

Abstract
The lower female reproductive tract is notoriously dominated by Lactobacillus species, among which Lactobacillus crispatus emerges for its protective and health‐promoting activities. Although previous comparative genome analyses highlighted genetic and phenotypic diversity within the L. crispatus species, most studies have focused on the presence/absence of accessory genes. Here, we investigated the variation at the single nucleotide level within protein‐encoding genes shared across a human‐derived L. crispatus strain selection, which includes 200 currently available human‐derived L. crispatus genomes as well as 41 chromosome sequences of such taxon that have been decoded in the framework of this study. Such data clearly pointed out the presence of intra‐species micro‐diversities that could have evolutionary significance contributing to phenotypical diversification by affecting protein domains. Specifically, two single nucleotide variations in the type II pullulanase gene sequence led to specific amino acid substitutions, possibly explaining the substantial differences in the growth performances and competition abilities observed in a multi‐strain bioreactor culture simulating the vaginal environment. Accordingly, L. crispatus strains display different growth performances, suggesting that the colonisation and stable persistence in the female reproductive tract between the members of this taxon is highly variable.
Within the lower female reproductive tract, Lactobacillus crispatus emerged for its protective and health‐promoting activities. The data presented here clearly pointed out the presence of intra‐species micro‐diversities that could have evolutionary significance contributing to phenotypical diversification by affecting protein domains.
INTRODUCTION
Bacteria evolved over millions of years to colonise different districts of the human body, for example skin, pulmonary, gastrointestinal and vaginal tracts, giving rise to complex and dynamic populations of microorganisms engaged in close relationships with the human host, referred to as the microbiota (Blum, 2017). In particular, the gut microbiota, with its vastity of microbial genera and species, has attracted increasing interest in the last decades for its ability to impact several aspects of human health, development and systemic physiology from infancy to adulthood (Sommer & Bäckhed, 2013; Strati & Facciotti, 2022; Tarracchini et al., 2022; Turroni, Rizzo et al., 2022; Turroni, van Sinderen et al., 2022; Warner, 2019). In contrast, the vaginal microbiome is typically manifested by a low degree of (bio)diversity and is commonly dominated by members of the Lactobacillus genus, such as Lactobacillus iners, L. gasseri, L. jensenii and L. crispatus. This latter is regarded as the primary determinant of vaginal health (Lepargneur, 2016; Tachedjian et al., 2017). Indeed, in healthy cervicovaginal microbiota, L. crispatus species prevails, producing D‐ and L‐lactic acid, hydrogen peroxide and bacteriocins, which prevent the overgrowth of possible pathogens, hence preventing upper genital tract infections in the host (Sanozky‐Dawes & Barrangou, 2022; Stapleton et al., 2011). For such reason, probiotic supplements based on L. crispatus are widely used as vehicles of health‐promoting strains in the vaginal environment (Bohbot et al., 2018; Cohen et al., 2020; Mändar et al., 2023).
Recently, the evolution of the genome sequences of L. crispatus species has been studied in relation to its adaptation to the human vaginal niche, underlining strain‐dependent efficiency to grow on glycogen as well as to inhibit pathogens (Abdelmaksoud et al., 2016; Argentini et al., 2022; Mendes‐Soares et al., 2014; Ojala et al., 2014; Puebla‐Barragan et al., 2021; van der Veer et al., 2019). Moreover, besides the human vaginal tract, L. crispatus have also been identified and isolated from various (sub)niches, ranging from healthy poultry gut to various districts of the human body, including the oral cavity, rectum, and urinary tract, highlighting within‐species genetic diversity and variegated metabolic capabilities (France et al., 2016; Mancabelli et al., 2021; Pan et al., 2020; Zhang et al., 2020). Taken together, this evidence suggests the presence of distinct evolutive trajectories underlying the observed phenotypic diversification within this species. However, comparative genomic analyses involving chromosome sequences of L. crispatus species have often focused on the relationship between the presence/absence of accessory genes and a particular phenotype (Pan et al., 2020; Zhang et al., 2020), overshadowing the importance of mutations to within‐species evolution (Juhas et al., 2011; Martínez‐Carranza et al., 2018; Rousset et al., 2021).
