

The study reveals that knocking out c-JUN improves cardiomyocyte generation in humans by increasing chromatin accessibility and H3K4me3 modification on key genes regulating heart development. These findings shed new light on c-JUN's unexpected role in human cardiogenesis.
Abstract
Loss of c-JUN leads to early mouse embryonic death, possibly because of a failure to develop a normal cardiac system. How c-JUN regulates human cardiomyocyte cell fate remains unknown. Here, we used the in vitro differentiation of human pluripotent stem cells into cardiomyocytes to study the role of c-JUN. Surprisingly, the knockout of c-JUN improved cardiomyocyte generation, as determined by the number of TNNT2+ cells. ATAC-seq data showed that the c-JUN defect led to increased chromatin accessibility on critical regulatory elements related to cardiomyocyte development. ChIP-seq data showed that the knockout c-JUN increased RBBP5 and SETD1B expression, leading to improved H3K4me3 deposition on key genes that regulate cardiogenesis. The c-JUN KO phenotype could be copied using the histone demethylase inhibitor CPI-455, which also up-regulated H3K4me3 levels and increased cardiomyocyte generation. Single-cell RNA-seq data defined three cell branches, and knockout c-JUN activated more regulons that are related to cardiogenesis. In summary, our data demonstrated that c-JUN could regulate cardiomyocyte cell fate by modulating H3K4me3 modification and chromatin accessibility and shed light on how c-JUN regulates heart development in humans.
Introduction
Numerous discoveries have led to an understanding of the regulatory network that governs cardiomyocyte cell fate decisions (1, 2, 3, 4, 5). However, the process in humans is obscured during early embryogenesis and is not amenable to genetic manipulations to uncover the regulatory mechanisms behind cardiomyocyte generation. Human embryonic stem cells (hESC) and human induced pluripotent stem cells can be differentiated into cardiomyocytes in vitro, thus providing a model system for studying the gene network regulating cardiomyocyte fate decisions and cardiomyocyte regeneration (6).
During cardiomyocyte formation, there is a complex transcriptional regulatory system involving multiple transcription factors (TFs) that control the progression through the hESC-cardiac mesoderm-cardiomyocyte stages. Particularly critical are key TFs, such as the GATA family/MESP1/ISLET1(ISL1) families of TFs. GATA family TFs such as GATA4/6 play essential and evolutionary conserved roles in heart development by regulating early mesoderm formation and the mesoderm-to-cardiac fate transition (7). Activating MESP1 in hESC promotes the epithelial-mesenchymal transition to generate mesoderm progenitors and ultimately cardiovascular cells (8, 9). ISL1 is expressed at the dorsal pharyngeal mesoderm and helps specify the second heart field (10, 11). When combined, overexpression of the key TFs GATA4/MEF2C/TBX5 and MESP1/BAF60C could reprogram cardiac fibroblasts toward an induced cardiomyocyte fate by remodeling chromatin structure (12, 13, 14). These data demonstrate those TFs play an important role in cardiac mesoderm and cardiomyocyte generation. However, a comprehensive understanding of the regulatory network in cardiomyocyte generation and maturation requires further study.
c-JUN/AP-1 TFs are essential for mouse embryonic development. Knockout of Jun (c-Jun) in mice leads to embryonic lethality around E13.5 (15, 16). Suggesting a fundamental role for c-JUN in embryonic development. In the adult mouse, tissue-specific mutation of c-JUN leads to heart disease such as dilated cardiomyopathy (17). Recent studies have implicated the c-JUN/AP-1 family of TFs in heart development, as repressing c-JUN/AP-1 TFs reduced cardiomyocyte regeneration in zebrafish (18). Interestingly, (19) reported that c-JUN/AP-1 binding motifs are needed for injury responses and cardiac regeneration in zebrafish and killifish, but mammals have lost (or never gained) this regenerative capability (19). During the differentiation of hESC to endoderm, c-JUN occupies hESC enhancers in partnership with pluripotent TFs and impedes the decommissioning of ESC enhancers, thus inhibiting hESC differentiation (20). However, it is unclear how c-JUN/AP-1 family TFs regulate human cardiomyocyte development.
Here, using an established cardiomyocyte differentiation protocol (21, 22), we found that the knockout of c-JUN significantly improved TNNT2+ cell generation to >90% purity. Interestingly, when we studied the mechanism behind how c-JUN inhibited cardiomyocyte generation, we found that the knockout of c-JUN activated RBBP5 and SETD1B, two members of the SET histone methylase complex, and elevated H3K4me3 levels. When analyzing the genome-wide H3K4me3 modification, we found that key factors that regulate cardiogenesis were more rapidly activated in response to the increased H3K4me3 and increased chromatin accessibility when c-JUN was knocked out. The c-JUN knockout effect could be phenocopied using the small molecule CPI-455, a histone lysine demethylase (KDM5) inhibitor, which caused increased H3K4me3 levels and accelerated the differentiation of hESC into cardiac mesoderm. Finally, using single-cell RNA-sequencing, we found 13 kinds of cell types that appeared during the hESC-to-cardiomyocyte transition and constructed three trajectories for the differentiating cells. These fates ended in cardiomyocyte, endothelial, and epicardial cells. Comparing the trajectory of hESC to cardiomyocytes, we found more key regulators related to cardiogenesis in the c-JUN KO than WT differentiation, such as EOMES, GATA6, and HEY. Overall, these results indicate that c-JUN impairs cardiomyocyte development by modulating the deposition of H3K4me3 on key genes.
Results
Differentiation of hESC into cardiomyocytes
hESC can be differentiated in vitro into cardiomyocytes (21, 22, 23, 24). Using a high-efficiency cardiomyocyte differentiation protocol (21), by day 15, ∼70% of cells were TNNT2+ and showed spontaneous contractions (Fig 1A and B and Video 1). During cardiomyocyte differentiation, there are three major developmental stages: pluripotent (hESC, marker gene: POU5F1 [OCT4]), cardiac mesoderm (marker gene: MESP1), and cardiomyocyte stage (marker gene: TNNT2) (Fig 1A). We harvested cells at D1, 3, 5, 7, and 15, along with hESC as control, and processed them for RNA-sequencing (RNA-seq). Pluripotent genes such as OCT4/SOX2 were quickly down-regulated after the cardiac mesoderm stages, while the cardiac mesoderm genes TBXT (T)/MESP1/DKK1 were immediately up-regulated during the early stages. Mature Cardiomyocyte marker genes, such as ISL1/NKX2-5/TNNT2/MYH6/ACTN2, were activated during the late stage, suggesting functional cardiomyocyte formation (Fig 1C). Immunofluorescence results show D15 cells expressed α-ACTININ and TNNT2 and had sarcomere-like structures (Fig 1D). To gain mechanistic insight into the cardiomyocyte generation, we divided the time-course RNA-seq data into three stages (C1–C3). The 2506 genes in the C3 group that were down-regulated from D3 onward were related to DNA replication and cell division (Fig 1E and F). The 519 genes expressed in C2 (cardiac mesoderm) were related to somitogenesis and mesoderm formation (Fig 1E and F). Finally, the 2,586 genes in C1 activated during the late cardiomyocyte stages were identified as involved in the regulation of heart development and sarcomere organization (Fig 1E and F). These results demonstrate an efficient cardiomyocyte generation system that is suitable for further study of the function of c-JUN in cardiomyocyte development.
Figure 1. Induction of human embryonic stem cells (hESC) differentiated into cardiomyocytes.

(A) Schematic for cardiomyocyte generation from human embryonic stem cells. Below is the cell morphology at different stages; D, day. (B) Flow cytometry quantification of cardiomyocyte differentiation efficiency according to the GiWi method (21). (C) RNA-seq data showing the expression profile of representative genes during hESC-to-cardiomyocyte differentiation. Data were from two replicates and are shown as the mean. (D) Immunostaining showed protein expression of the cardiomyocyte marker genes TNNT2 and α-ACTININ. Scale bar, 5 μm. The white arrow point to a sarcomere. (E) Heatmap shows the difference expressed genes during cardiomyocyte generation. According to the expression profile, genes expressing from low to high (C1, 2586) and high in the middle stage (C2, 519) and from high to low (C3, 2506) were clustered together. (F) Gene ontology analysis shows the different biology functions of the genes in C1∼C3 category.
Movie showing spontaneous contractions of cardiomyocytes from WT hESC in D15. Download video (5.8MB, mp4)
c-JUN is a barrier during cardiomyocyte differentiation
c-JUN is required for mouse embryonic development, especially hepatocyte formation (15, 16). The role of c-JUN in cardiomyocyte differentiation remains unknown. Here, we found c-JUN was expressed throughout the process of hESC to cardiomyocyte differentiation (Fig 2A). To test whether c-JUN could affect cardiomyocyte differentiation, we deleted the c-JUN CDS in hESC with CRISPR/Cas9, generated two c-JUN KO (#2 and #10) clones, and confirmed the loss of c-JUN by Western blot (Fig 2B).
Figure 2. c-JUN is a barrier in cardiomyocyte differentiation.
