

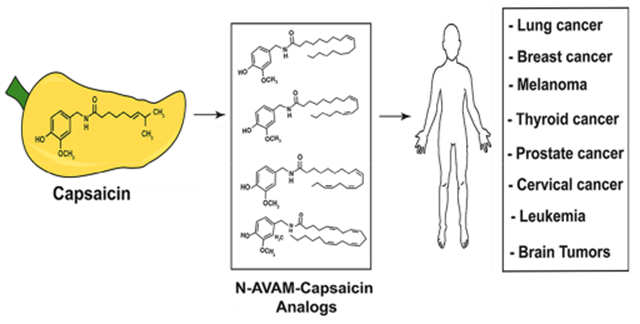
Abstract
Capsaicin displays robust growth-inhibitory activity in multiple human cancers. However, the feasibility of capsaicin as a clinically relevant anticancer drug is hampered by its adverse side effects. This concern has led to extensive research focused on the isolation and synthesis of second-generation nonpungent capsaicin analogues with potent antineoplastic activity. A major class of nonpungent capsaicin-like compounds belongs to the N-acyl-vanillylamide (N-AVAM) derivatives of capsaicin (hereafter referred as N-AVAM capsaicin analogues). This perspective discusses the isolation of N-AVAM capsaicin analogues from natural sources as well as their synthesis by chemical and enzymatic methods. The perspective describes the pharmacokinetic properties and anticancer activity of N-AVAM capsaicin analogues. The signaling pathways underlying the growth-inhibitory effects of N-AVAM capsaicin analogues have also been highlighted. It is hoped that the insights obtained in this perspective will facilitate the synthesis of a second generation of N-AVAM capsaicin analogues with improved stability and growth-suppressive activity in human cancer.
Graphical Abstract
1. INTRODUCTION
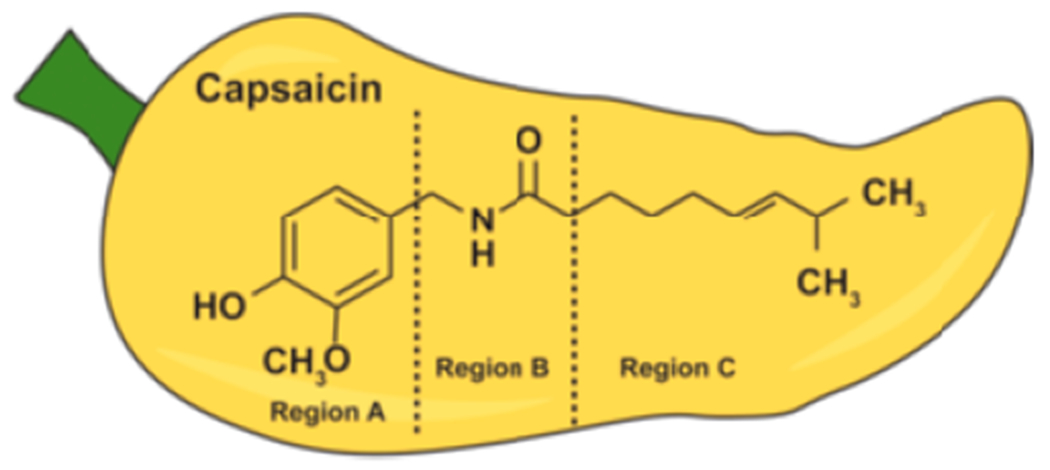
The nutritional compound capsaicin (trans-8-methyl-N-vanillyl-6-noneamide, Figure 1) is a strong pain-relieving agent, used in many over-the-counter creams (and lotions) to treat pain and inflammation associated with a variety of diseases.1,2 The pain-relieving activity of capsaicin is mediated by the transient receptor potential vanilloid (TRPV) receptor superfamily of ion-channel receptors on target cells. The TRPV family of receptors is comprised of six receptor subtypes (TRPV1–6).1 Capsaicin is a potent agonist of the TRPV1 receptor.3 The binding of capsaicin to TRPV1 results in a cascade of cellular signaling events which leads to eventual downregulation of Substance P, a neuropeptide involved in the nociceptive signals from nerve endings to the brain and the release of inflammatory cytokines.4,5 These molecular events lead to “defunctionalization” of nociceptor fibers and ablation of pain-sensation.6 The cloning and molecular characterization of TRPV1 has clarified our concept of the interactions between capsaicin and TRPV1. It has also spurred structure activity-relationship studies (SAR) and the discovery of capsaicin-like compounds possessing greater analgesic activity than capsaicin.
Figure 1.
Structure and pharmacophore of capsaicin.
Several lines of evidence show that capsaicin displays strong antineoplastic activity in several human cancers, both in vitro and in vivo.7–10 A surprising finding has been the fact that the anticancer activity of capsaicin (in the majority of human cancers) does not involve the TRPV1 receptor.11 The growth-suppressive properties of capsaicin and its related compounds are mediated by its ability to block cytoplasmic, mitochondrial, and metabolic survival pathways.7–10 Capsaicin recruits multiple growth-inhibitory signaling pathways including regulation of intracellular calcium, activation of the calpain family of apoptotic proteases, generation of reactive oxygen species (ROS), suppression of coenzyme Q (antioxidant and redox component in the respiratory chain), induction of apoptosis and autophagy, disruption of mitochondrial respiration, and inhibition of transcription factors like p53, STAT3, and NF-κB.7,8 An important antitumor mechanism of capsaicin is its ability to downregulate tumor angiogenesis.12,13 Capsaicin inhibits vital cancer-progression pathways like epithelial-mesenchymal transition (EMT), invasion, and metastasis.14 Finally, published data reveal that capsaicin sensitizes human cancer cells to the growth-suppressive effects of established cancer chemotherapy drugs and radiotherapy.15–18
The clinical applications of capsaicin as a viable anticancer drug are hampered by low aqueous solubility, poor bioavailability, and its unfavorable side effect profile. The administration of capsaicin causes skin redness, hyperalgesia, nausea, intense tearing in the eyes, conjunctivitis, blepharospasm (sustained, forced, involuntary closing of the eyelids), vomiting, abdominal pain, stomach cramps, bronchospasm, and burning diarrhea.6,19,20 Clinical trials investigating the analgesic activity of capsaicin have shown that such disagreeable side effects have led to patients discontinuing use of capsaicin.6,19,20 Another caveat of capsaicin (as an anticancer drug) is that it has been found to promote the growth of certain cancers like skin cancer, stomach cancer, colon cancer, and gastric cancers.21–23 Such findings have led to intense research focused on the identification of nonpungent capsaicin-analogues which possess improved growth-suppressive activity. Studies from several research laboratories have described a plethora of natural and synthetic capsaicin-mimetics which display enhanced pharmacological activity, improved selectivity, and a longer biological half-life than capsaicin.15,24,25 Out of all the nonpungent capsaicin-analogues, compounds belonging to the N-acyl vanillylamide (N-AVAM) family of capsaicin mimetics are some of one of the most extensively studied in terms of their functional activity, specificity (and selectivity), pharmacokinetics, and bioavailability.15 Published data show that N-AVAM capsaicin analogues display greater pain-relieving activity than capsaicin.24,26,27 However, only a handful of studies have reported the antineoplastic activity of this class of compounds. Most importantly, N-AVAM capsaicin analogues do not promote the growth of the cancer cell in vitro and in athymic mouse models. The objective of the present perspective is to describe the synthesis strategies, anticancer activity, pharmacokinetics, and biological half-life of N-AVAM capsaicin analogues. In addition, we will also discuss the signaling mechanisms underlying the anticancer activity of these compounds. Molecular modeling studies and high-throughput virtual screening experiments will pave the way to a second generation of N-AVAM capsaicin analogues with better bioactivity, stability, and therapeutic index.
2. PHARMACOPHORE OF CAPSAICIN
Structure activity relationship (SAR) studies have shown that the chemical structure of capsaicin is comprised of three distinct motifs (Figure 1). Region A encompasses the aromatic moiety, region B includes the amide group, and region C consists of the alkyl hydrophobic side chain.8,9,24
The N-AVAM capsaicin analogues contain long alkyl side chains in region C of capsaicin. Initial studies showed that side chains containing short acyl groups, short acyl groups with polar structures, and short branched acyl groups were inactive or showed weak pain-relieving activity.15,23 The presence of a saturated long chain alkyl group in region C gave rise to compounds with moderate analgesic activity. However, the incorporation of unsaturated long chain fatty acyl side groups yielded nonpungent, nontoxic, orally active capsaicin analogues with extremely high biological activity.15,24 The pharmacological activity of these capsaicin-analogues was measured by the uptake of radiolabeled calcium (45Ca) into dorsal root ganglia neurons in culture. The antinociceptive and antiinflammatory activity of these compounds was measured using mouse hot tail flick and croton-oil inflamed mouse ear models.15,24
Recent studies have examined the anticancer activity of N-AVAM capsaicin analogues. A majority of these publications have investigated the growth-suppressive activity of a specific N-AVAM capsaicin analogue such as 5, 8, or 10. There is currently a lack of high-throughput screening studies to test the growth-inhibitory activity of large numbers of N-AVAM capsaicin analogues. The anticancer activity of capsaicin is correlated to its ability to activate the functional activity of pro-apoptotic calpain proteases in human breast epithelial cells and small cell lung cancer (SCLC) cells. Data from our laboratory reveal that the growth-suppressive activity of N-AVAM capsaicin analogues in human SCLCs correlate with activation of calpain1 and calpain2.28 Based on these findings and observations (described later), it is tempting to speculate that N-AVAM capsaicin analogue-induced activation of calpain proteases may be a useful indicator of their growth-inhibitory activity.28 Thus, measuring the functional activity of calpain-1 and calpain-2 activity by N-AVAM capsaicin analogues may form the basis of a novel high-throughput drug screening strategy to discover improved N-AVAM capsaicin analogues with robust growth-suppressive activity in human cancers.
3. PREPARATION OF N-AVAM CAPSAICIN ANALOGUES
N-AVAM capsaicin analogues have been obtained from both natural sources as well as synthetic routes. Figure 2 shows the structure of N-AVAM capsaicin analogues whose anticancer activity has been investigated in cell culture and mouse models. The present section describes the techniques used for isolation of N-AVAM capsaicin analogues from natural sources and the synthetic routes by which they have been generated.
Figure 2.
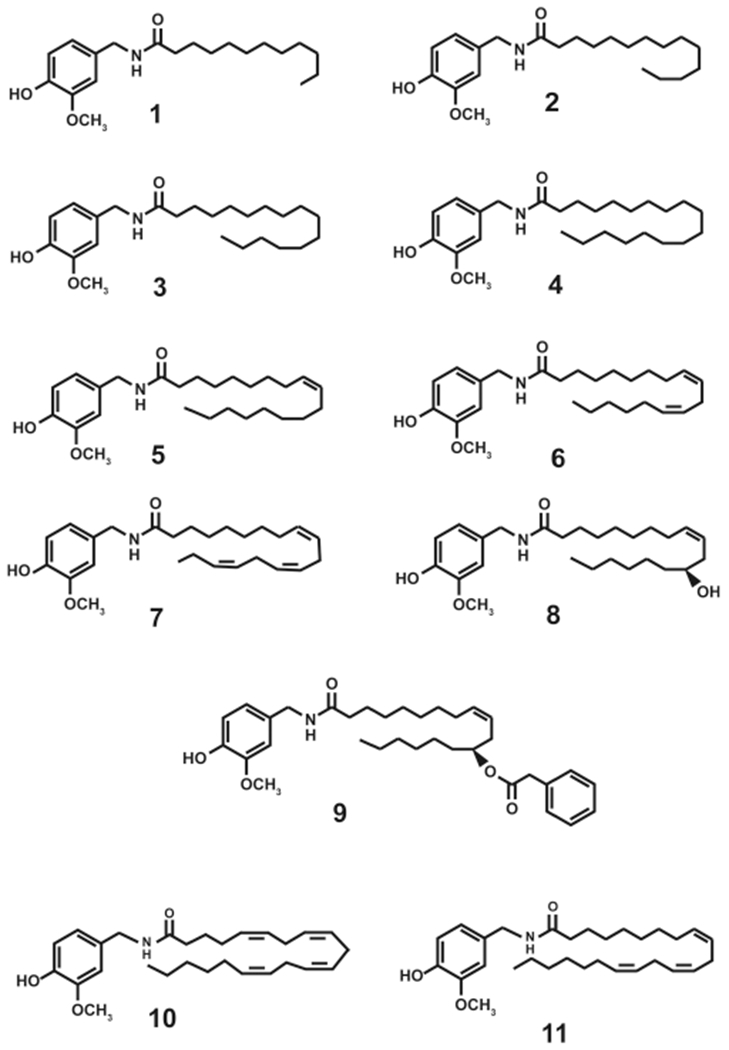
N-AVAM capsaicin analogues which have been investigated for their growth-suppressive activity in cell culture or mice models.
3.1. Isolation of N-AVAM Capsaicin Analogues from Natural Sources.
The N-AVAM capsaicin analogues 1–6 have been isolated from chili peppers (habanero and Takansosume peppers) as well as from Capsicum oleoresin.29 The term Capsicum oleoresin refers to an oily organic resin derived from the plants belonging to the Capsicum genus, namely, as chili peppers. The oleoresin is generated by ethanolic extraction of finely ground chili peppers. After extraction, the oleoresin is dried. This oleoresin is commonly used as a culinary seasoning agent in food. Kobata et al. isolated a panel of N-AVAM capsaicin analogues from Capsicum oleoresin (1–6, Scheme 1) and habanero and takansosume peppers (2–6, Scheme 2).29 The authors used three kinds of Capsicum oleoresin obtained from a Chinese market. The resins were extracted with methanol and were fractioned by silica gel column chromatography. The elution of the methanolic extracts by with stepwise elution of a mixture of 50% n-hexane and 50% ethyl acetate yielded 15 distinct fractions. The N-AVAM capsaicin analogues were present in fractions 12 and 13. Fraction 12 was purified by medium pressure liquid chromatography (MPLC) using a reversed phase silica gel column. Elution with 80–90% methanol yielded N-AVAM capsaicin compounds 1–6 represented in Scheme 1. The N-AVAM capsaicin analogues 1–6 were characterized by GC-MS analysis, APCI-MS, and NMR techniques.29 The compounds 3–6 were present in robust amounts in Capsicum oleoresin, whereas only minute amounts of compounds 1, 2, and 7 were detected in the methanolic oleoresin extract.
Scheme 1.