In this framework, the aim of this study is to evaluate genome sequence variations at the single nucleotide level within protein‐encoding genes shared across non‐identical L. crispatus chromosomes, providing a close‐up view of genetic (micro)diversity, which can contribute significantly to strain diversification within this species. In addition, to investigate the possible implications of the identified genetic differences in the L. crispatus intra‐species competition within the vaginal microbiota, we performed in vitro experiments consisting of carbohydrate growth assays involving L. crispatus multi‐strain co‐cultivation in a bioreactor simulating the vaginal tract.
Our findings revealed inter‐strain genotypic variation and phenotypic differences between L. crispatus strains, highlighting distinct evolutionary developments that may provide this species with differential abilities to long persist and predominate in the human vaginal tract.
EXPERIMENTAL PROCEDURES
Isolation of L. crispatus strains and retrieval of publicly available genome sequences
Candidate Lactobacillus strains were obtained from an isolation effort performed in a framework a previous study.
Identification of newly isolated L. crispatus strains was achieved through the Matrix‐Assisted Laser Desorption/Ionization Time‐of‐Flight Mass Spectrometry (MALDI‐TOF MS) Biotyper Sirius (Bruker) using the manufacturer's software FlexControl and the MALDI‐Biotyper software (MBT). In detail, a single bacterial colony grown on MRS agar was transferred onto a spot of the MSP 96 target polished steel BC MALDI target plate (Bruker). Subsequently, the bacterial sample was overlaid with 1 μL of matrix solution containing 10 mg/mL HCCA (a‐cyano‐4‐hydroxycinnamic acid, Sigma‐Aldrich) resolved in 50% acetonitrile (Carlo Erba) and 2.5% TFA (trifluoro‐acetic acid, Carlo Erba) and air‐dried (Schulthess et al., 2014; Werner et al., 2012). The MALDI target plate was then introduced into the spectrometer for automated measurement and data interpretation. The mass spectra were processed with the MALDI Biotyper 3.0 software package (Bruker) containing reference spectra, including different lactobacilli species. According to the criteria recommended by the manufacturer, a score of ≥2.000 indicates a significant similarity between the obtained spectrum and the database entry. Each sample was analysed in duplicate (two spots for each sample).
The default parameter settings are as follows: positive linear mode, laser frequency 200 Hz, ion source 1 = 19.84 kV, ion source 2 = 18.07 kV, Bruker's MBT_FC and MBT_AutoX methods, mass range: 2000–20,000 Da. Moreover, before analysis, calibration was performed with a bacterial test standard (Bruker) containing an extract of Escherichia coli DH5 alpha.
A total of 34 L. crispatus strains were identified and taken forward for whole genome sequencing. In addition, seven L. crispatus strains isolated from human biological samples were purchased from international bacterial collections. To perform chromosomal DNA extraction, the 41 L. crispatus strains were cultivated in MRS broth supplemented with 0.05% (wt/vol) L‐cysteine hydrochloride in an anaerobic atmosphere at 37°C (2.99% [vol/vol] H2, 17.01% [vol/vol] CO2, and 80% [vol/vol] N2) for 12 h. Subsequently, cells from 10 mL of the culture were harvested by centrifugation at 6000 rpm for 8 min, and the obtained cell pellet was used for DNA extraction using the GenElute bacterial genomic DNA kit (Sigma‐Aldrich) following the manufacturer's guide.
Furthermore, 200 L. crispatus genome assemblies with completeness >95% derived from strains isolated from biological material of human subjects were retrieved from the NCBI database.
Genome sequencing, assembly, and annotation
The DNA extracted from the 41 L. crispatus strains was subjected to whole‐genome sequencing using MiSeq (Illumina) at GenProbio srl, Parma, Italy (www.genprobio.com) according to the supplier's protocol (Illumina). Individual genome libraries were generated using the Nextera XT preparation kit and loaded into a 600‐cycle (250‐bp paired‐ends) flow cell version 3 (Illumina). Raw DNA sequence reads (fastq files) obtained from genome sequencing were assembled using the MEGAnnotator pipeline (Lugli et al., 2016). Briefly, SPAdes software was used for de novo assembly of the genome sequences with the pipeline option “‐‐carefull” and a list of k‐mer sizes of 21; 33; 55; 77; 99; 127 (Bankevich et al., 2012) and protein‐encoding genes were predicted for contig greater than 1000 bp using Prodigal (Hyatt et al., 2010). Functionally, annotation of the predicted genes was achieved through RAPSearch2 (cut‐off E value, 1 × 10−5; minimum alignment length, 20 amino acids) (Zhao, Tang et al., 2012) and hidden Markov model (HMM) profile searches (cut‐off E value, 1 × 10−10; http://hmmer.org/) performed against the NCBI nr database and the manually curated Pfam‐A database, respectively. Moreover, tRNA genes were determined using tRNAscan‐SE version 1.4 (Lowe & Eddy, 1997), and rRNA loci were identified with RNAmmer version 1.2 (Lagesen et al., 2007).
