

Abstract
Background and Aims:
Alcohol-associated liver disease (ALD) accounts for 70% of liver-related deaths in Europe, with no effective approved therapies. Although mitochondrial dysfunction is one of the earliest manifestations of alcohol-induced injury, restoring mitochondrial activity remains a problematic strategy due to oxidative stress. Here, we identify methylation-controlled J protein (MCJ) as a mediator for ALD progression and hypothesize that targeting MCJ may help in recovering mitochondrial fitness without collateral oxidative damage.
Approach and Results:
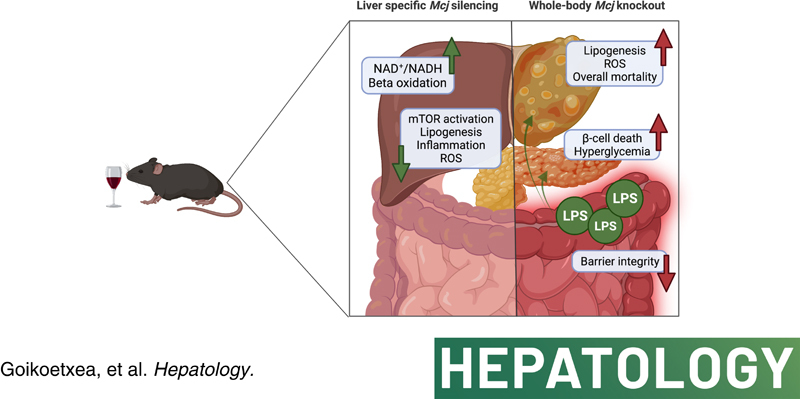
C57BL/6 mice [wild-type (Wt)] Mcj knockout and Mcj liver-specific silencing (MCJ-LSS) underwent the NIAAA dietary protocol (Lieber-DeCarli diet containing 5% (vol/vol) ethanol for 10 days, plus a single binge ethanol feeding at day 11). To evaluate the impact of a restored mitochondrial activity in ALD, the liver, gut, and pancreas were characterized, focusing on lipid metabolism, glucose homeostasis, intestinal permeability, and microbiota composition. MCJ, a protein acting as an endogenous negative regulator of mitochondrial respiration, is downregulated in the early stages of ALD and increases with the severity of the disease. Whole-body deficiency of MCJ is detrimental during ALD because it exacerbates the systemic effects of alcohol abuse through altered intestinal permeability, increased endotoxemia, and dysregulation of pancreatic function, which overall worsens liver injury. On the other hand, liver-specific Mcj silencing prevents main ALD hallmarks, that is, mitochondrial dysfunction, steatosis, inflammation, and oxidative stress, as it restores the NAD+/NADH ratio and SIRT1 function, hence preventing de novo lipogenesis and improving lipid oxidation.
Conclusions:
Improving mitochondrial respiration by liver-specific Mcj silencing might become a novel therapeutic approach for treating ALD.
INTRODUCTION
Alcohol-associated liver disease (ALD) contributes significantly to the global burden of liver diseases1,2 and represents the second most common indication for liver transplantation (LT) worldwide.3 Currently, permanent abstinence at early stages and LT at more advanced stages are the unique options for these patients.4 Therefore, a better understanding of the mechanisms mediating the initiation and progression of this disease is vital, enabling the development of a targeted therapy to treat or prevent it.
One of the earliest manifestations of hepatocyte injury caused by alcohol is morphological and functional changes in the mitochondria.5 Following the use of alcohol, the mitochondria increase oxygen consumption, in part as an adaptive response to oxidize the toxic metabolite acetaldehyde more rapidly and to increase NAD+ supply for alcohol metabolism.5 However, these results in the formation of reactive oxygen species (ROS) that alter mitochondrial activity and other signaling pathways, leading to fat accumulation and inflammation.6 Consequently, preventing mitochondrial dysfunction may be the firewall to delay or even stop ALD progression.
Although alcohol abuse mainly affects the liver, ALD is indeed a multisystemic disease.7 Acetaldehyde alters the gut barrier integrity and facilitates the translocation of bacterial products to the organism,8 which may affect other organs, such as the pancreas, even in those patients with early ALD.7 This highlights the relevance of avoiding imbalances in alcohol metabolism and maintaining mitochondrial homeostasis.
In this context, methylation-controlled J protein (MCJ), also known as DnaJC15, is an endogenous negative regulator of mitochondrial activity, as it interacts with and inhibits the mitochondrial complex I.9 Its absence leads to increased respiration and ATP synthesis, and stimulates the formation of respiratory supercomplexes, thereby limiting the production of ROS.9–11 MCJ seems dispensable under homeostatic conditions, and no altered phenotype is observed in Mcj knockout (MCJ-KO) mice.10 We have previously shown that silencing of MCJ by specific GalNAc-siRNA molecules enhances mitochondrial activity and ATP synthesis and results in decreased ROS generation and cell damage in NASH and DILI.10,12
In this study, we aimed to determine whether a lack of MCJ would alleviate alcohol-induced liver injury and halt its progression by preventing mitochondrial dysfunction. Surprisingly, following the NIAAA model developed by Gao et al,13 whole-body MCJ-KO mice showed increased mortality due to a worst systemic ethanol affection and pancreatic dysfunction caused by endotoxemia. However, MCJ liver-specific silencing (MCJ-LSS) proved to be hepatoprotective, as it reduced hepatic injury and facilitated liver regeneration. Altogether, results obtained from liver-specific Mcj silencing further support the possibility of targeting mitochondrial dysfunction as a therapeutic approach to ameliorate ALD and accompanying systemic complications.
METHODS
Human samples
A public data repository was used to analyze the expression of MCJ in patients. Patient data were included in the study by Argemi et al, (2019).14 Patients (n=61) were divided into different clinically relevant stage groups: (1) patients with early alcohol-associated steatohepatitis (ASH), who were nonobese with high alcohol intake, and presented mild elevation of transaminases and histologic criteria of steatohepatitis (Early ASH, N=11); (2) patients with histologically confirmed nonsevere alcohol-associated hepatitis (AH) who were biopsied before any treatment (Nonsevere AH, N=11); (3) patients with histologically confirmed AH who were biopsied before any treatment (Severe AH, N=18); and (4) explants from patients with AH who underwent early transplantation (Explant AH, N=11). Samples from these groups were compared with fragments of non-diseased human livers (Control N=10). Patients with malignancies were excluded from the study. Clinical characteristics of this cohort are described in the study by Argemi et al.14
Experimental design
The animal procedures were performed in accordance with the European Research Council for animal care and use and the National Institute of Health Guide For Care and Use of Laboratory Animals. The maximum authority of the Country Council of Bizkaia and the Institutional Animal Care and Use Committee of CIC bioGUNE approved the animal procedures (REGA 48/901/000/6106) (P-CBG-CBBA-218). MCJ-KO and wild-type (Wt) mice were bred at the CIC bioGUNE AAALAC-accredited animal facility. Adult (3-mo-old) male C57BL/6J mice were acquired from Charles River Laboratories and accommodated into the AALAC-accredited CIC bioGUNE animal facilities. All mice were maintained at 21°C, 45% humidity, and a 12/12 h light/dark cycle. Animal maintenance was based on ad libitum access to water and the respective diet. Experiments in this study employed the National Institute on Alcohol abuse and Alcoholism (NIAAA)13 experimental model of ALD (Supplementary Methods, http://links.lww.com/HEP/C649).
RESULTS
MCJ expression in human liver with severe AH
Although mitochondrial dysfunction is known to play a key role in the pathogenesis of ALD, to date, no studies have investigated MCJ/DNAJC15 in ALD patients. To this end, we compared MCJ expression in liver biopsies from patients with early ASH, nonsevere and severe AH, and explants undergoing LT. Our results showed that, compared to healthy individuals, expression of MCJ was downregulated in the early stages and significantly increased in the explants of AH patients undergoing LT (Figure 1A).
FIGURE 1.
