

Abstract
The first total synthesis and structure validation of an arenimycin/SF2446 type II polyketide is described, as represented by de novo construction of SF2446 B3, the aglycone shared by this family of type II polyketides. Ruthenium-catalyzed α-ketol-benzocyclobutenone [4+2] cycloaddition, which occurs via successive stereoablation-stereoregeneration, affects a double dynamic kinetic asymmetric transformation (DYKAT) wherein two racemic starting materials combine to form the congested angucycline bay region with control of regio-, diastereo- and enantioselectivity. This work represents the first application of transfer hydrogenative cycloaddition and enantioselective intermolecular metal-catalyzed C–C bond activation in target-oriented synthesis.
Graphical Abstract

Introduction
Arenimycin A is a secondary metabolite of the marine actinomycete Salinispora arenicola (strain CNR-647) that was initially obtained from the Grand Bahama Island mangrove tunicate Ecteinascidia turbinate,1a and is coproduced with arenimycin B from Salinispora arenicola (strain CNB-527).1b,2 Arenimycins C and D were isolated through “mining” and heterologous expression of soil metagenomes.1c The closely related bacterial metabolites SF2446 A1, A2, B1–B3 were isolated from the soil actinomycete Streptomyces SF2446.3 Type II polyketides of the angucycline family,4 arenimycins A-D and SF2446 A1, A2, B1–B3 possess a highly uncommon benzo[a]naphthacene quinone ring system,5 and are most notably differentiated by their N-glycoside moieties (Figure 1).6 Arenimycins A-D display potent activity against rifampin- and methicillin-resistant Staphylococcus aureus (MRSA).1 Despite their promising antibacterial activity, the exceedingly low natural abundance of the arenimycins and SF2446 congeners has prohibited elucidation of their mechanism of action, yet their isolation from both marine and terrestrial microbes suggest the fundamental importance of their pharmacophore. The structural assignment of the arenimycins and SF2446 family type II polyketides has not been corroborated by total synthesis and efforts toward their de novo construction remain absent in the literature.
Figure 1.
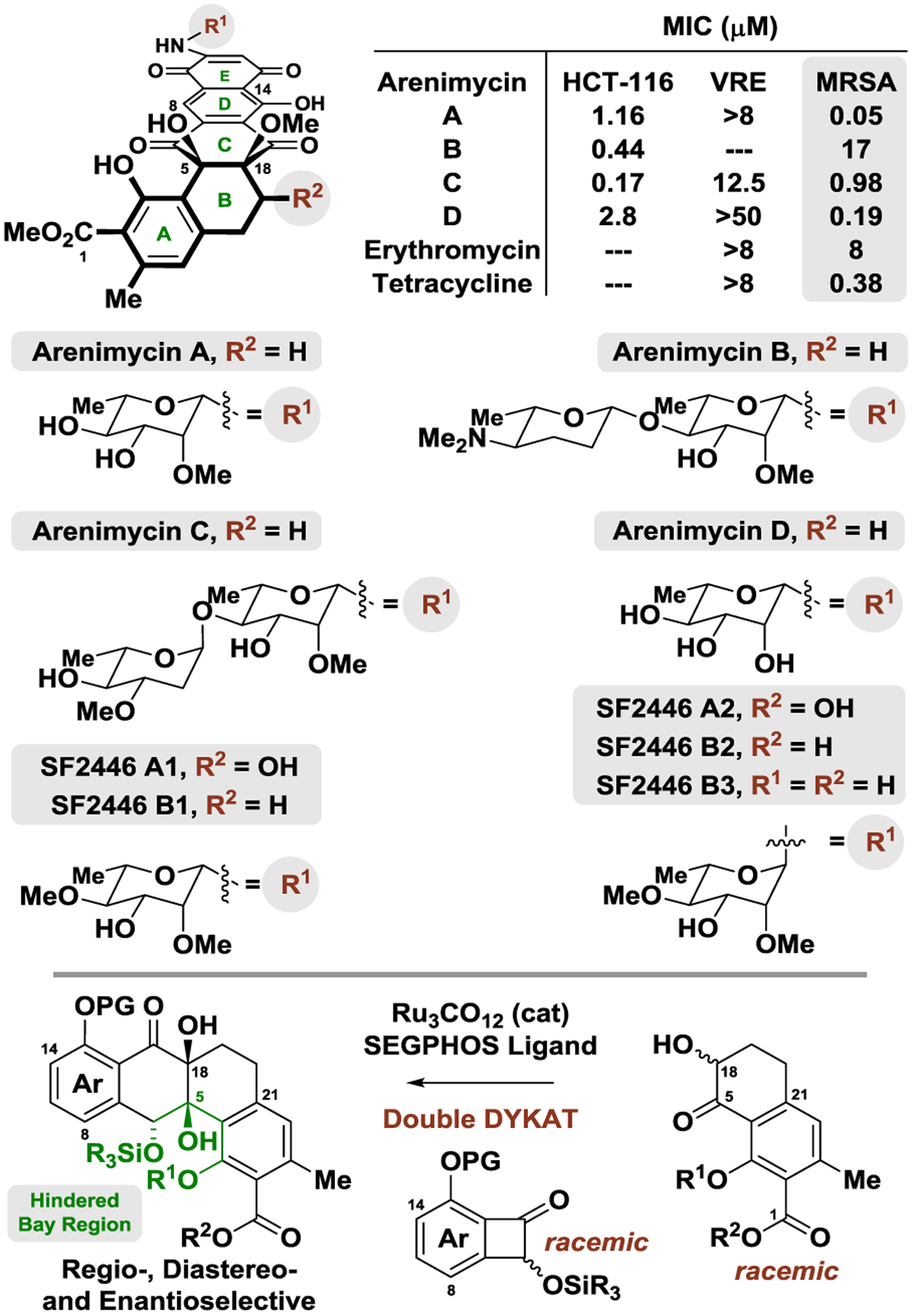
Arenimycins A-D and SF2446 A1, A2, B1–B3 and α-ketol-benzocyclobutenone [4+2] cycloaddition.