Furthermore, to obtain comparable quality standards for the analysed genomes, all the 200 L. crispatus genomes retrieved from the NCBI database were re‐annotated employing the same approach based on MEGAnnotator pipeline used for the 41 L. crispatus genomes decoded in the current study.
Pangenome analyses and phylogenomic tree reconstruction
All pangenome calculations were performed using PGAP [PanGenomes Analysis Pipeline, (Zhao, Wu et al., 2012)] as described previously (Lugli et al., 2019; Tarracchini et al., 2020). In detail, orthologous protein sequences were identified in genome sequences using BLAST analysis (cut‐off E‐value = 1 × 10−5; 50% identity over at least 80% of sequence coverage) and then organised into functional Clusters of Orthologous Groups (COGs) through the MCL algorithm (graph‐based Markov clustering algorithm) using the gene family (GF) method. Pangenome profiles were produced through an optimised procedure integrated into the PGAP software, based on a presence/absence matrix including all COGs identified in the given genomes. The concatenated protein sequences of core genes were aligned using Mafft v7.453 (Katoh et al., 2002) and then employed to build correspondent phylogenomic trees through the neighbour‐joining method in ClustalW version 2.1. Visual core genome‐based phylogenomic trees were developed using FigTree software (http://tree.bio.ed.ac.uk/software/figtree/).
Single‐nucleotide polymorphism identification
The species‐specific level of polymorphisms within the Lactobacillus genus was assessed exploiting the identified core gene set shared between L. crispatus and seven different Lactobacillus species. In detail, 159 core‐shaping genes were concatenated and aligned using the multiple genome aligner Mafft v7.453 (Katoh et al., 2002). Nucleotide variants at each sequence position were then extracted through the SNP‐sites program (version 2.5.1) (Page et al., 2016). Assuming that, unlike sequencing errors, real genetic variants should be observed in a quite number of independent genomes assembly, we considered only sequence positions in which two or more alternatives were observed in at least 20% of genome collection. The number of intra‐species SNPs obtained for each Lactobacillus species was converted into SNPs per Mbp to account for variation in genome length.
In a similar fashion, both concatenated and individual gene nucleotide sequences comprised within the L. crispatus core genome were aligned with Mafft v7.453 and parsed with the SNP‐site software. For these analyses, the genome sequence of the most divergent L. crispatus strain (assembly number GCF_015669875.1) was used as reference sequence. Synonymous and non‐synonymous nucleotide variations were discriminated using the ParaAT 2.0 software (Zhang et al., 2012) combined with KaKs Calculator 3.0 toolkit (Zhang, 2022). Whole‐genome SNPs were extracted by combining the short‐reads aligner BWA and the VarScan tool (version 2.3.6).
Carbohydrate growth assay
In vitro growth assays with different carbon sources, such as starch, amylopectin, pullulan, maltodextrin, glycogen and mucin, were performed on selected L. crispatus strains, that is LB97, LMG11440, LMG18200 and LMG11415. In detail, the four L. crispatus strains were cultivated overnight on a semisynthetic MRS medium supplemented with 0.05% (w/vol) L‐cysteine hydrochloride at 37°C under anaerobic conditions. Subsequently, cells were diluted in MRS without glucose to obtain an OD600 nm = 1, and 15 μL of the diluted cells were inoculated in 135 μL of MRS without glucose supplemented with 1% (wt/vol) of a particular sugar in a 96‐well microtiter plate and incubated in an anaerobic cabinet. Specifically, each carbohydrate was dissolved in MRS without glucose previously sterilised by autoclaving at 121°C for 15 min. Subsequently, the obtained solutions were sterilised using a 0.2 μm filter size before use. Cell growth was evaluated by monitoring the OD at 600 nm using a plate reader (Biotek). Each plate was read in discontinuous mode, with absorbance readings performed thrice at 3‐min intervals after 48 h of growth, and each reading was ahead of 30s of shaking at medium speed. Cultures were performed in triplicates for each strain, and the resulting growth data were expressed as the average OD600nm of these independent biological replicates. Carbohydrates tested in this study were purchased from Merck and Fisher Scientific, ACROS Organics and include soluble starch from potato, amylopectin from maize, pullulan, maltodextrin, glycogen from beef liver, mucin from porcine stomach. The semisynthetic MRS medium was used as the control medium.
