

Abstract
Background and Aims:
Cholestasis is characterized by intrahepatic accumulation of bile constituents, including bile acids (BAs), which promote liver damage. The apical sodium-dependent BA transporter (ASBT) plays an important role in BA reabsorption and signaling in ileum, bile ducts, and kidneys. Our aim was to investigate the pharmacokinetics and pharmacological activity of A3907, an oral and systemically available ASBT inhibitor in experimental mouse models of cholestasis. In addition, the tolerability, pharmacokinetics, and pharmacodynamics of A3907 were examined in healthy humans.
Approach and Results:
A3907 was a potent and selective ASBT inhibitor in vitro. In rodents, orally administered A3907 distributed to the ASBT-expressing organs, that is, ileum, liver, and kidneys, and dose dependently increased fecal BA excretion. A3907 improved biochemical, histological, and molecular markers of liver and bile duct injury in Mdr2 −/− mice and also had direct protective effects on rat cholangiocytes exposed to cytotoxic BA concentrations in vitro. In bile duct ligated mice, A3907 increased urinary BA elimination, reduced serum BA levels, and prevented body weight loss, while improving markers of liver injury. A3907 was well tolerated and demonstrated target engagement in healthy volunteers. Plasma exposure of A3907 in humans was within the range of systemic concentrations that achieved therapeutic efficacy in mouse.
Conclusions:
The systemic ASBT inhibitor A3907 improved experimental cholestatic disease by targeting ASBT function at the intestinal, liver, and kidney levels, resulting in marked clearance of circulating BAs and liver protection. A3907 is well tolerated in humans, supporting further clinical development for the treatment of cholestatic liver diseases.
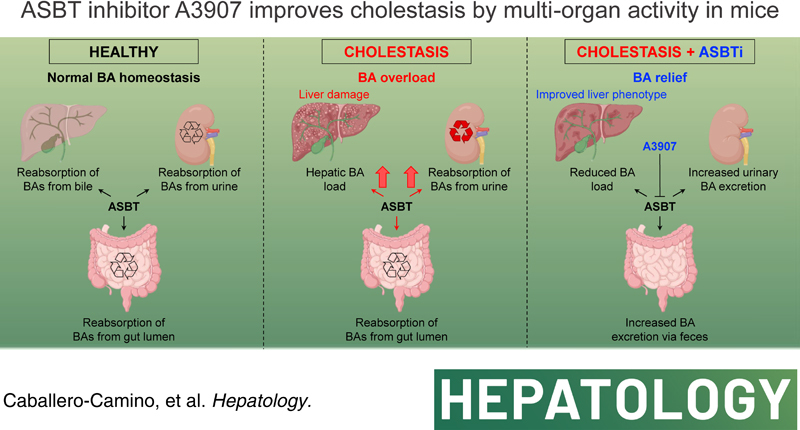
INTRODUCTION
Cholestasis is a severe clinical manifestation of adult and pediatric cholangiopathies of diverse etiologies, including genetic, obstructive, drug-induced, or immune-mediated biliary diseases.1,2 Cholestasis is characterized by decreased or obstructed bile flow, leading to hepatic accumulation of bile acids (BAs) and other bile-derived toxic substances, frequently inducing pruritus.3,4 Cholestatic liver diseases lack effective treatments and can progress to advanced stages (ie, cirrhosis), increasing the risk for developing hepatobiliary malignancies and liver failure.5,6
In human cholangiopathies, alterations in BA synthesis, biotransformation, and/or transport may result in pathological conditions, including severe cholestasis and end-stage liver disease, where liver transplantation remains as the only potential curative option. Under physiological conditions, a major fraction of the BAs reaching the intestine (~95%) is efficiently recovered and recirculated to the liver via the portal vein. This occurs through a complex process involving several BA transporters expressed in different cell types.7,8 The apical sodium-dependent BA transporter (ASBT), highly expressed in the apical membrane of ileal enterocytes, plays a major role in the reabsorption of BAs from the intestinal lumen, and thus for the regulation of BA dynamics and homeostasis.7 Interestingly, ASBT is also expressed in the bile ducts and renal proximal tubuli.9 However, the biliary and renal role of ASBT is less clear and may involve cholehepatic shunting and/or cholangiocyte signaling, as well as renal reabsorption of BAs from filtered primary urine.10
Serum BAs are typically elevated in cholestasis, reflecting hepatic BA overload.11 The high levels of BAs in cholestatic liver diseases lead to damage in the liver parenchyma, which may ultimately also extend to extrahepatic tissues. In this regard, highly efficient ASBT-driven reabsorption of BAs in the intestine and kidneys significantly contributes to disease progression and severity. ASBT inhibitors (ASBTi) with restricted intestinal activity in humans have therefore been developed and found useful in several cholestatic conditions by reducing BA load.12–16 However, because of the minimal systemic bioavailability of currently available ASBTis, the therapeutic potential of multiorgan ASBT inhibition for the treatment of different forms of cholestasis has not been explored.
Herein, we hypothesize that multiorgan targeting of ASBT, including the ileum, bile ducts, and renal proximal tubuli, may halt or reverse the progression of cholestatic diseases. This approach might be potentially superior to locally acting ASBTis in severe cholestatic conditions where enterohepatic circulation of BAs is severely compromised. Therefore, the current study aimed to investigate the molecular and in vivo pharmacology of the novel orally bioavailable ASBTi A3907 in well-characterized experimental models of cholestatic liver disease [ie, Mdr2 −/− mice and mice subjected to bile duct ligation (BDL)]17 and investigate its tolerability, oral bioavailability, and pharmacodynamics in healthy human subjects.
METHODS
A3907 activity and selectivity in vitro
Mouse and human ASBT and Na+-taurocholate cotransport polypeptide (NTCP) transporters were experimentally overexpressed with recombinant plasmids in CHOK1 hamster ovary cells obtained from the American Type Culture Collection (ATCC). Further experimental details are provided in Supplemental Material, http://links.lww.com/HEP/F6.
Quantitative whole-body autoradiography
Quantitative whole-body autoradiography (QWBA) in phosphor screens was performed using male rats administrated a single oral dose of radiolabeled [14C]-A3907 or a gut-restricted ASBT inhibitor [14C]-A330918 with 10 and 7.8 MBq/kg radioactive doses, respectively. Experimental details are provided in Supplemental Material, http://links.lww.com/HEP/F6.
A3907 pharmacokinetics and biodistribution in mice and rats
The oral pharmacokinetic profile of A3907 in normal animals was analyzed in 3 rodent studies (Envigo Research Laboratories): (1) male Hsd:ICR (CD-1) adult mice, (2) male C57BL/6J adult mice, and (3) male adult Wistar rats. The study protocol was approved by Institutional Animal Ethics Committee (IAEC) (SYNGENE/IAEC/ 800/01-2017 and SYN/IAEC/937/05-2018). A3907 biodistribution was analyzed by liquid chromatography tandem mass spectrometry in samples from male C57BL/6J mice (7 weeks of age) (Janvier Labs) treated with daily doses of A3907 (3, 10, or 30 mg/kg; n = 10 per group) by oral gavage for 7 days. The experiments were covered by a license issued by the Danish committee for animal research (2013-15-2934-00784). The oral pharmacokinetic profile of A3907 and A3309 was analyzed at Syngene International Ltd. (Bangalore, India) in male adult C57BL/6 mice (Hylasco, Hyderabad) subjected to BDL surgery. Fecal BA content following 7-day treatment of A3907, A3309, or vehicle was also analyzed. The study protocol was approved by Institutional Animal Ethics Committee (IAEC) (SYNGENE/IAEC/1253/03-2021). Further experimental details are provided in Supplemental Material, http://links.lww.com/HEP/F6.
Pharmacodynamic studies in normal wild-type mice
Pharmacodynamic studies were performed in adult (13–14 weeks) C57BL/6N mice (Hylasco, Hyderabad, India) administered daily doses of A3907 or A3309 at 3 or 10 mg/kg/d, or vehicle [0.5% carboxymethylcellulose (CMC) + 0.1% Tween 80] by oral gavage for 8 days. The study was approved by the Institutional Animal Ethics Committee (IAEC) [Protocol No: Syngene/ IAEC /1064/07 -2019]. Detailed information provided in Supplemental Material, http://links.lww.com/HEP/F6.
Toxicology studies of A3907 in normal rats
Systemic toxicity was evaluated in Wistar Han rats (Charles River). Herein, 7- to 10-week-old male (n = 76) or female (n = 76) rats were administered daily oral doses of A3907 (25, 150, and 1000 mg/kg/d) for 13 weeks by oral gavage. Animals were euthanized, and samples from a standard set of organs and tissues were collected and examined by a certified pathologist following hematoxylin & eosin (H&E) staining.
