

Abstract
The biomimetic two-phase strategy employing polyene cyclization and subsequent oxidation/substitution is an effective approach for divergent syntheses of [6-6-6]-tricyclic diterpenes. However, this strategy requires lengthy sequences for syntheses of oxygenated tricyclic aromatic abietane/podocarpane diterpenes owing to the many linear oxidation/substitution steps after cyclization. Here, we present a new synthetic route based on a convergent reverse two-phase strategy employing a reverse radical cyclization approach, which enabled the unified short syntheses of four aromatic abietane/podocarpane diterpenes and the divergent short syntheses of other related diterpenes. Oxygenated and substituted precursors for cyclization were convergently prepared through Friedel-Crafts acylation and rhodium-catalyzed site-selective iodination. Radical redox cyclization using an iridium photoredox catalyst involving neophyl rearrangement furnished the thermodynamically favored 6-membered ring preferentially. (±)-5,6-Dehydrosugiol, salvinolone, crossogumerin A, and Δ5-nimbidiol were synthesized in only 8 steps. An oxygenated cyclized intermediate was also useful for divergent derivatization to sugiol, ferruginol, saprorthoquinone, cryptomeriololide, and salvinolone.
Subject terms: Natural product synthesis, Synthetic chemistry methodology, Photocatalysis
Synthesis of the bioactive [6-6-6]-tricyclic diterpene skeleton has been developed using a biomimetic cyclization-oxidation strategy, however, the long oxidation steps hinder the total efficiency of the synthesis. Here, the authors develop a radical cyclization initiated from the C-ring using an oxidation-cyclization strategy in order to shorten the synthesis.
Introduction
Many diterpenes having a tricyclic [6-6-6]-fused skeleton with an aromatic C-ring (Fig. 1) such as aromatic abietanes have been isolated from natural resources1,2. Due to their unique carbon skeletons and attractive biological activities2–5, chemists have devoted themselves to developing syntheses of these tricyclic diterpenes6,7. In these synthetic approaches, a biomimetic strategy employing polyene cyclization followed by the introduction of functional groups by oxidation and substitution has been successful to produce many cyclic aromatic diterpenes8–15. This type of approach can be classified as a “two-phase strategy” consisting of the first, a cyclase phase, and the second, an oxidase phase, as recently described by Baran for terpenoid syntheses16–18.
Fig. 1. Naturally occurring tricyclic diterpenes possessing enone group in B-ring and substituted phenols in C-ring.

Tricyclic aromatic diterpene skeleton and a common structure (blue) of 5,6-dehydrosugiol (1a), crossogumerin A (1b), ∆5-nimbidiol (1c) and salvinolone (1d).
Among the family of tricyclic aromatic diterpenes, a certain number of diterpenoids including 5,6-dehydrosugiol (1a)19–21, crossogumerin A (1b)22, ∆5-nimbidiol (1c)23, and salvinolone (1d)24–26 possess a common oxygenated structure: 1) an enone group in the B-ring and 2) a substituted phenol in the C-ring (Fig. 1). These diterpenes exhibit a variety of bioactivities such as antitumor27–29, antibacterial30, antitermitic31, and antioxidant activities32. One total synthesis of 1a and 1d has been reported as a result of the above-mentioned two-phase strategy30. Two total syntheses also constructed the B-ring by cyclization between A- and C-rings followed by the formation of the enone goup33,34. Tada et al. synthesized abietane diterpenoids including 1a and 1d by cationic polyene cyclization forming a common tricyclic intermediate and its divergent functionalization (Fig. 2a), which was successful in producing various derivatives for evaluation of the antibacterial activities30,35. However, focusing on the total efficiency of the syntheses of 1a and 1d, the functionalization after cyclization required 8-11 linear steps, for a total of 11-14 steps, respectively. Salvinolone (1d) has also been prepared semisynthetically from dehydroabietic acid36.
Fig. 2. Two-phase strategy and reverse two-phase strategy for syntheses of tricyclic diterpenes.

a Syntheses of 5,6-dehydrosugiol (1a) and salvinolone (1d) using two-phase strategy by Tada et al. b (I) Problem in the cationic or radical cyclization initiated from the A-ring in reverse two-phase strategy; (II) This work: Cyclization initiated from the C-ring in convergent reverse two-phase strategy.
A strong candidate for the realization of shorter syntheses of the family of 1a–d would be the convergent preparation of oxygenated and substituted precursors possessing a carbonyl group at the B-ring moiety and subsequent cyclization in a “reverse two-phase strategy”37–39 (Fig. 2b (I)). The carbonyl group works as an electron-withdrawing group for the C-ring, while the C-ring possesses electron-donating alkoxyl and alkyl substituents. MacMillan et al. reported that radical cyclization of precursors with electron-withdrawing cyano, ester, or ketone groups on the aromatic ring proceeded40. On the other hand, Vanderwal et al. reported that electron-poor aromatic rings with cyano, trifluromethyl, or ester groups were not suitable in their radical cyclization41. Zhao and co-workers reported that cationic cyclization of substrates with strong electron-withdrawing cyano or nitro group did not take place42. Chandrasekhar et al. attempted a cationic cyclization of a similar skeleton to our substrates with a carbonyl group at the corresponding B-ring moiety and alkoxy groups at the aromatic ring43. However, cyclization did not proceed, while cyclization took place in the absence of the carbonyl group. Although we also initially attempted cationic or radical cyclization with precursors with a carbonyl group at the B-ring moiety and alkoxyl/alkyl substituents at the C-ring, all attempts failed to afford desired cyclized products (Fig. 2b (I)).
To solve this problem, we envisioned a reverse cyclization approach for oxygenated tricyclic aromatic diterpenes using a reverse two-phase strategy, in which radical cyclization is initiated from the C-ring (Fig. 2b (II)) in contrast to the biomimetic polyene cyclization initiated from the A-ring moiety. Oxygenated and substituted cyclization precursors with a halogen atom are prepared convergently from substituted phenol derivatives and acid chlorides followed by site-selective halogenation. Employing a photoredox catalyst, the carbon radical generated by scission of the halogen atom of the aromatic ring reacts with the alkene. The cyclization is then expected to directly furnish the tricyclic skeleton of 1a–d with an enone group in the B-ring and substituents on the C-ring. By changing the substituted phenols, 1a–d could be synthesized by a unified synthetic route. Herein, we report the unified eight-step synthesis of oxygenated tricyclic aromatic diterpenes (±)-1a–d by the reverse radical cyclization with the reverse two-phase strategy. The usefulness of the cyclized intermediate for divergent derivatization to other diterpenes is also reported.
