

Abstract
Myhre syndrome is a connective tissue disorder characterized by congenital cardiovascular, craniofacial, respiratory, skeletal, and cutaneous anomalies as well as intellectual disability and progressive fibrosis. It is caused by germline variants in the transcriptional co-regulator SMAD4 that localize at two positions within the SMAD4 protein, I500 and R496, with I500V/T/M variants more commonly identified in individuals with Myhre syndrome. Here we assess the functional impact of SMAD4-I500V variant, identified in two previously unpublished individuals with Myhre syndrome, and provide novel insights into the molecular mechanism of SMAD4-I500V dysfunction. We show that SMAD4-I500V can dimerize, but its transcriptional activity is severely compromised. Our data show that SMAD4-I500V acts dominant-negatively on SMAD4 and on receptor-regulated SMADs, affecting transcription of target genes. Furthermore, SMAD4-I500V impacts the transcription and function of crucial developmental transcription regulator, NKX2–5. Overall, our data reveal a dominant-negative model of disease for SMAD4-I500V where the function of SMAD4 encoded on the remaining allele, and of co-factors, are perturbed by the continued heterodimerization of the variant, leading to dysregulation of TGF and BMP signaling. Our findings not only provide novel insights into the mechanism of Myhre syndrome pathogenesis but also extend the current knowledge of how pathogenic variants in SMAD proteins cause disease.
Keywords: Myhre syndrome, SMAD4, TGF/BMP signaling, transcriptional regulation, dominant-negative, NKX2–5
1. Introduction
Myhre syndrome (OMIM 139210) defines a rare, phenotypically variable multisystem congenital disorder characterized by short stature, neurodevelopmental abnormalities, craniofacial dysmorphism, skeletal deformities, restricted joint mobility, skin thickening and congenital heart defects (Myhre et al. 1981; Le Goff et al. 2011; Caputo et al. 2012; Lin et al. 2016). In addition to the congenital and connective tissue anomalies, affected individuals may develop progressive cardiac, respiratory, and gastrointestinal fibrosis, which can lead to severe complications. To date, over 60 individuals with Myhre syndrome have been described in the literature with de novo missense variants affecting two key residues of the transcription factor SMAD family member 4 (SMAD4) (Le Goff et al. 2011; Caputo et al. 2012; Lin et al. 2016; Lin et al. 2020). Pathogenic variants at I500 (I500V/T/M) have so far been de novo, though a recent case of Myhre syndrome caused by the SMAD4-R496C variant was reported as the first case of autosomal dominant Myhre syndrome (Meerschaut et al. 2019). As Myhre syndrome cases numbers rise, understanding the relevant molecular etiology is becoming increasingly pertinent.
SMAD4 is referred to as common-mediator SMAD (co-SMAD) due to its involvement in both transforming growth factor (TGF) and bone morphogenic pathway (BMP) signaling (Ross and Hill 2008). Receptor-regulated SMADs (R-SMADs), SMAD family member 1 (SMAD1), SMAD2, SMAD3, SMAD5, and SMAD9, are phosphorylated following receptor activation by TGF or BMP ligands and trimerize with SMAD4 prior to nuclear localization and subsequent transcriptional regulation (Ross and Hill 2008). The transcriptional network associated with SMAD4 is further extended and modified by its association with transcriptional co-factors, such as NK2 homeobox 5 (NKX2–5) or inhibitory SMADs (I-SMADs), SMAD6, and SMAD7 (Imamura et al. 1997; Hata et al. 1998; Brown et al. 2004; Kang et al. 2012; Hu et al. 2021).
Deletion of Smad4 in mice leads to embryonic death prior to gastrulation, and heterozygous-null mice develop duodenal and stomach polyposis (Sirard et al. 1998; Yang et al. 1998; Chu et al. 2004; Alberici et al. 2006). Similar observations have been made in individuals with germline heterozygous loss-of-function (LOF) variants in SMAD4, indicating haploinsufficiency as a causal mechanism of juvenile polyposis syndrome (JPS, OMIM 174900) and JPS-hereditary hemorrhagic telangiectasia (JPS-HHT, OMIM 175050) (Gallione et al. 2010). In addition, individuals with somatic LOF variants in SMAD4 manifest pancreatic or colorectal carcinomas (OMIM 260350) (Miyaki and Kuroki 2003; Bardeesy et al. 2006). The non-overlapping phenotypes seen between individuals with Myhre syndrome and JPS-HHT caused by SMAD4 protein dysfunction suggests different aspects of SMAD4 function are perturbed. Several previous studies have examined the molecular consequences of the two hotspot SMAD4 variants that cause Myhre syndrome. Using cells from affected individuals, reduced SMAD4 ubiquitylation, increased SMAD4 protein expression, and a general downregulation of TGF/BMP-dependent target genes were observed (Le Goff et al. 2011; Piccolo et al. 2014). These observations have led to the current consensus that SMAD4-I500V/T/M and SMAD4-R496C pathogenic variants are gain-of-function (GOF), which distinguishes them from the LOF variants that cause JP, JPS-HHT, and cancer.
We here present two individuals with Myhre syndrome carrying the de novo SMAD4-I500V variant. Our functional assessment reveals that the SMAD4-I500V variant functions in a dominant-negative manner. We propose that the unique phenotypes of individuals with Myhre syndrome are caused by disruption of wild-type SMAD4 function as well as that of SMAD4 binding partners and aberrant activation of TGF/BMP signaling.
2. Materials and Methods
2.1. Patient recruitment
Patient 1 was recruited on IRB approved protocol for the genetic analysis of vertebral malformations. The study was conducted in accordance with the Declaration of Helsinki, and ethical approval for this study was obtained from the Sydney Children’s Hospital Network Human Research Ethics Committee (approval number HREC/18/SCHN/222). Informed, written consent was obtained from all participants.
2.2. Whole exome sequencing
Whole exome sequencing, variant calling, annotation and prioritization of patient 1 were performed as per (Martin et al. 2020). Patient 2 had gene panel sequencing (curated syndromic and non-syndromic intellectual disability gene list) performed at Fulgent (fulgentgenetics.com).
2.3. Plasmids and mutagenesis
pDONR223-SMAD2-WT, pDONR223-SMAD4-WT and pDONR223-SMAD5-WT plasmids were purchased from Addgene (#81830, #82216, #82217), then subcloned into the pCMX-FLAG vector via Gateway cloning (ThermoFisher Scientific). The SMAD4-I500V variant was generated by site directed mutagenesis using the following primers: F 5’ GACCTTCGTCGCTTATGCGTACTCAGGATGAGTTTTG 3’, R 5’ CAAAACTCAT CCTGAGTACGCATAAGCGACGAAGGTC 3’. pCMV5-SMAD1-HA was purchased from Addgene (#14956). Xvent2-luc reporter (Rastegar et al. 1999) was a kind gift from Dr. Craig A. Harrison (Hudson Institute of Medical Research, Clayton, Vic, Australia). SBE(4)-luc reporter (Zawel et al. 1998) was acquired from Addgene (#16495). pDONR221-NKX2–5-WT was purchased from DNASU plasmid repository (www.dnasu.org) and subcloned in to pcDNA3.1-HA vector via Gateway cloning. pGL4.24 Snai2-luc and pGL4.24 Rad50-luc reporters have been described previously (Bouveret et al. 2015).
2.4. Cell culture and transfections
HEK293T cells were obtained from ATCC (www.atcc.org) and maintained in DMEM medium containing 10% FCS in a humidified incubator at 37°C and 10% CO2. 80,000 cells were seeded in 12-well plates for luciferase assays and 100,000 cells were seeded in 6-well plates for RNA extraction or protein extraction. BMP4 ligand (Gibco, #PHC9534) was added to cells at 100 ng/mL and TGFβ1 (Miltenyi Biotec, #130–095-067) was added to cells at 10 ng/mL. Transfections were carried out using Lipofectamine 3000 Reagent (ThermoFisher Scientific) according to manufacturer’s instructions.
2.5. Luciferase assays
HEK293T cells were transfected with 100 ng of luciferase reporter constructs, 150 ng of expression vectors or empty vector, and 2.5 ng TK-Renilla. In dominant negative assays 150 ng, 75 ng, and 35 ng of SMAD4-I500V were used with 150 ng of wild-type constructs. Cells were treated with ligands overnight on the day of transfection. Assays were performed 24 hours after transfection. Dual luciferase assays were performed as per manufacturer’s instructions (Promega, #E1980). Firefly luciferase activity was normalized to Renilla luciferase activity.
2.6. Co-IP and western blotting
HEK293T cells were transfected with 1 μg of SMAD4-WT, SMAD4-I500V or SMAD4-WT/SMAD4-I500V. Cells were treated with ligands for 40 minutes prior to protein extraction. Cells were lysed 48 hours after transfection using whole cell extract buffer (20 mM HEPES, 420 mM NaCl, 0.5% NP-40, 25% glycerol, 0.2 mM EDTA, 1.5 mM MgCl2, 1 mM PMSF and protease inhibitors). Cells were washed in PBS and lysed with 250 μl WCE buffer for 10 minutes on ice. Lysed cells were scraped off the 6-well plates and homogenized with a 25 Gauge needle 10 times. The lysates were centrifuged for 30 minutes at 4°C. Supernatant was precleared with Protein G (Thermo Fisher Scientific) for 1 hr at 4°C, then incubated with anti-FLAG M2 antibody (Sigma Aldrich, #F1804, RRID AB_262044) at 1:150 dilution or an equal amount of mouse IgG overnight at 4°C. The lysates were incubated with Protein G beads for 2 hours at 4°C, then washed in WCE buffer 4 times. Protein was eluted in 4x sample buffer (Biorad) for 5 minutes at 95°C and loaded onto TGX stain-free precast gels (Biorad). Western blots were carried out using the following antibodies: anti-FLAG (1:1000, Cell Signaling Technology, #14793, RRID AB_2572291), anti-HA (1:1000, Cell Signaling Technology, C29F4, RRID AB_1549585), anti-pSMAD1,5,9 (1:1000, Cell Signaling Technology, #13820, RRID AB_2493181), anti-pSMAD2 (1:500, Cell Signaling Technology, #18338, RRID AB_2798798), anti-NKX2–5 (1:1000, Santa Cruz Biotechnology, sc-14033, RRID AB_650281).
