

Abstract

A less-studied halogen-free variation of the Hirao reaction involving the coupling of arylboronic acids with >P(O)H reagents, such as diarylphosphine oxides, diethyl phosphite, and ethyl phenyl-H-phosphinate, was investigated in detail using Pd(OAc)2 as the catalyst precursor and applying some excess of the P-reagent to supply the ligand via its trivalent tautomeric (>P–OH) form. The optimum conditions (1.2 equiv of the P-reagent, 135–150 °C, and air) were explored for the synthesis of diaryl-phenylphosphine oxides, aryl-diphenylphosphine oxides, diethyl arylphosphonates, ethyl diphenylphosphinate, and two bisphosphinoyl derivatives. In the reaction of 4-chlorophenyl- or 3-chlorophenylboronic acid with Ph2P(O)H, triphenylphosphine oxide was also formed as a byproduct. Theoretical calculations suggested that the catalytic cycle of the P–C coupling of PhB(OH)2 with Ph2P(O)H is different from that of the usual cross-coupling reactions. It comprises the addition of a phenyl anion and then the tautomeric form >P–OH of the >P(O)H reagent to the Pd2+ catalyst complex. This is then followed by reductive elimination affording Ph3PO that is accompanied with the conversion of Pd2+ to Pd0. There is a need for a subsequent stoichiometric oxidation of Pd(0) by molecular oxygen. The spontaneous formation of the self-assembling ligands around the Pd2+ center from the >P(O)H reactant plays a crucial role in the mechanism and promotes the efficiency of the catalyst.
Introduction
A heteroatomic variation of the cross-coupling reactions is the C–P coupling discovered by Hirao in 1980.1,2 Since then, this new reaction between bromoarenes and dialkyl phosphites or secondary phosphine oxides providing dialkyl arylphosphonates or tertiary phosphine oxides has become an important method.3−5 The original Pd(PPh3)4 catalyst was replaced by Pd(II) salts used together with mono- and bidentate P-ligands, and the scope of the reaction components was extended.6,7 The mechanism was evaluated.6,8 Efforts were made to perform the P–C coupling under green chemical conditions.9,10 Later on, Ni- and Cu-catalyzed versions were also elaborated.3,4,11,12
The Hirao reaction of arylboronic acids with different >P(O)H reagents is a less-studied field. The coupling of arylboronic acids with diphenylphosphine oxide, ethyl phenyl-H-phosphinate, and diethyl phosphite was best performed at 90 °C for 24 h applying Pd(OAc)2/bis(diphenylphosphino)butane (dppb) as the catalyst system, K2CO3 as the base in 1,4-dioxane as the solvent with the use of air as an oxidant. The corresponding tertiary phosphine oxides, phosphinates, and phosphonates were obtained in 48–94% yields after a reaction time of 24 h.13 The second example for the Pd(II) salt-catalyzed case is the microwave (MW)-assisted reaction of arylboronic acids with diethyl phosphite in the presence of Pd(OAc)2/2,9-dimethyl-1,10-phenantroline (dmphen) in dimethylformamide. After an irradiation at 100 °C for 20–30 min, the dialkyl arylphosphonates were obtained in yields of 51–90%. The couplings were promoted by the addition of p-benzoquinone as an oxidating agent.14 A series of pyridylboronic acids also underwent the coupling reaction with dialkyl phosphites at 100 °C, in this case, using PdCl2/PPh3 as the catalyst precursor in N-,N-dimethylacetamide, in the presence of Ag2O as an additive. After a 2 h reaction time, the yields fell in the range of 51–95%.15 A NiBr2/pyridine-catalyzed coupling of arylboronic acids and mainly diarylphosphine oxides carried out at 100 °C for 24 h in dichloroethane in the presence of K2CO3 furnished the corresponding triarylphosphine oxides in variable 5–99% yields.16 Last but not least, Cu2O/1,10-phenantrolin (phen) and a special Cu(II) complex were also applied as catalysts at 26 °C for 24–72 h in the reaction under discussion to provide dialkyl arylphosphonates in variable yields (47–96%) using diisopropylethylamine in acetonitrile and KOAc in tetrahydrofuran, respectively.17,18 The disadvantages of the coupling reactions with arylboronic acids include the need, in most cases, for bidentate P-ligands or N-ligands, variable yields, and often long reaction times. In the Pd(II)/(bidentate) phosphine-catalyzed cases, the involvement of an oxidizing agent (even air) may seem somewhat unusual, as in the P–C coupling of bromoarenes, there was no need for this actor.
In the past few years, we developed a P–C coupling, where it was not necessary to add the usual mono- or bidentate P-ligands. Under MW conditions, the reaction between bromoarenes and >P(O)H reagents (dialkyl phosphites and secondary phosphine oxides) took place if the catalyst precursor, Pd(OAc)2 was combined with some excess of the P-reagent.19 The point was that the trivalent tautomeric form (>P–OH) of the >P(O)H species may act as a ligand to Pd(0). Upon applying 10% of the Pd(II) salt, there was a need for 30% extra quantity of the P-reagent. One equivalent of the >P(O)H species to the precursor ensured the Pd(II) → Pd(0) reduction, while the remaining two equivalent quantities served as the P-ligands to Pd(0).20 The detailed mechanism for the “P-ligand-free” Hirao reaction was explored by high-level quantum chemical calculations.20,21 It was found that the rate-determining step is the reagent insertion into the Pd complex or the Pd–C bond formation that may be promoted by MW assistance. Our procedure meant a green approach, as there was no need to add the usual expensive P-ligands; hence, cost and environmental burden can be saved. Regarding atomic efficiency, starting from arylboronic acids means an advantage over the variation applying bromoarenes, not speaking about the fact that the former protocol is halogen-free. It was a challenge for us to develop a new protocol combining the halogen-free option with our MW-assisted method lacking the use of the usual P-ligands. It was another challenge to justify the need for an oxidizing agent and to evaluate the mechanism.
Results and Discussion
Preparative Results
The coupling of phenylboronic acid with diphenylphosphine oxide was chosen as the basic model of the MW-assisted Pd-catalyzed process. 10 mol % Pd(OAc)2 was applied as the catalyst precursor at 135 °C using different bases in a quantity of 1.1 equiv and acetonitrile or ethanol as the solvent. The P-reagent was measured in a 1.2 equiv quantity in order to provide the two Ph2POH ligands. We worked at aerobic conditions (Table 1). The crude reaction mixtures were analyzed by 31P NMR and liquid chromatography–mass spectrometry (LC–MS). Performing the reaction in the presence of Cs2CO3 in acetonitrile, after a 2.5 h irradiation the conversion was 80% (Table 1, entry 1). The next run carried out in ethanol was complete after 1.5 h (Table 1, entry 2). In this case, 5% of ethyl diphenylphosphinate could be detected as a byproduct. 1,4-Dioxane was also a suitable solvent under similar conditions (Table 1, entry 3). Changing for K2CO3, a treatment at 135 °C for 2.5 h in acetonitrile also led to almost quantitative (96%) conversion (Table 1, entry 4). However, the accomplishment in ethanol was more efficient, as required a shorter irradiation time of 1.5 h (Table 1, entry 5). Only 3% of (EtO)Ph2PO contaminated the product (1a). The use of triethylamine as the base led, after a treatment of 1.5 h, in both solvents to low (11/22%) conversions (Table 1, entries 6 and 7). From the best experiments (marked by entries 2–4), triphenylphosphine oxide (1a) was obtained in 78–81% yields after flash column chromatography. Under an inert atmosphere, the P–C coupling did not take place.
Table 1. Optimization of the Coupling Reaction between Phenylboronic Acid and Diphenylphosphine Oxide.

| product
composition (%)a,b |
|||||||
|---|---|---|---|---|---|---|---|
| entry | base | solvent | t (h) | conversion (%)a | 1a | (EtO)Ph2PO | yield (%) of 1a |
| 1 | Cs2CO3 | MeCN | 2.5c | 80 | 80 | ||
| 2c | Cs2CO3 | EtOH | 1.5 | 100 | 95 | 5 | 83 |
| 3 | Cs2CO3 | dioxane | 1.5 | 100 | 100 | 79 | |
| 4 | K2CO3 | MeCN | 2.5d | 96 | 96 | 78 | |
| 5 | K2CO3 | EtOH | 1.5 | 100 | 97 | 3 | 80 |
| 6 | Et3N | MeCN | 1.5 | 11 | 11 | ||
| 7 | Et3N | EtOH | 1.5 | 22 | 22 | ||
On the basis of relative 31P NMR intensities.
The average of two or three parallel experiments.
The coupling was also performed on a larger scale applying 1.0 mmol of PhB(OH)2 and 1.2 mmol of Ph2P(O)H. In this case, the yield of 1a was 87%.
The extrapolated reaction time is 3 h.
In general, the couplings were performed using 0.41 mmol of the phenylboronic acid. Carrying out the reaction on a 1.0 mmol scale, the yield of Ph3PO was 87% (footnote “c” of Table 1/entry 2). As a comparison, 10% Pd(PPh3)4 was also tested in the coupling reaction of phenylboronic acid with Ph2P(O)H at 135 °C for 1.5 h using 1.1 equiv of Cs2CO3 in ethanol. In this case, the reaction was not entirely clear-cut, and the yield of Ph3PO was 65%.