(A) Bar plot shows c-JUN expression pattern during cardiomyocyte generation. Data were from two replicates and shown as the mean ± SEM; D, day. (B) Western blot for c-JUN in WT and c-JUN knockout hESC clones. (C) Morphology of cardiomyocytes differentiated from WT and c-JUN knockout hESC. D, day; Scale bars, 200 μm. (D) Flow cytometry analysis of TNNT2 expression in cardiomyocytes differentiated from hESC. Percent of cell TNNT2+ is indicated at each condition; WT, induce cardiomyocytes from WT hESC; c-JUN KO, induce cardiomyocytes from c-JUN knockout hESC; D, day. (D, E) Flow cytometry quantification of differentiation efficiency from (panel (D)). Knockout of c-JUN enhances the number of TNNT2+ cells on D15 of the cardiomyocyte differentiation. Data were from six biological replicates in three independent experiments and are shown as the mean ± SEM. *** P-value < 0.001, two-way ANOVA with Sidak correction between the WT control and c-JUN KO groups. (F) Flow cytometry quantification of cardiomyocyte differentiation efficiency with or without JNK inhibitor (SP600125, 2 μm). JNKi was added to the culture medium from D0 to D15. Data were from seven biological replicates in two independent experiments and are shown as the mean ± SEM. *** P-value < 0.001, two-way ANOVA with Sidak correction between the control (DMSO) and JNK inhibitor groups. (G) Immunostaining showing knockout c-JUN promotes expression of cardiomyocyte marker genes TNNT2 and α-ACTININ. Scale bar, 10 μm. The white box marks a sarcomere. (H) Counts of the number of sarcomeres in each image. Data were from four to six replicates and are shown as the mean ± SEM. *** P-value < 0.001, ** P-value < 0.01, two-way ANOVA with Sidak correction between the WT and c-JUN KO groups.
Source data are available for this figure.

We then differentiated the hESC into cardiomyocytes in vitro and surprisingly found that WT and c-JUN KO cells were morphologically different (Fig 2C). Interestingly, we observed that spontaneous contractions of c-JUN KO cardiomyocytes appeared to be increased compared with WT cells (Video 1 and Video 2). Next, we quantified the level of the cardiomyocyte marker TNNT2 and showed that c-JUN KO cells have a higher proportion of TNNT2+ cells than WT, ∼95% versus ∼74% (Fig 2D and E). To test whether c-JUN activation is a barrier during hESC-to-cardiomyocyte transition, we repressed c-JUN N-terminal kinase (JNK) activity by small molecular SP600125; the result showed that the inhibition of JNK also enhanced cardiomyocyte generation (Fig 2F). However, the DMSO appears to impair cardiomyocyte differentiation, as the number of TNNT2+ cells in the control group was lower compared with untreated cells (Fig 2E and F). On the contrary, overexpressing c-JUN could inhibit normal cardiomyocyte morphology and severely inhibit TNNT2+ cell generation (Fig S1A–C). Nonetheless, reduced c-JUN, either through the knockout or inhibition of JNK, led to increased numbers of TNNT2+ cells. Immunofluorescence supported these observations as both TNNT2 and α-ACTININ were lower in WT cells compared with c-JUN KO cells, and the c-JUN KO cells also had clearer sarcomere-like structures (Fig 2G and H). Together, these findings suggest that c-JUN behaves as a barrier to cardiomyocyte generation.
Figure S1. Overexpress c-JUN inhibits TNNT2+ cell differentiation.
(A) TetOn c-JUN plasmid was transferred into hESC and then selected the dox-induced c-JUN hESC with puromycin. c-JUN was induced with 1 μg/ml dox and compared with control hESC by Western blot. (B) Overexpress c-JUN inhibits normal cardiomyocyte morphology. (C) Overexpress c-JUN inhibits TNNT2+ cell generation. Data were from 7–11 biological replicates in three independent experiments and are shown as the mean ± SEM. *** P-value < 0.001, unpaired t test between the control and OE c-JUN groups.
Source data are available for this figure.

Movie showing spontaneous contractions of cardiomyocytes from c-JUN KO hESC in D15. Download video (8.7MB, mp4)
Chromatin dynamics during cardiogenesis
To understand how c-JUN restrains cardiomyocyte differentiation, we performed RNA-seq and ATAC-seq (25, 26) on c-JUN KO samples, including c-JUN KO hESC (D0), D1, D3, D5, D7, and D15 (Fig 3A). We clustered the open/closed loci into three groups based on the chromatin changes during differentiation (Fig 3B). The first group contains pluripotent genes such as OCT4 that is Open in the pluripotency stage and Closed in the later stage (OC) (Fig 3A). The second group contains genes such as MESP1, that is closed in the pluripotency stage and cardiomyocyte stage but Transiently Open (TO) in the cardiac mesoderm (Fig 3A). Third group contains mature cardiac markers, such as TNNT2, that are Closed in the early stage but Open at the final cardiomyocyte stage (CO) (Fig 3A). Using these definitions, a clear pattern of chromatin dynamics appeared during the hESC-to-cardiomyocyte transition. The largest group of chromatin changes was the OC group (38437 loci), followed by the CO (11618 loci), and finally the TO group (8356 loci) (Fig 3C). In agreement with our previous reports (27), the chromatin signature of the starting cells closed rapidly upon differentiation, while the target cell type chromatin structure was slower to emerge.
Figure 3. c-JUN regulates chromatin opening during cardiomyocyte differentiation.
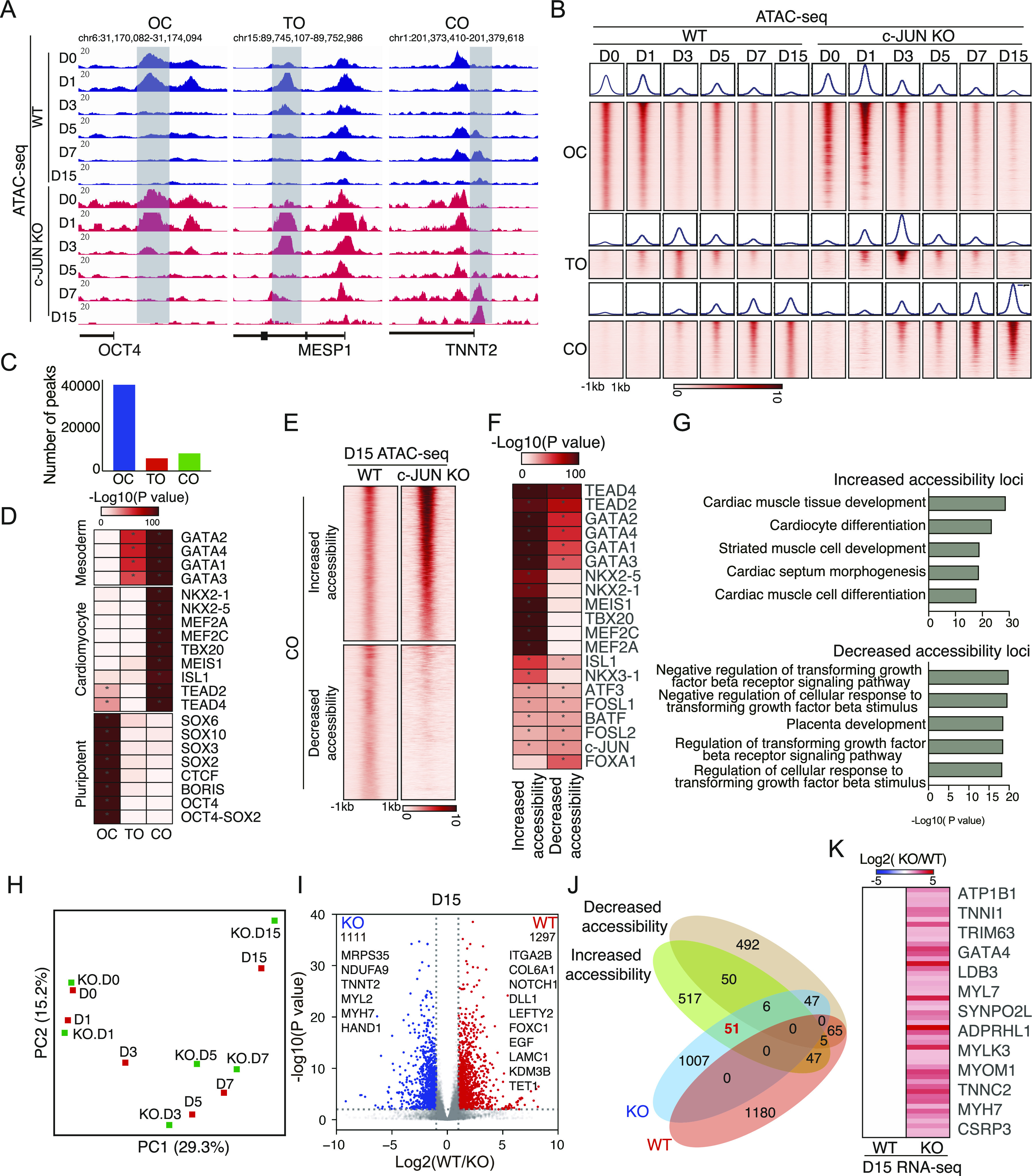
(A) Genome view of the ATAC-seq data for Open to Close (OC), Transition Open (TO), and Close to Open (CO), represented by the pluripotent gene OCT4, mesoderm gene MESP1, and cardiomyocyte marker gene TNNT2. (B) Heatmap and pileup showing the ATAC-seq chromatin accessibility for D0 to D15 cells at OC/TO/CO loci. The heatmap and pileups are centered on the ATAC-seq peaks center. (C) Number of peaks in each OC/TO/CO category. (D) TF motifs significantly enriched at least > 1.5-fold for OC/TO/CO categories of ATAC-seq peaks. * Indicates a P-value < 1e-20. (E) Heatmap showing knockout c-JUN promotes half of the CO loci more accessibility (Increased accessibility) and closed half of the CO loci (Decreased accessibility). (E, F) TF motifs were significantly enriched at least > 1.5-fold for Increased and Decreased loci as defined in panel (E). * Indicates a P-value < 1e-20. (G) Gene ontology (GO) analysis of the significantly enriched biological process terms related to Increased accessibility and Decreased accessibility loci. (H) Principal component analysis of the time courses of gene expression of cardiomyocytes generated from WT and c-JUN knockout hESC. (I) Volcano plot showing the differentially expressed genes on Day 15 (D15) cardiomyocytes generated from WT and c-JUN KO hESC. (I, J) Venn diagram showing the overlap of genes regulated by Increased and Decreased loci with differentially expressed genes (defined in (I)) in WT and c-JUN KO cells. (J, K) Heatmap showing the fold-change of gene expression of the 51 genes defined in panel (J).