Our laboratory has developed a suite of catalytic enantioselective C–C bond formations that convert lower alcohols to higher alcohols.7 The redox-economy and site-selectivity of these hydrogen auto-transfer processes has allowed diverse type I polyketide natural products to be prepared in significantly fewer steps than previously possible.8 In more recent work, this reactivity mode was used to inform parallel protocols for convergent type II polyketide construction, resulting in the development of several unique ruthenium-catalyzed [4+2] cycloadditions.9,10 In particular, a powerful ruthenium-catalyzed [4+2] cycloaddition of 1,2-diols, ketols or diones with benzocyclobutenones was developed,9d,e which provides entry to type II polyketides bearing bridgehead diol motifs, which are often challenging to install.11 Furthermore, these cycloadditions are capable of forming angucycline ring systems bearing highly congested “bay regions” with control of regio-, diastereo- and enantioselectivity.9g In the first example of enantioselective intermolecular metal-catalyzed C–C bond activation in natural product total synthesis,12 we herewith report the total synthesis and structure validation of SF2446 B3, the aglycone common to the arenimycins and SF2446 family type II polyketides, via catalytic asymmetric α-ketol-benzocyclobutenone [4+2] cycloaddition.
Results and Discussion
Given the nuanced requirements of the α-ketol and arylcyclobutenone partners, their selection in connection with the present synthesis of SF2446 B3 is herewith described in detail (Table 1). In prior work on the development of the catalytic asymmetric α-ketol-benzocyclobutenone [4+2] cycloaddition,9g enantioselectivity was not only dependent on the chiral ligand (DM-SEGPHOS), but was also influenced by structural features of both the benzocyclobutenone and the tetralone-derived ketol. As illustrated (Table 1, entries 1–4),9g an essentially racemic reaction could be rendered highly enantioselective upon successive introduction of alkoxy substituents flanking the ketone moieties of the benzocyclobutenone (7a to 7b, Table 1, entries 1 and 2) and tetralone-derived α-ketol (12a to 12c, Table 1, entry 4). In the optimal case, benzocyclobutenone 7b and the α-ketol 12c, which incorporates an ortho-benzyl ethyl moiety, were converted to cycloadduct 13bc in 96% yield as 97:3 ratio of enantiomers (Table 1, entry 4). Based on these data, it was posited that the structurally related benzyl ether 12d required to access arenimycin/SF2446 core would be a competent partner for cycloaddition with benzocyclobutenone 7b. To our surprise it was not (Table 1, entry 5). As the corresponding methyl ether-containing α-ketol 12e participates in cycloaddition with benzocyclobutenone 7b (Table 1, entry 6), it became apparent the C3 benzyl ether is not well tolerated in the presence of the adjacent carbomethoxy group. The methyl ether of α-ketol 12e, however, is not an ideal protecting group for the C3 phenol of the arenimycin/SF2446 aglycone, so the related 1,3-dioxinone-containing α-ketol 12f was explored and, to our delight, could be converted to cycloadduct 13bf in good yield with high levels of regio- and stereocontrol (Table 1, entry 7).
Table 1.
Ruthenium-catalyzed [4+2] cycloaddition of benzocyclobutenones 7a-h with α-ketols 12a-g.a

|
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary-phase HPLC analysis. Diastereoselectivities were determined by 1H NMR analysis of crude reaction mixtures.
Diol (200 mol%).
Ru3(CO)12 (10 mol%), (R)-DM-SEGPHOS (30 mol%), 140 °C, 48 hr.
Ru3(CO)12 (3.3 mol%), (R)-DM-SEGPHOS (10 mol%), 140 °C. ND = Not determined. See Supporting Information for further details.