Animal models of cholestasis
Experiments were performed in age-matched male mice on C57BL/6J genetic background (BDL mouse model)19 at the Biodonostia Health Research Institute (BHRI) and at Syngene International Ltd. Experiments were performed under the approval of the Animal Experimentation Ethics Committee of BHRI (PRO-AE-SS-223), and the Institutional Animal Ethics Committee at Syngene (SYNGENE/IAEC/1253/03-2021) respectively. (Bangalore, India) (pharmacokinetic profile) and on Abcb4 tm1Bor genetic background (Mdr2 −/− mouse)20,21 at Gubra (Hoersholm, Denmark). These experiments were covered by a license issued by the Danish committee for animal research (2013-15-2934-00784). Extended information provided in Supplemental Material, http://links.lww.com/HEP/F6.
Liver histology
For Mdr2 −/− mice, livers were fixed overnight in 4% paraformaldehyde, paraffin embedded, and sectioned (3 µm thickness). Tissue slides were stained with H&E or used for immunohistochemistry staining.
In the BDL mouse study, liver histology analysis and scoring were conducted as described.19 Briefly, formalin-fixed and paraffin-embedded slides were stained with H&E (MERCK) for the analysis of tissue morphology. Scores detailed in Supplemental Table S1 (http://links.lww.com/HEP/F6). Further details are included in Supplemental Material, http://links.lww.com/HEP/F6.
Serum, plasma and liver biochemistry
Blood was collected from the tail vein (Mdr2 −/− mice) or by cardiac puncture (BDL mice) for biochemical analysis. For liver biochemistry, whole liver was dissected and weighed, and a biopsy (~200 mg, <0.7×0.5 cm) was resected from the left lateral lobe. Detailed information of the methodology is provided in Supplemental Material, http://links.lww.com/HEP/F6.
BA and C4 analysis
BAs, and their intermediate/precursor 7α-hydroxy-4-cholesten-3-one (C4), were determined in plasma by liquid chromatography tandem mass spectrometry as described.22–24 In C57BL/6N wild-type (WT) mouse experiments, total BA levels were analyzed in serum (Cell Biolabs), stools (Cell Biolabs), and urine (Biovision) using commercial kits following manufacturer’s instructions. Further details are provided in Supplemental Material, http://links.lww.com/HEP/F6.
Cholangiocyte functional studies (apoptosis by flow cytometry)
Normal rat cholangiocytes were isolated from liver tissue of Wistar rats (Charles River Laboratories) at the BHRI (San Sebastian, Spain), and then cultured and characterized as we described.25 Cell apoptosis in vitro was assessed by flow cytometry as described in Supplemental Material, (http://links.lww.com/HEP/F6).
Immunoblotting
Analysis of ASBT expression in normal rat cholangiocytes was performed as described in Supplemental Material, (http://links.lww.com/HEP/F6).
RNA isolation and quantitative real-time PCR (RT-qPCR)
RNA was extracted from mouse liver and ileum tissues using the Maxwell RSC Instrument (Promega) following the manufacturer’s instructions and subjected to reverse transcription followed by RT-qPCR analysis in a CFX384 Touch Real-Time PCR Detection System (BioRad). Primer sequences employed for the RT-qPCR are indicated in Supplemental Table S2 (http://links.lww.com/HEP/F6). Details are outlined in Supplemental Material (http://links.lww.com/HEP/F6).
RNAseq analysis
RNA sequencing (RNAseq) was performed on RNA extracts from terminal liver samples (15 mg fresh tissue), as described in detail elsewhere.26 Untreated WT mice for analysis of Mdr2 −/− mouse data as control correspond to stock liver samples from age-matched mice at Gubra. Additional details are provided in Supplemental Material (http://links.lww.com/HEP/F6).
Safety, tolerability, pharmacokinetics, and pharmacodynamic studies in healthy subjects
This phase I study (Protocol Reference Number: A3907-001, Covance Study Number: 8445784, EudraCT Number: 2020‐004423‐17) was sponsored and designed by Albireo AB, Sweden, and conducted in accordance with both the Declarations of Helsinki and Istanbul and approved by the North-East - York Research Ethics Committee (Newcastle-upon-Tyne, UK). Informed consent was given in writing by all subjects. The study was conducted at a single site in the UK (Principal Investigator: Ashley Brooks, MB ChB, Associate Medical Director, Labcorp Clinical Research Unit Ltd., UK). Besides standard safety parameters, additional pharmacodynamic analyses included LDLc, serum BAs (total and individual), plasma C4 and FGF-19, and urine BAs. See Supplemental Material (http://links.lww.com/HEP/F6) for further information.
Statistical analysis
GraphPad Prism 8 (GraphPad Software) was used to perform the statistical analysis. Normality of the data set was assessed by Shapiro-Wilk test. For comparisons of 2 independent groups, parametric Student t test or nonparametric Mann-Whitney test were employed. When more than 2 related groups were compared, parametric 1-way ANOVA with Dunnett post hoc test or nonparametric Kruskal-Wallis test followed by Dunn multiple comparison test were used. For statistical analysis of the time-course data, 2-way ANOVA with Tukey multiple comparisons post hoc test was applied. Gene set analysis was conducted with the R package PIANO version 1.18.1 using the Stouffer method, andp-values were corrected for multiple testing using the Benjamini-Hochberg method (False Discovery Rate, p < 0.05). Data in bar graphs are expressed as mean ± SEM. Significance was defined as p < 0.05.
RESULTS
A3907 is a potent, selective, and orally available ASBT inhibitor that distributes to the intestine, liver, and kidneys in rodents
A3907 is a potent and selective inhibitor of both mouse and human ASBT, with similar potency and selectivity as the gut-restricted ASBT inhibitor A330918 (Figure 1A). Oral administration of A3907 (10 mg/kg) to rodents resulted in high exposure in plasma (Supplemental Figure S1A, http://links.lww.com/HEP/F6). A3907 pharmacokinetic parameters in different species, when dosed with 10 mg/kg, were Cmax 558-655 (ng/mL), Tmax 2.67–5.33 (h), AUCinf 4300-8250 (h*ng/mL), 30%–60% oral bioavailability, and terminal half-life (t 1/2) 4.49–7.4 (h). Moreover, oral administration of incrementing doses of A3907 (3, 10, and 30 mg/kg) in mice demonstrated a dose-dependent increase of compound concentrations in feces, serum, bile, kidneys, and liver, whereas low levels of A3907 were present in urine and detected only at the highest dose (Supplemental Figure S1B, http://links.lww.com/HEP/F6).
FIGURE 1.
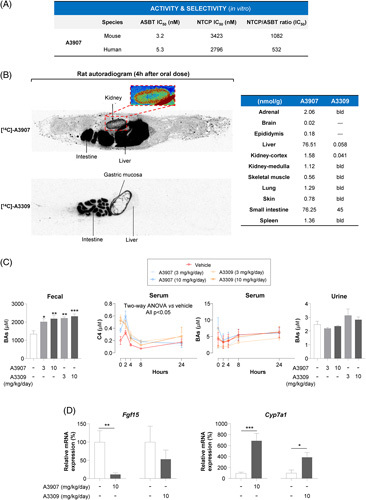
Activity, selectivity, biodistribution, and pharmacodynamics of A3907 and A3309 in healthy rodents. (A) Human or mouse ASBT and NTCP transporters were independently overexpressed in CHOK1 cells. A3907 was tested at 10 different concentrations to calculate the average half maximal inhibitory concentration (IC50) value for each transporter. (B) Representative images of whole-body autoradiogram 4 hours after a single oral administration of [14C]-A3907 (10 MBq/kg) or [14C]-A3309 (7.8 MBq/kg) to male rats (separate experiments), enlarged autoradiogram section of the kidney (black or red/pink regions represent areas with higher concentrations of radioactivity), and quantitative analysis of the concentration of each compound in different tissues. bld, below limit of detection. (C) Fecal BA concentrations in feces collected between days 6 and 7 of treatment, time profile of serum BA and C4 concentrations in C57BL/6N mice after 7 days of daily oral treatment (samples collected at 0, 2, 4, 8, and 24 hours after compound administration), and urinary BA concentrations 2 hours after dose administration on day 8 (terminal). (D) Ileal Fgf15 and hepatic Cyp7a1 expressions measured by RT-qPCR in C57BL/6NTac mice exposed to daily oral doses of A3907, A3309, or vehicle. All statistical data represent comparison versus vehicle group. For statistical analysis of fecal or urine BA concentrations, parametric 1-way ANOVA with Dunnett post hoc test was used. For statistical analysis of RT-qPCR data, Student t test or Mann-Whitney test was applied depending on the distribution of the data. Statistical significance of serum C4 and BA concentrations overtime was assessed by 2-way ANOVA with Tukey multiple comparisons post hoc test. *p < 0.05; **p < 0.01 and ***p < 0.001. Abbreviations: ASBT, apical sodium-dependent bile acid transporter; BAs, bile acids; NTCP, Na+-taurocholate cotransport polypeptide.
