

Abstract
Background and Aims
The ProCID study evaluated the efficacy and safety of three doses of a 10% liquid intravenous immunoglobulin (IVIg) preparation (panzyga®) in patients with chronic inflammatory demyelinating polyneuropathy (CIDP). This report describes the safety findings.
Methods
Patients were randomised to receive a 2.0 g/kg induction dose followed by maintenance doses of either 0.5, 1.0 or 2.0 g/kg IVIg every 3 weeks over 24 weeks.
Results
All 142 enrolled patients were included in the safety analyses. In total, 286 treatment-emergent adverse events (TEAEs) were reported in 89 patients, of which 173 (60.5%) were considered treatment-related. Most TEAEs were of mild severity. Eleven serious TEAEs were reported in 6 patients. Two serious TEAEs in one patient (headache and vomiting) were considered related to treatment, which resolved without study discontinuation. No treatment-related thrombotic events, haemolytic transfusion reactions or deaths occurred. One patient discontinued the study due to a TEAE (allergic dermatitis) probably related to IVIg. Headache was the only dose-dependent TEAE, with incidences ranging from 2.9 to 23.7%, the incidence of all other TEAEs was similar across treatment groups. Most TEAEs were associated with the induction dose infusion, and the rate of TEAEs decreased thereafter. The median (IQR) daily IVIg dose was 78 (64–90) g, and 94.4% of patients tolerated the maximal infusion rate of 0.12 ml/kg/min without pre-medication.
Interpretation
Infusions of 10% IVIg at doses up to 2.0 g/kg with high infusion rates were safe and well tolerated in patients with CIDP.
Clinical trial numbers
EudraCT 2015-005443-14, NCT02638207.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40264-023-01326-z.
Key Points
| Infusions of IVIg 0.5, 1.0 or 2.0 g/kg every 3 weeks for up to 24 weeks and daily doses of up to 128 g were safe and well tolerated in CIDP patients. |
| Headache was the only dose-dependent adverse event. |
Introduction
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a treatable peripheral neuropathy. Its course can be chronic progressive or relapsing, and while its aetiology remains to be fully elucidated, it is thought to be an autoimmune disorder [1]. Three treatments have been shown to be effective for CIDP: immunoglobulin both intravenous (IVIg) and subcutaneous (SCIg); corticosteroids; and plasma exchange [2, 3]. Where available, IVIg is frequently the first choice treatment and has been shown to improve patient outcomes rapidly and persistently [4–10]. The most commonly used treatment regimen for IVIg in patients with CIDP consists of a 2.0 g/kg induction dose followed by 1.0 g/kg maintenance doses every 3 weeks [3].
The Progress in Chronic Inflammatory Demyelinating polyneuropathy (ProCID) study investigated the efficacy and safety of IVIg (panzyga®) treatment by randomising patients to a standard induction dose of 2.0 g/kg followed by one of three maintenance doses: 0.5, 1.0 or 2.0 g/kg IVIg (ratio 1:2:1). The main results of the ProCID study were published and showed in the study population a response rate of 80% in the 1.0 g/kg treatment group and indicated that a lower or higher maintenance dose may be as beneficial in some patients [10]. Here, the detailed safety findings of the study are reported.
Materials and Methods
Study Design and Participants
The design of the ProCID study (EudraCT 2015-005443-14, NCT02638207) and the efficacy and main safety results have been published [10, 11]. In brief, all patients were above 18 years of age and diagnosed with CIDP according to the EFNS/PNS 2010 Guidelines [12]. The main inclusion criteria were active disease and ongoing treatment with corticosteroids or immunoglobulins. The main exclusion criteria were failure to respond to immunoglobulin treatment, recent treatment with immunomodulatory or chemotherapeutic agents, and clinical evidence of peripheral neuropathy of another cause. The protocol was approved by the ethics committees of all participating centres, and informed written consent was obtained from all patients prior to any study-related procedures.
Study Drug
IVIg (panzyga®, Octapharma Pharmazeutika Produktionsges.m.b.H., Vienna, Austria; previously called NewGam) is an intravenous human 10% immunoglobulin [13, 14] with demonstrated efficacy and safety in patients with primary immunodeficiency [15], thrombocytopenic purpura [16] and CIDP [10].
Procedures
Detailed study procedures were described previously [11]. Briefly, after an initial screening phase, eligible patients underwent a wash-out phase in which their current medication was reduced in a stepwise manner over a period of up to 12 weeks. Patients whose condition deteriorated during the wash-out phase were considered as having active CIDP. Deterioration was determined based on the Patients’ Global Impression of Change scale, the Inflammatory Neuropathy Cause and Treatment (INCAT) score, grip strength, or the Inflammatory Rasch-built Overall Disability Scale (I-RODS) [17]. Patients with active CIDP were enrolled in the dose-evaluation phase and randomised (1:2:1) to the three study arms with maintenance doses of 0.5, 1.0 or 2.0 g/kg IVIg. Following an initial induction dose of 2.0 g/kg IVIg, patients received maintenance doses every 3 weeks for 24 weeks. All dosing was based on the patient’s actual body weight without a maximal total amount of IVIg per day and delivered over 2 days. The initial infusion rate for all infusions was 0.01 mL/kg/min for the first 30 min. If tolerated, the infusion rate was increased every 30 min in the following steps: 0.02, 0.04, 0.08 and 0.12 mL/kg/min for the remainder of the infusion. From the third infusion on, the 30-min interval for the 0.02–0.08 mL/kg/min infusion rates could be shortened to 15 min at the investigator’s discretion. Pre-medication to alleviate potential tolerability problems was not permitted except for patients who experienced 2 consecutive infusion-related adverse events (AEs) that were likely to be prevented by pre-medication.
Safety and Tolerability Outcomes
AEs with a particular emphasis on thromboembolic events (TEE) and haemolytic transfusion reactions, vital signs (blood pressure, heart rate, body temperature and respiratory rate), and laboratory parameters (haematology and clinical chemistry) were recorded throughout the study. A physical examination was performed at screening and every 12 weeks starting at week 0 and in cases of potential AEs additionally if needed. Viral safety was assessed at baseline and at the end of the Extension Period by means of viral marker testing in plasma specimens.
AEs were classified by system organ class and preferred term as defined by the Medical Dictionary for Regulatory Activities (MedDRA® Version 20). Onset, duration, time to the AE occurrence from last dose, causality, dosage, severity, seriousness and actions taken were documented. An AE was defined as a treatment-emergent AE (TEAE) if first onset or worsening occurred after the start of the first IVIg infusion. TEAEs were considered temporally related if they occurred within 72 h after ending infusion. The intensity of TEAEs and their relationship to IVIg were evaluated by the investigator on a case-by-case basis and according to international standards for causality assessment. An AE was considered probably related if good reasons and sufficient documentation supported the assumption of a causal relationship, such as temporal sequence, expected response pattern, or dose-dependency. AEs for which causality was deemed not impossible and not unlikely, but the connection with the study drug remained uncertain or doubtful, were rated as possibly related to the study drug. AEs which were not following a reasonable temporal sequence from administration of the study drug were assessed as unlikely to be related. Events for which sufficient information existed to conclude that the aetiology is unrelated to drug safety were rated as not related/unrelated. Transient TEAEs causing discomfort without interfering with routine activities were classified as mild, TEAEs sufficiently discomforting to interfere with routine activities (but still possible) were considered moderate, and any TEAE incapacitating the patient and preventing the pursuit of routine activities was considered severe. Life-threatening TEAEs and TEAEs resulting in hospitalisation, persistent or significant disability, or death of the patient were classified as serious TEAEs.