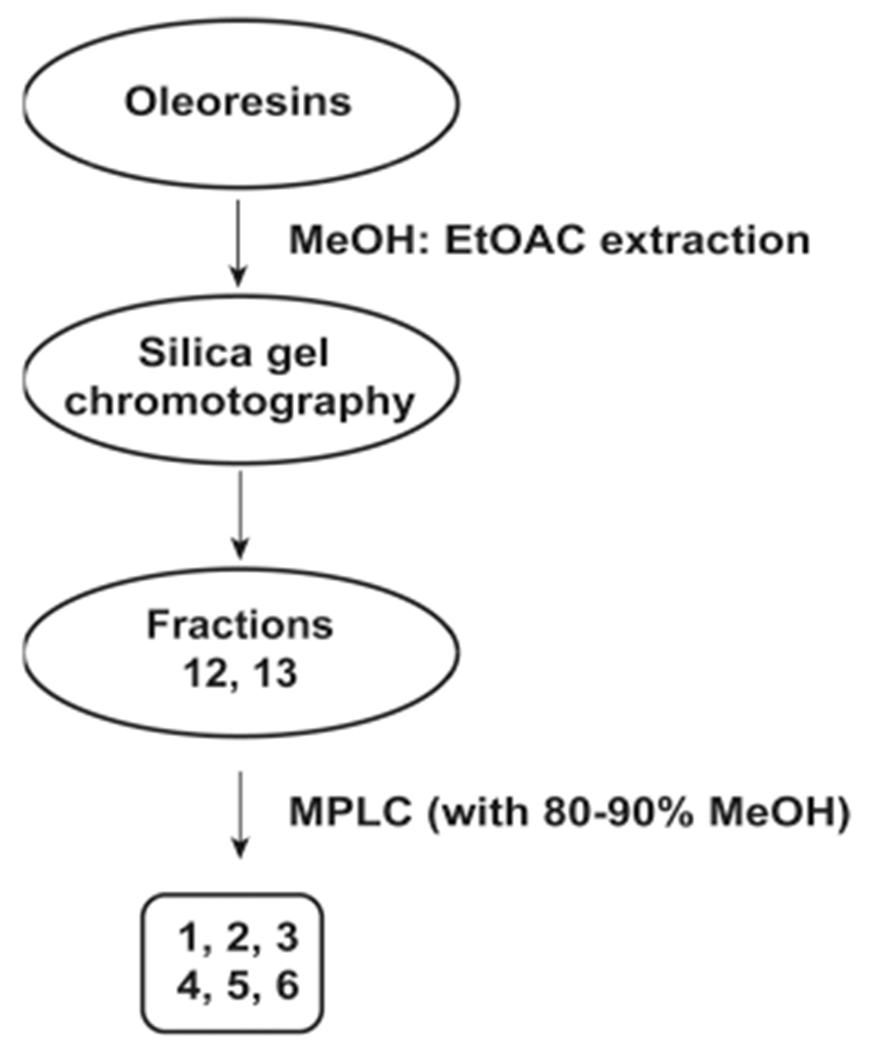
Extraction of N-AVAM Capsaicin Analogues from Capsicum oleoresin
Scheme 2.
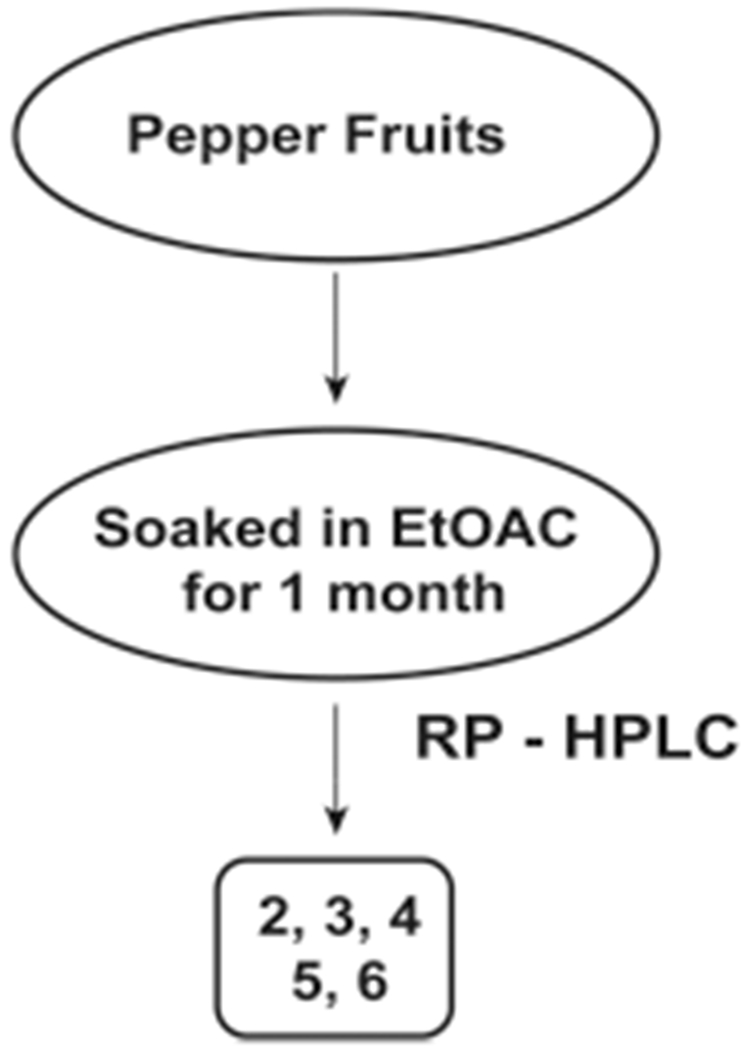
Extraction of Capsaicin from Habernero and Takanosume Chili Peppers
The isolation of N-AVAM capsaicin analogues 2–6 from habernero and takanosume peppers was accomplished by a slightly different method (Scheme 2). The peppers were freeze-dried, ground, and soaked with ethyl acetate (for 1 month) to obtain the fraction containing N-AVAM capsaicin analogues.29 The ethyl acetate fraction was purified by reverse-phase HPLC to obtain the purified N-AVAM capsaicin analogues. The compounds were characterized by APCI-mass spectrometry (Scheme 2). The compounds 1 and 7 were not detected in the habernero or takanosume peppers. 2 was present in miniscule amounts only in the habernero peppers.29 The peppers contained abundant amounts of 3, 5, and 6. A low concentration of 4 was found in the pepper fruits.
3.2. Generation of N-AVAM Capsaicin Analogues from Plant Oils.
N-AVAM capsaicin analogues have been generated by the nucleophilic amidation reaction using vanillylamine (12) and plant oils.30 The yield of the N-AVAM capsaicin analogues depended upon the fatty acid composition of the plant oil used in the reaction. The compound 12 was mixed with olive oil and the resultant mixture was heated to 180 °C for 1 h to generate a mixture of N-AVAM capsaicin analogues (3–7, Scheme 3).30 This reaction mixture was purified on a reverse phase silica gel column. The reaction product contained approximately 10% 3, 5% 4, 70% 5, 10% 6, and less than 1% 7. When vanillylamine was mixed with soybean oil, reaction products were comprised of 2% 2, 10% 3, 5% 4, 30% 5, 48% 6, and 5% 7 (Scheme 4). It is probable that natural N-AVAM capsaicin analogues in Capsaicum oleoresin were evolutionarily generated from the reaction of 12 and the oils found in the oleoresin.29,30
Scheme 3.
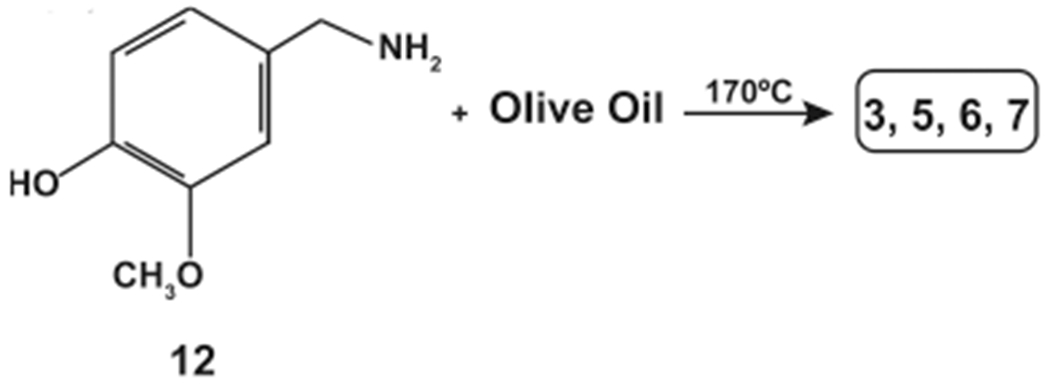
Synthesis of N-AVAM Capsaicin Analogues from Olive Oil
Scheme 4.
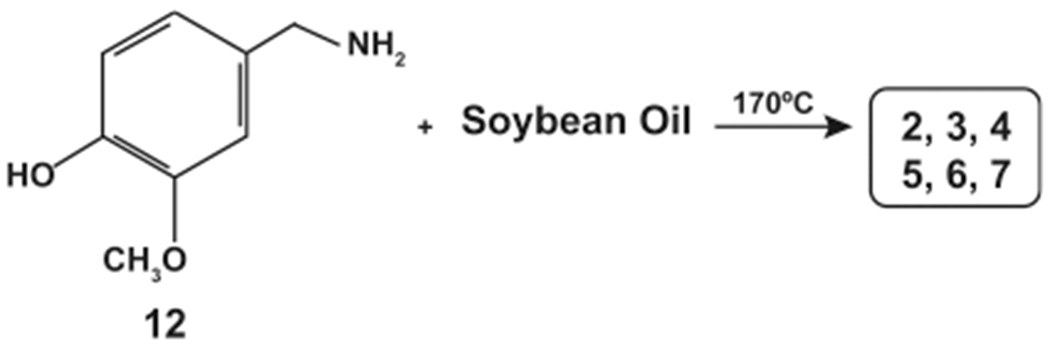
Synthesis of N-AVAM Capsaicin Analogues from Soybean Oil
3.3. Chemical Synthesis of N-AVAM Capsaicin Analogues.
Januscz et al. (1993) described six possible synthesis strategies to obtain N-AVAM capsaicin analogues (Figure 3).31 The key reactant chemical in Schemes 4–6 was 13 (3-methoxy 4-alkoxy vanillylamine derivatives, Figure 3).Scheme 4 involved the reaction of 13 (in dimethylformamide) with the corresponding fatty acyl chlorides (solubilized in ether), as represented by Figure 3. Scheme 5 was identical to Scheme 4 except that the reaction was performed using a two-phase system of water and ether (Figure 3). Scheme 6 involved the reaction of 13 with the relevant fatty acid using N,N-dicyclohexylcarbodiimide (DCC) as the coupling agent and 4-(dimethylamino)pyridine (DMAP) as the catalyst (Figure 3).
Figure 3.
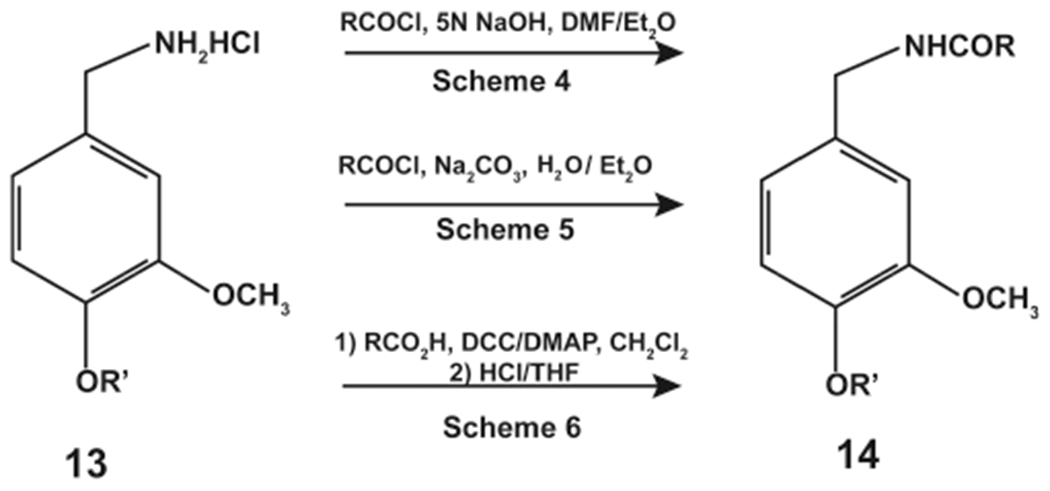
Synthesis of N-AVAM capsaicin analogues by Schemes 4–6.
The products obtained in Schemes 4–6 (Figure 3) were hydrolyzed to yield the desired N-AVAM capsaicin analogues (14).31 Schemes 7 and 8 involved the reaction of 15 (4-acetylhomovanillic acid chloride) with the desired amine to yield N-AVAM capsaicin analogues (16, Figure 4). In Scheme 8, 1 equiv of 15 was treated with 2 equiv of the appropriate amine in the presence of 1 equiv of trimethylamine (Figure 4). Scheme 8 involved the reaction of 1 equiv of 15 with 1 equiv of the desired amine and 1 equiv of trimethylamine (Figure 4, middle reaction).31 Takao et al. (2015) made a subtle variation in Scheme 7 that was used to synthesize 5 (Figure 4).32 The authors reacted 15 (oleoyl acid chloride) with vanillylamine hydrochloride in the presence of ethanolamine and dicholoro-methane to obtain 5.32
Figure 4.