QWBA analysis in rats orally administered with [14C]-A3907 revealed distribution to the intestine, liver, and kidney, which are the main ASBT-expressing organs. By contrast, independent QWBA experiments performed with [14C]-A3309 confirmed preferential accumulation to the intestine, with only trace amounts found in the liver and kidney (Figure 1B). Daily oral administration of up to 1000 mg/kg/d of A3907 to rats was well tolerated, and no adverse A3907-related findings were observed after the 13-week treatment period (Supplemental Figure S1C, http://links.lww.com/HEP/F6).
The pharmacodynamic profile of A3907 and A3309 was compared in normal mice. Both compounds were administered orally to nonfasted mice once daily for 7 days (3 and 10 mg/kg/d). Similar inhibition of ileal ASBT was observed for both compounds, resulting in significant elevation of fecal BA levels, while urine BA levels did not change in response to A3907 or A3309 (Figure 1C). As expected, ASBT inhibition led to transcriptional Fgf15 repression in the ileum and subsequent hepatic upregulation of Cyp7A1, particularly evident in A3907-exposed mice, yielding increased circulating C4 levels (reflecting BA synthesis) compared with controls (Figure 1C and D). As a result, serum BA levels in normal noncholestatic mice remained fairly constant in all experimental groups (Figure 1C).
A3907 reduces liver injury and markers of cholestasis in Mdr2−/− mice
The potential therapeutic effect of A3907 was investigated in Mdr2 −/− mice, which are characterized by spontaneous and progressive development of cholestasis and sclerosing cholangitis.20,21 Mdr2 −/− mice were treated with different doses of A3907 (1, 3, 10, and 30 mg/kg) once a day by oral gavage for 4 weeks (Figure 2A).
FIGURE 2.
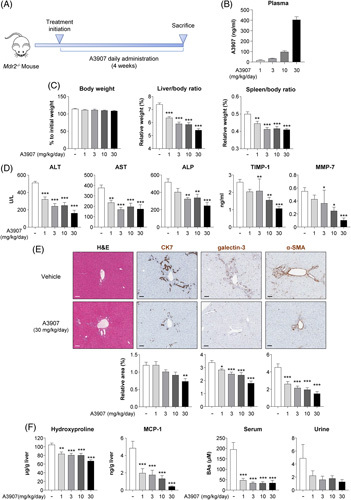
Therapeutic and pharmacodynamic effects of A3907 in Mdr2 −/− mice. (A) Schematic representation of the experimental design (n = 11 on each group). (B) Circulating levels of A3907 in plasma 2 hours after administration at the fourth week of treatment. (C) Comparison of the terminal body weight relative to treatment initiation and liver- and spleen-to-body weight ratios of the animals at sacrifice. (D) Plasma levels of circulating liver enzymes. (E) Representative hematoxylin-eosin (H&E) and immunohistochemistry (IHC) images of liver sections and comparison of the relative area of cytokeratin-7 (CK7), galectin-3, and α-smooth muscle actin (α-SMA); Scale bar: 100 µm. (F) Hepatic content of hydroxy-proline and monocyte chemoattractant protein-1 (MCP-1), and quantification of total BA concentration in serum and urine of the Mdr2 −/− mice. All statistical data represent comparison versus vehicle group. Parametric 1-way ANOVA with Dunnett post hoc test or nonparametric Kruskal-Wallis test followed by Dunn multiple comparison test was used to assess significance depending on the distribution of the data. *p < 0.05; **p < 0.01, and ***p < 0.001. Abbreviations: α-SMA, α-smooth muscle actin; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; MCP-1, monocyte chemoattractant protein-1.
A dose-dependent increase in A3907 levels was found in the plasma of Mdr2 −/− mice, with mean values of 17, 34, 97, and 405 ng/mL, respectively, measured 2 hours after dosing (Figure 2B). A3907 did not affect mice body weight but dose dependently decreased the liver-to-body and spleen-to-body weight ratios compared with vehicle controls (Figure 2C). A3907 dose dependently reduced plasma biomarkers of liver injury (alanine aminotransferase and aspartate aminotransferase) and cholestasis (alkaline phosphatase) (Figure 2D). Similarly, the plasma markers of sclerosing cholangitis (ie, biliary injury and fibrosis, TIMP-1,27 and MMP-728) were decreased in animals treated with A3907 compared with vehicle controls (Figure 2D). A3907 dose dependently decreased histological markers of ductular reaction (cytokeratin-7 (CK7)], portal inflammation (H&E and galectin-3), and fibrogenesis (α-smooth muscle actin) compared with controls (Figure 2E). In addition, whole-liver tissue levels of the proinflammatory chemokine MCP-1 and the fibrosis marker hydroxyproline were also lower in A3907-treated Mdr2 −/− mice compared with vehicle controls (Figure 2F). Furthermore, A3907 markedly reduced serum total BA levels at all doses investigated, whereas no significant changes in urinary BA and serum C4 concentrations were observed (Figure 2F; Supplemental Figure S2 and S3, http://links.lww.com/HEP/F6). As observed in normal mice, A3907 treatment led to ileal Fgf15 downregulation and consequent Cyp7a1 upregulation in the liver (Supplemental Figure S4, http://links.lww.com/HEP/F6).
Collectively, our data indicate that A3907 protects Mdr2 −/− mice from BA-induced ductular reaction and liver injury. The fact that all doses of A3907 similarly reduced serum BA levels, while the effects on ductular reaction were progressively and dose dependently improved suggests that normalizing BA load (which can often be achieved with intestinally restricted ASBT inhibitors) is not the only contributing factor to the improved ductular/liver protection. This may point toward direct target engagement of ASBT on cholangiocytes as a potential additive effect of A3907.
A3907 prevents BA-induced cholangiocyte apoptosis in vitro
Expression of ASBT in cultured cholangiocytes was confirmed by immunoblotting in normal rat cholangiocytes (Supplemental Figure S5, http://links.lww.com/HEP/F6). To mimic a context of cholestasis and sclerosing cholangitis, cholangiocytes were incubated with toxic concentrations of glycochenodeoxycholic acid (GCDCA) (1 mM) in vitro. Flow cytometry experiments indicated that coincubation of cholangiocytes with A3907 prevented BA-induced apoptosis. By contrast, and as expected, the apoptotic effect of the antibiotic puromycin (non-ASBT substrate) on cholangiocytes was not affected by A3907 exposure (Figure 3A).
FIGURE 3.
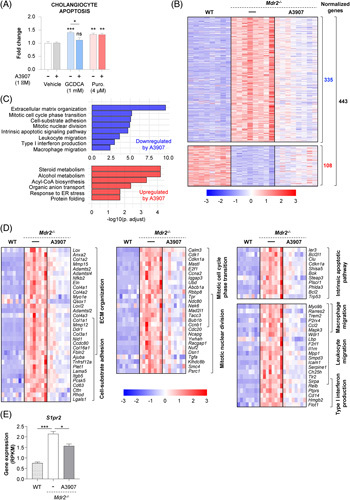
Hepatic transcriptome signatures in Mdr2 −/− mice after A3907 treatment and cytoprotective effects of A3907 in cultured cholangiocytes. (A) Quantification of the amount of apoptotic cell death analyzed by flow cytometry with Annexin V and To-PRO-3 dual staining after incubating normal rat cholangiocytes (NRCs) with vehicle, glycochenodeoxycholic acid [GCDCA (1 mM)], puromycin (4 μM), A3907 (1 μM), or combinations for 48 hours. (B) Heatmap displaying the hepatic expression of genes significantly normalized (compared with WT mice) by A3907 (3 mg/kg/d) (108 upregulated and 335 downregulated by A3907 compared with vehicle) in Mdr2 −/− mice by RNAseq analysis. (C) GO enrichment analysis of genes significantly modulated by A3907. (D) Heatmap representation of the differentially expressed genes annotated for each biological process. (E) Bar graph representation showing A3907-induced suppression of hepatic S1pr2 overexpression in Mdr2 −/− mice. For statistical analysis of flow cytometry data parametric 1-way ANOVA with Dunnett post hoc test was used to assess significance. *p < 0.05; **p < 0.01 and ***p < 0.001. Gene set analysis was conducted with the R package PIANO version 1.18.1 using the Stouffer method, and p-values were corrected for multiple testing using the Benjamini-Hochberg method (False Discovery Rate, p < 0.05). Abbreviation: WT, wild type.