In the next series of experiments, the reaction of phenylboronic acid with different >P(O)H reagents was investigated at 135 °C applying 10 mol % Pd(OAc)2 as the catalyst precursor. The base/solvent combinations included Cs2CO3/EtOH and K2CO3/MeCN (Table 2). The coupling with bis(4-methylphenyl)phosphine oxide was somewhat slower than that with Ph2P(O)H, no matter if it was carried out using Cs2CO3 in EtOH or K2CO3 in MeCN (Table 2, entries 3 and 4 vs entries 1 and 2). After an irradiation of 2 and 3 h, respectively, the conversion was complete. Beyond the 85/81% proportion of the expected bis(4-methylphenyl)-phenylphosphine oxide (1b), there was 15/19% of tris(4-methylphenyl)phosphine oxide as the byproduct (Table 2, entries 3 and 4). A similar situation was observed, when bis(3,5-dimetylphenyl)phosphine oxide was the reagent; however, in this instance, the coupling became even slower, as the complete conversion required a reaction time of 2.5 and 4 h, respectively (Table 2, entries 5 and 6). Diaryl-phenylphosphine oxide 1c was formed in 88 and 79%, respectively. The corresponding tris(3,5-dimethylphenyl)phosphine oxide was present in the mixture as a minor (12/21%) byproduct. From the best experiments, the diaryl-phenylphosphine oxides 1a–c were isolated in yields of 74–83% after chromatography. Changing for diethyl phosphite, the coupling with phenylboronic acid was slower at 135 °C, as the completion took 4 h (Table 2, entry 7). However, at 150 °C, an irradiation of 0.5 h was enough (Table 2, entry 8). In both cases, the reaction was clear-cut, and diethyl phenylphosphonate (1d) was obtained in 84/82% yield after purification. The combination of K2CO3/MeCN was, in this case, less efficient: after an irradiation at 135 °C for 4 h, the conversion was only 63% (Table 2, entry 9). However, at 150 °C/2 h, phosphonate 1d was the only product prepared in 73% yield (Table 2, entry 10). The next >P(O)H reagent, ethyl phenyl-H-phosphinate behaved similarly as (EtO)2P(O)H, and complete conversion could be attained after 4 h at 135 °C or 0.5 h at 150 °C (Table 2, entries 11 and 12). In the latter instance, 9% of (EtO)2PhPO also appeared in the crude mixture. Ethyl diphenylphosphinate (1e) was isolated in 82/86% yields.
Table 2. Coupling Reaction between Phenylboronic Acid and Different >P(O)H Reagents.

| product
composition (%)a,b |
||||||||
|---|---|---|---|---|---|---|---|---|
| entry | Y1, Y2 | base | solvent | t (h) | conversion (%)a | 1 | Ar3PO | yield (%) |
| 1 | Ph | Cs2CO3 | EtOH | 1.5 | 100b | 95c | 83 (1a) | |
| 2 | Ph | K2CO3 | MeCN | 2.5 | 96 | 96 | 78 (1a) | |
| 3 | 4-MeC6H4 | Cs2CO3 | EtOH | 2 | 100 | 85 | 15 | 78 (1b) |
| 4 | 4-MeC6H4 | K2CO3 | MeCN | 3 | 100 | 81 | 19 | 72 (1b) |
| 5 | 3,5-diMeC6H3 | Cs2CO3 | EtOH | 2.5 | 100 | 88 | 12 | 74 (1c) |
| 6 | 3,5-diMeC6H3 | K2CO3 | MeCN | 4 | 100 | 79 | 21 | 56 (1c) |
| 7 | EtO | Cs2CO3 | EtOH | 4 | 100 | 100 | 84 (1d) | |
| 8 | EtO | Cs2CO3 | EtOH | 0.5d | 100 | 100 | 82 (1d) | |
| 9 | EtO | K2CO3 | MeCN | 4 | 63 | 63 | ||
| 10 | EtO | K2CO3 | MeCN | 2d | 100 | 100 | 73 (1d) | |
| 11 | EtO, Ph | Cs2CO3 | EtOH | 4 | 100 | 100 | 82 (1e) | |
| 12 | EtO, Ph | Cs2CO3 | EtOH | 0.5d | 100 | 91e | 86 (1e) | |
On the basis of relative 31P NMR intensities.
The average of two or three parallel experiments.
5% of (EtO)Ph2PO as the byproduct.
The temperature was 150 °C.
9% of (EtO)2PhPO as the byproduct.
In the next round, Ph2P(O)H was reacted with a series of arylboronic acids, in the first approach, under the conditions applied above using Cs2CO3 in EtOH (Table 3). The coupling of 4-Me, 2-Me, 3-Me, and even the 4-F-substituted phenylboronic acids took place similarly as that with the unsubstituted PhB(OH)2. Complete conversions were attained after an irradiation of 1.5 h at 135 °C. The proportion of the expected products (2f–i) was 71–87%, while that of the (EtO)Ph2PO was 5–13%. Compounds 2f–i were prepared in 67–80% yield (Table 3, entries 2–5). It was surprising that the similar reaction of 4-chlorophenylboronic acid with 1.2 equiv of Ph2P(O)H at 135 °C/1.5 h afforded 1,4-bis(diphenylphosphinoyl)benzene 3A. The crude mixture contained 49% of bisphosphinoyl compound 3A and 51% of Ph3PO (Table 3, entry 6). There was no expected 4-chlorophenyl-diphenylphosphine oxide 2j in the mixture. Product 3A was isolated by chromatography in a yield of 23%. To promote the formation of the phosphinoyl-chlorobenzene 2j, the coupling was performed at a lower temperature of 90 °C for 4 h. Sure enough, the mixture comprised 46% of the monophosphinoyl product 2j, 5% of bisphosphinoyl 3A, 11% of (EtO)Ph2PO, and 38% of Ph3PO formed probably by dechlorination of 4-chlorophenyl-diphenylphosphine oxide (2j) (Table 3, entry 7). The small quantity of bisphosphinoyl compound 3A among the components referred to the crucial role of temperature on the selectivity toward the mono- and bisphosphinoylated products (2j and 3A). Repeating the previous P–C coupling on conventional heating at 90 °C/4 h, the conversion remained incomplete (77%), and the proportion of species 2j, 3A, and Ph3PO was 12, 41, and 18%, respectively, suggesting that the competitive dechlorination of 4-chlorophenyl-diphenylphosphine oxide (2j) was suppressed, and hence, more bis(Ph2P(O)) product (3A) could be formed (Table 3, entry 8). Returning to the result of the experiment marked by Table 3, entry 6, the low yield of 23% of the selective phosphinoylation may be explained by the fact that the 1.2 equiv quantity of Ph2P(O)H was not enough. Considering that a quantitative bisphosphinoylation would require 2.4 equiv of Ph2P(O)H, an experiment was carried out using this higher amount of the P-reactant. However, under such conditions, the proportion of bis-product 3A was not increased, and the excess of Ph2P(O)H was involved in side reactions. In respect of the 3-chlorophenylboronic acid, the outcome of the P–C coupling with Ph2P(O)H was similar to the reaction of the 4-chlorophenyl reagent. After an irradiation at 135 °C for 1.5 h, only 53% of bisphosphinoyl derivative 3B, 6% of (EtO)Ph2PO, and 41% of Ph3PO were present (Table 3, entry 10). The major component (3B) was isolated in a yield of 25%. Repeating the Hirao reaction at 90 °C for 4 h, the monophosphinoyl compound 2k was also present in 35%, beyond the 22% proportion of bis derivative 3B (Table 3, entry 11), and hence, the significant effect of the temperature was confirmed. In the thermal variation carried out, this occasion at 90 °C for 22 h, the proportion of 1,3-bis(diphenylphosphinoyl)benzene (3B) was increased to 44% (Table 3, entry 12). It can be noted that on MW irradiation, the statistically occurring local overheatings22 may cause the more intensive dechlorination of the primarily formed chlorophenyl-diphenylphosphine oxides (2j and 2k).
Table 3. P–C Coupling of Substituted Arylboronic Acids with Diphenylphosphine Oxide.

| product
composition (%)a,b |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | Z | T | t (h) | conversion (%)a | 2 | 3 | (EtO)Ph2PO | Ph3PO (1a) | yield (%) |
| 1 | H | 135 | 1.5 | 100b | 95 (1a) | 5 | 83 (1a) | ||
| 2 | 4-Me (f) | 135 | 1.5 | 100 | 81 | 5 | 14 | 73 (2f) | |
| 3 | 2-Me (g) | 135 | 1.5 | 100 | 71 | 13 | 16 | 67 (2g) | |
| 4 | 3-Me (h) | 135 | 1.5 | 100 | 87 | 8 | 5 | 80 (2h) | |
| 5 | 4-F (i) | 135 | 1.5 | 100 | 75 | 10 | 15 | 70 (2i) | |
| 6 | 4-Cl (j) | 135 | 1.5 | 100 | 49 (3A) | 51 | 23 (3A) | ||
| 7 | 4-Cl (j) | 90 | 4 | 100 | 46 | 5 | 11 | 38 | (2j)c |
| 8 | 4-Cl (j) | 90d | 4 | 77 | 12 | 41 (3A) | 6 | 18 | |
| 9 | 4-Cl (j) | 135e | 1 | 100 | 23 (3A) | 5 | 72 | ||
| 10 | 3-Cl (k) | 135 | 1.5 | 100 | 53 (3B) | 6 | 41 | 25 (3B) | |
| 11 | 3-Cl (k) | 90 | 4 | 96 | 35 | 22 (3B) | 11 | 28 | (2k)f |
| 12 | 3-Cl (k) | 90c | 22 | 100 | 33 | 44 (3B) | 4 | 19 | |
On the basis of relative 31P NMR intensities of the P-components.
The average of two or three parallel experiments.
31P NMR (CDCl3) δ 28.5, δP lit.23 (CDCl3) 28.2; HRMS (m/z): calcd for C18H15OPCl [M + H]+, 313.0549; found, 313.0542.
On conventional heating.
2.4 equiv of Ph2P(O)H was used.
31P NMR (CDCl3) δ 28.1, δP lit.24 (CDCl3) 28.1; HRMS (m/z): calcd for C18H15OPCl [M + H]+, 313.0549; found, 313.0538.
Finally, the substituted arylboronic acids were reacted with diethyl phosphite measured in a 1.2 equiv quantity using Cs2CO3 as the base and EtOH as the solvent (Table 4). The couplings with 4-MePh-, 2-MePh-, 3-MePh-, and 4-FPh-boronic acids were efficient on irradiation at 150 °C for 30 min. The diethyl arylphosphonates (1d and 4f–i) were isolated in yields of 77–83% (Table 4, entries 1–7). The similar reactions of the 4-ClPh- and 3-ClPh-boronic acids carried out under similar conditions were accompanied by the formation of 19% and 16% of (EtO)2PhPO (1d), respectively, and the expected products 4j and 4k could be obtained in 66/71% yields. The reductive dechlorination was a side reaction also in this case.
Table 4. Hirao Reaction between Substituted Arylboronic Acids and Diethyl Phosphite.