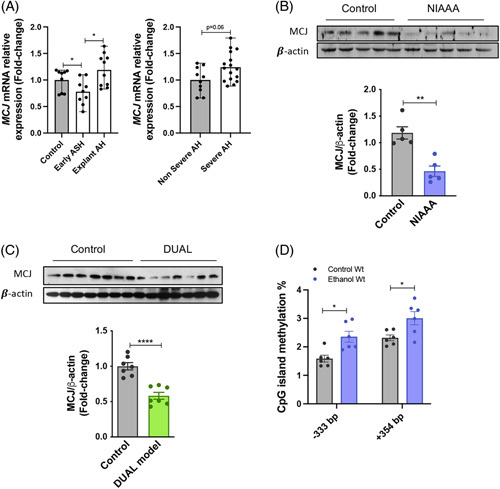
MCJ expression is regulated in alcohol-associated liver injury. (A) Relative MCJ mRNA levels in liver biopsies from patients with Early ASH (n=11), Nonsevere AH (n=11), Severe AH (n=18), and Explanted AH (n=11), together with healthy control individuals (n=10). Values represented as median ± range. U test was used to compare 2 groups. (B) Western blotting and densitometric quantification of MCJ in Wt liver extracts with control (n=5) and NIAAA diet (n=5) and (C) with control (n=7) and DUAL (n=7) diet. β-actin as a loading control. (D) CpG islands methylation status analyzed by bisulfite pyrosequencing. *p<0.05, **p<0.01, and ****p<0.0001 versus control.
On the other hand, while none of the current animal models can reproduce all major features of human ALD, they remain a very useful tool to study this disease.15 Following the NIAAA dietary model (Supplemental Figure 1a, http://links.lww.com/HEP/C648), which is used to study early stages of alcohol steatohepatitis,13 we measured downregulated hepatic MCJ protein levels (Figure 1B). Dysregulation of MCJ was further confirmed using the DUAL dietary model by WB and gene expression (Figure1C, Supplemental Figure 1b, http://links.lww.com/HEP/C648).16
In light of these results, we aimed to understand the mechanism by which the expression of Mcj is regulated after alcohol consumption. Mcj has been previously reported to be epigenetically regulated in NAFLD.12 Therefore, we used bisulfite pyrosequencing to analyze the DNA methylation status of a region of n base pairs spanning its transcriptional start site. Absolute DNA methylation level was heterogeneous among individual CpG sites (Figure 1D). Of note, data analysis identified 2 CpG sites in which DNA methylation is higher in mice exposed to alcohol than in control mice (Figure 1D), which suggests that DNA methylation might play a role in the downregulation of Mcj in response to alcohol exposure.
Whole-body knockout of MCJ increased ethanol consumption-induced liver injury
Based on previous studies that proved hepatoprotection in MCJ-KO mice,10,12,17,18 it was surprising to find increased mortality in MCJ-KO mice (55%) compared to Wt mice (15%) that were subjected to the NIAAA protocol (Figure 2A). The histopathological evaluation showed increased liver injury with more severe steatosis, inflammation, and a final injury score of 3.7 in ethanol-fed MCJ-KO versus 2.3 obtained by ethanol-fed Wt mice (Figure 2B). Significantly increased necrotic areas, cleaved caspase-3, and TUNEL staining levels were also observed in ethanol-fed MCJ-KO mice (Figure2C and Supplemental Figure 2a, http://links.lww.com/HEP/C648), although no significant changes were found for proapoptotic and antiapoptotic genes (Supplemental Figure 2b, http://links.lww.com/HEP/C648). Besides, serum ALT and bilirubin levels tended to increase in MCJ-KO mice (Figure 2D, Supplemental Fig 2c, http://links.lww.com/HEP/C648), while AST remained unchanged (data not shown). In sum, ethanol-fed MCJ-KO mice exhibit mildly increased hepatocellular injury compared with ethanol-fed Wt mice.
FIGURE 2.
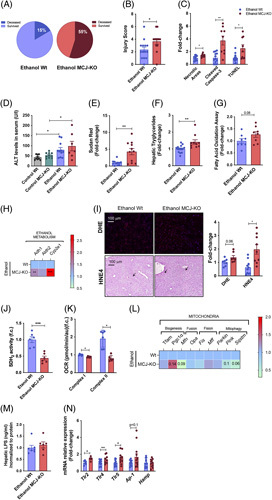
Whole body MCJ-KO show increased liver injury after ethanol consumption. (A) Survived and Deceased Wt (Left panel) and MCJ-KO (Right panel) mice following the NIAAA model. (B) Hepatic histopathological evaluation. (C) Quantification of Necrotic Areas (based on HE staining), Cleaved Caspase-3 and TUNEL stained liver sections. (D) ALT and AST plasmatic levels. (E) Quantification of Sudan Red stained liver sections. (F) Hepatic triglyceride content. (G) Hepatic fatty acid oxidation assay. (H) Heatmap showing the liver mRNA relative expression of genes involved in alcohol metabolism: Adh1, Aldh2, and Cyp2e1. (I) Quantification and representative DHE and 4HNE stained liver sections. (J) Hepatic SDH2 activity. (K) Oxygen consumption rate of mitochondrial complex I and complex II in freshly isolated hepatic mitochondria. (L) Heatmap showing the liver mRNA relative expression of mitochondrial quality control genes: Tfam, Pgc1a, Mfn, Opa1, Fis1, Mff, Prkn, Pink1, and Sqstm. (M) Hepatic LPS content. (N) Hepatic mRNA relative expression of genes involved in LPS recognition: Tlr, Ap-1, and Hamp. *p<0.05, **p<0.01, and ***p<0.001 versus Wt.
The study of lipid metabolism showed increased hepatic steatosis in ethanol-fed MCJ-KO mice (Figure2E, Supplemental Figure 2d, 2F, http://links.lww.com/HEP/C648), together with an upregulated expression of genes participating in lipid beta-oxidation and de novo lipogenesis/accumulation, depicting an overall altered metabolism (Supplemental Figure 2e, http://links.lww.com/HEP/C648). Moreover, significantly increased Glut2 expression levels were found in MCJ-KO mice (Supplemental Figure 2d, http://links.lww.com/HEP/C648). In line with previous results,12 hepatic beta-oxidation was significantly enhanced in MCJ-KO mice (Figure 2G).
Inflammation is another hallmark of ethanol-induced liver injury.19 No significant changes were observed regarding the number or the composition of liver infiltrating immune cells based on F4/80 staining (Supplemental Figure 2d, http://links.lww.com/HEP/C648) and flow cytometry (Supplemental Figure 2f, http://links.lww.com/HEP/C648). In this line, NK cells, B cells, CD8+ T, and CD4+T cells were the most represented populations, with no evident changes between Wt and MCJ-KO mice. Analysis of the expression of inflammatory cytokines just revealed significantly increased Ccr2 levels in ethanol-fed MCJ-KO mice (Supplemental Figure 2g. http://links.lww.com/HEP/C648). Finally, we found no signs of liver fibrosis in MCJ-KO mice (Supplemental Figure 2d, http://links.lww.com/HEP/C648).
The metabolism of high concentrations of alcohol results not only in acetaldehyde but also in the production of ROS that alter mitochondrial activity and other signaling pathways.6 Analysis of the main enzymes related to alcohol metabolism revealed significantly increased Adh1 and Cyp2e1 mRNA levels in MCJ-KO mice (Figure 2H), with a slight tendency towards a reduced hepatic accumulation of both ethanol and acetaldehyde (Supplemental Figure 2h, http://links.lww.com/HEP/C648). As for ROS, MCJ-KO showed higher levels of DHE staining, along with augmented levels of 4HNE staining, a marker for lipid peroxidation, as a consequence of increased hepatic steatosis and ROS production (Figure 2I). We observed no differences in the reduced (GSH)/oxidized (GSSG) glutathione ratio, a potent antioxidant (Supplemental Figure 2i, http://links.lww.com/HEP/C648). Finally, the analysis of hepatic succinate dehydrogenase (SDH2) activity (Figure 2J) and the oxygen consumption rate of both complex I and complex II in isolated mitochondria (Figure 2K) confirmed reduced mitochondrial function on ethanol-fed MCJ-KO mice, together with downregulated expression of genes participating on mitochondria quality control mechanisms, specifically fusion, and mitophagy (Figure 2L).