Chromatin accessibility is at least partially driven by the activity of specific TFs, so we searched for the DNA-binding motifs of TFs that were enriched in the OC/TO/CO categories (28). As expected, the OC group was generally dominated by key pluripotency-related factors, such as SOX and OCT TFs. Motifs representing the GATA family of TFs were enriched in TO and CO groups, representing the emergence of the definitive mesoderm (Fig 3D). Finally, loci in the CO category were enriched for binding motifs for ISL1/NKX2-5/MEF2A/MEF2C TFs, which are reported to play a critical function in cardiogenesis (11, 12, 14, 29, 30, 31, 32) (Fig 3D). Overall, these results suggest that the specific chromatin signature of cardiomyocytes has been established.
c-JUN inhibits cardiogenesis chromatin open
Next, to study how c-JUN regulates cardiomyocyte differentiation, we compared the ATAC-seq data between WT and c-JUN KO conditions and found that the loss of c-JUN led to increased chromatin accessibility within the CO as early as in D3 (Fig 3A and B). The TO groups also opened earlier and were more intense (Fig 3B). To understand the impact of the loss of c-JUN on chromatin accessibility, we divided the CO peaks into increased accessibility and decreased accessibility groups based upon the difference between the WT and c-JUN KO at D15 (Fig 3E). DNA-binding motifs for key cardiogenesis TFs TEAD/GATA and MEF were enriched in the c-JUN KO increased accessibility CO loci (Fig 3F). Surprisingly, motifs for AP-1 TFs were enriched in both increased and decreased CO loci (Fig 3F). This suggests the AP-1 binding loci are more available for remodeling in the absence of c-JUN. Supporting an enhanced differentiation, gene ontology analysis showed that the increased accessibility of CO loci was related to cardiac muscle tissue development and cardiocyte differentiation (Fig 3G). And decreased CO loci were related to transforming growth factor signaling pathway and placenta development (Fig 3G).
Chromatin remodeling regulates and often precedes gene expression programs (27). Analysis of transcriptome dynamics by principal component analysis indicated a similar overall trajectory for the WT and KO cells (Fig 3H). However, the KO cells appeared to be accelerated, particularly around D3–D5 (Fig 3H). This agrees with the faster and more intense changes in chromatin accessibility seen in the ATAC-seq data (Fig 3B). Differentially expressed genes (DEG) detected on D15 showed that cardiomyocyte-related genes TNNT2/HAND1/MYH7 were significantly up-regulated in c-JUN KO cells, whereas other cell fate-related genes, such as LEFTY2, COL6A1, FOXC1, and TET1, were up-regulated in WT cells (Fig 3I). To identify whether those DEGs were regulated by increased or decreased accessibility CO loci, we found 51 genes up-regulated in c-JUN KO cells and also regulated by increased accessibility loci (Fig 3J), including cardiac muscle contraction-related genes ATP1B1, TNNI1, MYH7, and sarcomere organization-related genes CSRP3, SYNPO2L, LDB3, and MYLK3 (Fig 3K). These results suggest that c-JUN inhibits cardiomyocyte differentiation by repressing chromatin opening and activating non-cardiogenesis genes.
c-JUN prevents H3K4me3 modification by repressing SET complex activation
As c-JUN is blocking chromatin opening, we next explored if c-JUN was mediating this function through histone modifications. Western blot of H3K4me3 and H3K27ac in WT and c-JUN KO hESC showed that H3K4me3 was up-regulated in c-JUN KO cells (Fig 4A). To explore how c-JUN affects H3K4me3 modification, we performed qRT-PCR to detect the expression of genes that are part of the H3K4 methyltransferase SET/MLL complex. Data showed that the knockout of c-JUN promoted the expression of RBBP5 and SETD1B, core components of the SET complex (33) (Figs 4B and S2A and B). Cleavage under targets and tagmentation (CUT&Tag) data also showed c-JUN is bound to the promoter regions of RBBP5 and SETD1B (Fig 4C). This suggests that c-JUN represses SET complex activation by inhibiting RBBP5 and SETD1B expression, thus influencing the deposition of H3K4me3.
Figure 4. c-JUN inhibits H3K4me3 modification during cardiomyocyte generation.
(A) Western blot showing the levels of H3K4me3 and H3K27ac in WT and knockout c-JUN hESC. (B) Relative expression of the indicated SET complex genes in WT and c-JUN KO hESC. Data were from eight biological replicates in four independent experiments and are shown as the mean ± SEM. *** P-value < 0.001, Unpaired t test between the WT and KO (c-JUN KO) groups. (C) Genome view of the ATAC-seq data, H3K4me3 ChIP-seq, and c-JUN CUT&Tag data at the SET complex genes RBBP5 and SETD1B loci. (D) Heatmap showing the read density for H3K4me3 ChIP-seq data during the time course of WT and c-JUN KO hESC differentiation into cardiomyocytes. c-JUN binding profile was generated from hESC by CUT&Tag. The data were arranged into Lost, Dynamic, and Gain categories, based on the behavior of H3K4me3 in WT cells. When overlapped with c-JUN KO data, the Gain H3K4me3 loci were further subdivided into P1 and P2 subgroups. P1 loci have H3K4me3 modification in both WT and c-JUN KO cells. P2 loci have no H3K4me3 modification in c-JUN KO cells. (E) Venn diagram showing the overlap of genes regulated by P1 and P2 loci with DEGs (defined in Fig 3I) in WT and c-JUN KO cells. (F) Genome view of the ATAC-seq data, H3K4me3 ChIP-seq, and c-JUN binding data for the key factors in cardiogenesis. (G) Genome view of the ATAC-seq data, H3K4me3 ChIP-seq, and c-JUN binding data for the key factors of cardiomyocytes. (H) The RNA expression pattern for the respective genes is shown in line plot. RNA-seq expression units are normalized tag counts. (I, J) Immunostaining showing the protein levels of the cardiac progenitor marker genes ISL1 and NKX2-5 (panel (I)) and cardiomyocyte marker genes TNNT2 and α-ACTININ (panel (J)). Scale bar, 20 μm.
Source data are available for this figure.
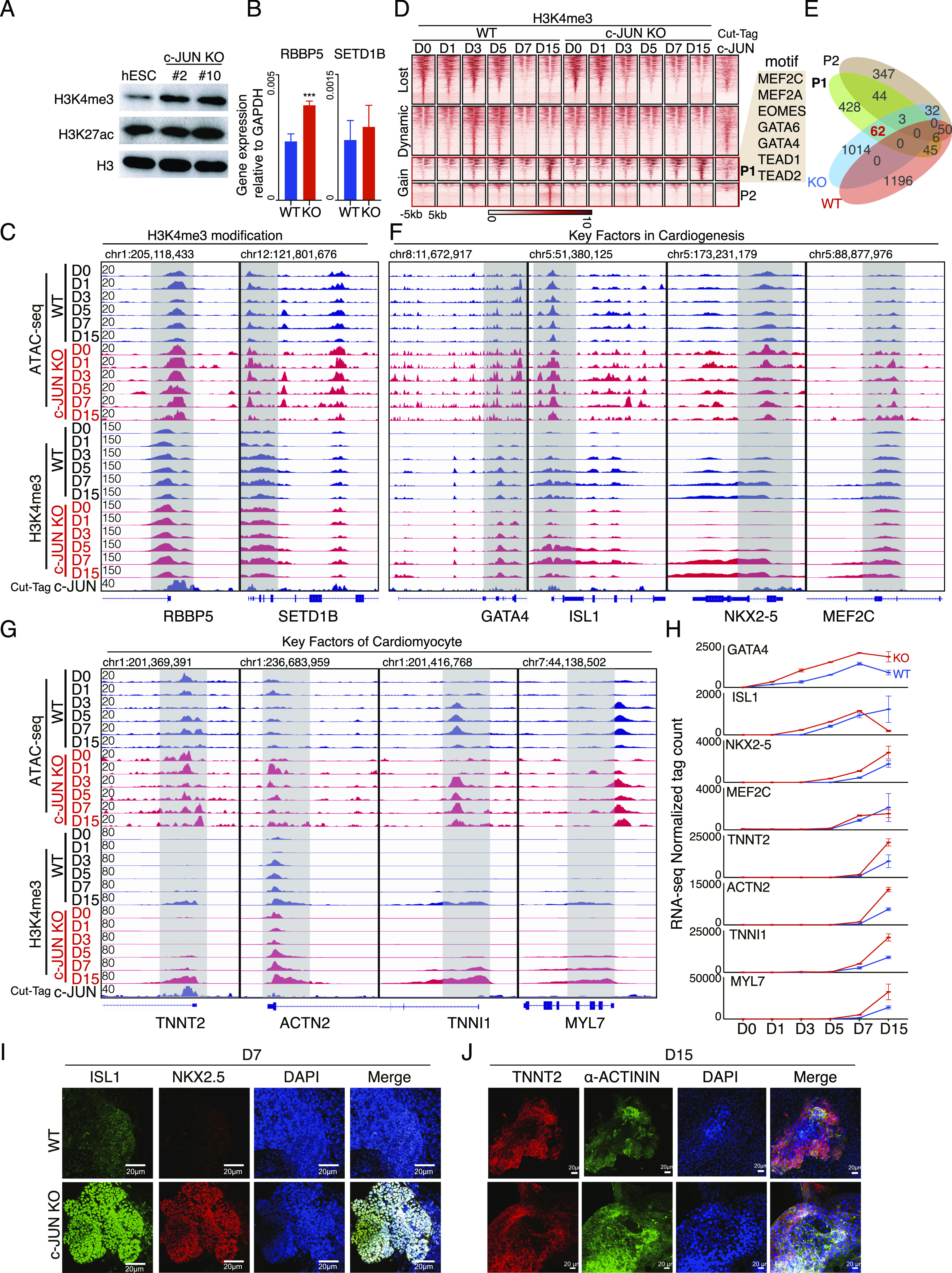
Figure S2. Gene expression pattern of RBBP5 and SETD1B during hESC-to-cardiomyocyte differentiation.

