

Abstract
In 1979, an H1N1 avian influenza virus crossed the species barrier, establishing a new lineage in European swine. Because there is no direct or serologic evidence of previous H1N1 strains in these pigs, these isolates provide a model for studying early evolution of influenza viruses. The evolutionary rates of both the coding and noncoding changes of the H1N1 swine strains are higher than those of human and classic swine influenza A viruses. In addition, early H1N1 swine isolates show a marked plaque heterogeneity that consistently appears after a few passages. The presence of a mutator mutation was postulated (C. Scholtissek, S. Ludwig, and W. M. Fitch, Arch. Virol. 131:237–250, 1993) to account for these observations and the successful establishment of an avian H1N1 strain in swine. To address this question, we calculated the mutation rates of A/Mallard/New York/6750/78 (H2N2) and A/Swine/Germany/2/81 (H1N1) by using the frequency of amantadine-resistant mutants. To account for the inherent variability of estimated mutation rates, we used a probabilistic model for the statistical analysis. The resulting estimated mutation rates of the two strains were not significantly different. Therefore, an increased mutation rate due to the presence of a mutator mutation is unlikely to have led to the successful introduction of avian H1N1 viruses in European swine.
Influenza viruses undergo rapid variation in nature, thereby limiting prevention of epidemics and pandemics (20). Therefore, questions regarding their evolution remain important, and the answers may yield information useful for predicting further antigenic changes and for explaining the occurrence of new pandemic strains. In 1979, an H1N1 influenza virus of avian origin was transmitted to pigs in northern Europe, thereby introducing a new stable lineage (22, 26). Because there is no direct or serologic evidence of previous H1N1 strains in these pigs (35), we do not need to extrapolate the point of introduction from evolutionary changes over time. Therefore, the European swine influenza H1N1 viruses are a model for studying the early evolution of influenza virus strains in a new host after their introduction in the absence of reassortment.
Several characteristics of these H1N1 swine viruses make them particularly interesting for studying the evolution of influenza viruses. Analysis of phylogenic data on their HA, NP, M, and NS genes revealed that the evolutionary rates of both the coding and noncoding changes of these strains are up to 54% higher than those of human and classic swine viruses (18, 33). Such higher evolutionary rates might be due to positive Darwinian selection in avian-like swine influenza viruses, reflecting adaptation to a new host; to differences in sampling frequencies (10) of the viral lineages compared; or to a higher mutation rate. The elevated rate of noncoding changes might be due to fixation of linked coding changes. In addition, early H1N1 swine viruses show a marked plaque heterogeneity and an unusually high escape rate in the presence of various monoclonal antibodies (18). These observations suggest that at least one other factor, independent from positive selection of advantageous variants, must have influenced the evolution of the swine H1N1 isolates since 1979. One candidate is the presence of a mutator mutation (24). An initial variant could have a mutation in its polymerase complex that caused it to be more error prone and to generate a broader spectrum of variants. Such a polymerase might be detrimental for an established strain but advantageous under stress conditions such as adaptation to a new environment. After that, variants with a less error-prone polymerase might again become predominant in the population (18).
We wanted to determine whether increased mutation rates due to the presence of a mutator mutation might have contributed to the establishment of the European swine H1N1 viruses. To this end, we calculated the mutation rates of A/Swine/Germany/2/81, a well-characterized early H1N1 swine isolate, and A/Mallard/New York/6750/78 (H2N2), a well-established avian isolate; the evolution of avian influenza viruses is considerably slower than that of classic swine and human isolates (16). An avian H1N1 precursor virus was not available for study. To accommodate the inherent high variability of estimated mutation rates, we developed a new probabilistic model (see Appendix) to calculate the mutation rate and its standard deviation (SD) and screened several parallel clones. To facilitate rapid screening of parallel cultures, we evaluated the frequency with which amantadine-resistant mutants developed. Our results suggest that the mutation rates of A/Swine/Germany/2/81 and A/Mallard/New York/6750/78 are not significantly different and fail to reflect their very different evolutionary backgrounds.
MATERIALS AND METHODS
Viruses.
The influenza viruses A/Mallard/New York/6750/78 (H2N2) and A/Swine/Germany/2/81 (H1N1) were plaque purified twice by using monolayers of Madin-Darby canine kidney (MDCK) cells. Culture was with minimal essential medium (MEM) (Sigma, St. Louis, Mo.), 0.9% agar (Difco Laboratories, Detroit, Mich.) overlays containing 0.5 g of TPCK (tolylsulfonyl phenylalanyl chloromethyl ketone)-trypsin (Worthington Biochemical Corporation, Freehold, N.J.) per ml, and a 3-day incubation at 37°C. Virus populations were then propagated individually from single well-isolated plaques in the allantoic cavities of 11-day embryonated chicken eggs by incubating for 2 days at 37°C; the eggs were stored overnight at 4°C before the allantoic fluid of each egg was harvested individually and used in the plaque inhibition assay.
Plaque inhibition assay with amantadine.
The plaque assay was performed on MDCK cell monolayers, which were grown in MEM and stained with crystal violet after 3 days as described by Ho et al. (14). Briefly, cells were inoculated with phosphate-buffered saline-diluted virus, left for 30 min at 37°C to allow adsorption, and then washed twice with phosphate-buffered saline, overlaid with MEM (Sigma) containing 0.9% agar (Difco Laboratories), and incubated at 37°C. After 3 days, the plaques were visualized by crystal violet (0.1% in 10% formaldehyde) staining.
For calculation of the frequency of amantadine-resistant mutants, the numbers of PFU per milliliter were determined for each clone in the absence and presence of 1 μg of amantadine hydrochloride (DuPont, Wilmington, Del.) per ml in the agar overlay.
Calculation of mutation rate and SD and statistical analysis.