Statistical Analysis
Safety was analysed in all randomised patients who received at least part of one IVIg infusion. Statistical analyses were performed using SAS Software version 9.4. Data for all endpoints are presented descriptively.
Results
Demographic and Baseline Characteristics
Patients were enrolled between 9 August 2017 and 5 September 2019. Out of 171 screened patients, 142 eligible patients with active disease were randomised to the three study arms. All randomised patients received at least one IVIg dose and are included in the safety analysis set: 35 (24.6%) patients in the 0.5 g/kg group; 69 (48.6%) in the 1.0 g/kg group, and 38 (26.8%) in the 2.0 g/kg group.
The demographics and clinical characteristics of the patients in the safety analysis set were similar across the dose groups (Table 1). The median age was 59 years, and 58 patients were female (40.8%). The patients’ weight ranged from 48 to 122 kg, with body mass index (BMI) ranging from 16.6 to 39.9 kg/m2. Most patients (91.5%) had typical CIDP. Prior to study enrolment, 87.3% of patients had been treated with corticosteroids, and 12.7% with immunoglobulins.
Table 1.
Baseline characteristics
| Characteristic | Treatment group | All patients | ||
|---|---|---|---|---|
| 0.5 g/kg (n = 35) | 1.0 g/kg (n = 69) | 2.0 g/kg (n = 38) | (n = 142) | |
| Female, n (%) | 13 (37.1) | 31 (44.9) | 14 (36.8) | 58 (40.8) |
| Age, years | 56 (26–73) | 59 (18–83) | 63 (30–83) | 59 (18–83) |
| Body weight, kg | 83 (56–120) | 80 (49–122) | 76 (48–122) | 79 (48–122) |
| Body mass index, kg/m2 | 27 (19–40) | 27 (17–39) | 25 (19–40) | 27 (17–40) |
| EFNS/PNS criteria, n (%) | ||||
| Definite CIDP | 35 (100) | 68 (98.6) | 38 (100) | 141 (99.3) |
| Probable CIDP | 0 | 1 (1.4) | 0 | 1 (0.7) |
| Type of CIDP, n (%) | ||||
| Typical | 34 (97.1) | 62 (89.9) | 34 (89.5) | 130 (91.5) |
| Atypical | 1 (2.9) | 7 (10.1) | 4 (10.5) | 12 (8.5) |
| Prior treatment, n (%) | ||||
| Corticosteroids | 30 (85.7) | 60 (87.0) | 34 (89.5) | 124 (87.3) |
| Immunoglobulins | 5 (14.2) | 9 (13.0) | 4 (10.5) | 18 (12.7) |
All values are the median (range) unless otherwise stated
n number of patients, CIDP chronic inflammatory demyelinating polyneuropathy, EFNS/PNS European Federation of Neurological Societies/Peripheral Nerve Society
Exposure
A total of 982 infusion cycles were administered with 105 patients (73.9%) receiving all their infusions on schedule. Including the induction dose of 2.0 g/kg, the median (range) IVIg dose per infusion cycle was 70 (38–169) g in the 0.5 g/kg group, 101 (57–208) g in the 1.0g/kg group, and 155 (96–237) g in the 2.0 g/kg group, corresponding to 0.69, 1.13 and 2.00 g/kg, respectively. Across the three groups, the daily IVIg doses ranged from 11 g to 128 g with a median (IQR) dose of 78 (64–90) g. Of the 982 infusions, 182 were given at a dose of 0.5 g/kg, 398 at 1.0 g/kg and 402 at 2.0 g/kg. The allowed maximum infusion rate of 0.12 mL/kg/min was reached at least once in 971 out of 982 infusion cycles (98.9%) and 94.4% of patients tolerated the maximal infusion rate of 0.12 ml/kg/min without pre-medication. Most patients (97.9%) had 2 consecutive infusion days with a total median (range) infusion duration of 320 (166–593) minutes for each infusion cycle. Three patients (2.1%) had only one infusion per cycle. Eleven patients (7.7%) received pre-medication (mostly antihistamines) to alleviate adverse events, as allowed by the protocol.
Safety
Of the 142 patients in the study, 89 patients (62.7%) experienced a total of 286 TEAEs. The proportion of patients with TEAEs was similar across dose groups (Table 2). The majority of TEAEs were mild in intensity (226, 79.0%), 18.2% were of moderate intensity and 2.8% of severe intensity. Seven of the severe TEAEs were reported in 3 patients in the 1.0 g/kg group: 1 patient experienced osteomyelitis; 1 patient experienced unilateral deafness; and 1 patient experienced respiratory arrest, decubitus ulcer, pneumonia, cardio-respiratory arrest and aspiration. One patient in the 2.0 g/kg group had a severe TEAE: encephalitis. None of the severe TEAEs were considered related to IVIg treatment.
Table 2.
Summary of TEAEs per treatment group
| TEAE category | Treatment group | Total | ||
|---|---|---|---|---|
| 0.5 g/kg (n = 35) n (%) N |
1.0 g/kg (n = 69) n (%) N |
2.0 g/kg (n = 38) n (%) N |
All patients (n = 142) n (%) N |
|
| TEAEs | 20 (57.1%) 54 | 45 (65.2%) 153 | 24 (63.2%) 79 | 89 (62.7%) 286 |
| Mild | 16 (45.7%) 48 | 28 (40.6%) 115 | 18 (47.4%) 63 | 62 (43.7%) 226 |
| Moderate | 4 (11.4%) 6 | 14 (20.3%) 31 | 5 (13.2%) 15 | 23 (16.2%) 52 |
| Severe | 0 (0.0%) 0 | 3 (4.3%) 7 | 1 (2.6%) 1 | 4 (2.8%) 8 |
| TEAEs related to IVIg treatment | 16 (45.7%) 37 | 32 (46.4%) 80 | 20 (52.6%) 56 | 68 (47.9%) 173 |
| Serious TEAEs | 1 (2.9%) 1 | 4 (5.8%) 9 | 1 (2.6%) 1 | 6 (4.2%) 11 |
| Related serious TEAEs | 0 (0.0%) 0 | 1 (1.4%) 2 | 0 (0.0%) 0 | 1 (0.7%) 2 |
| TEAEs leading to discontinuation of IVIg treatment | 2 (5.7%) 2 | 1 (1.4%) 2 | 2 (5.3%) 2 | 5 (3.5%) 6 |
| TEAEs leading to death | 0 (0.0%) 0 | 1 (1.4%) 1 | 1 (2.6%) 1 | 2 (1.4%) 2 |
n number of patients, N number of events, IVIg intravenous immunoglobulin, TEAE treatment-emergent adverse event
Of the 286 TEAEs, 173 in 68 patients were considered related to IVIg treatment (Table 2). The most frequent related TEAEs were headache, pyrexia and dermatitis (Table 3 and Supplementary Table 1). Overall, the profile of TEAEs related to IVIg treatment was similar across the dose groups (Supplementary Table 1).