Results and discussion
Synthesis of 5,6-dehydrosugiol (1a)
Our first target was the abietane diterpene, 5,6-dehydrosugiol (1a). Common A-ring segment 3 for 1a–d was prepared from β-homocyclocitral in 76% yield through oxidation and chlorination by following a known procedure44. Methyl ether 2a of the C-ring segment was prepared by methylation45 of commercially available 2-isopropylphenol in 96% yield (Fig. 3). Friedel-Crafts acylation between 2a and 3 in the presence of AlCl3 at −40 °C, accompanied by hydration, furnished alcohol 4a as a single diastereomer. The stereochemistry of 4a was not determined. The alcohol 4a was dehydrated by a catalytic amount of AgSbF6 (0.1 eq.) in 1,2-dichloroethane at 60 °C, giving alkene 5a. Site-selective halogenation of 5a was attempted under rhodium-catalyzed conditions reported by Glorius and co-workers46, but desired 6a (Br) was not obtained. Investigation of the conditions and products indicated that the double bond of 6a reacted with NBS, causing decomposition.
Fig. 3. Convergent synthesis of ketone 5a and attempted Rh-catalyzed bromination.
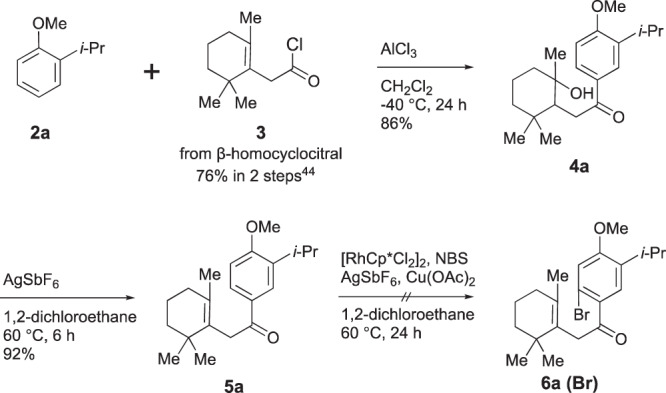
Cp*: pentamethylcyclopentadienyl, NBS: N-bromosuccinimide.
To avoid degradation, we screened conditions for the halogenation of alcohol 4a (Table 1). The conditions of Glorius using [RhCp*Cl2]2 (2.5 mol%), AgSbF6 (10 mol%), and Cu(OAc)2 (1.2 eq.) in dichloroethane gave a similar result to the case of 5a (Table 1, entry 1). In the presence of AgSbF6 at 60 °C, dehydration from 4a to 5a occurred preferentially. Reported transition metal-catalyzed halogenations using a carbonyl group as a directing group required temperatures higher than 60 °C47,48. However, Wu et al. and Du et al. reported that an ionic liquid promoted rhodium-catalyzed C-H cyanation and alkenylation with other directing groups at room temperature49,50. Thus, we applied the mixture of an ionic liquid BMIM•NTf2 and chloroform as the solvent to the halogenation of 4a (entry 2), in which the low solubility of 4a in BMIM•NTf2 was improved by chloroform. Under this condition, degradation was not observed, and desired 7a (Br) was obtained in a 24% yield. Bromination proceeded only at the C9 position, and other regioisomers were not observed. Through examination of brominating reagents, silver catalysts, and carboxylates (entries 2–8), the combination of NBS, AgNTf2, and AgOAc was the best, furnishing 7a (Br) in 55% yield (entry 4). Dehydration of 7a (Br) under the same conditions as 4a gave 6a (Br) an 86% yield (Fig. 4).
Table 1.
Screening of rhodium-catalyzed halogenation of 4aa.
 | ||||
|---|---|---|---|---|
| Entry | NXS | Silver cat. | Carboxylate | Yield (%) |
| 1b | NBS | AgSbF6 | Cu(OAc)2 | - |
| 2 | NBS | AgNTf2 | Cu(OAc)2 | 24 |
| 3 | NBS | AgNTf2 | PivOH | - |
| 4 | NBS | AgNTf2 | AgOAc | 55 |
| 5 | NBS | AgSbF6 | AgOAc | 51 |
| 6 | NBS | AgOTf | AgOAc | 38 |
| 7 | NBP | AgNTf2 | AgOAc | 45 |
| 8 | DBH | AgNTf2 | AgOAc | - |
| 9 | NIS | AgNTf2 | AgOAc | 26 |
aReaction conditions: 11a (0.20 mmol), [RhCp*Cl2]2 (2.5 mol%), NXS (1.5 eq.), silver cat. (10 mol%), carboxylate (1.2 eq.), BMIM•NTf2/CHCl3 = 1/2 (0.33 M).
![]()
b1,2-Dichloroethane was used instead of BMIM•NTf2/CHCl3 at 60 °C.
Fig. 4. Syntheses of precursors 6a for radical cyclization.

Cp*: pentamethylcyclopentadienyl, NIS: N-iodosuccinimide. AgNTf2: silver bis(trifluoromethanesulfonyl)imide, BMIM · NTf2: 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide.
Iodination with NIS under similar conditions as bromination afforded 7a (I), but the yield was decreased to 26% (Table 1, entry 9). To improve the yield, the directing group was altered from a carbonyl group to an oxime ether, which is known to form a stronger interaction with a rhodium catalyst (Fig. 4)51. Thus, oxime ether 8a was obtained from 4a in 94% yield through condensation with methoxy amine. Under the same reaction conditions as bromination, Rh-catalyzed iodination of 8a with NIS proceeded smoothly, giving only desired 9a in 98% yield without regioisomers. Palladium-catalyzed halogenation using an oxime ether as a directing group was also reported but did not work well for this substrate 8a52–54. We also attempted to reuse the rhodium and silver catalysts in the ionic liquid and it was found they could be recycled up to three times with almost the same yields (Supplementary Information). The reaction mechanism of rhodium-catalyzed halogenation was examined by DFT calculations by Glorius and co-workers55, and we assume that this halogenation proceeded by the same mechanism.
Deprotection of oxime ether 9a was first attempted by hydrochloric acid, but the reaction resulted in a complex mixture. The conversion was achieved by treatment with Meerwein reagent56,57. Methylation to the oxime salt, deprotection, and dehydration took place in one pot under mild conditions, giving 6a (I) in 84% yield. The reaction possibly proceeded through intramolecular addition of the hydroxy group to the oxime salt followed by elimination (Supplementary Fig. 1).