(A) RNA-seq data showing the expression of RBBP5 during hESC to cardiomyocyte differentiation between WT and c-JUN KO cells. (B) RNA-seq data showing the expression of SETD1B during hESC-to-cardiomyocyte differentiation between WT and c-JUN KO cells.
To gain a genome-wide view of the H3K4me3 modification dynamics, we performed ChIP-seq for H3K4me3 and clustered the peaks into three categories. Loci with H3K4me3 at the early stage but lost during cardiogenesis (Lost), loci only with H3K4me3 at the middle stage (Dynamic), and loci that gain H3K4me3 at the late stage (Gain) (Fig 4D). We further subdivided the Gain group into P1 (those that had gained H3K4me3 in both WT and KO cells) and P2 (loci that had no H3K4me3 in the c-JUN KO cells) (Fig 4D). Interestingly, the Gain H3K4me3 modification loci (P1) are also enriched with key TFs that relate to cardiogenesis such as MEF families, GATA families, and TEAD families (Fig 4D). To explore whether c-JUN represses cardiomyocyte differentiation by regulate H3K4me3 modification, we analyzed the DEGs associated with P1 and P2 loci. In total, 62 genes were activated in c-JUN KO and had increased H3K4me3 (Fig 4E), including key factors involved in cardiogenesis, such as GATA4, ISL1, NKX2-5, and MEF2C, and key factors of cardiomyocytes, such as TNNT2, ACTN2, TNNI1, and MYL7 (Fig 4F and G). In addition, these genes were higher expressed in the c-JUN KO cells (Fig 4H–J). Consistent with the motif analysis in Fig 3, c-JUN KO promoted the opening of loci enriched with DNA-binding motifs for GATA/ISL1/NKX2-5/MEF2C/MEF2A (Fig 3E and F).
Together, our data showed that c-JUN binds to and represses RBBP5 and SETD1B expression, thus controlling H3K4me3 modification and chromatin accessibility dynamics and inhibiting the activation of the key factors which regulate cardiogenesis.
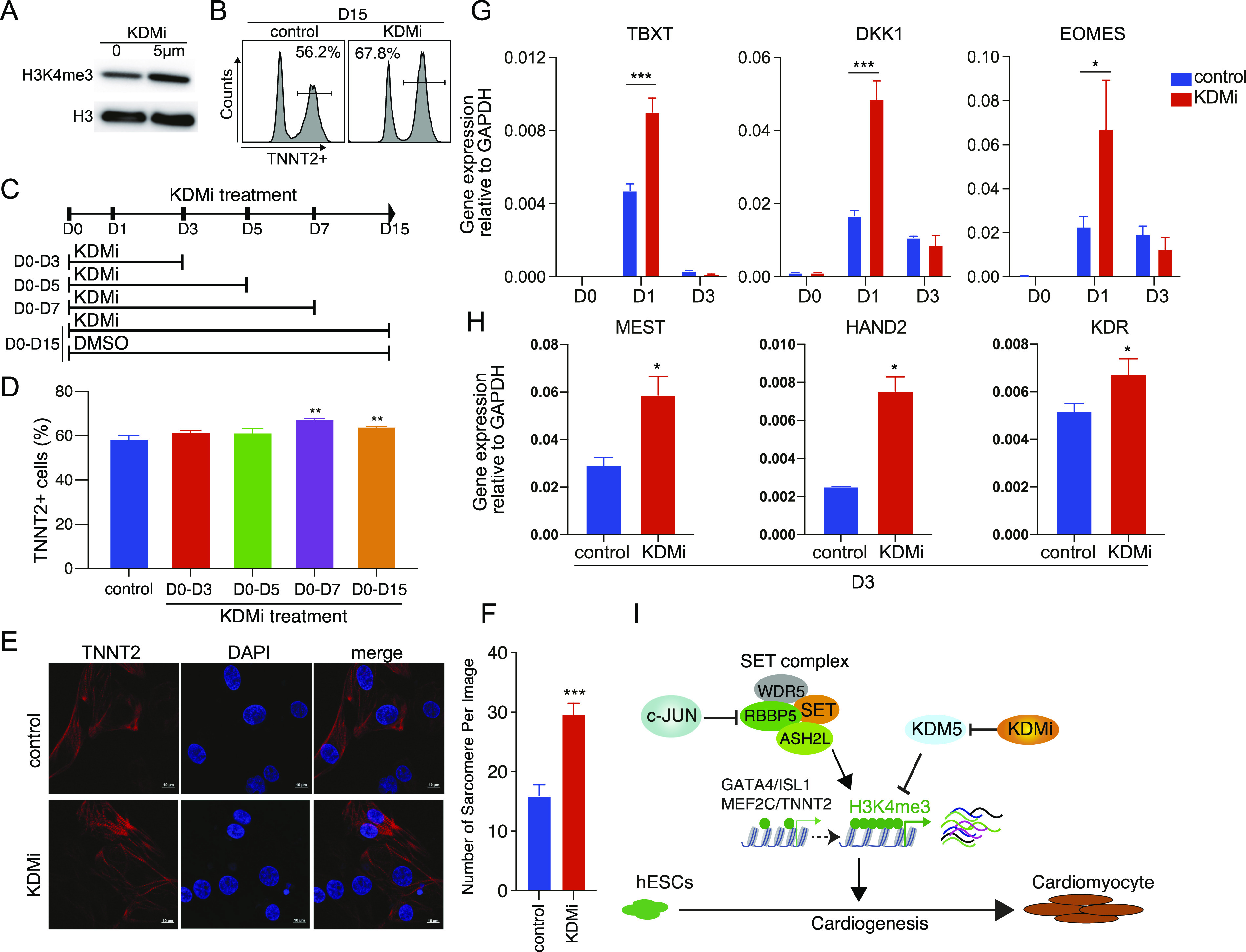
Inhibition of H3K4me3 demethylation promotes cardiomyocyte differentiation
To investigate whether H3K4me3 modification itself is critical for cardiomyocyte differentiation, we used the small molecule CPI-455, a specific inhibitor of histone demethylases 5 (KDM5) (KDMi). Western blot showed that the level of H3K4me3 was increased during KDMi treatment (Fig 5A). And consistent with our hypothesis, when KDMi was added to the cardiomyocyte differentiation medium, the spontaneous contractions of cardiomyocytes in the KDMi group were observed to be stronger than those of control cells (Video 3 and Video 4). Flow cytometry analysis also recorded an increase in the cardiomyocytes marker TNNT2 in KDMi-treated cells compared with the control (67% versus 56%; Fig 5B). Interestingly, the window experiment shows 7 d KDMi treatment (D0–D7) could generate a higher differentiation efficiency (Fig 5C and D). Immunofluorescence results showed that KDMi could increase TNNT2 protein levels and enhance the number of sarcomeres in cardiomyocytes (Fig 5E and F).
Figure 5. KDM inhibitor promotes hESC differentiation into cardiomyocytes.
(A) Western blot showing the H3K4me3 protein level in hESC treated with 0 μm and 5 μm KDMi. (B) Flow cytometry analysis of TNNT2 expression in cardiomyocytes differentiated from hESC. Percent of cells TNNT2+ is indicated for each condition, control: induced cardiomyocytes from WT hESC with DMSO, KDMi: induce cardiomyocytes from hESC with DMSO-dissolved KDM inhibitor (CPI-455, 5 μm). KDMi was added into the culture medium from D0 to D15. (C) Schematic of the time window of KDMi treatment. (C, D) Flow cytometry quantification of cardiomyocyte differentiation efficiency, based on the percent of TNNT2+ cells in different groups in (C). Data were from eight biological replicates in three independent experiments and are shown as the mean ± SEM. ** P-value < 0.01, two-way ANOVA with Sidak correction between the control and KDMi groups. (E) Immunostaining showing the level of TNNT2 in cardiomyocytes that generated with DMSO or 7-D KDMi treatement (D0–D7). Scale bar, 10 μm. (F) Counts of the number of sarcomeres in each image. Data were from seven to nine replicates and are shown as the mean ± SEM. *** P-value < 0.001, two-way ANOVA with Sidak correction between the control and KDMi groups. (G) Relative expression of the indicated mesoderm marker genes during the differentiation of hESC into mesoderm cells with or without KDMi. Data were from six biological replicates in three independent experiments and are shown as the mean ± SEM. *** P-value < 0.001, * P-value < 0.05, two-way ANOVA with Sidak correction between the control and KDMi groups. (H) Relative expression of the indicated cardiac mesoderm marker genes during differentiation of hESC into cardiac mesoderm cells with or without KDMi. Data were from six biological replicates in three independent experiments and are shown as the mean ± SEM. * P-value < 0.05, Unpaired t test between the control and KDMi groups. (I) Schematic for the role of c-JUN in modulating chromatin accessibility and H3K4me3 modification during hESC to cardiomyocyte differentiation.
Source data are available for this figure.
Movie showing spontaneous contractions of cardiomyocytes from WT hESC without KDMi in D15. Download video (6MB, mp4)
Movie showing spontaneous contractions of cardiomyocytes from WT hESC with KDMi treatment in D15. Download video (6.7MB, mp4)
To validate whether KDMi could accelerate the key factors in regulating cardiogenesis, we performed a quantitative polymerase chain reaction (qRT-PCR) to detect cardiac mesoderm marker gene expression. The results show TBXT (T), DKK1, EOMES, MEST, HAND2, and KDR, key markers of cardiac mesoderm, were quickly activated on D1 or D3 in response to KDMi treatment (Fig 5G and H). These results indicate H3K4me3 plays an important role in cardiac mesoderm and cardiomyocyte development. Overall, either knocking out c-JUN or using a KDM inhibitor could up-regulate H3K4me3 and speed up cardiac mesoderm and cardiomyocyte formation (Fig 5I).