2.7. Quantitative real-time PCR
HEK293T cells were transfected as per co-IP and treated with ligands for either 2 hours or 48 hours. Cells were lysed 48 hours after transfection and RNA extracted using PureLink RNA Mini kit (Thermo Fisher Scientific). cDNA synthesis and qPCR were performed as per (Alankarage et al. 2019). The following primers were used: SMAD4 For 5’ CTGCTGCTGGAATTGGTGTT 3’, Rev 5’ GTCTAAAGGTTGTGGGTCTGC 3’; ID1–3 For 5’ CATCTCCAACGACAAAAGGAG 3’, Rev 5’ CTTCCGGCAGGAGAGGTT 3’; COL1A1 For 5’ CCCCGAGGCTCTGAAGGT 3’, Rev 5’ GCAATACCAGGAGCACCATTG 3’; SMAD6 For 5’ GGGCCCGAATCTCCGC 3’, Rev 5’ ACATGCTGGCGTCTGAGAAT 3’; SMAD7 For 5’ TTCAGGACCAAACGATCTGC 3’, Rev 5’ GGGATGGTGGTGACCTTTG 3’; CTGF For 5’ CTCCTGCAGGCTAGAGAAGC 3’, Rev 5’ GATGCACTTTTTGCCCTTCTT 3’; NKX2–5 For 5’ AAGTGTGCGTCTGCCTTTCC 3’, Rev 5’ GCGCGCACAGCTCTTTCTTT 3; HSP90AA1 For 5’ CGTTTCTGAGAAGCAGGGCA 3’, Rev 5’ GGGAATGCAGAGACGTGGAA 3’.
2.8. Statistical analysis
Data were analyzed using Graphpad Prism software (9.2.0). Luciferase and qPCR data were analyzed using ANOVA and presented as mean ± SD.
3. Results
3.1. SMAD4-I500V has greatly reduced ability to activate TGFβ1- and BMP4-responsive promoters and inhibits wild-type SMAD4
SMAD4-I500V variant was identified in two previously unpublished individuals, who presented with phenotypes consistent with Myhre syndrome (case reports in supplementary data, Fig. S1). SMAD4 is the common-mediator SMAD, acting downstream of both TGF and BMP signaling. In order to assess whether SMAD4 carrying the I500V variant (SMAD4-I500V) affects the signaling pathways differently to wild-type SMAD4, two promoters were used to test transcriptional activation: (1) the SBE(4)-luc reporter which contains four consecutive SMAD binding elements and is strongly activated by TGFβ1(Zawel et al. 1998); and (2) the Xvent2-luc reporter which contains the BMP4-responsive element from the Xenopus Xvent-2B gene that regulates patterning of the ventral mesoderm (Rastegar et al. 1999; Henningfeld et al. 2000). After confirming that SMAD4-I500V was able to translocate to the nucleus similar to its wild-type counterpart (Fig. S2), HEK293T cells were transfected with WT-SMAD4, SMAD4-I500V, or empty vector alongside either SBE(4)-luc or Xvent2-luc. Luciferase activity was measured in untreated cells or cells treated with TGFβ1 or BMP4 ligands.
WT-SMAD4 activated the SBE(4)-luc reporter above vector levels in untreated conditions, and more significantly in TGFβ1-treated conditions (Fig. 1A). SMAD4-I500V was able to activate the promoter above vector levels in basal conditions, although activity was 1.8-fold lower than WT-SMAD4- transfected cells. In TGFβ1-treated cells, the activity of the variant fell below that of empty vector-transfected cells and was 1.9-fold lower than WT-SMAD4 transfected cells. Overall, SMAD4-I500V showed reduced capacity to activate the SBE(4)-luc reporter compared to WT-SMAD4 in both basal and ligand activated conditions. WT-SMAD4 was not able to activate the Xvent2-luc above vector levels in both untreated and BMP4-treated cells (Fig. 1B). However, SMAD4-I500V could not reproduce WT-SMAD4 level activity in either condition, and its activity was 3-fold lower than vector or WT-SMAD4-transfected cells.
Fig. 1. SMAD4-I500V shows greatly reduced ability to activate target promoters and acts dominant-negatively on WT-SMAD4.
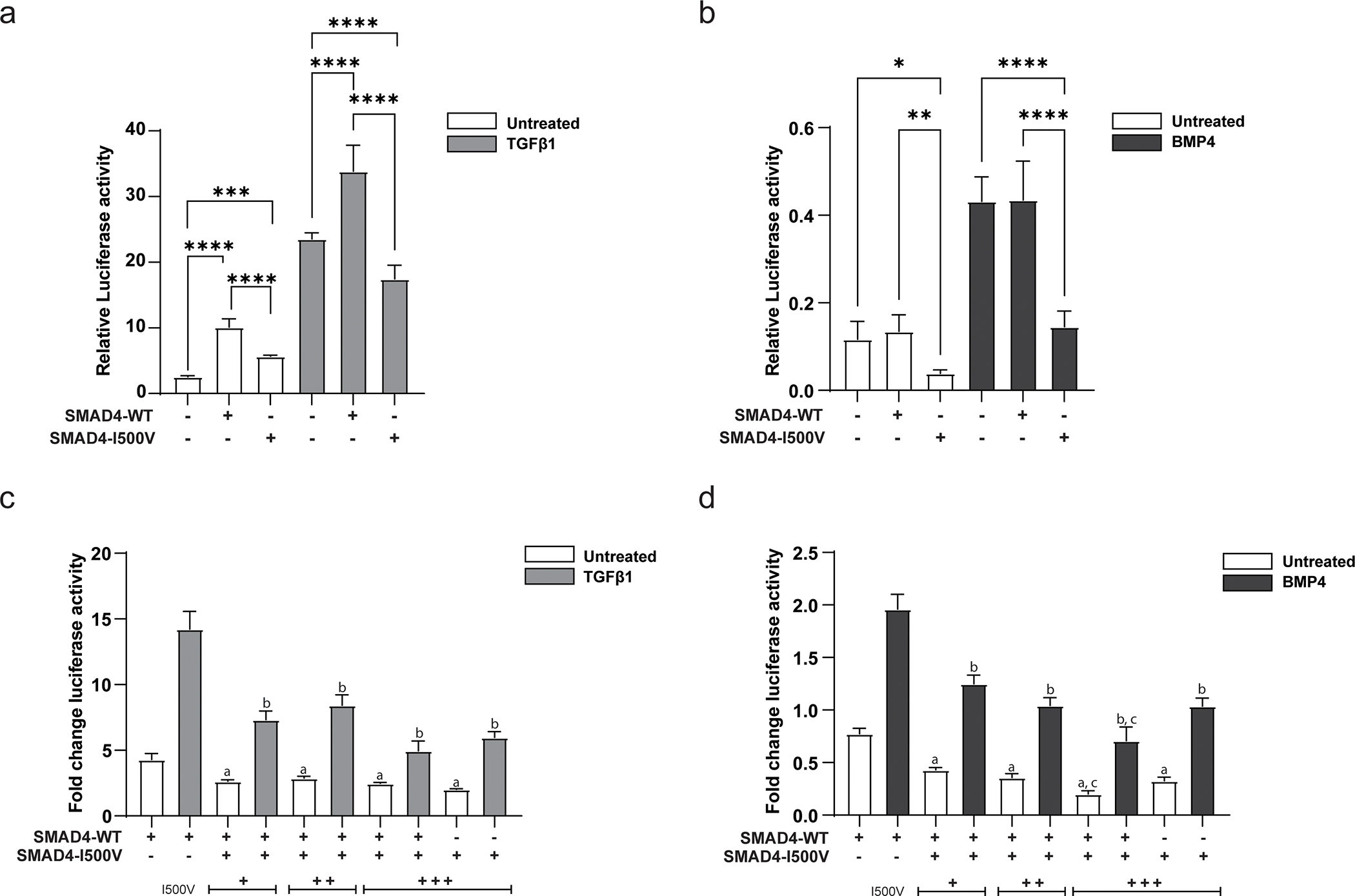
(A) Impact of the I500V variant on basal and TGFβ1-activated SBE(4)-luc was assessed by luciferase assays, ***<0.001, **** < 0.0001, n = 9–12. (B) Impact of the I500V variant on basal and BMP4-activated Xvent2-luc was assessed by luciferase assays, * < 0.05, ** < 0.01, **** < 0.0001, n = 9–12. Luciferase activity was normalized to Renilla and analyzed by one-way ANOVA, presented as mean ± SD. (C) Dominant-negative response of the I500V variant on WT-SMAD4 action on SBE(4)-luc in basal and TGFβ1-treated cells. (D) Dominant-negative response of the I500V variant on WT-SMAD4 action on Xvent2-luc in basal and BMP4-treated cells, a: comparison between WT-SMAD4- and WT/I500V- or I500V-transfected cells in basal conditions, p < 0.0001; b: comparison between WT-SMAD4- and WT/I500V- or I500V-transfected cells in TGFβ1/BMP4-treated cells, p < 0.001–0.0001; c: comparison between WT/I500V- and I500V-transfected cells in basal and BMP4-treated cells, p < 0.001–0.01. Luciferase activity was normalized to vector transfected cells within each treatment group, analyzed by one-way ANOVA, presented as mean ± SD, n = 6.
Given that the variant could not reach vector level of activity with either promoter, the ability of the variant to act dominant-negatively on WT-SMAD4 was tested. Increasing amounts of SMAD4-I500V were titrated with a constant amount of WT-SMAD4. On both SBE(4)-luc and Xvent2-luc reporters, SMAD4-I500V was able to downregulate WT-SMAD4 activity in untreated and ligand-activated conditions (Fig. 1C,D). These results show that in addition to reduced ability to activate target promoters, SMAD4-I500V also decreases the activity of WT-SMAD4.
3.2. SMAD4-I500V interferes with promoter activation by SMAD1, SMAD2, and SMAD5
Dominant-negative function of the SMAD4-I500V variant was further investigated by assessing its impact on the activity of SMAD1, SMAD2, and SMAD5. Again, SMAD4-I500V was added in increasing amounts with wild-type expression constructs (Fig. S3a). The ability of SMAD4-I500V to influence SMAD1 and SMAD5 directed signaling was tested on the Xvent2-luc reporter with BMP4 activation while the variant’s effect on SMAD2 was assessed on the SBE(4)-luc reporter with TGFβ1 treatment.
In cells co-transfected with WT-SMAD4 and SMAD1 with the Xvent2-luc reporter, SMAD1 activity was significantly increased in the presence of SMAD4 under basal conditions (Fig. 2A). There was no difference in SMAD1 activity between SMAD1/WT-SMAD4 and SMAD1-only transfected cells, however, when treated with BMP4. Unlike WT-SMAD4, the SMAD4-I500V variant did not increase the activity of SMAD1 in either of the transfected conditions, and instead significantly downregulated the activation by BMP4.
Fig. 2. Dominant-negative activity of SMAD4-I500V inhibits function of R-SMADs.