With the two precursors 6a (Br) and 6a (I) in hand, we investigated their radical cyclization to enone 10a. Cyclization of 6a could afford a six-membered ring 10a and a five-membered ring 11a depending on the bond-forming position of the double bond (Table 2). Generally, 5-exo radical cyclization is kinetically favored over 6-endo radical cyclization58, but the equilibrium between five- and six-membered rings through neophyl rearrangement is known to alter their ratio59–61. Blakey and co-workers reported that the ratio of five- and six-membered rings was controlled by the concentration of hydrogen atom donors that quench radical species after the cyclization process, which is known as reductive radical cyclization62. In contrast, we investigated the formation of a six-membered ring over a 5-membered ring under the conditions of redox radical cyclization using photoredox catalysts without hydrogen atom donors, in which oxidative quenching of radical species would furnish the enone 10a directly as the product.
Table 2.
Screening of radical cyclization of 6aa.
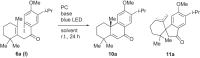 | |||||
|---|---|---|---|---|---|
| Entry | PC | Base | Solvent | Yield (%)b | |
| 10a | 11a | ||||
| 1c | Ir-1 | KH2PO4 | MeCN | 0 | 0 |
| 2 | Ir-1 | KH2PO4 | MeCN | 21 | 51 |
| 3 | Ir-1 | 2,6-lutidine | MeCN | 22 | 66 |
| 4 | Ir-1 | TMEDA | MeCN | 16 | 69 |
| 5 | Ir-1 | DBU | MeCN | 33 | 64 |
| 6 | Ir-1 | TMEDA/DBU | MeCN | 0 | 88 |
| 7 | Ru | DBU | MeCN | 0 | 0 |
| 8 | Ir-2 | DBUd | MeCN | 42 | 46 |
| 9 | Ir-2 | DBUd | Benezene | 11 | trace |
| 10 | Ir-2 | DBUd | MeOH | 33 | 64 |
| 11 | Ir-2 | DBUd | DMF | 0 | 0 |
| 12 | Ir-2 | DBUd | DMSO | 45 | 28 |
| 13 | Ir-2 | DBUe | DMSO | 55 (50) | 29 |
| 14 | Ir-2 | DBUf | DMSO | 52 | 29 |
| 15g | Ir-2 | DBUd | DMSO | 0 | 0 |
| 16 | Ir-2 | KH2PO4 | DMSO | 0 | 0 |
aReaction conditions: 6a (0.10 mmol), PC (5.0 mol%), base (2 eq.), solvent (0.05 M).
bYields were estimated by 1H NMR using an internal standard.
c6a (Br).
d3 eq.
e9 eq.
f15 eq.
gIn the dark.
First, 6a (Br) was treated with fac-Ir(ppy)3 (Ir-1, 0.05 eq.)63,64 and K2HPO4 (2 eq.) in acetonitrile under irradiation of blue LED. However, radical cyclization did not proceed at all (Table 2, entry 1). It was estimated that the reduction potential of Ir-1 [Ir(III)*/Ir(IV) = − 1.73 V vs. SCE]63,64 was insufficient for the reduction with bromoarene 6a (Br) (Br-Ph, −2.07 V)65. Therefore, the substrate was changed to 6a (I) (I-Ph, −1. 59 V)65. The reaction of 6a (I) with Ir-1 and K2HPO4 furnished desired six-membered 10a in 21% yield as well as five-membered 11a (the mixture of regioisomers of a double bond) in 51% yield as by-products (entry 2). Since desired 10a was a minor product, the reaction conditions were screened further. Among the investigated bases, KH2PO4, 2,6-lutidine, TMEDA, DBU, and TMEDA/DBU (entries 2–6), the best yield (33%) was obtained using DBU (entry 5). With DBU, we then examined photoredox catalysts. The reaction did not proceed at all with Ru(bpy)3Cl2 possessing relatively high redox potentials [Ru(II)*/Ru(III) = − 0.81 V, Ru(II)*/Ru(I) = +0.77 V]63,64 (entry 7). Using Ir[dF(CF3)ppy]2(dtbpy)PF6 (Ir-2) with a reduction potential (−0.89 V) close to Ru(bpy)3Cl2 but with a lower oxidation potential (+1.21 V)64, 10a and 11a were furnished in almost equal amounts, 42% and 46% yields (entry 8). Next, with Ir-2, solvents such as benzene, methanol, DMF, and DMSO (entries 8-12) were examined. The reaction in DMSO afforded the highest 45% yield of 10a and a 28% yield of 11a. When the amount of DBU was increased to 9 eq., the yield of 10a was further improved to 55% (50% isolated yield) (entry 13). With 15 eq. of DBU, the yield was similar to that with 9 eq. (entry 14). In the dark without irradiation of light, the reaction did not proceed (entry 15). No reaction with K2HPO4 (entry 16) implied that DBU was not only a base but was involved in the redox reaction (vide infra).
Finally, (±)-5,6-dehydrosugiol (1a) was synthesized by deprotection of 10a using dodecanethiol according to Tada’s method30,66,67. The total yield of 1a was 31% in 6 steps from 2a (Fig. 5) and 24% in 8 steps from β-homocyclocitral, which was much improved from the reported 13% overall yield in 11 steps30,35.
Fig. 5. Syntheses of tricyclic aromatic diterpenes (±)-1a–d with different substitution patterns on C-ring.

Cp*: pentamethylcyclopentadienyl, NIS: N-iodosuccinimide. AgNTf2: silver bis(trifluoromethanesulfonyl)imide, BMIM∙NTf2: 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide, Ir-2: (4,4’-Di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-κN)phenyl-κC]iridium(III) hexafluorophosphate.