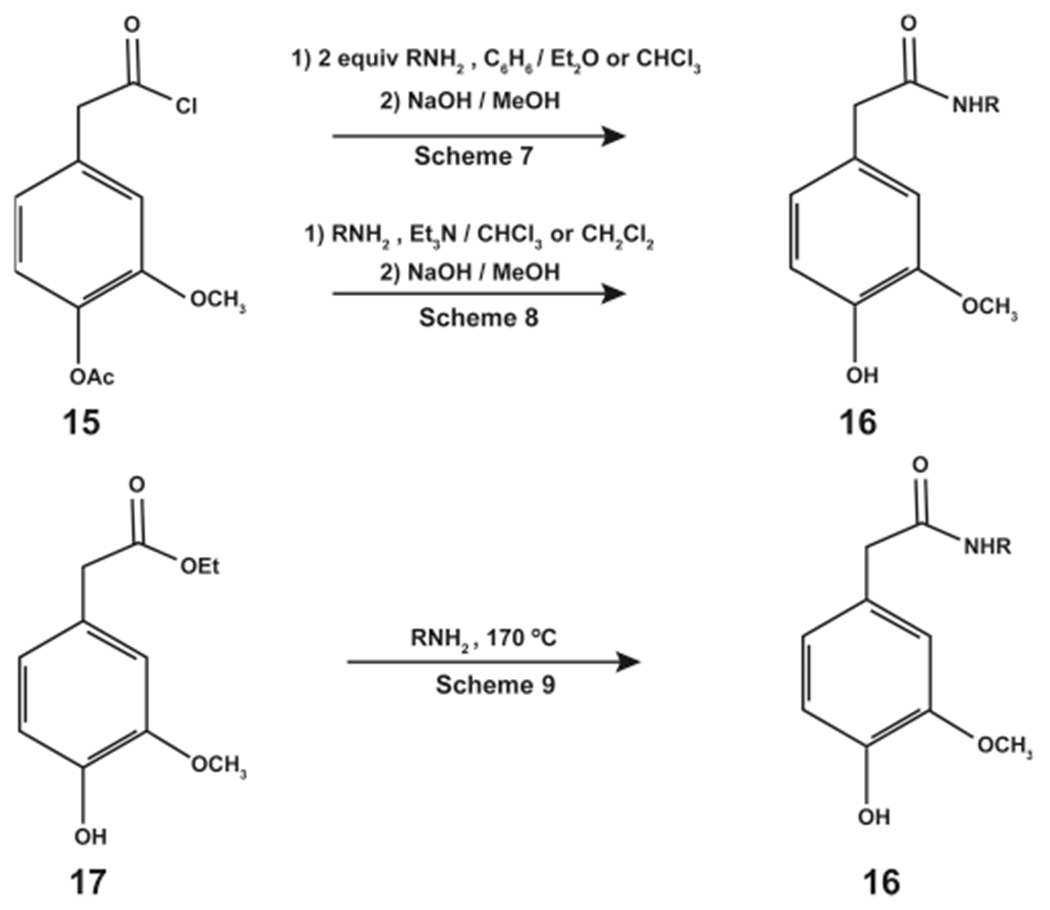
Synthesis of N-AVAM capsaicin analogues by Schemes 7–9.
Scheme 9 used ethyl homovanillate (17) as the starting material for the reaction (Figure 4). 17 was reacted with the desired amine at an elevated temperature (170 °C). Method F did not require any tertiary amines or carbodiimides to catalyze the reaction. The N-AVAM capsaicin compounds were purified by RP-HPLC and characterized by gas chromatography, mass spectrometry, and NMR techniques.31 Most interestingly, the reaction products obtained from Schemes 7–9 are reverse amides of the reaction products from Schemes 4–6.
The synthesis strategy outlined in Schemes 4–6 suffered from a few drawbacks. Schemes 4–6 use fatty acid acyl chlorides, which may emulsify, especially with long chain fatty acids. The formation of fatty acid acyl chlorides from simple unfunctionalized acids is a standard procedure; however, carefully controlled conditions are required to obtain acyl chlorides from polyunsaturated acids. The use of carbodiimide condensing agents (DCC) required protection of the phenolic hydroxyl group of vanillylamine of 13.31 All these considerations underscored the need for a single step synthesis procedure coupled with an easy isolation protocol to obtain N-AVAM capsaicin analogues.
Appendino et al. (2006) designed a single step synthetic route to generate N-AVAM capsaicin analogues using vanillylamine hydrochloride as the starting material (Scheme 10).33 The starting compound for the synthesis of 10 was arachidonic acid (18).33 18 was reacted with the hydrochloride salt of 12 (in the presence of DEPC and trimethylamine) under an inert nitrogen environment for 90 min.33 Volatile reaction products were removed under reduced pressure, and the nonvolatile residues were purified by silica gel open chromatography to obtain 95% pure arvanil (10, Scheme 10). The synthesis of 8, 9, and 11 was accomplished using similar conditions with ricinoleic acid, phenylacetylricinoleic acid, and 4,7,10,13,16,19-docosahexanoic acid being used as the starting materials (Table 1).
Scheme 10.
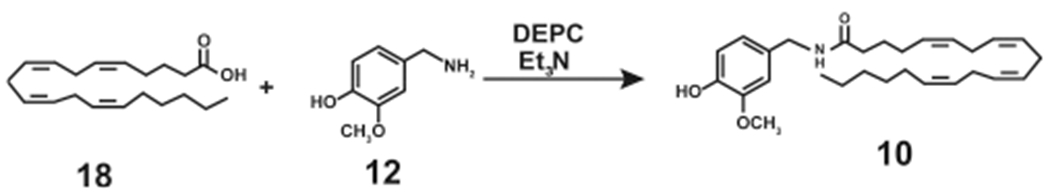
Single Step High-Yielding Synthesis of Arvanil (10)
Table 1.
Fatty Acids Used to Synthesize N-AVAM Capsaicin Analogues
| fatty acid | N-AVAM capsaicin analogue |
|---|---|
| ricinoleic acid | 8 |
| phenylacetylricinoleic acid | 9 |
| 4,7,10,13,16,19 docohexanoic acid | 11 |
Dasse et al. (2000) used an innovative synthesis strategy to generate N-AVAM derivatives of 10 (Scheme 11).31 They synthesized the intermediate compound methyl-14-hydroxy-(all-cis)-5,8,11-tetradecatrienoate (19) from the commercially available hex-5-ynoic acid.34 19 was converted into the corresponding phosphonium iodide, 20. Subsequently, 20 was reacted with the desired aldehyde in a Witting reaction to generate the ester (22, Scheme 11). Hydrolysis of the ester followed by treatment with oxalyl chloride and vanillylamine generated 10 and its related N-AVAM analogues.34
Scheme 11.
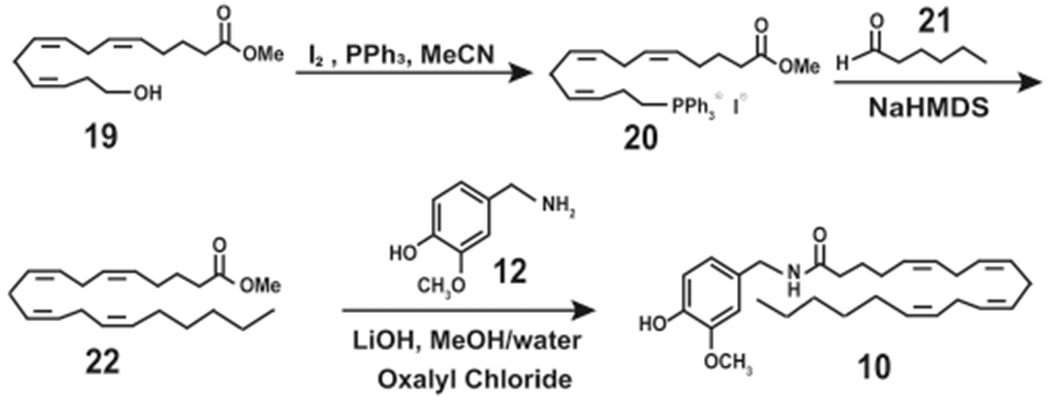
High Yield Synthesis of Arvanil (10) from Methyl-14-hydroxy-(all-cis)-5,8,11-tetradecatrienoate
Carpino (1993) described using HATU (hexafluorophosphate azabenzotriazole tetramethyl uronium) as a coupling agent which facilitated the rapid, high-yield acyl amidation reaction of carboxylic acids with nucleophilic amines (Scheme 12).35 Moriello et al. (2018) used the HATU reagent to obtain N-AVAM capsaicin analogues.36 The starting material for the synthesis was the relevant fatty acyl methyl esters. The fatty acyl methyl esters were hydrolyzed to yield the corresponding fatty acid, which was then reacted with 12 (in the presence of DIPEA in anhydrous DMF) using the azabezotriazole based coupling agent HATU to obtain robust yields of N-AVAM capsaicin analogues.36 Scheme 12 shows the schema of this coupling reaction using oleic acid (23) as an example reactant for the reaction. The reaction of 23 with 4-hydroxy-3-vanillylamine (in the presence of HATU) yields 5.
Scheme 12.
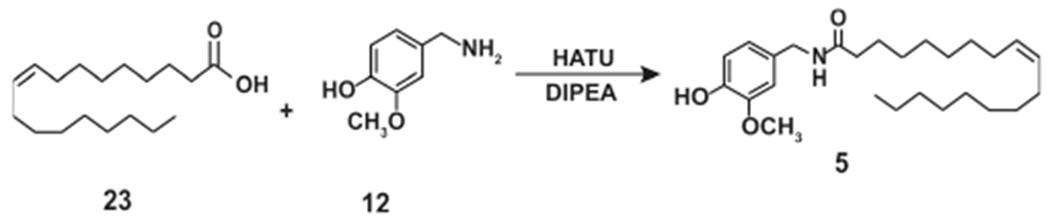
Synthesis of Olvanil (5) Using HATU Coupling Agent Methodology
3.4. Enzyme-Based Synthesis of N-AVAM Capsaicin Analogues.
Kobata et al. (2010) were the first to use biocatalysts from acetone powder of a liver extract to obtain capsaicin analogues.29 Subsequently, they extended these studies to synthesize N-AVAM capsaicin analogues from vanillylamine hydrochloride (24) and fatty acid esters in a two-phase system using the enzyme lipase AK or lipase PS (dissolved in organic solvents). The addition of N,N-diisopropylethylamine (DIPEA) was to release 12 from its hydrochloride salt. Such lipase catalyzed amidation was used to obtain moderate yields (40–59%) of 1, 5, and 7 (Scheme 13). In a later published report, Kobata et al. (2010) reacted 12 with natural oils, namely, safflower oil, perilla oil, and olive oil (using lipase enzymes as catalysts), to obtain a mixture of N-AVAM capsaicin analogues (Scheme 13).29 The reaction of 12 with olive oil (catalyzed by lipase B, lipase D, lipase R, or Novozym435) yielded 5.30 When safflower oil was used in the above reaction, the product obtained was a mixture of 5 and 6. Similarly, lipase-catalyzed reaction of 12 with perilla oil generated 7.30 The rate limiting step of this lipase-catalyed synthesis of 5 was the release of 12 from its hydrochloride salt (Scheme 13). Reyes-Duarte et al. increased the efficacy of the lipase-catalyzed reaction by adding DIPEA along with vanillylamine hydrochloride salt (24).37 The addition of DIPEA enabled efficient release of the vanillylamine (from its hydrochloride salt) which was treated with oleic acid (23) in the presence of lipase B to yield 5. Furthermore, they preincubated the 24 with a 12-fold molar excess of DIPEA (for 30 min) to achieve an almost complete conversion of 24 to 12.37 The 12 (generated in situ) was reacted with 23 (in the presence of lipase B) to obtain 5 (Scheme 13).
Scheme 13.
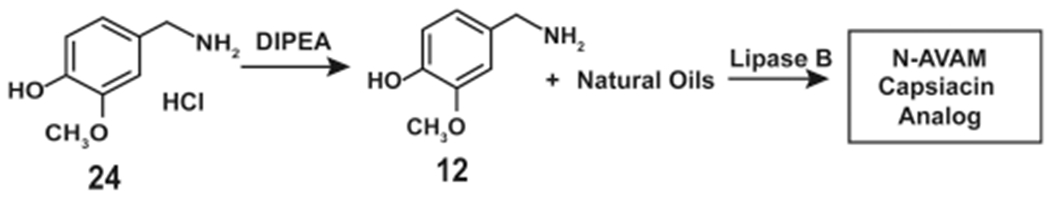
Enzymatic Synthesis of Olvanil Using Lipase B
The use of enzymes (in their free form) for industrial scale synthesis is often hindered by low product yields, low stability of the enzyme, and poor recovery of the enzyme after one round of synthesis. Such drawbacks may be circumvented by the use of immobilized enzymes for synthesis reactions.38,39 The immobilized enzyme is more stable than the free enzyme due to an increase in protein rigidity, which prevents conformational changes that can potentially lead to inactivation. The immobilized enzymes can be easily recovered after the reaction, which yields a purer product.38,39 The reuse of immobilized enzymes also decreases the economic cost of the synthesis process. Reyes-Duarte et al. immobilized recombinant Candida antarctica lipase B (CALB) on acrylic resin (called Novozym435) to obtain extremely high yields of 5 (~80%).37 More recently, Diaz-Vidal et al. immobilized recombinant Candida antarctica lipase B (CALB) by cross-linked enzyme aggregate (CLEA) techniques to obtain enzyme aggregates.40 These CALB-CLEA enzyme aggregates were used to synthesize 5. The authors preincubated 24 with DIPEA to obtain the free base. The compound 12 so obtained was mixed with 23 and CALB-CLEA in anhydrous 2-methyl butanol for 72 h to obtain 5 (Scheme 14). 5 was purified by high-pressure thin layer chromatography (HPTLC) and characterized by electron spray ionization (ESI) mass spectrometry.40 Other N-AVAM capsaicin analogues obtained using immobilized lipase B (Novozym435) include 2, 6, and 11.41
Scheme 14.
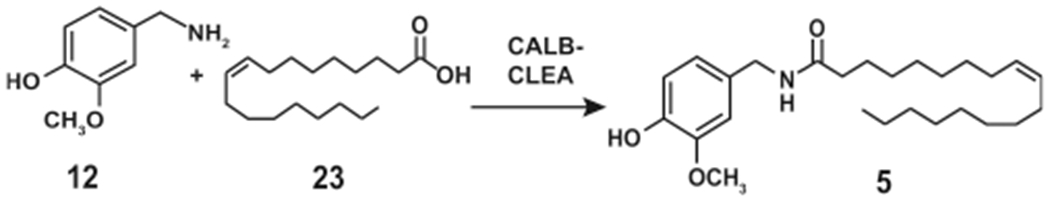
Enzymatic Synthesis of Olvanil Using Recombinant Candida antarctica Lipase B (CALB) Immobilized by Cross-Linked Enzyme Aggregate (CLEA) Techniques
The selectivity of lipases for substrates can be improved by an innovative technique called “Bioimprinting”.42,43 This method involves generating the transition state intermediate of the bound enzyme and the substrate in organic solvents. This technique can be used for substrate analogues, additives, and inhibitors, which are referred to as the “bioimprint molecule”. The transition-state intermediate is cross-linked, precipitated, and lyophilized to lock the active conformation of the enzyme with the bioimprint molecule. Such bioimprinting has been shown to improve enzyme specificity, selectivity, catalytic activity, and the yields of the product (Figure 5).42,43 For example, the yield of 5 obtained from CALB-CLEA is about 16% over 72 h.
Figure 5.