Cell trajectory during hESC to cardiomyocytes differentiation
To uncover how c-JUN alters the cell trajectory of hESC to cardiomyocytes, we performed single-cell RNA-seq experiments and obtained 52,695 cells from hESC (D0), D3, D7, and D15 of WT and c-JUN knockout cells. In agreement with the bulk RNA-seq data, single-cell RNA-seq data also showed TNNT2 was highly expressed in D7 and D15 in c-JUN KO conditions (Figs 4H and S3A). Knockout of c-JUN led to an enhanced percentage of TNNT2+ cells on D7 and a small decrease on D15 (Fig S3B). Potentially, the discrepancy between the single-cell RNA-seq data and FACS data may be caused by slower RNA transcription and reduced protein degradation at the cardiomyocyte stage. Next, we integrated the WT and c-JUN KO data sets and visualized the cell clusters through a t-SNE projection (Fig 6A). After cell type annotation, we found cell heterogeneity in D3 to D15 in both WT and c-JUN KO conditions (Figs 6A–C and S3C and D). Interestingly, knockout of c-JUN inhibited non-cardiomyocyte-related expression of TNNT2 (Fig S3E). From the single-cell distributions, c-JUN KO and WT hESC clustered closely together, which suggests c-JUN KO hESC were similar to WT hESC, and c-JUN KO had only a minimal impact overall on hESC (Fig 6A). In agreement with previous results, the pluripotent marker OCT4 was down-regulated quicker in KO than WT cells (Fig 6D). Cardiac mesoderm marker genes TBXT (T) and HAND1 were activated earlier in KO cells, and TNNT2 was highly expressed in both WT and KO cardiomyocytes (Fig 6D). For trajectory analysis, c-JUN KO and WT cells both appeared to develop three branches, including hESC to cardiomyocytes, epicardial, and endothelial cells (Fig 6E–G). Those results suggest c-JUN plays a role in regulating pluripotency (20) and support a role for c-JUN in inhibiting cardiomyocyte formation by repressing key factors involved in regulating cardiogenesis, in agreement with the H3K4me3 and ATAC-seq data.
Figure S3. Single-cell RNA-seq illustrates cell dynamics during hESC-to-cardiomyocyte differentiation.

(A) Single-cell RNA-seq data shows read count of TNNT2 during hESC-to-cardiomyocyte transition between WT and c-JUN KO cells. (B) Percentage of TNNT2+ cell in D0-D15 between WT and c-JUN KO cells. (C) t-SNE map shows the cell distribution of WT and c-JUN KO conditions. (D) Percentage of each cell type in each day during hESC-to-cardiomyocyte transition. (E) Violin plot shows the expression of TNNT2 in each cell type in D15.
Figure 6. Single-cell transcriptome analysis showing the trajectories of cardiomyocyte differentiation.
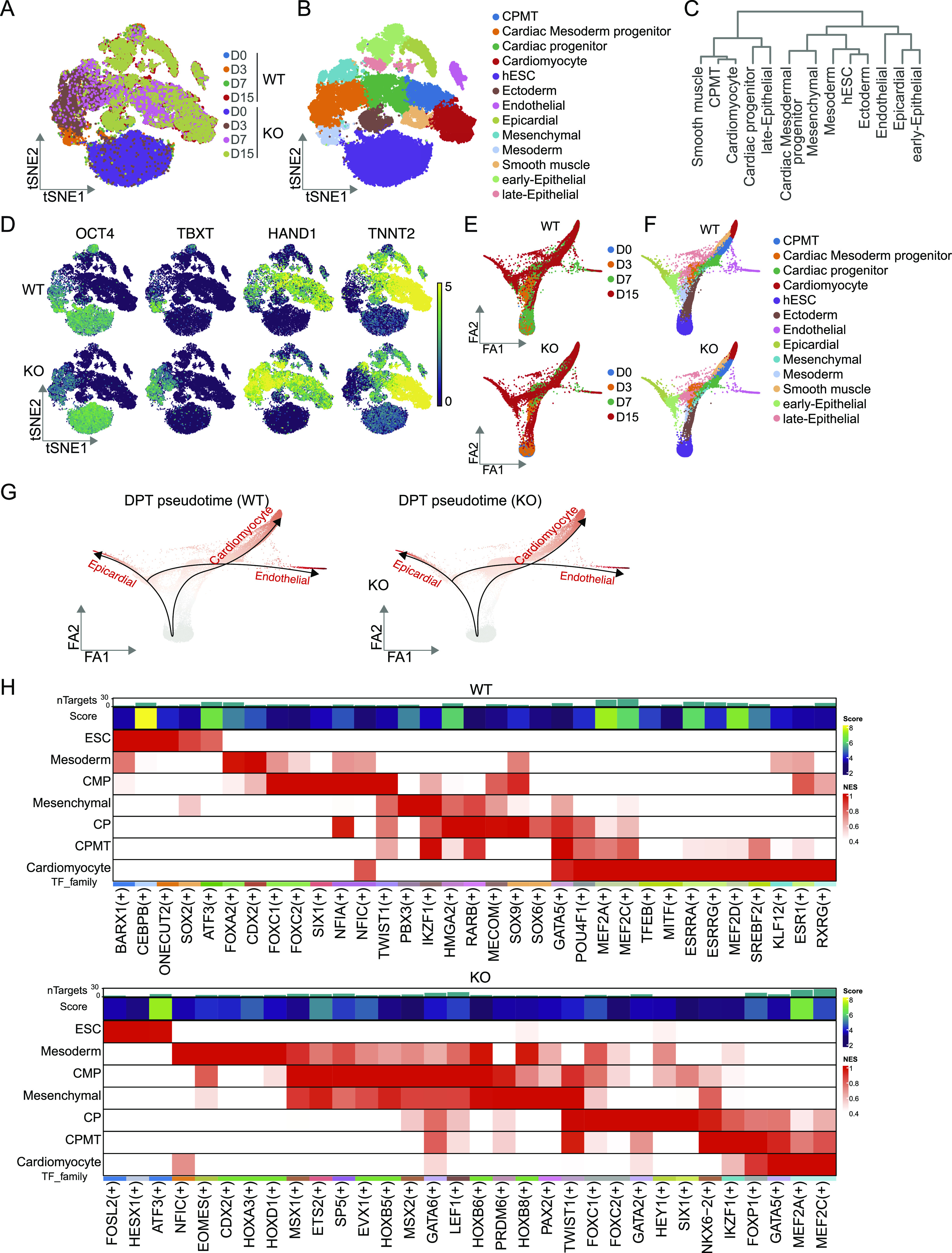
(A, B) A t-SNE showing the single-cell RNA-seq data of WT and c-JUN KO cells during hESC differentiation into cardiomyocytes. (B) 13 cell clusters were defined according to the specific marker genes. (C) Dendrogram plot shows the correlation of the 13 cell clusters. (D) t-SNE colored by the expression level of representative marker genes during the induction of hESC into cardiomyocytes. Each dot represents one cell, and RNA level is colored by selected marker intensities from high (yellow) to low (purple). (E, F) Cell differentiation trajectory calculated using the ForceAtlas2 algorithm. (E, F) WT and c-JUN KO cells are shown in separated plots (E) based on time and (F) based on the cell types. (G) Diffusion pseudotime shows the differentiation trajectory of WT and c-JUN KO cells. (H) SCENIC results on the WT and c-JUN KO condition, master regulators are color-matched with the cell types.
c-JUN inhibits key regulons activity during cardiogenesis
To investigate how c-JUN perturbs cardiomyocyte cell fate, we analyzed the gene regulatory network using SCENIC (34, 35). We analyzed the hESC-to-cardiomyocyte branch and used SCENIC to predict the TFs used in each cell type in WT and c-JUN KO conditions. The results showed 24 TFs, including EOMES, HOX, GATA, and FOX families, were enriched in the mesoderm-to-cardiomyocyte stages in c-JUN KO cells (Fig 6H). However, neural ectoderm genes SOX9/SOX6 were enriched in WT cells. Consistent with the ATAC-seq data, WT cells opened loci related to placental development (Figs 3E–G and 6H). Meantime, H3K4me3 modification loci were also enriched with EOMES/GATA/MEF TFs that are predicted by SCENIC, indicating transcriptome, epigenetic modification, and chromatin dynamics were synergistic remodeling to reshape the cell fate to cardiomyocyte (Fig 4D). These results illustrate the differential gene regulatory network used in WT and c-JUN KO cells and indicate c-JUN plays an important role in cardiac mesoderm and cardiomyocyte cell fate decisions.
Together, our data suggest that c-JUN controls cardiogenesis by inhibiting chromatin opening and repressing the expression of key factors that regulate cardiogenesis and cardiomyocyte formation. Furthermore, c-JUN was bound to the loci of and repressed the expression of the SET complex genes RBBP5 and SETD1B, thus leading to a lower level of H3K4me3 on the promoters of key cardiogenesis factors to inhibit cardiomyocyte differentiation.
Discussion
c-JUN was previously reported as a key TF for embryonic development (15, 16). During mouse embryonic development, knockout of c-Jun leads to the failure to form a correct heart structure (36). However, the mechanism of c-JUN in embryonic development remains unknown, especially in humans. In this work, we used an in vitro hESC-to-cardiomyocyte differentiation system to study c-JUN’s function and found that c-JUN acts as a barrier to cardiomyocyte differentiation. The small molecular JNK inhibitor (SP600125) could phenocopy the c-JUN KO phenotype and release the negative impact of c-JUN in cardiomyocyte generation (Fig 2F). Further study revealed that c-JUN inhibits the hESC-to-cardiomyocyte transition by maintaining the chromatin state and affecting the modification of H3K4me3 by inhibiting SET complex activation.