Syntheses of 1b–d
The established synthetic method for (±)-1a was applied to the syntheses of (±)-1b–d with different substitution patterns on the C-ring (Fig. 5 and Table 3). (±)-Crossogumerin A (1b), a podocarpane diterpene, was synthesized by the same synthetic route in 26% overall yield from known methyl ether 2b68, in which the yield of each step was similar to that of 1a. In the synthesis of (±)-Δ5-nimbidiol (1c), Friedel-Crafts acylation of commercially available 2c with 3 was carried out at 0 °C owing to the lower reactivity of 2c. The yield of 4c was lowered to 51% accompanied by the formation of by-product 5c without hydration (alkene 5 in Fig. 3). The yields of steps 2 to 5 were similar to those of 1a. Our rhodium-catalyzed conditions allowed the site-selective iodination of 9c in 97% yield, although a methoxy group was reported to be a sterically insufficient substituent to control the reaction site in the rhodium-catalyzed halogenation at high temperature46. Deprotection of 10c was performed by the combination of AlCl3 and dodecanethiol reported by Matsumoto et al.36, giving 1c in 60% yield. The total yield of Δ5-nimbidiol (1c) was 12% from 2c. (±)-Salvinolone (1d), an abietane diterpene, was also synthesized in 1.9% overall yield from our previously synthesized 2d68,69. In the synthesis of 1d, the yields of steps 1 to 4 were similar to those of 1a. It is worth noting that our rhodium-catalyzed iodination conditions could distinguish the sterically close C4 and C6 positions completely and iodination proceeded only at the less sterically hindered C4 position. The yield of radical cyclization was slightly lowered because cyclic precursor 6d (I) underwent deiodination due to the increased steric hindrance on the C-ring. The lower total yield of 1d compared to those of 1a–c was due to the low yield of deprotection of the acetal on the C-ring. The enone group of the B-ring was reported to be sensitive to acidic conditions70. Even after screening various conditions, the deprotection yield of 1d by trifluoroacetic acid and H2O at 90 °C with microwave heating was only 11% with unidentified polar by-products.
Table 3.
Yields for each step from 2a–d to 1a–d.
| 1st | 2nd | 3rd | 4th | 5th | 6th | |
|---|---|---|---|---|---|---|
| 4 | 8 | 9 | 6 (I) | 10 | 1 | |
| 1a | 86 | 94 | 98 | 84 | 50 | 94 |
| 1b | 77 | 96 | 88 | 84 | 51 | 94 |
| 1c | 51 | 97 | 97 | 89 | 46 | 60 |
| 1d | 81 | 93 | 93 | 84 | 36 | 11 |
Derivatization to other diterpenes
Tricyclic intermediate (±)-10a for 1a was also useful for divergent syntheses of other diterpenes (Fig. 6) due to its easily convertible structure. Modification of the B-ring afforded (±)-sugiol (12)21,71 and (±)-ferruginol (13)72. Using Na2S2O4 in H2O/EtOH at reflux73, 1,4-reduction of the enone group in the B-ring proceeded selectively. When Pd(OH)2 was used under a hydrogen atmosphere in ethyl acetate, reduction of the carbonyl group proceeded in addition to 1,4-reduction74,75. These reductions proceeded diastereoselectively. Sugiol (12) and ferruginol (13) were obtained after demethylation in 94% and 75% yields in 2 steps, respectively. 10a was also reported as a precursor for saprorthoquinone (14)24,36 and cryptomeriololide (15)76. We succeeded in the site-selective hydroxylation on the C-ring of 1a using stabilized IBX in methanol41,68,77, giving (±)-salvinolone (1d) in 84% yield from 1a. While the total yield of 1d was 1.9% in 6 steps from 2d in the above synthesis owing to the low yield of deprotection, the total yield of 1d was much improved to 27% in 7 steps from 2a.
Fig. 6. Derivatization of intermediate (±)-10a to other diterpenes.

SIBX: stabilized 2-iodoxybenzoic acid with benzoic acid and isophthalic acid.
Mechanistic consideration of cyclization
To understand the reaction profile of radical cyclization, DFT calculations at the UM06-2X/6-311 + + G(d,p) level of theory with SMD (DMSO) were performed (Fig. 7). The activation barrier (TSA-B) of 6-endo cyclization from radical intermediate A to six-membered B was 2.1 kcal/mol, and the energy of intermediate B was −32.4 kcal/mol. The activation barrier [TSA-C)] of 5-exo cyclization from A to five-membered C was 1.8 kcal/mol, which was 0.3 kcal/mol lower than that of 6-endo cyclization. The energy of C was −30.8 kcal/mol, which was 1.6 kcal/mol higher than that of six-membered B. These calculations indicated that 5-exo cyclization was kinetically favored over 6-endo cyclization, while six-membered B was thermodynamically favored over five-membered C. Neophyl rearrangement from C to B was calculated to go through TSC-D, intermediate D, and TSD-B. The activation barrier from C to B was 17.6 kcal/mol, which was reasonable for the rearrangement to occur at the experimental temperature of cyclization (room temperature).
Fig. 7. Calculated energy profile of 6-endo, 5-exo cyclizations, and neophyl rearrangement.

UM06-2X/6-311 + + G(d,p) with SMD (DMSO).
Based on these experimental and calculated results, a mechanism of radical cyclization from 6a (I) to 10a and 11a with Ir[dF(CF3)ppy]2(dtbpy)PF6 and DBU is proposed (Fig. 8). Judging from the redox potentials of excited Ir[dF(CF3)ppy]2(dtbpy)PF6 [Ir(III)*/Ir(IV) = − 0.89 V, Ir(III)*/Ir(II) = +1.21 V vs. SCE]64 by light, I-Ph (−1.59 V)65, and DBU (+1.28 V)63, Ir(III)* does not reduce 6a (I) but oxidizes DBU to form Ir(II) and DBU•+. Iodide 6a (I) is reduced by Ir(II) to give radical intermediate A with regeneration of Ir(III). The lack of reaction with K2HPO4 (Table 2, entry 16) also supports that Ir(III)* does not directly reduce 6a (I). Cyclization of intermediate A gives kinetically favored 5-membered C as the major product and 6-membered B as the minor product. The equilibrium between C and B through neophyl rearrangement is shifted to the thermodynamically favored 6-membered B. Finally, the abstraction of hydrogen from intermediates B and C by DBU•+ 78,79 gives 10a and 11a. The ratio of 10a and 11a would be determined by the combination of the first kinetic ratio of B and C, the rate of neophyl rearrangement from C to B, and the rate of oxidation from B or C to 10a or 11a depending on the reaction conditions. Although the details are not clear, it is supposed that the slow oxidative quench from C to 11a and the shift from C to B allowed the preferential formation of 10a over 11a under the optimized conditions.
Fig. 8. Proposed reaction mechanism of radical cyclization of 6a (I).