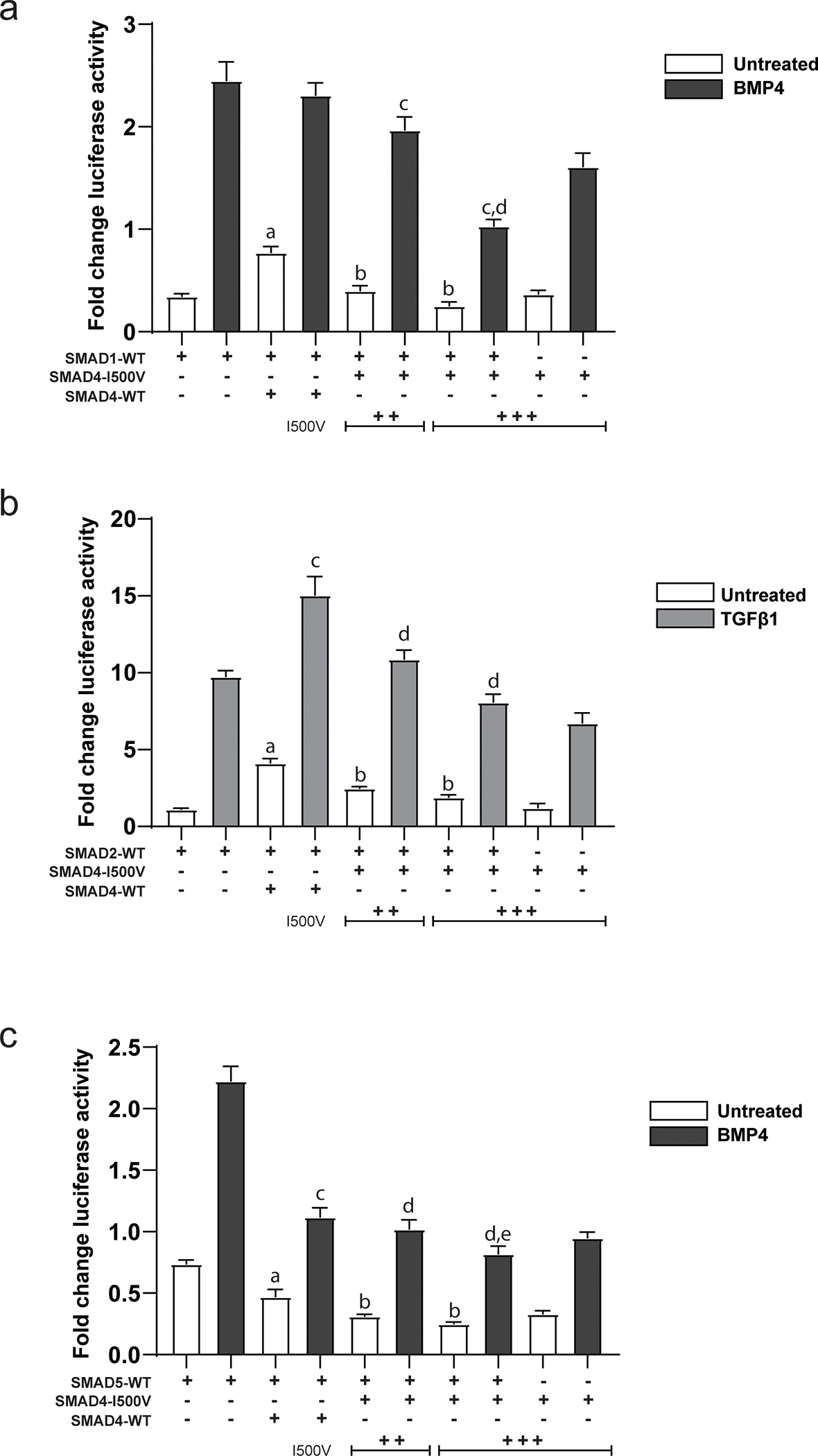
(A) Action of the I500V variant on SMAD1 activity on the Xvent2-luc reporter in basal and BMP4-treated cells was assessed, a: comparison between SMAD1- and SMAD1/WT-SMAD4- transfected cells in basal conditions, p < 0.0001; b: comparison between SMAD1/WT-SMAD4- and SMAD1/SMAD4-I500V- transfected cells in basal conditions, p < 0.0001; c: comparison between SMAD1/WT-SMAD4- and SMAD1/SMAD4-I500V- transfected cells in BMP4-treated cells, p < 0.0001; d: comparison between SMAD4-I500V- and SMAD1/SMAD4-I500V- transfected cells in BMP4-treated cells, p < 0.0001. (B) Action of the I500V variant on SMAD2 activity on the SBE(4)-luc reporter in basal and TGFβ1-treated cells was assessed, a: comparison between SMAD2- and SMAD2/WT-SMAD4-transfected cells in basal conditions, p < 0.0001; b: comparison between SMAD2/WT-SMAD4- and SMAD2/SMAD4-I500V-transfected cells in basal conditions, p < 0.0001; c: comparison between SMAD2- and SMAD2/WT-SMAD4- transfected cells in TGFβ1-treated conditions, p < 0.0001; d: comparison between SMAD2/WT-SMAD4- and SMAD2/ SMAD4-I500V-transfected cells in TGFβ1-treated conditions, p < 0.0001. (C) Action of the I500V variant on SMAD5 activity on the Xvent2-luc reporter in basal and BMP4-treated cells was assessed, a: comparison between SMAD5- and SMAD5/WT-SMAD4- transfected cells in basal conditions, p < 0.0001; b: comparison between SMAD5/WT-SMAD4- and SMAD5/SMAD4-I500V-transfected cells in basal conditions, p < 0.0001; c: comparison between SMAD5- and SMAD5/WT-SMAD4- transfected cells in BMP4-treated conditions, p < 0.0001; d: comparison between SMAD5/WT-SMAD4- and SMAD5/SMAD4-I500V- transfected cells in BMP4-treated conditions, p < 0.05, 0.0001; e: comparison between SMAD5/SMAD4-I500V- and SMAD4-I500V- transfected cells in BMP4-treated cells, p < 0.001. Luciferase activity was normalized to vector transfected cells and analyzed by one-way ANOVA, presented as mean ± SD, n = 6–9.
Addition of WT-SMAD4 increased the activation of the SBE(4)-luc promoter by SMAD2 in both basal and TGFβ1-treated conditions (Fig. 2B). This activation was lost when SMAD2 was co-transfected with SMAD4-I500V in both basal and ligand-treated conditions.
In contrast to SMAD1 and SMAD2 observations, activation of the Xvent2-luc reporter by SMAD5 was negated by both WT-SMAD4 and SMAD4-I500V in both basal and BMP4-treated cells (Fig. 2C). However, SMAD5/SMAD4-I500V co-transfected cells had lower reporter activity than cells co-transfected with SMAD5/WT-SMAD4 in both basal and BMP4-treated conditions.
These results show that in addition to inhibiting WT-SMAD4, SMAD4-I500V also reduces the ability of R-SMADs to transcriptionally activate promoters by dominant-negative inhibition, and thus more profoundly affect TGFβ and BMP signaling.
3.3. SMAD4-I500V binds pSMAD1,5,9 and pSMAD2
Given the I500V variant resides in the Mad homology domain 2 (MH2) that dictates SMAD4 interaction with R-SMADs, whether protein-protein interactions were affected by the SMAD4 variant was tested using co-immunoprecipitation assays. HEK293T cells were transfected with FLAG-tagged WT-SMAD4 or SMAD4-I500V or co-transfected with WT/I500V, then assessed in untreated, TGFβ1-treated or BMP4-treated conditions. SMAD4 (WT or I500V) was pulled down using an anti- FLAG antibody, then probed with antibodies against phosphorylated (p) SMAD1,5,9 or phosphorylated SMAD2.
The SMAD4-I500V variant bound to pSMAD1,5,9 in untreated (Fig. 3A, lanes 2,3), TGFβ1-treated (Fig. 3A, lanes 5,6) and BMP4-treated (Fig. 3A, lanes 8,9) cells. Similarly, SMAD4-I500V bound to pSMAD2 in all three treatment conditions (Fig. 3B, lanes 2,3,5,6,8,9). Interestingly, in untreated cells and in cells treated with ligand, SMAD4-I500V bound more pSMADs than WT-SMAD4. These results show that heterodimerization of the SMAD4-I500V variant is unperturbed, retaining the capacity to bind activated SMADs during basal cell conditions as well as during active ligand signaling.
Fig. 3. SMAD4-I500V binds phosphorylated R-SMADs.
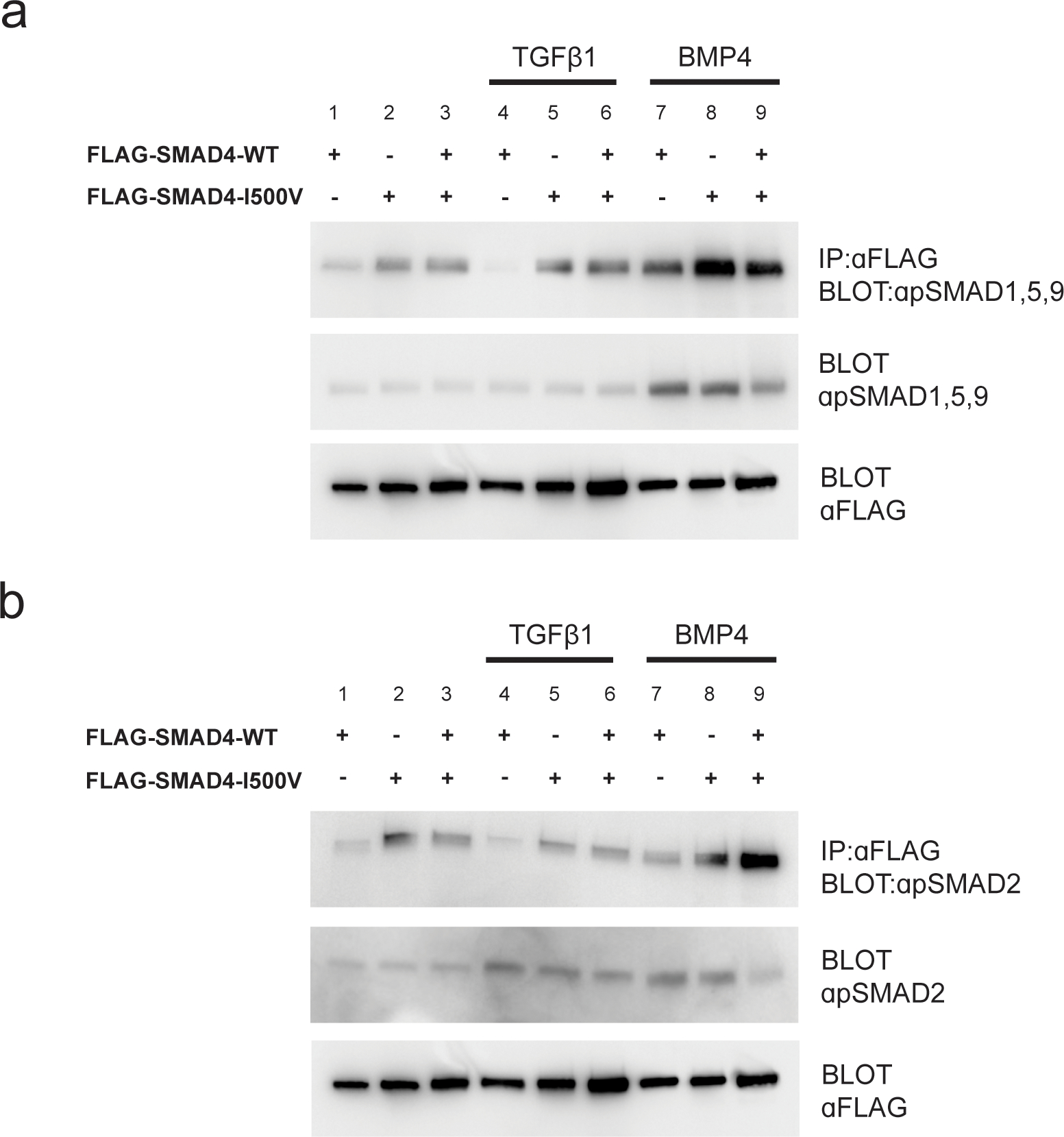
(A) Immunoprecipitation of WT-SMAD4 or SMAD4-I500V with anti-FLAG and detection of phospho-SMAD1,5,9. (B) Immunoprecipitation of WT-SMAD4 or SMAD4-I500V with anti-FLAG and detection of phospho-SMAD2. FLAG-WT-SMAD4, FLAG-SMAD4-I500V, or FLAG-WT-SMAD4/FLAG-SMAD4-I500V were immunoprecipitated from cell lysates at basal, TGFβ1-treated, or BMP4-treated conditions, and blotted for phosphorylated SMAD1,5,9 or phosphorylated SMAD2.