A3907 normalizes hepatic gene signatures of inflammation, regeneration, damage, and fibrosis in Mdr2−/− mice
RNAseq analysis of liver samples from Mdr2 −/− mice revealed important biological processes modulated by A3907. Vehicle-dosed Mdr2 −/− mice displayed marked perturbations in the liver transcriptome as indicated by an excessive number of differentially expressed genes (DEGs, n = 9047) compared with WT mice. Among them, the levels of 443 DEGs were normalized in Mdr2 −/− mice treated with 3 mg/kg/d A3907 (108 upregulated and 335 downregulated following A3907 treatment; Figure 3B). Gene ontology enrichment analysis of these 443 DEGs normalized by A3907 revealed the influence of this compound on several important biological processes, including fibrosis, inflammation, cell proliferation and apoptosis, endoplasmic reticulum stress, and steroid metabolism (Figure 3C, D; Supplemental Figure S6, http://links.lww.com/HEP/F6). Of note, the increased expression levels of S1pr2 observed in Mdr2 −/− mice, a receptor closely related to cholangiocyte proliferation and cholestatic injury in response to conjugated BAs,29 were reduced by A3907 (Figure 3E).
A3907 enhances renal BA clearance and halts liver disease progression in experimental obstructive cholestasis
Proximal tubuli epithelial cells lining the urinary tract represent another cell target for A3907. Here, apical ASBT reclaims renally filtered BAs.9 Although our data in Mdr2 −/− mice suggest that inhibition of intestinal and biliary ASBT could be sufficient to reverse increased plasma BA levels, we investigated whether renal ASBT inhibition could be a novel mechanism to ameliorate cholestasis during biliary obstructive conditions. Thus, we evaluated the efficacy of A3907 in a model of obstructive cholestasis (BDL mice), where enterohepatic circulation of BAs is blocked and BAs can therefore only be excreted via renal clearance. We also used locally acting A3309 as a comparator.
Three days after surgery, BDL mice were treated with different daily doses of A3907 (3, 10, and 30 mg/kg) or A3309 (3 mg/kg) by oral gavage for 11 days (Figure 4A). Twenty-four hours after the last A3907 administration, a dose-dependent increase in A3907 levels was observed in serum (mean values of 34, 48, and 197 ng/mL, respectively) and in bile (mean values of 75, 251, and 1358 ng/mL, respectively) (Figure 4B), suggesting that A3907 reaches ASBT on the apical surface of cholangiocytes at pharmacologically relevant concentrations. BDL mice receiving vehicle demonstrated pronounced body weight loss, as compared with sham-operated control mice, and this progressive body weight loss was reversed by all A3907 doses (Figure 4C). Liver histopathological analyses indicated lower hepatic inflammatory cell infiltration and fewer necrotic areas in A3907-treated BDL mice compared with vehicle (Figure 4D). A3907 markedly enhanced the excretion of urinary BAs, predominantly the hydrophilic TMCAs and TCA, by up to 90-fold, compared with vehicle controls (Figure 4E; Supplemental Figure S7, http://links.lww.com/HEP/F6). This led to reduced serum and bile levels of total BAs compared with control BDL mice. In agreement, an inverse correlation between serum versus urine BA levels was observed in BDL mice (Figure 4E). As observed for Mdr2 −/− mice, no changes in serum C4 concentrations were found in the A3907-treated BDL mice (Supplemental Figure S3, http://links.lww.com/HEP/F6). Compared with vehicle, A3907 also reduced BDL-induced increases in serum levels of transaminases (aspartate aminotransferase and alanine aminotransferase), bilirubin (total, direct, and indirect) and urea, as well as total and LDL cholesterol, while counteracting the decreased serum levels of albumin, HDL, and glucose (Figure 4F). In contrast, alkaline phosphatase levels remained unchanged following A3907 treatment (Figure 4F).
FIGURE 4.
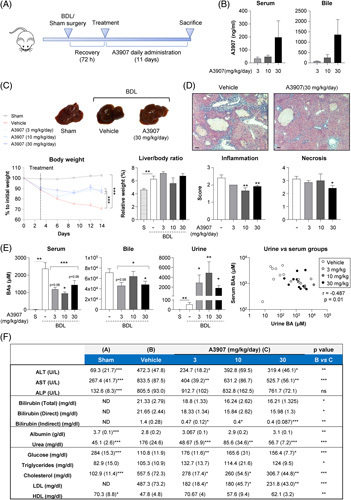
Therapeutic and pharmacodynamic effects of A3907 in mice subjected to bile duct ligation (BDL). (A) Schematic representation of the experimental design (n = 24 for vehicle and A3907 30 mg/kg groups, n = 14 for A3907 3 mg/kg group, and n = 12 for A3907 10 mg/kg group). (B) Terminal serum and bile concentrations of A3907 24 hours after administration of the last dose. (C) Representative whole-liver images and comparative body weight changes overtime, and terminal liver-to-body weight ratio. (D) Representative H&E images of liver sections and blinded scoring of inflammation and necrosis performed by an independent expert pathologist; Scale bar: 100 µm. (E) Total serum, bile, and urine BA levels at sacrifice in mice subjected to BDL after daily treatment with A3907 for 11 days, and correlation analysis of serum versus urine BA levels in individual animals. Dashed lines represent comparison between A3907 grouped doses and vehicle-administered BDL mice. (F) Serum biochemical parameters at sacrifice. Values are represented as mean (SEM). For statistical analysis of the histopathological scoring, Mann-Whitney test was applied. Statistical analysis of body weight change over time was performed by two-way ANOVA with Tukey multiple comparisons post hoc test. For statistical analysis of individual doses versus vehicle, parametric one-way ANOVA with Dunnett post hoc test or nonparametric Kruskal-Wallis test followed by Dunn multiple comparison test was used depending on the normality of the data. For comparisons between A3907 group versus vehicle and sham versus vehicle, Student t test or Mann-Whitney test was applied depending on the distribution of the data. For correlation analysis, Spearman test was applied. *p < 0.05; **p < 0.01 and ***p < 0.001. Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; BAs, bile acids; BDL, bile duct ligated; ND, not detected; S, sham; -, vehicle.
Altogether, these results suggest that experimental obstructive cholestasis can be ameliorated by A3907-induced ASBT inhibition in the kidneys, enhancing renal BA clearance to reduce the BA load and liver toxicity and improving liver health.
Next, we compared the pharmacokinetic and therapeutic effects of A3907 with the intestinally restricted ASBTi A3309 in the BDL model. For that purpose, we first characterized the systemic exposure of both compounds in BDL mice orally administered with daily doses of each compound (3 or 10 mg/kg) for 7 days (Figure 5A). As expected, A3907 displayed high systemic bioavailability, reaching mean serum concentration peaks of 193 and 936 ng/mL in BDL mice treated once daily with 3 or 10 mg/kg, respectively (Figure 5A). Interestingly, A3309 was also detected in serum of the BDL mice after 7 days of treatment. These plasma exposures were ~6-fold higher than exposures reached in normal WT mice under the same experimental conditions (data not shown). This discrepancy is probably explained by a general increase in intestinal permeability in response to the BDL procedure. However, serum levels of A3309 were significantly lower than A3907 (Figure 5A). In this regard, although both compounds induced urinary BA excretion to some extent, only A3907 was able to significantly reduce both serum and biliary BAs levels in BDL mice, while A3309 attenuated BA levels in a nonsignificant fashion (Figure 5B). By contrast, no changes in fecal BA content were observed in any of the experimental groups (Supplemental Figure S8, http://links.lww.com/HEP/F6). The superior efficacy of A3907 in relieving BA overload was mirrored by its capacity to prevent BDL-associated progressive body weight loss as well as to reduce the exacerbated levels of transaminases and urea in the BDL mice (Figure 5C, D). Furthermore, A3907, but not A3309, significantly reduced serum LDL concentrations in the BDL mice (Supplementary Table S3, http://links.lww.com/HEP/F6). Hence, the mild effects of A3309 together with its lower but still present systemic bioavailability seem to reinforce the importance of systemic ASBTi in a biliary obstructive context.
FIGURE 5.