Having identified α-ketol 12f as a competent partner for ruthenium-catalyzed [4+2] cycloaddition, we turned our attention to the evaluation of potential cyclobutenone partners. The naphthoquinone-containing cyclobutenones 7c and 7d would be ideal as they incorporate native structural features of SF2446 B3. However, cycloadditions of 7c and 7d were not successful due to competing catechol formation from the transient 1,2-dione derived from α-ketol 12e (which was used due to its availability at the time) (Table 1, entries 8 and 9). Speculating that the quinone moiety of 7c and 7d may be incompatible with intervening ruthenium hydrides, the cycloaddition of the corresponding hydroquinone methyl ether 7e was attempted. However, under a variety of conditions, including temperatures as high as 200 °C, hydroquinone 7e failed to react and could be recovered in high yield from the reaction mixture (Table 1, entry 10). These data suggested that the greater loss of resonance stabilization energy may preclude cycloreversion of naphthocyclobutenone 7e or may shorten the lifetime of the ketene methide that is reversibly formed to a prohibitive extent; an hypothesis corroborated by the successful cycloaddition of the angular naphthocyclobutenone 7f with cyclohexane diol 12g (Table 1, entry 11)13 and DFT calculations (Figure 2). For this reason, the tetrahydronaphthocyclobutenones 7g and 7h were evaluated. The cycloadditions of 7g with α-ketols 12c-12e again revealed that the C3 benzyl ether is not well tolerated in the presence of the C1 carbomethoxy group (Table 1, entries 12–14). Finally, as aromatization of the oxa-bicycle moiety of cycloadducts derived from 7g was problematic, the tetralone-derived cyclobutenone 7h was subjected to cycloaddition with α-ketol 12f. The cycloadduct 13hf was formed in 74% yield with high levels of regio-, diastereo-, and enantioselectivity (Table 1, entry 15).
Figure 2.

Cycloreversion and loss of resonance stabilization energy vis-à-vis ruthenaindanone formation.a
aSee Supporting Information for further details.
The synthesis of benzocyclobutenone 7h begins with the palladium-catalyzed hydroxylation14 of 6-methoxy-α-tetralone 1 followed by hydrobromic acid mediated demethylation of the phenolic ether (Scheme 1).15 Chemoselective iodination16 of the resulting resorcinol 2 provides the iodide 3, which upon regioselective triflation17 delivers sulfonate 4. Successive exposure of 4 to sodium borohydride and dibromomethane provides the aryne precursor 5.18 Finally, [2+2] cycloaddition of the aryne derived from 5 with the silyl ketene acetal 6 delivers the benzocyclobutenone 7h in a total of 8 steps (longest linear sequence, LLS) from 6-methoxy-α-tetralone 1.19
Scheme 1.

Synthesis of tetrahydronaphthocyclobutenone 7h via aryne-mediated [2+2] cycloaddition.a
aYields of material isolated by silica gel chromatography. See Supporting Information for further details.
The preparation of α-ketol 12 begins with condensation of dimethyl oxoglutarate and acetylacetone to provide (after O-benzylation) the isophthalate 9 (Scheme 2).20 Staunton-Weinreb-type annulation of 9 using methyl acrylate followed by concomitant acid-mediated decarboxylation and debenzylation provides the 8-hydroxy-α-tetralone 10.21 Conversion of the 8-hydroxy-α-tetralone 10 to the 1,3-dioxinone 1122 followed by Rubottom-type oxidation23 delivered α-ketol 12f in a total of 8 steps (LLS) from dimethyl oxoglutarate and acetylacetone.
Scheme 2.

Synthesis of α-ketol 12f.a
aYields of material isolated by silica gel chromatography. See Supporting Information for further details.
The conversion of benzocyclobutenone 7h and α-ketol 12f to the penultimate synthetic intermediate en route to SF2446 B3, quinone 16, is highlighted in Scheme 3. Ruthenium-catalyzed cycloaddition9f of tetralone-derived cyclobutenone 7h with α-ketol 12f convergently assembles 13hf, which incorporates the complete carbon skeleton of the aglycone, including the congested angucycline bay region, with high levels of regio-, diastereo-, and enantioselectivity. Selective mono-methylation of the bridgehead 3°,3°-diol at C5,C18 by way of the tin ketal occurred exclusively at the less hindered C18-hydroxyl group.24 Exposure of the methyl ether to hydrochloric acid in organic media resulted in concomitant hydrolysis of the 1,3-dioxane at C13,C15 and elimination of the C13 hydroxyl to form the C12,C13 olefin 14. In a 3-step sequence, the C6 TIPS ether of 14 was removed, the C12,C13 olefin was converted to the bromohydrin and the C6 and C13 hydroxyls were simultaneously oxidized to form the triketone 15. Dehydrobromination mediated by DBU provided the highly insoluble phenol, which was directly subjected to copper-catalyzed oxidation to deliver the brick-red quinone 16.25
Scheme 3.