3.4. SMAD4-I500V perturbs target gene regulation by TGFβ1 and BMP4
To assess the impact of the variant on gene transcription, genes that have previously been assessed (Le Goff et al. 2011) as TGF/BMP target genes were selected, in addition to SMAD4 and SMAD7. Gene expression was assessed by quantitative real-time PCR, in several conditions: untransfected cells that were untreated, TGFβ1-treated or BMP4-treated (Fig. 4A); WT-SMAD4-, SMAD4-I500V-, or WT/I500V-transfected cells that were untreated (Fig. 4B); transfected cells that were TGFβ1-treated for 2 hours or 48 hours (Fig. 4C,D); and transfected cells that were BMP4-treated for 2 hours or 48 hours (Fig. 4E,F).
Fig. 4. SMAD4-I500V leads to transcript expression changes in genes downstream of TGFβ1 and BMP4 signaling.
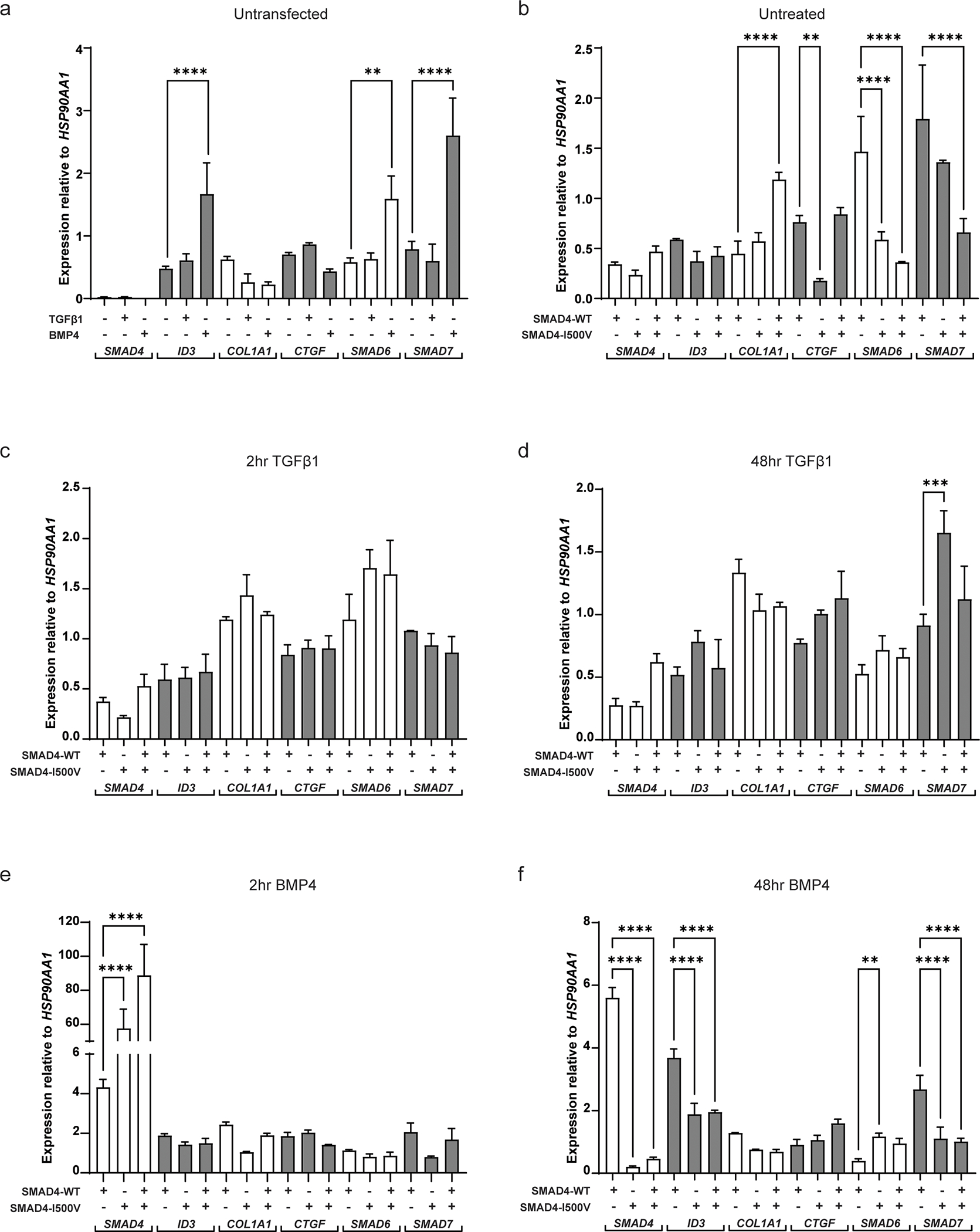
(A) Gene expression of SMAD4, ID3, COL1A1, CTGF, SMAD6 and SMAD7 in untransfected cells treated with or without TGFβ1 or BMP4. (B) Gene expression in cells transfected with WT-SMAD4, SMAD4-I500V or WT/I500V. (C) Gene expression in cells treated with TGFβ1 for 2 hours in cells transfected with WT-SMAD4, SMAD4-I500V, or WT/I500V. (D) Gene expression in cells treated with TGFβ1 for 48 hours in cells transfected with WT-SMAD4, SMAD4-I500V, or WT/I500V. (E) Gene expression in cells treated with BMP4 for 2 hours in cells transfected with WT-SMAD4, SMAD4-I500V, or WT/I500V. (F) Gene expression in cells treated with BMP4 for 48 hours in cells transfected with WT-SMAD4, SMAD4-I500V, or WT/I500V. Differences in expression between WT-SMAD4, SMAD4-I500V, or WT/I500V -transfected cells were statistically analyzed by one-way ANOVA and presented as mean ± SD, ** p < 0.01, *** p < 0.001, **** p < 0.0001; n = 3.
Endogenous expression of SMAD4 was barely detectable regardless of treatment condition (Fig. 4A). SMAD4 expression increased in transfected cells with no significant differences between WT-SMAD4-, SMAD4-I500V-, or WT/I500V-transfected cells, indicating equal transfection between conditions (Fig. 4B). SMAD4 expression remained without any significant changes in transfected cells treated with TGFβ1 for 2 or 48 hours (Fig. 4C,D). In comparison to WT-SMAD4-transfected-BMP4-treated cells, at 2 hours, SMAD4 transcript levels in SMAD4-I500V-transfected cells and in WT/I500V-transfected cells were 14-fold and 21-fold increased, respectively (Fig. 4E). After 48 hours of BMP4 treatment, SMAD4 expression in WT-SMAD4-transfected cells remained at similar levels to 2 hours of BMP4 treatment (Fig. S4a). However, compared to WT-SMAD4, SMAD4 expression in I500V-transfected cells had dropped by 28-fold and 12-fold in SMAD4-I500V- and WT/I500V-transfected cells, respectively (Fig. 4F), and by 281-fold and 192-fold compared to transcript levels at 2 hours of BMP4 treatment (Fig. S4a).
Expression of inhibitor of DNA binding 3 (ID3), a transcriptional inhibitor downstream of BMP signaling, increased significantly in BMP4-treated, untransfected cells (4-fold increase) compared to untreated, untransfected cells (Fig. 4A). No differences in ID3 expression were observed between WT-SMAD4- or SMAD4-I500V-transfected cells in untreated cells (Fig. 4B). Significant differences in ID3 expression were observed at 48 hours of BMP4 treatment, where expression in WT-SMAD4-transfected cells had increased to 8-fold higher than untreated cells and 2-fold higher than 2 hours of BMP4 treatment (Fig. S4b). At 48 hours of BMP4 treatment, ID3 gene expression in SMAD4-I500V- and WT/I500V-transfected cells were 2-fold lower than WT-SMAD4-transfected cells (Fig. 4F).
The gene expression of collagen type 1, α1 (COL1A1), an extracellular matrix component, increased by 3-fold in untreated WT/I500V-transfected cells compared to WT-SMAD4-transfected cells (Fig. 4B). COL1A1 expression after 2 hours of BMP4 treatment in WT-SMAD4-transfected cells was 5-fold higher than untreated WT-SMAD4-transfected cells (Fig. S4c). However, in SMAD4-I500V-transfected cells, COL1A1 transcript was 2.5-fold lower than in WT-transfected cells. At 48 hours, there was a slight reduction of COL1A1 gene expression in SMAD4-I500V-transfected cells in TGFβ1-treated cells (1.3-fold) and a more significant reduction of expression in BMP4-treated cells (2-fold) (Fig. 4D,F; Fig. S4c).
Transcript levels of connective tissue growth factor (CTGF), another extracellular matrix component, decreased by 3-fold in untreated SMAD4-I500V-transfected cells compared to WT-SMAD4-transfected levels (3-fold) (Fig. 4B). After 2 hours of BMP4 treatment, CTGF transcript in WT-SMAD4-transfected cells was 2-fold higher than untreated levels. BMP4 also rescued CTGF expression in SMAD4-I500V-transfected cells, which was at similar levels to WT-SMAD4-transfected cells (Fig. S4d). In contrast, CTGF expression in WT/I500V-transfected cells was lower (1.5-fold) than the WT- or SMAD4-I500V-transfected cells (Fig. 4E; Fig. S4d). At 48 hours of BMP4 treatment, CTGF transcript in WT/I500V-transfected cells remained at similar levels to 2 hours of BMP4 treatment whereas transcript levels in WT-SMAD4- and SMAD4-I500V-transfected cells had dropped to levels similar to untreated conditions (Fig. 4E; Fig. S4d).