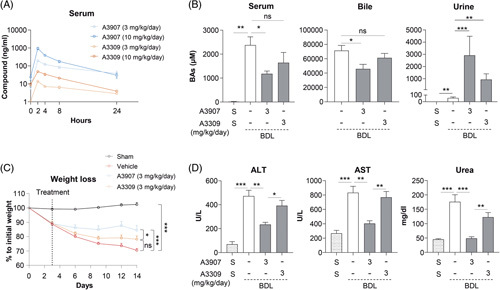
Comparative analysis of the pharmacokinetics, pharmacodynamics, and therapeutic effects of A3907 and A3309 in BDL mice. (A) Pharmacokinetic analysis of A3907 and A3309 in BDL mice administered with 3 and 10 mg/kg daily oral doses for 7 days. (B) Total BA levels at sacrifice in serum, bile, and urine after daily treatment with either A3907 or A3309 (3 mg/kg/d) or vehicle for 11 days in mice subjected to BDL. (C) Body weight loss over time in sham and BDL mice receiving A3907 or A3309 (3 mg/kg/d) or vehicle. (D) Levels of transaminases (ALT and AST) and urea in the different experimental groups at sacrifice. For statistical analysis Student t test or Mann-Whitney test was applied depending on the distribution of the data. Statistical analysis of body weight loss over time was performed by 2-way ANOVA with Tukey multiple comparisons post hoc test. *p < 0.05; **p < 0.01 and ***p < 0.001. Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; BAs, bile acids; BDL, bile duct ligated; S, sham; -, vehicle.
A3907 normalizes the dysregulated transcriptional profile of BDL mouse livers
Similar to observations in Mdr2 −/− mice, transcriptomic analysis of liver samples from BDL mice revealed substantial perturbations in hepatic global gene expression (5790 DEGs) compared with sham-operated control mice. A3907 (3 mg/kg/d) normalized the expression of 303 dysregulated genes (126 upregulated and 177 downregulated following A3907 treatment) in BDL mice (Figure 6A), while only 15 genes were normalized by A3309 (3 mg/kg/d). Gene ontology enrichment analysis of genes normalized by A3907 revealed genes participating in important processes related to BA and liver homeostasis, as well as to cell growth, oxidative stress, lipid metabolism and homeostasis, cell transport, xenobiotic metabolism, coenzyme metabolism, and TOR signaling (Figure 6B, C). Increased expression levels of the genes encoding for the nuclear receptor PXR and the basolateral transporter organic solute transporter beta (OST-β) mice (proteins involved in hepatocyte basolateral BA efflux)30 were found upregulated in BDL mice, compared with sham, and were specifically normalized following A3907 treatment (Figure 6D).
FIGURE 6.
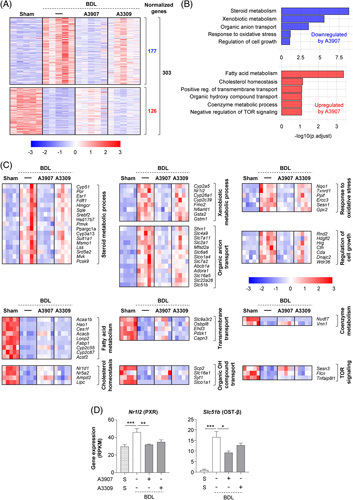
Comparison of hepatic transcriptome signatures of A3907 and A3309 treatment in BDL mice. (A) Heatmap illustrating genes significantly normalized by A3907 (3 mg/kg/day) (126 upregulated and 177 downregulated by A3907). (B) GO enrichment analysis of genes significantly normalized by A3907. (C) Heatmaps representing the expression of individual genes annotated for each biological process. (D) Hepatic expression of Nr1I2 and Slc51b analyzed by RNAseq. Gene set analysis was conducted with the R package PIANO version 1.18.1 using the Stouffer method, and p-values were corrected for multiple testing using the Benjamini-Hochberg method (False Discovery Rate, p < 0.05). Abbreviation: BDL, bile duct ligated.
Phase I study of A3907 safety, tolerability, oral bioavailability, and pharmacodynamic effects in healthy humans
Considering the positive preclinical effects of A3907 in experimental models of cholestatic liver disease, we investigated the tolerability, pharmacokinetics, and pharmacodynamic effects of A3907 in healthy human subjects in a single-ascending-dose study (data not shown), followed by a multiple-ascending-dose study (Figure 7A). The single-ascending-dose study revealed that A3907 exposure increased proportionally to doses from 1 to 81 mg. The multiple-ascending-dose clinical trial indicated that daily oral administration of A3907 for 7 days was well tolerated in the dose range investigated (9, 27, and 67.5 mg), showing no serious treatment-emergent adverse events (Supplemental Table S4, http://links.lww.com/HEP/F6). The most frequent adverse effects were mild, that is, diarrhea and headache. Oral administration of 67.5 mg A3907 resulted in significant compound exposure in plasma (Figure 7B), with a Cmax of 78.3 ng/mL, a Tmax of 8 hours, an AUCinf of 810 h*ng/mL, and a t 1/2 of 8.4 hours (Figure 7B). A similar pharmacokinetic profile was obtained on days 1 and 7 after daily oral administration (Supplemental Table S5, http://links.lww.com/HEP/F6).
FIGURE 7.
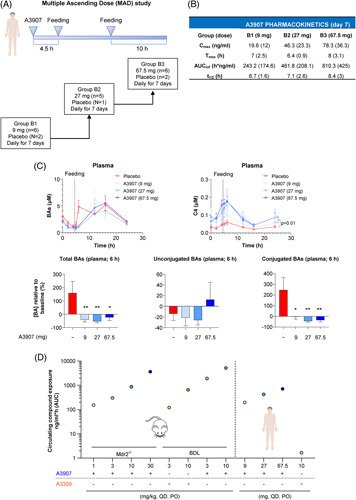
Pharmacokinetic profile and pharmacological effects of A3907 in healthy subjects subjected to MAD. (A) MAD study design. Healthy subjects were randomized into 3 different groups [group B1: 9 mg/d (A3907 n = 6, placebo n = 2); group B2: 27 mg/d (A3907 n = 5, placebo n = 1); group B3: 67.5 mg/d (A3907 n = 6, placebo n = 2)] and were administered one single oral dose of A3907 every 24 hour for 7 days. Daily dose was administered after 10 hour of fasting, and first meal was given 4.5 hour after A3907 administration. (B) Plasma pharmacokinetic profile in healthy subjects at day 7. Values are expressed as mean ± SD. (C) Pharmacological effect of A3907. Time profile of plasma total BA and C4 concentrations over the last 24-hour dosing interval. Postprandial (6 h) plasma concentrations of total, unconjugated, and conjugated BAs relative to baseline. (D) Comparison of circulating levels of A3907 in Mdr2 −/− and BDL mice and human healthy subjects, as well as A3309 in BDL mice and human healthy subjects. AUC values in the Mdr2 −/− mice are approximations calculated from pharmacokinetic data in C57BL/6J and CD-1 mice assuming comparable ADME profile. Statistical analysis of plasma BAs and C4 levels over time was performed by two-way ANOVA with Tukey multiple comparisons post hoc test. For statistical analysis of postprandial BA levels, parametric 1-way ANOVA with Dunnett post hoc test was used. *p < 0.05; **p < 0.01 and ***p < 0.001. Abbreviations: AUCinf, AUC from the time of dosing to the last measurable concentration and extrapolated to infinity; Cmax, maximum plasma concentrations; t 1/2, terminal half-life; Tmax, time to reach the maximum concentrations.
Functionally, A3907 significantly decreased postprandial plasma total BA levels relative to baseline in healthy humans (Figure 7C). Although levels of unconjugated BAs [cholic acid (CA), lithocholic acid (LCA), deoxycholic acid (DCA), chenodeoxycholic acid (CDCA), and ursodeoxycholic acid (UDCA)] remained unaltered, A3907 significantly decreased the postprandial rise in plasma levels of conjugated BAs (GCA, GLCA, GDCA, GCDCA, GUDCA, TCA, TLCA, TDCA, TCDCA, and TUDCA) as compared with baseline (Figure 7C). Similar to normal mice, BAs were gradually normalized in healthy human subjects administered A3907 potentially due to compensatory increases in liver BA synthesis as indicated by increased circulating C4 levels, compared with controls (Figure 7C). Importantly, plasma exposure of A3907 in healthy subjects was within the range of plasma concentrations that achieved anticholestatic efficacy in Mdr2 −/− and BDL mice (Figure 7D), supporting potential translation to human cholestatic liver disease. By contrast, the systemic A3309 exposure observed in humans at the therapeutically relevant dose18 was several-fold lower than levels demonstrating signs of efficacy in BDL mice in the current study (Figure 7D). This questions the human translational relevance of the preclinical findings with A3309 presented in the current study.