The pathophysiology of ALD is closely related to the effect of ethanol and its metabolites not only on the liver but also on other organs such as the gut.19 We found significantly increased hepatic lipopolysaccharide (LPS) concentrations, a Gram-negative bacteria-derived harmful product (Figure 2M) that significantly elevated the expression of Toll-like receptors (Figure 2N) involved in recognition of LPS in ethanol-fed MCJ-KO mice, suggesting an increased gut injury and presumably endotoxemia.
Altogether, MCJ-KO mice showed increased mortality, liver injury, impaired mitochondrial respiration, and an apparent gut injury following ethanol consumption.
Augmented intestinal permeability and translocation of bacterial products in ethanol-fed MCJ-KO mice
Alcohol leads to changes in the composition of the gut microbiome, the disruption of the gut barrier, and increased intestinal permeability, facilitating the translocation of microbial products.20 Intestinal expression of Mcj remained unchanged following the NIAAA model (Figure 3A).
FIGURE 3.
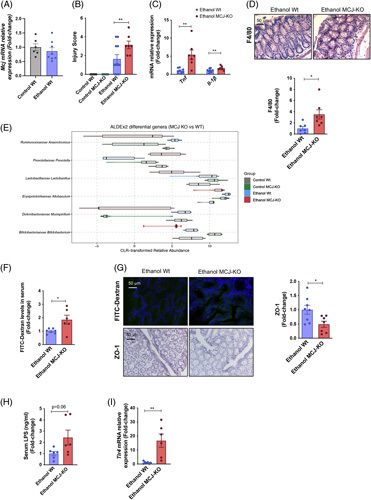
Increased intestinal permeability and translocation in ethanol-fed MCJ-KO mice. (A) Relative Mcj mRNA expression in gut tissue from control and ethanol-fed Wt mice. (B) Gut histopathological evaluation. (C) Gut mRNA relative expression of Tnf and Il-1b. (D) Quantification and representative F4/80 stained gut sections. (E) Central Log-Ratio (CLR) transformed abundance for the significant genera (FDR <10%) found for the comparison between the gut microbiome of MCJ-KO mice versus Wt mice. For each genus, the distribution of the CLR-transformed is represented for each of the 4 study groups using a combination of a violin plot for the general distribution and a boxplot for the summary distribution, colored accordingly. (F) Serum FITC-Dextran levels. (G) Immunohistochemical representation of intestinal FITC-Dextran levels and quantification and representative Zonula occludens stained gut sections. (H) Serum LPS content. (I) Gut mRNA relative expression of Tlr4. *p<0.05, **p<0.01, and ***p<0.001 versus Wt.
The histopathological evaluation of the colon showed neither injury nor differences between control-fed Wt and MCJ-KO mice, as shown21 (Figure 3B). However, it depicted focal ulceration, mononuclear cell infiltration, and edema in the mucosa and the submucosa of MCJ-KO mice, who displayed a mean injury score value of 3.42 versus 1.63 of Wt mice, indicating a more severe lesion at the colon level (Figure 3B). Consistently, the elevated expression of Tnf and Il-1ß (Figure 3C), together with increased F4/80 staining levels in ethanol-fed MCJ-KO mice (Figure 3D), confirmed that whole-body lack of MCJ boosts a proinflammatory response in the gut.
The V3-V4 regions of 16S rRNA amplicon sequencing identified alterations of the gut microbiota composition in ethanol-fed MCJ-KO mice, suggestive of a dysbiosis event following ethanol consumption (Figure 3E). Compared to Wt mice, higher levels of Prevotella, a bacterium known to degrade mucin, leading to gut barrier integrity disruption, and lower abundance of Bifidobacteriaceae, Lactobacillaceae, and Ruminococcaceae, which maintain mucosal barrier integrity, were identified in ethanol-fed MCJ-KO mice. The hypothesis of a dysbiotic event following ethanol consumption is further supported by the reduction of Ruminococcaceae and the increase of Bifidobacteriaceae found in the gut microbiome of control MCJ-KO mice compared to control Wt.
Indeed, evaluation of intestinal permeability with FITC-labelled dextran showed higher serum levels in ethanol-fed MCJ-KO mice (Figure 3F). Besides, reduced levels of tight junction proteins (Zonula occludens) detected by immunohistochemistry (Figure 3G) confirmed augmented intestinal permeability and decreased junctional integrity in these mice. In fact, higher serum levels of LPS were measured in ethanol-fed MCJ-KO mice (Figure 3H), along with an increased intestinal expression of Toll-like receptors (Figure 3I).
Altogether, ethanol consumption caused a significant dysbiosis event in ethanol-fed MCJ-KO mice, inducing intestinal immune dysregulations, increasing intestinal permeability, and facilitating the translocation of bacterial endotoxins. This “leaky” gut might aggravate the hepatic injury and affect other organs.
Dysregulation of pancreatic beta-cells and hyperglycemia in ethanol-fed MCJ-KO mice
Chronic pancreatitis and endocrine dysfunctions are common among individuals with alcoholism.22,23 Increased hepatic Glut2 expression (Supplemental Figure 2d, http://links.lww.com/HEP/C648) along with altered glucose levels in ethanol-fed MCJ-KO mice suggested a possible pancreatic injury (Supplemental Figure 3a, http://links.lww.com/HEP/C648).
Expression of Mcj was confirmed in both human (Supplemental Figure 3b, http://links.lww.com/HEP/C648) and mouse pancreatic islets (Supplemental Figure 3c, http://links.lww.com/HEP/C648), without evident changes after alcohol insult. Following the ethanol bolus at day 11, we performed an IP glucose tolerance test (IPGTT), which revealed no differences between control Wt and MCJ-KO mice. However, ethanol-fed MCJ-KO mice were unable to handle their blood glucose, reaching ≥600 mg/dL concentrations (Figure 4A), which was accompanied by the death of these animals. To elucidate whether MCJ-KO mice already showed chronic pancreatic injury, an IPGTT was performed at day 6, before the bolus. Ethanol-fed MCJ-KO mice showed significantly higher glucose levels, and they already exhibited worse glycemic control (Supplemental. Figure 3d, http://links.lww.com/HEP/C648). Accordingly, defective insulin secretion was identified in ethanol-fed MCJ-KO mice compared with Wt mice since insulin levels dropped significantly at 90 minutes of the IPGTT, matching with the start of the hyperglycemic event (Figure 4B).
FIGURE 4.
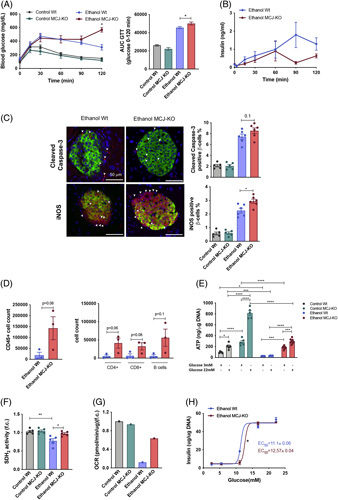
Augmented pancreatic injury and hyperglycaemia in ethanol-fed MCJ-KO mice. (A) Blood glucose levels and the resulting AUC during the IPGTT (B) Insulin levels during the IPGTT. (C) Quantification and representative Cleaved caspase-3 and iNOS stained pancreatic beta cell sections. (D) Pancreatic infiltrating total CD45+ cells and further characterization of CD4+, CD8+, and B cells populations using FACS. (E) ATP content in isolated pancreatic islets. (F) Pancreatic SDH2 activity (G) A representative measurement of the oxygen consumption rate of freshly isolated pancreatic islets. (H) In vitro static insulin release assay, using freshly isolated pancreatic islets. *p<0.05, **p<0.01, and ****p<0.0001 versus Wt.