Expression of the I-SMADs, SMAD6 and SMAD7, were also assessed in our cell culture system. SMAD6 responded strongly to BMP4 and to WT-SMAD4 independently, and there was significantly lower gene expression in SMAD4-I500V- and WT/I500V-transfected cells (Fig. 4A,B). In TGFβ1-treated cells at 2 hours, a trend of increased SMAD6 expression was observed in I500V-transfected cells which was maintained at 48 hours although overall levels of transcript were lower across all three transfection conditions (Fig. 4C,D; Fig. S4e). BMP4 treatment for 2 hours revealed a slight decrease in SMAD6 transcript levels in SMAD4-I500V- and WT/I500V-transfected cells compared to WT-SMAD4-transfected cells (Fig. 4E). Interestingly, at 48 hours, transcript levels in WT-SMAD4-transfected cells had decreased by 2.5-fold compared to expression levels in SMAD4-I500V- or double-transfected cells (Fig. 4F). Furthermore, after 48 hours of TGFβ1 and BMP4 treatment, SMAD6 transcript levels in WT-SMAD4-transfected cells were decreased compared to 2 hours of ligand treated WT-SMAD4-transfected cells (Fig. S4e). The results show that SMAD6 is not maintained as an inhibitor in cells exposed to WT-SMAD4-BMP4 for longer periods of time, whereas its expression is maintained in I500V-BMP4 (48 hr) cells.
SMAD7 transcript increased significantly in BMP4-treated, untransfected cells compared to untreated, untransfected cells (Fig. 4A). 48 hours of TGFβ1 treatment saw an increase in SMAD7 transcript in SMAD4-I500V-transfected cells while levels in WT-SMAD4-transfected cells remained similar to levels at 2 hours of treatment (Fig. 4C,D; Fig. S4f). In WT-SMAD4-transfected cells and in WT/I500V-transfected cells, 2 hours of BMP4 treatment led to similar SMAD7 expression levels, whereas SMAD4-I500V-transfected cells could not maintain equivalent levels of gene expression (Fig. 4E). In cells treated with 48 hours of BMP4, SMAD7 expression was at similarly high levels as 2 hours of BMP4 treatment in WT-SMAD4-transfected cells. In contrast, in SMAD4-I500V-transfected cells, as well as in WT/I500V-transfected cells, SMAD7 levels were 2.3-fold and 2.6-fold lower than in WT-SMAD4 transfected cells (Fig. 4F), respectively. These results show that BMP4 regulates SMAD7 transcription, which exerts its inhibitory function on TGFβ1 signaling.
Overall, gene expression responded positively to ligand treatment (ID3, SMAD6, SMAD7) or WT-SMAD4 (SMAD6, SMAD7), or both (SMAD6, SMAD7). Transfection with SMAD4-I500V or WT/I500V lead to downregulation of transcription in most tested genes (ID3, COL1A1, SMAD7) and up-regulation of I-SMADs in TGFβ1- (SMAD7) or BMP4- (SMAD6) treated cells.
3.5. SMAD4-I500V decreases NKX2–5 transcription, binds and negatively impacts promoter activation by NKX2–5
Several previous studies have investigated the relationship between SMAD4 and crucial cardiac developmental transcription factor, NKX2–5 (Lien et al. 2002; Brown et al. 2004; Hu et al. 2021). Most recently, it was shown that SMAD4 directly binds NKX2–5, thereby regulating its transcription and cellular localization (Hu et al. 2021). Heart abnormalities, both congenital and post-natal, are a common occurrence in individuals with Myhre syndrome (Lin et al. 2016). To assess whether SMAD4-I500V differentially affects regulation of NKX2–5, transcriptional regulation of NKX2–5 by SMAD4, direct binding between SMAD4 and NKX2–5, and the effect of SMAD4-I500V on NKX2–5-mediated transcriptional regulation were tested.
NKX2–5 transcription was induced by WT-SMAD4 (2-fold over untransfected cells), whereas in WT/I500V-transfected cells, transcript remained at untransfected levels (Fig. 5A). Two hours of TGFβ1 treatment saw a general upregulation of NKX2–5 transcript in all transfected conditions. BMP4 treatment at 2 hours did not induce a transcriptional response above untransfected cells in any transfected condition. At 48 hours, NKX2–5 gene expression in TGFβ1-treated WT-SMAD4-transfected cells remained at similar levels to 2-hour treatment, however, SMAD4-I500V- and WT/I500V-transfected cells no longer maintained NKX2–5 expression, which dropped down to basal conditions. After 48 hours of BMP4 treatment saw a 1.7-fold and 2.3-fold decrease in NKX2–5 transcript levels in SMAD4-I500V- and WT/I500V-transfected cells, respectively, compared to WT-SMAD4-transfected cells.
Fig. 5. SMAD4-I500V reduces SMAD4-mediated NKX2–5 transcription, binds to NKX2–5, and perturbs NKX2–5-mediated transcriptional regulation.
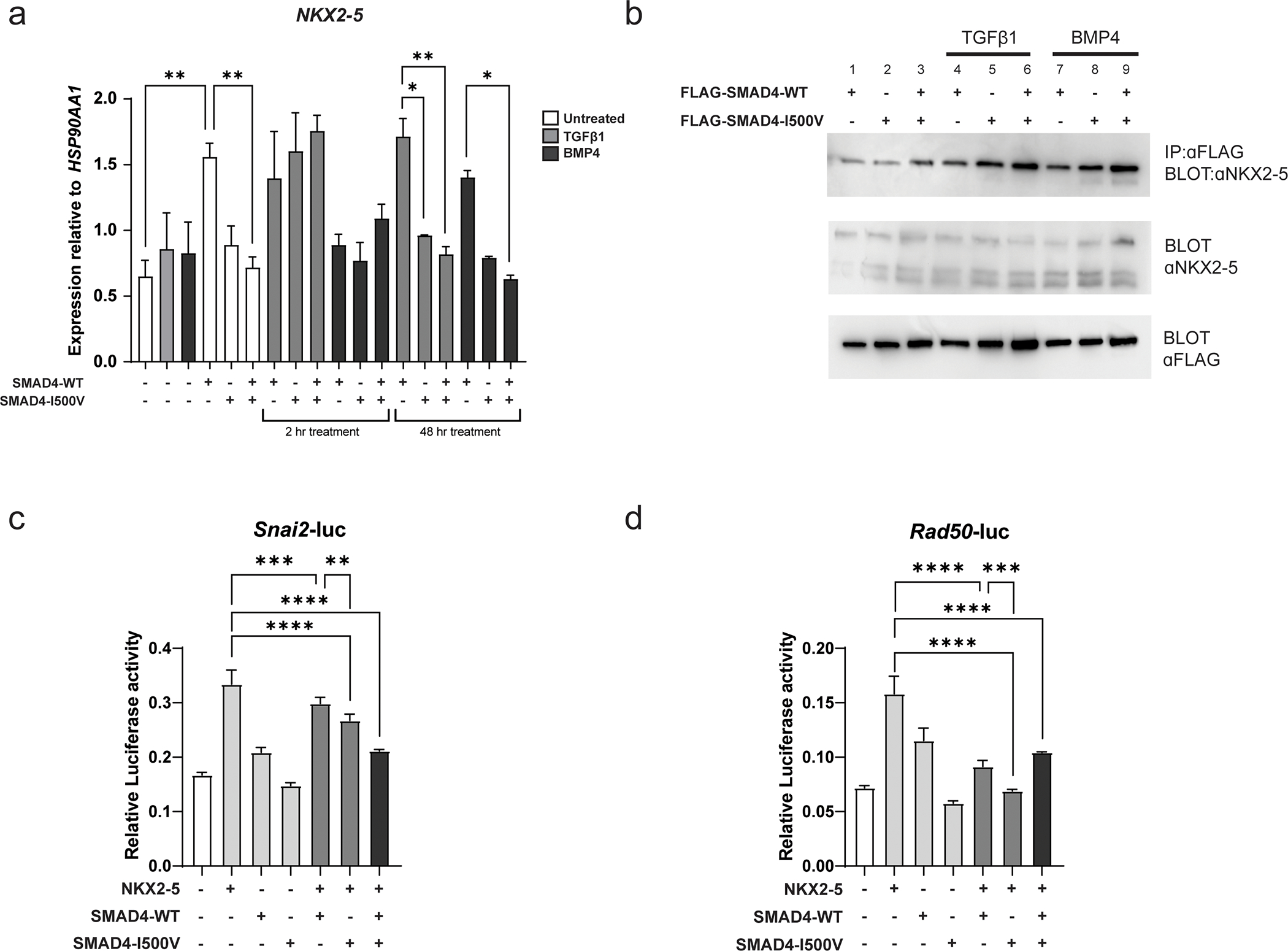
(A) NKX2–5 gene expression was measured in untransfected and transfected cells in basal, TGFβ1-, and BMP4- treated conditions at 2 hours and 48 hours. Differences in expression between WT-SMAD4 and SMAD4-I500V-transfected cells in different treatment groups were statistically analyzed by two-way ANOVA and presented as mean ± SD, *p < 0.05, **p < 0.01; n = 3. (B) FLAG-WT-SMAD4, FLAG-SMAD4-I500V or FLAG-WT-SMAD4/FLAG-SMAD4-I500V were immunoprecipitated from cell lysates at basal, TGFβ1-, or BMP4- treated conditions, and blotted for NKX2–5. (C) Impact of SMAD4-I500V on NKX2–5-mediated activation of the Snai2-luc reporter. (d) Impact of SMAD4-I500V on NKX2–5-mediated activation of the Rad50-luc reporter. Data was analyzed by one-way ANOVA and presented as mean ± SD, **p < 0.01, ***p < 0.001, ****p < 0.0001; n = 3–6.
Co-immunoprecipitation assays confirmed that WT-SMAD4 bound to NKX2–5 in untreated (Fig. 5B, lane 1), TGFβ1- (Fig. 5B, lane 4), and BMP4- (Fig. 5B, lane 7) treated conditions and revealed that SMAD4-I500V maintained binding to NKX2–5 in all treatment conditions (Fig. 5B, lanes 2,3,5,6,8,9).
Whether SMAD4-I500V could interfere with the activation of target promoters by NKX2–5 was then assessed using Snai2-luc and Rad50-luc reporters, both of which are targets of NKX2–5 (Bouveret et al. 2015) (Fig. 5C,D). On both reporters, NKX2–5 was able to activate luciferase transcription as previously observed (Bouveret et al. 2015). Co-transfection of NKX2–5 with SMAD4-I500V or with WT/I500V was able to significantly decrease the activity of NKX2–5 on both reporters.