Conversion of benzocyclobutenone 7h and α-ketol 12f to the penultimate synthetic intermediate, quinone 16.a
aYields of material isolated by silica gel chromatography. See Supporting Information for further details.
Quinone 16 is a potential gateway to diverse arenimycins and SF2446 family type II polyketides. Direct aminoglycosylation via addition of β−1-amino-l-rhamnose 18 to quinone 16 followed by aerobic oxidation of the resulting hydroquinone was envisioned. While related oxidative amine additions to symmetric naphthoquinones have been reported,26 to our knowledge, the formation of N-glycosidic linkages in this manner has not been described. Potential selectivity issues associated with this strategy involved anomerization of 1-aminorhamnose and oxidative aminoglycosylation at C11 versus C12. A model system to probe the issue of regioselective oxidative aminoglycosylation was found in juglone 17. Exposure of juglone 17 to β−1-amino-l-rhamnose 18 in the presence of a copper catalyst in a reaction vessel under an atmosphere of air gave no reaction (Scheme 4). Under microwave conditions at higher loadings of catalyst (20 mol%), the N-glycoside formed but underwent cleavage in situ to form mixtures of 2- and 3-amino juglone. Gratifyingly, at lower loadings of the copper catalyst (5 mol%), glycosidic cleavage could be suppressed and the regioisomeric C2 and C3 N-glycosides 19a and 19b could be isolated in 85% yield as a 3:1 mixture. Despite the low reaction temperatures, microwave irradiation was required to promote conversion of juglone to adducts 19a and 19b. Single crystal X-ray diffraction analysis confirmed the structural assignment of 19a as the β-rhamnoside. Having established a favorable regiochemical bias in the model system of juglone 17, the optimized conditions for direct regioselective oxidative aminoglycosylation were applied to quinone 16. However, attempted methanolysis of the 1,3-dioxinone moiety under a variety of conditions resulted in only partial cleavage of the 1,3-dioxinone moiety to form the surprisingly stable formaldehyde hemiacetal.27 As 1,3-dioxinone methanolysis occurred smoothly on α-ketol 12f, it appeared that the hindered bay region of adducts derived from 16 impeded deprotection. However, under more forcing conditions, quinone 16 could be reacted with 18 to deliver the aglycone SF2446 B3 in 21% yield over the 2-step sequence as a single regioisomer. The NMR spectral data and optical rotation of synthetic SF2446 B3 matched that of the naturally occurring material, thus validating its structural assignment.
Scheme 4.

Development of direct regioselective oxidative aminoglycosylation and total synthesis of SF2446-B3.a
aYields of material isolated by silica gel chromatography. See Supporting Information for further details.
Conclusions
In summary, we report the first total synthesis and structure validation of an arenimycin/SF2446 type II polyketide, SF2446 B3; the aglycone shared by this family of type II polyketides. Whereas classical convergent type II polyketide total syntheses typically rely on fragment union via Hauser-Kraus-type annulations28 or aryne mediated cycloadditions,29 we demonstrate the utility of a unique enantioselective ruthenium-catalyzed cycloaddition for convergent type II polyketide construction: the [4+2] cycloaddition of α-ketols and benzocyclobutenones. This cycloaddition occurs via successive stereoablation-stereoregeneration, enabling a double dynamic kinetic asymmetric transformation (DYKAT) in which two racemic starting materials combine to construct the congested angucycline bay region with control of regio-, diastereo- and enantioselectivity. The synthetic challenges posed by the arenimycin/SF2446 type II polyketides also led us to develop of a novel method for the direct copper-catalyzed aerobic oxidative aminoglycosylation of quinones that occurs with high levels of regiocontrol. These studies and prior work from our laboratory contribute to a growing lexicon of catalytic methods for polyketide construction beyond premetalated reagents.
Supplementary Material
ACKNOWLEDGMENT
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM093905) are acknowledged for partial support of this research. We thank Dr. Thomas Wurm for kindly conducting DFT calculations. Undergraduates Jeremy J. Nicolai, Weston Bonnet and Lan Do provided skillful technical assistance. This work is dedicated to Professor Masahiro Murakami on the occasion of his retirement.
Footnotes
Supporting Information. Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra and HPLC traces for racemic and enantiomerically enriched compounds. Single-crystal X-ray diffraction data for compound 19a.
