liver damage in obese children: an alternative way to highlight the CB2 hepatoprotective properties. Hepatology 54:1102, author reply 1102–1103. [DOI] [PubMed] [Google Scholar]
- Rossi G, Gioacchini G, Pengo G, Suchodolski JS, Jergens AE, Allenspach K, Gavazza A, Scarpona S, Berardi S, Galosi L, et al. (2020) Enterocolic increase of cannabinoid receptor type 1 and type 2 and clinical improvement after probiotic administration in dogs with chronic signs of colonic dysmotility without mucosal inflammatory changes. Neurogastroenterol Motil 32:e13717. [DOI] [PubMed] [Google Scholar]
- Rousseaux C, Thuru X, Gelot A, Barnich N, Neut C, Dubuquoy L, Dubuquoy C, Merour E, Geboes K, Chamaillard M, et al. (2007) Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat Med 13:35–37. [DOI] [PubMed] [Google Scholar]
- Rouzer CA, Marnett LJ (2011) Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem Rev 111:5899–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin A, Lemberger L, Warrick P, Crabtree RE, Sullivan H, Rowe H, Obermeyer BD (1977) Physiologic disposition of nabilone, a cannabinol derivative, in man. Clin Pharmacol Ther 22:85–91. [DOI] [PubMed] [Google Scholar]
- Russo EB (2018) Cannabis therapeutics and the future of neurology. Front Integr Nuerosci 12:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo EB, Jiang HE, Li X, Sutton A, Carboni A, del Bianco F, Mandolino G, Potter DJ, Zhao YX, Bera S, et al. (2008) Phytochemical and genetic analyses of ancient cannabis from Central Asia. J Exp Bot 59:4171–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghatelian A, McKinney MK, Bandell M, Patapoutian A, Cravatt BF (2006) A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry 45:9007–9015. [DOI] [PubMed] [Google Scholar]
- Saleh N, Hucke O, Kramer G, Schmidt E, Montel F, Lipinski R, Ferger B, Clark T, Hildebrand PW, Tautermann CS (2018) Multiple binding sites contribute to the mechanism of mixed agonistic and positive allosteric modulators of the cannabinoid CB1 receptor. Angew Chem Int Ed Engl 57:2580–2585. [DOI] [PubMed] [Google Scholar]
- Sanson B, Wang T, Sun J, Wang L, Kaczocha M, Ojima I, Deutsch D, Li H (2014) Crystallographic study of FABP5 as an intracellular endocannabinoid transporter. Acta Crystallogr D Biol Crystallogr 70:290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarott RC, Westphal MV, Pfaff P, Korn C, Sykes DA, Gazzi T, Brennecke B, Atz K, Weise M, Mostinski Y, et al. (2020) Development of high-specificity fluorescent probes to enable cannabinoid type 2 receptor studies in living cells. J Am Chem Soc 142:16953–16964. [DOI] [PubMed] [Google Scholar]
- Sasso O, Moreno-Sanz G, Martucci C, Realini N, Dionisi M, Mengatto L, Duranti A, Tarozzo G, Tarzia G, Mor M, et al. (2013) Antinociceptive effects of the N-acylethanolamine acid amidase inhibitor ARN077 in rodent pain models. Pain 154:350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasso O, Pontis S, Armirotti A, Cardinali G, Kovacs D, Migliore M, Summa M, Moreno-Sanz G, Picardo M, Piomelli D (2016) Endogenous N-acyl taurines regulate skin wound healing. Proc Natl Acad Sci USA 113:E4397–E4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasso O, Summa M, Armirotti A, Pontis S, De Mei C, Piomelli D (2018) The N-acylethanolamine acid amidase inhibitor ARN077 suppresses inflammation and pruritus in a mouse model of allergic dermatitis. J Invest Dermatol 138:562–569. [DOI] [PubMed] [Google Scholar]
- Saumell-Esnaola M, Barrondo S, García Del Caño G, Goicolea MA, Sallés J, Lutz B, Monory K (2021) Subsynaptic distribution, lipid raft targeting and G protein-dependent signalling of the type 1 cannabinoid receptor in synaptosomes from the mouse hippocampus and frontal cortex. Molecules 26:6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafroth MA, Carreira EM (2017) Synthesis of phytocannabinoids. Prog Chem Org Nat Prod 103:37–59. [DOI] [PubMed] [Google Scholar]
- Schiano Moriello A, López Chinarro S, Novo Fernández O, Eras J, Amodeo P, Canela-Garayoa R, Vitale RM, Di Marzo V, De Petrocellis L (2018) Elongation of the hydrophobic chain as a molecular switch: discovery of capsaicin derivatives and endogenous lipids as potent transient receptor potential vanilloid channel 2 antagonists. J Med Chem 61:8255–8281. [DOI] [PubMed] [Google Scholar]