4. Discussion
To date, at least 61 individuals with Myhre syndrome carrying SMAD4 pathogenic variants have been published, most with a de novo I500V/T/M variant (Lin et al. 2020). We identified the pathogenic SMAD4-I500V variant in two previously unreported individuals with Myhre syndrome. Although there is phenotypic variability between the two individuals, they fall within the spectrum of previously observed Myhre syndrome phenotypes. We assessed the functional impact of the SMAD4-I500V variant with the aim of understanding the underlying disease mechanism that may not only provide insights into Myhre syndrome pathogenesis but also reveal possible mechanisms of dysfunction in other SMAD proteins.
Our results show that the primary mechanism of SMAD4-I500V-mediated disease causality is the greatly reduced transcriptional activity of SMAD4. However, it is not a simple LOF model as made evident by the unique phenotypes of individuals with Myhre syndrome compared to those with JPS-HHT. Our findings suggest that an additional mechanism underlying disease causality of the SMAD4-I500V variant is the retained ability to dimerize. SMAD4 is able to homodimerize, which is a means to sequester inactive SMAD4 dimers in the cytoplasm (Hata et al. 1997; Shi et al. 1997; Qin et al. 1999). Thus, SMAD4-I500V may prevent WT-SMAD4 activity by direct protein-protein interaction. The direct interaction of the SMAD4-I500V variant with R-SMADs is also likely to exacerbate this loss of activity, where transcriptional activation by SMAD4-SMAD1 and SMAD4-SMAD2 is lost when the SMAD4-I500V variant replaces WT-SMAD4. The loss of transcriptional ability of SMAD4-I500V is the likely mechanism leading to the decrease in reporter activation in co-transfected cells, given that binding between SMAD4-I500V and R-SMADs remain undisturbed by the variant.
The perturbation of biological processes regulated by TGF and BMP signaling via the R-SMADs would be a major factor underlying the congenital and progressive phenotypic manifestations in Myhre syndrome. Individuals with pathogenic LOF SMAD2 variants present with congenital defects and connective tissue disorder reminiscent of Myhre syndrome (Granadillo et al. 2018), and Smad1, Smad2, and Smad5 knock-out mouse models exhibit similar skeletal, cardiac and gastrointestinal defects (Chang et al. 1999; Tremblay et al. 2001; Aubin et al. 2004; Monteiro et al. 2008; Wang et al. 2011), which suggests that SMAD1,SMAD2 and SMAD5 functions are perturbed by the SMAD4-I500V variant.
In our cell culture system, where both WT-SMAD4- and SMAD4-I500V- transfected cells are treated with equal amounts of ligands, we observe that SMAD4-I500V binds more phosphorylated R-SMADs than WT-SMAD4. We believe that aberrant ubiquitination of SMAD4 due to the I500V variation may underlie this observation. Monoubiquitylation of SMAD4 at K519 by TRIM33 has been demonstrated to negatively regulate TGF signaling by hindering trimerization of SMAD4 with R-SMADs (Dupont et al. 2009). The authors showed that deubiquitylation does not stabilize SMAD4, and instead regulates its ability to respond to TGF signaling, possibly by preventing homodimerization of SMAD4. Previous work has shown that SMAD4 is less ubiquitylated in individuals with the SMAD4-I500T variant; this loss of ubiquitylation is thought to underlie the increased SMAD4 protein observed in fibroblasts from individuals with SMAD4-I500V, SMAD4-I500T, and SMAD4-R496C pathogenic variants (Le Goff et al. 2011). In terms of the impact of the SMAD4-I500V variant on ubiquitylation, there are two possibilities. The first is that the SMAD4-I500V variant directly hinders the ubiquitylation of K519, leading to loss of ubiquitylation and perdurance of SMAD4-I500V that remains bound to R-SMADs and co-factors. The second possibility is that in response to loss of SMAD4 activity, the cell actively prevents SMAD4 ubiquitylation in order to rescue function, which increases the amount of non-ubiquitylated SMAD4 available for signal propagation. Non-ubiquitylated SMAD4-I500V is not dislodged from the R-SMAD trimers and remains bound (Dupont et al. 2009), leading to a build-up of phosphorylated R-SMADs. Furthermore, cells may also increase ligand activation of R-SMAD phosphorylation in an attempt to recover SMAD signaling, which may exacerbate SMAD4-I500V-R-SMAD buildup. Although we only tested the SMAD4-I500V variant, it is likely that the SMAD4-I500M and SMAD4-I500T variants also perturb TGFβ and BMP signaling in a similar manner, given the same build-up of SMAD4 and R-SMADs has been observed in SMAD4-I500M/T cases, and there are no phenotypic distinctions between individuals with any of the three SMAD4-I500 variants.
Our assessment of transcriptional misregulation by SMAD4-I500V showed a 200-fold upregulation of the SMAD4-I500V transcript in the BMP4-treated, SMAD4-I500V-transfected cells. As the variant has reduced transactivation ability, this unexpected increase in transcript most likely indicates a compensatory stabilization of the SMAD4-I500V transcript as an attempt by the cells to rescue lost SMAD4 activity. The lack of corresponding gene expression in the target genes analyzed confirms the loss of transcriptional activity of the SMAD4-I500V variant. Instead, expression of the majority of the target genes (ID3, COL1A1, SMAD7) were downregulated at 48 hours of BMP4 treatment in WT/I500V-transfected cells compared to cells transfected only with WT-SMAD4, and similar to observations in fibroblasts of individuals with Myhre syndrome (Le Goff et al. 2011). In contrast, we saw an increase in SMAD6 expression in SMAD4-I500V- transfected cells. Le Goff et al., assessed SMAD6 gene expression in Myhre syndrome fibroblasts, saw an increase in expression in cells of Myhre syndrome individuals compared to control cells (Le Goff et al. 2011). As an inhibitory SMAD, SMAD6 responds to increased BMP signaling, replacing SMAD4 in SMAD1-SMAD4 transcriptional complexes to inactivate SMAD1-mediated transcriptional regulation (Hata et al. 1998). This increase in SMAD6 could explain the decrease in luciferase activity in SMAD1/SMAD4-I500V-transfected cells, whereby the available SMAD1 is inactive because it is bound by both SMAD4-I500V and SMAD6. The higher expression of SMAD6 in Myhre syndrome fibroblasts is likely a response to the buildup of phosphorylated R-SMADs within the cell that is typically indicative of elevated ligand activity and requires maintenance of inhibitors such as SMAD6 and SMAD7. Collectively, our results establish the direct transcriptional impact of interrupted SMAD signaling due to SMAD4-I500V and validate WT/I500V transfected cells as an experimental condition similar to heterozygous individuals (Le Goff et al. 2011).
SMAD4-I500V resides within the MH2 domain of SMAD4 (321–530aa), which dictates transcriptional activation and dimerization. Our work establishes that the SMAD4-I500 residue is essential for the transcriptional activity of SMAD4. Whether this is a function directly attributable to SMAD4-I500 or whether the SMAD4-I500V variant nullifies the function of R502, R496, and R416 (previously shown to be crucial for SMAD4-mediated transcriptional activation (Chacko et al. 2001)) remains to be investigated. It is possible that pathogenic variants at SMAD4-I500 and SMAD4-R496 in Myhre syndrome individuals are observed because they are the only residues in the MH2 domain responsible for mediating the transcriptional activity of SMAD4 where certain mutations are compatible with life. Mutation of SMAD4-I500 or SMAD4-R496 to more damaging residues (I500R, I500K, R496P, R496G) or mutation of other residues within this region of the protein such as R515 (essential for heterodimerization), or K519 (ubiquitylated), which are absent from ClinVar (Landrum et al. 2016) and the gnomAD reference dataset (Karczewski et al. 2020), may cause embryonic lethality (Chacko et al. 2001; Dupont et al. 2009).
We believe that the SMAD4-I500V may contribute to disease causality in several ways: limited transcriptional activity of SMAD4-I500V, loss of WT-SMAD4 activity, sequestering of phosphorylated R-SMADs and other co-factors in inactive transcriptional complexes, and an increase in I-SMADs (Fig. 6). The high phenotypic variability in individuals with the identical underlying SMAD4-I500V variant may be explained by the effect of SMAD4-I500V upon the diverse co-factors associated with SMAD4-mediated transcriptional regulation of TGF/BMP targeted processes. Further interaction of these pathways with other transcriptional modifiers, such as hypoxia, which has been shown to alter gene expression during organ development and affect embryo survival, may also determine variant expressivity (Moreau et al. 2019).
Fig. 6. Schematic representation of SMAD4-I500V disrupting TGFβ and BMP signaling.
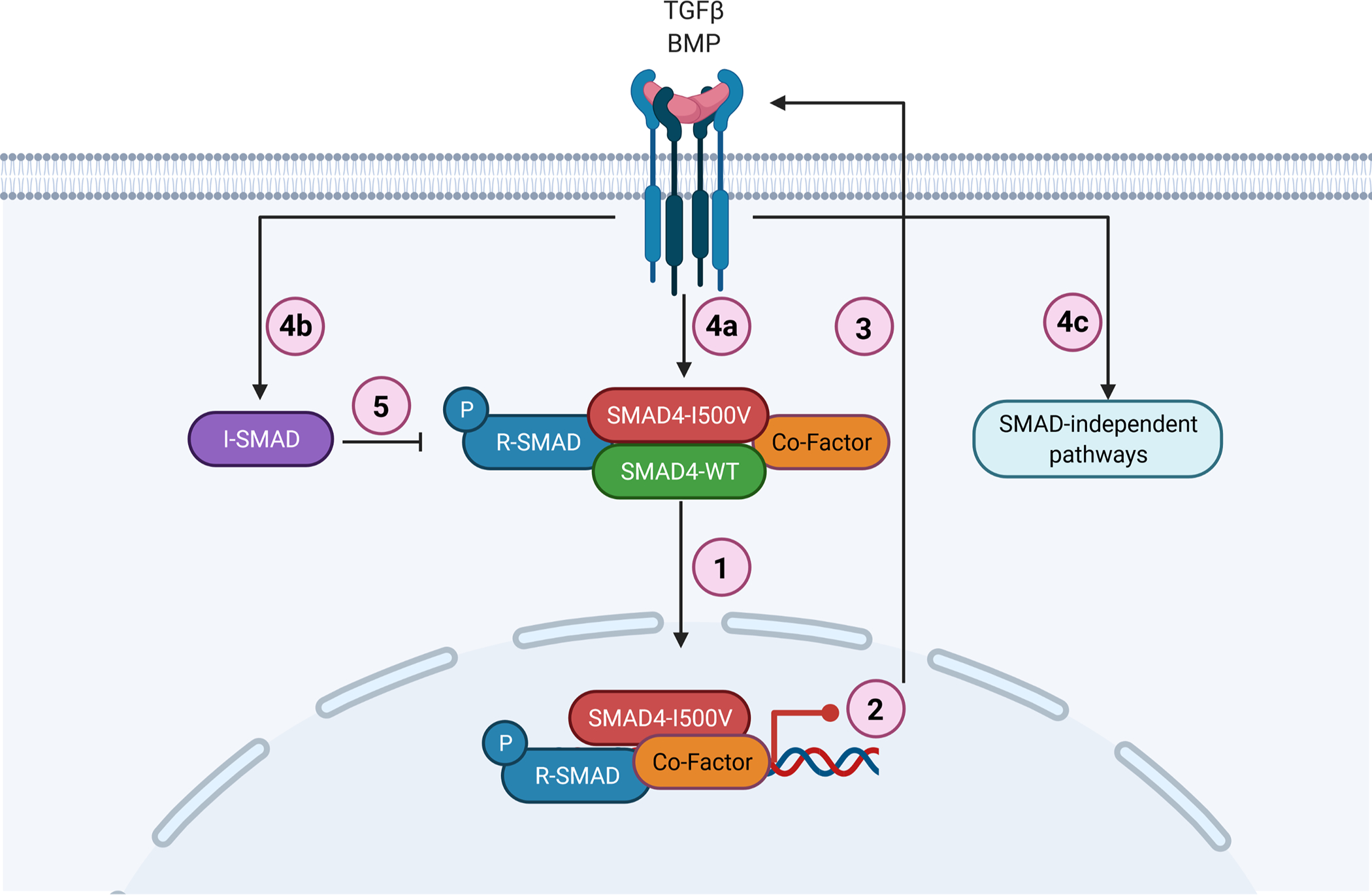
(1) SMAD4-I500V binds to SMAD4, R-SMADs, and co-factors such as NKX2–5. (2) SMAD4-I500V has reduced transcriptional activity and expression of downstream target genes is perturbed. (3) Loss of SMAD4 function activates TGF/BMP signaling. (4a) Active TGF/BMP signaling leads to increased phosphorylation of R-SMADs and buildup of SMAD4-I500V-R-SMADs. (4b) Activation of TGF/BMP signaling leads to increased I-SMADs. (4c) Activation of TGF/BMP signaling leads to initiation of SMAD-independent pathways. (5) Increased I-SMADs lead to further inhibition of SMAD-mediated transcriptional regulation. Image created with BioRender.