For calculating the estimated rate of mutation, we have developed a probabilistic model (see Appendix). The estimate of the mutation rate m is m̂ = 2M/(Nlog2 N), where M is the number of mutants in the population (i.e., the number of PFU of amantadine-resistant mutants) and N is the population size from one egg-grown clone. Both numbers are the products of the respective PFU per milliliter from the plaque inhibition assay and the total amount of allantoic fluid (milliliters). The SD of that estimate is 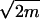 /log2 N).
/log2 N).
For amantadine at a concentration of 1 μg/ml, substitution at one of four amino acids (positions 27, 30, 31, and 34 in the M2 ion channel) leads to resistance (13); the sequences of A/Mallard/New York/6750/78 and A/Swine/Germany/2/81 are identical at these positions. From the comparison with the codons of the amino acid substitutions that lead to resistance (13), it can be concluded that one nucleotide substitution in each triplet, i.e., one of four possible nucleotides, causes the resistant phenotype in both strains. Therefore, m has to be divided by 4, and the formula for the estimated mutation rate (the number of nucleotide substitutions per site per replication) for the experimental approach described here is m̂ = 0.5 M/(Nlog2 N). We made no attempt to calculate a maximum mutation rate by considering more than one nucleotide substitution at each site, because transitions are more frequent than transversions (32). Therefore, the assumption of an equal probability of all changes is unlikely.
Because the generation at which a mutation occurs markedly influences the frequency of mutants (6), we grew multiple parallel cultures (C). Therefore, the estimated mutation rate for each viral strain is the arithmetic mean of the mutation rate from every clone,
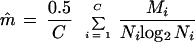 |
and the SD is
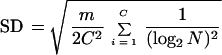 |
For comparison, the ad hoc SD was calculated as
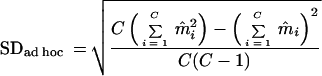 |
where m̂i is the determined mutation rate of the ith clone and C is the whole number of observed clones from one isolate.
For the statistical analysis, the null hypothesis (H0), mmall = msw, versus the alternative hypothesis (Ha), mmall ≠ msw, was tested by using the normal approximation Z = (m̂mall − m̂sw)/√SDmall2 + SDsw2, and the test claims that Ha is true if |Z| > 1.96 (97.5% quantile of the normal distribution).(mmall and msw are the mutation rates for strains A/Mallard/New York/6750/78 and A/Swine/Germany/2/81, respectively.)
The basic assumptions of our mathematical model are as follows.
(i) The mutation rate is the same for all observed sites and at all replications. For the present study, this means that loci related to amantadine resistance mutate at a rate which is representative of those for all other genome loci, including those involved in species transmission. Positions other than 27, 30, 31, and 34 in M2 (e.g., flanking sequences) could affect the generation of amantadine resistance, thus obscuring intrinsic differences in mutation rate. That is assumed to be negligible, because no such data supporting this view are available for influenza virus.
(ii) The mutation rate m is very small compared to 1 (10−4 to 10−6); therefore, higher orders of m such as m2, m3, and m4 (as would apply to two-, three-, or fourfold substitutions) can be ignored.
(iii) From the second assumption, it follows that reverse mutations are also negligible.
(iv) The model applies only to neutral mutants in an ideal way. Bean et al. showed that amantadine-resistant influenza virus in chickens is capable of competing with wild-type virus and does not revert back in the absence of amantadine over a period of >20 days, thus having no detectable biologic impairment compared to wild-type virus (1). Moreover, negative selection (elimination of deleterious mutation as soon as they occur) is assumed to be identical for the strains compared.
(v) Possible variations in polymerase (or other fidelity-determining proteins) upon virus growth in cell culture need not be considered, because the estimated mutation rate m̂ is defined as base substitutions per site per replication.
RESULTS
Evaluation of several independent clones.
We measured the frequencies of amantadine-resistant mutants of A/Mallard/New York/6750/78 (H2N2) (Table 1) and A/Swine/Germany/2/81 (H1N1) (Table 2) to indirectly assess the mutation rates of the two parent strains. Because the variability of mutant frequency is inherently high, we evaluated multiple parallel, independently grown viral stocks. Extensive plaque purification ensured that all amantadine-resistant variants occurred during growth in the plaque or allantoic fluid and did not exist prior to culture. A primary resistant plaque would grow to become a viral stock that was comprised almost entirely of resistant viruses and therefore would be recognized immediately.
TABLE 1.
Mutation rate ofA/Mallard/New York/6750/78
| Clone no. | Plaque inhibition assay result (PFU/ml)
|
Allantoic fluid (ml) | Population size (PFU)a | Mutant frequencyb | Mutation rate (bp/site/replication)c | Quotient (control/ amantadine) | Quotient × 4 | Quotient × 4/ genome length | |
|---|---|---|---|---|---|---|---|---|---|
| Control | Amantadine | ||||||||
| 1 | 1.6 × 107 | 1.2 × 104 | 7.0 | 1.1 × 108 | 7.5 × 10−4 | 1.4 × 10−5 | 1.3 × 103 | 5.3 × 103 | 0.4 |
| 2 | 4.0 × 107 | 3.2 × 104 | 9.1 | 3.6 × 108 | 8.0 × 10−4 | 1.4 × 10−5 | 1.3 × 103 | 5.0 × 103 | 0.4 |
| 3 | 1.0 × 107 | 1.2 × 104 | 7.1 | 7.1 × 107 | 1.2 × 10−3 | 2.3 × 10−5 | 8.3 × 102 | 3.3 × 103 | 0.2 |
| 4 | 2.9 × 107 | 1.2 × 104 | 7.8 | 2.3 × 108 | 4.1 × 10−4 | 7.5 × 10−6 | 2.4 × 103 | 9.7 × 103 | 0.7 |
| 5 | 2.0 × 107 | 4.4 × 104 | 8.5 | 1.7 × 108 | 2.2 × 10−3 | 4.0 × 10−5 | 4.5 × 102 | 1.8 × 103 | 0.1 |
| 6 | 9.6 × 106 | 1.6 × 104 | 13.3 | 1.3 × 108 | 1.7 × 10−3 | 3.1 × 10−5 | 6.0 × 102 | 2.4 × 103 | 0.2 |
| 7 | 6.8 × 107 | 8.0 × 103 | 9.3 | 6.3 × 108 | 1.2 × 10−4 | 2.0 × 10−6 | 8.5 × 103 | 3.4 × 104 | 2.5 |
| 8 | 8.4 × 107 | 4.0 × 104 | 10.4 | 8.7 × 108 | 4.8 × 10−4 | 8.0 × 10−6 | 2.1 × 103 | 8.4 × 103 | 0.6 |
| 9 | 3.2 × 107 | 3.2 × 105 | 7.9 | 2.5 × 108 | 1.0 × 10−2 | 1.8 × 10−4 | 1.0 × 102 | 4.0 × 102 | 0.0 |
| 10 | 1.1 × 108 | 4.0 × 103 | 13.6 | 1.5 × 109 | 3.6 × 10−5 | 6.0 × 10−7 | 2.8 × 104 | 1.1 × 105 | 8.1 |
| Average | 4.2 × 107 | 5.0 × 104 | 4.3 × 108 | 1.8 × 10−3 | 3.2 × 10−5 | 4.5 × 103 | 1.8 × 104 | 1.3 | |
Product of titer from control and amount of allantoic fluid.