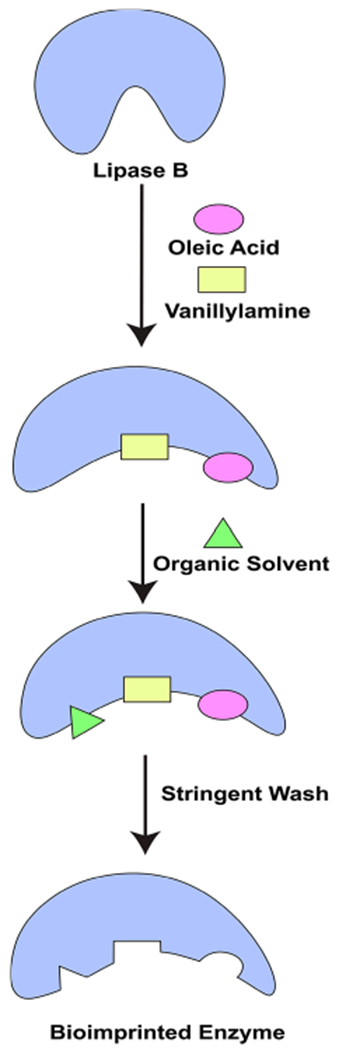
Schematic diagram showing the synthesis of olvanil by bioimprinting technology.
When CALB was bioimprinted with 5, the reaction yield was increased by 1.3-fold leading to a 25% yield of 5 over 72 h.40 An interesting observation was that CALB-CLEA bioimprinted with 12 or 23 gave poor yields of 5. When the enzyme was bioimprinted with 5, higher yields of the reaction product were obtained. These observations may be explained by the fact that the binding of 12 with lipase B induces a conformational change in the active site of the enzyme, which hinders the accessibility of the oleic aid to the active site of lipase B. Bioimprinting is a cutting edge technology which can increase the catalytic activity of lipases up to 18-fold.40 Therefore, it may be envisaged that bioimprinted lipases will pave the way to single-step synthesis of large quantities of highly pure N-AVAM capsaicin analogues.
4. STABILITY AND METABOLISM OF N-AVAM CAPSAICIN ANALOGUES
The nutritional compound capsaicin is an agonist of the TRPV1 receptor.3 The addition of an unsaturated long chain alkyl group to region C of capsaicin endows the derivative with low to moderate affinity for endocannabinoid receptors.27 Traditionally, the endocannabinoid system is comprised of two “classical” endocannabinoids, namely, N-arachidonoylethanol-amine (anandamide, AEA) and 2-arachidonoylglycerol (2-AG), and the endocannabinoid receptors CB1 and CB2. All N-AVAM capsaicin analogues are agonists of CB1 and CB2 endocannabinoid receptors. The N-AVAM capsaicin analogues display several characteristics of endocannabinoids including binding to endocannabinoid receptors, regulating the activity of the putative anandamide transporter (AMT), and inhibiting the uptake of anandamide (AEA) through lipophilic cell membranes.44,45 The degradation of endocannabinoids is mediated by the enzymes fatty acid amide hydroxylase (FAAH) and monoacylglycerol lipase (MAGL).46,47 The N-AVAM capsaicin analogue 10 is resistant to FAAH-mediated hydrolysis, which increases its biological half-life in the cellular microenvironment relative to other N-AVAM capsaicin analogues.
Studies by Janusz et al. (1993) show that the lipophilicity of N-AVAM capsaicin analogues was directly correlated with their biological activity.28 The lipophilicity of these N-AVAM capsaicin analogues was determined by the octanol number (log 1 – octanol/water partition index).48,49 They observed that the pain-relieving activity of these N-AVAM capsaicin analogues correlated directly to the lipophilicity of the compound.31 The authors concluded that short-chain N-AVAM capsaicin analogues would be rapidly metabolized by the liver leading to lower bioavailability of the active drug moiety.
The majority of pharmacokinetic studies with the N-AVAM capsaicin analogues have been performed with olvanil (5). Early studies from Sietsema et al. revealed that 5 displayed pain-relieving activity only with subcutaneous injection and not via oral administration in a mouse model using the hot plate antinociception assay.50 The authors performed a pharmacokinetic experiment with radiolabeled 5 in male CF-1 mice to test if the difference of bioactivity was related to variations in the plasma concentration of 5 following the two routes of administration. The plasma area under the curve (AUC) for all radioactive compounds was not different between subcutaneous and oral dosing, suggesting good oral absorption. Further evaluation specific for the levels of 5 (in the plasma) as a function of time indicated that the subcutaneous administration of 5 led to a rapid elevation of its concentration in the plasma within 4 h of dosing followed by a slow decline over 24 h. In contrast, when radioactive 5 was orally administered to the mice, there was a negligible amount of 5 detected in the blood within the first 2 h, which completely disappeared by 4 h.50 The AUC for 5 in mouse plasma was much higher following subcutaneous administration (8 ± 2.2 μg-h/g) compared to oral administration (0.1 ± 0.01 μg-h/g). These findings indicate that the lack of 5’s ability to produce analgesic effects after oral dosing is not due to lack of absorption of the drug but because of its rapid first pass metabolism, relative to subcutaneous 550 Such a first pass metabolism may initially occur in the gastrointestinal (GI) tract during the absorption process. After reaching the hepatic portal vein, the drug may be further metabolized in multiple tissues including the liver, lungs, and heart. No studies have identified the exact site of 5’s first pass metabolism in mice.
Several published reports have characterized the compounds generated from the first pass metabolism of capsaicin. Based on these findings Wehmeyer (1990) et al. hypothesized that the potential routes of metabolism of 5 are the hydrolysis of the amide bond and (ὡ-β)-oxidation of the side chain and conjugation of the phenolic group (Figure 6).47,51 The authors tested their hypothesis by labeling 5 with 14C at the benzylic carbon atom (represented by the black circle in Figure 6) or with 3H on the oleoyl side chain (represented by the black square in Figure 6).51 The metabolism of 14C-olvanil and 3H-olvanil was studied in vitro using isolated proteolytic enzymes, cell free intestinal and liver supernatants, isolated hepatocytes, enterocytes, and isolated intestinal perfusion systems (isolated from Sprague–Dawley rats). The treatment of 14C-olvanil with type VIII porcine liver protease and porcine intestinal protease yielded a metabolite which coeluted with 12.51 Similarly, the incubation of 14C-olvanil with cell-free liver supernatant resulted in the generation of 12. An interesting observation was that 5 was not metabolized by several common proteolytic enzymes like chymotrypsin, elastase, papain, cathepsin C, pepsin, leucine aminopeptidase, and porcine liver esterase.51
Figure 6.
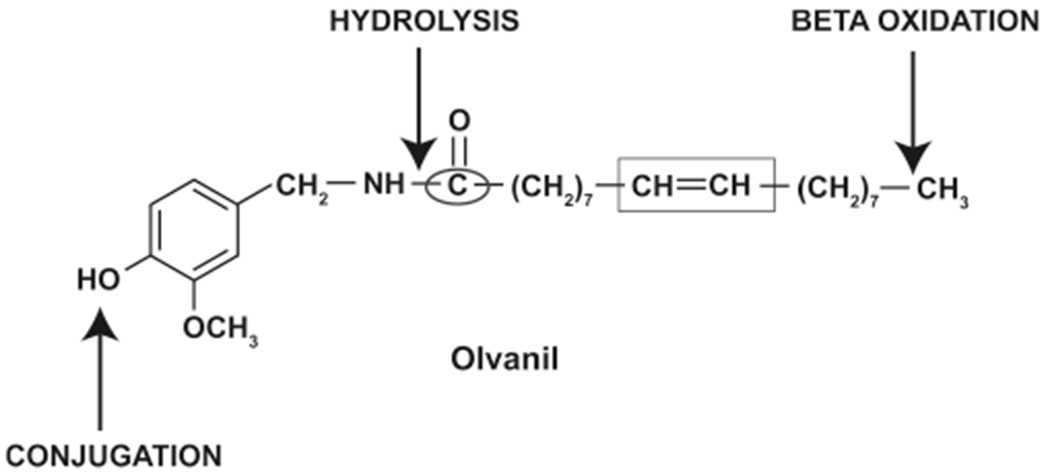
Putative sites if metabolism of olvanil.
Isolated enterocytes metabolized 14C-olvanil rapidly to generate 5 and vanillin. 14C-olvanil was primarily metabolized by enterocytes between 30 min to an hour incubation of the reactant. The amount of intact 14C-olvanil left after 1 h incubation with enterocytes was approximately 1%.51 It is unclear whether the enterocytes directly hydrolyzed 5 to 12 or whether the 12 was generated indirectly from metabolic reactions like beta-oxidation of 5. The incubation of 3H-olvanil with isolated enterocytes predominantly yielded oleic acid (74% of total metabolites generated).
The metabolism of 5 in isolated hepatocytes (isolated from male Sprague–Dawley rats) occurred much more rapidly relative to enterocytes. Within 30 min, the amount of intact 14C-olvanil remaining in isolated hepatocytes was only 4%. 12 was not detected after the metabolism of 14C-olvanil with rat hepatocytes. The metabolic products of 14C-olvanil in hepatocytes included two polar compounds which were not identified.51 Hepatocytes metabolized 3H-olvanil to yield oleic acid (24% of total metabolites generated). Such results demonstrate that hydrolysis is the dominant route of the metabolism of 5 in enterocytes and hepatocytes.
The above data suggest that the liver and intestine are the primary sites of metabolism of 5. This hypothesis was tested by incubating radiolabeled 5 with isolated perfused intestine. The radiolabeled 5 (14C-olvanil) was injected via the intraduodenal route, and the perfusate was collected using a portal cannula after 1 h. 14C-olvanil was almost completely metabolized (90% metabolized) by intestinal tissue to yield 12 and an unknown polar compound.51
The metabolism of 5 in vivo was explored in adult male Sprague–Dawley rats. The rats were administered 14C-olvanil (200 mg/kg) by oral gavage. After 3 h, the plasma was tested for metabolites of 5 using reversed-phase HPLC with sequential UV and online radiochemical detection (LC-RAD). Olvanil-O-glucuronide and an unknown polar compound were major metabolites detected in the plasma. The metabolism experiments in rat models show that glucuronidation of the phenolic group (to yield olvanil-O-glucuronide) may be a key route of the metabolism of 5 in vivo The treatment of the plasma with β-d-glucuronidase resulted in the appearance of 12. Intact 5 was also detected in the β-d-glucuronidase-treated plasma.51 The incubation of 5 in whole isolated blood (from Sprague–Dawley rats) does not induce first pass metabolism of 5. No published reports have investigated the metabolism of 5 in the lung. Taken together, the in vitro and in vivo metabolism experiments showed that the major route of metabolism of 5 is hydrolysis of the amide bond to yield 12, vanillin, and olvanil-O-glucuronide (Figure 6).
Capsaicin has been extensively used as a pain-relieving agent in topical formulations like creams and lotions. Kasting et al. (1997) compared the skin penetration ability of 5 in rat and human skin sections mounted on Franz diffusion cells.52 The steady state flux rates of 5 was measured between 7 and 48 h postincubation with radiolabeled 5. The permeability rate of 5 across rat skin (from SkH:Fz rat and CD:VAF rat) was higher than human skin. These observations were confirmed in dermal absorption studies performed in CD:VAF rats. The steady state flux of 5 across CD:VAF rat skin increased over time from 24- to 72 h post-treatment.52 The dermal metabolism of 5 was studied in SkH:Fz perfused rat skin 72 h after topical application. 5 is a highly lipophilic compound, so it was predicted to be efficiently absorbed into the skin. Surprisingly, only 3.6% of 5 was absorbed across the skin after 72 h, and a majority of 5 was excreted via the urine after dermal absorption.52 The major pathway for dermal metabolism of 5 was via hydrolysis of the amide moiety to yield 12 (Figure 6).
Although, all the pharmacokinetic studies of N-AVAM capsaicin analogues were performed using 5, a few important patterns were observed.50,52 The primary sites of N-AVAM capsaicin analogue metabolism were the liver, intestine, and the skin. 5 was primarily metabolized by direct/indirect hydrolysis to yield 12 and vanillin. The metabolite olvanil-O-glucuronide was detected in rat plasma after oral administration of 5 (Figure 7). Several unknown polar and nonpolar metabolites were detected by HPLC techniques.50,52 The identification and characterization of these compounds will be pave the way to precise metabolic profiling of N-AVAM capsaicin analogues in vivo. The elucidation of the “N-AVAM capsaicin analogue-metabolome” will facilitate the rational design of N-AVAM capsaicin analogues with improved pharmacokinetic properties and therapeutic indices.
Figure 7.
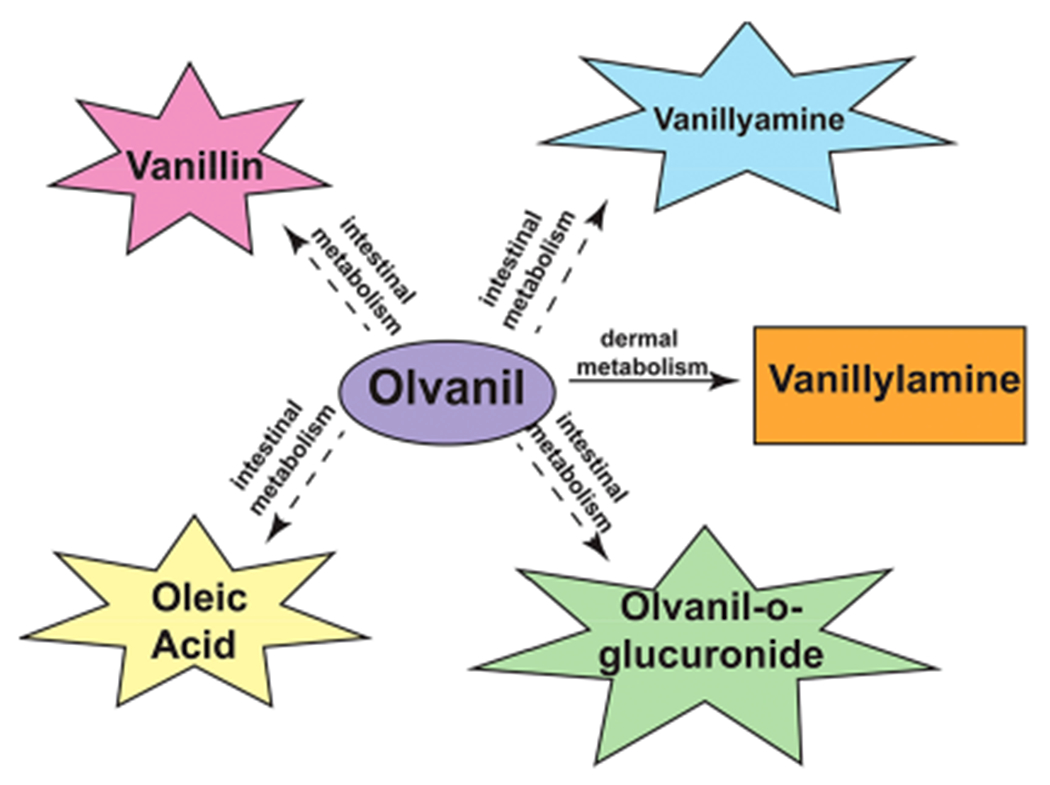
Schematic diagram showing the compounds generated by the intestinal metabolism and dermal metabolism of olvanil.