Accession Codes. CCDC 2234328 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).For isolation and antibacterial activity of arenimycins A-D, see:; (a) Asolkar RN; Kirkland TN; Jensen PR; Fenical W Arenimycin, an Antibiotic Effective Against Rifampin- and Methicillin-Resistant Staphylococcus aureus from the Marine Actinomycete Salinispora Arenicola. J. Antibiot 2010, 63, 37–39. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kersten RD; Ziemert N; Gonzalez DJ; Duggan BM; Nizet V; Dorrestein PC Moore BS Glycogenomics as a Mass Spectrometry-Guided Genome-Mining Method for Microbial Glycosylated Molecules. Proc. Nat. Acad. Sci. U.S.A 2013, 110, E4407–E4416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kang H-S; Brady SF Mining Soil Metagenomes to Better Understand the Evolution of Natural Product Structural Diversity: Pentangular Polyphenols as a Case Study. J. Am. Chem. Soc 2014, 136, 18111–18119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For a review of Salinispora-derived secondary metabolites, see:; Jensen PR; Moore BS; Fenical W The Marine Actinomycete Genus Salinispora: A Model Organism for Secondary Metabolite Discovery. Nat. Prod. Rep 2015, 32, 738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).For isolation and antibacterial activity of SF2446 A1, A2, B1–B3, see:; (a) Takeda U; Okada T; Takagi M; Gomi S; Itoh J; Sezaki M; Ito M; Miyahdoh S; Shomura T SF2446, New Benzo[a]naphthacene Quinone Antibiotics. I. Taxonomy and Fermentation of the Producing Strain, Isolation and Characterization of Antibiotics. J. Antibiot 1988, 41, 417–424. [DOI] [PubMed] [Google Scholar]; (b) Gomi S; Sasaki T; Itoh J; Sezaki M SF2446, New Benzo[a]naphthacene Quinone Antibiotics. II. The Structural Elucidation. J. Antibiot 1988, 41, 425–432. [DOI] [PubMed] [Google Scholar]
- (4).For selected reviews on angucycline natural products, see:; (a) Krohn K; Rohr J Angucyclines: Total Syntheses, New Structures, and Biosynthetic Studies of an Emerging New Class of Antibiotics. Top. Curr. Chem 1997, 188, 127–195. [Google Scholar]; (b) Carreño MC; Urbano A Recent Advances in the Synthesis of Angucyclines. Synlett 2005, 1–25. [Google Scholar]; (c) Kharel MK; Pahari P; Shepherd MD; Tibrewal N; Nybo SE; Shaaban KA; Rohr J Angucyclines. Biosynthesis, Mode-of-Action, New Natural Products, and Synthesis. Nat. Prod. Rep 2012, 29, 264–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Highly uncommon in nature, the arenimycin ring system is found in only a few other natural products, including collinone (ref. a), G-2N and G-2A (ref. b), KS-619 (ref. c) and BE-19412A (ref. d):; (a) Martin R; Sterner O; Alvarez MA; De Clercq E; Bailey JE; Minas W Collinone, A New Recombinant Angular Polyketide Antibiotic Made by an Engineered Streptomyces Strain. J. Antibiot 2001, 54, 239–249. [DOI] [PubMed] [Google Scholar]; (b) Gerber NN; Lechevalier MP Novel Benzo[a]naphthacene Quinone from an Actinomycete, Frankia G-2 (ORS 020604). Can J. Chem 1984, 62, 2818–2821. [Google Scholar]; (c) Yasuzawa T; Yoshida M; Shirahata K; Sano H Structure of a Novel Ca and Calmodulin-Dependent Cyclic Nucleotide Phospodiesterase Inhibitor KS-619–1. J. Antibiot 1987, 40, 1111–1114. [DOI] [PubMed] [Google Scholar]; (d) Sukamoto M; Nakajima S; Awakawa H; Sugiura Y; Suzuki H; Hirayama M; Kamiya S; Teshima Y; Kondo H; Kojiri K; Suda H A New Antitumor Antibiotic, BE-19412A, Produced by a Streptomycete. J. Antibiot 1998, 51, 908–914. [DOI] [PubMed] [Google Scholar]
- (6).The arenimycins present an excellent opportunity to study glycoside-dependent antibacterial activity:; (a) Křen V; Řezanka T Sweet Antibiotics - The Role of Glycosidic Residues in Antibiotic and Antitumor Activity and Their Randomization. FEMS Microbiol. Rev 2008, 32, 858–889. [DOI] [PubMed] [Google Scholar]; (b) Elshahawi Sherif I.; Shaaban Khaled A.; Kharel Madan K.; Thorson Jon S. A Comprehensive Review of Glycosylated Bacterial Natural Products. Chem. Soc. Rev 2015, 44, 7591–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Grynkiewicz G; Szeja W Synthetic Glycosides and Glycoconjugates of Low Molecular Weight Natural Products. Curr. Pharm. Des 2016, 22, 1592–1627. [DOI] [PubMed] [Google Scholar]