Quotient of titers from plaque inhibition assays with amantadine and control.
Rate for each clone calculated from mutant frequency and population size as described in Materials and Methods.
TABLE 2.
Mutation rate of A/Swine/Germany/2/81
| Clone no. | Plaque inhibition assay result (PFU/ml)
|
Allantoic fluid (ml) | Population size (PFU)a | Mutant frequencyb | Mutation rate (bp/site/replication)c | Quotient (control/ amantadine) | Quotient × 4 | Quotient × 4/ genome length | |
|---|---|---|---|---|---|---|---|---|---|
| Control | Amantadine | ||||||||
| 1 | 9.2 × 108 | 1.2 × 105 | 13.0 | 1.2 × 1010 | 1.3 × 10−4 | 1.9 × 10−6 | 7.7 × 103 | 3.1 × 104 | 2.3 |
| 2 | 6.0 × 108 | 2.6 × 105 | 9.0 | 5.4 × 109 | 4.3 × 10−4 | 6.7 × 10−6 | 2.3 × 103 | 9.2 × 103 | 0.7 |
| 3 | 2.3 × 108 | 1.8 × 105 | 13.0 | 3.0 × 109 | 7.8 × 10−4 | 1.2 × 10−5 | 1.3 × 103 | 5.1 × 103 | 0.4 |
| 4 | 6.0 × 108 | 2.6 × 105 | 11.0 | 6.6 × 109 | 4.3 × 10−4 | 6.6 × 10−6 | 2.3 × 103 | 9.2 × 103 | 0.7 |
| 5 | 1.0 × 109 | 2.0 × 105 | 12.0 | 1.2 × 1010 | 2.0 × 10−4 | 3.0 × 10−6 | 5.0 × 103 | 2.0 × 104 | 1.5 |
| 6 | 2.2 × 108 | 8.0 × 104 | 10.7 | 2.4 × 109 | 3.6 × 10−4 | 5.8 × 10−6 | 2.8 × 103 | 1.1 × 104 | 0.8 |
| 7 | 4.2 × 108 | 1.4 × 105 | 9.3 | 3.9 × 109 | 3.3 × 10−4 | 5.2 × 10−6 | 3.0 × 103 | 1.2 × 104 | 0.9 |
| 8 | 4.0 × 107 | 4.0 × 104 | 8.6 | 3.4 × 108 | 1.0 × 10−3 | 1.8 × 10−5 | 1.0 × 103 | 4.0 × 103 | 0.3 |
| 9 | 6.0 × 107 | 1.8 × 104 | 9.6 | 5.8 × 108 | 3.0 × 10−4 | 5.2 × 10−6 | 3.3 × 103 | 1.3 × 104 | 1.0 |
| 10 | 3.2 × 107 | 2.0 × 104 | 10.5 | 3.4 × 108 | 6.3 × 10−4 | 1.1 × 10−5 | 1.6 × 103 | 6.4 × 103 | 0.5 |
| 11 | 1.0 × 108 | 1.2 × 104 | 6.0 | 6.0 × 108 | 1.2 × 104 | 2.1 × 10−6 | 8.3 × 103 | 3.3 × 104 | 2.5 |
| 12 | 4.0 × 108 | 4.0 × 104 | 13.5 | 5.4 × 109 | 1.0 × 10−4 | 1.5 × 10−6 | 1.0 × 104 | 4.0 × 104 | 2.9 |
| 13 | 2.8 × 108 | 2.4 × 105 | 10.8 | 3.0 × 109 | 8.6 × 10−4 | 1.4 × 10−5 | 1.2 × 103 | 4.7 × 103 | 0.3 |
| 14 | 4.0 × 107 | 8.0 × 103 | 8.8 | 3.5 × 108 | 2.0 × 10−4 | 3.5 × 10−6 | 5.0 × 103 | 2.0 × 104 | 1.5 |
| 15 | 1.2 × 108 | 4.8 × 104 | 9.0 | 1.1 × 109 | 4.0 × 10−4 | 6.7 × 10−6 | 2.5 × 103 | 1.0 × 104 | 0.7 |
| 16 | 1.3 × 108 | 1.6 × 104 | 11.9 | 1.5 × 109 | 1.2 × 10−4 | 2.0 × 10−6 | 8.1 × 103 | 3.3 × 104 | 2.4 |
| 17 | 1.4 × 108 | 9.6 × 104 | 9.8 | 1.4 × 109 | 6.9 × 10−4 | 1.1 × 10−5 | 1.5 × 103 | 5.8 × 103 | 0.4 |
| Average | 3.1 × 108 | 1.0 × 105 | 3.5 × 109 | 4.2 × 10−4 | 6.8 × 10−6 | 3.9 × 103 | 1.6 × 104 | 1.2 | |
Product of titer from control and amount of allantoic fluid.