| entry | Z | conversion (%)a | proportion of 4 (%)a,b | yield (%) |
|---|---|---|---|---|
| 1 | H | 100 | 95 | 83 (1d) |
| 2 | 4-Me | 100 | 100 | 77 (4f) |
| 3 | 2-Me | 100 | 100 | 78 (4g) |
| 4 | 3-Me | 100 | 100 | 80 (4h) |
| 5 | 4-F | 100 | 100 | 77 (4i) |
| 6 | 4-Cl | 100c | 81 | 66 (4j) |
| 7 | 3-Cl | 100d | 84 | 71 (4k) |
On the basis of relative 31P NMR intensities.
The average of two or three parallel experiments.
19% of (EtO)2PhPO as the byproduct.
16% of (EtO)2PhPO as the byproduct.
The diaryl-phenylphosphine oxides (1a–c), aryl-diphenylphosphine oxides (2f–i), diethyl arylphosphonates (1d, 4f–k), ethyl diphenylphosphinate (1e), and bisphosphinoyl derivatives 3A and 3B, all together 17 compounds, were prepared by the method developed, and they were fully characterized by 31P, 13C and 1H NMR, as well as HRMS. The resulting phosphine oxides and related compounds may be utilized as P-ligands after deoxygenation.
Theoretical Calculations
There are only a few examples in the literature for Hirao reactions starting from arylboronic acids. There is no detailed mechanistic study for this version of the P–C coupling, and only a putative cycle was described.13 However, it occurred that an oxidant is needed to promote the reaction.13−15 Looking at the overall reaction, the +3 oxidation number of the P atom in the tautomeric form of diphenylphosphine oxide is increased to +5 in the product triphenylphosphine oxide (1a). At the same time, the oxidation numbers of the boron and carbon atoms of the boronic acid remains unchanged. The main process is exothermic (Figure 1). We confirm that during the course of the reaction, Pd2+ is reduced to Pd0 that is then oxidized back by the oxygen present. The O2 is reduced from the state 0 to −2 during the transformation.
Figure 1.

Overall transformation and reaction enthalpy (ΔHR), as well as Gibbs free energy (ΔGR) for the Hirao reaction of Ph2P(O)H with phenylboronic acid.
First, we explored a complex equilibrium of Pd(OAc)2 in the presence of excess of Ph2P(O)H as the ligand (L), including two-, three-, and tetraligations to afford species 5, 6, and 7, respectively. The tautomeric form Ph2POH of Ph2P(O)H is able to organize a strong hydrogen bond net around the Pd2+. An inorganic base (e.g., K2CO3) may deprotonate one or two Ph2POH units to provide complexes 8, 9, and 10, and as a consequence, the bonded Ph2POH pairs are stabilized, forming one (as in 8 or 9) or two (as in 10) quasi bidentate ligands (Figure 2). This spontaneous arrangement around the Pd2+ ion may be considered, as if a self-assembling bidentate P-ligand or two bidentate P-ligands were (indicated as Q; blue diamonds) around the central Pd2+ion. Formation of the tetraligated Pd2+ complex (10) is the most favorable in the series. This quasi bidentate anionic ligands activate the Pd2+ center for the coupling reaction.
Figure 2.
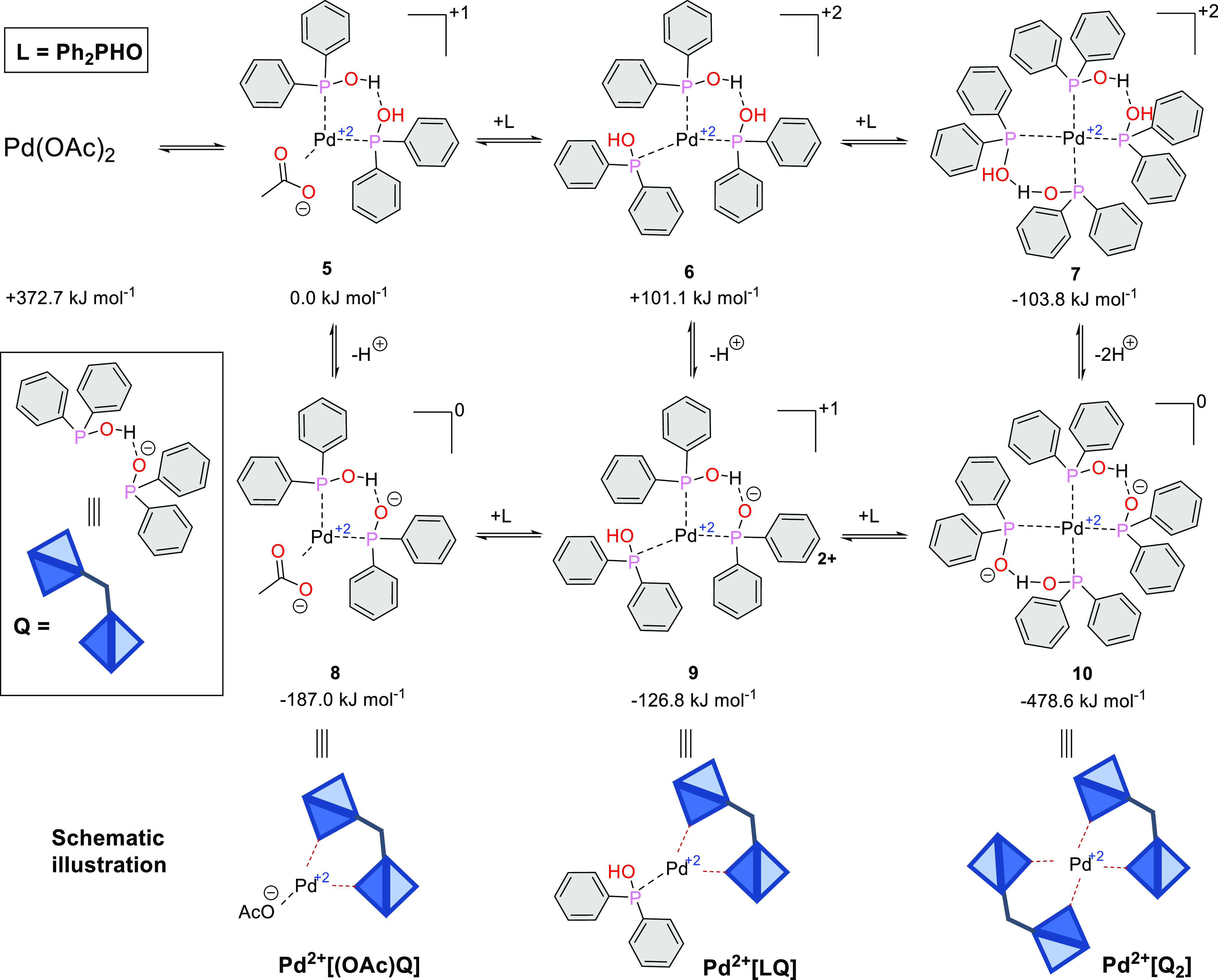
Equilibrium system of complexes formed from Pd2+ and Ph2P(O)H.
Then, we studied the catalytic cycle (Figure 3). In the first step, the boronic acid is deprotonated by the base present. Then, a weak π-complex (6) is formed between the Pd2+ center of the catalyst complex and the aryl ring of the boronic acid anion (ΔH = −11.6 kJ mol–1). The π-complex (11) is transformed to the σ-complex (13) via a well-determined TS (12) with a +117.9 kJ mol–1 enthalpy gap, which can be considered as a moderate barrier at elevated reaction temperature. The resulting σ-complex with an aryl anion (13) is of a high enthalpy content (106.2 kJ mol–1). The elimination of metaboric acid (HOBO) is favorable, but the entry of an additional Ph2POH ligand leading to complex 15 with an enthalpy of −7.3 kJ mol–1 is the real driving force. In the next step, the aryl anion is transferred from the central Pd2+ to the P atom of the adjacent ligand via TS 16 with a lower enthalpy content of 77.8 kJ mol–1 yielding the final tiphenylphosphine oxide (1a). During the reductive elimination step, the Pd2+ is converted to Pd0. Finally, the Pd0 is regenerated to Pd2+ by reaction with the O2 of air. Then, the catalytic cycle may start again. The enthalpy diagram for the P–C coupling under discussion can be seen in Figure 4. The suggested mechanism comprises two steps (involving transition states 12 and 16), which have impact on the overall reaction rate. As the first step leading to transition state 12 exhibits the higher enthalpy barrier, the route via species 12 is the rate-determining step. The overall reaction enthalpy follows a slightly exothermic run until the formation of the triphenylphosphine oxide (1a) that is connected with the elimination of Pd0 complex 17. However, the subsequent reoxidation of Pd0 complex 17 to Pd2+ catalyst 18 with the oxygen of air is rather exothermic, making the “quasi” catalytic process irreversible.
Figure 3.