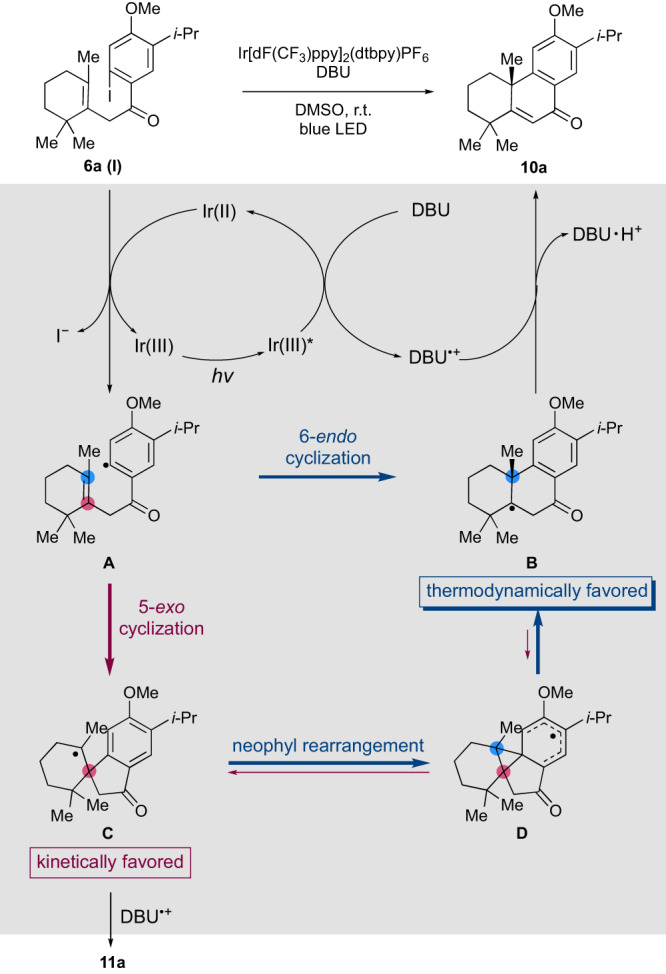
Ir[dF(CF3)ppy]2(dtbpy)PF6: (4,4’-Di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-κN)phenyl-κC]iridium(III) hexafluorophosphate, DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene, LED: light emitting diode.
Conclusion
We established a new synthetic route to oxygenated tricyclic aromatic diterpenes, (±)-5,6-dehydrosugiol (1a), (±)-crossogumerin A (1b), (±)-∆5-nimbidiol (1c), and (±)-salvinolone (1d), possessing an enone group in the B-ring and different substitution patterns of the C-ring, based on the reverse cyclization approach from the arene side in a convergent reverse two-phase strategy. The oxygenated and substituted precursors for cyclization were convergently prepared through Friedel-Crafts acylation between A- and C-ring moieties and site-selective iodination. The iodination with exclusive site selectivity was achieved using a rhodium catalyst on an oxime ether in an ionic liquid at room temperature under mild conditions. Radical redox cyclization using an iridium photoredox catalyst and DBU succeeded in furnishing the thermodynamically favored 6-membered product with an enone group in the B-ring preferentially through neophyl rearrangement over the kinetically favored 5-membered product, whose reaction profile was supported by DFT calculations. (±)-5,6-Dehydrosugiol (1a), (±)-crossogumerin A (1b), (±)-Δ5-nimbidiol (1c), and (±)-salvinolone (1d) were synthesized in only 8 steps from β-homocyclocitral by this synthetic route including deprotection. The syntheses of 1a and 1d were much improved from those of previous reports, and these are the first syntheses for 1b and 1c. The oxygenated cyclized intermediate 10a was also useful for divergent derivatization to diterpenes, (±)-sugiol (12), (±)-ferruginol (13), (±)-saprorthoquinone (14), (±)-cryptomeriololide (15), and (±)-salvinolone (1d). In addition to the biomimetic polyene cyclization in the two-phase strategy, this reverses radical cyclization in the convergent reverse two-phase strategy is expected to become a strong approach for the efficient syntheses of bioactive oxygenated tricyclic aromatic diterpenoids and derivatives.
Methods
General procedure for iodination
Oxime ether 8 was dissolved in BMIM∙NTf2 and anhydrous dichloromethane (1/1, 0.14 M), and [RhCp*Cl2]2 (2.5 mol%), silver bis(trifluoromethanesulfonyl)imide (0.1 eq.), silver acetate (1.1 eq.) and NIS (1.2 eq.) were added to the solution at room temperature under argon atmosphere. After stirring at room temperature for 24 h, the reaction mixture was extracted six times with ethyl acetate. The combined extracts were washed with saturated aqueous sodium thiosulfate, saturated aqueous sodium hydrogen carbonate, and brine, dried over anhydrous sodium sulfate, filtered through a cotton plug, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography with hexane/ethyl acetate to afford iodide 9.
General procedure for radical cyclization
Iodide 6 was dissolved in anhydrous DMSO (0.05 M) in a J. Young test tube, and Ir[dF(CF3)ppy]2(dtbpy)PF6 (5 mol%) and 1,8-diazabicyclo[5.4.0]undec-7ene (DBU, 9.0 eq.) were added to the solution. The mixture was degassed by three freeze-pump-thaw cycles. The tube was placed 6 cm away from 40 W blue LED lamps (Kessil A160WE Tuna Blue) with a cooling fan blowing air at room temperature to keep the reaction vessel at room temperature. After stirring for 24 h, the reaction was quenched by saturated aqueous ammonium chloride solution and ethyl acetate. The organic layer was separated, and the aqueous layer was extracted twice with ethyl acetate. The combined extracts were washed with water and brine, dried over anhydrous sodium sulfate, filtered through a cotton plug, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography with hexane/ethyl acetate to afford tricyclic compound 10.
Other experimental procedures, characterization data of compounds, NMR spectra, and reaction coordinates of calculations are included in Supplementary Methods in the Supplementary Information, and Supplementary Data 1 and 2.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP20K05499 (S.H.), JST SPRING Grant Number JPMJSP2123 (R.H.), the Fukuoka Naohiko Memorial Foundation (S.H.), and the Sumitomo Foundation (S.H.). The computations were performed using Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C230).
Author contributions
R.H. carried out the experiments and calculations. T.S., S.H., and K.H. supervised the research project and directed the experiments and calculations. All authors contributed to the writing of the manuscript.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
All data are included in this article, Supplementary Information, Supplementary Data 1 (NMR spectra), and Supplementary Data 2 (DFT calculations).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Riichi Hashimoto, Email: riichi8222hashimoto@keio.jp.