Table 3.
Related TEAEs per preceding dose
| System organ class Preferred term | Preceding dose | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 g/kg (n = 34) | 1.0 g/kg (n = 66) | 2.0 g/kg (n = 142) | All patients (n = 142) | |||||||||
| n (%) | n′ (%) | N | n (%) | n′ (%) | N | n (%) | n′ (%) | N | n (%) | n′ (%) | N | |
| Nervous system disorders | ||||||||||||
| Headache | 3 (4.6) | 3 (0.8) | 3 | 18 (12.7) | 22 (5.5) | 25 | 20 (14.1) | 25 (2.6) | 28 | |||
| Dizziness | 1 (2.9) | 1 (0.6) | 1 | 1 (0.7) | 1 (0.3) | 1 | 2 (1.4) | 2 (0.2) | 2 | |||
| Somnolence | 1 (2.9) | 1 (0.6) | 1 | 3 (2.1) | 3 (0.8) | 3 | 3 (2.1) | 4 (0.4) | 4 | |||
| General disorders and administration site conditions | ||||||||||||
| Pyrexia | 2 (5.9) | 3 (1.7) | 3 | 3 (4.6) | 3 (0.8) | 3 | 14 (9.9) | 14 (3.5) | 17 | 18 (12.7) | 20 (2.0) | 23 |
| Chills | 1 (2.9) | 2 (1.1) | 2 | 5 (3.5) | 5 (1.2) | 5 | 6 (4.2) | 7 (0.7) | 7 | |||
| Asthenia | 1 (0.7) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Chest pain | 1 (0.7) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Influenza like illness | 1 (1.5) | 1 (0.3) | 1 | 2 (1.4) | 2 (0.5) | 2 | 3 (2.1) | 3 (0.3) | 3 | |||
| Chest discomfort | 1 (0.7) | 2 (0.5) | 2 | 1 (0.7) | 2 (0.2) | 2 | ||||||
| Gastrointestinal disorders | ||||||||||||
| Abdominal pain | 1 (1.5) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Nausea | 1 (1.5) | 1 (0.3) | 1 | 5 (3.5) | 6 (1.5) | 7 | 5 (3.5) | 7 (0.7) | 8 | |||
| Vomiting | 1 (1.5) | 1 (0.3) | 1 | 3 (2.1) | 3 (0.8) | 3 | 3 (2.1) | 4 (0.4) | 4 | |||
| Diarrhoea | 1 (0.7) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Blood and lymphatic system disorders | ||||||||||||
| Leukopenia | 1 (2.9) | 2 (1.1) | 2 | 1 (1.5) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.3) | 1 | 3 (2.1) | 4 (0.4) | 4 |
| Anaemia | 1 (0.7) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Respiratory, thoracic and mediastinal disorders | ||||||||||||
| Cough | 2 (1.4) | 2 (0.5) | 2 | 2 (1.4) | 2 (0.2) | 2 | ||||||
| Tachypnoea | 1 (0.7) | 1 (0.3) | 4 | 1 (0.7) | 1 (0.1) | 4 | ||||||
| Dyspnoea | 1 (0.7) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Cardiac disorders | ||||||||||||
| Tachycardia | 4 (2.8) | 4 (1.0) | 6 | 4 (2.8) | 4 (0.4) | 6 | ||||||
| Skin and subcutaneous tissue disorders | ||||||||||||
| Rash | 1 (2.9) | 1 (0.6) | 1 | 1 (1.5) | 1 (0.3) | 1 | 1 (0.7) | 5 (1.2) | 5 | 3 (2.1) | 7 (0.7) | 7 |
| Dermatitis | 2 (5.9) | 2 (1.1) | 2 | 4 (6.0) | 8 (2.0) | 8 | 9 (6.3) | 10 (2.5) | 11 | 14 (9.9) | 20 (2.0) | 21 |
| Skin exfoliation | 1 (2.9) | 1 (0.6) | 1 | 2 (3.0) | 2 (0.5) | 3 | 1 (0.7) | 1 (0.3) | 1 | 4 (2.8) | 4 (0.4) | 5 |
| Urticaria | 1 (2.9) | 1 (0.6) | 1 | 3 (2.1) | 3 (0.8) | 3 | 3 (2.1) | 4 (0.4) | 4 | |||
| Pruritus | 2 (1.4) | 3 (0.8) | 3 | 2 (1.4) | 3 (0.3) | 3 | ||||||
| Erythema | 1 (2.9) | 2 (1.1) | 2 | 1 (0.7) | 2 (0.2) | 2 | ||||||
| Vascular disorders | ||||||||||||
| Hypertension | 2 (5.9) | 2 (1.1) | 2 | 2 (3.0) | 3 (0.8) | 3 | 4 (2.8) | 4 (1.0) | 5 | 7 (4.9) | 9 (0.9) | 10 |
| Hypotension | 1 (0.7) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Investigations | ||||||||||||
| Blood lactate dehydrogenase increased | 4 (6.0) | 4 (1.0) | 4 | 1 (0.7) | 1 (0.3) | 1 | 5 (3.5) | 5 (0.5) | 5 | |||
| Transaminases increased | 1 (2.9) | 1 (0.6) | 1 | 1 (1.5) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.3) | 1 | 3 (2.1) | 3 (0.3) | 3 |
| Alanine aminotransferase increased | 2 (3.0) | 2 (0.5) | 2 | 1 (0.7) | 1 (0.3) | 1 | 2 (1.4) | 3 (0.3) | 3 | |||
| Aspartate aminotransferase increased | 2 (3.0) | 2 (0.5) | 2 | 1 (0.7) | 1 (0.3) | 1 | 2 (1.4) | 3 (0.3) | 3 | |||
| Haemoglobin decreased | 1 (1.5) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Infections and infestations | ||||||||||||
| Nasopharyngitis | 1 (0.7) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
| Renal and urinary disorders | ||||||||||||
| Micturition urgency | 1 (0.7) | 1 (0.3) | 1 | 1 (0.7) | 1 (0.1) | 1 | ||||||
n number of patients, n′ number of infusions, N number of events, TEAE treatment-emergent adverse event
Headache was the only IVIg-related TEAE showing dose-dependency. The incidence per allocated treatment group was 2.9%, 14.5% and 23.7% in the 0.5, 1.0 and 2.0 g/kg groups, respectively (Supplementary Table 1). Dose dependency was also seen when analysed according to the preceding IVIg dose rather than allocated treatment group. No patients experienced headache after a dose of 0.5 g/kg, 3 patients (4.5%) reported headache after a dose of 1.0 g/kg, and 18 patients (12.7%) experienced headache after an induction or maintenance dose of 2.0 g/kg IVIg (Fig. 1, Table 3).