DISCUSSION
Previous preclinical and clinical studies have demonstrated that locally acting intestinal ASBTis are effective in reducing BA load. For instance, SC-43531 and A4250 (odevixibat)32 reduced BA levels in serum and bile and improved cholestatic liver disease in Mdr2 −/− mice. Clinically, odevixibat and maralixibat reduced BA load and decreased pruritus in pediatric indications such as progressive familial intrahepatic cholestasis16 and Alagille syndrome12, respectively. However, although locally acting ASBTis effectively reduced BA load and pruritus in adult cholestatic cholangiopathies such as primary biliary cholangitis and primary sclerosing cholangitis, they did not have a meaningful impact on biomarkers believed to reflect disease progression such as alkaline phosphatase or alanine aminotransferase /aspartate aminotransferase and were often associated with dose-limiting diarrhea.33–35 Furthermore, since the therapeutic outcome of intestinal ASBTis relies on intestinal inhibition of BA reabsorption, the efficacy of these drugs is likely limited in cholestatic situations where the bile flow is severely compromised, such as obstructive cholestasis. In the current study, we report (1) the discovery and characterization of A3907, the first oral systemically available ASBTi in clinical development; (2) its multimodal effects on BA reabsorption and signaling in cholangitis (Mdr2 −/− mouse) and obstructive cholestasis (BDL) mouse models; (3) a detailed assessment of its translational potential to humans; and (4) a summary of its tolerability, pharmacokinetics, and pharmacodynamics in healthy subjects.
In WT mice, oral A3907 got distributed to key ASBT-expressing organs such as ileum, liver, and kidney. Also, A3907 stimulated fecal excretion of BAs without affecting urinary BA levels, being in agreement with fecal BA excretion as the main route for eliminating BAs under physiological conditions.36 As expected, this effect was rapidly counterbalanced by intestinal Fgf15 reduction and subsequent increased BA synthesis, underscoring the tightly controlled BA homeostasis under normal conditions. As for normal mice, high systemic A3907 exposure was confirmed in both Mdr2 −/− and BDL mice.
Liver injury in Mdr2 −/− mice is caused by loss of canalicular biliary phospholipid secretion, resulting in excessive biliary accumulation of free BAs, which leads to cholangiocyte injury, pericholangitis, periductal fibrosis with ductular proliferation, and eventually progression to sclerosing cholangitis.20,21 A3907 dose dependently reduced liver-to-body and spleen-to-body weight ratios, plasma markers of liver injury, as well as improved several liver histological markers of inflammation and ductular reaction in Mdr2 /- mice. It is noteworthy that A3907 significantly lowered hepatic α-smooth muscle actin levels, a reliable marker of fibrogenic stellate cell activation,37,38 in addition to reduced plasma and liver biochemical markers of fibrosis. Moreover, RNAseq analysis of livers revealed that A3907 impacted important biological processes related to cholestatic liver injury, for instance normalizing the expression of key related genes like S1pr2, which is closely related to cholangiocyte proliferation and cholestatic injury in response to conjugated BAs. Interestingly, in Mdr2 /- mice, the lowest dose of A3907 tested (1 mg/kg/d) was sufficient to induce maximal serum BA reduction. However, higher doses of A3907 provided additional dose-dependent anticholestatic, anti-inflammatory, and anti-fibrogenic effects, suggesting that A3907 can also provide additional beneficial effects independent from its ability to decrease systemic BA overload. In this regard, targeting ASBT at the site of insult (cholangiocytes per se) and subsequent inhibition of local BA transport and ASBT-mediated signaling may contribute to the additional efficacy observed for A3907. In line with this, the high levels of A3907 found in bile, together with its capacity to attenuate cytotoxic effects of hydrophobic BAs in cultured cholangiocytes, provide support for the cholangiocyte-protective effect of A3907.
The robust hepatoprotective effects of A3907 were corroborated in BDL mice, a model of obstructive cholestasis where the enterohepatic circulation of BAs is blocked. A3907 evoked profound urinary excretion of BAs in BDL mice. We estimated that the urinary excretion of BAs following A3907 administration to BDL mice may reach levels of up to 10 µmol/d, which is reflected by reductions in serum and biliary BA levels. As a result of the marked renal clearance of BAs, hallmarks of severe progressive cholestatic liver injury in BDL mice, including acute obstructive jaundice and hepatic necroinflammation, were partially reversed following A3907 treatment. Furthermore, A3907 also suppressed the extreme elevations of circulating liver enzymes, BAs, total bilirubin, and cholesterol in this model. These results provide strong evidence about the promising translational potential of A3907 in conditions where the bile flow is physically blocked, like situations of common bile duct stenosis or distal cholangiocarcinomas.
Interestingly, measurable concentrations of A3309 were also detected in serum of BDL mice treated with 3 mg/kg/d dose of this compound, probably as a result of increased intestinal permeability following BDL surgery.39 However, A3309 proved significantly less effective in relieving systemic BA overload and improving pathophysiological hallmarks of cholestatic disease. Most importantly, the systemic exposures of A3309 needed for the observed effects in the BDL mice are far from the trace levels obtained in humans at therapeutically relevant doses,18 suggesting limited translational potential for the treatment of obstructive forms of cholestasis. Furthermore, as in Mdr2 −/− mice, transcriptomic analysis showed that only A3907 was able to modulate important hepatic-dysregulated processes during obstructive cholestasis (BDL), including cell growth, oxidative stress, lipid metabolism and homeostasis, and xenobiotic metabolism, among others. All these results are further supported by a previous work demonstrating that Asbt −/− mice are partially resistant to BDL-induced liver injury, linked to increased urinary BA excretion and consequently lower systemic BA levels.40
Phase I studies demonstrated that A3907 is well tolerated and shows favorable pharmacokinetics at pharmacologically active doses. Accordingly, A3907 reduced postprandial circulating levels of conjugated BAs and dose dependently increased serum C4 levels, a marker of BA synthesis downstream of CYP7A141 in healthy human subjects (and in normal mice). Increases in circulating C4 levels are an expected compensatory response to ASBT inhibition that has been observed for most gut-restricted ASBT inhibitors in healthy humans. However, unlike other inhibitors, well-tolerated oral doses of A3907 in humans resulted in plasma levels resembling those observed at therapeutically effective doses in experimental models of cholestasis.
ASBTis have emerged as promising drugs to expand the currently limited pharmacological toolbox for the treatment of cholestasis. Both locally acting31,32 and systemic ASBTis have shown to improve cholestatic liver injury in Mdr2 −/− mice. However, the therapeutic efficacies reported in these studies are not comparable due to methodological differences. Although A3907 and A4250 were investigated in adult (8 weeks) mice and treatment was extended over 4 weeks,32 experiments with SC-435 were conducted in younger animals (4 weeks) treated for a shorter period (2 weeks).31 Moreover, the range of doses employed was different between studies. Therefore, future investigations including direct comparisons between locally acting and systemic ASBTis in different experimental models of cholestasis (not only in BDL mice) will provide further insights on their therapeutic similarities and differences. Finally, the translation of preclinical results into human cholestatic situations should be interpreted with caution due to intrinsic limitations of experimental models to resemble the complex and heterogeneous scenario of human cholestatic disorders.42,43
In conclusion, A3907 is the first oral systemic ASBT inhibitor that acts at the level of the intestine, liver, and kidney to robustly attenuate cholestatic liver damage in experimental models. Our data indicate that the unique systemic nature of A3907, compared with intestinally restricted ASBTis, represents an opportunity for the treatment in clinical manifestations where the bile flow is physically blocked (eg, common bile duct stenosis or distal cholangiocarcinomas), providing new avenues for treatments. Furthermore, the cholangiocyte-targeting and protection capacity of A3907 may be valuable to improve outcomes for patients suffering from cholangiopathies such as primary biliary cholangitis and primary sclerosing cholangitis. Importantly, the A3907 exposures achieved in healthy subjects were comparable to those required for attaining therapeutic effects in animal models of cholestasis, suggesting translational potential. Further clinical studies will be needed to determine the efficacy, safety, and optimal dosing regimens of A3907 in patients with chronic cholestatic liver disease such as primary biliary cholangitis and primary sclerosing cholangitis.