- Schindler CW, Redhi GH, Vemuri K, Makriyannis A, Le Foll B, Bergman J, Goldberg SR, Justinova Z (2016a) Blockade of nicotine and cannabinoid reinforcement and relapse by a cannabinoid CB1-receptor neutral antagonist AM4113 and inverse agonist rimonabant in squirrel monkeys. Neuropsychopharmacology 41:2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler CW, Scherma M, Redhi GH, Vadivel SK, Makriyannis A, Goldberg SR, Justinova Z (2016b) Self-administration of the anandamide transport inhibitor AM404 by squirrel monkeys. Psychopharmacology (Berl) 233:1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, et al. (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13:1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ME, Liebowitz MR, Stein MB, Grunfeld J, Van Hove I, Simmons WK, Van Der Ark P, Palmer JA, Saad ZS, Pemberton DJ, et al. (2021) The effects of inhibition of fatty acid amide hydrolase (FAAH) by JNJ-42165279 in social anxiety disorder: a double-blind, randomized, placebo-controlled proof-of-concept study. Neuropsychopharmacology 46:1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurman LD, Carper MC, Moncayo LV, Ogasawara D, Richardson K, Yu L, Liu X, Poklis JL, Liu QS, Cravatt BF, et al. (2019) Diacylglycerol lipase-alpha regulates hippocampal-dependent learning and memory processes in mice. J Neurosci 39:5949–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurman LD, Lu D, Kendall DA, Howlett AC, Lichtman AH (2020) Molecular mechanism and cannabinoid pharmacology. Handb Exp Pharmacol 258:323–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GJ, Fu J, Astarita G, Li X, Gaetani S, Campolongo P, Cuomo V, Piomelli D (2008) The lipid messenger OEA links dietary fat intake to satiety. Cell Metab 8:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seillier A, Giuffrida A (2018) The cannabinoid transporter inhibitor OMDM-2 reduces social interaction: further evidence for transporter-mediated endocannabinoid release. Neuropharmacology 130:1–9. [DOI] [PubMed] [Google Scholar]
- Sellitto I, Le Bourdonnec B, Worm K, Goodman A, Savolainen MA, Chu GH, Ajello CW, Saeui CT, Leister LK, Cassel JA, et al. (2010) Novel sulfamoyl benzamides as selective CB(2) agonists with improved in vitro metabolic stability. Bioorg Med Chem Lett 20:387–391. [DOI] [PubMed] [Google Scholar]
- Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, Rosenbaum DM (2016) High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 540:602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey KA, Wiley JW (2016) The role of the endocannabinoid system in the brain-gut axis. Gastroenterology 151:252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Shen M, Zhao X, Zhu H, Yang Y, Lu S, Tan Y, Li G, Li M, Wang J, et al. (2017) Anti-obesity effect of capsaicin in mice fed with high-fat diet is associated with an increase in population of the gut bacterium Akkermansia muciniphila. Front Microbiol 8:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Snyder HW, Donvito G, Schurman LD, Fox TE, Lichtman AH, Kester M, Hsu KL (2018) Liposomal delivery of diacylglycerol lipase-beta inhibitors to macrophages dramatically enhances selectivity and efficacy in vivo. Mol Pharm 15:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Ware TB, Hsu KL (2020) DAGL-beta functions as a PUFA-specific triacylglycerol lipase in macrophages. Cell Chem Biol 27:314–321.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Bluett RJ, Ramikie TS, Báldi R, Hermanson DJ, Kingsley PJ, Marnett LJ, Winder DG, Colbran RJ, Patel S (2014) Genetic disruption of 2-arachidonoylglycerol synthesis reveals a key role for endocannabinoid signaling in anxiety modulation. Cell Rep 9:1644–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Stephenson JR, Marks CR, Colbran RJ (2020) Cyclic AMP-dependent protein kinase and D1 dopamine receptors regulate diacylglycerol lipase-α and synaptic 2-arachidonoyl glycerol signaling. J Neurochem 153:334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Wang X, Rose KL, Ramikie TS, Cavener VS, Rentz T, Baucum AJ 2nd, Jalan-Sakrikar N, Mackie K, Winder DG, et al. (2013) CaMKII regulates diacylglycerol lipase-α and striatal endocannabinoid signaling. Nat Neurosci 16:456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore DM, Reggio PH (2015) The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front Pharmacol 6:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showalter VM, Compton DR, Martin BR, Abood ME (1996) Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther 278:989–999. [PubMed] [Google Scholar]