Fig. 1.
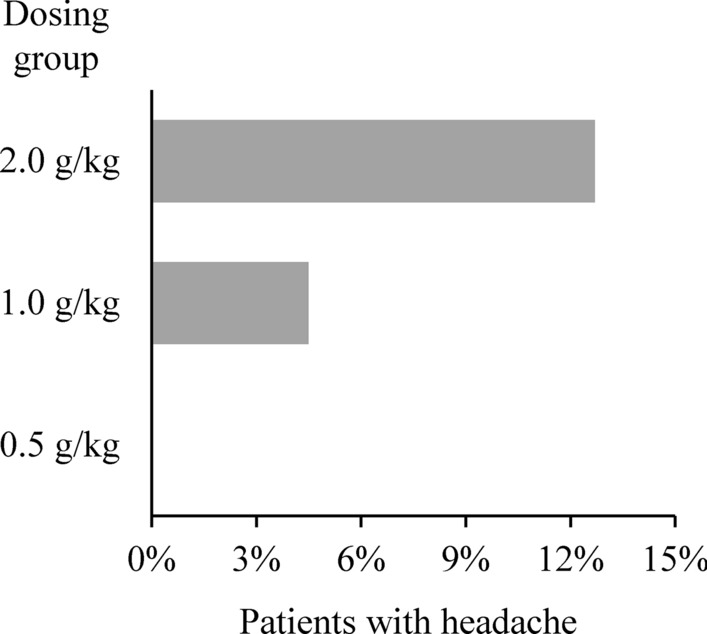
Incidence of treatment-related headache per preceding dose
Eleven serious TEAEs were reported in 6 patients (4.2%), two of which were considered related to IVIg treatment. Both events, headache and vomiting, developed in the same patient in the 1.0 g/kg group, were of moderate intensity and resolved without study discontinuation. The same patient withdrew from the study following a diagnosis of meningioma, which was surgically removed 2 months later. Two patients in the 1.0 g/kg group experienced serious TEAEs of osteomyelitis and unilateral deafness. Both events were rated as severe, resolved following treatment, and were considered unrelated to the study drug. For the patient with deafness, the diagnosis was concluded to be sudden right ear deafness during otorhinolaryngology consultation. There were no signs of aseptic or immune meningitis. The patient recovered and the deafness resolved. The patient continued in the study and completed the study 3 months later. One serious event of moderate osteonecrosis leading to study withdrawal is described below. Two patients experienced TEAEs leading to death. One patient in the 1.0 g/kg group developed fatal encephalitis (rhomboencephalitis, probably autoimmune), which was considered unrelated to IVIg treatment. One patient in the 2.0 g/kg group with a history of cardiovascular disease developed bronchopneumonia, which resulted in cardio-respiratory arrest. The aspiration and respiratory arrest were not related to IVIg, the patient had a history of coughing before receiving the study drug (this was only mentioned by the family after the aspiration, not before the IVIg infusion) and the event of aspiration happened after the IVIg infusion and immediately after the patient took a spoon of natural plantago syrup. The patient had a relevant medical history of hypertension, peripheral vascular disorder, peripheral arterial occlusive disease, adrenal adenoma, cholelithiasis, intervertebral disc protrusion, haemangioma of bone, and cough.
Five patients (3.5%) experienced 6 TEAEs that led to discontinuation of the study drug (Table 4). One event of allergic dermatitis in the patient in the 1.0 g/kg group was considered probably related to IVIg treatment. The same patient experienced a urinary tract infection, which was considered unrelated to IVIg treatment. One patient in the 0.5 g/kg group developed autoimmune hepatitis, which did not resolve despite treatment but was considered unrelated to the study drug. A second patient in the 0.5 g/kg group developed moderate osteonecrosis, which did not resolve. The event was rated as a serious TEAE and considered as unlikely to be related to IVIg treatment. In the 2.0 g/kg group, one patient had mildly increased fibrin D dimer but no signs and symptoms of a TEE. The increase was considered as unrelated to the study drug. A second patient in the 2.0 g/kg group developed a serious TEAE of encephalitis, which was considered not related to IVIg treatment (see above).
Table 4.
TEAEs leading to study discontinuation
| Treatment group | Dose type | Preferred term | Intensity | Outcome | Causality | SAE? |
|---|---|---|---|---|---|---|
| 0.5 g/kg | Induction | Autoimmune hepatitis | Moderate | Not resolved | Not related | No |
| 2.0 g/kg | Induction | Fibrin D dimer increased | Mild | Unknown | Unlikely | No |
| 0.5 g/kg | Maintenance | Osteonecrosis | Moderate | Not resolved | Unlikely | Yes |
| 2.0 g/kg | Maintenance | Encephalitis | Severe | Fatal | Not related | Yes |
| 1.0 g/kg |
Maintenance Maintenance |
Dermatitis allergic Urinary tract infection |
Moderate Moderate |
Resolved Resolved |
Probable Not related |
No No |
SAE serious adverse event, TEAE treatment-emergent adverse event
The median daily dose of IVIg was 78 g, and the incidence of related TEAEs was similar in patients receiving higher (> 78 g) or lower (≤ 78 g) daily doses of IVIg (Fig. 2 and Supplementary Table 2). In total, 96 related TEAEs were observed after 721 doses ≤ 78 g IVIg (13.3%), compared with 77 after 662 doses > 78 g IVIg (11.6%). In the lower IVIg daily dose group, the most frequent TEAEs were headache, followed by pyrexia and dermatitis. The most common TEAEs in patients receiving higher daily IVIg doses were dermatitis followed by pyrexia and headache.
Fig. 2.