Quotient of titers from plaque inhibition assays with amantadine and control.
Rate for each clone calculated from mutant frequency and population size as described in Materials and Methods.
Mutant frequencies and mutation rates.
As described in Materials and Methods, we performed plaque inhibition assays to determine the population size and the frequency of amantadine-resistant mutants, and then we calculated the average mutation rate for each strain and its SD (Tables 1 and 2). For A/Mallard/New York/6750/78, the mutation rate (m̂mall) is 3.2 × 10−5 base substitutions per site per replication and its SD (SDmall) is 4.5 × 10−5; for A/Swine/Germany/2/81, m̂sw = 6.8 × 10−6 base substitutions per site per replication and SDsw = 1.5 × 10−5. Table 3 shows that our data correspond well with two values previously published for the mutation rate of influenza virus.
TABLE 3.
Comparison of estimated mutation rates for various influenza virus strains
| Virus | Reference | Assay | Gene | Mutation rate (bp/site/replication) |
|---|---|---|---|---|
| A/Victoria/3/75 (H3N2) | 28 | Monoclonal antibody escape | HA | 1.0 × 10−5 |
| A/WSN/33 (H1N1) | 21;2 | Direct sequencing of RNA | NS | 0.84 × 10−5 |
| A/Mallard/New York/6750/78 (H2N2) | This study | Amantadine escape | M | 3.2 × 10−5 |
| A/Swine/Germany/2/81 (H1N1) | This study | Amantadine escape | M | 0.68 × 10−5 |
Dispersion of data.
The mutation rates of clones 9 and 10 of A/Mallard/New York/6750/78 (Table 1) demonstrate the problem of variability in estimates of mutation rate. The estimate for clone 9 was almost 6 times higher than the average mutation rate, whereas that for clone 10 was 50 times lower. Both values were included in our calculations, although one may consider them outliers. Interestingly, the calculation of the ad hoc SD showed a result for A/Mallard/New York/6750/78 similar to that obtained with our formula for SD when the mutation rates from clones 9 and 10 were included (SDad hoc = 5.3 × 10−5 and SD = 4.5 × 10−5), whereas for A/Swine/Germany/2/81, the values are SDad hoc = 5.2 × 10−6 and SD = 1.5 × 10−5. This finding indicates that our theoretical formula for SD is applicable to our data and describes the dispersion well. The mutation rates from clones 9 and 10 are very divergent from the mean, indicating the influence of the time point during growth of the viral stock at which mutational events leading to amantadine resistance occurred on the estimated mutation rate. These observations emphasize the randomness of mutational events in nature and demonstrate that measuring the mutation rate of a single clone can be misleading.
Minimum number of replication cycles.
In light of the small ratio of the number of sites at which changes lead to amantadine resistance (4 positions) to the genome length (almost 14,000 positions), mutation to resistance is highly improbable compared with a mutation at any other site in the entire viral genome. Therefore, such a mutational event within the viral population is improbable when the population size is considerably smaller than one-fourth of the genome length (for only one position, it would be improbable when the population size is considerably smaller than the entire genome). If all mutants are assumed to have developed from a single mutational event, then the ratio of the PFU per milliliter from plaque inhibition assays without amantadine (control) to that from assays with amantadine in the plaque overlay reveals a theoretical minimum population size in which the first mutant would have arisen. Multiplying the calculated minimum population size of A/Mallard/New York/6750/78 or A/Swine/Germany/2/81 by 4 reveals a value close to the genome length (Tables 1 and 2). The resulting value can be considered to be the minimum number of replication cycles necessary for each additional mutational step (requiring only a single-base substitution) during further growth. Therefore, in light of our results regarding amantadine, a population of approximately 5.0 × 103 PFU probably carries at least one mutated virion; for a mutated phenotype caused by a base substitution at only one possible position, this value would be approximately 2.0 × 104 PFU, and for two mutational steps in one viral genome, it would be approximately 4.0 × 108 PFU. These numbers indicate the importance of the size of a finite viral population in a host and the turnover of virions during infection for the probability of occurrence a particular virus mutant requiring several point mutations. That relationship has been discussed by Coffin (2) and has been described in a mathematical model, derived from a quasispecies equation, by Ribeiro et al. (23) in regard to human immunodeficiency virus population dynamics in view of the frequency of resistant mutant virus before antiviral therapy.
Statistical analysis.
The statistical analysis was performed as described in Materials and Methods. For the two-sided test with a significance level of 0.05, because (m̂mall − m̂sw)/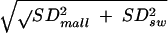 = 0.5 (a value smaller than 1.96, the 97.5% quantile of the normal distribution), the null hypothesis (mmall = msw) cannot be rejected. Therefore, we conclude that the mutation rates of A/Mallard/New York/6750/78 and A/Swine/Germany/2/81 do not differ significantly.
= 0.5 (a value smaller than 1.96, the 97.5% quantile of the normal distribution), the null hypothesis (mmall = msw) cannot be rejected. Therefore, we conclude that the mutation rates of A/Mallard/New York/6750/78 and A/Swine/Germany/2/81 do not differ significantly.