Shuhei Higashibayashi, Email: higashibayashi-sh@pha.keio.ac.jp.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-023-00979-2.
References
- 1.Jassbi AR, Zare S, Firuzi O, Xiao J. Bioactive phytochemicals from shoots and roots of Salvia species. Phytochem. Rev. 2016;15:829–867. [Google Scholar]
- 2.Gáborová M, Šmejkal K, Kubínová R. Abietane diterpenes of the genus Plectranthus sensu lato. Molecules. 2021;27:166. doi: 10.3390/molecules27010166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González MA. Aromatic abietane diterpenoids: their biological activity and synthesis. Nat. Prod. Rep. 2015;32:684–704. doi: 10.1039/c4np00110a. [DOI] [PubMed] [Google Scholar]
- 4.Grayer RJ, Paton AJ, Simmonds MSJ, Howes M-JR. Differences in diterpenoid diversity reveal new evidence for separating the genus Coleus from Plectranthus. Nat. Prod. Rep. 2021;38:1720–1728. doi: 10.1039/d0np00081g. [DOI] [PubMed] [Google Scholar]
- 5.Kuźma Ł, Gomulski J. Biologically active diterpenoids in the Clerodendrum genus—a review. Int. J. Mol. Sci. 2022;23:11001. doi: 10.3390/ijms231911001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.González MA. Aromatic abietane diterpenoids: total syntheses and synthetic studies. Tetrahedron. 2015;71:1883–1908. [Google Scholar]
- 7.Kang J, Quynh Le,T, Oh CH. Recent advances in abietane/icetexane synthesis. Tetrahedron Lett. 2022;108:154133. [Google Scholar]
- 8.Johnson W. Biomimetic polyene cyclizations: a review. Bioorg. Chem. 1976;5:51–98. doi: 10.1002/anie.197600091. [DOI] [PubMed] [Google Scholar]
- 9.Taylor SK. Biosynthetic, biomimetic and related epoxide cyclizations. A review. Org. Prep. Proced. Int. 1992;24:245–284. [Google Scholar]
- 10.Yoder RA, Johnston JN. A case study in biomimetic total synthesis: Polyolefin carbocyclizations to terpenes and steroids. Chem. Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett A, Ma T-K, Mies T. Recent developments in polyene cyclizations and their applications in natural product synthesis. Synthesis. 2019;51:67–82. [Google Scholar]
- 12.Ungarean CN, Southgate EH, Sarlah D. Enantioselective polyene cyclizations. Org. Biomol. Chem. 2016;14:5454–5467. doi: 10.1039/c6ob00375c. [DOI] [PubMed] [Google Scholar]
- 13.García-Pedrero O, Rodríguez F. Cationic cyclization reactions with alkyne terminating groups: A useful tool in biomimetic synthesis. Chem. Commun. 2022;58:1089–1099. doi: 10.1039/d1cc05826f. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Gill GB, Pattenden G. New radical mediated polyolefin cyclisations directed towards steroid ring synthesis. Tetrahedron Lett. 1994;35:2593–2596. [Google Scholar]
- 15.Handa S, Nair PS, Pattenden G. Novel regio-and stereoserective cascade 6-endo-trig cyclisations from polyene acyl radical intermediates leading to steroid-like pentacycles and heptacycles. Helv. Chim. Acta. 2000;83:2629–2643. [Google Scholar]
- 16.Chen K, Baran PS. Total synthesis of eudesmane terpenes by site-selective C-H oxidations. Nature. 2009;459:824–828. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- 17.Jørgensen L, et al. 14-Step synthesis of (+)-ingenol from (+)-3-carene. Science. 2013;341:878–882. doi: 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- 18.Kanda Y, Ishihara Y, Wilde NC, Baran PS. Two-phase total synthesis of taxanes: tactics and strategies. J. Org. Chem. 2020;85:10293–10320. doi: 10.1021/acs.joc.0c01287. [DOI] [PubMed] [Google Scholar]
- 19.Kupchan SM, Karim A, Marcks C. Tumor inhibitors. XLVIII. Taxodione and taxodone, two novel diterpenoid quinone methide tumor inhibitors from Taxodium distichum. J. Org. Chem. 1969;34:3912–3918. doi: 10.1021/jo01264a036. [DOI] [PubMed] [Google Scholar]
- 20.Kuo Y-H, Wu T-R, Cheng M-C, Wang Y. Five new compounds form the heartwood of Juniperus formosana HAYATA. Chem. Pharm. Bull. 1990;38:3195–3201. [Google Scholar]
- 21.Kuo Y-H, Yu MT. Dehydroabietane diterpenes from Juniperus formosana hay. var. concolor hay. Phytochemistry. 1996;42:779–781. [Google Scholar]
- 22.Miron-Lopez G, et al. Cytotoxic diterpenes from roots of Crossopetalum gaumeri, a Celastraceae species from Yucatan Peninsula. Bioorg. Med. Chem. Lett. 2014;24:2105–2109. doi: 10.1016/j.bmcl.2014.03.051. [DOI] [PubMed] [Google Scholar]
- 23.Wu J, Zhou Y, Wang L, Zuo J, Zhao W. Terpenoids from root bark of Celastrus orbiculatus. Phytochemistry. 2012;75:159–168. doi: 10.1016/j.phytochem.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 24.Lin L-Z, Blasko G, Cordell GA. Diterpenes of Salvia prionitis. Phytochemistry. 1989;28:177–181. [Google Scholar]
- 25.Kusumoto N, Ashitani T, Murayama T, Ogiyama K, Takahashi K. Antifungal abietane-type diterpenes from the cones of Taxodium distichum Rich. J. Chem. Ecol. 2010;36:1381–1386. doi: 10.1007/s10886-010-9875-2. [DOI] [PubMed] [Google Scholar]
- 26.Kadir A, et al. Structurally diverse diterpenoids from the roots of Salvia deserta based on nine different skeletal types. J. Nat. Prod. 2021;84:1442–1452. doi: 10.1021/acs.jnatprod.0c01180. [DOI] [PubMed] [Google Scholar]
- 27.Gil RR, Cordell GA, Topçu G, Ulubelen A. Montbretol and salvinolone and identical. J. Nat. Prod. 1994;57:181–185. [Google Scholar]
- 28.Li S, Wang P, Deng G, Yuan W, Su Z. Cytotoxic compounds from invasive giant salvinia (Salvinia molesta) against human tumor cells. Bioorg. Med. Chem. Lett. 2013;23:6682–6687. doi: 10.1016/j.bmcl.2013.10.040. [DOI] [PubMed] [Google Scholar]
- 29.Kusumoto N, Aburai N, Ashitani T, Takahashi K, Kimura K. Pharmacological prospects of oxygenated abietane-type diterpenoids from Taxodium distichum cones. Adv. Biol. Chem. 2014;4:109–115. [Google Scholar]
- 30.Yang Z, et al. Synthesis of variously oxidized abietane diterpenes and their antibacterial activities against MRSA and VRE. Bioorg. Med. Chem. 2001;9:347–356. doi: 10.1016/s0968-0896(00)00253-4. [DOI] [PubMed] [Google Scholar]
- 31.Kusumoto N, et al. Antitermitic activities of abietane-type diterpenes from Taxodium distichum cones. J. Chem. Ecol. 2009;35:635–642. doi: 10.1007/s10886-009-9646-0. [DOI] [PubMed] [Google Scholar]
- 32.Vo QV, et al. The antioxidant activity of natural diterpenes: Theoretical insights. RSC Adv. 2020;10:14937–14943. doi: 10.1039/d0ra02681f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, A. et al. Stereoselective synthesis of (+)-Δ5-dehydrosugiyl methyl ether. J. Chem. Res. (S) 328–329 (2001).