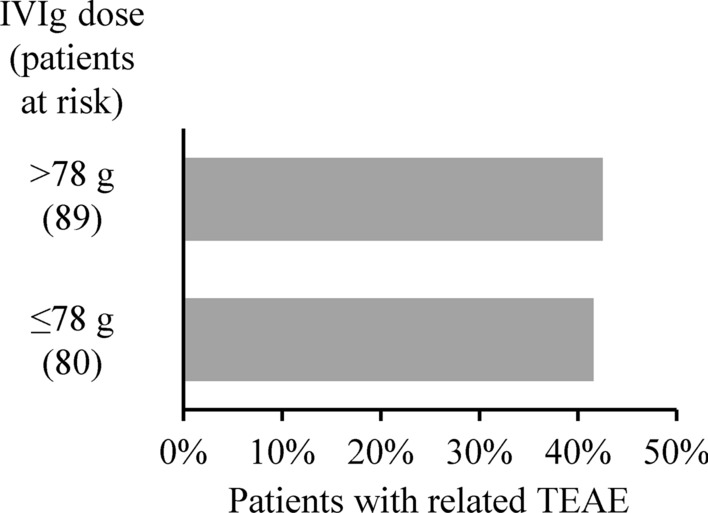
Incidence of related TEAEs after higher (> 78 g) and lower (≤ 78 g) total daily IVIg doses. TEAE treatment-emergent adverse event
The majority of related TEAEs occurred following infusion of the 2.0 g/kg induction doses (Fig. 3). Eighty-eight related TEAEs were reported for the 142 induction dose infusion cycles in the study (0.62 TEAEs/infusion) compared with 85 events after 840 maintenance doses (0.10 TEAEs/infusion, Supplementary Table 3). For all treatment groups, the incidence of related TEAEs per infusion decreased sharply after the induction dose and gradually declined further throughout the maintenance phase from 0.16 TEAEs per infusion for the first maintenance dose to 0.05 by the end of the study. Only 8 of 142 patients (5.6%) did not tolerate the highest infusion rate (0.12 mL/kg/min) due to the occurrence of TEAEs. Among these 8 patients, 3 never reached the highest infusion rate due to TEAEs at lower rates.
Fig. 3.

Rate of related TEAEs per infusion for induction and maintenance doses. TEAE treatment-emergent adverse event
The results of the laboratory assessments (haematology, clinical chemistry, urinalysis and viral markers) did not raise any safety concerns. There were no unusual observations on physical examination or in the patients’ vital signs. No signs of thromboembolism or haemolysis were recorded in this study.
Discussion
The ProCID study was the first randomised clinical trial comparing the efficacy and safety of different maintenance doses of IVIg treatment in patients with CIDP. TEAEs in CIDP patients were mostly mild in intensity with headache as the most frequent and the only dose-dependent TEAE. More than half of the events were associated with the induction dose infusion, and the incidence of TEAEs per infusion decreased considerably thereafter. The distribution of TEAEs was similar across dose groups with the exception of headache.
Although reactions to IVIg treatment are often mitigated by either pre-medication or a reduction in infusion rate [18], only 7.7% of patients needed pre-medication and the maximum infusion rate of 0.12 mL/kg/min was reached in almost all infusions in ProCID, thus confirming the good tolerability and safety of IVIg even at high infusion rates in patients with CIDP [15, 16]. These results also support earlier findings which showed that infusions of IVIg at infusion rates up to 0.14 mL/kg/min were well tolerated in CIDP patients [19]. Our study suggests that routine pre-medication even before high dosages of IVIg is not needed.
IVIg treatment is generally well-tolerated, but a number of adverse effects have been documented and are associated with specific IVIg preparations and individual differences [20]. The most frequent immediate TEAEs related to IVIg infusions are flu-like symptoms, e.g. headache, nausea, fever and chills, followed by dermatological effects such as urticaria or dermatitis. Flu-like symptoms account for more than 80% of IVIg-associated side effects [20, 21]. They usually occur within the first hour of infusion and are in most cases mild and transient [21].
An important aspect of this study was the inclusion of the 2.0 g/kg dose group who received 232 of infusions, not counting the induction dose which all dose groups received. The literature does contain likely hundreds of reports of patients who received 2.0 g/kg usually monthly for a variety of diseases (see Supplemental Table 4 Reference list, a partial list) but most are only small case series and evaluate high dose IVIg treatment for only 4–16 weeks. There are very few controlled studies in the literature of longitudinal dosing at this high dose [22–24]. These report either no adverse events or common ones such as headache or hypertension. One study described 6 patients who received very high dose IVIg, ranging from 2.0–9.0 g/kg/month, for periods up to 48 months [25]. The authors reported no IVIg-related adverse events.
IVIg treatment has been shown to cause thrombotic events, renal failure and haematologic disorders such as haemolysis and neutropenia [20]. While the incidence of renal impairment is generally rare, thromboembolic events and haemolysis affect approximately 1% of patients [20, 26]. Both events have been reported in patients with CIDP after IVIg administration and are usually associated with high IVIg doses [5, 8] or specific indications such as dermatomyositis [27–29]. Our study treated patients for up to 24 weeks and confirmed the safety and tolerability of the 2.0 g/kg dose given every 3 weeks. The single study most similar to ProCID is the recently published ProDERM study [30]. In that study, patients with dermatomyositis, the majority also on corticosteroids, were treated with IVIg 10% at a dose of 2.0 g/kg monthly for up to 16 weeks in the primary study. Patients could then join an open-label study that lasted up to 24 weeks. While the main safety and tolerability results, with headache and nausea being the most common related TEAEs, are similar to the 2.0 g/kg dose group in the ProCID study, the major difference was the occurrence of 6 related TEEs in 5 patients—one deep vein thrombosis, two pulmonary embolism, one hypoesthesia and two strokes. This is most likely due to the underlying condition, dermatomyositis, which alone predisposes to TEE [27–29]. A large UK study found that IVIg is a risk factor for TEE but only for those with prior TEE [31] supporting the hypothesis that not high IVIg dosing but rather additional risk factors such as prior TEE or specific clinical conditions (e.g. dermatomyositis) increase the risk of TEEs in patients receiving IVIg therapy [32, 33]. This is also supported by a study evaluating a second dose of IVIg in patients with severe Guillain-Barré syndrome. Patients receiving a second dose of IVIg experienced more TEEs than those receiving a single dose [34]. The immobility due to the severe disease and the older age of the second dose IVIg group (the authors state that the 2 groups were not evenly matched) represented independent risk factors for TEE. The ProCID study indicates that, contrary to dermatomyositis, CIDP by itself is not a risk factor for TEE. In the ProCID study, patients received daily doses of IVIg of up to 128 g IVIg without any reported thrombotic or haemolytic events or other unexpected safety signals.
The safety findings in our study are in line with those of previous trials with this IVIg treatment in patients with primary immunodeficiency [15] and chronic immune thrombocytopenia [16]. The efficacy and safety of IVIg in CIDP were also investigated in the ICE, PATH, PRIMA and PRISM studies at a maintenance dose of 1.0 g/kg. In the ICE study, the most frequent related TEAEs in the IVIg group were headache in 32% of patients, followed by pyrexia (13%) and hypertension (9%) [4]. In a combined analysis of the PATH and PRIMA studies, treatment-related TEAEs were reported in 45% of patients, with headache (17%), nausea (5%), hypertension and haemolysis (4% each) as the most frequent TEAEs [5]. In the PRIMA study, headache was reported in 29% of patients and hypertension and asthenia in 14% each [6]. At this dose, headache was documented in 14.5% of patients in ProCID, followed by pyrexia (11.6%), and hypertension, dermatitis and increased blood lactate dehydrogenase (7.3% each). In contrast to the above studies [4, 5, 8], the vast majority of patients (89.4%) in the ProCID study were previously treated with corticosteriods and were IVIg-naive. These data suggest that patients can be safely switched from corticosteroids to IVIg.