- (7).For selected reviews, see:; (a) Ketcham JM; Shin I; Montgomery TP; Krische MJ Catalytic Enantioselective C-H Functionalization of Alcohols by Redox-Triggered Carbonyl Addition: Borrowing Hydrogen, Returning Carbon. Angew. Chem. Int. Ed 2014, 53, 9142–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ Metal-Catalyzed Reductive Coupling of Olefin-Derived Nucleophiles: Reinventing Carbonyl Addition. Science 2016, 354, aah5133. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res 2017, 50, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Santana CG; Krische MJ From Hydrogenation to Transfer Hydrogenation to Hydrogen Auto-Transfer in Enantioselective Metal-Catalyzed Carbonyl Reductive Coupling: Past, Present and Future. ACS Catal. 2021, 11, 5572–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ortiz E; Shen W; Shezaf JZ; Krische MJ Ruthenium-Catalyzed Dehydrogenation: Historical Perspective and Survey of Enantioselective for Conversion of Lower Alcohols to Higher Alcohols. Chem. Sci 2022, 13, 12625–12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For selected reviews, see:; (a) Dechert-Schmitt A-MR; Schmitt DC; Gao X; Itoh T; Krische MJ Polyketide Construction via Hydrohydroxyalkylation and Related Alcohol CH Functionalizations: Reinventing the Chemistry of Carbonyl Addition. Nat. Prod. Rep 2014, 31, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Feng J; Kasun ZA; Krische MJ Enantioselective Alcohol C-H Functionalization for Polyketide Construction: Unlocking Redox-Economy and Site-Selectivity for Ideal Chemical Synthesis. J. Am. Chem. Soc 2016, 138, 5467–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis. Angew. Chem. Int. Ed 2019, 58, 14055–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Geary LM; Glasspoole BW; Kim MM; Krische MJ Successive C–C Coupling of Dienes to Vicinally Dioxygenated Hydrocarbons: Ruthenium Catalyzed [4+2] Cycloaddition across the Diol, Hydroxycarbonyl or Dione Oxidation Levels. J. Am. Chem. Soc 2013, 135, 3796–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saxena A; Perez F; Krische MJ Ruthenium(0) Catalyzed Endiyne-α-Ketol [4+2] Cycloaddition: Convergent Assembly of Type II Polyketide Substructures. J. Am. Chem. Soc 2015, 137, 5883–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Saxena A; Perez F; Krische MJ Ruthenium(0) Catalyzed [4+2] Cycloaddition of Acetylenic Aldehydes with α-Ketols: Convergent Construction of Angucycline Ring Systems. Angew. Chem. Int. Ed 2016, 55, 1493–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bender M; Turnbull BWH; Ambler BR; Krische MJ Ru-Catalyzed Insertion of Adjacent Diol Carbon Atoms into C–C bonds: Entry to Type II Polyketides. Science 2017, 357, 779–781. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ambler BR; Turnbull BWH; Suravarapu SR; Uteuliyev M; Huynh NO; Krische MJ Enantioselective Ruthenium-Catalyzed Benzocyclobutenone–Ketol Cycloaddition: Merging C–C Bond Activation and Transfer Hydrogenative Coupling for Type II Polyketide Construction. J. Am. Chem. Soc 2018, 140, 9091–9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For selected reviews, see:; (a) Sato H; Turnbull BWH; Fukaya K; Krische MJ Ruthenium(0) Catalyzed Cycloaddition of 1,2-Diols, Ketols or Diones via Alcohol-Mediated Hydrogen Transfer. Angew. Chem. Int. Ed 2018, 57, 3012–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Doerksen RS; Hodík T; Hu G; Huynh NO; Shuler WG; Krische MJ Ruthenium-Catalyzed Cycloaddition to Form Five-, Six-, and Seven-Membered Rings. Chem. Rev 2021, 121, 4045–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nicolaou KC; Hale CRH; Nilewski C; Ioannidou HA; ElMarrouni A; Nilewski LG; Beabout K; Wang TT Shamoo Y Total Synthesis of Viridicatumtoxin B and Analogues Thereof: Strategy Evolution, Structural Revision, and Biological Evaluation. J. Am. Chem. Soc 2014, 136, 12137–12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For selected reviews on metal-catalyzed C–C bond activation, see:; (a) Bishop KC III. Transition Metal Catalyzed Rearrangements of Small Ring Organic Molecules. Chem. Rev 1976, 76, 461–486. [Google Scholar]; (b) Kondo T; Mitsudo T.-a. Ruthenium-Catalyzed Reconstructive Synthesis of Functional Organic Molecules via Cleavage of Carbon–Carbon Bonds. Chem. Lett 2005, 34, 1462–1467. [Google Scholar]; (c) Murakami M; Matsuda T Metal-Catalyzed Cleavage of Carbon-Carbon Bonds. Chem. Comm 2011, 47, 1100–1105. [DOI] [PubMed] [Google Scholar]; (d) Nakao Y Catalytic C–CN Bond Activation. Top. Curr. Chem 2014, 346, 33–58. [DOI] [PubMed] [Google Scholar]; (e) Souillart L; Cramer N Catalytic C–C Bond Activations via Oxidative Addition to Transition Metals. Chem. Rev 2015, 115, 9410–9464. [DOI] [PubMed] [Google Scholar]; (f) Chen P.