5. ANTI-NEOPLASTIC ACTIVITY OF N-AVAM CAPSAICIN ANALOGUES
A large number of published reports have investigated the pain-relieving activity of N-AVAM capsaicin analogues.24,27,30 Structure activity-relationship studies showed that the introduction of long chain unsaturated fatty acids in region C generated nonpungent capsaicin analogues with extremely high pain-relieving activity. A similar trend has been observed in the growth-suppressive activity of N-AVAM capsaicin analogues (Table 2). The N-AVAM capsaicin analogues which contain a long chain unsaturated fatty acyl group (in region C) displayed greater growth-inhibitory activity than N-AVAM capsaicin analogues containing saturated fatty acyl side chains. The extent of unsaturation of the fatty acyl side chain correlated to the growth-suppressive activity of these compounds. The N-AVAM capsaicin analogues with few double bonds (in the carbon chain backbone in Region C) displayed lower growth-suppressive activity than the compounds with large number of double bonds in their fatty acyl side chain. Studies in our laboratory were the first to conduct systematic SAR experiments to delineate the contributions of “length of the fatty acyl side chain” and “number of double bonds in fatty acyl side chain” toward the growth-inhibitory activity of N-AVAM-capsaicin analogues (Figure 8) in human small cell lung cancer (SCLC).28 The impetus for these experiments were derived from published reports comparing the growth-suppressive activity of 3, 5, and 10. 3 is an N-AVAM capsaicin analogue which contains a 16-carbon atom acyl side chain in region C of capsaicin. 3 has no double bonds (C16:0). 5 has an acyl side chain of 18 carbon atoms and one double bond (C18:1). 10 has a side chain of 20 carbon atoms with four double bonds (C18:4). The growth-suppressive activity of 5 and 10 were tested in MCF-7, EFM-19, and T47D breast cancer cells.27 A survey of the IC50 values showed that the growth suppressive activity was 3 [IC50 (MCF-7) = 2.2 μM; IC50 (T47D) = 1.6 μM; IC50 (EFM-19) = 1 μM] was lower than 5 [IC50 (MCF-7) = 1.6 μM; IC50 (T47D) = 0.75 μM; IC50 (EFM-19) = 0.7 μM] which was lower than 10 [IC50 (MCF-7) = 0.4 μM; IC50 (T47D) = 0.35 μM; IC50 (EFM-19) = 0.55 μM] in the three breast cancer cell lines. Similarly, 10 induced about a 3-fold higher magnitude of apoptosis (60% apoptotic cells) in human peripheral blood mononuclear cells (PBMCs) than 5 (20.5% apoptotic cells).53 Although, these trends were observational, they motivated us to investigate whether increasing the acyl side chain or the number of double bonds could enhance the growth-inhibitory activity of N-AVAM capsaicin analogues. We examined the effect of a panel of N-AVAM capsaicin analogues (Figure 8) on the viability of DMS114 human SCLC after 24 h.28 MTT [[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assays showed that 3 and 4 had no growth inhibitory activity in DMS114 cells (Figure 8A). The growth-suppressive effects of the compounds were in the order, 5 (C18:1, IC50 > 50 μM) < 6 (C18:2, IC50 = 43 μM) < 7 (C18:3, IC50 = 15 μM) < 10 (C20:4, IC50 = 15 μM) (Figure 8A). Taken together this suggests that the growth-inhibition of these N-AVAM capsaicin analogues are directly proportional to the chain length of the fatty acyl group (in region C) and the number of double bonds present within this fatty acyl side chain.
Table 2.
Overview of the Anticancer Activity of N-AVAM Capsaicin Analogues
| compound | type of cancer | IC50 (in cell culture) | effect on normal cells | experimental models used | phenotypic effects | mechanism of action | ref |
|---|---|---|---|---|---|---|---|
| 5 | small cell lung cancer | ND | ND | cell culture | inhibition of cell invasion | inhibition of AMPK pathway | 73 |
| 5 | breast cancer | MCF-7: 1.6 μM T47D: 0.75 μM |
ND | cell culture | inhibition of cell proliferation | activation of TRPV1 and CB1 receptors | 25, 26, 55 |
| 10 | breast cancer | MCF-7: 0.4 μM EFM-19: 0.55 μM T47D: 0.35 μM |
ND | cell culture | inhibition of cell viability | activation of TRPV1 and CB1 receptors | 25, 26, 55 |
| 10 | small cell lung cancer | DMS114: 15 μM | no effect on the viability of normal lung epithelial cells | cell culture | inhibition of cell viability | activation of calpain pathway | 53 |
| 10 | rat thyroid carcinoma | ND | ND | athymic mouse model | inhibition of tumor growth in athymic mice | activation of CB1 receptor | 43 |
| 10 | high grade astrocytoma | ND | ND | orthotopic scid mouse model | inhibition of tumor growth in scid mice | activation of TRPV1 receptor | 61 |
| 10 | melanoma | ND | ND | cell culture (melanoma cell line derived from a patient) | decrease in cell viability | ND | 61 |
| 10 | T-cell leukemia | ND | no apoptosis of normal T-lymphocytes | cell culture | induction of apoptosis | activation of FADD/Caspase-8 pathway | 57 |
| 10 | prostate cancer | TSU: ~100 nM PPC-1: ~1 μM |
ND | cell culture | inhibition of cell viability | activation of CB1 receptors | 57 |
| 10 | epidermoid carcinoma | ND | ND | cell culture, | decrease in cell viability | ND | 61 |
| 10 | small cell lung cancer | ND | ND | cell culture | inhibition of cell invasion | inhibition of AMPK pathway | 66 |
| 6 | small cell lung cancer | DMS114: 43 μM | no effect on the viability of normal lung epithelial cells | cell culture | inhibition of cell viability | activation of calpain pathway | 53 |
| 7 | small cell lung cancer | DMS114: 15 μM | no effect on the viability of normal lung epithelial cells | cell culture | inhibition of cell viability | activation of calpain pathway | 53 |
| 8 | breast cancer | P388: 9 μg/mL J774:8 μg/mL WEHI: 3 μg/mL |
normal bone marrow, IC50 = 40 μg/mL | cell culture | inhibition of cell proliferation | apoptosis via the caspase pathway | 67 |
| 8 | cervical cancer | HeLa: 62 μg/mL CaSki: 91 μg/mL ViBo: 149 μg/mL |
no effect on the viability of normal human lymphocytes | cell culture | inhibition of cell proliferation | apoptosis via the caspase pathway | 68 |
| 9 | leukemia | P388: 49 μg/mL J774: 10 μg/mL WEHI: 31 μg/mL |
normal bone marrow, IC50 = 73 μg/mL | cell culture | inhibition of cell proliferation | apoptosis via the caspase pathway | 67 |
| 9 | cervical cancer | HeLa: 57 μg/mL CaSki: 122 μg/mL ViBo: 74 μg/mL |
PhAR decreased the viability of normal human lymphocytes. No selectivity for tumor cells was observed. | cell culture | inhibition of cell proliferation | apoptosis via the caspase pathway | 68 |
| 11 | cervical cancer | HeLa: ~50 μM Taxol-resistant HeLa: ~40 μM |
ND | cell culture | inhibition of cell viability | ND | 40, 70 |
| 11 | histocytic leukemia | U937: ~25 μM | ND | cell culture | inhibition of cell viability | ND | 40 |
| 11 | melanoma | B16F10: ~10 μM | normal mouse fibroblast IC50 = 65 μM | cell culture | inhibition of cell viability | ND | 40 |
| 11 | breast cancer | MCF-7: ~25 μM | ND | cell culture | inhibition of cell viability | apoptosis via caspase pathway | 69 |
Figure 8.
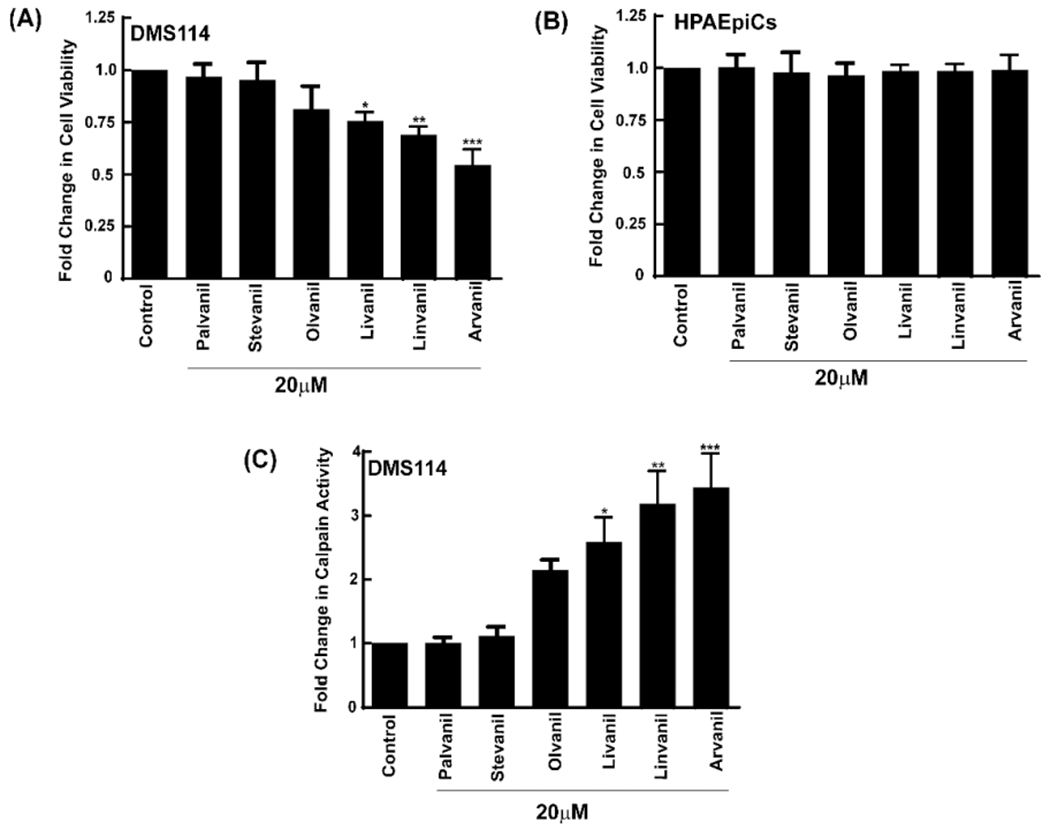
(A) MTT assays show that the growth-suppressive activity of N-AVAM capsaicin analogues increases with increased unsaturation in the compounds. (B) N-AVAM capsaicin analogues do not impact the viability of HPAEpiCs. (C) N-AVAM capsaicin analogues stimulated the activity of the calpain-1, calpain-2 class of apoptotic proteolytic enzymes. Values represented by the symbol * are statistically significant relative to the control (P ≤ 0.05).
The N-AVAM capsaicin compounds did not affect the viability of normal pulmonary alveolar epithelial cells (HPAEpiCs; Figure 8B).28 Therefore, our data suggested that N-AVAM capsaicin analogues selectively suppressed the growth of lung cancer cells and spared normal cells. The growth-suppressive activity of these N-AVAM capsaicin compounds correlated with their ability to increase the activity of the pro-apoptotic enzymes calpains 1 and 2 (Figure 8C).28 It may be possible that the basal calpain enzyme activity in normal human lung epithelial cells is lower than human SCLC cells. This may at least, in part, explain the observation that these N-AVAM capsaicin analogues only kill human SCLC cells and not normal lung epithelial cells. The N-AVAM capsaicin analogues with longer fatty acyl side chains and a larger number of double bonds (within the fatty acyl side chain) showed higher calpain activity than the N-AVAM capsaicin analogues with short acyl chains or a saturated long acyl chain. Such findings may form the basis of a new method to screen the growth suppressive activity of N-AVAM capsaicin analogues based on their ability to induce calpain activity.28
Jacobsson et al. (2001) compared the antiproliferative activity of 5 and capsaicin in C6 rat glioma cells.54 They incubated C6 glioma cells with varying concentrations of 5 and capsaicin for 4 days. They observed that 10 μM of capsaicin decreased the proliferation of C6 glioma cells by ~20%, whereas 10 μM of 5 suppressed the proliferation of C6 glioma cells by 95%. Similarly, Marzo et al. showed that 5 displayed greater antiproliferative activity in human breast cancer cell lines MCF-7 and T47D relative to capsaicin.55 The IC50 for capsaicin in MCF-7 and T47D cells (IC50 ~ 100 μM) were approximately 100-fold higher than 5 (IC50 ~ 1 μM). These observations show that the growth-suppressive activity of N-AVAM capsaicin analogues is higher than capsaicin in breast cancer and glioma cells.
An innovative study by Marquez et al. (2006) compared the effects of olvanil (5), iodo-olvanil (25), arvanil (10), and iodoarvanil (26) on the proliferation of human PBMCs.53 Tritiated thymidine assays revealed that 10 potently suppressed staphyloccal enterotoxin (SEB) induced proliferation of human PBMCs. 5 had a modest cytostatic effect on SEB-induced proliferation of human PBMCs.53 The iodination of 5 caused 3-fold higher apoptosis in SEB-treated human PBMCs compared to the parent compound (Figure 9). In contrast, 26 induced modestly improved pro-apoptotic activity (~1.3-fold) relative to 10 in human PBMCs. This data aligns well with the findings of Malfitano et al. (2006) who observed that 10 robustly inhibited the proliferation of activated human PBMCs.56
Figure 9.
Structures of iodoolvanil and iodoarvanil.