AUTHOR CONTRIBUTIONS
Francisco J. Caballero-Camino, Per-Göran Gillberg, Ellen Strängberg, Fredrik Wångsell, Jan P. Mattsson, Erik Lindström, Peter Åkerblad, and Jesus M. Banales: study concept and design, analysis and interpretation of data, drafting of the manuscript, review, and editing. Anna Wallebäck: monitoring the clinical study. Henrik H. Hansen, Bo Angelin, Sara Straniero, and IS: analysis and interpretation of data, drafting of the manuscript, review, and editing. Francisco J. Caballero-Camino, Pedro M. Rodrigues, Aloña Agirre-Lizaso, Paula Olaizola, Laura Izquierdo-Sanchez, Maria J. Perugorria, and Luis Bujanda: acquisition of data. Francisco J. Caballero-Camino, Jesus M. Banales: statistical analysis. Erik Lindström, Peter Åkerblad, Bo Angelin, Sara Straniero, and Jesus M. Banales: obtained funding. All authors have read and agreed to the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Professor Paul Dawson from the Emory University School of Medicine in Atlanta for generously providing the primary antibody used to assess ASBT expression in NRC; Ingela Arvidsson and Jennifer Härdfeldt at Karolinska Institutet at Karolinska University Hospital Huddinge, Stockholm (Sweden), for skillful analyses at the mass spectrometry laboratory; Simon Evers, study director on the Mdr2 −/− study, and Michael Feigh, for contributing to study design and interpretation of data of the Mdr2 −/− study at Gubra, Denmark; Judit Bartis and Ben Kohn at Albireo for leading the manufacturing of A3907; and Santosh Kulkarni, Runa Pal, Shivendra Singh, Ramesh Kangarajan, and Ashwani Gaur from Syngene International Ltd., Bangalore, India, for, in collaboration with Albireo, manufacturing of the compound and in vitro and in vivo evaluation of A3907.
FUNDING INFORMATION
Spanish Carlos III Health Institute (ISCIII) [Jesus M. Banales (FIS PI18/01075, PI21/00922, and Miguel Servet Program CPII19/00008); Maria J. Perugorria (FIS PI14/00399, PI17/00022, and PI20/00186); Pedro M. Rodrigues (Sara Borrell CD19/00254 and Miguel Servet Program CP22/00073)] cofinanced by “Fondo Europeo de Desarrollo Regional” (FEDER); “Instituto de Salud Carlos III” [CIBERehd: Jesus M. Banales, Maria J. Perugorria, Pedro M. Rodrigues and Luis Bujanda], Spain; Department of Health of the Basque Country (Maria J. Perugorria: 2019111024 and Jesus M. Banales: 2021111021), “Euskadi RIS3” (Jesus M. Banales: 2019222054, 2020333010, and 2021333003), and Department of Industry of the Basque Country (Jesus M. Banales: Elkartek: KK-2020/00008); La Caixa Scientific Foundation (Jesus M. Banales: HR17-00601); European Union Horizon 2020 Research and Innovation Program (Jesus M. Banales: grant number 825510, ESCALON). Maria J. Perugorria was funded by the Spanish Ministry of Economy and Competitiveness (MINECO: “Ramón y Cajal” Programme RYC-2015-17755), Aloña Agirre-Lizaso by the Basque Government (PRE_2018_1_0184), and Francisco J. Caballero-Camino by the Spanish Ministry of Universities and European Union-Next Generation EU (MARSA21/17, University of Basque Country). IKERBASQUE, Basque foundation for science (to Jesus M. Banales and Pedro M. Rodrigues); “Diputación Foral Gipuzkoa” (2020-CIEN-000067-01 and 2021-CIEN-000029-04-01 to Pedro M. Rodrigues). Project support from the Swedish Heart-Lung Foundation, Stockholm Region/Karolinska Institutet (ALF, CIMED) (to Bo Angelin), and the Swedish Foundation for Strategic Research (to Sara Straniero).
CONFLICTS OF INTEREST
Fredrik Wångsell consults for Albireo. Bo Angelin consults and received grants from AstraZeneca and Albireo. He received grants from Amgen. Sara Straniero received grants from Albireo. Anna Wallebäck is employed by and owns stock in Albireo. Ingemar Starke is employed by and owns stock in Albireo. Per-Göran Gillberg was previously employed at Albireo. Ellen Strängberg is employed by and owns stock in Albireo. Britta Bonn is employed by and owns stock in Albireo. Jan P. Mattsson is employed by and owns stock in Albireo. Martin R. Madsen is employed by Gubra. Henrik H. Hansen is employed by Gubra. Erik Lindström is employed by and owns stock in Albireo. Peter Åkerblad is employed by and owns stock in Albireo. Jesus M. Banales consults for and received grants from Albireo. He received lecture fees and grants from Incyte. He consults for QED Therapeutics, OWL Metabolomics, CYMABAY Therapeutics, and Ikan Biotech. He received lecture fees from Intercept. The remaining authors have no conflicts to report.
Supplementary Material
Footnotes
Abbreviations: α-SMA, α-smooth muscle actin; ALP, alkaline phosphatase; ALT, alanine aminotransferase; ASBT, apical sodium-dependent bile acid transporter; ASBTi, ASBT inhibitor; ATCC, American Type Culture Collection; AUCinf, AUC from the time of dosing to the last measurable concentration and extrapolated to infinity; BAs, bile acids; BDL, bile duct ligated; C4, 7α-hydroxy-4-cholesten-3-one; CA, cholic acid; CDCA, chenodeoxycholic acid; CK7, cytokeratin-7; CMC, carboxymethylcellulose; Cyp7a1, cytochrome P450 family 7 subfamily A member 1; d, day; DCA, deoxycholic acid; DEGs, differentially expressed genes; GCDCA, glycochenodeoxycholic acid; GO, gene ontology; H&E, hematoxylin-eosin; IC50, half maximal inhibitory concentration; LCA, lithocholic acid; LC-MS/MS, liquid chromatography tandem mass spectrometry; MCA, muricholic acids; MCP-1, monocyte chemoattractant protein-1; MDR3, multidrug resistance protein 3; matrix metalloproteinase 7 (MMP-7), matrix metalloproteinase 7; NRC, normal rat cholangiocytes; NTCP, Na+-taurocholate cotransport polypeptide; OSTα/OSTβ, organic solute transporter alpha/beta; QWBA, quantitative whole-body autoradiography; RNAseq, RNA sequencing; RT-qPCR, quantitative real-time PCR; SAD, single-ascending-dose; t 1/2, terminal half-life; UDCA, ursodeoxycholic acid; WT, wild type.
Erik Lindström, Peter Åkerblad, and Jesus M. Banales shared senior authorship.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com.
Contributor Information
Francisco J. Caballero-Camino, Email: javier.caballero@biodonostia.org.
Pedro M. Rodrigues, Email: pedro.rodrigues@biodonostia.org.
Fredrik Wångsell, Email: fredrikwangsell.contractor@albireopharma.com.
Aloña Agirre-Lizaso, Email: alona.agirre@biodonostia.org.
Paula Olaizola, Email: paula.olaizola@biodonostia.org.
Laura Izquierdo-Sanchez, Email: laura.izquierdo@biodonostia.org.
Maria J. Perugorria, Email: matxus.perugorria@biodonostia.org.
Luis Bujanda, Email: luis.bujandafernandezdepierola@osakidetza.eus.
Bo Angelin, Email: bo.angelin@ki.se.
Sara Straniero, Email: Sara.straniero@ki.se.
Anna Wallebäck, Email: anna.walleback@albireopharma.com.
Ingemar Starke, Email: ingemar.Starke@albireopharma.com.
Per-Göran Gillberg, Email: gillbergpergoran@gmail.com.
Ellen Strängberg, Email: Ellen.Strangberg@albireopharma.com.
Britta Bonn, Email: britta.bonn@albireopharma.com.
Jan P. Mattsson, Email: Jan.Mattsson@albireopharma.com.
Martin R. Madsen, Email: mam@gubra.dk.
Henrik H. Hansen, Email: hbh@gubra.dk.
Erik Lindström, Email: erik.lindstrom@albireopharma.com.
Peter Åkerblad, Email: peter.akerblad@albireopharma.com.
Jesus M. Banales, Email: JESUS.BANALES@biodonostia.org.