The risk of adverse reactions to IVIg treatment is considered to be higher in obese patients, who generally require high total doses of IVIg and frequently present with multiple cardiovascular risk factors [35]. In the ProCID study, all dosing was based on actual body weight, and there was no limit on the total daily amount of IVIg. High total amounts of IVIg were not associated with an increase in the incidence of TEAEs, suggesting that dosing based on actual body weight does not necessarily put patients at an increased risk of TEAEs. Moreover, the incidence of TEAEs associated with maintenance infusions of 2.0 g/kg IVIg was similar to those observed for 0.5 g/kg and 1.0 g/kg doses.
The favourable safety profile of IVIg at high daily (up to 128 g/day) and total doses (up to 2 g/kg) and high infusion rates (up to 0.12 mL/kg/min) in this carefully selected patient population suggests that there is no general need to limit daily or maintenance dosing or infusion rates for safety reasons, as long as patients are monitored for possible risk factors for adverse events (such as TEEs).
The strengths of this study include the multiple and high IVIg doses administered, the large size and age range of the patient population and the limited prior exposure to IVIg. The main limitation of this safety study was the duration of 24 weeks which provides limited information on longer-term side-effects associated with IVIg treatment.
Conclusions
The evaluation of safety and tolerability parameters showed that administration of IVIg in the ProCID study was safe in patients with CIDP. In a largely IVIg naïve patient population, the profile of TEAEs was as expected for IVIg products. Headache was the only dose-dependent TEAE, and very few patients required premedication to mitigate TEAEs. IVIg infusions were well tolerated at high daily and total doses and at high infusion rates, with maximal infusion rates of 0.12 mL/kg/min reached at least once in 99% of infusion cycles.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This study was sponsored by Octapharma Pharmazeutika Produktionsges.m.b.H. (Vienna, Austria) who thank investigators, trial personnel and patients for their participation. Medical writing assistance was provided by nspm ltd, Meggen, Switzerland, and funded by Octapharma. The members of ProCID study investigators: Bulgaria S Kastrev and V Rizova, Multiprofile Hospital for Active Treatment Puls AD, Blagoevgrad; I Milanov, University Neurological Hospital St. Naum, Sofia. Canada R Massie, Montreal Neurological Hospital, Montreal. Czech Republic R Taleb and M Bednar, Private Neurology Practice, Hradec Kralove; P Ridzon, Thomayers Hospital, Prague. Germany J Schmidt and J Zschüntzsch, University Medical Center Göttingen. Hungary R Csilla, Jahn Ferenc Del‐pesti Hospital, Budapest; L Vécsei, University of Szeged, Szeged. Poland K Rejdak, Medical University of Lublin; M Koszewicz and S Budrewicz, Wroclaw Medical University, Wroclaw. Romania A Docu-Axelerad, Spitalul Clinic Judetean de Urgenta “Sf. Apostol Andrei”, Constanta; A Dulamea and M Marian, University of Medicine and Pharmacy "Carol Davila" Fundeni Clinical Institute, Bucharest; A. Kadar and L Zecheru-Lapusneanu, Teo Health Brasov, Brasov. Russia V Mikhailov and D Zakharov, Bekhterev National Research Medical Center for Psychiatry and Neurology, St. Petersburg; N Suponeva and M Piradov, Russian Academy of Sciences Research Center of Neurology, Moscow. Ukraine N Smolko and D Smolko, Vinnytsya National Medical University, Vinnytsya.
Declarations
Funding
This study was funded, and open access fees paid for by Octapharma Pharmazeutika Produktionsges.m.b.H. (Vienna, Austria).
Conflict of interest
D.R.C. reports consulting for Amgen Inc., Annexon Biosciences, Boehringer Ingelheim, Grifols S.A., Johnson & Johnson, Neura Bio, Novartis, Octapharma AG, Pfizer Inc., Roche, Seattle Genetics Inc., Valenzabio. D.R.C. is on the Data Safety Monitoring Board for the following: PledPharma, Hansa Medical, and Mitsubishi Tanabe Pharma Corporation. D.R.C. is on the Scientific Advisory Board for Sinomab, Algotherapeutics, Nervosave. D.R.C. received royalties for technology licensing from Worldwide Clinical Trials, Inc., CMIC, MedImmune Ltd., RWS Life Sciences, Levicept, AstraZeneca Pharmaceuticals, LP, Genentech, Inc., Chiesi Farmaceutici S.p.A., Beijing 3E-Regenacy Pharmaceuticals Co., Passage Bio, and Disarm Therapeutics, outside the submitted work. P.A.v.D. reports grants from Sanquin Blood Supply Prinses Beatrix Spierfonds, and Takeda during the conduct of the study; and consulting fees from Annexion, Argenx, Octapharma, and Hansa, all outside the submitted work. All grants and consulting fees are transferred to the Erasmus MC Research fund. H.P.H. reports consulting for CSL Behring, Sanofi Genzyme, and UCB. H.P.H. received payments or honoraria from CSL Behring and Octapharma. I.S.J.M. reports grants from Talecris Talents program, GBS/CIDP Foundation International and FP7 EU program, outside the submitted work. Furthermore, a research foundation at the University of Maastricht received honoraria on behalf of him for participation in steering committees of the Talecris Immune Globulin Intravenous For Chronic Inflammatory Demyelinating Polyneuropathy Study, Commonwealth Serum Laboratories, Behring, Octapharma, LFB, Novartis, Union Chimique Belge, Johnson & Johnson, Argenx, outside the submitted work. And Octapharma during the conduct of the study. H.D.K. is on Steering Committees for Octapharma and Sanofi Genzyme. H.D.K. reports travel support and consulting fees from Octapharma in relation to a study design advisory board. H.D.K. reports consulting for UCB, Terumo, Akcea, Alnylam, CSL Behring, Merz, Pfizer, Roche and Dyne. H.D.K. is on Data Safety Monitoring Boards or Advisory Boards for UCB, Syneos and Octapharma. H.D.K. received a grant from Takeda for investigator-initiated research. D.H. and E.C. are employees of Octapharma PPG, Vienna, Austria.
Availability of data and material
The ProCID study protocol and key results are available at https://clinicaltrials.gov/ct2/show/results/NCT02638207. To safeguard patient privacy, individual patient data will not be shared.
Code availability
Not applicable.