Co‐culture using a bioreactor system
The four selected L. crispatus strains (reported above) were grown anaerobically at 37°C for 24 h in simulated vaginal fluid (SVF) (Pan et al., 2020) to adapt the microorganisms to the medium. Next, revitalised cells were inoculated in a bioreactor system (Solaris Biotech Solutions) to obtain a final concentration per bacterial strain of 5 × 106 cells/mL in 400 mL of SVF. The co‐culture of the four L. crispatus strains was performed with a non‐continuous supply of the growth medium for the first 12 h to stabilise the microbial community. Subsequently, the cultivation was shifted to a continuous mode to provide fresh SVF medium and continued for 48 h under anaerobic conditions at 37°C with a mechanical agitation set at 180 rpm. In addition, the pH was maintained at 4.5 by adding 2.5 M NaOH to mimic the pH of the human vaginal environment (Pan et al., 2020). During bacterial growth, culture aliquots were collected at 10, 24, and 48.
DNA extraction and shotgun metagenomic sequencing
Each aliquot collected from bioreactor cultivation was subjected to DNA extraction using the ZymoBIOMICS DNA Miniprep Kit (Zymo Research, D4300) following the manufacturer's instructions. Then, after assessing DNA concentration and purity using a BioPhotometer D30 (Eppendorf, Germany), each DNA sample was sequenced by GenProbio srl, Parma, Italy (www.genprobio.com) employing next‐generation sequencing technique (shotgun metagenomic sequencing). The preparation of DNA libraries was performed using the Nextera XT DNA sample preparation kit (Illumina) according to the manufacturer's instructions, using 1 ng of DNA from each metagenomic sample. The isolated DNA underwent fragmentation, adapter ligation and purification. The ready‐to‐go libraries were pooled equimolarly and diluted to a sequencing concentration of 650 pM. On‐board DNA denaturation and sequencing were performed on a NextSeq 2000 instrument (Illumina), according to the manufacturer's instructions, using the 2 × 150 bp NextSeq 1000/2000 P2 Reagents (300 Cycles) v3 and spike‐in of 1% PhiX control library. Whole‐metagenome shotgun (WMGS) sequencing of the three bioreactor culture aliquots produced an average of 21,861,838 ± 7,995,330 paired‐end 150 bp reads per sample. Raw metagenomic sequencing reads were trimmed and quality filtered with fastq‐mcf software supplied by Illumina Inc (minimum mean quality score, 20; window size, 5 bp; and minimum length, 100 bp). Following quality filtering, an average of 18,662,746 ± 5,671,267 quality‐filtered microbial reads per sample were retained (Table S8).
Lactobacillus crispatus strain‐level profiling of the bioreactor‐derived cultures
To disentangle the different L. crispatus strains in the co‐culture aliquots collected at different time points (10, 24, and 48 h) during bioreactor growth, the filtered metagenomic reads obtained from each shotgun sequencing effort were mapped against specific distinctive regions of every L. crispatus genome using the software BBMap (https://sourceforge.net/projects/bbmap/) with 100% homology (perfect mode = t flag). Notably, to reduced multi‐mapped metagenomic reads, we ensured that the selected L. crispatus genomes returned pairwise ANI values <98%, as advised in a previous qualified study (Olm, 2021) (https://drep.readthedocs.io/en/latest/choosing_parameters.html). In detail, to identify suitable discriminative genes, the whole set of genes unique to each L. crispatus strain detected in the PGAP analysis were mapped against the combined genomes of all strains using the Bowtie2 ‐‐very‐sensitive mode (Langmead & Salzberg, 2012). Genes that did not return any hits other than those corresponding to the genome to which they belong were retained as candidate strain‐specific marker genes. This selection was then manually inspected to exclude genes corresponding to transposases, phage genes and genes located alongside the contig ends. This procedure identified a set of roughly ten unique marker genes for each L. crispatus strain that were used in downstream analyses on the bioreactor‐derived metagenomic reads (Table S7). A proxy measure of each strain abundance was calculated by normalising the mapped read count on the corresponding marker gene length and library size using the RPKM mathematical formula [(109 * Number of mapped reads to a gene)/(Total mapped reads * gene length in base‐pairs]).