An important finding from our assessment is the impact of SMAD4-I500V upon NKX2–5. This early developmental transcription factor is essential for cardiac development and pathogenic variants can cause congenital heart defects (Schott et al. 1998; Benson et al. 1999). SMAD4 transcriptionally regulates NKX2–5 and was recently shown to directly interact with NKX2–5 (Brown et al. 2004; Hu et al. 2021). In our assays, transcription of NKX2–5 itself is downregulated in SMAD4-I500V- transfected cells, indicating that the expression of this transcription factor would be decreased in affected individuals. We show that NKX2–5 is bound to SMAD4-I500V, similar to what we observe with the phosphorylated R-SMADs, thereby affecting the ability of NKX2–5 to activate its target promoters. The dysregulation of NKX2–5 by direct binding and reduced transcription may underlie certain phenotypes observed in individuals with Myhre syndrome (Lin et al. 2016). Hu et al., 2021 (Hu et al. 2021) showed that loss of SMAD4 did not completely abolish NKX2–5 expression, indicating that its expression can be maintained independently of SMAD4, which may explain the absence of heart defects in 30% of cases with Myhre syndrome (Lin et al. 2016).
Pathogenic LOF variants of SMAD4 are found in JPS/HHT caused by germline SMAD4 variants and, in cancer, caused by somatic SMAD4 variants (Miyaki and Kuroki 2003; Gallione et al. 2010). The mechanism of protein dysfunction of the Myhre syndrome variants was believed to be distinct from the LOF variants due to the lack of cancer development in Myhre syndrome and non-overlapping phenotypes from JPS/HHT. Recently, the same germline variants that cause Myhre syndrome were identified in somatic tissue of individuals with gastrointestinal cancers, including most recently the observation of neoplasia or endometrial cancer in individuals with Myhre syndrome (Ashktorab et al. 2017; Lin et al. 2020). These novel observations are consistent with our disease model and the dominant-negative action of the SMAD4-I500V variant.
SMAD proteins acting dominant-negatively have been previously observed, including functional characterization of SMAD2 variants identified in somatic cancer (Mucsi and Goldberg 1997; Goto et al. 1998; Hoodless et al. 1999; Ho et al. 2007). Dominant-negative function of SMAD4 has been previously studied using deletion constructs that showed reduced expression of COL1A1 (Tsuchida et al. 2003). However, to our knowledge, this is the first report of a dominant-negative SMAD4 variant linked to human disease. Furthermore, we expect that as more patients are reported, damaging variants in the MH2 domain of R-SMADs in residues that functionally correspond to SMAD4-I500 which affect transcriptional activity but not heterodimerization, will yield unique phenotypes that are distinct from those caused by LOF variants or missense variants in the other domains of these related genes.
Our results confirm that addition of ligands is unable to overcome the dominant-negative activity of SMAD4-I500V. In addition, our transcriptional findings in ligand treated, WT/I500V transfected cells are similar to transcriptional data from Myhre syndrome fibroblasts, which suggest that TGFβ and BMP ligands are highly active in the affected individuals. This is likely to be an attempt at rectifying loss of SMAD4-I500V-mediated transcription but may instead lead to pathological induction of SMAD4-independent TGF/BMP processes that may partially underlie Myhre syndrome phenotypes (Imamichi et al. 2005; Aashaq et al. 2021). Promisingly, a recent trial of losartan, a TGFβ inhibitor, showed signs of improving the fibrosis-related phenotypes associated with Myhre syndrome, thereby establishing activated TGF signaling as a disease mechanism in Myhre syndrome (Cappuccio et al. 2021). Their findings suggest that attenuation of TGF may be a viable approach for managing at least the fibrotic phenotypes observed in Myhre syndrome.
We conclude that the congenital and progressive fibrotic defects observed in Myhre syndrome patients may be explained by the culmination of dysregulated signaling, wherein active TGF/BMP signaling is unable to compensate for loss of canonical SMAD4 function, thereby over-activating SMAD4-independent cellular processes. We propose that similar variants identified in R-SMADs in patients with phenotypes discordant from those expected be given sufficient consideration with respect to their contribution towards disease causality. Finally, we expect that our findings may facilitate consideration of novel modes of therapeutic interventions for individuals with Myhre syndrome.
Supplementary Material
Acknowledgments:
We thank the patients and families for participation in this study. The authors also thank Terry Foss and Marshfield Clinic research foundation. The authors thank Dr. Justin Szot for manuscript editing. We gratefully acknowledge the support of the Malika Ray, Asok K. Ray, FRCS/(Edin) Initiative for Child Health.
Funding:
This work was supported by the National Health and Medical Research Council (NHMRC) (Project Grant ID1044543 to S.L.D, D.B.S. and E.L.D and Fellowship ID1135886 and ID1042002 to S.L.D.), the Office of Health and Medical Research New South Wales (NSW) Government to S.L.D., Research reported in this publication was also supported in part by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number R03HD099516 to P.F.G.
Footnotes
Ethics approval and consent: This study was conducted in accordance with the Declaration of Helsinki, and ethical approval for this study was obtained from the Sydney Children’s Hospital Network Human Research Ethics Committee (approval number HREC/18/SCHN/222). Informed, written consent was obtained from all participants.
Conflicts of interest: None.
Supplementary data: The following are available online. Supplementary methods, case reports for patients 1 and 2, and supplementary figures. Fig S1: Pedigree (a), Sanger sequencing (b), X-ray of the cervical vertebra showing C2-C3 segmentations defects (c) of patient 1, and pedigree (d) and photograph indicating the facial dysmorphism (e) of patient 2, Fig S2: Expression of SMAD4-WT and SMAD4-I500V in nuclear and cytoplasmic cell compartments, Fig S3: Transfection controls for figures 2 and 4, Fig S4: Gene expression analysis of SMAD4, ID3, COL1A1, CTGF, SMAD6 and SMAD7 grouped by individual genes in all assessed conditions.