In agreement with the increased H3K4me3 level by knocking out c-JUN to promote cardiomyocyte differentiation, manipulating H3K4me3 modification by KDMi also activated mesoderm/cardiac mesoderm genes, thus constantly pushing the cells toward a CM cell fate. Recently, a study published in Exp Mol Med suggested that inhibiting KDM5A could ameliorate pathological cardiac fibrosis (37). However, another study submitted to BioRxiv (38 Preprint) suggests KDM5A could regulate cardiomyocyte maturation by promoting fatty acid oxidation, oxidative phosphorylation, and myofibrillar organization. Those studies indicate a complex role for KDM5 in regulating cardiomyocyte cell fate decision and maturation.
Our study contradicts previous in vivo studies of the c-Jun KO in mice which were post-implantation lethal at around E13.5 because of cardiac problems (36). Our work shows, conversely, that c-JUN is a barrier to cardiomyocyte differentiation. However, it is important to highlight critical differences between the in vivo knockout of c-Jun and the in vitro differentiation system used here. Potentially, an accelerated generation of cardiomyocytes may be deleterious for proper heart generation because of reduced numbers of support cells. Indeed, our single-cell RNA-seq data indicate that the cardiomyocyte population expands at the expense of epicardial cells. Potentially, in the even more complex in vivo development of the heart, other cell types may also be defective, leading to a failure to establish the correct balance of cells required for a functioning heart organ.
c-JUN is a somatic key factor and could repress somatic cell reprogramming and induce pluripotency exit quickly when overexpressed in mouse ESC (39). However, in human ESC, c-JUN helps maintain the hESC in a pluripotent state; these contradictory characteristics of c-JUN may be caused by the context-dependent nature of c-JUN, as in somatic cells, c-JUN co-binds with TFs to inhibit cell fate transitions (27, 40), but in hESC, c-JUN is co-binding with pluripotent TFs to inhibit differentiation (20). This conflicting role of c-JUN in different cell types shows how c-JUN can function as a guardian to maintain cell fate identity.
Our work demonstrates how c-JUN can impact chromatin accessibility, and inhibit cell fate transitions (27). Here, we use the cardiomyocyte generation model to study c-JUN’s function and find that c-JUN could inhibit chromatin opening and limit chromatin accessibility. This showed that c-JUN has a powerful ability to modulate chromatin structure. Previous studies have shown that c-JUN/AP-1 could interact with cell type-specific TFs and bind to nucleosomes to facilitate recruitment of BAF complex and increase chromatin accessibility (40). We also found c-JUN could regulate H3K4me3 by binding to and repressing RBBP5 and SETD1B transcriptional activation, the core components of the H3K4 methyltransferase SET complex. Indeed, H3K4me3 is an open (active) chromatin marker that has been reported to play a key role in mesoderm and cardiomyocyte cell fate decisions (29, 41) and even play an important role in the development of cardiovascular disease (42, 43, 44). Overall, our work uncovered a new function of c-JUN in regulating chromatin accessibility and H3K4me3 modification which play an important role in cardiomyocyte cell fate decision.
Finally, c-JUN/AP-1 families play complex roles in embryonic development (15, 16), tissue regeneration (19), somatic cell reprogramming, hESC differentiation (20, 27, 39), and chromatin structure remodeling (27, 40, 45, 46). However, the role of c-JUN in cell fate decisions and epigenetic regulation remains unclear; indeed, opposing roles for c-JUN have been reported in some conditions (20, 39). For a more comprehensive study of the function of c-JUN, new model systems such as early embryonic development and new methods such as single-cell ATAC-seq and spatial single-cell technology should be used to explore the novel role of c-JUN in cardiomyocyte cell fate decisions.
Materials and Methods
Differentiation of human ESC to cardiac
Differentiation toward cardiomyocyte lineage was induced using small molecules to modulate the WNT signaling pathway and was optimized according to a small molecule-based monolayer method as previously described (21). Briefly, hESC cultured in mTeSR1 medium were digested into single cells by Accutase (Sigma-Aldrich) and plated onto Matrigel-coated culture dishes at a density of 4 x 105 cells/12 well in mTeSR1 medium with ROCKi inhibitor (Y-27632; Selleck) for 24 h. After 1 d, Y-27632 was withdrawn from the medium, and Cells were then induced with mTeSR1 medium for 48 h. On day 0, cells were treated with cardiac differentiation medium for 24 h, consisting of RPMI/1640 and CHIR99021 (4 μM; Selleck). On day 1, the medium was exchanged by fresh RPMI/1640, and cells were induced for another 48 h. Subsequently, on day 3, the medium was changed to RPMI/1640 medium containing IWP2 (5 μM; Selleck) for 2 d. After 2 d (day 5), the medium was exchanged for RPMI/1640 medium for another 2 d. For maintenance of the obtained cardiomyocytes, at day 7, the medium was changed to RPMI/1640 containing insulin (10 μg/ml) and Vitamin C (200 μg/ml), and half the medium was exchanged every day until day 15. Contracting cells were seen from day 9 to day 15.
Generation of knockout hESC
c-JUN-/-hESC was generated by CRISPR/Cas9, and pairs of sgRNA were used to delete the target exons. sgRNA1: acaagtttcggggccgcaac; sgRNA2: gagaacttgacaagttgcga.
Flow cytometry
Cells were dissociated with Accutase (Sigma-Aldrich) and filtered through a 70-μm cell strainer (BD Biosciences) to get a single-cell suspension. Single cells were fixed with 4% paraformaldehyde for 20 min and washed twice in PBS for at least 5 min each. Then, cells were incubated in 1% BSA for 30 min, permeabilized in 0.2% Triton X-100 for 10 min, and washed three times with PBS containing 0.05% Triton X-100. Subsequently, cells were stained with 647-conjugated primary antibodies (1:200; cTnT) for 1 h at room temperature. After PBS washing steps, cells were analyzed using an Accuri C6 Plus flow cytometer (BD Biosciences), and data were processed using FlowJo 10.4 software.
Immunofluorescence
Cells growing on coverslips were washed three times with PBS, then fixed with 4% PFA for 30 min, and subsequently, cell membranes were penetrated with 0.1% Triton X-100 and blocked with 3% BSA for 30 min at room temperature. Cells were then incubated with c-JUN antibody for 1 h. After three washes in PBS, followed by 1 h of incubation in secondary antibodies, cells were incubated in DAPI for 1 min. Then the coverslips were mounted on the slides for observation on a confocal microscope (Leica). The following antibody was used in this project: anti-c-JUN (no. 9165, 1:100; CST).
Western blot analysis
hESC or differentiated cardiomyocytes were lysed in RIPA buffer (Thermo Fisher Scientific). The extracted proteins were quantified by a BCA Protein Assay Kit (Thermo Fisher Scientific), respectively. Total protein was subjected to SDS–PAGE, then transferred to a polyvinylidene fluoride membrane (PVDF) and probed with the specific primary antibodies at 4°C overnight. The PVDF membrane was washed with PBST three times, and then the HRP-conjugated secondary antibody was incubated for 1 h at room temperature. Proteins were detected by enhanced chemiluminescent substrates (ECL Kit; Thermo Fisher Scientific). The antibodies used are shown in Table S1.
Table S1 Antibodies for Western blotting, immunofluorescence, and ChIP-seq. (15.2KB, docx)
ATAC-seq
ATAC-seq was performed as previously described (25). Briefly, 50,000 cells were washed with 50 μl cold PBS and resuspended in 50 μl lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.2% [vol/vol] IGEPAL CA-630). The suspension of nuclei was then centrifuged for 10 min at 500g at 4°C, followed by the addition of 50 μl transposition reaction mix (25 μl TD buffer, 2.5 μl Tn5 transposase, and 22.5 μl nuclease-free H2O) from the Nextera DNA library Preparation Kit (96 samples) (FC-121-1031; Illumina). Samples were then PCR amplified and incubated at 37°C for 30 min. DNA was isolated using a MinElute Kit (QIAGEN). ATAC-seq libraries were first subjected to five cycles of pre-amplification. To determine the suitable number of cycles required for the next round of PCR, the libraries were assessed by quantitative PCR as described (26), and the libraries were then PCR amplified for the appropriate number of cycles according to the qRT-PCR results. Libraries were purified with a Qiaquick PCR (QIAGEN) column, and the library’s concentration was measured using a KAPA Library Quantification Kit (KK4824) according to the manufacturer’s instructions. Finally, the ATAC libraries were sequenced on the NextSeq 500 sequencing platform using a NextSeq 500 High Output Kit v2 (150 cycles) (FC-404-2002; Illumina) according to the manufacturer’s instructions.
ChIP-seq
H3K4me3 ChIP was performed as described previously (39). Briefly, 1 × 109 cells were fixed in 8.75 ml DMEM/F12 with 1% formaldehyde for 15 min at room temperature with rotation and then followed by the reaction with 0.125 M glycine. Cells were then lysed in ChIP-buffer A (50 mM HEPES-KOH, 140 mM NaCl, 1 mM EDTA [pH 8.0], 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, 50 mM Tris–HCl [pH 8.0], and protease inhibitor cocktail) for 10 min at 4°C. Samples were centrifuged at 1,400g for 5 min at 4°C. Pellets were lysed in ChIP-buffer B (1% SDS, 50 mM Tris–HCl [pH 8.0], 10 mM EDTA, and protease inhibitor cocktail) for 5 min at 4°C. The DNA was fragmented to 100–500 bp by sonication and then centrifuged at 12,000g for 2 min. The supernatant was diluted with ChIP IP buffer (0.01% SDS, 1% Triton X-100, 2 mM EDTA, 50 mM Tris–HCl [pH 8.0], 150 mM NaCl, and protease inhibitor cocktail). Immunoprecipitation was performed with 2 μg rabbit anti-H3K4me3 antibody (ab8580; Abcam) coupled to Dynabeads with protein A/G overnight at 4°C. Beads were washed, eluted, and reverse cross-linked. DNA was extracted with phenol/chloroform for sequencing. The ChIP DNA library was constructed with VAHTS Universal DNA Library Prep Kit for Illumina (Nd604; Vazyme Biotech) according to the manufacturer’s instructions. The DNA libraries were quantified and tested by qRT-PCR with positive primers to assess the quality of the library. Then, libraries were sequenced on an Illumina NextSeq 500 instrument using 75-bp paired-end reads.