- Sigel E, Baur R, Rácz I, Marazzi J, Smart TG, Zimmer A, Gertsch J (2011) The major central endocannabinoid directly acts at GABA(A) receptors. Proc Natl Acad Sci USA 108:18150–18155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihag J, Di Marzo V (2022) (Wh)olistic (e)ndocannabinoidome-microbiome-axis modulation through (n)utrition (WHEN) to curb obesity and related disorders. Lipids Health Dis 21:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Archambault A-S, Lavoie JC, Dumais É, Di Marzo V, Flamand N (2022) Biosynthesis and metabolism of endocannabinoids and their congeners from the monoacylglycerol and N-acyl-ethanolamine families. Biochem Pharmacol 205:115261. [DOI] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF (2006) Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J Biol Chem 281:26465–26472. [DOI] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF (2008) Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J Biol Chem 283:9341–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Oyagawa CRM, Macdonald C, Grimsey NL, Glass M, Vernall AJ (2019) Chromenopyrazole-based high affinity, selective fluorescent ligands for cannabinoid type 2 receptor. ACS Med Chem Lett 10:209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sink KS, McLaughlin PJ, Wood JAT, Brown C, Fan P, Vemuri VK, Peng Y, Olszewska T, Thakur GA, Makriyannis A, et al. (2008) The novel cannabinoid CB1 receptor neutral antagonist AM4113 suppresses food intake and food-reinforced behavior but does not induce signs of nausea in rats. Neuropsychopharmacology 33:946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sink KS, Segovia KN, Sink J, Randall PA, Collins LE, Correa M, Markus EJ, Vemuri VK, Makriyannis A, Salamone JD (2010) Potential anxiogenic effects of cannabinoid CB1 receptor antagonists/inverse agonists in rats: comparisons between AM4113, AM251, and the benzodiazepine inverse agonist FG-7142. Eur Neuropsychopharmacol 20:112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sionov RV, Steinberg D (2022) Anti-microbial activity of phytocannabinoids and endocannabinoids in the light of their physiological and pathophysiological roles. Biomedicines 10:631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe JC, Chiang K, Gerber AL, Beutler E, Cravatt BF (2002) A missense mutation in human fatty acid amide hydrolase associated with problem drug use. Proc Natl Acad Sci USA 99:8394–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavik R, Grether U, Müller Herde A, Gobbi L, Fingerle J, Ullmer C, Krämer SD, Schibli R, Mu L, Ametamey SM (2015) Discovery of a high affinity and selective pyridine analog as a potential positron emission tomography imaging agent for cannabinoid type 2 receptor. J Med Chem 58:4266–4277. [DOI] [PubMed] [Google Scholar]
- Slivicki RA, Iyer V, Mali SS, Garai S, Thakur GA, Crystal JD, Hohmann AG (2020) Positive allosteric modulation of CB1 cannabinoid receptor signaling enhances morphine antinociception and attenuates morphine tolerance without enhancing morphine-induced dependence or reward. Front Mol Neurosci 13:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivicki RA, Xu Z, Kulkarni PM, Pertwee RG, Mackie K, Thakur GA, Hohmann AG (2018) Positive allosteric modulation of cannabinoid receptor type 1 suppresses pathological pain without producing tolerance or dependence. Biol Psychiatry 84:722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PB, Compton DR, Welch SP, Razdan RK, Mechoulam R, Martin BR (1994) The pharmacological activity of anandamide, a putative endogenous cannabinoid, in mice. J Pharmacol Exp Ther 270:219–227. [PubMed] [Google Scholar]
- Snider NT, Nast JA, Tesmer LA, Hollenberg PF (2009) A cytochrome P450-derived epoxygenated metabolite of anandamide is a potent cannabinoid receptor 2-selective agonist. Mol Pharmacol 75:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soethoudt M, Grether U, Fingerle J, Grim TW, Fezza F, de Petrocellis L, Ullmer C, Rothenhäusler B, Perret C, van Gils N, et al. (2017) Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity. Nat Commun 8:13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soethoudt M, Stolze SC, Westphal MV, van Stralen L, Martella A, van Rooden EJ, Guba W, Varga ZV, Deng H, van Kasteren SI, et al. (2018a) Selective photoaffinity probe that enables assessment of cannabinoid CB2 receptor expression and ligand engagement in human cells. J Am Chem Soc 140:6067–6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soethoudt M, Hoorens MWH, Doelman W, Martella A, van der Stelt M, Heitman LH (2018b) Structure-kinetic relationship studies of cannabinoid CB2 receptor agonists reveal substituent-specific lipophilic effects on residence time. Biochem Pharmacol 152:129–142. [DOI] [PubMed] [Google Scholar]
- Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, Mor M, Russo R, Maccarrone M, Antonietti F, et al. (2009) Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci USA 106:20966–20971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solymosi K, Köfalvi A (2017) Cannabis: a treasure trove or Pandora’s box? Mini Rev Med Chem 17:1223–1291. [DOI] [PubMed] [Google Scholar]
- Somvanshi RK, Zou S, Kadhim S, Padania S, Hsu E, Kumar U (2022) Cannabinol modulates neuroprotection and intraocular pressure: a potential multi-target therapeutic intervention for glaucoma. Biochim Biophys Acta Mol Basis Dis 1868:166325. [DOI] [PubMed] [Google Scholar]
- Stasiulewicz A, Lesniak A, Setny P, Bujalska-Zadrożny M, Sulkowska JI (2022) Identification of CB1 ligands among drugs, phytochemicals and natural-like compounds: virtual screening and in vitro verification. ACS Chem Neurosci 13:2991–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N (2023) THC and CBD: similarities and differences between siblings. Neuron 111:302–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticht MA, Rock EM, Limebeer CL, Parker LA (2015) Endocannabinoid mechanisms influencing nausea. Int Rev Neurobiol 125:127–162. [DOI] [PubMed] [Google Scholar]
- Storr MA, Bashashati M, Hirota C, Vemuri VK, Keenan CM, Duncan M, Lutz B, Mackie K, Makriyannis A, Macnaughton WK, et al. (2010) Differential effects of CB(1) neutral antagonists and inverse agonists on gastrointestinal motility in mice. Neurogastroenterol Motil 22:787–796, e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Wilson S, Corey W, Dvorakova M, Bosquez T, Tracey J, Wilkowski C, Ho K, Wager-Miller J, Mackie K (2021) An evaluation of understudied phytocannabinoids and their effects in two neuronal models. Molecules 26:5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K (1995) 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 215:89–97. [DOI] [PubMed] [Google Scholar]
- Sulsky R, Magnin DR, Huang Y, Simpkins L, Taunk P, Patel M, Zhu Y, Stouch TR, Bassolino-Klimas D, Parker R, et al. (2007) Potent and selective biphenyl azole inhibitors of adipocyte fatty acid binding protein (aFABP). Bioorg Med Chem Lett 17:3511–3515. [DOI] [PubMed] [Google Scholar]
- Sun Y, Alexander SP, Kendall DA, Bennett AJ(2008) Involvement of fatty acid binding proteins in the transport of endocannabinoids to peroxisome proliferator activated receptors, in 18th ICRS Annual Symposium (International Cannabinoid Research Society ed) p 17, Burlington. [Google Scholar]
- Suzuki N, Suzuki M, Murakami K, Hamajo K, Tsukamoto T, Shimojo M (2012) Cerebroprotective effects of TAK-937, a cannabinoid receptor agonist, on ischemic brain damage in middle cerebral artery occluded rats and non-human primates. Brain Res 1430:93–100. [DOI] [PubMed] [Google Scholar]
- Szymanski D, Papanastasiou M, Pandarinathan L, Zvonok N, Janero DR, Pavlopoulos S, Vouros P, Makriyannis A (2018) Aliphatic azides as selective cysteine labeling reagents for integral membrane proteins. J Med Chem 61:11199–11208. [DOI] [PubMed] [Google Scholar]
- Szymanski DW, Papanastasiou M, Melchior K, Zvonok N, Mercier RW, Janero DR, Thakur GA, Cha S, Wu B, Karger B, et al. (2011) Mass spectrometry-based proteomics of human cannabinoid receptor 2: covalent cysteine 6.47(257)-ligand interaction affording megagonist receptor activation. J Proteome Res 10:4789–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, Szanda G, Mukhopadhyay B, Chedester L, Liow JS, et al. (2012) Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab 16:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J, Vemuri VK, Liu J, Bátkai S, Mukhopadhyay B, Godlewski G, Osei-Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, et al. (2010) Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest 120:2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, et al. (2010) The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 65:320–327. [DOI] [PubMed] [Google Scholar]
- Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D (2003) Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J Med Chem 46:2352–2360. [DOI] [PubMed] [Google Scholar]
- Tepper MA, Zurier RB, Burstein SH (2014) Ultrapure ajulemic acid has improved CB2 selectivity with reduced CB1 activity. Bioorg Med Chem 22:3245–3251. [DOI] [PubMed] [Google Scholar]
- Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, et al. (2005) Virtual computational chemistry laboratory--design and description. J Comput Aided Mol Des 19:453–463. [DOI] [PubMed] [Google Scholar]