DISCUSSION
The influenza pandemic of 1918 was caused by an H1N1 influenza virus (31). This isolate was probably closely related to later classic swine viruses and is thought to have been transmitted previously from birds into pigs without reassortment (24, 33). In 1979, an influenza virus of avian origin was introduced as a whole into swine of northern Europe (26), leading to the development of a new virus lineage. Before that time, H1N1 influenza viruses did not exist in pigs in northern Europe (22, 35). Therefore, we can hypothesize that the 1979 event is analogous to that with the 1918 isolate. Use of the northern European isolates as a model for studying the early evolution of influenza viruses is likely to improve our understanding of the conditions and circumstances that surround the emergence of an entirely new pandemic strain.
Evolution of influenza viruses occurs, in various degrees, by Darwinian selection and random drift, including mutation. Elevated rates of mutation, caused by a mutator mutation, may have accelerated adaptation of the H1N1 swine isolates (24). To test this theory, we determined the mutation rates of A/Swine/Germany/2/81 (H1N1), an early representative of the northern European swine influenza lineage, and A/Mallard/New York/6750/78 (H2N2). The latter strain, a well-established avian isolate, likely does not have an elevated mutation rate, because the evolution rates of other avian isolates were very slow (16) or even static (11, 12), and no available information suggests that A/Mallard/New York/6750/78 underwent several host changes in multiple avian species. Our results show that the mutation rates of A/Swine/Germany/2/81 and A/Mallard/New York/6750/78 do not differ significantly. This finding indicates that Darwinian selection rather than an increased mutation rate contributed to the early adaptation and high evolutionary rates of European swine influenza H1N1 viruses.
Our results further demonstrate that directly inferring the rates of mutation of avian-like swine influenza viruses from phylogenic data is difficult. This is because the evolutionary rate (i.e., number of base substitutions per year per site) is also influenced by fluctuations of the number of replication cycles (i.e., the actual population size over time) and random sampling effects (5) in the field during the observed time span, in contrast to the mutation rate (i.e., number of base substitutions per site per replication), which is determined under clear lab conditions. Therefore, the evolution rate reflects only cumulated changes of viral sequences year by year in the field. Even if noncoding changes were counted as silent mutations and not approximately as changes of the third position of each codon independent of whether an amino acid replacement occurred there (as most software does), so that fixation of noncoding changes by linked coding changes and selection would be excluded, such a resulting rate would not be a clear correlate to the mutation rate. In light of our results and these considerations, we suggest that, in addition to Darwinian selection, random sampling effects (5) and an increased number of replication cycles (reflecting the effects of factors such as the number of infected animals, the yields of various strains in swine, a greater number of infected target cells in swine than in birds, and the burst size per infected cell) contributed to the high evolutionary rates of European swine influenza H1N1 viruses. Witte et al. discussed that the rapid exhaustive expansion of the avian-like swine influenza viruses from 1979 to 1981 in northern Europe is possibly due to large-scale animal husbandry, in contrast to the few infections observed in Germany in 1957 (35).
The estimated mutation rate has considerable inherent variability because the measured frequency of mutants reflects progeny from one or more mutation events. Further, the specific time point at which a specific mutational event occurs is due to change, and because of the exponential characteristics of virus growth, this contributes greatly to the observed variability among estimated mutation rates (6, 19). To accommodate these factors, we applied a probabilistic model (see Appendix) that enabled us to determine the mutation rate at a given accuracy and to perform a statistical analysis. Our formula for the SD supports the predicted high variability, which was confirmed by the comparison with the ad hoc SD for A/Mallard/New York/6750/78. In addition, we needed to screen several parallel cultures, and we chose amantadine for the indirect determination of the mutation rate because the sites for resistance-inducing mutations are known, the inhibitory effect is complete for both isolates in plaque inhibition assay, and this compound facilitates rapid screening.
In contrast the techniques that we used, Suarez et al. (27) determined mutational rates by using the fluctuation test (19) in the presence of a monoclonal antibody against the HA1 portion of the hemagglutinin. The fluctuation test requires many parallel cultures and therefore is very labor-intensive. Parvin et al. (21) sequenced the NS genes of 108 clones that were derived from a single well-isolated plaque of A/WSN/33. This labor-intensive approach led to the most exact determination of the mutant spectrum, but, as those authors stated, because the mutation rate was evaluated from only one plaque, no discussion of the variability of the estimate of the mutation rate was possible. Table 3 shows that our data are in good accordance with the values for the mutation rate of influenza virus from both groups despite the use of three different virus genes from four different isolates and three different methods. This comparison indicates that differences in mutation rate cannot account for the evolutionary rates of various virus genes. The similarity of the mutation rates for the highly variable HA1 region and the highly conserved ion channel of the M2 gene again emphasizes that a high mutation rate cannot directly account for a high rate of evolution. Therefore, another explanation remains possible, regarding the potential role of a mutator mutation in the early evolution of the northern European swine influenza virus. For a mutator mutation to have contributed to this process, it must have emerged prior to 1981, possibly in 1979 immediately after transmission of the virus to the new host or even in the avian host before transmission to swine. Simulation studies for finite clonal asexual populations under directional selection (30) have suggested that mutator mutants increase in frequency if they are abundant in the population and associated with a favorable mutation which prevents their extinction (hitchhiking effect) but that they are kept at low frequency when their primary number is low. A mutator mutant could become dominant during a bottleneck event that dramatically reduces the population size; such bottlenecks are probably frequent in the life cycles of RNA viruses because of their airborne or droplet transmission (5). In such situations, an otherwise rare mutator mutant might accelerate adaptation for a brief transitional period until reverting back. To revert easily, the mutator mutation should comprise only a few (at most) point mutations if located in only one segment or should be disrupted by reassortment. For established strains at an equilibrium of mutation and selection, the mutation rate is close to Eigens error threshold (7) (Table 3).