- 34.Tian, Y. et al. The total synthesis of salvinolone. J. Chem. Res. 33–33 (1997).
- 35.Tada, M. et al. Synthesis of (+)- and (−)-ferruginol via asymmetric cyclization of a polyene. J. Chem. Soc., Perkin Trans. 1, 2657–2664 (2000).
- 36.Matsumoto T, Tanaka Y, Terao H, Takeda Y, Tada M. The synthesis of salvinolone, saprorthoquinone, and 4-hydroxysapriparaquinone from (+)-dehydroabietic acid. Bull. Chem. Soc. Jpn. 1993;66:3053–3057. [Google Scholar]
- 37.Trost BM, Min C. Total synthesis of terpenes via palladium-catalysed cyclization strategy. Nat. Chem. 2020;12:568–573. doi: 10.1038/s41557-020-0439-y. [DOI] [PubMed] [Google Scholar]
- 38.Vrubliauskas D, Gross BM, Vanderwal CD. Stereocontrolled radical bicyclizations of oxygenated precursors enable short syntheses of oxidized abietane diterpenoids. J. Am. Chem. Soc. 2021;143:2944–2952. doi: 10.1021/jacs.0c13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson LK, Niman SW, Vrubliauskas D, Vanderwal CD. Stereocontrolled synthesis and structural revision of plebeianiol A. Org. Lett. 2021;23:9569–9573. doi: 10.1021/acs.orglett.1c03791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rendler S, MacMillan DWC. Enantioselective polyene cyclization via organo-SOMO catalysis. J. Am. Chem. Soc. 2010;132:5027–5029. doi: 10.1021/ja100185p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vrubliauskas D, Vanderwal CD. Cobalt‐catalyzed hydrogen‐atom transfer induces bicyclizations that tolerate electron‐rich and electron‐deficient intermediate alkenes. Angew. Chem. Int. Ed. 2020;59:6115–6121. doi: 10.1002/anie.202000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan L, et al. Enantioselective polyene cyclization catalyzed by a chiral Brønsted acid. Angew. Chem. Int. Ed. 2018;57:2115–2119. doi: 10.1002/anie.201711603. [DOI] [PubMed] [Google Scholar]
- 43.Nagaraju K, Chegondi R, Chandrasekhar S. Expanding diversity without protecting groups: (+)-Sclareolide to indolosesquiterpene alkaloid mycoleptodiscin A and analogues. Org. Lett. 2016;18:2684–2687. doi: 10.1021/acs.orglett.6b01145. [DOI] [PubMed] [Google Scholar]
- 44.Branca SJ, Lock RL, Smith AB. Exploitation of the vinylogous Wolff rearrangement. An efficient. total synthesis of (±)-mayurone, (±)-thujopsene, and (±)-thujopsadiene. J. Org. Chem. 1977;42:3165–3168. [Google Scholar]
- 45.Seong C, Kang J, Chai U, Mac DH, Oh CH. Total synthesis of 1-oxomiltirone and arucadiol. Synlett. 2020;31:1953–1956. [Google Scholar]
- 46.Schröder N, Wencel-Delord J, Glorius F. High-yielding, versatile, and practical [Rh(III)Cp*]-catalyzed ortho bromination and iodination of arenes. J. Am. Chem. Soc. 2012;134:8298–8301. doi: 10.1021/ja302631j. [DOI] [PubMed] [Google Scholar]
- 47.Collins KD, Glorius F. Employing a robustness screen: Rapid assessment of rhodium(III)-catalysed C-H activation reactions. Tetrahedron. 2013;69:7817–7825. [Google Scholar]
- 48.Sun X, Shan G, Sun Y, Rao Y. Regio- and chemoselective C-H chlorination/bromination of electron-deficient arenes by weak coordination and study of relative directing-group abilities. Angew. Chem. Int. Ed. 2013;52:4440–4444. doi: 10.1002/anie.201300176. [DOI] [PubMed] [Google Scholar]
- 49.Lv S, et al. Rhodium-catalyzed direct C-H bond cyanation in ionic liquids. Org. Lett. 2018;20:4994–4997. doi: 10.1021/acs.orglett.8b01952. [DOI] [PubMed] [Google Scholar]
- 50.Yao T, Du K. Temperature-controlled mono-and diolefination of arene using Rh(III)/RTIL as an efficient and recyclable ctalytic system. ACS Sustain. Chem. Eng. 2019;7:6068–6077. [Google Scholar]
- 51.Bolotin DS, Bokach NA, Demakova MY, Kukushkin VY. Metal-involving synthesis and reactions of oximes. Chem. Rev. 2017;117:13039–13122. doi: 10.1021/acs.chemrev.7b00264. [DOI] [PubMed] [Google Scholar]
- 52.Kalyani D, Dick AR, Anani WQ, Sanford MS. Scope and selectivity in palladium-catalyzed directed C-H bond halogenation reactions. Tetrahedron. 2006;62:11483–11498. [Google Scholar]
- 53.Dubost E, Fossey C, Cailly T, Rault S, Fabis F. Selective ortho-bromination of substituted benzaldoximes using Pd-catalyzed C-H Activation: Application to the synthesis of substituted 2-bromobenzaldehydes. J. Org. Chem. 2011;76:6414–6420. doi: 10.1021/jo200853j. [DOI] [PubMed] [Google Scholar]
- 54.Lou S-J, Xu D-Q, Xu Z-Y. Mild and versatile nitrate-promoted C-H Bond Fluorination. Angew. Chem. Int. Ed. 2014;53:10330–10335. doi: 10.1002/anie.201404423. [DOI] [PubMed] [Google Scholar]
- 55.Lied F, Lerchen A, Knecht T, Mück-Lichtenfeld C, Glorius F. Versatile Cp*Rh(III)-catalyzed selective ortho-chlorination of arenes and heteroarenes. ACS Catal. 2016;6:7839–7843. [Google Scholar]