In accordance with reduced insulin levels, ethanol consumption significantly decreased the number of pancreatic islets, although we observed no differences between ethanol-fed groups (Supplemental Figure 3e, http://links.lww.com/HEP/C648). Interestingly, the histological evaluation of the pancreatic islets showed an increasing trend in cleaved caspase-3 staining and significantly increased iNOS levels in ethanol-fed MCJ-KO mice (Figure 4C and Supplemental. Figure 3f, http://links.lww.com/HEP/C648). We confirmed increased levels of cleaved caspase-3 in pancreatic acinar cells following alcohol consumption, without changes between ethanol-fed groups (Supplemental Figure 3g, http://links.lww.com/HEP/C648). Moreover, we found increased inflammatory infiltration of immune CD4+T, CD8+T, and B cells in ethanol-fed MCJ-KO mice compared with Wt mice (Figure 4D).
At the endpoint of the model, the pancreatic islets were isolated, allowing the in vitro testing of their functionality. As insulin secretion depends on ATP production and Ca+2 signaling,24 intracellular ATP concentrations were measured. MCJ-KO islets produced significantly higher ATP levels in response to glucose in basal and after ethanol MCJ-KO, although these were lower in both groups following ethanol consumption (Figure 4E). In line with this, the activity of mitochondrial SDH2 was increased in the islets of ethanol-fed MCJ-KO mice (Figure 4F), together with the oxygen consumption rate (Figure 4G). However, the static insulin release assay highlighted the reduced glucose-sensing capacity of MCJ-KO islets, as higher glucose levels were needed to release the same amounts of insulin (Figure 4H).
Altogether the pancreas of ethanol-fed MCJ-KO mice suffered 3 main insults: oxidative stress, increased serum LPS, and inflammatory infiltration. All these affect pancreatic islet’s function, causing a decreased glucose-stimulated insulin secretion sensitivity, defective insulin secretion, and thus hyperglycemia.
MJC-LSS ameliorates liver injury and avoids lipid accumulation following ethanol use
We have seen that lack of MCJ results in increased intestinal inflammation, permeability, and LPS leakage, worsening the hepatic injury and causing the failure of pancreatic beta-cells following alcohol consumption, showing that ALD is a systemic affection. However, based on previous studies where liver-specific MCJ silencing proved to be hepatoprotective,10,12,17,18 treatment with liver-specific MCJ siRNA (MCJ-LSS) was used to study liver damage following chronic and acute ethanol abuse. Three-month-old Wt mice were treated by i.v. tail vein injection with MCJ-LSS or siCtrl at day 5 of the NIAAA model (Supplemental. Figure 4a, http://links.lww.com/HEP/C648), and the efficient knockdown of MCJ protein expression was confirmed at the end of the model (Supplemental. Figure 4b, http://links.lww.com/HEP/C648). We also measured the expression of Mcj in both hepatocytes and liver resident macrophages, or Kupffer cells (KCs) which allowed us to confirm the hepatocyte specificity of MCJ-LSS (Supplemental. Figure 4c, http://links.lww.com/HEP/C648).
Surprisingly, we obtained a 100% survival percentage in both ethanol-fed siCtrl and MCJ-LSS treated groups (Figure 5A). Importantly, ethanol-fed MCJ-LSS mice showed reduced liver injury and increased hepatic regeneration, as documented by significantly reduced necrotic areas and lower cleaved caspase-3 and TUNEL staining levels, along with increased PCNA positive immunostainings (Figure 5B and Supplemental. Figure 5a, http://links.lww.com/HEP/C648). The histopathological analysis revealed that ethanol-fed siCtrl mice show more severe hepatic lesions (steatosis and inflammation), with a final score of 2 versus 1.3 obtained by ethanol-fed MCJ-LSS mice (Figure 5C). In addition, proapoptotic BAX protein levels were significantly reduced in MCJ-LSS mice, and although antiapoptotic BCL2 and BCL-XL showed opposite regulation, regenerative PCNA was significantly augmented in MCJ-LSS mice (Supplemental. Figure 5b, http://links.lww.com/HEP/C648). No changes were observed in serum ALT and AST levels; however, ethanol-fed MCJ-LSS mice showed reduced bilirubin and increased prothrombin and albumin levels (Supplemental. Figure 5c, http://links.lww.com/HEP/C648), proving improved hepatic function.
FIGURE 5.
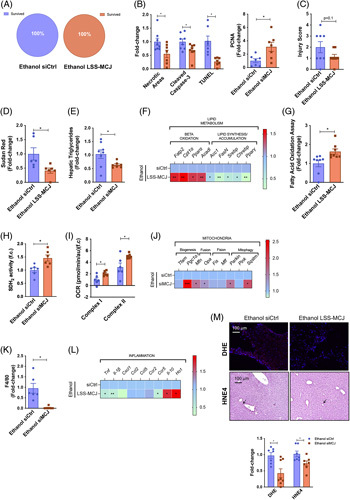
Liver specific silencing of Mcj reduces liver damage and steatosis following the NIAAA model. (A) Survived and Deceased siCtrl (Left panel) and MCJ-LSS (Right panel) mice. (B) Quantification of Necrotic Areas (based on HE staining), Cleaved Caspase-3, TUNEL, and PCNA stained liver sections. (C) Hepatic histopathological evaluation. (D) Quantification of Sudan Red stained liver sections. (E) Hepatic triglyceride content. (F) Heatmap showing the liver mRNA relative expression of genes involved in lipid metabolism: Fatp2, Cpt1a, Ppara, Acadl, Acc, FasN, Srebp, Chrebp, and Pparg. (G) Hepatic fatty acid oxidation assay. (H) Hepatic SDH2 activity. (I) Oxygen Consumption Rate of mitochondrial Complex I and Complex II in freshly isolated mitochondria. (J) Heatmap showing the liver mRNA relative expression of mitochondrial quality control genes: Tfam, Pgc1a, Mfn, Opa1, Fis1, Mff, Prkn, Pink1, and Sqstm. (K) Quantification of F4/80 stained liver sections. (L) Heatmap showing the liver mRNA relative expression of genes involved in inflammation: Tnf, Il-1b, Cxcl1, Ccl2, Ccl5, Ccr2, Ccr5, Il-10, and Ho-1. (M) Quantification and representative DHE and 4HNE stained liver sections. *p<0.05, **p<0.01, and ***p<0.001 versus Wt.
In line with previous studies, liver-specific MCJ silencing avoided lipid accumulation following ethanol abuse (Figure 5D, Supplemental. 5d, (http://links.lww.com/HEP/C648) and Figure 5E). Compared to siCtrl, MCJ-LSS mice showed increased expression of the genes involved in fatty acid oxidation and reduced expression of those involved in de novo lipogenesis and accumulation (Figure 5F). Increased fatty acid oxidation activity in MCJ-LSS mice confirmed that metabolism is oriented towards lipid catabolism (Figure 5G). Additionally, improved mitochondrial function was observed in MCJ-LSS mice, with increased SDH2 activity (Figure 5H) and mitochondrial respiration (Figure 5I), together with higher expression of genes coordinating mitochondrial biogenesis, fusion, and mitophagy (Figure 5J).
The inflammatory response was also reduced in MCJ-LSS mice based on reduced hepatic monocyte infiltration (Figure 5K and Supplemental Figure 5d, http://links.lww.com/HEP/C648), decreased levels of the proinflammatory cytokines Tnf, Il-1b, and Ccr5, and increased expression of antiinflammatory Il-10 and Ho-1 compared to Wt mice (Figure 5L). No fibrotic markers were detected (Supplemental. Figure 5d, http://links.lww.com/HEP/C648).
Additionally, MCJ-LSS mice showed no differences related to ethanol accumulation and a slight tendency towards reduced acetaldehyde content (Supplemental Figure 5e, http://links.lww.com/HEP/C648). No significant changes were observed in mRNA levels of the main enzymes related to alcohol metabolism (Supplemental. Figure 5f, http://links.lww.com/HEP/C648). Importantly, the lack of MCJ avoided excessive ROS production with significantly lower DHE and 4HNE staining levels (Figure 5M). We could not observe any differences in the GSH/GSSG ratio (Supplemental Figure 5g, http://links.lww.com/HEP/C648).