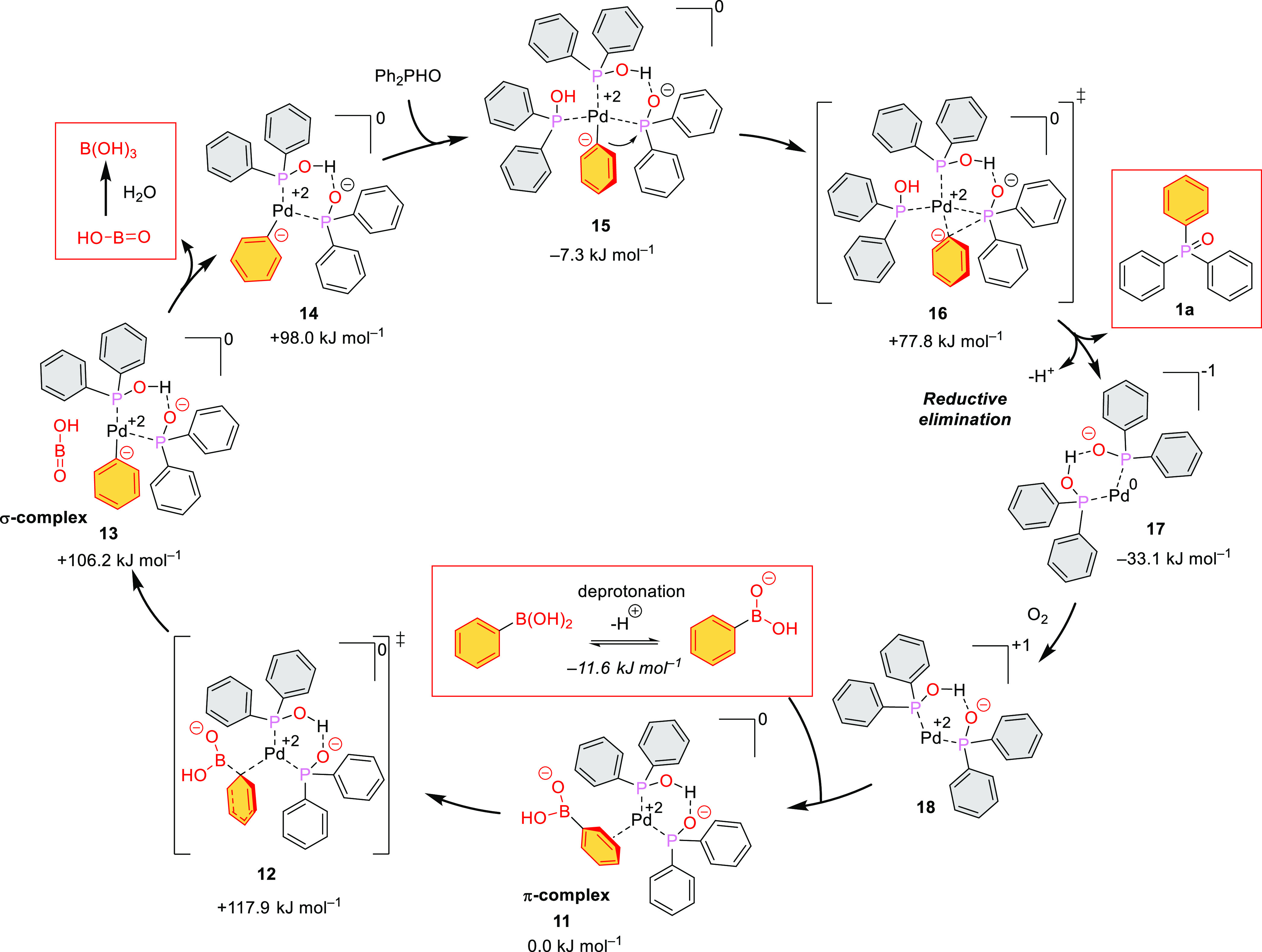
Formation of the desired triphenylphosphine oxide (1a) via a Pd-catalyzed reaction cycle.
Figure 4.

Enthalpy diagram for the Pd(OAc)2-catalyzed P–C coupling reaction of phenylboronic acid and diphenylphosphine oxide.
During the P–C coupling of Ph2P(O)H and arylboronic acids, a significant amount of triphenylphosphine oxide (1a) was also formed beside the expected aryl-diphenylphosphine oxide (2f–k) as discussed above (Table 3). In the case of 4-chlorophenyl- and 3-chlorophenylboronic acid, the dehalogenation by the Pd catalyst may also be a reasonable side reaction; however, for the 4-, 2- and 3-methylphenyl- and 4-fluorophenylboronic acid derivatives, the question arises how the triphenylphosphine oxide (1a) byproduct may be formed. Well, catalyst complex 18 may take up a Ph2POH ligand to give complex 9 (Figure 5). Migration of the phenyl ring from the Pd2+ to the adjacent P atom via transition state 19 leads to key intermediate 20, which then enters into the previously discussed catalytic process, yielding triphenylphosphine oxide (1a) without the involvement of the arylboronic acid. The activation enthalpy for the 9 → 20 transformation is ca 200 kJ mol–1 that is significantly higher than the activation barrier of the rate-determining step of the main reaction (117.9 kJ mol–1). For this, the side reaction remains a minor component. The other byproduct, Ph2P(O)OEt may be formed by the P–C coupling of Ph(OEt)(OH) (22) with Ph2P(O)H. Species 22 is the result of the interaction of intermediate 21 and ethanol.
Figure 5.

Proposed route for the formation of the triphenylphosphine oxide (1a) and ethyl diphenylphosphine oxide side products.
Conclusions
In summary, the less applied, halogene-free Hirao reaction of arylboronic acids and a series of >P(O)H reagents, such as diarylphosphine oxides, diethyl phosphite, and ethyl phenyl-H-phosphinate, was studied in detail. Another green aspect was that not traditional (cost-meaning and environment-burdening) mono- or bidante P-ligands were used to serve the P-ligand to the Pd(OAc)2 precursor (10%) but the excess (20%) of the >P(O)H reagent via its >P–OH trivalent tautomer form. MW assistance enhanced the P–C couplings that were optimized for the different substituted cases. Theoretical calculations supported that there is no oxidative addition step, as the primary Pd2+/>P–OH···–O–P< complex remains unchanged during the addition of the aryl anion and the addition of the tautomer form of the P-reagent. In the last reductive elimination step, the central Pd2+ ion of the catalyst is reduced to Pd0. For this, it is necessary to apply a stoichiometric oxidant, that is, in our case, air. The ligation of Pd2+ with two >P(O)H units that form a dimer-like associate enhances the reactivity of the central Pd2+ in complexation with the aryl anion. Formation of the typical byproducts, e.g., (EtO)Ph2PO and Ph3PO, the latter formed also by dechlorination, was also explained.
Experimental Section
General Information
The reactions were carried out in a CEM Discover Model SP (300 W) focused microwave reactor (CEM Microwave Technology Ltd., Buckingham, U.K.) equipped with a stirrer and a pressure controller using 80–100 W irradiation under isothermal conditions. The reaction mixtures were irradiated in sealed glass vessels (with a volume of 10 mL) available from the supplier of CEM. The reaction temperature was monitored by an external IR sensor.
The 31P, 13C, and 1H NMR spectra were taken on a Bruker Avance 300/Avance 500 spectrometer (Rheinstetten, Germany) operating at 121.5/202.4, 75.5/125.7, and 300/500 MHz, respectively, in CDCl3 solution. The 31P chemical shifts are downfield relative to H3PO4, while the 13C and 1H chemical shifts are downfield relative to TMS. The couplings are given in Hz. The exact mass measurements were performed using an Agilent 6545 Q-TOF mass spectrometer (Santa Clara, CA) in high-resolution, positive electrospray mode. The melting points of products 1a, 1b, 1c, 2f, 2g, 2h, 2i, and 3A were determined using a Setaram differential scanning calorimetry 92 device.
General Procedure for the P–C Coupling of Phenylboronic Acid and >P(O)H Reagents (Table 2, Entries 1, 3, 5, 8, and 12)
To a MW glass vessel were added 1 mL of ethanol, 0.038 mmol (0.0085 g) of Pd(OAc)2, 0.41 mmol (0.050 g) of phenylboronic acid, 0.49 mmol of diarylphosphine oxide [diphenylphosphine oxide: 0.10 g, bis(4-methylphenyl)phosphine oxide: 0.11 g, bis(3,5-dimethylphenyl)phosphine oxide: 0.13 g], 0.49 mmol (0.063 mL) of diethyl phosphite, or 0.49 mmol (0.074 mL) of ethyl phenyl phosphinate, and 0.45 mmol (0.15 g) of cesium carbonate. Then, the vial was closed and irradiated in the MW reactor isothermally at 135–150 °C for 0.5–2.5 h (Table 2, entries 1, 3, 5, 8, and 12). The reaction mixture was diluted with 3 mL of EtOH, filtrated, and the residue obtained after evaporation of the filtrate was filtered through a thin (2–3 cm) layer of silica gel using ethyl acetate as the eluant. The crude mixture was analyzed by 31P NMR spectroscopy. Then, if the sample was relevant, it was purified further by column chromatography (silica gel, ethyl acetate–hexane 1:1 as the eluant) to afford products 1a–e.
This procedure was repeated on a larger scale starting from 1.0 mmol (0.12 g) of phenylboronic acid, 1.2 mmol (0.24 g) of diphenylphosphine oxide, 1.1 mmol (0.36 g) of Cs2CO3, 0.10 mmol (0.022 g) of Pd(OAc)2 and 3.5 ml of ethanol. After an irradiation at 135 °C for 1.5 h, the work-up was similar as above to furnish 0.24 g (87%) of Ph3PO.
Following Compounds Were Thus Prepared
Triphenylphosphine Oxide (1a) (Table 2, Entry 1)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.087 g (83%) obtained as white crystals; mp. 156–157 °C, mp lit.25 156.6–157.4 °C; 31P{1H} NMR (121.5 MHz, CDCl3) δ 29.1, δP lit.25 (162 MHz, CDCl3) 29.5; 13C{1H} NMR (75.5 MHz, CDCl3) δ 132.7 (d, J = 103.8 Hz), 132.2 (d, J = 9.9 Hz,), 132.0 (d, J = 2.8 Hz), 128.6 (d, J = 12.1 Hz), δC lit.25 (100 MHz, CDCl3) 132.8 (d, J = 104.6 Hz), 132.5 (d, J = 9.9 Hz), 131.9 (d, J = 2.2 Hz), 128.4 (d, J = 12.1 Hz); 1H NMR (300 MHz, CDCl3) δ 7.72–7.59 (m, 6H), 7.56–7.48 (m, 3H), 7.48–7.38 (m, 6H), δH lit.25 (400 MHz, CDCl3) 7.70–7.64 (m, 6H), 7.56–7.52 (m, 3H), 7.48–7.43 (m, 6H); HRMS (m/z): calcd for C18H16OP [M + H]+, 279.0939; found, 279.0941.
Bis(4-methylphenyl)phenylphosphine Oxide (1b) (Table 2, Entry 3)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.090 g (78%) obtained as white crystals; mp. 76 °C, mp lit.11 78–79 °C; 31P{1H} NMR (121.5 MHz, CDCl3) δ 27.8, δP lit.26 (162 MHz, CDCl3) 30.5; 13C{1H} NMR (75.5 MHz, CDCl3) δ 142.4 (d, J = 2.8 Hz), 133.1 (d, J = 104.1 Hz), 132.1 (d, J = 10.3 Hz), 132.1 (d, J = 9.8 Hz), 131.8 (d, J = 2.7 Hz), 129.4 (d, J = 106.6 Hz), 129.3 (d, J = 12.5 Hz), 128.5 (d, J = 12.1 Hz), 21.6 (s), δC lit.26 (100 MHz, CDCl3) 142.6 (d, J = 2.9 Hz), 133.0 (d, J = 102.5 Hz), 132.2 (d, J = 10.2 Hz), 132.0 (d, J = 8.7 Hz), 131.9 (d, J = 3.2 Hz), 129.4 (d, J = 106.9 Hz), 129.4 (d, J = 12.6 Hz), 128.6 (d, J = 11.8 Hz), 21.7; 1H NMR (300 MHz, CDCl3) δ 7.73–7.61 (m, 2H), 7.61–7.47 (m, 5H), 7.47–7.37 (m, 2H), 7.32–7.18 (m, 4H), 2.39 (s, 6H); δH lit26 (400 MHz, CDCl3) 7.68–7.62 (m, 2H), 7.53 (dd, J1 = 11.8 Hz, J2 = 8.0 Hz, 4H), 7.48 (m, 1H), 7.24 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 4H), 2.38 (s, 6H); HRMS (m/z): calcd for C20H20OP [M + H]+, 307.1252; found, 307.1252.