- Thakur GA, Tichkule R, Bajaj S, Makriyannis A (2009) Latest advances in cannabinoid receptor agonists. Expert Opin Ther Pat 19:1647–1673. [DOI] [PubMed] [Google Scholar]
- Tham M, Yilmaz O, Alaverdashvili M, Kelly MEM, Denovan-Wright EM, Laprairie RB (2019) Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br J Pharmacol 176:1455–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG (2007) Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol 150:613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang CC, Giudice MG (2016) Nabilone for the management of pain. Pharmacotherapy 36:273–286. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Okamoto Y, Ikematsu N, Inoue M, Shimizu Y, Uyama T, Wang J, Deutsch DG, Burns MP, Ulloa NM, et al. (2011) Enzymatic formation of N-acylethanolamines from N-acylethanolamine plasmalogen through N-acylphosphatidylethanolamine-hydrolyzing phospholipase D-dependent and -independent pathways. Biochim Biophys Acta 1811:565–577. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Okamoto Y, Rahman IA, Uyama T, Inoue T, Tokumura A, Ueda N (2015) Glycerophosphodiesterase GDE4 as a novel lysophospholipase D: a possible involvement in bioactive N-acylethanolamine biosynthesis. Biochim Biophys Acta 1851:537–548. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Tai T, Yamashita R, Ali H, Watanabe T, Uyama T, Okamoto Y, Kitakaze K, Takenouchi Y, Go S, et al. (2021) Involvement of acid ceramidase in the degradation of bioactive N-acylethanolamines. Biochim Biophys Acta Mol Cell Biol Lipids 1866:158972. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Takezaki N, Ueda N (2007a) The N-acylethanolamine-hydrolyzing acid amidase (NAAA). Chem Biodivers 4:1914–1925. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Uyama T, Okamoto Y, Ueda N (2018) Endocannabinoids and related N-acylethanolamines: biological activities and metabolism. Inflamm Regen 38:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K, Zhao LY, Okamoto Y, Araki N, Ueno M, Sakamoto H, Ueda N (2007b) Predominant expression of lysosomal N-acylethanolamine-hydrolyzing acid amidase in macrophages revealed by immunochemical studies. Biochim Biophys Acta 1771:623–632. [DOI] [PubMed] [Google Scholar]
- Tuo W, Leleu-Chavain N, Spencer J, Sansook S, Millet R, Chavatte P (2017) Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N-acylethanolamine acid amidase inhibitors. J Med Chem 60:4–46. [DOI] [PubMed] [Google Scholar]
- Turner SE, Williams CM, Iversen L, Whalley BJ (2017) Molecular pharmacology of phytocannabinoids. Prog Chem Org Nat Prod 103:61–101. [DOI] [PubMed] [Google Scholar]
- Tutunchi H, Saghafi-Asl M, Ostadrahimi A (2020) A systematic review of the effects of oleoylethanolamide, a high-affinity endogenous ligand of PPAR-α, on the management and prevention of obesity. Clin Exp Pharmacol Physiol 47:543–552. [DOI] [PubMed] [Google Scholar]
- Ueda N, Tsuboi K, Uyama T (2010) N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog Lipid Res 49:299–315. [DOI] [PubMed] [Google Scholar]
- Uyama T, Ikematsu N, Inoue M, Shinohara N, Jin XH, Tsuboi K, Tonai T, Tokumura A, Ueda N (2012) Generation of N-acylphosphatidylethanolamine by members of the phospholipase A/acyltransferase (PLA/AT) family. J Biol Chem 287:31905–31919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyama T, Tsuboi K, Ueda N (2017) An involvement of phospholipase A/acyltransferase family proteins in peroxisome regulation and plasmalogen metabolism. FEBS Lett 591:2745–2760. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, van Kuik JA, Bari M, van Zadelhoff G, Leeflang BR, Veldink GA, Finazzi-Agrò A, Vliegenthart JFG, Maccarrone M (2002) Oxygenated metabolites of anandamide and 2-arachidonoylglycerol: conformational analysis and interaction with cannabinoid receptors, membrane transporter, and fatty acid amide hydrolase. J Med Chem 45:3709–3720. [DOI] [PubMed] [Google Scholar]
- van Egmond N, Straub VM, van der Stelt M (2021) Targeting endocannabinoid signaling: FAAH and MAG lipase inhibitors. Annu Rev Pharmacol Toxicol 61:441–463. [DOI] [PubMed] [Google Scholar]
- Vandevoorde S, Fowler CJ (2005) Inhibition of fatty acid amide hydrolase and monoacylglycerol lipase by the anandamide uptake inhibitor VDM11: evidence that VDM11 acts as an FAAH substrate. Br J Pharmacol 145:885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga K, Lake K, Martin BR, Kunos G (1995) Novel antagonist implicates the CB1 cannabinoid receptor in the hypotensive action of anandamide. Eur J Pharmacol 278:279–283. [DOI] [PubMed] [Google Scholar]