For the study of systemic changes induced by ethanol, we first confirmed that MCJ-LSS did not affect the expression of Mcj in both intestine and pancreatic islets (Supplemental 6a, b, http://links.lww.com/HEP/C648). In contrast to MCJ-KO, MCJ-LSS did not increase intestinal permeability (Figure 6A, B) and serum LPS levels (Figure 6C), thus avoiding further systemic effects. Indeed, both groups showed similar glucose levels (Figure 6D), glycemic control (Figure 6E), and pancreatic mitochondrial function (Figure 6F, G).
FIGURE 6.
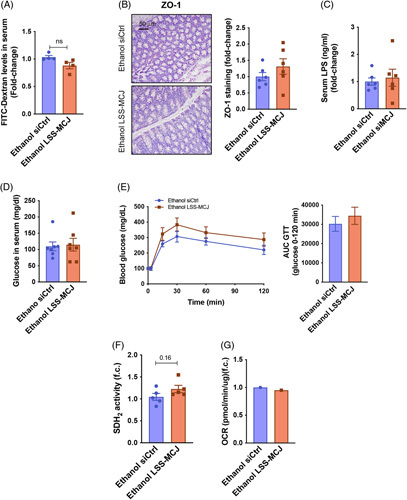
Liver-specific silencing of Mcj does not induce a systemic alcohol injury. (A) Serum FITC-Dextran levels. (B) Quantification and representative Zonula occludens stained gut sections. (C) Serum LPS content. (D) Serum glucose levels. (E) Blood glucose levels and the resulting AUC during the IPGTT. (F) Pancreatic SDH2 activity. (G) A representative measurement of the oxygen consumption rate of freshly isolated pancreatic islets.
Thus, MCJ-LSS is hepatoprotective following chronic and acute ethanol consumption, and it could be posed as a new therapeutic approach to treat ALD.
MCJ-LSS inhibits mTOR activation avoiding de novo lipogenesis
We aimed at understanding the exact mechanism by which MCJ-LSS improves hepatic steatosis and ameliorates ALD. Thus, we performed high-throughput proteomics liquid chromatography-mass spectrometry-based analyses in ethanol-fed siCtrl and MCJ-LSS mice. Ingenuity pathway analysis was used to identify the major canonical pathways involved in the hepatoprotective effect of MCJ-LSS in ALD, suggesting mTOR signaling and its downstream pathways, which play an essential role in regulating lipid metabolism, fatty acid oxidation, and de novo lipogenesis25–27 to be altered (Figure 7A). All the identified hepatic proteins were exhibited in a Volcano Plot (Figure 7B). Among the 69 significantly differentially expressed proteins highlighted within the plot, we focused on mTOR interactors. Interestingly, we observed proteins whose expression is upregulated due to mTOR activation in siCtrl mice (COX1, SERPINA), and MCJ-LSS mice show proteins that are positively regulated when the mTOR pathway is inhibited (ATP2B, DMD).
FIGURE 7.
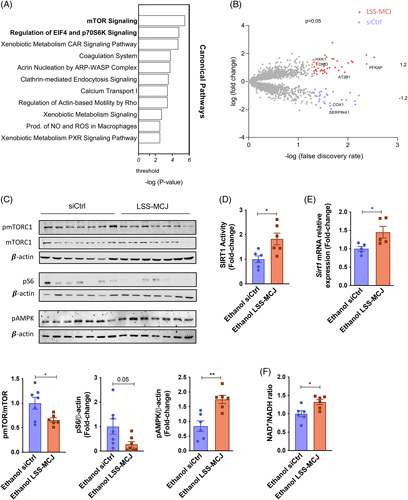
Silencing of MCJ inhibits mTOR activation via increased NAD+ and improved Sirt1 activity. (A) Ingenuity pathway analysis of top canonical pathways. (B) Volcano plot showing all the identified hepatic proteins. Statistically significant proteins are shown in the corresponding colors, and the highlighted proteins were identified as mTOR interactors. (C) Activated and total hepatic mTOR protein levels, together with activated S6 protein levels, by western blotting and densitometric quantification. ß-actin as a loading control. (D) Hepatic SIRT1 activity. (E) Sirt1 hepatic mRNA relative expression. (F) Hepatic NAD+/NADH ratio. *p<0.05 versus Wt.
To confirm these results, we studied the activation levels of the mTOR pathway and its downstream interactors in both siCtrl and MCJ-LSS mice by western blot (Figure 7C). MCJ-LSS significantly reduced mTORC1 phosphorylation and, therefore, its activation compared to siCtrl mice. Following mTOR inhibition, the phosphorylation of S6 protein (pS6), a downstream target of mTOR, was also reduced in MCJ-LSS mice. mTORC1 regulates de novo lipogenesis by increasing the expression of the main transcriptional factors for the enzymes participating in lipid synthesis.28 Expression of transcriptional factors Srebp1 and Chrebp was previously measured, and it was significantly reduced in MCJ-LSS mice (Figure 5E). Moreover, increased activation of AMPK (Figure 7C), whose downstream signaling inhibits de novo lipogenesis29 and enhanced fatty acid oxidation (Figure 5G) were observed in MCJ-LSS mice.
Aberrant activation of mTORC1 has been previously linked to defects in SIRT1, which appears downregulated in ALD patients.28,30 Silencing of MCJ significantly augmented both hepatic SIRT1 activity (Figure 7D) and expression (Figure 7E) compared to siCtrl mice. Besides, since SIRT1 activity is NAD+ dependent, a significantly higher NAD+/NADH ratio was found in MCJ-LSS mice compared to siCtrl mice (Figure 7F).
Overall, our study shows that Mcj silencing improves mitochondrial activity and helps recover an appropriate NAD+/NADH ratio, which enhances lipid beta-oxidation and by means of SIRT1 also avoids mTORC1 activation and the subsequent de novo lipogenesis. Thus, targeting mitochondrial dysfunction prevents alcohol-mediated hepatic steatosis and ALD progression, posing it as a potential therapeutic approach for treating this disease.
DISCUSSION
Excessive alcohol consumption is the primary cause of liver-related mortality in Western countries.31 Based on the recent data showing that lack of MCJ, a negative regulator of the complex I in the mitochondrial electron transport chain, boosts mitochondrial activity without collateral ROS,10,12,17,18 we aimed to study its effect in ALD. Herein, we have shown both sides of the coin. On the one hand, the whole body's lack of MCJ worsens the systemic effects of ALD through intestinal inflammation and altered permeability, increased endotoxemia, and dysregulated pancreatic function, which overall exacerbates liver injury. On the other hand, liver-specific Mcj silencing reduces liver damage, accelerates regeneration, and ameliorates main ALD hallmarks: steatosis, inflammation, and ROS, thereby halting its progression. Our data suggest that a lack of hepatic MCJ prevents mitochondrial dysfunction and metabolic alterations by improving mitochondrial activity and restoring NAD+ levels together with SIRT1 function without any additional ROS production.
Mitochondrial dysfunction is one of the earliest indicators of alcohol-induced injury.5 Our work has attempted for the first time to study the implication of MCJ in the initiation and pathophysiology of ALD, and to do so, we chose the NIAAA model, developed by Gao and colleagues, and the DUAL model, developed by Benedé-Ubieto et al13, both used to study the early stages of ALD. In these mice, hepatic expression of Mcj was decreased, seemingly due to hypermethylation of the CpG islands found in the transcription starting site, as Barbier-Torres et al12 had observed. Based on decreased Mcj expression, even in 23 and 52 weeks of the DUAL model, new animal models are required to further investigate the role of MCJ in advanced stages of ALD. Interestingly, ALD patients exhibited differential expression patterns depending on the severity and chronicity of the disease, downregulated at early stages and overexpressed in the later stages. In contrast to our previous works where we have linked MCJ overexpression with liver damage,10,12,17,32 we hypothesize that hepatic downregulation of MCJ at early ALD stages might be an adaptive response to metabolize the increasing alcohol intake.