Bis(3,5-dimethylphenyl)phenylphosphine Oxide (1c) (Table 2, Entry 5)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.094 g (74%) obtained as white crystals; mp. 159 °C, mp. lit.26 158.6–159.2 °C; 31P{1H} NMR (121.5 MHz, CDCl3): δ 29.6, δP lit.26 (162 MHz, CDCl3) 30.9; 13C{1H} NMR (75.5 MHz, CDCl3) δ 138.1 (d, J = 12.7 Hz), 133.7 (d, J = 2.8 Hz), 133.1 (d, J = 103.1 Hz), 132.4 (d, J = 105.3 Hz), 132.1 (d, J = 9.9 Hz), 131.7, 129.7 (d, J = 9.8 Hz), 128.4 (d, J = 12.0 Hz), 21.4 (s), δC lit.26 (100 MHz, CDCl3) 138.3 (d, J = 12.2 Hz), 133.9 (d, J = 2.3 Hz), 133.1 (d, J = 102.7 Hz), 132.4 (d, J = 102.6 Hz), 132.3 (d, J = 9.7 Hz), 131.9 (d, J = 2.2 Hz), 129.8 (d, J = 10.0 Hz), 128.6 (d, J = 11.7 Hz), 21.56; 1H NMR (300 MHz, CDCl3) δ 7.73–7.62 (m, 2H), 7.55–7.39 (m, 3H), 7.28 (d, J = 12.2 Hz, 4H), 7.15 (s, 2H), 2.31 (s, 12H), δH lit.26 (400 MHz, CDCl3) 7.68–7.63 (m, 2H), 7.55–7.51 (m, 1H), 7.47–7.42 (m, 2H), 7.26 (d, J = 12.4 Hz, 4H), 7.15 (s, 2H), 2.31 (s, 12H); HRMS (m/z): calcd for C22H24OP [M + H]+, 335.1565; found, 335.1566.
Diethyl Phenylphosphonate (1d) (Table 2, Entry 8)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.068 g (84%) obtained as colorless oil; 31P{1H} NMR (121.5 MHz, CDCl3) δ 18.9, δP lit.9 (162 MHz, CDCl3) 18.8; 13C{1H} NMR (75.5 MHz, CDCl3) δ 132.5 (d, J = 3.0 Hz), 131.9 (d, J = 9.8 Hz), 128.6 (d, J = 15.0 Hz), 128.5 (d, J = 188.0 Hz), 62.2 (d, J = 5.4 Hz), 16.4 (d, J = 6.5 Hz), δC lit.9 (100 MHz, CDCl3) 132.3 (d, J = 2.7 Hz), 131.7 (d, J = 9.2 Hz), 128.44 (d, J = 15.2 Hz), 128.41 (d, J = 187.6 Hz), 62.0 (d, J = 5.9 Hz), 16.3 (d, J = 6.5 Hz); 1H NMR (300 MHz, CDCl3) δ 7.84–7.71 (m, 2H), 7.56–7.37 (m, 3H), 4.21–3.97 (m, 4H), 1.29 (t, J = 7.1 Hz, 6H), δH lit.9 (400 MHz, CDCl3) 7.82 (m, 2H), 7.55 (∼tq, J1 = 7.5 Hz, J2 = 1.4 Hz, 1H), 7.47 (m, 2H), 4.12 (m, 4H), 1.32 (td, J1= 7.0 Hz, J2 = 0.5 Hz, 6H); HRMS (m/z): calcd for C10H16O3P [M + H]+, 215.0837; found, 215.0835.
Diphenyl Ethylphosphinate (1e) (Table 2, Entry 12)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.080 g (86%) obtained as colorless oil; 31P{1H} NMR (121.5 MHz, CDCl3) δ 32.2, δP lit.27 (120 MHz, CDCl3) 30.8; 13C{1H} NMR (75.5 MHz, CDCl3) δ 132.0 (d, J = 2.8 Hz), 131.7 (d, J = 137.0 Hz), 131.6 (d, J = 10.1 Hz), 128.4 (d, J = 13.1 Hz), 61.1 (d, J = 5.9 Hz), 16.5 (d, J = 6.6 Hz), δC lit.27 (75 MHz, CDCl3) 139.9 (d, J = 11.1 Hz), 133.7 (d, J = 143.4 Hz), 130.9 (d, J = 12.9 Hz), 127.0, 59.0, 16.7; 1H NMR (300 MHz, CDCl3) δ 7.88–7.75 (m, 4H), 7.58–7.39 (m, 6H), 4.16–4.01 (m, 2H), 1.36 (t, J = 7.1 Hz, 3H), δH lit.27 (300 MHz, CDCl3) 7.70–7.63 (m, 4H), 7.43–7.31 (m, 6H), 4.16–4.09 (m, 2H), 1.30 (t, J = 7.3 Hz, 3H); HRMS (m/z): calcd for C14H16O2P [M + H]+, 247.0888; found, 247.0889.
General Procedure for the P–C Coupling of Arylboronic Acids and Diphenylphosphine Oxide or Diethyl Phosphite (Table 3, Entries 2–6 and 10 and Table 4, Entries 2–7)
To 0.038 mmol (0.0085 g) of Pd(OAc)2 in 1 mL of ethanol were added 0.41 mmol of the arlylboronic acid [4-, 2-, or 3-methylphenylboronic acid: 0.056 g, 4-fluorophenylboronic acid: 0.057 g, 4- or 3-chlorophenylboronic acid: 0.064 g], 0.49 mmol (0.10 g) of diphenylphosphine oxide or 0.49 mmol (0.063 mL) of diethyl phosphite, and 0.45 mmol (0.15 g) of cesium carbonate. Then, the resulting mixture was irradiated in a closed vial in the MW reactor isothermally at 135 or 150 °C for 1.5 or 0.5 h (Table 3, entries 2–6 and 10 and Table 4). The reaction mixture was diluted with 3 mL of EtOH, filtrated, and the residue obtained after evaporation of the filtrate was filtered through a thin (2–3 cm) layer of silica gel using ethyl acetate as the eluant. The crude mixture obtained was analyzed by 31P NMR spectroscopy. Then, if the sample was relevant, it was purified further by column chromatography (silica gel, ethyl acetate–hexane 1:1 as the eluant) to afford products 2f–4k.
Following Compounds Were Thus Prepared
(4-Methylphenyl)diphenylphosphine Oxide (2f) (Table 3, Entry 2)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.081 g (73%) obtained as a white solid; mp 118–119 °C, mp. lit.13 129.5–130.2 °C; 31P{1H} NMR (121.5 MHz, CDCl3) δ 29.3, δP lit.13 (162 MHz, CDCl3) 29.1; 13C{1H} NMR (125.7 MHz, CDCl3) δ 142.4 (d, J = 2.8 Hz), 132.8 (d, J = 105.9 Hz), 132.1 (d, J = 10.2 Hz), 132.0 (d, J = 9.9 Hz), 131.8 (d, J = 2.7 Hz), 129.2 (d, J = 12.6 Hz), 129.1 (d, J = 106.4 Hz), 128.4 (d, J = 12.1 Hz), 21.6 (s), δC lit.13 (100 MHz, CDCl3) 142.5 (d, J = 2.6 Hz), 132.9 (d, J = 104.1 Hz), 132.2 (d, J = 10.2 Hz), 132.1 (d, J = 10.0 Hz), 131.9 (d, J = 2.8 Hz), 129.3 (d, J = 12.5 Hz), 129.2 (d, J = 106.4 Hz), 128.5 (d, J = 11.9 Hz), 21.6; 1H NMR (500 MHz, CDCl3) δ 7.80–7.40 (m, 12H), 7.29–7.11 (m, 2H), 2.40 (s, 3H), δH lit.13 (400 MHz, CDCl3) 7.67–7.62 (m, 4H), 7.56–7.48 (m, 4H), 7.44–7.40 (m, 4H), 7.26–7.23 (m, 2H), 2.37 (s, 3H); HRMS (m/z): calcd for C19H18OP [M + H]+, 293.1095; found, 293.1101.
(2-Methylphenyl)diphenylphosphine Oxide (2g) (Table 3, Entry 3)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.074 g (67%) obtained as white crystals; mp. 120–121 °C, mp. lit.28 121.5–122.9 °C; 31P{1H} NMR (202.4 MHz, CDCl3) δ 31.8, δP lit.28 (202 MHz, CDCl3) 31.8; 13C{1H} NMR (125.7 MHz, CDCl3) δ 143.4 (d, J = 8.0 Hz), 133.5 (d, J = 12.9 Hz), 132.8 (d, J = 103.6 Hz), 132.1 (d, J = 2.6 Hz), 132.0 (d), 131.8 (d, J = 2.7 Hz), 130.8 (d, J = 103.2 Hz), 128.6 (d, J = 12.0 Hz), 125.2 (d, J = 12.8 Hz), 21.7 (d, J = 4.7 Hz), δC lit.28 (125 MHz, CDCl3) 143.2 (d, J = 8.2 Hz), 133.4 (d, J = 12.4 Hz), 132.6 (d, J = 102.6 Hz), 132.0 (d, J = 2.7 Hz), 131.8 (d, J = 10.0 Hz), 131.7 (d, J = 2.7 Hz), 130.7 (d, J = 103.5 Hz), 128.5 (d, J = 12.7 Hz), 125.1 (d, J = 12.7 Hz), 21.6 (d, J = 5.4 Hz); 1H NMR (500 MHz, CDCl3) δ 7.66 (m, 4H), 7.55 (tq, J1 = 7.3 Hz, J2 = 1.6 Hz, 2H), 7.47 (td, J1 = 7.7 Hz, J2 = 2.8 Hz, 4H), 7.42 (tt, J1 = 7.6 Hz, J2 = 1.6 Hz, 1H), 7.28 (dd, J1 = 7.6 Hz, J2 = 4.1 Hz, 1H), 7.13 (td, J1 = 7.6 Hz, J2 = 2.9 Hz, 1H), 7.03 (ddd, J1 = 14.1 Hz, J2 = 7.8 Hz, J3 = 1.1 Hz, 1H), 2.45 (s, 3H), δH lit.28 (500 MHz, CDCl3) 7.68–7.62 (m, 4H), 7.57–7.52 (m, 2H), 7.49–7.45 (m, 4H), 7.44–7.39 (m, 1H), 7.30–7.26 (m, 1H), 7.15–7.10 (m, 1H), 7.05–7.00 (m, 1H), 2.45 (s, 3H); HRMS (m/z): calcd for C19H18OP [M + H]+, 293.1095; found, 293.1094.