Ethics approval
The ethics committees of all participating centres approved the study protocol in accordance with the 1964 Helsinki Declaration. The study was approved in Canada by the NEUPSY Research Ethics Board of the McGill University Health Care centers (#2018-4058), in Bulgaria by the Ministry of Health Ethics Committee for Multicenter Trials (#KH-63/21.07.17), in Hungary by the Medical Research Council Ethics Committee for Clinical Pharmacology (ECCP; # ETT File No. 24931-0/2017-EKL), in Ukraine by the ethics board of the Municipal Institution “Zaporizhzhia Regional Clinical Hospital” (05.05.2017: #91), by the Ethics Committee at Ivano-Frankivsk Regional Clinical Hospital (Committee Meeting #28, 24.04.2017), by the Ethics Committee at Volyn Regional Clinical Hospital (Committee Meeting #108 28.04.2017), by the Ethics Committee of Kyiv City Clinical Hospital No. 9 (Committee Meeting #145/02), by the Ethics Committee at Municipal Institution “O.I. Yushchenko Vinnytsia Regional Psychoneurological Hospital” (Agreed Opinion 04.05.2017: #09.17), in Romania by the Academy of Medical Sciences National Bioethics Committee for Medicine and Medical Devices (No. 15S/1-4/13.04.2017, 685A-3.05.2017, 875A-10.07.2017), in Czech Republic by the Ethics Committee, University Hospital Hradec Kralove (CEC Reference Number: 201705 D06M, LEC: 532/21.04.2017), by the Ethics Committee of the Institute of Clinical and Experimental Medicine and Thomayer Hospital (CEC Reference Number: 201705 D06M, LEC: 666/17 + 905/17), by the Ethics Committee Oblastni nemocnice Nachod a.s. (CEC Reference Number: 201705 D06M, LEC: S0065/2017), in Poland by the Bioethics Committee at the Warmia and Mazury Medical Chambers in Olszyn (Resolution No 12/2017/VI), in Germany by the Ethics Committee of the University Medical Center Göttingen (#14/2/17; 28.04.2017), in Russia by the Central Ethics Committee of the Ministry of Health of the Russian Federation, Council of Ethics (Vn. No 51693) and the Local Ethics committees of the Nizhny Novgorod Regional Clinical Hospital n.a. N.A. Semashko, City Multi-Field Hospital No. 2 Budgetary Public Health Facility of Saint-Petersburg, Republican Clinical Neurological Centre Kazan, Independent Ethics Committee at FSBI "Saint Petersburg V.M. Bekhterev Psychoneurological Research Institute", Ethics Committee at Federal State-Financed Research Institution Neurology Research Center.
Consent to participate
All patients gave written informed consent prior to any study-related procedures.
Consent for publication
Not applicable.
Author contributions
D.R.C., P.A.v.D. and H.D.K. contributed substantially to the conception and design of the study. D.R.C. drafted the manuscript, which was revised critically by all other authors. All authors read and approved the final manuscript before submission.
Footnotes
The members of ProCID study investigators are listed in the acknowledgements.
Contributor Information
David R. Cornblath, Email: dcornbl@jhmi.edu
the ProCID Investigators:
S. Kastrev, V. Rizova, I. Milanov, R. Massie, R. Taleb, M. Bednar, P. Ridzon, J. Schmidt, J. Zschüntzsch, R. Csilla, L. Vécsei, K. Rejdak, M. Koszewicz, S. Budrewicz, A. Docu-Axelerad, A. Dulamea, M. Marian, A. Kadar, L. Zecheru-Lapusneanu, V. Mikhailov, D. Zakharov, N. Suponeva, M. Piradov, N. Smolko, and D. Smolko
References
- 1.Koike H, Katsuno M. Pathophysiology of chronic inflammatory demyelinating polyneuropathy: insights into classification and therapeutic strategy. Neurol Ther. 2020;9:213–227. doi: 10.1007/s40120-020-00190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunschoten C, Jacobs BC, van den Bergh PYK, Cornblath DR, van Doorn PA. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol. 2019;18:784–794. doi: 10.1016/S1474-4422(19)30144-9. [DOI] [PubMed] [Google Scholar]
- 3.van den Bergh PYK, van Doorn PA, Hadden RDM, Avau B, Vankrunkelsven P, Allen JA, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint Task Force-Second revision. Eur J Neurol. 2021;28:3556–3583. doi: 10.1111/ene.14959. [DOI] [PubMed] [Google Scholar]
- 4.Hughes RAC, Donofrio P, Bril V, Dalakas MC, Deng C, Hanna K, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7:136–144. doi: 10.1016/S1474-4422(07)70329-0. [DOI] [PubMed] [Google Scholar]
- 5.Merkies ISJ, van Schaik IN, Léger J-M, Bril V, van Geloven N, Hartung H-P, et al. Efficacy and safety of IVIG in CIDP: combined data of the PRIMA and PATH studies. J Peripher Nerv Syst. 2019;24:48–55. doi: 10.1111/jns.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Léger J-M, de Bleecker JL, Sommer C, Robberecht W, Saarela M, Kamienowski J, et al. Efficacy and safety of Privigen(®) in patients with chronic inflammatory demyelinating polyneuropathy: results of a prospective, single-arm, open-label Phase III study (the PRIMA study) J Peripher Nerv Syst. 2013;18:130–140. doi: 10.1111/jns5.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nobile-Orazio E, Pujol S, Kasiborski F, Ouaja R, Della Corte G, Bonek R, et al. An international multicenter efficacy and safety study of IqYmune in initial and maintenance treatment of patients with chronic inflammatory demyelinating polyradiculoneuropathy: PRISM study. J Peripher Nerv Syst. 2020;25:356–365. doi: 10.1111/jns.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuwabara S, Mori M, Misawa S, Suzuki M, Nishiyama K, Mutoh T, et al. Intravenous immunoglobulin for maintenance treatment of chronic inflammatory demyelinating polyneuropathy: a multicentre, open-label, 52-week phase III trial. J Neurol Neurosurg Psychiatry. 2017;88:832–838. doi: 10.1136/jnnp-2017-316427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belmokhtar C, Lozeron P, Adams D, Franques J, Lacour A, Godet E, et al. Efficacy and safety of Octagam® in patients with chronic inflammatory demyelinating polyneuropathy. Neurol Ther. 2019;8:69–78. doi: 10.1007/s40120-019-0132-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornblath DR, van Doorn PA, Hartung H-P, Merkies ISJ, Katzberg HD, Hinterberger D, Clodi E. Randomized trial of three IVIg doses for treating chronic inflammatory demyelinating polyneuropathy. Brain. 2022 doi: 10.1093/brain/awab422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornblath DR, Hartung H-P, Katzberg HD, Merkies ISJ, van Doorn PA. A randomised, multi-centre phase III study of 3 different doses of intravenous immunoglobulin 10% in patients with chronic inflammatory demyelinating polyradiculoneuropathy (ProCID trial): Study design and protocol. J Peripher Nerv Syst. 2018;23:108–114. doi: 10.1111/jns.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van den Bergh PYK, Hadden RDM, Bouche P, Cornblath DR, Hahn A, Illa I, et al. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. Eur J Neurol. 2010;17:356–363. doi: 10.1111/j.1468-1331.2009.02930.x. [DOI] [PubMed] [Google Scholar]