The metabolism of high concentrations of alcohol results not only in acetaldehyde, which has cellular toxic effects, but also in the decreased NAD+/NADH ratio, altering critical metabolic pathways.6 Following the use of alcohol, the mitochondria need to augment oxygen consumption as an adaptive response to oxidize the toxic metabolite acetaldehyde more rapidly and to increase NAD+ supply both for alcohol metabolism and restore hepatic metabolism. This phenomenon is named Swift Increase in Alcohol Metabolism.5 Thus, the downregulation of MCJ in the initial ALD states would enhance mitochondrial respiration and help metabolize the high amounts of alcohol. However, if alcohol consumption becomes chronic, mitochondrial overactivation may lead to ROS overproduction. Thus, repression of mitochondrial respiration by MCJ overexpression may seem to be a mechanism to reduce ROS production; however, it also reduces NAD+ generation, an essential cofactor for alcohol and aldehyde dehydrogenases. Therefore, ethanol in this condition would be primarily metabolized through CYP2E1, which consumes NADPH, generates ROS, and leads to toxic acetaldehyde accumulation, driving disease progression.33,34 Therefore, avoiding mitochondrial dysfunction and ROS overproduction by silencing Mcj may be the firewall to delay or even prevent ALD patients’ downfall.
Following the NIAAA model, whole-body MCJ-KO mice showed nearly 4 times increased mortality when compared to WT mice. It needs to be highlighted that alcohol causes systemic disease.7 However, the effect of acetaldehyde and ROS lie far beyond the liver, affecting other organs.8 We did find slightly increased hepatic injury in ethanol-fed MCJ-KO mice, but it was moderate, mainly caused by mitochondrial dysfunction, altered lipid metabolism, and augmented fatty acid deposition.
Gut injury turned out to be the aggravating factor. Translocation of bacterial endotoxins due to increased immune infiltration, dysbiosis, and augmented intestinal permeability contributed to hepatic apoptosis. Our results confirm what Pascual-Itoiz et al21 already described; under inflammatory conditions, lack of MCJ plays a detrimental role in the gut following the dextran sulfate sodium model. In our study, ethanol-fed MCJ-KO exhibits metabolic endotoxemia, a diet-induced, 2–3-fold increase in plasma LPS levels, associated with low-grade systemic inflammation and worsening of the disease.35,36
Ethanol-fed MCJ-KO mice also showed pancreatic affliction. Chronic pancreatitis is common in people suffering from alcoholism,22 and metabolic endotoxemia has been associated with diabetes mellitus.37 Consistently, the study of the endocrine pancreatic function highlighted a worsened glycemic regulation in MCJ-KO mice, along with defective insulin secretion. Metabolic endotoxemia and ROS production seem to have blunted the glucose-sensing capacity of MCJ-KO pancreatic beta-cells, leading to increased blood glucose levels that, in the worst cases, ended in fatal hyperglycemia. Moreover, significantly increased expression of hepatic glucose transporter Glut2 and several genes implicated in the de novo lipogenesis, specially Chrebp, whose expression is upregulated by substrate supply, suggest that ethanol-fed MCJ-KO livers are taking high glucose amounts and turning them into fatty acids, hence, the augmented fatty acid deposition. Therefore, following ethanol consumption, whole-body MCJ-KO mice do not die due to their hepatic damage but because of systemic injury, metabolic endotoxemia, and hyperglycemia.
On the contrary, MCJ-LSS did show hepatoprotective effects. Following the NIAAA model, lack of MCJ significantly reduced hepatic injury, augmented liver regeneration, and prevented mitochondrial dysfunction, steatosis, inflammation, and ROS production. Besides, MCJ-LSS shockingly increased the survival rate to 100%. No differences were found in blood LPS and glucose levels when compared to siCtrl mice, confirming the deleterious role of MCJ in nonhepatic tissues following ALD.
MCJ-LSS exerts hepatoprotective functions, enhancing lipid beta-oxidation, as already published by our lab,12 but also modulates de novo lipogenesis, shifting lipid metabolism towards catabolism. Alcohol-induced hepatic steatosis happens at early ALD stages, and it is reversible, highlighting the opportunity for therapeutic intervention to prevent ALD. In silico analysis of liquid chromatography-mass spectrometry results showed decreased enrichment of the mTOR and its downstream pathways in MCJ-LSS mice. mTORC1 signaling plays a vital role in regulating lipid metabolism, such as de novo lipogenesis by augmenting the transcription of SREBP1.38 Chen et al28 already showed that mTORC1 activity is enhanced in experimental animals and ALD patients.28 Interestingly, aberrant activation of mTORC1 has been linked to defects in NAD+-dependent SIRT1.28,30 Ethanol exposure downregulates the expression of SIRT1,28 and its deacetylase activity is sensitive to NADH redox state, so alterations in NAD+/NADH ratio caused by alcohol metabolism also blunt its function.39 Thus, increased NAD+, by silencing Mcj, restores both SIRT1 expression and activity, avoiding mTOR activation and subsequent de novo lipogenesis. Moreover, SIRT1 also activates AMPK,29 whose downstream signaling inhibits ATP-consuming processes, such as de novo lipogenesis, by phosphorylating and inhibiting ACC, ChREBP, and SREBP1, key lipogenic enzymes, and transcription factors.
Altogether, our study highlights the need for targeted, specific therapeutic approaches. Liver-specific Mcj silencing ameliorates one of the earliest indicators and possible drivers of ALD, mitochondrial dysfunction, posing it as a potential therapeutic approach for treating this disease. However, the use of a drug that would modulate MCJ levels in a broad range would be deleterious. Importantly, Barbier-Torres et al12, used FDA-approved nanoparticle-formulated and GalNAc-formulated siRNA to efficiently target hepatic MCJ, laying the foundation for a direct, fast, and already accessible therapeutic approach that would halt, ameliorate, and even prevent the progression of ALD.
AUTHOR CONTRIBUTIONS
Naroa Goikoetxea-Usandizaga, Miren Bravo, Franz Martín, and María Luz Martínez-Chantar: conception or designing of the work, drafting, and substantively revising the work. Naroa Goikoetxea-Usandizaga, Miren Bravo, Leire Egia-Mendikute, Leticia Abecia, Marina Serrano-Macia, Rocio G Urdinguio, Marc Clos-Garcia, Ruben Rodriguez-Agudo, Raquel Araujo-Legido, Lucia Lopez-Bermudo, Teresa C Delgado, Irene Gonzalez-Recio, Sofia Lachiondo-Ortega, Claudia Gil-Pitarch, Ainize Pena-Cearra, Jorge Simon, Raquel Benede-Ubieto, Silvia Arino, Jose M. Herranz, Mikel Azkargorta, Julio Salazar-Bermeo, Nuria Marti, Marta Varela-Rey, Juan M. Falcon-Perez, Oscar Lorenzo, Ruben Nogueiras, Felix Elortza, Yulia A Nevzorova, Francisco J Cubero, Domingo Saura, Luis Alfonso Martinez-Cruz, Guadalupe Sabio, Asis Palazon, Pau Sancho-Bru, Natalia Elguezabal, Mario F. Fraga, Matias A. Avila, Ramon Bataller, Jose J. G. Marin, Franz Martin, and Maria Luz Martinez-Chantar: acquisition, analysis, or interpretation of data.
ACKNOWLEDGMENTS
The authors thank Begoña Rodríguez Iruretagoyena for the technical support provided.