Several convergent studies show that N-AVAM capsaicin analogues suppressed the growth of C6 mouse glioma cells, Jurkat human T-cell leukemia cells, rat thyroid carcinoma (KiMol cells), human breast cancer cells (MCF-7, T47D and EFM-19 cell lines), prostate cancer cells (PPC-1, and TSU cell lines), and epidermoid carcinoma cells (JWF2, A431 cell lines).27,44,53,54,57–60 The growth inhibitory activity of N-AVAM capsaicin analogues has been examined in several human cancer cell lines. Stock et al. (2012) explored the growth-inhibitory activity of 10 using an ex vivo organotypic culture model.61 They observed that 10 suppressed the growth of HG-astrocytoma cells organotypically grown in mouse brain slices. 10 suppressed the growth of HG-astrocytoma at a relatively low concentration of 50 nM. Stock et al. (2012) also examined the effect of murine astrocytoma tumors implanted orthotopically in mouse models.61 They found that 10 at a dose of 1 mg/kg body weight strongly decreased the growth rate of astrocytoma tumors in mice models.61 Another study which investigated the anticancer activity of 10 in vivo was by Bifulcoet al. (2002), who observed that the administration of 10 at a dose of 1 mg/kg body weight potently suppressed the growth of thyroid carcinoma tumors xenotransplanted in immunodeficient mice.44
SCLC is a neuroendocrine tumor characterized by rapid doubling time, an aggressive clinical course, and a dismal 5-year survival rate62,63 The invasion of neoplastic cells into the adjacent blood/lymphatic vessels is a vital step for their metastasis to distant organs.64,65 5 and 10 suppressed the invasion of human small cell lung cancer cells at 20-fold lower concentrations relative to capsaicin.66 Such findings suggest that N-AVAM capsaicin analogues may display antimetastatic activity in human lung cancers.
Luviano et al. (2014) studied the growth-inhibitory activity of rinvanil (8) and phenylacetylrinvanil (PhAR, 9, Figure 2) in J774, P388, and WEHI-3 mouse leukemic cell lines.67 The compound 8 showed improved growth inhibitory activity ([IC50(P388) = 9 μg/mL; IC50(J774) = 8 μg/mL; IC50(WEHI) = 3 μg/mL] relative to 9 ([IC50(P388) = 49 μg/mL; IC50(J774) = 10 μg/mL; IC50(WEHI) = 31 μg/mL] in all the cell lines studied (Table 2). 9 displayed some selectivity for leukemic cell lines relative to normal mouse bone marrow bone marrow cells.67 The antiproliferative and pro-apoptotic activity of 8 and 9 were also explored in a panel of human cervical cancer cell lines (HeLa, CaSki, and ViBo).68 9 was more potent in suppressing the proliferation of ViBo human cervical carcinoma cells [IC50 (ViBo) = 74 μg/mL] than rinvanil ([IC50 (ViBo) = 149 μg/mL]. The researchers observed that 8 showed selective growth-inhibitory effects on the cervical cancer cells relative to normal lymphocytes, whereas 9 showed no selectivity between normal and tumor cells.68 Such variance in results may be attributed to the nature of the cancer, species specific differences (human cell lines versus mouse cell lines), and the disparity in the methodology used in the two studies. Whereas the studies performed by Luviano et al. (2014) studied the growth-inhibitory effects of PhAR and rinvanil by the Sulforhodamine B assay,67 Sanchez-Sanchez et al. (2015) used the lactate dehydrogenase assay to evaluate the effect of 8 and 9 on normal lymphocytes.67,68
The N-AVAM capsaicin analogue 11 (Figure 2) induced a greater magnitude of apoptosis in MCF-7 human breast cancer cells than capsaicin in vitro.69 11 also showed increased growth-suppressive activity in melanoma, leukemia, and human cervical carcinoma cells and Taxol-resistant human cervical carcinoma cells compared to capsaicin.41,70 11 displayed substantial selectivity for human cancer cells versus normal cells. The growth-inhibitory activity of 11 in normal human fibroblast cells was observed at 3-fold higher concentrations (~100 μM) than in melanoma, leukemia, and human cervical carcinoma cells (~30 μM).41
An exciting development in the field of N-AVAM capsaicin analogues is that they have been found to sensitize human cancer cells to the growth-suppressive activity of chemotherapeutic drugs. Stock et al. observed that the combination of 10 and temozolomide showed an increase in survival times in mice bearing orthotopic astrocytoma tumors when compared to either agent administered alone or a mice administered vehicle only.61 Similarly, the combination of 11 and Taxol displayed higher growth-inhibitory activity in Taxol-resistant HeLa human carcinoma cells relative to either drug alone.70 These results demonstrate that N-AVAM capsaicin analogues may be useful for the treatment of both classical cancers and drug-resistant cancers.
6. SIGNALING PATHWAYS UNDERLYING THE GROWTH SUPPRESSIVE EFFECTS OF N-AVAM CAPSAICIN ANALOGUES
There are only a few studies which have investigated the signaling pathways underlying the anticancer activity of N-AVAM capsaicin analogues. Capsaicin functions as a strong agonist of the TRPV1 receptor.3 In contrast, N-AVAM-capsaicin analogues are agonists at both the TRPV1 and the endocannabinoid receptors CB1 and CB2.33,71,72 Studies show that the role of TRPV receptors or CB1/CB2 receptors in mediating the growth-suppressive activity of N-AVAM-capsaicin analogues may depend on the nature of the cancer and the structural features of the N-AVAM capsaicin analogues. Stock et al. showed that 10 triggered robust apoptosis in high-grade astrocytoma cells via the TRPV1 receptor.58 The knockdown of TRPV1 in astrocytoma cells ameliorated the pro-apoptotic activity of 10 in both cell culture and mouse models. On the other hand, the cytostatic activity of 5 and 10 in human breast cancer cells was jointly mediated by both the TRPV receptors and the CB1 receptors.27 The antiproliferative activity of 10 in prostate cancer cells and thyroid carcinoma cells required only the function of CB1 receptors,44 not vanilliod receptors, whereas the pro-apoptotic effects of 10 in human T-cell leukemia cells was independent of both TRPV and endocannabinoid receptors.57
The growth-suppressive effects of N-AVAM capsaicin analogues are mediated via divergent mechanisms in normal and cancer cells (Table 2). N-AVAM capsaicin compounds like 5, 8, 9, and 11 induced apoptosis in breast cancer, cervical carcinoma, glioma, and leukemia cells via the caspase family of proteases.54,67–69 10 triggered a 4–5-fold increase in apoptosis in Jurkat cells (human T-cell leukemia) in a cell cycle independent manner by the inhibition of protein kinase C, which was promoted by the recruitment of the Fas-associated death domain (FADD) death signaling complex followed by activation of caspase-8.57 10 was also found to induce reactive oxygen species (ROS) in human leukemic cells, but the ROS pathway plays a peripheral role in 10 induced apoptosis of Jurkat cells.57
The ability of 10 to block the growth and activation of normal peripheral blood mononuclear cells PBMCs and T-cells plays a vital role in its ability to inhibit inflammation.53 10 does not inhibit the proliferation of CD4+ T cells.56 Its growth-suppressive effects on human PBMCs and normal T-cells is mediated by the combination of cell cycle arrest (at the G1/S phase) and apoptosis. The cytostatic effects of 10 required the activation of the p21/Waf-1/Cip-1 and inhibition of the Akt pathway.56 The pro-apoptotic activity of 10 in human PBMCs occurs via inhibition of the NF-kappa-B signaling pathway.53
Data from our laboratory show that 5 and 10 inhibit the invasion of human SCLC cells.67 The anti-invasive activity of 5 and 10 was independent of both TRPV and cannabinoid receptor pathways. 5 and 10 activated the 5′ AMP-activated protein kinase (AMPK) pathway to inhibit the invasion of human SCLC.67
All the signal transduction studies involving N-AVAM capsaicin analogues have been performed in cell culture models. These observations underscore the importance of confirming the data from cell culture systems in animal models. Several studies show that some of the N-AVAM capsaicin analogues are selective for cancer cells and do not kill normal cells. The basis of such selectivity of N-AVAM capsaicin analogues is yet to be elucidated. Most of the published reports have focused on the downstream mechanism of N-AVAM capsaicin analogue-induced apoptosis in cells. There are very few studies which have explored the mechanisms by which these compounds communicate to the cell cycle machinery or apoptotic signaling networks inside the nucleus/mitochondria of the cells. Recent studies have shown that N-AVAM capsaicin analogues function as chemosensitizers and improve the pro-apoptotic activity of conventional chemotherapeutic drugs like temozolomide and Taxol.61,70 The mechanism of such chemosensitization activity of N-AVAM capsaicin analogues is not known. All these observations define the arena where in-depth studies are urgently required to clarify the molecular mechanisms underlying the growth-suppressive activity of N-AVAM capsaicin analogues in normal and neoplastic cells.
7. CONCLUSIONS AND FUTURE DIRECTIONS
Capsaicin displays robust antineoplastic effects in multiple human cancers. However, the application of capsaicin as a clinically useful anticancer drug has been limited by its unpleasant side effects. A method to circumvent this drawback is to identify nonpungent capsaicin-mimetics with potent anticancer activity. A promising class of nonpungent capsaicin-mimetics are long-chain unsaturated N-AVAM capsaicin analogues. Several lines of evidence show that N-AVAM capsaicin analogues display improved growth-suppressive activity in human cancers, relative to capsaicin. An advantage of N-AVAM capsaicin analogues is that they suppress the growth of human cancer cells and do not harm normal cells. The growth-inhibitory activity of some N-AVAM capsaicin analogues has been predominantly demonstrated in cell culture systems and not in animal models. Such data underline the importance of examining the antineoplastic effects of different types of N-AVAM capsaicin analogues in athymic mouse and patient-derived xenograft (PDX) models. It must be remembered that the efficacy of an anticancer drug is dependent on its concentration at the target tissues. There is a paucity of studies exploring the pharmacokinetics of N-AVAM-capsaicin analogues in animal models. The elucidation of pathways governing the metabolism of N-AVAM capsaicin analogues will pave the way to designing of novel N-AVAM capsaicin analogues with greater stability and bioavailability in vivo. A promising strategy to improve the bioavailability of capsaicin has been to design sustained release formulations of capsaicin. Capsaicin nanoparticles display greater anticancer activity and stability than the parent compound.73–75 The development of N-AVAM capsaicin analogue-nanoparticle formulations may revolutionize their applications as an analgesic and as an anticancer drug in patients.
An exciting finding is that the N-AVAM capsaicin analogues enhance the growth-suppressive activity of conventional chemotherapy in both classical and drug resistant cancers. Certain cancers like small cell lung cancer (SCLC) are known to relapse within a few months, and these relapsed tumors are usually resistant to chemotherapy and radiation.76,77 The combination of N-AVAM capsaicin analogues with chemotherapeutic drugs may provide new strategies to combat relapse and drug-resistance of cancers.
The antiangiogenic and antimetastatic activity of capsaicin have been observed in several cancers.14 However, no studies have examined the effect of N-AVAM capsaicin analogues on tumor angiogenesis and metastasis. The anticancer activity of N-AVAM capsaicin analogues are mediated via multiple signaling networks. The majority of studies have analyzed downstream effectors which play a vital role in the apoptotic activity of the N-AVAM capsaicin analogues. An important question in the field of N-AVAM capsaicin analogue biology is whether the growth-suppressive activity of these compounds requires the TRPV receptors or the cannabinoid receptors or both of these receptors or none of these receptors to exert their anticancer activity. It is hoped that future studies will shed light on the mechanisms by which these drugs link to the intracellular apoptosis or cell-cycle arrest pathways inside cells. The development of second generation nonpungent N-AVAM capsaicin analogues with improved pharmacokinetic properties and anticancer activity will foster the hopes of novel N-AVAM capsaicin analogue-based combination therapies for multiple human cancers.
ACKNOWLEDGMENTS
We acknowledge Dr. S. Chellappan and his laboratory for their continuous support. S.D.R is a recipient of NSF-SURE and WV-NASA Space Consortium undergraduate fellowships. P.D. and M.A.V. are supported by a National Institutes of Health R15 Academic Research Enhancement Award (Grants 1R15CA161491-01A1 and 2R15CA161491-02). This work was supported in part by the West Virginia IDeA Network of Biomedical Research Excellence (WV-INBRE) grant (NIH Grant P20GM103434; PI, Dr. G. O. Rankin), the National Institute of General Medical Sciences of the National Institutes of Health under the Award Number P30GM122733.
ABBREVIATIONS USED
- TRPV
transient receptor potential vanilloid
- SAR
structure activity relationship studies
- N-AVAM
N-acyl vanillyl acylamide
- ROS
reactive oxygen species
- EMT
epithelial-mesenchymal transition
- MPLC
medium pressure liquid chromatography
- HPLC
high-pressure liquid chromatography
- RP-HPLC
reverse phase-high-pressure liquid chromatography
- APCI-MS
atmospheric-pressure chemical ionization mass spectrometry
- NMR
nuclear magnetic resonance
- DCC
N,N-dicyclohexylcarbodiimide
- DMAP
4-(dimethylamino)-pyridine
- DEPC
diethyl pyrocarbonate
- NaHMDS
sodium bis(trimethylsilyl)amide
- PhAR
phenylacetylrinvanil
- DMF
dimethylformamide
- DIPEA
N,N-diisopropylethylamine
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uranium
- CALB
Candida antarctica lipase b
- CLEA
cross-linked enzyme aggregate
- FAAH
fatty acid amide hydroxylase
- MAGL
monoacylglycerol lipase
- AUC
area under the curve
- MTT
[[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
- HPAEpiCs
human pulmonary alveolar epithelial cells
- PBMC
peripheral blood mononuclear cells
- SEB
Staphyloccal enterotoxin
- FADD
Fas-associated death domain
- SCLC
small cell lung cancer
- AMPK
5′ AMP-activated protein kinase
Biographies
Biographies
Stephen D. Richbart obtained his undergraduate degree at Marshall University, and he majored in Biology. He is currently performing research in the laboratory of Dr. Piyali Dasgupta. His research projects involve nutritional therapies for human lung cancers.
Jamie R. Friedman received her Bachelor’s degree in Biochemistry from Bethany College in 2015. She received her Ph.D. in Biomedical Sciences in 2019 from Marshall University. Her doctoral research focused on the application of capsaicinoids in the treatment of lung cancer. Currently she is a Scientist II at Bioagilytix Laboratories, a contract research lab that focuses on the development and validation of immunogenicity assays for a variety of pharmaceutical companies.
Kathleen C. Brown studied at Marshall University where she received both her Bachelor’s degree in Biology and Master’s degree in Forensic Science with emphases in forensic chemistry and digital forensics. She is currently performing research in the laboratory of Dr. Monica Valentovic at the Joan C. Edwards School of Medicine at Marshall University.
Rama S. Gadepalli is Senior Scientist in Medicinal Chemistry and the Core Research Facility Manager of the NIH-COBRE supported Chemistry Core in the Department of BioMolecular Sciences at the University of Mississippi. He received his Master’s degree in Pharmaceutical Chemistry from Jadavpur University in 1989 and Ph.D. in Medicinal Chemistry from Kakatiya University in 1994 supervised by Professor V. Malla Reddy. After 8 years of teaching and research in India and a short stay as Visiting Scientist at the University of Regensburg, Germany, he joined the University of Mississippi in 2001. His research interests are in synthetic medicinal chemistry and the design and development of bioactive molecules.