REFERENCES
- 1.Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol. 2019;16:269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feldman AG, Sokol RJ. Neonatal cholestasis: emerging molecular diagnostics and potential novel therapeutics. Nat Rev Gastroenterol Hepatol 2019 166. 2019;16:346–60. [DOI] [PubMed] [Google Scholar]
- 3.de Vries E, Beuers U. Management of cholestatic disease in 2017. Liver Int. 2017;37(suppl 1):123–9. [DOI] [PubMed] [Google Scholar]
- 4.De Vloo C, Nevens F. Cholestatic pruritus : an update. Acta Gastroenterol Belg. 2019;82:75–82. [PubMed] [Google Scholar]
- 5.Dyson JK, Beuers U, Jones DEJ, Lohse AW, Hudson M. Primary sclerosing cholangitis. Lancet (London, England). 2018;391:2547–59. [DOI] [PubMed] [Google Scholar]
- 6.Yokoda RT, Carey EJ. Primary biliary cholangitis and primary sclerosing cholangitis. Am J Gastroenterol. 2019;114:1593–605. [DOI] [PubMed] [Google Scholar]
- 7.Slijepcevic D, Van De Graaf SFJ. Bile acid uptake transporters as targets for therapy. Dig Dis. 2017;35:251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Ciaula A, Garruti G, Baccetto RL, Molina-Molina E, Bonfrate L, Wang DQH, et al. Bile acid physiology. Ann Hepatol. 2017;16:s4–14. [DOI] [PubMed] [Google Scholar]
- 9.St-Pierre MV, Kullak-Ublick GA, Hagenbuch B, Meier PJ. Transport of bile acids in hepatic and non-hepatic tissues. J Exp Biol. 2001;204:1673–86. [DOI] [PubMed] [Google Scholar]
- 10.Durník R, Šindlerová L, Babica P, Jurček O. Bile acids transporters of enterohepatic circulation for targeted drug delivery. Molecules. 2022;27:2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trottier J, Białek A, Caron P, Straka RJ, Heathcote J, Milkiewicz P, et al. Metabolomic profiling of 17 bile acids in serum from patients with primary biliary cirrhosis and primary sclerosing cholangitis: a pilot study. Dig Liver Dis. 2012;44:303–10. [DOI] [PubMed] [Google Scholar]
- 12.Gonzales E, Hardikar W, Stormon M, Baker A, Hierro L, Gliwicz D, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet. 2021;398:1581–92. [DOI] [PubMed] [Google Scholar]
- 13.Yang N, Dong YQ, Jia GX, Fan SM, Li SZ, Yang SS, et al. ASBT(SLC10A2): a promising target for treatment of diseases and drug discovery. Biomed Pharmacother. 2020;132:110835. [DOI] [PubMed] [Google Scholar]
- 14.Al-Dury S, Marschall HU. Ileal bile acid transporter inhibition for the treatment of chronic constipation, cholestatic pruritus, and NASH. Front Pharmacol. 2018;9:931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graffner H, Gillberg PG, Rikner L, Marschall HU. The ileal bile acid transporter inhibitor A4250 decreases serum bile acids by interrupting the enterohepatic circulation. Aliment Pharmacol Ther. 2016;43:303–10. [DOI] [PubMed] [Google Scholar]
- 16.Thompson RJ, Arnell H, Artan R, Baumann U, Calvo PL, Czubkowski P, et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2022;7:830–42. [DOI] [PubMed] [Google Scholar]
- 17.Gijbels E, Pieters A, De Muynck K, Vinken M, Devisscher L. Rodent models of cholestatic liver disease: a practical guide for translational research. Liver Int. 2021;41:656–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong BS, Camilleri M. Elobixibat for the treatment of constipation. Expert Opin Investig Drugs. 2013;22:277–84. [DOI] [PubMed] [Google Scholar]
- 19.Labiano I, Agirre-Lizaso A, Olaizola P, Echebarria A, Huici-Izagirre M, Olaizola I, et al. TREM-2 plays a protective role in cholestasis by acting as a negative regulator of inflammation. J Hepatol. 2022;77:991–1004. [DOI] [PubMed] [Google Scholar]
- 20.Smit JJM, Schinkel AH, Elferink RPJO, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–62. [DOI] [PubMed] [Google Scholar]
- 21.Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Weiglein AH, Lammert F, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123:1238–51. [DOI] [PubMed] [Google Scholar]
- 22.Huang J, Bathena SPR, Csanaky IL, Alnouti Y. Simultaneous characterization of bile acids and their sulfate metabolites in mouse liver, plasma, bile, and urine using LC-MS/MS. J Pharm Biomed Anal. 2011;55:1111–19. [DOI] [PubMed] [Google Scholar]
- 23.Al-Khaifi A, Straniero S, Voronova V, Chernikova D, Sokolov V, Kumar C, et al. Asynchronous rhythms of circulating conjugated and unconjugated bile acids in the modulation of human metabolism. J Intern Med. 2018;284:546–59. [DOI] [PubMed] [Google Scholar]
- 24.Straniero S, Laskar A, Savva C, Härdfeldt J, Angelin B, Rudling M. Of mice and men: murine bile acids explain species differences in the regulation of bile acid and cholesterol metabolism. J Lipid Res. 2020;61:480–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banales JM, Arenas F, Rodríguez-Ortigosa CM, Sáez E, Uriarte I, Doctor RB, et al. Bicarbonate-rich choleresis induced by secretin in normal rat is taurocholate-dependent and involves AE2 anion exchanger. Hepatology. 2006;43:266–75. [DOI] [PubMed] [Google Scholar]
- 26.Hansen HH, Ægidius HM, Oró D, Evers SS, Heebøll S, Eriksen PL, et al. Human translatability of the GAN diet-induced obese mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol. 2020;20:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ueno T, Tamaki S, Sugawara H, Inuzuka S, Torimura T, Sata M, et al. Significance of serum tissue inhibitor of metalloproteinases-1 in various liver diseases. J Hepatol. 1996;24:177–84. [DOI] [PubMed] [Google Scholar]
- 28.Lam S, Singh R, Dillman JR, Trout AT, Serai SD, Sharma D, et al. Serum matrix metalloproteinase 7 is a diagnostic biomarker of biliary injury and fibrosis in pediatric autoimmune liver disease. Hepatol Commun. 2020;4:1680–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Aoki H, Yang J, Peng K, Liu R, Li X, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology. 2017;65:2005–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol. 2013;58:155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miethke AG, Zhang W, Simmons J, Taylor AE, Shi T, Shanmukhappa SK, et al. Pharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology. 2016;63:512–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baghdasaryan A, Fuchs CD, Österreicher CH, Lemberger UJ, Halilbasic E, Påhlman I, et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol. 2016;64:674–81. [DOI] [PubMed] [Google Scholar]
- 33.Hegade VS, Kendrick SFW, Dobbins RL, Miller SR, Thompson D, Richards D, et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet (London, England). 2017;389:1114–23. [DOI] [PubMed] [Google Scholar]
- 34.Mayo MJ, Pockros PJ, Jones D, Bowlus CL, Levy C, Patanwala I, et al. A randomized, controlled, phase 2 study of maralixibat in the treatment of itching associated with primary biliary cholangitis. Hepatol Commun. 2019;3:365–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levy C, Kendrick S, Bowlus CL, Tanaka A, Jones D, Kremer AE, et al. GLIMMER: a randomized phase 2b dose-ranging trial of linerixibat in primary biliary cholangitis patients with pruritus. Clin Gastroenterol Hepatol. 2022;S1542-3565:01021–7. [DOI] [PubMed] [Google Scholar]
- 36.Hofmann AF. Biliary secretion and excretion in health and disease: current concepts. Ann Hepatol. 2007;6:15–27. [PubMed] [Google Scholar]
- 37.Nouchi T, Tanaka Y, Tsukada T, Sato C, Marumo F. Appearance of alpha-smooth-muscle-actin-positive cells in hepatic fibrosis. Liver. 1991;11:100–5. [DOI] [PubMed] [Google Scholar]
- 38.Carpino G, Morini S, Ginanni Corradini S, Franchitto A, Merli M, Siciliano M, et al. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig Liver Dis. 2005;37:349–56. [DOI] [PubMed] [Google Scholar]
- 39.Cabrera-Rubio R, Patterson AM, Cotter PD, Beraza N. Cholestasis induced by bile duct ligation promotes changes in the intestinal microbiome in mice. Sci Rep. 2019;9:12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kunst RF, Waart DR, de, Wolters F, Duijst S, Vogels EW, Bolt I, et al. Systemic ASBT inactivation protects against liver damage in obstructive cholestasis in mice. JHEP Reports. 2022;4:100573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sauter G, Berr F, Beuers U, Fischer S, Paumgartner G. Serum concentrations of 7alpha-hydroxy-4-cholesten-3-one reflect bile acid synthesis in humans. Hepatology. 1996;24:123–6. [DOI] [PubMed] [Google Scholar]
- 42.Fickert P. Time to say goodbye to the drug or the model ?—why do drugs fail to live up to their promise in bile duct ligated mice ? J Hepatol. 2014;60:12–5. [DOI] [PubMed] [Google Scholar]
- 43.Fickert P, Pollheimer MJ, Beuers U, Lackner C, Hirschfield G, Housset C, et al. Characterization of animal models for primary sclerosing cholangitis (PSC). J Hepatol. 2014;60:1290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]