- Veldhuis WB, van der Stelt M, Wadman MW, van Zadelhoff G, Maccarrone M, Fezza F, Veldink GA, Vliegenthart JFG, Bär PR, Nicolay K, et al. (2003) Neuroprotection by the endogenous cannabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: role of vanilloid receptors and lipoxygenases. J Neurosci 23:4127–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeckx KC, Voortman T, Balvers MG, Hendriks HFM, M Wortelboer H, Witkamp RF (2011) Presence, formation and putative biological activities of N-acyl serotonins, a novel class of fatty-acid derived mediators, in the intestinal tract. Biochim Biophys Acta 1811:578–586. [DOI] [PubMed] [Google Scholar]
- Viader A, Ogasawara D, Joslyn CM, Sanchez-Alavez M, Mori S, Nguyen W, Conti B, Cravatt BF (2016) A chemical proteomic atlas of brain serine hydrolases identifies cell type-specific pathways regulating neuroinflammation. eLife 5:e12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel Z, Barg J, Levy R, Saya D, Heldman E, Mechoulam R (1993) Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J Neurochem 61:352–355. [DOI] [PubMed] [Google Scholar]
- Wagenlehner FME, van Till JWO, Houbiers JGA, Martina RV, Cerneus DP, Melis JHJM, Majek A, Vjaters E, Urban M, Ramonas H, et al. (2017) Fatty acid amide hydrolase inhibitor treatment in men with chronic prostatitis/chronic pelvic pain syndrome: an adaptive double-blind, randomized controlled trial. Urology 103:191–197. [DOI] [PubMed] [Google Scholar]
- Wang B, Zhou Z, Li L (2022) Gut microbiota regulation of AHR signaling in liver disease. Biomolecules 12:1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Okamoto Y, Morishita J, Tsuboi K, Miyatake A, Ueda N (2006) Functional analysis of the purified anandamide-generating phospholipase D as a member of the metallo-beta-lactamase family. J Biol Chem 281:12325–12335. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu J, Harvey-White J, Zimmer A, Kunos G (2003) Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc Natl Acad Sci USA 100:1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangensteen T, Akselsen H, Holmen J, Undlien D, Retterstøl L (2011) A common haplotype in NAPEPLD is associated with severe obesity in a Norwegian population-based cohort (the HUNT study). Obesity (Silver Spring) 19:612–617. [DOI] [PubMed] [Google Scholar]
- Ward A, Holmes B (1985) Nabilone. a preliminary review of its pharmacological properties and therapeutic use. Drugs 30:127–144. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B (2003) Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424:434–438. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Kayano Y, Matsunaga T, Yamamoto I, Yoshimura H (1996) Inhibition of anandamide amidase activity in mouse brain microsomes by cannabinoids. Biol Pharm Bull 19:1109–1111. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yamaori S, Funahashi T, Kimura T, Yamamoto I (2007) Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci 80:1415–1419. [DOI] [PubMed] [Google Scholar]
- Watson JE, Kim JS, Das A (2019) Emerging class of omega-3 fatty acid endocannabinoids & their derivatives. Prostaglandins Other Lipid Mediat 143:106337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF (2006) A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem 281:36569–36578. [DOI] [PubMed] [Google Scholar]
- Weichert D, Gmeiner P (2015) Covalent molecular probes for class A G protein-coupled receptors: advances and applications. ACS Chem Biol 10:1376–1386. [DOI] [PubMed] [Google Scholar]
- Westphal MV, Sarott RC, Zirwes EA, Osterwald A, Guba W, Ullmer C, Grether U, Carreira EM (2020) Highly selective, amine-derived cannabinoid receptor 2 probes. Chemistry 26:1380–1387. [DOI] [PubMed] [Google Scholar]
- Wilkerson JL, Bilbrey JA, Felix JS, Makriyannis A, McMahon LR (2021) Untapped endocannabinoid pharmacological targets: pipe dream or pipeline? Pharmacol Biochem Behav 206:173192. [DOI] [PubMed] [Google Scholar]