CUT&Tag
c-JUN CUT&Tag data was generated by the Hyperactive Universal CUT&Tag Assay Kit (Vazyme) according to the manufacturer’s instructions. H1 cells (1 × 105) were used for the CUT&Tag experiment, and pA-Tn5 transposase was used to cut the genome and add a special adaptor sequence to build a library. The enrichment of target sites in the library was detected using qPCR. The c-JUN antibody was purchased from Cell Signaling Technology (#9165; Cell Signaling Technology).
Single-cell sequencing
Cells at hESC (D0) and differentiated to cardiomyocyte (D3, D7, and D15) time points were collected and resuspended in DPBS with 0.04% BSA. Then, cell suspensions (500–1,000 cells/µl) were loaded on a Chromium Single Cell Controller (10x Genomics) to obtain single-cell gel beads in emulsion (GEMs) by using Single Cell 3′ Library and Gel Bead Kit V2 (120237; 10x Genomics). Captured cDNAs were lysed, and the released RNA was barcoded through reverse transcription in singular GEMs. Barcoded cDNAs were pooled and cleaned by DynaBeads MyOne Silane Beads (37002D; Invitrogen). Single-cell RNA-seq libraries were prepared by Single Cell 3′ Library Gel Bead Kit V2 (120237; 10x Genomics) following the manufacturer’s instructions. Sequencing was operated on an Illumina HiSeq X 10 with pair-end of 150 bp (PE150).
RNA-seq and gene expression analysis
Total RNA was prepared with TRIzol. For quantitative PCR, cDNAs were synthesized with ReverTra Ace (Toyobo) and oligo-dT (Takara) and then analyzed by qRT-PCR with Premix Ex Taq (Takara). For RNA-seq, TruSeq RNA Sample Prep Kit (RS-122-2001; Illumina) was used for library construction, and the sequencing was performed using a NextSeq 500 High Output Kit v2 (75 cycles) (FC-404-1005; Illumina), according to the manufacturer’s instructions. RNA-seq was processed as previously described (47, 48); briefly, reads were aligned to a transcriptome index generated from the Ensembl annotations, using RSEM (49), Bowtie2 (50), and normalized using EDASeq (51). RNA-seq data are expressed in units of GC-normalized tag counts. DEGs were analyzed by DESeq2 (52).
ATAC-seq bioinformatic analysis and peak calling
All sequencing data were mapped to the hg38 human genome assembly using Bowtie2 with the options --very-sensitive. samtools was used to remove low-quality mapped reads with option view –q 35, and unique reads were kept. We removed mitochondrial sequences using “grep –v ‘chrM.’’ Biological replicates were merged, and peaks were called using dfilter (53) (with the settings: –bs = 100 –ks = 60 –refine –std = 5). BigWig files were produced using genomeCoverageBed from bedtools (scale=107/<one sample’s total unique reads>) and then bedGraphToBigWig. Gene ontology and gene expression measures were called by first collecting all transcription start sites within 10 kb of an ATAC-seq peak and then performing GO analysis with goseq (54), or measuring gene expression. Other analysis was performed using GLBase (47).
Recalling weak peaks from the ATAC-seq data
Peak recalling is based on the method we previously described (27). Briefly, when dfilter is used to discover peaks, as described above, it is generally conservative and will not call a weak peak. Hence, we “re-call” all peaks by measuring the sequence tag density in all ATAC-seq libraries for all possible peaks in any other ATAC-seq library, irrespective of which library the peak was called in by dfilter (53). Then we get a superset of all possible peaks in any library, and based on our previous analysis, we used an arbitrary minimum threshold of 0.2734 to filter out false peaks; if the ATAC-seq is below this value, it is annotated as “closed” and above “open.” All downstream analysis was based on this new peak list.
ChIP-seq data analysis
Reads from ChIP-seq experiments were mapped to the mouse genome (hg38) using Bowtie2 (--very-sensitive), as described for ATAC-seq data, and only the uniquely mapped reads were kept for further analysis. Peaks were called using MACS2 (55) software with the default parameters.
Computational analysis of TF binding sites and open chromatin regions
Motif analysis was performed by HOMER 28 with default settings. Motifs were only kept if the P-value was <0.01 and (<percent of target>/<percent of background>) was >1.5. Annotation of the ChIP-seq/ATAC-seq peaks/open chromatin to genes was performed by HOMER using default settings.
Single-cell RNA-seq data analysis
Fastq reads were aligned to the genome using STAR (56) with the setting ' --soloType Droplet --soloFeatures Gene --runThreadN 20 --soloCBstart 1 --soloCBlen 16 --soloUMIstart 17 --soloUMIlen 12 --soloBarcodeReadLength 0 --readFilesCommand zcat --outSAMtype BAM SortedByCoordinate --outSAMattributes NH HI AS nM CR CY UR UY'. The count matrix was lightly filtered to exclude cell barcodes with low numbers of counts: Cells with <2,000 UMIs, <500 genes, or >20% fraction of mitochondrial counts were removed. The filtered matrix was normalized according to Scanpy tutorials. The top 3,000 most highly variable genes were used for principal component analysis, and the first six PCs (principal components) were used for downstream analysis with Scanpy. Gene regulatory network inference with SCENIC was used (35).
Data Availability
The raw sequence data reported in this work have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in the National Genomics Data Center (Nucleic Acids Res 2021), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: HRA003227) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
Supplementary Material
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (92068201, 32270574, 32150710521), the National Key Research and Development Program of China (2018YFA0108700 and 2017YFA0105602), the Guangdong Science and Technology Project (2020B1212060052, 2022B1212010010), and Science and Technology Projects in Guangzhou (2023A03J0045, 2023A04J0156). Guangzhou Key Laboratory of Biological Targeting Diagnosis and Therapy (202201020379) and the Youth Innovation Promotion of the Chinese Academy of Sciences (2019348) to D Li. Guangdong Cardiovascular Institute (2020XXG002), High-level Hospital Construction Project (DFJHBF202110, DFJHBF202111), and Plan on Enhancing Scientific Research in GMU.
Author Contributions
H Zhong: validation and methodology.
R Zhang: resources, validation, methodology, and project administration.
G Li: resources and validation.
P Huang: resources and validation.
Y Zhang: resources and validation.
J Zhu: resources and validation.
J Kuang: resources.
AP Hutchins: resources.
D Qin: resources and funding acquisition.
P Zhu: funding acquisition.
D Pei: funding acquisition.
D Li: conceptualization, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, project administration, and writing—original draft, review, and editing.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References
- 1.Kwon C, Qian L, Cheng P, Nigam V, Arnold J, Srivastava D (2009) A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol 11: 951–957. 10.1038/ncb1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Junion G, Spivakov M, Girardot C, Braun M, Gustafson EH, Birney E, Furlong EE (2012) A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148: 473–486. 10.1016/j.cell.2012.01.030 [DOI] [PubMed] [Google Scholar]
- 3.Zhou Y, Liu Z, Welch JD, Gao X, Wang L, Garbutt T, Keepers B, Ma H, Prins JF, Shen W, et al. (2019) Single-cell transcriptomic analyses of cell fate transitions during human cardiac reprogramming. Cell Stem Cell 25: 149–164.e9. 10.1016/j.stem.2019.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad SM, Busser BW, Huang D, Cozart EJ, Michaud S, Zhu X, Jeffries N, Aboukhalil A, Bulyk ML, Ovcharenko I, et al. (2014) Machine learning classification of cell-specific cardiac enhancers uncovers developmental subnetworks regulating progenitor cell division and cell fate specification. Development 141: 878–888. 10.1242/dev.101709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lui KO, Zangi L, Silva EA, Bu L, Sahara M, Li RA, Mooney DJ, Chien KR (2013) Driving vascular endothelial cell fate of human multipotent Isl1+ heart progenitors with VEGF modified mRNA. Cell Res 23: 1172–1186. 10.1038/cr.2013.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshida Y, Yamanaka S (2017) Induced pluripotent stem cells 10 Years later: For cardiac applications. Circ Res 120: 1958–1968. 10.1161/CIRCRESAHA.117.311080 [DOI] [PubMed] [Google Scholar]
- 7.Song M, Yuan X, Racioppi C, Leslie M, Stutt N, Aleksandrova A, Christiaen L, Wilson MD, Scott IC (2022) GATA4/5/6 family transcription factors are conserved determinants of cardiac versus pharyngeal mesoderm fate. Sci Adv 8: eabg0834. 10.1126/sciadv.abg0834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindsley RC, Gill JG, Murphy TL, Langer EM, Cai M, Mashayekhi M, Wang W, Niwa N, Nerbonne JM, Kyba M, et al. (2008) Mesp1 coordinately regulates cardiovascular fate restriction and epithelial-mesenchymal transition in differentiating ESCs. Cell Stem Cell 3: 55–68. 10.1016/j.stem.2008.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bondue A, Blanpain C (2010) Mesp1: A key regulator of cardiovascular lineage commitment. Circ Res 107: 1414–1427. 10.1161/CIRCRESAHA.110.227058 [DOI] [PubMed] [Google Scholar]
- 10.Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S (2003) Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 5: 877–889. 10.1016/s1534-5807(03)00363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren J, Miao D, Li Y, Gao R (2021) Spotlight on Isl1: A key player in cardiovascular development and diseases. Front Cell Dev Biol 9: 793605. 10.3389/fcell.2021.793605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stone NR, Gifford CA, Thomas R, Pratt KJB, Samse-Knapp K, Mohamed TMA, Radzinsky EM, Schricker A, Ye L, Yu P, et al. (2019) Context-specific transcription factor functions regulate epigenomic and transcriptional dynamics during cardiac reprogramming. Cell Stem Cell 25: 87–102.e9. 10.1016/j.stem.2019.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel-Duby R, et al. (2013) Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci U S A 110: 5588–5593. 10.1073/pnas.1301019110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Londono P, Cao Y, Sharpe EJ, Proenza C, O’Rourke R, Jones KL, Jeong MY, Walker LA, Buttrick PM, et al. (2015) High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling. Nat Commun 6: 8243. 10.1038/ncomms9243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilberg F, Howells NA, Wagner EF (1993) c-jun is essential for normal mouse development and hepatogenesis. Nature 365: 179–181. 10.1038/365179a0 [DOI] [PubMed] [Google Scholar]