- 56.Takikawa H, Takada A, Hikita K, Suzuki K. Formation of α‐hydroxy‐β‐diketones through hydroxylation of isoxazolium salts: Stereoselective approach to angular cis‐diols in polycyclic systems. Angew. Chem. Int. Ed. 2008;47:7446–7449. doi: 10.1002/anie.200801586. [DOI] [PubMed] [Google Scholar]
- 57.González-Nogal AM, Calle M. Silylated azolium salts and their applications in the synthesis of azolines and β-enaminoketones bearing allyl-, vinyl-, and acylsilane or α-silylketone units. Tetrahedron. 2009;65:5472–5483. [Google Scholar]
- 58.Beckwith ALJ, O’Shea DM. Kinetics and mechanism of some vinyl radical cyclisations. Tetrahedron Lett. 1986;27:4525–4528. [Google Scholar]
- 59.Stork G, Baine NH. Cyclization of vinyl radicals: a versatile method for the construction of five- and six-membered rings. J. Am. Chem. Soc. 1982;104:2321–2323. [Google Scholar]
- 60.Beckwith ALJ, Schiesser CH. Regio- and stereo-selectivity of alkenyl radical ring closing: a theoretical study. Tetrahedron. 1985;41:3925–3941. [Google Scholar]
- 61.Ishibashi H, Kobayashi T, Nakashima S, Tamura O. Regiochemistry in aryl radical cyclization onto methylenecycloalkanes. J. Org. Chem. 2000;65:9022–9027. doi: 10.1021/jo001086h. [DOI] [PubMed] [Google Scholar]
- 62.Maust MC, Hendy CM, Jui NT, Blakey SB. Switchable regioselective 6-endo or 5-exo radical cyclization via photoredox catalysis. J. Am. Chem. Soc. 2022;144:3776–3781. doi: 10.1021/jacs.2c00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roth H, Romero N, Nicewicz D. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett. 2015;27:714–723. [Google Scholar]
- 64.Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fry AJ, Krieger RL. Electrolyte effects upon the polarographic reduction of alkyl halides in dimethyl sulfoxide. J. Org. Chem. 1976;41:54–57. [Google Scholar]
- 66.Li A, She X, Zhang J, Wu T, Pan X. Synthesis of C-7 oxidized abietane diterpenes from racemic ferruginyl methyl ether. Tetrahedron. 2003;59:5737–5741. [Google Scholar]
- 67.Chae J. Practical demethylation of aryl methyl ethers using an odorless thiol reagent. Arch. Pharm. Res. 2008;31:305–309. doi: 10.1007/s12272-001-1156-y. [DOI] [PubMed] [Google Scholar]
- 68.Hashimoto R, Hanaya K, Sugai T, Higashibayashi S. 1,2-Rearrangement from o-quinols to multisubstituted catechols via retro Diels-Alder reaction of o-quinol dimers. Bull. Chem. Soc. Jpn. 2022;95:663–672. [Google Scholar]
- 69.Pramanik C, et al. Commercial manufacturing of propofol: Simplifying the isolation process and control. on related substances. Org. Process Res. Dev. 2014;18:152–156. [Google Scholar]
- 70.Matsumoto T, Tanaka Y, Terao, Takeda Y, Wada M. Rearrangement of the angular methyl group in dehydroabietic acid derivatives. Chem. Pharm. Bull. 1993;41:1960–1964. [Google Scholar]
- 71.Su W-C, Fang J-M, Cheng Y-S. Abietanes and kauranes from leaves of Cryptomeria japonica. Phytochemistry. 1994;35:1279–1284. [Google Scholar]
- 72.Brandt, C. W. & Neubauer, L. G. Miro Resin. Part I. Ferruginol. J. Chem. Soc. 1031–1037 (1939).
- 73.Dhillon RS, Singh RP, Kaur D. Selective 1,4-reduction of conjugated aldehydes and ketones in the presence of unconjugated aldehydes and ketones with sodium dithionite. Tetrahedron Lett. 1995;36:1107–1108. [Google Scholar]
- 74.Shibanuma Y, Okamoto T. Synthetic approach to diterpene alkaroids: Construction of the bridged azabicyclic ring system of kobusine. Chem. Pharm. Bull. 1985;33:3187–3194. [Google Scholar]
- 75.Kametani, T. Synthesis of (±)-pisiferin, (±)-pisiferol, and related compounds by Intramolecular [4 + 2]cycloaddition. J. Chem. Soc. Perkin Trans 1. 5–10 (1990).
- 76.Feng L, et al. A pair of enantiomeric bis-seco-abietane diterpenoids from Cryptomeria fortunei. J. Nat. Prod. 2018;81:2667–2672. doi: 10.1021/acs.jnatprod.8b00482. [DOI] [PubMed] [Google Scholar]
- 77.Tada M, Ohkanda T, Kurabe J. Syntheses of carnosic acid and carnosol, anti-oxidants in rosemary, from pisiferic acid, the major constituent of Sawara. Chem. Pharm. Bull. 2010;58:27–29. doi: 10.1248/cpb.58.27. [DOI] [PubMed] [Google Scholar]
- 78.Tucker JW, Narayanam JMR, Krabbe SW, Stephenson CRJ. Electron transfer photoredox catalysis: Intramolecular radical addition to indoles and pyrroles. Org. Lett. 2010;12:368–371. doi: 10.1021/ol902703k. [DOI] [PubMed] [Google Scholar]
- 79.Amador AG, Sherbrook EM, Yoon TP. Enantioselective photocatalytic [3+2] cycloadditions of aryl cyclopropyl ketones. J. Am. Chem. Soc. 2016;138:4722–4725. doi: 10.1021/jacs.6b01728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
All data are included in this article, Supplementary Information, Supplementary Data 1 (NMR spectra), and Supplementary Data 2 (DFT calculations).