- Wilkerson JL, Donvito G, Grim TW, Abdullah RA, Ogasawara D, Cravatt BF, Lichtman AH (2017) Investigation of diacylglycerol lipase alpha inhibition in the mouse lipopolysaccharide inflammatory pain model. J Pharmacol Exp Ther 363:394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JL, Ghosh S, Bagdas D, Mason BL, Crowe MS, Hsu KL, Wise LE, Kinsey SG, Damaj MI, Cravatt BF, et al. (2016) Diacylglycerol lipase β inhibition reverses nociceptive behaviour in mouse models of inflammatory and neuropathic pain. Br J Pharmacol 173:1678–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold EA, Chen J, Cunningham KA, Zhou J (2019) Allosteric modulation of class A GPCRs: targets, agents, and emerging concepts. J Med Chem 62:88–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhams SG, Chapman V, Finn DP, Hohmann AG, Neugebauer V (2017) The cannabinoid system and pain. Neuropharmacology 124:105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward DF, Wang JW, Poloso NJ (2013) Recent progress in prostaglandin F2α ethanolamide (prostamide F2α) research and therapeutics. Pharmacol Rev 65:1135–1147. [DOI] [PubMed] [Google Scholar]
- Wortley MA, Adcock JJ, Dubuis ED, Maher SA, Bonvini SJ, Delescluse I, Kinloch R, McMurray G, Perros-Huguet C, Papakosta M, et al. (2017) Targeting fatty acid amide hydrolase as a therapeutic strategy for antitussive therapy. Eur Respir J 50:1700782. [DOI] [PubMed] [Google Scholar]
- Wu S, Liao D, Li X, Liu Z, Zhang L, Mo FM, Hu S, Xia J, Yang X (2021) Endogenous oleoylethanolamide crystals-loaded lipid nanoparticles with enhanced hydrophobic drug loading capacity for efficient stroke therapy. Int J Nanomedicine 16:8103–8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Li Y, Xu S, Zhou P, Yang L, Xu Y, Qiu Y, Yang Y, Li Y (2022) Genetic blockade of NAAA cell-specifically regulates fatty acid ethanolamides (FAEs) metabolism and inflammatory responses. Front Pharmacol 12:817603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing C, Zhuang Y, Xu TH, Feng Z, Zhou XE, Chen M, Wang L, Meng X, Xue Y, Wang J, et al. (2020) Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell 180:645–654.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu Y, Wang K, Chen J, Zhuo Z, Xiu Y (2020) Liposomal N-acylethanolamine-hydrolyzing acid amidase (NAAA) inhibitor F96 as a new therapy for colitis. RSC Advances 10:34197–34202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaka N (2007) Mammalian glycerophosphodiester phosphodiesterases. Biosci Biotechnol Biochem 71:1811–1818. [DOI] [PubMed] [Google Scholar]
- Yang L, Li L, Chen L, Li Y, Chen H, Li Y, Ji G, Lin D, Liu Z, Qiu Y (2015) Potential analgesic effects of a novel N-acylethanolamine acid amidase inhibitor F96 through PPAR-α. Sci Rep 5:13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, van Veldhoven JPD, Offringa J, Kuiper BJ, Lenselink EB, Heitman LH, van der Es D, IJzerman AP (2019) Development of covalent ligands for G protein-coupled receptors: a case for the human adenosine A3 receptor. J Med Chem 62:3539–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wang X, Xu Z, Wu C, Zhou Y, Wang Y, Lin G, Li K, Wu M, Xia A, et al. (2022) Molecular mechanism of allosteric modulation for the cannabinoid receptor CB1. Nat Chem Biol 18:831–840. [DOI] [PubMed] [Google Scholar]
- Yao BB, Hsieh G, Daza AV, Fan Y, Grayson GK, Garrison TR, El Kouhen O, Hooker BA, Pai M, Wensink EJ, et al. (2009) Characterization of a cannabinoid CB2 receptor-selective agonist, A-836339 [2,2,3,3-tetramethyl-cyclopropanecarboxylic acid [3-(2-methoxy-ethyl)-4,5-dimethyl-3H-thiazol-(2Z)-ylidene]-amide], using in vitro pharmacological assays, in vivo pain models, and pharmacological magnetic resonance imaging. J Pharmacol Exp Ther 328:141–151. [DOI] [PubMed] [Google Scholar]
- Yao BB, Hsieh GC, Frost JM, Fan Y, Garrison TR, Daza AV, Grayson GK, Zhu CZ, Pai M, Chandran P, et al. (2008) In vitro and in vivo characterization of A-796260: a selective cannabinoid CB2 receptor agonist exhibiting analgesic activity in rodent pain models. Br J Pharmacol 153:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates ML, Barker EL (2009) Organized trafficking of anandamide and related lipids. Vitam Horm 81:25–53. [DOI] [PubMed] [Google Scholar]
- Yeliseev A, Iyer MR, Joseph TT, Coffey NJ, Cinar R, Zoubak L, Kunos G, Gawrisch K (2021) Cholesterol as a modulator of cannabinoid receptor CB2 signaling. Sci Rep 11:3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Myczek K, Penzes P (2021) cAMP signaling-mediated phosphorylation of diacylglycerol lipase α regulates interaction with ankyrin-G and dendritic spine morphology. Biol Psychiatry 90:263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Levi L, Casadesus G, Kunos G, Noy N (2014) Fatty acid-binding protein 5 (FABP5) regulates cognitive function both by decreasing anandamide levels and by activating the nuclear receptor peroxisome proliferator-activated receptor β/δ (PPARβ/δ) in the brain. J Biol Chem 289:12748–12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan CY, Zhou V, Sauber G, Stollenwerk T, Komorowski R, López A, Tolón RM, Romero J, Hillard CJ, Drobyski WR (2021) Signaling through the type 2 cannabinoid receptor regulates the severity of acute and chronic graft-versus-host disease. Blood 137:1241–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Jiang C, Wang J, Chen CJ, Hao Y, Zhao G, Feng Z, Xie XQ (2022) In silico prediction and validation of CB2 allosteric binding sites to aid the design of allosteric modulators. Molecules 27:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Shen H, Jordan CJ, Liu QR, Gardner EL, Bonci A, Xi ZX (2019) CB2 receptor antibody signal specificity: correlations with the use of partial CB2-knockout mice and anti-rat CB2 receptor antibodies. Acta Pharmacol Sin 40:398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Shao P, Ling X, Yang L, Hou W, Thorne SH, Beaino W, Anderson CJ, Ding Y, Bai M (2015b) In vivo inflammation imaging using a CB2R-targeted near infrared fluorescent probe. Am J Nucl Med Mol Imaging 5:246–258. [PMC free article] [PubMed] [Google Scholar]
- Zhang YM, Greco MN, Macielag MJ, Teleha CA, DesJarlais RL, Tang Y, Ho G, Hou C, Chen C, Zhao S, et al. (2018) 6-Benzhydryl-4-amino-quinolin-2-ones as potent cannabinoid type 1 (CB1) receptor inverse agonists and chemical modifications for peripheral selectivity. J Med Chem 61:10276–10298. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang W, Zhong P, Liu SJ, Long JZ, Zhao L, Gao HQ, Cravatt BF, Liu QS (2015a) Blockade of 2-arachidonoylglycerol hydrolysis produces antidepressant-like effects and enhances adult hippocampal neurogenesis and synaptic plasticity. Hippocampus 25:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LY, Tsuboi K, Okamoto Y, Nagahata S, Ueda N (2007) Proteolytic activation and glycosylation of N-acylethanolamine-hydrolyzing acid amidase, a lysosomal enzyme involved in the endocannabinoid metabolism. Biochim Biophys Acta 1771:1397–1405. [DOI] [PubMed] [Google Scholar]
- Zhao P, Abood ME (2013) GPR55 and GPR35 and their relationship to cannabinoid and lysophospholipid receptors. Life Sci 92:453–457. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Elmes MW, Sweeney JM, Joseph OM, Che J, Hsu HC, Li H, Deutsch DG, Ojima I, Kaczocha M, et al. (2019a) Identification of fatty acid binding protein 5 inhibitors through similarity-based screening. Biochemistry 58:4304–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Xiang L, Yang Y, Wu Y, Hu T, Liu X, Lin F, Xiu Y, Wu K, Lu C, et al. (2019b) N-acylethanolamine acid amidase (NAAA) inhibitor F215 as a novel therapeutic agent for osteoarthritis. Pharmacol Res 145:104264. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Howell FV, Glebov OO, Albrecht D, Williams G, Doherty P (2016) Regulated endosomal trafficking of Diacylglycerol lipase alpha (DAGLα) generates distinct cellular pools; implications for endocannabinoid signaling. Mol Cell Neurosci 76:76–86. [DOI] [PubMed] [Google Scholar]
- Zhu YF, Linher-Melville K, Niazmand MJ, Sharma M, Shahid A, Zhu KL, Parzei N, Sidhu J, Haj C, Mechoulam R, et al. (2020) An evaluation of the anti-hyperalgesic effects of cannabidiolic acid-methyl ester in a preclinical model of peripheral neuropathic pain. Br J Pharmacol 177:2712–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindell R, Walker ER, Scott J, Amouzegh P, Wu L, Ermann M, Thomson D, Fisher MB, Fullenwider CL, Grbic H, et al. (2011) Aryl 1,4-diazepane compounds as potent and selective CB2 agonists: optimization of drug-like properties and target independent parameters. Bioorg Med Chem Lett 21:4276–4280. [DOI] [PubMed] [Google Scholar]
- Ziring D, Wei B, Velazquez P, Schrage M, Buckley NE, Braun J (2006) Formation of B and T cell subsets require the cannabinoid receptor CB2. Immunogenetics 58:714–725. [DOI] [PubMed] [Google Scholar]
- Zoja C, Locatelli M, Corna D, Villa S, Rottoli D, Nava V, Verde R, Piscitelli F, Di Marzo V, Fingerle J, et al. (2016) Therapy with a selective cannabinoid receptor type 2 agonist limits albuminuria and renal injury in mice with type 2 diabetic nephropathy. Nephron 132:59–69. [DOI] [PubMed] [Google Scholar]
- Zurier RB, Rossetti RG, Lane JH, Goldberg JM, Hunter SA, Burstein SH (1998) Dimethylheptyl-THC-11 oic acid: a nonpsychoactive antiinflammatory agent with a cannabinoid template structure. Arthritis Rheum 41:163–170. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V, Julius D, Högestätt ED (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400:452–457. [DOI] [PubMed] [Google Scholar]