Sarah L. Miles received her Doctorate degree in Biomedical Science from the Joan C. Edwards School of Medicine at Marshall University, Huntington, WV, where she is currently a Research Assistant Professor in the Department of Biomedical Sciences. Her research interests include identifying the causative serum borne factor and molecular mechanism of the paraneoplastic syndrome Bilateral Diffuse Uveal Melanocytic Proliferation (BDUMP). Her studies focus on identifying critical targets to develop clinical molecular diagnostic and therapeutic procedures for BDUMP patients. Her additional research involves the use of vitamin C and other natural compounds as adjuvant or supplemental therapeutics and identify the mechanisms by which they may work to increase the efficacy of chemotherapeutic drugs, reduce side effects, and improve patient response and tolerance to chemotherapy.
John M. Rimoldi is Professor of Medicinal Chemistry & Environmental Toxicology and Research Professor of The Research Institute of Pharmaceutical Sciences in the School of Pharmacy at The University of Mississippi. He serves as Director of Research and Graduate Affairs and Director of the NIH-COBRE supported Chemistry and DM/PK Core Facility in the Department of BioMolecular Sciences. He obtained his B.S. in Chemistry from the University of Pittsburgh and Ph.D. in Chemistry from Virginia Tech and was a postdoctoral fellow at the Peters Center for the Study of Parkinson’s Disease and Disorders of the CNS. His research activities include programs in drug discovery and development, natural products chemistry, environmental chemistry, and drug metabolism.
Gary O. Rankin received his Doctorate degree in Medicinal Chemistry in 1976 from the University of Mississippi. He is currently Professor and Chair of the Department of Biomedical Sciences and Vice Dean for Basic Sciences in the Joan C. Edwards School of Medicine at Marshall University. His research interests are primarily focused on the nephrotoxicity induced by halogenated aromatic hydrocarbons and the use of natural products as cancer therapeutics. He is also contributing to understanding how pharmacogenetics contributes to overdose deaths from narcotic analgesics.
Monica A. Valentovic obtained a Bachelor of Science degree in Chemistry from Michigan Technological University in 1978. She obtained a Master of Science in Pharmacology from the University of Toledo in 1980 and a Ph.D. from the University of Kentucky School of Pharmacy in 1983. Dr. Valentovic is currently a Professor in the Department of Biomedical Sciences, Joan C. Edwards School of Medicine at Marshall University. She is also the Toxicology Research Cluster Coordinator. Her research interests are focused on examining the mechanisms of drug and environmental chemical mediated renal and hepatic toxicity. Her research also examines the impact of natural products on reducing the adverse effects of cancer chemotherapy agents.
Maria T. Tirona earned her medical degree from the University of the East Ramon Magsaysay Memorial Medical Center School in the Philippines. She completed her Internal Medicine Residency training at Mercy Hospital in Buffalo, NY, and Hematology-Oncology Fellowship training at Emory University in Atlanta, GA. She is currently the Section Chief of the Division of Hematology-Oncology and Professor of Medicine at Marshall University School of Medicine in Huntington, WA. She has at least 30 years Cancer Clinical Trials/Research experience and currently serves as one of the Site Principal Investigators of The ALLIANCE (a major cancer clinical trials cooperative group in the U.S.). Her main interest is in breast cancer management and research.
Paul T. Finch earned his Medical degree at Temple University School of Medicine in Philadelphia, PA. He completed his residency in Pediatrics at the University of Tennessee Medical Center (at LeBonheur Children’s Hospital) in Memphis, TN, and his fellowship training at the Children’s Hospital of Pittsburgh, UPMC, Pittsburgh, PA. He is currently the Head of Pediatric Hematology-Oncology section of the Department of Oncology at the Edward’s Comprehensive Cancer Center and Hoops’ Family Children’s Hospital at Marshall University School of Medicine in Huntington, WV. He is the Lead Investigator of the Children’s Oncology Group (COG) subsite of Nationwide Children’s Hospital, responsible for the oversight of clinical trials. His main interest is in improving the access to clinical trials for local patients.
Joshus A. Hess earned his Medical degree at Marshall University’s Joan C. Edwards School of Medicine in Huntington, WV. He completed his residency in Internal Medicine and Pediatric at the Marshall University Medical Center in Huntington, WV, and his fellowship training in Pediatric Hematology-Oncology at the St. Jude Children’s Research Hospital in Memphis, TN. He is currently with the Pediatric Hematology-Oncology section of the Department of Oncology at the Edward’s Comprehensive Cancer Center and Hoops’ Family Children’s Hospital as well as Assistant Professor of Pediatrics at Marshall University Joan C. Edwards School of Medicine in Huntington, WV. His research interests involve cancer metabolism and pharmacology.
Piyali Dasgupta is an Associate Professor in the Department of Biomedical Sciences, Joan C. Edwards School of Medicine at Marshall University. She received her Bachelor’s degree in Chemistry from the University of Delhi, India, in 1992 and Master’s degree in Chemistry from the Indian Institute of Technology, India, in 1994. Thereafter, she obtained her Ph.D. degree in Life Sciences from the National Institute of Immunology, Delhi, India. Her research interests include studying the anticancer activity of nutritional compounds in human lung cancer.
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c01679
Contributor Information
Stephen D. Richbart, Department of Biomedical Sciences, Toxicology Research Cluster, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States
Jamie R. Friedman, BioAgilytix Inc., Durham, North Carolina 27713, United States
Kathleen C. Brown, Department of Biomedical Sciences, Toxicology Research Cluster, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States
Rama S. Gadepalli, Department of Biomolecular Sciences, School of Pharmacy, Thad Cochran Research Center, University of Mississippi, University, Mississippi 38677, United States
Sarah L. Miles, Department of Biomedical Sciences, Toxicology Research Cluster, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States
John M. Rimoldi, Department of Biomolecular Sciences, School of Pharmacy, Thad Cochran Research Center, University of Mississippi, University, Mississippi 38677, United States
Gary O. Rankin, Department of Biomedical Sciences, Toxicology Research Cluster, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States
Monica A. Valentovic, Department of Biomedical Sciences, Toxicology Research Cluster, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States
Maria T. Tirona, Department of Hematology-Oncology, Edwards Cancer Center, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States
Paul T. Finch, Department of Oncology, Edwards Cancer Center, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States
Joshua A. Hess, Department of Oncology, Edwards Cancer Center, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States
Piyali Dasgupta, Department of Biomedical Sciences, Toxicology Research Cluster, Joan C. Edwards School of Medicine, Marshall University, Huntington, West Virginia 25755, United States.
REFERENCES
- (1).Blair NT; Carvacho I; Chaudhuri D; Clapham DE; DeCaen P; Delling M; Doerner JF; Fan L; Ha K; Jordt SE; Julius D; Kahle KT; Liu B; McKemy D; Nilius B; Oancea E; Owsianik G; Riccio A; Sah R; Stotz SC; Tian J; Tong D; Van den Eynde C; Vriens J; Wu L-J; Xu H; Yue L; Zhang X; Zhu MX Transient receptor potential channels. (version 2019.4) in the IUPHAR/BPS guide to pharmacology database. IUPHAR/BPS Guide to Pharmacology CITE 2019, DOI: 10.2218/gtopdb/F78/2019.4. [DOI] [Google Scholar]
- (2).Frias B; Merighi A Capsaicin, nociception and pain. Molecules 2016, 21 (6), 797–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Caterina MJ; Schumacher MA; Tominaga M; Rosen TA; Levine JD; Julius D The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [DOI] [PubMed] [Google Scholar]
- (4).Benitez-Angeles M; Morales-Lazaro SL; Juarez-Gonzalez E; Rosenbaum T TRPV1: structure, endogenous agonists, and mechanisms. Int. J. Mol. Sci 2020, 21 (10), 3421–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fattori V; Hohmann MS; Rossaneis AC; Pinho-Ribeiro FA; Verri WA Capsaicin: current understanding of its mechanisms and therapy of pain and other pre-clinical and clinical uses. Molecules 2016, 21 (7), 844–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hall OM; Broussard A; Range T; Carroll Turpin MA; Ellis S; Lim VM; Cornett EM; Kaye AD Novel agents in neuropathic pain, the role of capsaicin: pharmacology, efficacy, side effects, different preparations. Current Pain and Headache Reports 2020, 24 (9), 53–65. [DOI] [PubMed] [Google Scholar]
- (7).Chapa-Oliver AM; Mejia-Teniente L Capsaicin: from plants to a cancer-suppressing agent. Molecules 2016, 21 (8), 931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Clark R; Lee SH Anticancer properties of capsaicin against human cancer. Anticancer Res. 2016, 36 (3), 837–843. [PubMed] [Google Scholar]
- (9).Diaz-Laviada I; Rodriguez-Henche N The potential antitumor effects of capsaicin. Prog. Drug Res 2014, 68, 181–208. [DOI] [PubMed] [Google Scholar]
- (10).Srinivasan K Biological activities of red pepper (Capsicum annuum) and its pungent principle capsaicin: a review. Crit. Rev. Food Sci. Nutr 2016, 56 (9), 1488–1500. [DOI] [PubMed] [Google Scholar]
- (11).Lau JK; Brown KC; Dom AM; Dasgupta P Capsaicin: Potential Applications in Cancer Therapy. In Nutrition and Cancer; Claudio PP, Niles RM, Eds.; Bentham Press Inc.: London, U.K., 2012; pp 15–25. [Google Scholar]
- (12).Min JK; Han KY; Kim EC; Kim YM; Lee SW; Kim OH; Kim KW; Gho YS; Kwon YG Capsaicin inhibits in vitro and in vivo angiogenesis. Cancer Res. 2004, 64 (2), 644–651. [DOI] [PubMed] [Google Scholar]
- (13).Pyun BJ; Choi S; Lee Y; Kim TW; Min JK; Kim Y; Kim BD; Kim JH; Kim TY; Kim YM; Kwon YG Capsiate, a nonpungent capsaicin-like compound, inhibits angiogenesis and vascular permeability via a direct inhibition of Src kinase activity. Cancer Res. 2008, 68 (1), 227–235. [DOI] [PubMed] [Google Scholar]
- (14).Friedman JR; Richbart SD; Merritt JC; Brown KC; Denning KL; Tirona MT; Valentovic MA; Miles SL; Dasgupta P Capsaicinoids: Multiple effects on angiogenesis, invasion and metastasis in human cancers. Biomed. Pharmacother 2019, 118, 109317–109326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Friedman JR; Nolan NA; Brown KC; Miles SL; Akers AT; Colclough KW; Seidler JM; Rimoldi JM; Valentovic MA; Dasgupta P Anticancer activity of natural and synthetic capsaicin analogs. J. Pharmacol. Exp. Ther 2018, 364 (3), 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Arzuman L; Beale P; Yu JQ; Huq F Synthesis of tris(quinoline)monochloroplatinum(II) chloride and its activity alone and in combination with capsaicin and curcumin in human ovarian cancer cell lines. Anticancer Res. 2016, 36 (6), 2809–2818. [PubMed] [Google Scholar]
- (17).Venier NA; Colquhoun AJ; Sasaki H; Kiss A; Sugar L; Adomat H; Fleshner NE; Klotz LH; Venkateswaran V Capsaicin: a novel radio-sensitizing agent for prostate cancer. Prostate 2015, 75 (2), 113–125. [DOI] [PubMed] [Google Scholar]
- (18).Huh HC; Lee SY; Lee SK; Park NH; Han IS Capsaicin induces apoptosis of Cisplatin-resistant stomach cancer cells by causing degradation of cisplatin-inducible Aurora-a protein. Nutr. Cancer 2011, 63 (7), 1095–1103. [DOI] [PubMed] [Google Scholar]
- (19).Hammer J Effect of repeated capsaicin ingestion on intestinal chemosensation and mechanosensation. Aliment. Pharmacol. Ther 2006, 24 (4), 679–686. [DOI] [PubMed] [Google Scholar]
- (20).Drewes AM; Schipper KP; Dimcevski G; Petersen P; Gregersen H; Funch-Jensen P; Arendt-Nielsen L Gut pain and hyperalgesia induced by capsaicin: a human experimental model. Pain 2003, 104 (1–2), 333–341. [DOI] [PubMed] [Google Scholar]
- (21).Bley K; Boorman G; Mohammad B; McKenzie D; Babbar S A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol. Pathol 2012, 40 (6), 847–873. [DOI] [PubMed] [Google Scholar]
- (22).Georgescu SR; Sarbu MI; Matei C; Ilie MA; Caruntu C; Constantin C; Neagu M; Tampa M Capsaicin: friend or foe in skin cancer and other related malignancies? Nutrients 2017, 9 (12), 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Du Y; Lv Y; Zha W; Hong X; Luo Q Chili consumption and risk of gastric cancer: a meta-analysis. Nutr. Cancer 2020, 2, 1–10. [DOI] [PubMed] [Google Scholar]
- (24).Huang XF; Xue JY; Jiang AQ; Zhu HL Capsaicin and its analogues: structure-activity relationship study. Curr. Med. Chem 2013, 20 (21), 2661–2672. [DOI] [PubMed] [Google Scholar]
- (25).Thomas KC; Ethirajan M; Shahrokh K; Sun H; Lee J; Cheatham TE 3rd; Yost GS; Reilly CA Structure-activity relationship of capsaicin analogs and transient receptor potential vanilloid 1-mediated human lung epithelial cell toxicity. J. Pharmacol. Exp. Ther 2011, 337 (2), 400–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Di Marzo V; Bisogno T; De Petrocellis L; Melck D; Martin BR Cannabimimetic fatty acid derivatives: the anandamide family and other endocannabinoids. Curr. Med. Chem 1999, 6 (8), 721–744. [PubMed] [Google Scholar]
- (27).Melck D; Bisogno T; De Petrocellis L; Chuang H; Julius D; Bifulco M; Di Marzo V Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem. Biophys. Res. Commun 1999, 262 (1), 275–284. [DOI] [PubMed] [Google Scholar]
- (28).Friedman JR; Gadapalli RS; Akers AT; Nolan NA; Brown KC; Colclough KW; Miles SL; Rimoldi JM; Dasgupta P Anti-cancer Activity of Non-pungent Capsaicin Analogs: a Structure-Activity Study. In Experimental Biology Conference, San Diego, CA, April 21–25, 2018; Abstract No. 407.1. [Google Scholar]
- (29).Kobata K; Saito K; Tate H; Nashimoto A; Okuda H; Takemura I; Miyakawa K; Takahashi M; Iwai K; Watanabe T Long-chain N-vanillyl-acylamides from capsicum oleoresin. J. Agric. Food Chem 2010, 58 (6), 3627–3631. [DOI] [PubMed] [Google Scholar]
- (30).Kobata K; Takemura I; Tago G; Moriya T; Kubota K; Nakatani S; Wada M; Watanabe T Formation of long-chain N-vanillyl-acylamides from plant oils. Biosci., Biotechnol., Biochem 2014, 78 (7), 1242–1245. [DOI] [PubMed] [Google Scholar]
- (31).Janusz JM; Buckwalter BL; Young PA; LaHann TR; Farmer RW; Kasting GB; Loomans ME; Kerckaert GA; Maddin CS Vanilloids. 1. Analogs of capsaicin with antinociceptive and antiinflammatory activity. J. Med. Chem 1993, 36 (18), 2595–2604. [DOI] [PubMed] [Google Scholar]
- (32).Takao K; Noguchi K; Hashimoto Y; Shirahata A; Sugita Y Synthesis and evaluation of fatty acid amides on the N-oleoylethanolamide-like activation of peroxisome proliferator activated receptor alpha. Chem. Pharm. Bull 2015, 63 (4), 278–285. [DOI] [PubMed] [Google Scholar]
- (33).Appendino G; Minassi A; Morello AS; De Petrocellis L; Di Marzo V N-Acylvanillamides: development of an expeditious synthesis and discovery of new acyl templates for powerful activation of the vanilloid receptor. J. Med. Chem 2002, 45 (17), 3739–3745. [DOI] [PubMed] [Google Scholar]
- (34).Dasse O; Mahadevan A; Han L; Martin BR; Marzo VD; Razdan RK The synthesis of N-Vanillyl-arachidonoyl-amide (arvanil) and its analogs: an improved procedure for the synthesis of the key synthon methyl-14-hydroxy-(all-cis-)-5,8,11-tetradecatrienoate. Tetrahedron 2000, 56 (47), 9195–9202. [Google Scholar]
- (35).Carpino M 1-Hydroxy-7-azabenzotrizole: An efficient peptide coupling agent. J. Am. Chem. Soc 1993, 115 (10), 4397–4399. [Google Scholar]
- (36).Schiano Moriello A; Lopez Chinarro S; Novo Fernandez O; Eras J; Amodeo P; Canela-Garayoa R; Vitale RM; Di Marzo V; De Petrocellis L Elongation of the hydrophobic chain as a molecular switch: discovery of capsaicin derivatives and endogenous lipids as potent transient receptor potential vanilliod channel 2 antagonists. J. Med. Chem 2018, 61 (18), 8255–8281. [DOI] [PubMed] [Google Scholar]
- (37).Reyes-Duarte D; Castillo E; Martinez R; Lopez-Munguia A Lipase-catalyzed synthesis of olvanil in organic solvents. Biotechnol. Lett 2002, 24, 2057–2061. [Google Scholar]
- (38).Basso A; Serban S Industrial applications of immobilized enzymes. Mol. Catalysis 2019, 479, 110607. [Google Scholar]
- (39).Homaei AA; Sariri R; Vianello F; Stevanato R Enzyme immobilization: an update. J. Chem. Biol 2013, 6 (4), 185–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Diaz-Vidal T; Armenta-Perez VP; Rosales-Rivera LC; Mateos-Diaz JC; Rodriguez JA Cross-linked enzyme aggregates of recombinant Candida antarctica lipase B for the efficient synthesis of olvanil, a nonpungent capsaicin analogue. Biotechnol. Prog 2019, 35 (4), No. e2807. [DOI] [PubMed] [Google Scholar]
- (41).Takahata K Anti-tumor Pharmaceutical Composition comprising N-vanillyl Fatty Acid Amide. U.S. Patent Application 20040110844 A1, April 25, 2006.