References
- Aashaq S, Batool A, Mir SA, Beigh MA, Andrabi KI, Shah ZA (2021) TGF-β signaling: A recap of SMAD-independent and SMAD-dependent pathways. J Cell Physiol [DOI] [PubMed] [Google Scholar]
- Alankarage D, Szot JO, Pachter N, Slavotinek A, Selleri L, Shieh JT, Winlaw D, Giannoulatou E, Chapman G, Dunwoodie SL (2019) Functional characterisation of a novel PBX1 de novo missense variant identified in a patient with syndromic congenital heart disease. Hum Mol Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberici P, Jagmohan-Changur S, De Pater E, Van Der Valk M, Smits R, Hohenstein P, Fodde R (2006) Smad4 haploinsufficiency in mouse models for intestinal cancer. Oncogene 25:1841–1851 [DOI] [PubMed] [Google Scholar]
- Ashktorab H, Mokarram P, Azimi H, Olumi H, Varma S, Nickerson ML, Brim H (2017) Targeted exome sequencing reveals distinct pathogenic variants in Iranians with colorectal cancer. Oncotarget 8:7852–7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin J, Davy A, Soriano P (2004) In vivo convergence of BMP and MAPK signaling pathways: impact of differential Smad1 phosphorylation on development and homeostasis. Genes Dev 18:1482–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, DePinho RA (2006) Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 20:3130–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD (1999) Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest 104:1567–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouveret R, Waardenberg AJ, Schonrock N, Ramialison M, Doan T, de Jong D, Bondue A, Kaur G, Mohamed S, Fonoudi H, Chen CM, Wouters MA, Bhattacharya S, Plachta N, Dunwoodie SL, Chapman G, Blanpain C, Harvey RP (2015) NKX2–5 mutations causative for congenital heart disease retain functionality and are directed to hundreds of targets. Elife 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CO 3rd, Chi X, Garcia-Gras E, Shirai M, Feng XH, Schwartz RJ (2004) The cardiac determination factor, Nkx2–5, is activated by mutual cofactors GATA-4 and Smad1/4 via a novel upstream enhancer. J Biol Chem 279:10659–10669 [DOI] [PubMed] [Google Scholar]
- Cappuccio G, Caiazza M, Roca A, Melis D, Iuliano A, Matyas G, Rubino M, Limongelli G, Brunetti-Pierri N (2021) A pilot clinical trial with losartan in Myhre syndrome. Am J Med Genet A 185:702–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo V, Cianetti L, Niceta M, Carta C, Ciolfi A, Bocchinfuso G, Carrani E, Dentici ML, Biamino E, Belligni E, Garavelli L, Boccone L, Melis D, Andria G, Gelb BD, Stella L, Silengo M, Dallapiccola B, Tartaglia M (2012) A restricted spectrum of mutations in the SMAD4 tumor-suppressor gene underlies Myhre syndrome. Am J Hum Genet 90:161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko BM, Qin B, Correia JJ, Lam SS, de Caestecker MP, Lin K (2001) The L3 loop and C-terminal phosphorylation jointly define Smad protein trimerization. Nat Struct Biol 8:248–253 [DOI] [PubMed] [Google Scholar]
- Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A (1999) Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 126:1631–1642 [DOI] [PubMed] [Google Scholar]
- Chu GC, Dunn NR, Anderson DC, Oxburgh L, Robertson EJ (2004) Differential requirements for Smad4 in TGFbeta-dependent patterning of the early mouse embryo. Development 131:3501–3512 [DOI] [PubMed] [Google Scholar]
- Coulter ME, Musaev D, DeGennaro EM, Zhang X, Henke K, James KN, Smith RS, Hill RS, Partlow JN, Muna A-S, Kamumbu AS, Hatem N, Barkovich AJ, Aziza J, Chassaing N, Zaki MS, Sultan T, Burglen L, Rajab A, Al-Gazali L, Mochida GH, Harris MP, Gleeson JG, Walsh CA (2020) Regulation of human cerebral cortical development by EXOC7 and EXOC8, components of the exocyst complex, and roles in neural progenitor cell proliferation and survival. Genet Med 22:1040–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick A, Meier A, Hammerschmidt M (1999) Smad1 and Smad5 have distinct roles during dorsoventral patterning of the zebrafish embryo. Dev Dyn 216:285–298 [DOI] [PubMed] [Google Scholar]
- Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L, Inui M, Moro S, Modena N, Argenton F, Newfeld SJ, Piccolo S (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 136:123–135 [DOI] [PubMed] [Google Scholar]
- Gallione C, Aylsworth AS, Beis J, Berk T, Bernhardt B, Clark RD, Clericuzio C, Danesino C, Drautz J, Fahl J, Fan Z, Faughnan ME, Ganguly A, Garvie J, Henderson K, Kini U, Leedom T, Ludman M, Lux A, Maisenbacher M, Mazzucco S, Olivieri C, Ploos van Amstel JK, Prigoda-Lee N, Pyeritz RE, Reardon W, Vandezande K, Waldman JD, White RI Jr., Williams CA, Marchuk DA (2010) Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am J Med Genet A 152a:333–339 [DOI] [PubMed] [Google Scholar]
- Goto D, Yagi K, Inoue H, Iwamoto I, Kawabata M, Miyazono K, Kato M (1998) A single missense mutant of Smad3 inhibits activation of both Smad2 and Smad3, and has a dominant negative effect on TGF-beta signals. FEBS Lett 430:201–204 [DOI] [PubMed] [Google Scholar]
- Granadillo JL, Chung WK, Hecht L, Corsten-Janssen N, Wegner D, Nij Bijvank SWA, Toler TL, Pineda-Alvarez DE, Douglas G, Murphy JJ, Shimony J, Shinawi M (2018) Variable cardiovascular phenotypes associated with SMAD2 pathogenic variants. Hum Mutat 39:1875–1884 [DOI] [PubMed] [Google Scholar]
- Hata A, Lagna G, Massagué J, Hemmati-Brivanlou A (1998) Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev 12:186–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Lo RS, Wotton D, Lagna G, Massagué J (1997) Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature 388:82–87 [DOI] [PubMed] [Google Scholar]
- Henningfeld KA, Rastegar S, Adler G, Knöchel W (2000) Smad1 and Smad4 are components of the bone morphogenetic protein-4 (BMP-4)-induced transcription complex of the Xvent-2B promoter. J Biol Chem 275:21827–21835 [DOI] [PubMed] [Google Scholar]
- Ho J, Chen H, Lebrun JJ (2007) Novel dominant negative Smad antagonists to TGFbeta signaling. Cell Signal 19:1565–1574 [DOI] [PubMed] [Google Scholar]
- Hoodless PA, Tsukazaki T, Nishimatsu S, Attisano L, Wrana JL, Thomsen GH (1999) Dominant-negative Smad2 mutants inhibit activin/Vg1 signaling and disrupt axis formation in Xenopus. Dev Biol 207:364–379 [DOI] [PubMed] [Google Scholar]
- Hu W, Dong A, Karasaki K, Sogabe S, Okamoto D, Saigo M, Ishida M, Yoshizumi M, Kokubo H (2021) Smad4 regulates the nuclear translocation of Nkx2–5 in cardiac differentiation. Sci Rep 11:3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamichi Y, Waidmann O, Hein R, Eleftheriou P, Giehl K, Menke A (2005) TGF beta-induced focal complex formation in epithelial cells is mediated by activated ERK and JNK MAP kinases and is independent of Smad4. Biol Chem 386:225–236 [DOI] [PubMed] [Google Scholar]
- Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K (1997) Smad6 inhibits signalling by the TGF-beta superfamily. Nature 389:622–626 [DOI] [PubMed] [Google Scholar]
- Kang YJ, Shin JW, Yoon JH, Oh IH, Lee SP, Kim SY, Park SH, Mamura M (2012) Inhibition of erythropoiesis by Smad6 in human cord blood hematopoietic stem cells. Biochem Biophys Res Commun 423:750–756 [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ, MacArthur DG (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44:D862–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, Afenjar A, Destrée A, di Rocco M, Héron D, Jacquemont S, Marlin S, Simon M, Tolmie J, Verloes A, Casanova JL, Munnich A, Cormier-Daire V (2011) Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet 44:85–88 [DOI] [PubMed] [Google Scholar]
- Lien CL, McAnally J, Richardson JA, Olson EN (2002) Cardiac-specific activity of an Nkx2–5 enhancer requires an evolutionarily conserved Smad binding site. Dev Biol 244:257–266 [DOI] [PubMed] [Google Scholar]
- Lin AE, Alali A, Starr LJ, Shah N, Beavis A, Pereira EM, Lindsay ME, Klugman S (2020) Gain-of-function pathogenic variants in SMAD4 are associated with neoplasia in Myhre syndrome. Am J Med Genet A 182:328–337 [DOI] [PubMed] [Google Scholar]
- Lin AE, Michot C, Cormier-Daire V, L’Ecuyer TJ, Matherne GP, Barnes BH, Humberson JB, Edmondson AC, Zackai E, O’Connor MJ, Kaplan JD, Ebeid MR, Krier J, Krieg E, Ghoshhajra B, Lindsay ME (2016) Gain-of-function mutations in SMAD4 cause a distinctive repertoire of cardiovascular phenotypes in patients with Myhre syndrome. Am J Med Genet A 170:2617–2631 [DOI] [PubMed] [Google Scholar]
- Martin E, Enriquez A, Sparrow DB, Humphreys DT, McInerney-Leo AM, Leo PJ, Duncan EL, Iyer KR, Greasby JA, Ip E, Giannoulatou E, Sheng D, Wohler E, Dimartino C, Amiel J, Capri Y, Lehalle D, Mory A, Wilnai Y, Lebenthal Y, Gharavi AG, Krzemień GG, Miklaszewska M, Steiner RD, Raggio C, Blank R, Baris Feldman H, Milo Rasouly H, Sobreira NLM, Jobling R, Gordon CT, Giampietro PF, Dunwoodie SL, Chapman G (2020) Heterozygous loss of WBP11 function causes multiple congenital defects in humans and mice. Hum Mol Genet 29:3662–3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerschaut I, Beyens A, Steyaert W, De Rycke R, Bonte K, De Backer T, Janssens S, Panzer J, Plasschaert F, De Wolf D, Callewaert B (2019) Myhre syndrome: A first familial recurrence and broadening of the phenotypic spectrum. Am J Med Genet A 179:2494–2499 [DOI] [PubMed] [Google Scholar]
- Miyaki M, Kuroki T (2003) Role of Smad4 (DPC4) inactivation in human cancer. Biochem Biophys Res Commun 306:799–804 [DOI] [PubMed] [Google Scholar]
- Monteiro RM, de Sousa Lopes SM, Bialecka M, de Boer S, Zwijsen A, Mummery CL (2008) Real time monitoring of BMP Smads transcriptional activity during mouse development. Genesis 46:335–346 [DOI] [PubMed] [Google Scholar]
- Moreau JLM, Kesteven S, Martin E, Lau KS, Yam MX, O’Reilly VC, Del Monte-Nieto G, Baldini A, Feneley MP, Moon AM, Harvey RP, Sparrow DB, Chapman G, Dunwoodie SL (2019) Gene-environment interaction impacts on heart development and embryo survival. Development 146 [DOI] [PubMed] [Google Scholar]
- Mucsi I, Goldberg HJ (1997) Dominant-negative SMAD-3 interferes with transcriptional activation by multiple agonists. Biochem Biophys Res Commun 232:517–521 [DOI] [PubMed] [Google Scholar]
- Myhre SA, Ruvalcaba RH, Graham CB (1981) A new growth deficiency syndrome. Clin Genet 20:1–5 [DOI] [PubMed] [Google Scholar]
- Piccolo P, Mithbaokar P, Sabatino V, Tolmie J, Melis D, Schiaffino MC, Filocamo M, Andria G, Brunetti-Pierri N (2014) SMAD4 mutations causing Myhre syndrome result in disorganization of extracellular matrix improved by losartan. Eur J Hum Genet 22:988–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B, Lam SS, Lin K (1999) Crystal structure of a transcriptionally active Smad4 fragment. Structure 7:1493–1503 [DOI] [PubMed] [Google Scholar]
- Rastegar S, Friedle H, Frommer G, Knöchel W (1999) Transcriptional regulation of Xvent homeobox genes. Mech Dev 81:139–149 [DOI] [PubMed] [Google Scholar]
- Ross S, Hill CS (2008) How the Smads regulate transcription. Int J Biochem Cell Biol 40:383–408 [DOI] [PubMed] [Google Scholar]
- Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG (1998) Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science 281:108–111 [DOI] [PubMed] [Google Scholar]
- Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP (1997) A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 388:87–93 [DOI] [PubMed] [Google Scholar]
- Sirard C, de la Pompa JL, Elia A, Itie A, Mirtsos C, Cheung A, Hahn S, Wakeham A, Schwartz L, Kern SE, Rossant J, Mak TW (1998) The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev 12:107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay KD, Dunn NR, Robertson EJ (2001) Mouse embryos lacking Smad1 signals display defects in extraembryonic tissues and germ cell formation. Development 128:3609–3621 [DOI] [PubMed] [Google Scholar]
- Tsuchida K, Zhu Y, Siva S, Dunn SR, Sharma K (2003) Role of Smad4 on TGF-beta-induced extracellular matrix stimulation in mesangial cells. Kidney Int 63:2000–2009 [DOI] [PubMed] [Google Scholar]
- Wang M, Jin H, Tang D, Huang S, Zuscik MJ, Chen D (2011) Smad1 plays an essential role in bone development and postnatal bone formation. Osteoarthritis Cartilage 19:751–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li C, Xu X, Deng C (1998) The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A 95:3667–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE (1998) Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell 1:611–617 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.