(3-Methylphenyl)diphenylphosphine Oxide (2h) (Table 3, Entry 4)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.089 g (80%) obtained as a white solid; mp. 122–123 °C, mp. lit.26 123.7–124.2 °C; 31P{1H} NMR (202.4 MHz, CDCl3) δ 29.3, δP lit.29 (162 MHz, CDCl3) 29.5; 13C{1H} NMR (125.7 MHz, CDCl3) δ 138.5 (d, J = 12.1 Hz), 132.8 (d, J = 3.0 Hz), 132.7 (d, J = 104.0 Hz), 132.5 (d, J = 9.5 Hz), 132.3 (d, J = 96.7 Hz), 132.1 (d, J = 9.9 Hz), 131.9 (d, J = 2.6 Hz), 129.2 (d, J = 10.2 Hz), 128.5 (d, J = 12.0 Hz), 128.3 (d, J = 12.9 Hz), 21.4 (s), δC lit.29 (100 MHz, CDCl3) 138.4 (d, J = 15.9 Hz), 133.1, 132.8 (d, J = 2.4 Hz), 132.5 (d, J = 9.5 Hz), 132.2 (d, J = 103.4 Hz), 132.0 (d, J = 9.8 Hz), 131.8 (d, J = 2.5 Hz), 129.2 (d, J = 10.2 Hz), 128.5 (d, J = 12.1 Hz), 128.2, 21.5; 1H NMR (500 MHz, CDCl3) δ 7.72–7.67 (m, 4H), 7.61–7.55 (m, 3H), 7.50–7.47 (m, 4H), 7.42–7.34 (m, 3H), 2.39 (s, 3H), δH lit.29 (400 MHz, CDCl3) 7.64 (dd, J1 = 11.6 Hz, J2 = 7.6 Hz, 4H), 7.56–7.49 (m, 3H), 7.44–7.43 (m, 4H), 7.32 (m, 3H), 2.33 (s, 3H); HRMS (m/z): calcd for C19H18OP [M + H]+, 293.1095; found, 293.1097.
(4-Fluorophenyl)diphenylphosphine Oxide (2i) (Table 3, Entry 5)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.079 g (70%) obtained as pale yellow crystals; mp. 134–135 °C, mp.30 134–135 °C; 31P{1H} NMR (121.5 MHz, CDCl3) δ 28.5, δP lit.23 (162 MHz, CDCl3) 28.3; 13C{1H} NMR (125.7 MHz, CDCl3) δ 165.0 (dd, J1 = 3.2 Hz, J2 = 253.6 Hz), 134.5 (dd, J1 = 11.3 Hz, J2 = 8.8 Hz), 132.3 (d, J = 105.0 Hz), ∼132.1 (d, J ∼ 3.0 Hz), 132.0 (d, J = 12.2 Hz), 128.53 (d, J = 12.2 Hz), 128.52 (dd, J1 = 106.5 Hz, J2 = 3.4 Hz), 115.8 (dd, J1 = 13.2 Hz, J2 = 21.4 Hz), δC lit.23 (100 MHz, CDCl3) 165.0 (dd, J1 = 254.2 Hz, J2 = 3.2 Hz), 134.6 (dd, J1 = 11.3 Hz, J2 = 8.9 Hz), 132.5 (d, J = 105.2 Hz), 132.1 (d, J = 4.1 Hz), 132.0, 128.7 (dd, J1 = 106.6 Hz, J2 = 3.1 Hz), 128.6 (d, J = 12.5 Hz), 115.9 (dd, J1 = 22.6 Hz, J2 = 13.3 Hz); 1H NMR (500 MHz, CDCl3) δ 7.75–7.37 (m, 12H), 7.20–7.06 (m, 2H), δH lit.23 (400 MHz, CDCl3) 7.67–7.60 (m, 6H), 7.53–7.49 (m, 2H), 7.44–7.41 (m, 4H), 7.11 (t, J = 8.5 Hz, 2H); HRMS (m/z): calcd for C18H15FOP [M + H]+, 297.0845; found, 297.0850.
1,4-Phenylenebis(diphenylphosphine oxide) (3A) (Table 3, Entry 6)
In this case, dichloromethane–methanol 97:3 was used as the eluant. Yield: 0.021 g (23%) obtained as white crystals; mp. 300–301 °C, mp. lit.25 >300 °C; 31P{1H} NMR (202.4 MHz, CDCl3) δ 28.6, δP lit.25 (162 MHz, CDCl3) 30.5; 13C{1H} NMR (125.7 MHz, CDCl3) δ 137.0 (dd, J1 = 100.6 Hz, J2 = 2.7 Hz), 132.3, 132.06 (d, J = 10.1 Hz), 132.06, 131.7 (d, J = 105.2 Hz), 128.7 (d, J1 = 12.8 Hz), δC lit.25 (100 MHz, CDCl3) 136.9 (d, J = 100.2 Hz), 132.3, 132.0 (d, J = 13.9 Hz), 131.9, 131.0, 128.7 (d, J = 12.2 Hz); 1H NMR (500 MHz, CDCl3) δ 7.76 (m, 4H), 7.66 (ddd, J1 = 12.1 Hz, J2 = 8.4 Hz, J3 = 1.3 Hz, 8H), 7.57 (tq, J1 = 7.4 Hz, J2 = 1.5 Hz, 4H), 7.48 (td, J1 = 7.8 Hz, J2 = 2.8 Hz, 8H), δH lit.25 (400 MHz, CDCl3) 7.77–7.74 (m, 4H), 7.69–7.64 (m, 8H), 7.58–7.55 (m, 4H), 7.50–7.46 (m, 8H); HRMS (m/z): calcd for C30H25O2P2 [M + H]+, 479.1330; found, 479.1320.
1,3-Phenylenebis(diphenylphosphine oxide) (3B) (Table 3, Entry 10)
In this instance, dichloromethane–methanol 97:3 was used as the eluant. Yield: 0.023 g (25%) obtained as colorless oil; 31P{1H} NMR (202.4 MHz, CDCl3) δ 28.5, δP lit.25 (162 MHz, CDCl3) 30.5; 13C{1H} NMR (125.7 MHz, CDCl3) δ 135.5 (dd, J1 = 10.1 Hz, J2 = 3.1 Hz), 135.4 (t, J = 11.2 Hz), 133.6 (dd, J1 = 101.8 Hz, J2 = 10.7 Hz), 132.2 (d, J = 2.3 Hz), 132.0 (d, J = 10.2 Hz), 131.7 (d, J = 105.1 Hz), 129.0 (t, J = 11.3 Hz), 128.6 (d, J = 12.5 Hz), δC lit.25 (100 MHz, CDCl3) 135.4–135.2 (m, 2C), 135.1, 133.5 (dd, J1 = 101.7 Hz, J2 = 10.7 Hz), 132.0, 131.8 (d, J = 10.3 Hz), 131.5 (d, J = 104.9 Hz), 128.8 (t, J = 11.2 Hz), 128.4 (d, J = 12.6 Hz), 127.1; 1H NMR (500 MHz, CDCl3) δ 7.96 (ddm, J1 = 12.5 Hz, J2 = 7.7 Hz, 2H), 7.69 (tt, J1 = 11.7 Hz, J2 = 1.5 Hz, 1H), 7.62 (tt, J1 = 7.7 Hz, J2 = 2.5 Hz, 1H), 7.58 (dd, J1 = 12.1 Hz, J2 = 7.9 Hz, 8H), 7.53 (t, J = 7.4 Hz, 4H), 7.41 (td, J1 = 7.7 Hz, J2 = 2.8 Hz, 8H), δH lit.25 (400 MHz, CDCl3) 7.98–7.93 (m, 2H), 7.71 (t, J = 11.7 Hz, 1H), 7.63–7.50 (m, 13H), 7.43–7.38 (m, 8H); HRMS (m/z): calcd for C30H25O2P2 [M + H]+, 479.1330; found, 479.1323.
Diethyl (4-Methylphenyl)phosphonate (4f) (Table 4, Entry 2)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.067 g (77%) obtained as colorless oil; 31P{1H} NMR (121.5 MHz, CDCl3) δ 20.5, δP lit.17 (162 MHz, CDCl3) 19.5; 13C{1H} NMR (75.5 MHz, CDCl3) δ 142.8 (d, J = 3.1 Hz), 131.7 (d, J = 10.3 Hz), 129.1 (d, J = 15.4 Hz), 125.0 (d, J = 190.1 Hz), 61.9 (d, J = 5.3 Hz), 21.6 (d, J = 1.15 Hz), 16.2 (d, J = 6.5 Hz), δC lit.17 (100 MHz, CDCl3) 142.9 (d, J = 3.3 Hz), 131.8 (d, J = 10.4 Hz), 129.3 (d, J = 15.4 Hz), 125.0 (d, J = 190.2 Hz), 61.9 (d, J = 5.3 Hz), 21.6, 16.3 (d, J = 6.3 Hz); 1H NMR (300 MHz, CDCl3) δ 7.75–7.63 (m, 2H), 7.25–7.20 (m, 2H), 4.19–3.94 (m, 4H), 2.37 (s, 3H), 1.28 (t, J = 7.1 Hz, 6H), δH lit.17 (400 MHz, CDCl3) 7.63 (dd, J1 = 12.9 Hz, J2 = 7.8 Hz, 2H), 7.24–7.16 (m, 2H), 4.12–3.93 (m, 4H), 2.33 (s, 3H), 1.24 (t, J = 6.9 Hz, 6H); HRMS (m/z): calcd for C11H18O3P [M + H]+, 229.0994 found 229.0996.