- (42).Brandao LMS; Barbosa MS; Souza RL; Pereira MM; Lima AS; Soares CMF Lipase activation by molecular bioimprinting: the role of interactions between fatty acids and enzyme active site. Biotechnol. Prog 2020, No. e3064. [DOI] [PubMed] [Google Scholar]
- (43).Yang H; Zhang W Surfactant imprinting hyperactivated immobilized lipase as efficient biocatalyst for biodiesel production from waste cooking oil. Catalysts 2019, 9 (11), 914–927. [Google Scholar]
- (44).Bifulco M; Di Marzo V Targeting the endocannabinoid system in cancer therapy: a call for further research. Nat. Med 2002, 8, 547–550. [DOI] [PubMed] [Google Scholar]
- (45).De Petrocellis L; Bisogno T; Ligresti A; Bifulco M; Melck D; Di Marzo V Effect on cancer cell proliferation of palmitoylethanolamide, a fatty acid amide interacting with both the cannabinoid and vanilloid signalling systems. Fundam. Clin. Pharmacol 2002, 16 (4), 297–302. [DOI] [PubMed] [Google Scholar]
- (46).de Lago E; Urbani P; Ramos JA; Di Marzo V; Fernandez-Ruiz J Arvanil, a hybrid endocannabinoid and vanilloid compound, behaves as an antihyperkinetic agent in a rat model of Huntington’s disease. Brain Res. 2005, 1050 (1–2), 210–216. [DOI] [PubMed] [Google Scholar]
- (47).Di Marzo V; Griffin G; De Petrocellis L; Brandi I; Bisogno T; Williams W; Grier MC; Kulasegram S; Mahadevan A; Razdan RK; Martin BR A structure/activity relationship study on arvanil, an endocannabinoid and vanilloid hybrid. J. Pharmacol. Exp. Ther 2002, 300 (3), 984–991. [DOI] [PubMed] [Google Scholar]
- (48).Klopman G; Zhu H Recent methodologies for the estimation of N-octanol/water partition coefficients and their use in the prediction of membrane transport properties of drugs. Mini-Rev. Med. Chem 2005, 5 (2), 127–133. [DOI] [PubMed] [Google Scholar]
- (49).Kuwata M; Lee W-C 1-octanol-water partitioning as a classifier of water soluble organic matters: Implication for solubility distribution. Aerosol Sci. Technol 2017, 51 (5), 602–613. [Google Scholar]
- (50).Sietsema WK; Berman EF; Farmer RW; Maddin CS The antinociceptive effect and pharmacokinetics of olvanil following oral and subcutaneous dosing in the mouse. Life Sci. 1988, 43 (17), 1385–1391. [DOI] [PubMed] [Google Scholar]
- (51).Wehmeyer KR; Kasting GB; Powell JH; Kuhlenbeck DL; Underwood RA; Bowman LA Application of liquid chromatography with on-line radiochemical detection to metabolism studies on a novel class of analgesics. J. Pharm. Biomed. Anal 1990, 8 (2), 177–183. [DOI] [PubMed] [Google Scholar]
- (52).Kasting GB; Francis WR; Bowman LA; Kinnett GO Percutaneous absorption of vanilloids: in vivo and in vitro studies. J. Pharm. Sci 1997, 86 (1), 142–146. [DOI] [PubMed] [Google Scholar]
- (53).Marquez N; De Petrocellis L; Caballero FJ; Macho A; Schiano-Moriello A; Minassi A; Appendino G; Munoz E; Di Marzo V Iodinated N-acylvanillamines: potential “multiple-target” anti-inflammatory agents acting via the inhibition of T-cell activation and antagonism at vanilloid TRPV1 channels. Mol. Pharmacol 2006, 69 (4), 1373–1382. [DOI] [PubMed] [Google Scholar]
- (54).Jacobsson SO; Wallin T; Fowler CJ Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J. Pharmacol. Exp. Ther 2001, 299 (3), 951–959. [PubMed] [Google Scholar]
- (55).Di Marzo V; Melck D; De Petrocellis L; Bisogno T Cannabimimetic fatty acid derivatives in cancer and inflammation. Prostaglandins Other Lipid Mediators 2000, 61 (1–2), 43–61. [DOI] [PubMed] [Google Scholar]
- (56).Malfitano AM; Matarese G; Pisanti S; Grimaldi C; Laezza C; Bisogno T; Di Marzo V; Lechler RI; Bifulco M Arvanil inhibits T lymphocyte activation and ameliorates autoimmune encephalomyelitis. J. Neuroimmunol 2006, 171 (1–2), 110–119. [DOI] [PubMed] [Google Scholar]
- (57).Sancho R; de la Vega L; Appendino G; Di Marzo V; Macho A; Munoz E The CB1/VR1 agonist arvanil induces apoptosis through an FADD/caspase-8-dependent pathway. Br. J. Pharmacol 2003, 140 (6), 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Guindon J; Hohmann AG The endocannabinoid system and cancer: therapeutic implication. Br. J. Pharmacol 2011, 163 (7), 1447–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Morris A; Soliman E; Vvan Dross R; Burns C Structural modification of the chemotherapeutic anandamide: designing anti-cancer agents and investigating their COX-2 metabolic products. In American Association of Cancer Research, Washington, DC, April 1–5, 2017; Abstract No. 2193. [Google Scholar]
- (60).Li W; Moore BM The effect of arvanil on on prostate cancer cells studied by whole cell high resolution magic angle spinning NMR. Mod. Chem. Appl 2014, 2, 1000119. [Google Scholar]
- (61).Stock K; Kumar J; Synowitz M; Petrosino S; Imperatore R; Smith ES; Wend P; Purfurst B; Nuber UA; Gurok U; Matyash V; Walzlein JH; Chirasani SR; Dittmar G; Cravatt BF; Momma S; Lewin GR; Ligresti A; De Petrocellis L; Cristino L; Di Marzo V; Kettenmann H; Glass R Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med 2012, 18, 1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Wang S; Zimmermann S; Parikh K; Mansfield AS; Adjei AA Current diagnosis and management of small-cell lung cancer. Mayo Clin. Proc 2019, 94 (8), 1599–1622. [DOI] [PubMed] [Google Scholar]
- (63).Yang S; Zhang Z; Wang Q Emerging therapies for small cell lung cancer. J. Hematol Oncol 2019, 12, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Jiang WG; Sanders AJ; Katoh M; Ungefroren H; Gieseler F; Prince M; Thompson SK; Zollo M; Spano D; Dhawan P; Sliva D; Subbarayan PR; Sarkar M; Honoki K; Fujii H; Georgakilas AG; Amedei A; Niccolai E; Amin A; Ashraf SS; Ye L; Helferich WG; Yang X; Boosani CS; Guha G; Ciriolo MR; Aquilano K; Chen S; Azmi AS; Keith WN; Bilsland A; Bhakta D; Halicka D; Nowsheen S; Pantano F; Santini D Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin. Cancer Biol 2015, 35 (Suppl), S244–S275. [DOI] [PubMed] [Google Scholar]
- (65).Krakhmal NV; Zavyalova MV; Denisov EV; Vtorushin SV; Perelmuter VM Cancer invasion: patterns and mechanisms. Acta. Naturae 2015, 7 (2), 17–28. [PMC free article] [PubMed] [Google Scholar]
- (66).Hurley JD; Akers AT; Friedman JR; Nolan NA; Brown KC; Dasgupta P Non-pungent long chain capsaicin-analogs arvanil and olvanil display better anti-invasive activity than capsaicin in human small cell lung cancers. Cell Adh. Migr 2017, 11 (1), 80–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Luviano A; Aguiniga-Sanchez I; Demare P; Tiburcio R; Ledesma-Martinez E; Santiago-Osorio E; Regla I Antineoplastic activity of rinvanil and phenylacetylrinvanil in leukaemia cell lines. Oncol. Lett 2014, 7 (5), 1651–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Sanchez-Sanchez L; Alvarado-Sansininea JJ; Escobar ML; Lopez-Munoz H; Hernandez-Vazquez JM; Monsalvo-Montiel I; Demare P; Regla I; Weiss-Steider B Evaluation of the antitumour activity of rinvanil and phenylacetylrinvanil on the cervical cancer tumour cell lines HeLa, CaSKi and ViBo. Eur. J. Pharmacol 2015, 758, 129–136. [DOI] [PubMed] [Google Scholar]
- (69).Tuoya; Baba N; Shimoishi Y; Murata Y; Tada M; Koseki M; Takahata K Apoptosis induction by dohevanil, a DHA substitutive analog of capsaicin, in MCF-7 cells. Life Sci. 2006, 78 (13), 1515–1519. [DOI] [PubMed] [Google Scholar]
- (70).Jin Y; Ishihata K; Kajiyama S; Fukusaki E; Kobayashi A; Baba N; Takahata K Effect of capsaicin and N-docosahexaenoyl-vanillylamide on growth of taxol-tolerant HeLa cells. Jpn. J. Food Chem 2002, 9 (2), 50–53. [Google Scholar]
- (71).Appendino G; Cascio MG; Bacchiega S; Moriello AS; Minassi A; Thomas A; Ross R; Pertwee R; De Petrocellis L; Di Marzo V First “hybrid” ligands of vanilloid TRPV1 and cannabinoid CB2 receptors and non-polyunsaturated fatty acid-derived CB2-selective ligands. FEBS Lett. 2006, 580 (2), 568–574. [DOI] [PubMed] [Google Scholar]
- (72).Muller C; Morales P; Reggio PH Cannabinoid ligands targeting TRP channels. Front. Mol. Neurosci 2019, 11, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Han J; Zhang S; Liu X; Xiao C Fabrication of capsaicin emulsions: improving the stability of the system and relieving the irritation to the gastrointestinal tract of rats. J. Sci. Food Agric 2020, 100 (1), 129–138. [DOI] [PubMed] [Google Scholar]
- (74).Lu M; Chen C; Lan Y; Xiao J; Li R; Huang J; Huang Q; Cao Y; Ho CT Capsaicin-the major bioactive ingredient of chili peppers: bio-efficacy and delivery systems. Food Funct. 2020, 11 (4), 2848–2860. [DOI] [PubMed] [Google Scholar]
- (75).Zhang S; Wang D; Huang J; Hu Y; Xu Y Application of capsaicin as a potential new therapeutic drug in human cancers. J. Clin. Pharm. Ther 2020, 45 (1), 16–28. [DOI] [PubMed] [Google Scholar]
- (76).Arrieta O; Lara-Mejia L; Zatarain-Barron ZL Carboplatin plus etoposide or topotecan for small-cell lung cancer. Lancet Oncol. 2020, 21 (9), 1132–1134. [DOI] [PubMed] [Google Scholar]
- (77).Qin A; Kalemkerian GP Treatment options for relapsed small-cell lung cancer: what progress have we made? J. Oncol. Pract 2018, 14 (6), 369–370. [DOI] [PubMed] [Google Scholar]