An alternative mechanism for crossing the species barrier may exist in the absence of a mutator mutation. Perhaps the avian virus undergoes several mutations (at the normal mutation rate) in the primary host that are necessary for successful establishment in the new host. The product of the theoretical minimal number of replication cycles (see e.g., Tables 2 and 3) of all necessary mutations would have to be less than the possible finite population size in the avian host. This scenario is likely to apply to mutations that are detrimental or even deadly in avian hosts, whereas changes that also accommodate functional constraints of the new host often occur in multiple avian hosts before the transmission of the virus. From the purpose of quasispecies, to suppress a newly arisen, even superior mutant progeny (4), the new host should be free of another already well-established strain.
An example of the alternate mechanism might be the recent emergence of an H5N1 influenza virus in humans (3). The antecedent strain is a highly pathogenic H5N1 chicken isolate (29). Virulent avian H5 isolates (15) and the new human H5N1 isolate (29) are pantropic in chickens. Such viruses have greater finite population sizes in each chicken than do apathogenic isolates. Therefore, the mutations required for establishment in humans might occur in chickens without the need for an intermediate host (e.g., swine). Both mechanisms (mutator mutation and the alternate mechanism) could coexist in nature, and the one that is used would depend on the number of required adaptive mutations, i.e., on the ratio of the frequency of a mutator mutant to that of an already adapted mutant within the viral population in the avian host. If only a few adaptive mutations were necessary, then a large population size would be sufficient; if multiple changes were required, then a mutator phenotype might be necessary.
Suarez et al. (27) isolated variants of A/Victoria/3/75 whose mutation rates were increased three- to fourfold and estimated that 13% of the viral population had similarly elevated mutation rates. Notwithstanding the ever-present problem of obtaining cultures that have very high mutant frequencies because the mutational events occurred early during the growth of population (thereby mimicking mutator mutants), even those reported values seem to indicate minimal acceleration on adaptation according to simulation studies. According to both Coffin (2) and Taddei et al. (30), the mutation rate should have been at least 10-fold higher. However, Suarez et al. (27) elegantly showed that the mutation rate is a weighted average over the population, and their finding does not exclude the possibility that mutator mutants (with their associated high mutation rates) might have occurred at very low frequencies. Whether such variants actually contributed to the stable host change seen in 1979 is unknown; however, in light of the data that are available (from 1979 to 1985), changes in mutation rate cannot account for the early evolution of northern European swine influenza viruses. Whether the mutation rates fluctuated during growth of the initial avian influenza H1N1 virus in the earliest-infected pigs or in the avian host and whether those fluctuations in mutation rates were a prerequisite for successful establishment remain to be investigated further.
Ludwig et al. (18) demonstrated that all northern European swine isolates prior to 1985 had a highly heterogeneous plaque morphology, which persisted even after several plaque passages and which was taken as evidence of relatively high genetic instability. Because an increased mutational rate can now be excluded as the basis of this observation, we have to find another explanation, and the one we propose may also be a possible mechanism by which an avian influenza virus can pass the species barrier into swine. Relatively stable partial heterozygotes can be formed after double infection with two different influenza A viruses (25) or by introduction of an additional segment by reverse genetics (8) if bidirectional selection maintains both allelic segments so that they can complement each other’s weaknesses. Suppose an influenza virus gains a mutation that enables it to multiply better in swine but that is detrimental for replication to high titers. Such a virus might be able to cross the species barrier if it is partially heterozygous with respect to this gene (RNA segment). Once in the new species, selection pressure for this partial heterozygote persists until suppressor mutations overcome the otherwise detrimental mutation, and one segment gets lost; this process might take several years. Such partial heterozygotes might also be responsible for the observed plaque heterogeneity and for higher frequencies of monoclonal antibody escape mutants, too, if the required mutations are located in the same segment, e.g., the hemagglutinin gene. This alternate mechanism would allow an influenza virus to cross the species barrier to form a stable lineage and would accounts for the observation described by Ludwig et al. (18).
ACKNOWLEDGMENTS
We thank Krisna Wells and Scott Krauss for excellent advice and expert technical assistance. We thank Amy L. B. Frazier for critical comments and editing the manuscript.
This work was supported in part by Public Health Service research grants AI08831 and AI29680 and by the American Lebanese Syrian Associated Charities (ALSAC).
Appendix
The number of virions in culture during maturation is known to grow exponentially (9, 34) and can be described as N = ect. From that exponential function, one can derive an equation, N = ect = 2g, reflecting an ideal population grown as a binary tree, where each virion gives rise to exactly two progenies of the next generation and all viruses at a given time point t belong to the same generation g. Such a binary tree allows us to consider the number of unmutated viruses in the virus multiplication as a branching process. Using the branching process (17), we have derived estimates of the mutation rate and its variance by counts of mutated and unmutated viruses at a given generation which is log2 of the size of such an ideal population. The resulting formula for the mutation rate is similar to the formula described by Drake (6), but the possibility of calculating a variance allows statistical testing. The model agrees with our understanding that during the first generations, mutation to amantadine resistance is very improbable because the number of loci involved (4 loci) is very small in relation to the genome length (∼14,000 bases), so that in our approach the mutation frequency is determined many generations after clonal infection during the maturation phase, e.g., the above-mentioned exponential growth, and not during the eclipse phase. Therefore, the contribution of mutants arising during the eclipse phase to the mutant frequency is negligible.
Notation
m = mutation rate (probability of mutation per nucleotide per copy).
mvir = mutation rate of virus, where the mutants can be defined according to a given criterion. For example, a mutant is defined as a virus that has at least one mutated nucleotide in given sites of the nucleotide sequence.
M = number of mutants in the culture.
N = population size of the culture.
f = frequency of mutants in the population, defined as M/N.
k = number of sites in the sequence of nucleotides where mutation to amantadine resistance is possible.
Estimation of mutation rate. We are interested in m, the mutation rate (probability of mutation per nucleotide per replication), but it cannot be directly estimated. Therefore, we first estimate mvir from M and N and then derive the estimate of m from that of mvir by using the relationship between m and mvir.