- 16.Johnson RS, van Lingen B, Papaioannou VE, Spiegelman BM (1993) A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev 7: 1309–1317. 10.1101/gad.7.7b.1309 [DOI] [PubMed] [Google Scholar]
- 17.Windak R, Muller J, Felley A, Akhmedov A, Wagner EF, Pedrazzini T, Sumara G, Ricci R (2013) The AP-1 transcription factor c-Jun prevents stress-imposed maladaptive remodeling of the heart. PLoS One 8: e73294. 10.1371/journal.pone.0073294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beisaw A, Kuenne C, Guenther S, Dallmann J, Wu CC, Bentsen M, Looso M, Stainier DYR (2020) AP-1 contributes to chromatin accessibility to promote sarcomere disassembly and cardiomyocyte protrusion during zebrafish heart regeneration. Circ Res 126: 1760–1778. 10.1161/CIRCRESAHA.119.316167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W, Hu CK, Zeng A, Alegre D, Hu D, Gotting K, Ortega Granillo A, Wang Y, Robb S, Schnittker R, et al. (2020) Changes in regeneration-responsive enhancers shape regenerative capacities in vertebrates. Science 369: eaaz3090. 10.1126/science.aaz3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li QV, Dixon G, Verma N, Rosen BP, Gordillo M, Luo R, Xu C, Wang Q, Soh CL, Yang D, et al. (2019) Genome-scale screens identify JNK-JUN signaling as a barrier for pluripotency exit and endoderm differentiation. Nat Genet 51: 999–1010. 10.1038/s41588-019-0408-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP (2013) Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc 8: 162–175. 10.1038/nprot.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lian X, Bao X, Zilberter M, Westman M, Fisahn A, Hsiao C, Hazeltine LB, Dunn KK, Kamp TJ, Palecek SP (2015) Chemically defined, albumin-free human cardiomyocyte generation. Nat Methods 12: 595–596. 10.1038/nmeth.3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mummery CL, Zhang J, Ng ES, Elliott DA, Elefanty AG, Kamp TJ (2012) Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: A methods overview. Circ Res 111: 344–358. 10.1161/CIRCRESAHA.110.227512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burridge PW, Keller G, Gold JD, Wu JC (2012) Production of de novo cardiomyocytes: Human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 10: 16–28. 10.1016/j.stem.2011.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 109: 21 29 1–21.29.9. 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, Qin Y, Guo L, He J, Yu S, et al. (2017) Chromatin accessibility dynamics during iPSC reprogramming. Cell Stem Cell 21: 819–833.e6. 10.1016/j.stem.2017.10.012 [DOI] [PubMed] [Google Scholar]
- 28.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong SH, Rampalli S, Lee JB, McNicol J, Collins T, Draper JS, Bhatia M (2011) Cell fate potential of human pluripotent stem cells is encoded by histone modifications. Cell Stem Cell 9: 24–36. 10.1016/j.stem.2011.06.002 [DOI] [PubMed] [Google Scholar]
- 30.Jia G, Preussner J, Chen X, Guenther S, Yuan X, Yekelchyk M, Kuenne C, Looso M, Zhou Y, Teichmann S, et al. (2018) Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat Commun 9: 4877. 10.1038/s41467-018-07307-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, et al. (2016) Complex interdependence regulates heterotypic transcription factor distribution and coordinates cardiogenesis. Cell 164: 999–1014. 10.1016/j.cell.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen JX, Krane M, Deutsch MA, Wang L, Rav-Acha M, Gregoire S, Engels MC, Rajarajan K, Karra R, Abel ED, et al. (2012) Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ Res 111: 50–55. 10.1161/CIRCRESAHA.112.270264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith E, Lin C, Shilatifard A (2011) The super elongation complex (SEC) and MLL in development and disease. Genes Dev 25: 661–672. 10.1101/gad.2015411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017) SCENIC: Single-cell regulatory network inference and clustering. Nat Methods 14: 1083–1086. 10.1038/nmeth.4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q, et al. (2020) A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat Protoc 15: 2247–2276. 10.1038/s41596-020-0336-2 [DOI] [PubMed] [Google Scholar]
- 36.Eferl R, Sibilia M, Hilberg F, Fuchsbichler A, Kufferath I, Guertl B, Zenz R, Wagner EF, Zatloukal K (1999) Functions of c-Jun in liver and heart development. J Cell Biol 145: 1049–1061. 10.1083/jcb.145.5.1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang B, Tan Y, Zhang Y, Zhang S, Duan X, Jiang Y, Li T, Zhou Q, Liu X, Zhan Z (2022) Loss of KDM5B ameliorates pathological cardiac fibrosis and dysfunction by epigenetically enhancing ATF3 expression. Exp Mol Med 54: 2175–2187. 10.1038/s12276-022-00904-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deogharia M, Agrawal A, Shi M, Jain AK, McHugh KJ, Altamirano F, Marian AJ, Gurha P (2023) Histone demethylase KDM5 regulates cardiomyocyte maturation by promoting fatty acid oxidation, oxidative phosphorylation, and myofibrillar organization. BioRxiv. 10.1101/2023.04.11.535169 (Preprint posted May 28, 2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Han Q, Peng T, Peng M, Wei B, Li D, Wang X, Yu S, Yang J, Cao S, et al. (2015) The oncogene c-Jun impedes somatic cell reprogramming. Nat Cell Biol 17: 856–867. 10.1038/ncb3193 [DOI] [PubMed] [Google Scholar]
- 40.Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, Greenberg ME (2017) AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol Cell 68: 1067–1082.e12. 10.1016/j.molcel.2017.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao N, Huang Y, Zheng J, Spencer CI, Zhang Y, Fu JD, Nie B, Xie M, Zhang M, Wang H, et al. (2016) Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science 352: 1216–1220. 10.1126/science.aaf1502 [DOI] [PubMed] [Google Scholar]
- 42.Shi Y, Zhang H, Huang S, Yin L, Wang F, Luo P, Huang H (2022) Epigenetic regulation in cardiovascular disease: Mechanisms and advances in clinical trials. Signal Transduct Target Ther 7: 200. 10.1038/s41392-022-01055-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W, Song M, Qu J, Liu GH (2018) Epigenetic modifications in cardiovascular aging and diseases. Circ Res 123: 773–786. 10.1161/CIRCRESAHA.118.312497 [DOI] [PubMed] [Google Scholar]
- 44.Qin J, Guo N, Tong J, Wang Z (2021) Function of histone methylation and acetylation modifiers in cardiac hypertrophy. J Mol Cell Cardiol 159: 120–129. 10.1016/j.yjmcc.2021.06.011 [DOI] [PubMed] [Google Scholar]
- 45.Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, et al. (2011) Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43: 145–155. 10.1016/j.molcel.2011.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol I, Love MI, Aiden EL, Bassik MC, Snyder MP (2017) Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol Cell 67: 1037–1048.e6. 10.1016/j.molcel.2017.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hutchins AP, Jauch R, Dyla M, Miranda-Saavedra D (2014) glbase: A framework for combining, analyzing and displaying heterogeneous genomic and high-throughput sequencing data. Cell Regen 3: 1. 10.1186/2045-9769-3-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hutchins AP, Yang Z, Li Y, He F, Fu X, Wang X, Li D, Liu K, He J, Wang Y, et al. (2017) Models of global gene expression define major domains of cell type and tissue identity. Nucleic Acids Res 45: 2354–2367. 10.1093/nar/gkx054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li B, Dewey CN (2011) RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Risso D, Schwartz K, Sherlock G, Dudoit S (2011) GC-content normalization for RNA-Seq data. BMC Bioinformatics 12: 480. 10.1186/1471-2105-12-480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar V, Muratani M, Rayan NA, Kraus P, Lufkin T, Ng HH, Prabhakar S (2013) Uniform, optimal signal processing of mapped deep-sequencing data. Nat Biotechnol 31: 615–622. 10.1038/nbt.2596 [DOI] [PubMed] [Google Scholar]
- 54.Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010) Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol 11: R14. 10.1186/gb-2010-11-2-r14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008) Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S, et al. (2020) Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367: 405–411. 10.1126/science.aax0249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie showing spontaneous contractions of cardiomyocytes from WT hESC in D15. Download video (5.8MB, mp4)
Source Data for Figure 2LSA-2023-02121_SdataF2.ai (5.3MB, ai)
Movie showing spontaneous contractions of cardiomyocytes from c-JUN KO hESC in D15. Download video (8.7MB, mp4)
Source Data for Figure S1LSA-2023-02121_SdataFS1.ai (2MB, ai)
Source Data for Figure 4LSA-2023-02121_SdataF4.ai (6.1MB, ai)
Source Data for Figure 5LSA-2023-02121_SdataF5.ai (2.1MB, ai)
Movie showing spontaneous contractions of cardiomyocytes from WT hESC without KDMi in D15. Download video (6MB, mp4)
Movie showing spontaneous contractions of cardiomyocytes from WT hESC with KDMi treatment in D15. Download video (6.7MB, mp4)
Table S1 Antibodies for Western blotting, immunofluorescence, and ChIP-seq. (15.2KB, docx)
Data Availability Statement
The raw sequence data reported in this work have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in the National Genomics Data Center (Nucleic Acids Res 2021), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: HRA003227) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.