-h.; Dong G Cyclobutenones and Benzocyclobutenones: Versatile Synthons in Organic Synthesis. Chem. Eur. J 2016, 22, 18290–18315. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Murakami M; Ishida N Potential of Metal-Catalyzed C–C Single Bond Cleavage for Organic Synthesis. J. Am. Chem. Soc 2016, 138, 13759–13769. [DOI] [PubMed] [Google Scholar]; (h) Chen P.-h.; Billet BA; Tsukamoto T; Dong G “Cut and Sew” Transformations via Transition-Metal-Catalyzed Carbon–Carbon Bond Activation. ACS Catal. 2017, 7, 1340–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Fumagalli G, Stanton S, Bower JF, Recent J Methodologies That Exploit C–C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes. Chem. Rev 2017, 117, 9404–9432. [DOI] [PubMed] [Google Scholar]
- (13).For preparation of naphtho[a]- and naphtho[b]cyclobutenes and enhanced reactivity of the latter angular regioisomer, see:; (a) Cava MP; Shirley RL Condensed Cyclobutane Aromatic Compounds. X. Naphtho[b]cyclobutene. J. Am. Chem. Soc 1960, 82, 654–656. [Google Scholar]; (b) Cava MP; Shirley RL Condensed Cyclobutane Aromatic Compounds. XVII. Naphtho[a]cyclobutene. J. Org. Chem 1962, 27, 755–757. [Google Scholar]; Also see MNDO calculations in the following study,; (c) Mayer A; Meier H 1H-Naphtho[2,1-b]thiete and 2H-Naphtho[2,3-b]thiete - Synthesis and Reactivity. Tetrahedron Lett. 1994, 35, 2161–2164. [Google Scholar]
- (14).(a) Shan G; Yang X; Ma L; Rao Y Pd-Catalyzed C-H Oxygenation with TFA/TFAA: Expedient Access to Oxygen-Containing Heterocycles and Late-Stage Drug Modification. Angew. Chem. Int. Ed 2012, 51, 13070–13074. [DOI] [PubMed] [Google Scholar]; (b) Mo F; Trzepkowski LJ; Dong G Synthesis of ortho-Acylphenols through the Palladium-Catalyzed Ketone-Directed Hydroxylation of Arenes. Angew. Chem. Int. Ed 2012, 51, 13075–13079. [DOI] [PubMed] [Google Scholar]; (c) Choy PY; Kwong FY Palladium-Catalyzed ortho-CH-Bond Oxygenation of Aromatic Ketones. Org. Lett 2013, 15, 270–273. [DOI] [PubMed] [Google Scholar]
- (15).(a) Haberland G Versuche zur Synthese Natürlicher Sterine, I. Mitteil.: Ringschlußmöglichkeiten Verschieden Hydrierter Naphthyl-buttersäuren und ein Bequemer Weg zur Gewinnung des 7-Methoxy-1-keto-1.2.3.4-tetrahydro-phenanthrens. Chem. Ber 1936, 69, 1380–1386. [Google Scholar]; (b) Almansa C; Carceller E; Bartrolí J; Forn J A Short, Efficient Synthesis of 6-Cyano-1-Tetralones. Synth. Commun 1993, 23, 2965–2971. [Google Scholar]
- (16).Thomsen I; Torssell KBG Iodination of Resorcinol, 5-Methoxyresorcinol, Phloroglucinol and Resorcyclic Acid. Acta Chem. Scand 1991, 45, 539–542. [Google Scholar]
- (17).Matsumoto T; Yamaguchi H; Suzuki K C-Glycosyl Juglone in Angucycline Synthesis: Total Synthesis of Galtamycinone, Common Aglycon of C-Glysosyl Naphthacenequinone-Type Angucylines. Tetrahedron 1997, 53, 16533–16544. [Google Scholar]
- (18).Castro MA; del Corral JMM; Gordaliza M; Gómez-Zurita MA; de la Puente ML; Betancur-Galvis LA; Sierra J; San Feliciano A Synthesis, Cytotoxicity and Antiviral Activity of Podophyllotoxin Analogues Modified in the E-Ring. Eur. J. Med. Chem 2003, 38, 899–911. [DOI] [PubMed] [Google Scholar]
- (19).(a) Hamura T; Hosoya T; Yamaguchi H; Kuriyama Y; Tanabe M; Miyamoto M; Yasui Y; Matsumoto T; Suzuki K Facile Access to Versatile Polyaromatic Building Blocks: Selectively Protected Benzocyclobutenedione Derivatives via Regioselective [2+2] Cycloaddition of α-Alkoxybenzyne and Ketene Silyl Acetal. Helv. Chim. Acta 2002, 85, 3589–3604. [Google Scholar]; (b) Takemura I; Imura K; Matsumoto T; Suzuki K Concise Three-Component Synthesis of Defucogilvocarcin M. Org. Lett 2004, 6, 2503–2505. [DOI] [PubMed] [Google Scholar]
- (20).Bertz SH An Improved Synthesis of Some Highly Substituted Phenols – The Prelog Condensation with 2,4,6-Heptanetrione. Synthesis 1980, 708–710. [Google Scholar]
- (21).(a) Tarnchompoo B; Thebtaranonth C; Thebtaranonth Y Synthesis of 8-Methoxy-1-Tetralones. Synthesis 1986, 18, 785–786. [Google Scholar]; (b) Caron B; Brassard P An Integrated Approach to the Synthesis of Contiguously Substituted Xanthopurpurins, Pachybasins and Purpurins. Tetrahedron 1993, 49, 771–784. [Google Scholar]
- (22).Cui X; Lin F; Song Q; Gao Y Method for synthesizing 4H-1,3-benzodioxin-4-one compound. CN103709137 A, April 9, 2014.