Diethyl (2-Methylphenyl)phosphonate (4g) (Table 4, Entry 3)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.067 g (78%) obtained as colorless oil; 31P{1H} NMR (202.4 MHz, CDCl3) δ 19.5, δP lit.17 (162 MHz, CDCl3) 19.4; 13C{1H} NMR (125.7 MHz, CDCl3) δ 141.8 (d, J = 10.1 Hz), 133.9 (d, J = 10.4 Hz), 132.4 (d, J = 2.8 Hz), 131.2 (d, J = 15.0 Hz), 126.8 (d, J = 184.0 Hz), 125.4 (d, J = 14.8 Hz), 61.9 (d, J = 5.5 Hz), 21.2 (d, J = 3.7 Hz), 16.3 (d, J = 6.6 Hz), δC lit.17 (100 MHz, CDCl3) 141.8 (d, J = 10.4 Hz), 133.9 (d, J = 10.3 Hz), 132.4 (d, J = 2.9 Hz), 131.1 (d, J = 15.5 Hz), 127.0 (d, J = 184.4 Hz), 125.3 (d, J = 14.9 Hz), 61.8 (d, J = 5.6 Hz), 21.2 (d, J = 3.7 Hz), 16.3 (d, J = 6.6 Hz); 1H NMR (500 MHz, CDCl3) δ 7.96–7.90 (m, 1H), 7.47–7.43 (m, 1H), 7.32–7.25 (m, 2H), 4.24–4.05 (m, 4H), 2.60 (s, 3H), 1.35 (t, J = 7.1 Hz, 6H), δH lit.17 (400 MHz, CDCl3) 7.95–7.88 (m, 1H), 7.45–7.40 (m, 1H), 7.29–7.23 (m, 2H), 4.20–4.03 (m, 4H), 2.58 (s, 3H), 1.33 (t, J = 7.0 Hz, 6H); HRMS (m/z): calcd for C11H18O3P [M + H]+, 229.0994; found, 229.0993.
Diethyl (3-Methylphenyl)phosphonate (4h) (Table 4, Entry 4)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.069 g (80%) obtained as colorless oil; 31P{1H} NMR (202.4 MHz, CDCl3) δ 19.5, δP lit.17 (162 MHz, CDCl3) 19.2; 13C{1H} NMR (125.7 MHz, CDCl3) δ 138.3 (d, J = 14.9 Hz), 133.2 (d, J = 3.1 Hz), 132.3 (d, J = 10.0 Hz), 128.8 (d, J = 9.6 Hz), 128.4 (d, J = 15.8 Hz), 128.1 (d, J = 187.6 Hz), 62.1 (d, J = 5.4 Hz), 21.3 (s), 16.3 (d, J = 6.5 Hz), δC lit.17 (100 MHz, CDCl3) 138.2 (d, J = 14.8 Hz), 133.1 (d, J = 3.0 Hz), 132.2 (d, J = 9.7 Hz), 128.7 (d, J = 9.7 Hz), 128.3 (d, J = 15.4 Hz), 128.0 (d, J = 187.0 Hz), 61.9 (d, J = 5.2 Hz), 21.2, 16.3 (d, J = 6.2 Hz); 1H NMR (500 MHz, CDCl3) δ 7.67–7.57 (m, 2H), 7.38–7.34 (m, 2H), 4.19–4.03 (m, 4H), 2.40 (s, 3H), 1.33 (t, J = 7.1 Hz, 6H), δH lit.17 (400 MHz, CDCl3) 7.63–7.52 (m, 2H), 7.33–7.27 (m, 2H), 4.16–3.97 (m, 4H), 2.34 (s, 3H), 1.27 (t, J = 7.1 Hz, 6H); HRMS (m/z): calcd for C11H18O3P [M + H]+, 229.0994; found, 229.0992.
Diethyl (4-Fluorophenyl)phosphonate (4i) (Table 4, Entry 5)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.068 g (77%) obtained as colorless oil; 31P{1H} NMR (121.5 MHz, CDCl3) δ 18.7, δP lit.17 (162 MHz, CDCl3) 17.7; 13C{1H} NMR (75.5 MHz, CDCl3) δ 165.2 (dd, J1 = 3.9 Hz, J2 = 253.4 Hz), 134.2 (dd, J1 = 11.2 Hz, J2 = 8.9 Hz), 124.4 (dd, J1 = 192.7 Hz, J2 = 3.4 Hz), 115.6 (dd, J1 = 16.3 Hz, J2 = 21.4 Hz), 62.0 (d, J = 5.4 Hz), 16.2 (d, J = 6.4 Hz), δC lit.17 (100 MHz, CDCl3) 165.3 (dd, J1 = 253.1 Hz, J2 = 4.1 Hz), 134.5 (dd, J1 = 11.2 Hz, J2 = 8.8 Hz), 124.4 (d, J = 192.7 Hz), 115.8 (dd, J1 = 22.1 Hz, J2 = 16.2 Hz), 62.1 (d, J = 5.4 Hz), 16.3 (d, J = 6.2 Hz); 1H NMR (300 MHz, CDCl3) δ 7.81–7.70 (m, 2H), 7.11–7.00 (m, 2H), 4.15–3.93 (m, 4H), 1.32–1.15 (m, 6H), δH lit.17 (400 MHz, CDCl3) 7.80 (ddd, J1 = 14.0 Hz, J2 = 7.4 Hz, J3 = 6.9 Hz, 2H), 7.17–7.09 (m, 2H), 4.19–4.00 (m, 4H), 1.30 (t, J = 7.2 Hz, 6H); HRMS (m/z): calcd for C10H15FO3P [M + H]+, 233.0743; found, 233.0742.
Diethyl (4-Chlorophenyl)phosphonate (4j) (Table 4, Entry 6)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.062 g (66%) obtained as colorless oil; 31P{1H} NMR (121.5 MHz, CDCl3) δ 18.5, δP lit.31 (162 MHz, CDCl3) 18.1; 13C{1H} NMR (75.5 MHz, CDCl3) δ 139.0 (d, J = 4.1 Hz), 133.2 (d, J = 10.7 Hz), 128.9 (d, J = 15.6 Hz), 127.1 (d, J = 190.9 Hz), 62.3 (d, J = 5.5 Hz), 16.4 (d, J = 6.4 Hz), δC lit.31 (62.5 MHz, CDCl3) 138.5, 134.5, 129.2, 123.3, 58.0, 16.8; 1H NMR (300 MHz, CDCl3) δ 7.81–7.65 (m, 2H), 7.51–7.40 (m, 2H), 4.21–4.00 (m, 4H), 1.32 (t, J = 7.0 Hz, 6H), δH lit.31 (250 MHz, CDCl3) 7.69–7.61 (m, 2H), 7.38–7.33 (m, 2H), 4.04–3.95 (m, 4H), 1.22 (t, 6H); HRMS (m/z): calcd for C10H15ClO3P [M + H]+, 249.0447; found, 249.0446.
Diethyl (3-Chlorophenyl)phosphonate (4k) (Table 4, Entry 7)
Eluant: ethyl acetate–hexane 1:1; Yield: 0.067 g (71%) obtained as colorless oil; 31P{1H} NMR (121.5 MHz, CDCl3) δ 17.4, δP lit.32 (202.4 MHz, CDCl3) 16.7; 13C{1H} NMR (125.7 MHz, CDCl3) δ 134.7 (d, J = 20.3 Hz), 132.4 (d, J = 3.0 Hz), 131.6 (d, J = 10.7 Hz), 130.7 (d, J = 187.9 Hz), 129.74 (d, J = 16.3 Hz), 129.69 (d, J = 9.2 Hz), 62.3 (d, J = 5.5 Hz), 16.2 (d, J = 6.4 Hz), δC lit.32 (125.7 MHz, CDCl3) 134.8 (d, J = 20.3 Hz), 132.5 (d, J = 3.0 Hz), 131.6 (d, J = 10.7 Hz), 130.7 (d, J = 188.2 Hz), 130.0 (d, J = 16.2 Hz), 129.8 (d, J = 9.1 Hz), 62.5 (d, J = 5.5 Hz), 16.3 (d, J = 6.4 Hz); 1H NMR (500 MHz, CDCl3) δ 7.81–7.59 (m, 2H), 7.51–7.43 (m, 1H), 7.40–7.31 (m, 1H), 4.20–3.96 (m, 4H), 1.30 (t, J = 7.1 Hz, 6H), δH lit.32 (500 MHz, CDCl3) 7.78–7.37 (m, 4H), 4.18–4.02 (m, 4H), 1.31 (t, J = 7.0 Hz, 6H); HRMS (m/z): calcd for C10H15ClO3P [M + H]+, 249.0447; found, 249.0448.
Computational Methods
All computations were carried out with the Gaussian16 program package (G16),33 using standard convergence criteria for the gradients of the root-mean-square (RMS) force, maximum force, and RMS displacement and maximum displacement vectors (3.0 × 10–4, 4.5 × 10–4, 1.2 × 10–3, and 1.8 × 10–3). Computations were carried out at the M06-2X level of theory.34 The basis set of 6-31G(d,p) was applied for C, H, O, P, N, and B and SDD/MWB28 for Pd.35 The vibrational frequencies were computed at the same levels of theory, in order to confirm properly all structures as residing at minima on their potential energy hypersurfaces (PESs). Thermodynamic functions U, H, G, and S were computed at 398.15 K. Besides the vacuum calculations, the IEFPCM method was also applied to model the solvent effect, by using the default settings of G16, setting the ε = 24.852 (for ethanol).36 See the Supporting Information for details.
Acknowledgments
This project was supported by the National Research, Development and Innovation Office (K134318). The authors are grateful to Dr András Simon (Department of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Hungary) for his advice during NMR assignments. Z.M. is grateful for supports from BO/799 and ÚNKP-22-5-ME-3, and also indepted to Dr. Ervin Kovács for making available the ELKH Cloud under the project “Development and mechanistic study of DNA dyes”.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.3c01269.