FUNDING INFORMATION
This work was supported by grants from Ministerio de Ciencia e Innovación, Programa Retos-Colaboración RTC2019-007125-1 (for Jorge Simon and Maria Luz Martinez-Chantar); Ministerio de Economía, Industria y Competitividad, Retos a la Sociedad AGL2017-86927R (for F.M.); Instituto de Salud Carlos III, Proyectos de Investigación en Salud DTS20/00138 and DTS21/00094 (for Jorge Simon and Maria Luz Martinez-Chantar, and Asis Palazon. respectively); Instituto de Salud Carlos III, Fondo de Investigaciones Sanitarias co-founded by European Regional Development Fund/European Social Fund, “Investing in your future” PI19/00819, “Una manera de hacer Europa” FIS PI20/00765, and PI21/01067 (for Jose J. G. Marin., Pau Sancho-Bru,. and Mario F. Fraga respectively); Departamento de Industria del Gobierno Vasco (for Maria Luz Martinez-Chantar); Asturias Government (PCTI) co-funding 2018-2023/FEDER IDI/2021/000077 (for Mario F. Fraga.); Ministerio de Ciencia, Innovación y Universidades MICINN: PID2020-117116RB-I00, CEX2021-001136-S PID2020-117941RB-I00, PID2020-11827RB-I00 and PID2019-107956RA-100 integrado en el Plan Estatal de Investigación Científica y Técnica y Innovación, cofinanciado con Fondos FEDER (for Maria Luz Martinez-Chantar, Francisco J Cubero., Yulia A Nevzorova and Asis Palazon); Ayudas Ramón y Cajal de la Agencia Estatal de Investigación RY2013-13666 and RYC2018-024183-I (for Leticia Abecia and Asis Palazon); European Research Council Starting Grant 804236 NEXTGEN-IO (for Asis Palazon); The German Research Foundation SFB/TRR57/P04, SFB1382-403224013/A02 and DFG NE 2128/2-1 (for Francisco J Cubero and Yulia A Nevzorova); National Institute of Health (NIH)/National Institute of Alcohol Abuse and Alcoholism (NIAAA) 1U01AA026972-01 (For Pau Sancho-Bru); Junta de Castilla y León SA074P20 (for Jose J. G. Marin); Junta de Andalucía, Grupo PAIDI BIO311 (for Franz Martin); CIBERER Acciones Cooperativas y Complementarias Intramurales ACCI20-35 (for Mario F. Fraga); Ministerio de Educación, Cultura y Deporte FPU17/04992 (for Silvia Ariño); Fundació Marato TV3 201916-31 (for Jose J. G. Marin.); Ainize Pena-Cearra is a fellow of the University of the Basque Country (UPV/EHU); BIOEF (Basque Foundation for Innovation and Health Research); Asociación Española contra el Cáncer (Maria Luz Martinez-Chantar and Teresa C. Delgado.); Fundación Científica de la Asociación Española Contra el Cáncer (AECC Scientific Foundation) Rare Tumor Calls 2017 (for Maria Luz Martinez-Chantar); La Caixa Foundation Program (for Maria Luz Martinez-Chantar); Proyecto Desarrollo Tecnologico CIBERehd (for Maria Luz Martinez-Chantar); Ciberehd_ISCIII_MINECO is funded by the Instituto de Salud Carlos III.
CONFLICTS OF INTEREST
María Luz Martínez-Chantar advises for Mitotherapeutix LLC. Guadalupe Sabio received grants from Mitotherapeutix LLC. The remaining authors have nothing to report.
Supplementary Material
Footnotes
Abbreviations: 4HNE, 4-hydroxynonenal; APAP, Acetaminophen); Acc, Acetyl-CoA carboxylase; Ap-1, Activator protein-1; Acadl, Acyl-CoA Dehydrogenase Long Chain; ALT, Alanine aminotransferase; Adh1, Alcohol dehydrogenase; AH, alcohol-associated hepatitis; ALD, alcohol-associated liver disease; ASH, alcohol-associated steatohepatitis; Aldh2, Aldehyde dehydrogenase 2; AMPK, AMP-activated protein kinase; AST, Aspartate aminotransferase; Chrebp, Carbohydrate-responsive element-binding protein; Cpt1a, Carnitine Palmitoyltransferase 1A; Ccl2, C-C Motif Chemokine Ligand 2; Ccl5, C-C Motif Chemokine Ligand 5; CLR, Central Log-Ratio; Cxcl1, C-X-C Motif Chemokine Ligand 1; Cyp2e1, Cytochrome P450 Family 2 Subfamily E Member 1; DHE, Dihydroethidium; Dnm1l, Dynamin like 1; FasN, Fatty acid synthase; Fatp2, Fatty acid transport protein 2; FITC, Fluorescein isothiocyanate; GTT, Glucose tolerance test; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; Ho-1, Heme oxygenase-1; Hamp, Hepcidin antimicrobial peptide; iNOS, Inducible nitric oxide synthase; IPA, Ingenuity pathway analysis; Il-1b, Interleukin 1 beta; Il-10, Interleukin-10; IPGTT, Intraperitoneal glucose tolerance test; LPS, Lipopolysaccharide; LT, Liver transplantation; mTOR, Mammalian target of rapamycin; MCJ-KO, Mcj knockout; MCJ-LSS, Mcj liver-specific silencing; MCJ, Methylation-controlled J protein; Mff, Mitochondrial fission factor; Fis1, Mitochondrial fission protein 1; Mfn, Mitofusin; NIAAA, National Institute on Alcohol abuse and Alcoholism; OPA1, mitochondrial dynamin like GTPase (Opa1); Prkn, Parkin; Ppara, Peroxisome proliferator-activated receptor alpha; Pparg, Peroxisome proliferator-activated receptor gamma; Pgc1a, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; Pink1, PTEN induced kinase 1; ROS, Reactive oxygen species; Sqstm1, Sequestosome1; Sirt1, Sirtuin 1; Srebp, Sterol regulatory element-binding protein 1; SDH2, Succinate dehydrogenase 2; TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling; Tlr, Toll-Like receptor; Tlr4, Toll-Like receptor 4; Tfam, Transcription factor A, mitochondrial; Tnf, Tumor necrosis factor; Wt, Wild-type; ZO-1, Zonula occludens
Naroa Goikoetxea-Usandizaga and Miren Bravo are joint first authors.
Current address: Marc Clos-García, LEITAT Technological Center, Terrassa 08225, Spain
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com.
Contributor Information
Naroa Goikoetxea-Usandizaga, Email: ngoikoetxea@cicbiogune.es.
Miren Bravo, Email: mbravo@cicbiogune.es.
Leire Egia-Mendikute, Email: legia@cicbiogune.es.
Leticia Abecia, Email: labecia@cicbiogune.es.
Marina Serrano-Maciá, Email: mserrano@cicbiogune.es.
Rocío G. Urdinguio, Email: rgurdinguio@gmail.com.
Marc Clos-García, Email: mclos@leitat.org.
Rubén Rodríguez-Agudo, Email: rrodriguez@cicbiogune.es.
Raquel Araujo-Legido, Email: raquel.araujo@cabimer.es.
Lucía López-Bermudo, Email: lucia.lopez@cabimer.es.
Teresa C. Delgado, Email: tcardoso@cicbiogune.es.
Sofía Lachiondo-Ortega, Email: slachiondo@cicbiogune.es.
Irene González-Recio, Email: irecio@cicbiogune.es.
Clàudia Gil-Pitarch, Email: cgil@cicbiogune.es.
Ainize Peña-Cearra, Email: ainize.zearra@hotmail.com.
Jorge Simón, Email: jsimon.ciberehd@cicbiogune.es.
Raquel Benedé-Ubieto, Email: rabenede@ucm.es.
Silvia Ariño, Email: SARINOM@CLINIC.CAT.
Jose M. Herranz, Email: jherranz.1@alumni.unav.es.
Mikel Azkargorta, Email: mazkargorta@cicbiogune.es.
Julio Salazar-Bermeo, Email: jsalazar@mitrasoltech.com.
Nuria Martí, Email: nmarti@umh.es.
Marta Varela-Rey, Email: martavarela.rey@usc.es.
Juan M. Falcón-Pérez, Email: jfalcon@cicbiogune.es.
Óscar Lorenzo, Email: OLorenzo@fjd.es.