- 13.Mersich C, Ahrer K, Buchacher A, Ernegger T, Kohla G, Kannicht C, et al. Biochemical characterization and stability of immune globulin intravenous 10% liquid (Panzyga®) Biologicals. 2017;45:33–38. doi: 10.1016/j.biologicals.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Radomski KU, Lattner G, Schmidt T, Römisch J. Pathogen Safety of a New Intravenous Immune Globulin 10% Liquid. BioDrugs. 2017;31:125–134. doi: 10.1007/s40259-017-0212-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borte M, Melamed IR, Pulka G, Pyringer B, Knutsen AP, Ochs HD, et al. Efficacy and safety of human intravenous immunoglobulin 10% (Panzyga®) in Patients with primary immunodeficiency diseases: a two-stage, multicenter, prospective, Open-Label Study. J Clin Immunol. 2017;37:603–612. doi: 10.1007/s10875-017-0424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arbach O, Taumberger AB, Wietek S, Cervinek L, Salama A. Efficacy and safety of a new intravenous immunoglobulin (Panzyga® ) in chronic immune thrombocytopenia. Transfus Med. 2019;29:48–54. doi: 10.1111/tme.12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Draak THP, Vanhoutte EK, van Nes SI, Gorson KC, van der Pol WL, Notermans NC, et al. Changing outcome in inflammatory neuropathies: Rasch-comparative responsiveness. Neurology. 2014;83:2124–2132. doi: 10.1212/WNL.0000000000001044. [DOI] [PubMed] [Google Scholar]
- 18.Orbach H, Katz U, Sherer Y, Shoenfeld Y. Intravenous immunoglobulin: adverse effects and safe administration. Clin Rev Allergy Immunol. 2005;29:173–184. doi: 10.1385/CRIAI:29:3:173. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y, Mendoza M, Sarpong E, Mannan S, Ng E, Katzberg H, et al. Efficacy and safety of high infusion rate IVIG in CIDP. Muscle Nerve. 2020;62:637–641. doi: 10.1002/mus.27044. [DOI] [PubMed] [Google Scholar]
- 20.Guo Y, Tian X, Wang X, Xiao Z. Adverse effects of immunoglobulin therapy. Front Immunol. 2018;9:1299. doi: 10.3389/fimmu.2018.01299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bichuetti-Silva DC, Furlan FP, Nobre FA, Pereira CTM, Gonçalves TRT, Gouveia-Pereira M, et al. Immediate infusion-related adverse reactions to intravenous immunoglobulin in a prospective cohort of 1765 infusions. Int Immunopharmacol. 2014;23:442–446. doi: 10.1016/j.intimp.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Mignogna MD, Fortuna G, Ruoppo E, Adamo D, Leuci S, Fedele S. Variations in serum hemoglobin, albumin, and electrolytes in patients receiving intravenous immunoglobulin therapy: a real clinical threat? Am J Clin Dermatol. 2007;8:291–299. doi: 10.2165/00128071-200708050-00004. [DOI] [PubMed] [Google Scholar]
- 23.Göttfried I, Seeber A, Anegg B, Rieger A, Stingl G, Volc-Platzer B. High dose intravenous immunoglobulin (IVIG) in dermatomyositis: clinical responses and effect on sIL-2R levels. Eur J Dermatol. 2000;10:29–35. [PubMed] [Google Scholar]
- 24.Jordan SC, Tyan D, Stablein D, McIntosh M, Rose S, Vo A, et al. Evaluation of intravenous immunoglobulin as an agent to lower allosensitization and improve transplantation in highly sensitized adult patients with end-stage renal disease: report of the NIH IG02 trial. J Am Soc Nephrol. 2004;15:3256–3262. doi: 10.1097/01.ASN.0000145878.92906.9F. [DOI] [PubMed] [Google Scholar]
- 25.Kapoor M, Reilly MM, Manji H, Lunn MP, Aisling SC. Dramatic clinical response to ultra-high dose IVIg in otherwise treatment resistant inflammatory neuropathies. Int J Neurosci. 2022;132:352–361. doi: 10.1080/00207454.2020.1815733. [DOI] [PubMed] [Google Scholar]
- 26.Bonilla FA. Adverse effects of immunoglobulin G therapy: thromboembolism and haemolysis. Clin Exp Immunol. 2014;178:72–74. doi: 10.1111/cei.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carruthers EC, Choi HK, Sayre EC, Aviña-Zubieta JA. Risk of deep venous thrombosis and pulmonary embolism in individuals with polymyositis and dermatomyositis: a general population-based study. Ann Rheum Dis. 2016;75:110–116. doi: 10.1136/annrheumdis-2014-205800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung W-S, Lin C-L, Sung F-C, Lu C-C, Kao C-H. Increased risk of venous thromboembolism in patients with dermatomyositis/polymyositis: a nationwide cohort study. Thromb Res. 2014;134:622–626. doi: 10.1016/j.thromres.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 29.Selva-O'Callaghan A, Fernández-Luque A, Martínez-Gómez X, Labirua-Iturburu A, Vilardell-Tarrés M. Venous thromboembolism in patients with dermatomyositis and polymyositis. Clin Exp Rheumatol. 2011;29:846–849. [PubMed] [Google Scholar]
- 30.Aggarwal R, Charles-Schoeman C, Schessl J, Bata-Csörgő Z, Dimachkie MM, Griger Z, et al. Trial of intravenous immune globulin in dermatomyositis. N Engl J Med. 2022;387:1264–1278. doi: 10.1056/NEJMoa2117912. [DOI] [PubMed] [Google Scholar]
- 31.Kapoor M, Hunt I, Spillane J, Bonnett LJ, Hutton EJ, McFadyen J, et al. IVIg-exposure and thromboembolic event risk: findings from the UK Biobank. J Neurol Neurosurg Psychiatry. 2022 doi: 10.1136/jnnp-2022-328881. [DOI] [PubMed] [Google Scholar]
- 32.Neeman E, Liu V, Mishra P, Thai KK, Xu J, Clancy HA, et al. Trends and risk factors for venous thromboembolism among hospitalized medical patients. JAMA Netw Open. 2022 doi: 10.1001/jamanetworkopen.2022.40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson FA, Spencer FA. Risk factors for venous thromboembolism. Circulation. 2003;107:I9–16. doi: 10.1161/01.CIR.0000078469.07362.E6. [DOI] [PubMed] [Google Scholar]
- 34.Walgaard C, Jacobs BC, Lingsma HF, Steyerberg EW, van den Berg B, Doets AY, et al. Second intravenous immunoglobulin dose in patients with Guillain–Barré syndrome with poor prognosis (SID-GBS): a double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2021;20:275–283. doi: 10.1016/S1474-4422(20)30494-4. [DOI] [PubMed] [Google Scholar]
- 35.Hodkinson JP. Considerations for dosing immunoglobulin in obese patients. Clin Exp Immunol. 2017;188:353–362. doi: 10.1111/cei.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.