(i) One culture. Let N be the number of viruses in a culture of virus, and let M be the number of mutants. Then the estimate of mvir is
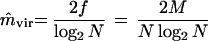 |
A1 |
which was derived by using the theory of branching processes (17). The variance of the estimate is
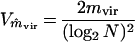 |
A2 |
In the experiment using amantadine, N is the number of viruses in the culture in the absence of amantadine and M is the number of survivors in the presence of amantadine in the culture. In the presence of amantadine, the viruses survive or die according to whether there is any mutation at the k specific sites in the nucleotide sequence of the virus.
When mvir and m are close to 0 and k is not large (e.g., when m = 10−5 and k ≤ 100), then m ≈ mvir/k. From this relation and equations A1 and A2, the estimate of m is
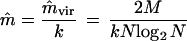 |
A3 |
and the variance of the estimate is
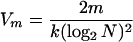 |
A4 |
In the discussion above, m̂vir, m̂, and M are random variables, while mvir, m, and N are constants. From equation A4, the SD for the estimate of m is
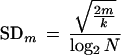 |
A5 |
which is much larger than m when m is close to 0. For example, let m = 10−5 and N = 108, which are usual in virus cultures. Then, from equation A5, we have SD = 8.4 × 10−5, which implies that the SD of the estimate is 8.4-fold the rate m. An estimate with such a large variability cannot be used for hypothesis testing.
(ii) Multiple cultures. To compare the mutation rates of two types of viruses, we must use multiple cultures of viruses to reduce the errors of estimates. Denote C = total number of parallel cultures, Mi = number of mutants in the ith culture, and Ni = population size in the ith culture, where i = 1,...,C. We have two types of estimates; one is a simple average of estimates from each culture, and the other is a weighted average of those estimates.
(a) Simple average. By taking the average of estimates from equation A3 for each culture, we have the estimate of m:
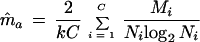 |
A6 |
By equations A3, A4, and A6, the variance of ma is
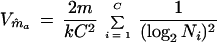 |
A7 |
Since m in equation A7 is unknown, the variance Vm̂a must be estimated by using equation A7 and by replacing m by m̂a from equation A6.
(b) Weighted average. If there are large differences among the Nis, simple averaging is not a good way to combine estimates from each culture. Obviously from equation A4, the estimate in equation A3 from a culture with a larger population has smaller variance than the one from a culture with a smaller population. Thus, the former should be weighted more than the latter. With optimal weighting, the estimate is derived as
 |
A8 |
where
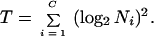 |
The variance ofm̂w is
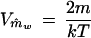 |
A9 |
which was minimized by allocating weights according to the inverse of the individual variance. Vm̂w in equation A9 contains m, which is unknown. Replacing m in equation A9 by its estimate in equation A8, the variance in equation A9 is estimated by
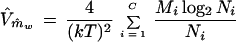 |
A10 |
Comparison of mutation rates. Assuming that there are two types of viruses with mutation rates mmall and msw, we are interested in testing the hypothesis H0(mmall = msw) versus Ha(mmall ≠ msw) for a fixed α level (e.g., 0.001, 0.05, or 0.1). Let m̂mall and m̂sw be the estimates of mmall and msw, respectively. The estimates of mutation rates, m̂mall and m̂sw can be computed either by the simple average in equation A6 or by the weighted average in equation A8. Accordingly, the estimated variances V̂mall and V̂sw of the estimates are computed either by equation A7 or by equation A10. Using the normal approximation, we formulate the test statistic as
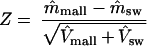 |
H0 is rejected if |Z| > Zα/2, where Zα is the upper α quantile of the normal distribution (e.g., Z0.025 = 1.96).
REFERENCES
- 1.Bean W J, Threlkeld St C, Webster R G. Biologic potential of amantadine-resistant influenza A virus in an avian model. J Infect Dis. 1989;159:1050–1056. doi: 10.1093/infdis/159.6.1050. [DOI] [PubMed] [Google Scholar]
- 2.Coffin J M. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 3.de Jong J C, Osterhaus A D M E, Webster R G, Lim W L. A pandemic warning? Nature. 1997;389:554. doi: 10.1038/39218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de la Torre J C, Holland J J. RNA virus quasispecies populations can suppress vastly superior mutant progeny. J Virol. 1990;64:6278–6281. doi: 10.1128/jvi.64.12.6278-6281.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domingo E, Escarmis C, Sevilla N, Moya A, Elena S F, Quer J, Novella I S, Holland J J. Basic concepts in RNA virus evolution. FASEB J. 1996;10:859–864. doi: 10.1096/fasebj.10.8.8666162. [DOI] [PubMed] [Google Scholar]
- 6.Drake J W. The molecular basis of mutation. San Francisco, Calif: Holden-Day; 1970. [Google Scholar]
- 7.Eigen M, Schuster P. The hypercycle A principle of self organization part A: emergence of hypercycle. Naturwissenschaften. 1977;64:541–565. doi: 10.1007/BF00450633. [DOI] [PubMed] [Google Scholar]
- 8.Enami M, Sharma G, Benham C, Palese P. An influenza virus containing nine different RNA segments. Virology. 1991;185:291–298. doi: 10.1016/0042-6822(91)90776-8. [DOI] [PubMed] [Google Scholar]
- 9.Fields B N, Knipe D M. Virology. 2nd ed. New York, N.Y: Raven Press; 1990. [Google Scholar]