Rubén Nogueiras, Email: ruben.nogueiras@usc.es.
Félix Elortza, Email: felortza@cicbiogune.es.
Yulia A. Nevzorova, Email: yulianev@ucm.es.
Francisco J. Cubero, Email: fcubero@ucm.es.
Domingo Saura, Email: dsaura@umh.es.
Luis Alfonso Martínez-Cruz, Email: amartinez@cicbiogune.es.
Guadalupe Sabio, Email: guadalupe.sabio@cnic.es.
Asís Palazón, Email: apalazon@cicbiogune.es.
Pau Sancho-Bru, Email: PSANCHO@clinic.cat.
Natalia Elguezabal, Email: nelguezabal@neiker.eus.
Mario F. Fraga, Email: mffraga@cinn.es.
Matías A. Ávila, Email: maavila@unav.es.
Ramón Bataller, Email: bataller@pitt.edu.
José J.G. Marín, Email: jjgmarin@usal.es.
Franz Martín, Email: fmarber@upo.es.
María Luz Martínez-Chantar, Email: mlmartinez@cicbiogune.es.
REFERENCES
- 1.Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol. 2013;59:160–8. [DOI] [PubMed] [Google Scholar]
- 2.Pimpin L, Cortez-Pinto H, Negro F, Corbould E, Lazarus JV, Webber L, Sheron N. EASL HEPAHEALTH Steering Committe. Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies. J Hepatol. 2018;69:718–35. [DOI] [PubMed] [Google Scholar]
- 3.Mathurin P, Lucey MR. Liver transplantation in patients with alcohol-related liver disease: current status and future directions. Lancet Gastroenterol Hepatol. 2020;5:507–14. [DOI] [PubMed] [Google Scholar]
- 4.Teschke R. Alcoholic liver disease: alcohol metabolism, cascade of molecular mechanisms, cellular targets, and clinical aspects. Biomedicines. 2018;6:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong Z, Ramshesh VK, Rehman H, Liu Q, Theruvath TP, Krishnasamy Y, et al. Acute ethanol causes hepatic mitochondrial depolarization in mice: role of ethanol metabolism. PLoS One. 2014;9:e91308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World J Gastroenterol. 2014;20:17756–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.González-Reimers E. Alcoholism: a systemic proinflammatory condition. World J Gastroenterol. 2014;20:14660–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatle KM, Gummadidala P, Navasa N, Bernardo E, Dodge J, Silverstrim B, et al. MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol Cell Biol. 2013;33:2302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbier-Torres L, Iruzubieta P, Fernández-Ramos D, Delgado TC, Taibo D, Guitiérrez-de-Juan V, et al. The mitochondrial negative regulator MCJ is a therapeutic target for acetaminophen-induced liver injury. Nat Commun. 2017;8:2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navasa N, Martín I, Iglesias-Pedraz JM, Beraza N, Atondo E, Izadi H, et al. Regulation of oxidative stress by methylation-controlled j protein controls macrophage responses to inflammatory insults. J Infect Dis. 2015;211:135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbier-Torres L, Fortner KA, Iruzubieta P, Delgado TC, Giddings E, Chen Y, et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat Commun. 2020;11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8:627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Argemi J, Latasa MU, Atkinson SR, Blokhin IO, Massey V, Gue JP, et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun. 2019;10:3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nevzorova YA, Boyer-Diaz Z, Cubero FJ, Gracia-Sancho J. Animal models for liver disease—a practical approach for translational research. J Hepatol. 2020;73:423–40. [DOI] [PubMed] [Google Scholar]
- 16.Benedé‐Ubieto R, Estévez‐Vázquez O, Guo F, Chen C, Singh Y, Nakaya HI, et al. An experimental DUAL model of advanced liver damage. Hepatol Commun. 2021;5:1051–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iruzubieta P, Goikoetxea-Usandizaga N, Barbier-Torres L, Serrano-Maciá M, Fernández-Ramos D, Fernández-Tussy P, et al. Boosting mitochondria activity by silencing MCJ overcomes cholestasis-induced liver injury. JHEP Rep. 2021;3:100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goikoetxea‐Usandizaga N, Serrano‐Maciá M, Delgado TC, Simón J, Fernández Ramos D, Barriales D, et al. Mitochondrial bioenergetics boost macrophage activation, promoting liver regeneration in metabolically compromised animals. Hepatology. 2021;75:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avila MA, Dufour JF, Gerbes AL, Zoulim F, Bataller R, Burra P, et al. Recent advances in alcohol-related liver disease (ALD): summary of a Gut round table meeting. Gut. 2021;69:764–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bishehsari F, Magno E, Swanson G, Desai V, Voigt RM, Forsyth CB, et al. Alcohol and Gut-Derived Inflammation. Alcohol Res Curr Rev. 2013;38:163–71. [PMC free article] [PubMed] [Google Scholar]
- 21.Pascual-Itoiz MA, Peña-Cearra A, Martín-Ruiz I, Lavín JL, Simó C, Rodríguez H, et al. The mitochondrial negative regulator MCJ modulates the interplay between microbiota and the host during ulcerative colitis. Sci Rep. 2020;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren Z, Yang F, Wang X, Wang Y, Xu M, Frank JA, et al. Chronic plus binge ethanol exposure causes more severe pancreatic injury and inflammation. Toxicol Appl Pharmacol. 2016;308:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yue F, Zhang X, Zhang H, Jiang X, Gao L, Zhao J. Association of alcohol consumption with the impaired β-cell function independent of body mass index among chinese men. Endocr J. 2012;59:425–33. [DOI] [PubMed] [Google Scholar]
- 24.Klec C, Ziomek G, Pichler M, Malli R, Graier WF. Calcium Signaling in ß-cell Physiology and Pathology: A Revisit. Int J Mol Sci. 2019;20:6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19:R1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrano-Maciá M, Simón J, González-Rellan MJ, Azkargorta M, Goikoetxea-Usandizaga N, Lopitz-Otsoa F, et al. Neddylation inhibition ameliorates steatosis in NAFLD by boosting hepatic fatty acid oxidation via the DEPTOR-mTOR axis. Mol Metab. 2021;53:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricoult SJH, Manning BD. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013;14:242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H, Shen F, Sherban A, Nocon A, Li Y, Wang H, et al. DEPTOR Suppresses Lipogenesis and Ameliorates Hepatic Steatosis and Acute-on-Chronic Liver Injury in Alcoholic Liver Disease. Hepatology. 2019;68:496–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price NL, Gomes AP, Ling AJY, Duarte FV, Martin-Montalvo A, North BJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X, et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 2011;25:1664–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–42. [DOI] [PubMed] [Google Scholar]
- 32.Goikoetxea‐Usandizaga N, Serrano‐Maciá M, Delgado TC, Simón J, Fernández Ramos D, Barriales D, et al. Mitochondrial bioenergetics boost macrophage activation, promoting liver regeneration in metabolically compromised animals. Hepatology. 2022;75:550–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdelmegeed MA, Banerjee A, Jang S, Yoo S-H, Yun J-W, Gonzalez FJ, et al. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis and apoptosis. Free Radic Biol Med. 2013;65:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, Cederbaum AI. Cytochrome P450S and Alcoholic Liver Disease. Curr Pharm Des. 2018;24:1502–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boutagy NE, McMillan RP, Frisard MI, Hulver MW. Metabolic endotoxemia with obesity: is it real and is it relevant? Biochimie. 2016;124:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohammad S, Thiemermann C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front Immunol. 2021;11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomes JMG, Costa JA, Alfenas RCG. Metabolic endotoxemia and diabetes mellitus: a systematic review. Metabolism. 2017;68:133–44. [DOI] [PubMed] [Google Scholar]
- 38.Caron A, Richard D, Laplante M. The Roles of mTOR Complexes in Lipid Metabolism. Annu Rev Nutr. 2015;35:321–48. [DOI] [PubMed] [Google Scholar]
- 39.You M, Arteel GE. Effect of ethanol on lipid metabolism. J Hepatol. 2019;70:237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]