31P, 13C, and 1H NMR spectra and data of the theoretical calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hirao T.; Masunaga T.; Ohshiro Y.; Agawa T. Stereoselective synthesis of vinylphosphonate. Tetrahedron Lett. 1980, 21, 3595–3598. 10.1016/0040-4039(80)80245-0. [DOI] [Google Scholar]
- Hirao T.; Masunaga T.; Yamada N.; Ohshiro Y.; Agawa T. Palladium-catalyzed new carbon-phosphorus bond formation. Bull. Chem. Soc. Jpn. 1982, 55, 909–913. 10.1246/bcsj.55.909. [DOI] [Google Scholar]
- Jablonkai E.; Keglevich G. P–C bond formation by coupling reaction utilizing >P(O)H species as the reagents. Curr. Org. Synth. 2014, 11, 429–453. 10.2174/15701794113109990066. [DOI] [Google Scholar]
- Jablonkai E.; Keglevich G. Advances and new variations of the Hirao reaction. Org. Prep. Proced. Int. 2014, 46, 281–316. 10.1080/00304948.2014.922376. [DOI] [Google Scholar]
- Henyecz R.; Keglevich G. New developments on the Hirao reactions, especially from”green” point of view. Curr. Org. Synth. 2019, 16, 523–545. 10.2174/1570179416666190415110834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalek M.; Stawinski J. Pd(0)-catalyzed phosphorus–carbon bond formation. mechanistic and synthetic studies on the role of the palladium sources and anionic additives. Organometallics 2007, 26, 5840–5847. 10.1021/om700797k. [DOI] [Google Scholar]
- Kalek M.; Jezowska M.; Stawinski J. Preparation of arylphosphonates by Pd(0)-catalyzed cross-coupling in the presence of acetate additives. Synthetic and mechanistic studies. Adv. Synth. Catal. 2009, 351, 3207–3216. 10.1002/adsc.200900590. [DOI] [Google Scholar]
- Kalek M.; Stawinski J. Palladium-catalyzed C–P bond formation: Mechanistic studies on the ligand substitution and the reductive elimination. An intramolecular catalysis by the acetate group in PdII complexes. Organometallics 2008, 27, 5876–5888. 10.1021/om800641n. [DOI] [Google Scholar]
- Kalek M.; Ziadi A.; Stawinski J. Microwave-assisted palladium-catalyzed cross-coupling of aryl and vinyl halides with H-phosphonate diesters. Org. Lett. 2008, 10, 4637–4640. 10.1021/ol801935r. [DOI] [PubMed] [Google Scholar]
- Villemin D.; Jaffrès P.-A.; Siméon F. Rapid and efficient phosphonation of aryl halides catalysed by palladium under microwaves irradiation. Phosphorus, Sulfur Silicon Relat. Elem. 1997, 130, 59–63. 10.1080/10426509708033697. [DOI] [Google Scholar]
- Jablonkai E.; Balázs L. B.; Keglevich G. A P-ligand-free nickel-catalyzed variation of the Hirao reaction under microwave conditions. Curr. Org. Chem. 2015, 19, 197–202. 10.2174/1385272819666150114235413. [DOI] [Google Scholar]
- Huszár B.; Mucsi Z.; Szolga R.; Keglevich G. New data on the Hirao reaction; The use of Cu(II) salts as the catalyst precursor under microwave irradiation in the absence of added P-ligands. J. Organomet. Chem. 2022, 982, 122526 10.1016/j.jorganchem.2022.122526. [DOI] [Google Scholar]
- Fu T.; Qiao H.; Peng Z.; Hu G.; Wu X.; Gao Y.; Zhao Y. Palladium-catalyzed air-based oxidative coupling of arylboronic acids with H-phosphine oxides leading to aryl phosphine oxides. Org. Biomol. Chem. 2014, 12, 2895–2902. 10.1039/c3ob42470g. [DOI] [PubMed] [Google Scholar]
- Andaloussi M.; Lindh J.; Savmarker J.; Sjöberg P. J. R.; Larhed M. Microwave-promoted palladium(II)-catalyzed C–P bond formation by using arylboronic acids or aryltrifluoroborates. Chem. - Eur. J. 2009, 15, 13069–13074. 10.1002/chem.200901473. [DOI] [PubMed] [Google Scholar]
- Geng Z.; Zhang Y.; Zheng L.; Li J.; Zou D.; Wu Y.; Wu Y. Pd-catalyzed C–P coupling of heteroaryl boronic acid with H-phosphonate diester. Tetrahedron Lett. 2016, 57, 3063–3066. 10.1016/j.tetlet.2016.05.038. [DOI] [Google Scholar]
- Hu G.; Chen W.; Fu T.; Peng Z.; Qiao H.; Gao Y.; Zhao Y. Nickel-catalyzed C–P cross-coupling of arylboronic acids with P(O)H compounds. Org. Lett. 2013, 15, 5362–5365. 10.1021/ol402672e. [DOI] [PubMed] [Google Scholar]
- Zhuang R.; Xu J.; Cai Z.; Tang G.; Fang M.; Zhao Y. Copper-catalyzed C–P bond construction via direct coupling of phenylboronic acids with H-phosphonate diesters. Org. Lett. 2011, 13, 2110–2113. 10.1021/ol200465z. [DOI] [PubMed] [Google Scholar]
- Wan H.; Zhao Y.; Wang Q.; Zhang Y.; Li Y. The Cu-catalyzed C–P coupling of phosphonate esters with arylboronic acids. Russ. J. Gen. Chem. 2016, 86, 150–153. 10.1134/S1070363216010230. [DOI] [Google Scholar]
- Jablonkai E.; Keglevich G. P-Ligand-free, microwave-assisted variation of the Hirao reaction under solvent-free conditions; the P–C coupling reaction of >P(O)H species and bromoarenes. Tetrahedron Lett. 2013, 54, 4185–4188. 10.1016/j.tetlet.2013.05.111. [DOI] [Google Scholar]
- Keglevich G.; Henyecz R.; Mucsi Z.; Kiss N. Z. The palladium acetate-catalyzed microwave-assisted Hirao reaction without an added phosphorus ligand as a “green” protocol: A quantum chemical study on the mechanism. Adv. Synth. Catal. 2017, 359, 4322–4331. 10.1002/adsc.201700895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henyecz R.; Mucsi Z.; Keglevich G. Palladium-catalyzed microwave-assisted Hirao reaction utilizing the excess of the diarylphosphine oxide reagent as the P-ligand; a study on the activity and formation of the “PdP2” catalyst. Pure Appl. Chem. 2019, 91, 121–134. 10.1515/pac-2018-1004. [DOI] [Google Scholar]
- Keglevich G.; Mucsi Z.. Interpretation of the Rate Enhancing Effect of Microwaves. In Microwave Chemistry; Cravotto G.; Carnaroglio D., Eds.; De Gruyter: Berlin, 2017; pp 53–64. [Google Scholar]
- Xu J.; Zhang P.; Gao Y.; Chen Y.; Tang G.; Zhao Y. Copper-catalyzed P-arylation via direct coupling of diaryliodonium salts with phosphorus nucleophiles at room temperature. J. Org. Chem. 2013, 78, 8176–8183. 10.1021/jo4012199. [DOI] [PubMed] [Google Scholar]
- Huszár B.; Varga P. R.; Szűcs N. Á.; Simon A.; Drahos L.; Keglevich G. Pd-catalyzed Hirao P–C coupling reactions with dihalogenobenzenes without the usual P-ligands under MW conditions. Catalysts 2022, 12, 1080 10.3390/catal12101080. [DOI] [Google Scholar]
- Zhang X.; Liu H.; Hu X.; Tang G.; Zhu J.; Zhao Y. Ni(II)/Zn catalyzed reductive coupling of aryl halides with diphenylphosphine oxide in water. Org. Lett. 2011, 13, 3478–3481. 10.1021/ol201141m. [DOI] [PubMed] [Google Scholar]
- Shen C.; Yang G.; Zhang W. Nickel-catalyzed C–P coupling of aryl mesylates and tosylates with H(O)PR1R2. Org. Biomol. Chem. 2012, 10, 3500–3505. 10.1039/c2ob25225b. [DOI] [PubMed] [Google Scholar]
- Olszewski T. K.; Boduszek B. Synthesis of new thiazole-2, -4, and -5-yl-(amino)methylphosphonates and phosphinates: Unprecedented cleavage of thiazole-2 derivatives under acidic conditions. Tetrahedron 2010, 66, 8661–8666. 10.1016/j.tet.2010.09.026. [DOI] [Google Scholar]
- Stankevič M.; Włodarczyk A. Efficient copper(I)-catalyzed coupling of secondary phosphine oxides with aryl halides. Tetrahedron 2013, 69, 73–81. 10.1016/j.tet.2012.10.064. [DOI] [Google Scholar]
- Chen Y-X.; Zhang S.; Xue Y-J.; Mo L-P.; Zhang Z-H. Palladium anchored on a covalent organic framework as a heterogeneous catalyst for phosphorylation of aryl bromides. Appl. Organomet. Chem. 2022, 36, e6480 10.1002/aoc.6480. [DOI] [Google Scholar]
- Rayshys J. W.; Taft R. W.; Sheppard W. A. Electronic properties of tri-, tetra-, and pentacoordinate phosphorus substitutents. J. Am. Chem. Soc. 1968, 90, 5236–5243. 10.1021/ja01021a030. [DOI] [Google Scholar]
- Iranpoor N.; Firouzabadi H.; Moghadam K. R.; Motavalli S. First reusable ligand-free palladium catalyzed C–P bond formation of aryl halides with trialkylphosphites in neat water. RSC Adv. 2014, 4, 55732–55737. 10.1039/C4RA07680J. [DOI] [Google Scholar]
- Dargó G.; Bölcskei A.; Grün A.; Béni S.; Szántó Z.; Lopata A.; Keglevich G.; Balogh G. T.; Balogh G. T. Proton dissociation properties of arylphosphonates: Determination of accurate Hammett equation parameters. J. Pharmaceut. Biomed. 2017, 143, 101–109. 10.1016/j.jpba.2017.05.038. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford CT, 2016.
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Cao X. Y.; Dolg M. Segmented contraction scheme for small-core lanthanide pseudopotential basis sets. J. Mol. Struct.: THEOCHEM 2002, 581, 139–147. 10.1016/S0166-1280(01)00751-5. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.