- 10.Fitch W M, Bush R M, Bender C A, Cox N J. Long term trends in the evolution of H(3) HA1 human influenza type A. Proc Natl Acad Sci USA. 1997;94:7712–7718. doi: 10.1073/pnas.94.15.7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gammelin M, Altmüller A, Reinhardt U, Mandler J, Harley V R, Hudson P J, Fitch W M, Scholtissek C. Phylogenetic analysis of nucleoproteins suggests that human influenza A viruses emerged from a 19th-century avian ancestor. Mol Biol Evol. 1990;7:194–200. doi: 10.1093/oxfordjournals.molbev.a040594. [DOI] [PubMed] [Google Scholar]
- 12.Gorman O T, Bean W J, Webster R G. Evolutionary processes in influenza viruses: divergence, rapid evolution, and stasis. Curr Top Microbiol Immunol. 1992;176:75–97. doi: 10.1007/978-3-642-77011-1_6. [DOI] [PubMed] [Google Scholar]
- 13.Hay A J, Wolstenholme A J, Skehel J J, Smith M H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho P P K, Young A L, Truehaft M. Plaque formation with influenza viruses in dog kidney cells. J Gen Virol. 1976;33:143–145. doi: 10.1099/0022-1317-33-1-143. [DOI] [PubMed] [Google Scholar]
- 15.Horimoto T, Kawaoka Y. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza A virus. J Virol. 1994;68:3120–3128. doi: 10.1128/jvi.68.5.3120-3128.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito T, Gorman O T, Kawaoka Y, Bean W J, Webster R G. Evolutionary analysis of the influenza virus M gene with comparison of the M1 and M2 proteins. J Virol. 1991;65:5491–5498. doi: 10.1128/jvi.65.10.5491-5498.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlin S, Taylor H. A first course in stochastic processes. 2nd ed. New York, N.Y: Academic Press; 1975. [Google Scholar]
- 18.Ludwig S, Stitz L, Planz O, Van H, Fitch W M, Scholtissek C. European swine virus as a possible source for the next influenza pandemic? Virology. 1995;212:555–561. doi: 10.1006/viro.1995.1513. [DOI] [PubMed] [Google Scholar]
- 19.Luria S E, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palese P, Young J F. Variation of influenza A, B, and C viruses. Science. 1982;215:1468–1474. doi: 10.1126/science.7038875. [DOI] [PubMed] [Google Scholar]
- 21.Parvin J D, Moscona A, Pan W T, Leider J M, Palese P. Measurement of the mutation rates of animal viruses: influenza A virus and poliovirus type 1. J Virol. 1986;59:377–383. doi: 10.1128/jvi.59.2.377-383.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pensaert M, Ottis K, Vanderputte J, Kaplan M M, Bachmann P A. Evidence for the natural transmission of influenza A virus from wild duck to swine and its potential importance for man. Bull W H O. 1981;59:75–78. [PMC free article] [PubMed] [Google Scholar]
- 23.Ribeiro R M, Bonhoeffer S, Nowak M A. The frequency of resistant mutant virus before antiviral therapy. AIDS. 1998;12:461–465. doi: 10.1097/00002030-199805000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Scholtissek C, Ludwig S, Fitch W M. Analysis of influenza A virus nucleoproteins for the assessment of molecular genetic mechanisms leading to new phylogenetic virus lineages. Arch Virol. 1993;131:237–250. doi: 10.1007/BF01378629. [DOI] [PubMed] [Google Scholar]
- 25.Scholtissek C, Rohde W, Harms E, Rott R, Orlich M, Boschek C B. A possible partial heterozygote of an influenza A virus. Virology. 1978;89:506–516. doi: 10.1016/0042-6822(78)90192-7. [DOI] [PubMed] [Google Scholar]
- 26.Schultz U, Fitch W M, Ludwig S, Mandler J, Scholtissek C. Evolution of pig influenza viruses. Virology. 1991;183:61–73. doi: 10.1016/0042-6822(91)90118-u. [DOI] [PubMed] [Google Scholar]
- 27.Suarez P, Valcarcel J, Ortin J. Heterogeneity of the mutation rates of influenza A viruses: isolation of mutator mutants. J Virol. 1992;66:2491–2494. doi: 10.1128/jvi.66.4.2491-2494.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suarez-Lopez P, Ortin J. An estimation of the nucleotide substitution rate at defined positions in the influenza haemagglutinin gene. J Gen Virol. 1994;75:389–393. doi: 10.1099/0022-1317-75-2-389. [DOI] [PubMed] [Google Scholar]
- 29.Subbarao K, Klimov A, Katz J, Regenery H, Lim W, Hall H, Perdue M, Swayne D, Bender C, Huang J, Hamphill M, Rowe T, Shaw M, Xu X, Fukuda K, Cox N. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science. 1998;279:393–396. doi: 10.1126/science.279.5349.393. [DOI] [PubMed] [Google Scholar]
- 30.Taddei F, Radman M, Maynard-Smith J M, Toupance B, Gouyon P H, Godelle B. Role of mutator alleles in adaptive evolution. Nature. 1997;387:700–702. doi: 10.1038/42696. [DOI] [PubMed] [Google Scholar]
- 31.Taubenberger J K, Heid A H, Krafft A E, Bijwaard K E, Fanning T G. Initial genetic characterization of the 1918 Spanish influenza virus. Science. 1997;275:1793–1796. doi: 10.1126/science.275.5307.1793. [DOI] [PubMed] [Google Scholar]
- 32.Topal M D, Fresco J R. Complementary base pairing and the origin of substitution mutations. Nature. 1976;263:285–289. doi: 10.1038/263285a0. [DOI] [PubMed] [Google Scholar]
- 33.Webster R G, Bean W J, Gorman O T, Chambers T M, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White D O. Some aspects of influenza virus multiplication in the surviving allantois-on-shell system. Virology. 1960;10:21–28. doi: 10.1016/0042-6822(60)90003-9. [DOI] [PubMed] [Google Scholar]
- 35.Witte K H, Niendorf E, Ernst H, Schmidt U, Prager D. Erstmaliges Auftreten einer durch das Schweineinfluenzavirus verursachten Epizootie in Schweinebestaenden der Bundesrepublik Deutschland. Tieraerztl Umschau. 1981;36:591–606. [Google Scholar]