- (23).McCormick JP; Tomasik W; Johnson MW α-Hydroxylation of Ketones: Osmium Tetroxide/N-Methylmorpholine-N-Oxide Oxidation of Silyl Enol Ethers. Tetrahedron Lett. 1981, 22, 607–610. [Google Scholar]
- (24).Haque ME; Kikuchi T; Yoshimoto K; Tsuda Y Regioselective Monoalkylation of Non-protected Glycopyranosides by the Dibutyltin Oxide Method. Chem. Pharm. Bull 1985, 33, 2243–2255. [Google Scholar]
- (25).Jiang J-H; Boominathan SSK; Hu W-P; Chen C-Y; Vandavasi JK; Lin Y-T; Wang J-J Sequential, One-Pot Access to Arylated Benzoquinones/Naphthoquinones from Phenols/Naphthols. Eur. J. Org. Chem 2016, 2284–2289. [Google Scholar]
- (26).Lisboa C. d. S.; Santos VG; Vaz BG; de Lucas NC; Eberlin MN; Garden SJ C-H Functionalization of 1,4-Naphthoquinone by Oxidative Coupling with Anilines in the Presence of a Catalytic Quantity of Copper (II) Acetate. J. Org. Chem 2011, 76, 5264–5273. [DOI] [PubMed] [Google Scholar]
- (27).Formaldehyde-phenol hemiacetals are isolable and remarkably stable:; James R; Glen JB Synthesis, Biological Evaluation, and Preliminary Structure-Activity Considerations of a Series of Alkylphenols as Intravenous Anesthetic Agents. J. Med. Chem 1980, 23, 1350–1357. [DOI] [PubMed] [Google Scholar]
- (28).For selected reviews on Hauser-Kraus and related annulations, see:; (a) Mitchell AS; Russell RA Tetrahedron 1995, 51, 5207–. [Google Scholar]; (b) Rathwell K; Brimble MA Use of Stabilized Phthalide Anion Annulation Reactions in Synthesis: An Update. Synthesis 2007, 643–662. [Google Scholar]; (c) Mal D; Pahari P Recent Advances in the Hauser Annulation. Chem. Rev 2007, 107, 1892–1918. [DOI] [PubMed] [Google Scholar]; (d) de Koning CB; Georgiou KH, Michael JP; Rousseau AL Hauser–Kraus, Sammes, Staunton–Weinreb, and Tamura Annulations. Org. React 2021, 107, DOI: 10.1002/0471264180.or107.02. [DOI] [Google Scholar]; (e) Gartman JA; Tambar UK Recent Total Syntheses of Anthraquinone-Based Natural Products. Tetrahedron 2022, 105, 132501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).For selected examples of the use of aryne-mediated benzannulation in the synthesis of Type II polyketides, see:; (a) Hosoya T; Takashiro E; Matsumoto T; Total Synthesis of the Gilvocarcins. Suzuki, K. J. Am. Chem. Soc 1994, 116, 1004–1015. [Google Scholar]; (b) Matsumoto T; Sohma T; Yamaguchi H; Kurata S; Suzuki K Benzyne-Furan Cycloaddition Approach to the Angucyclines: First Total Synthesis of Antibiotic C104. Synlett 1995, 263–266. [Google Scholar]; (c) Chen C-L; Sparks SM; Martin SF C-Aryl Glycosides via Tandem Intramolecular Benzyne-Furan Cycloadditions. Total Synthesis of Vineomycinone B2 Methyl Ester. J. Am. Chem. Soc 2006, 128, 13696–13697. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) O’Keefe BM; Mans DM; Kaelin DE Jr.; Martin SF Total Synthesis of Isokidamycin. J. Am. Chem. Soc 2010, 132, 15528–15530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.