Moreover, the set of genes associated uniquely with each of the four co‐cultivated strains was functionally investigated to discover potential accessory protein‐encoding sequences conferring peculiar growth abilities in the cultivation medium. To this scope, we employed the MetaCyc database (https://metacyc.org/), which allowed us to assign a detailed functional annotation to each scrutinized gene. In addition, the Transporter Classification Database (TCDB) was exploited to characterise transport systems and identify their possible substrates (https://tcdb.org/).
Statistical analysis
The software SPSS version 25 and OriginPro version 2023 (www.ibm.com/software/it/analytics/spss/) (https://www.originlab.com/) were used for statistical data analyses and graphing. One‐way ANOVA with Bonferroni correction was used to determine the statistical significance of differences in the OD600 measures (growth assay) and normalised read counts (bioreactor‐based co‐cultivation experiment). AlphaFold (Jumper et al., 2021) and PyMOL software (https://pymol.org/2/) were used to observe SNPs within the predicted 3D protein structure of the pullulanase type II gene derived from the L. crispatus LB97.
RESULTS AND DISCUSSION
Identification of representative Lactobacillus crispatus genomes
To investigate the genomic differences between human L. crispatus strains, an extensive comparative genome analysis was performed on L. crispatus genomes recovered from human specimens of healthy donors, including faecal, vaginal, saliva and urine samples (Table S1). Specifically, seven L. crispatus strains were obtained from international bacteria culture collection (Table 1), and their genomes were sequenced along with those of 34 strains isolated from the human vaginal tract in the context of a previous study (Table 1). Additionally, with the aim of expanding the overview of the genetic variability of this taxon, 200 genome sequences (complete and draft) of L. crispatus strains isolated from human biological samples were selected from public repositories (Table S1). Following dereplication aimed at removing the genomic redundancy by grouping essentially identical genomes (using dRep tool, version 2.2.0, with average nucleotide identity >99%, [https://drep.readthedocs.io/en/latest/choosing_parameters.html]), 22 L. crispatus chromosomes with average completeness of 98.97% ± 0.14% were retained as representatives of the sequence variation observed in our genome repertoire and therefore used for comparative analysis (Table S2).
TABLE 1.
Genome features of the 22 representative Lactobacillus crispatus genomes.
| Assembly ID | Strain name | Genome size Mbp | CDS number | Genome completeness (%) | Isolation source |
|---|---|---|---|---|---|
| GCF_000162255.1 | 125‐2‐CHN | 2.30525 | 2032 | 99.03 | Human vagina |
| GCF_000162315.1 | MV‐3A‐US | 2.43708 | 2252 | 98.38 | Human vagina |
| GCF_002861805.1 | UMB0824 | 2.17405 | 2061 | 99.03 | Human urine |
| GCF_002861815.1 | UMB0085 | 2.17506 | 2081 | 99.03 | Human urine |
| GCF_009857395.1 | Indica2 | 2.20949 | 2028 | 99.03 | Human vagina |
| GCF_007713895.1 | NCK1350 | 2.04734 | 1932 | 99.03 | Human stool |
| GCF_013456995.1 | B4 | 2.03959 | 1902 | 99.03 | Human stool |
| GCF_000160515.1 | JV‐V01 | 2.2172 | 1992 | 98.03 | Human vagina |
| GCF_014654865.1 | BC5 | 2.06419 | 1901 | 99.03 | Human vagina |
| GCF_015669875.1 | D31t1 | 2.2782 | 2120 | 99.03 | Human stool |
| GCF_018987235.1 | ATCC 33820 | 2.23909 | 2020 | 99.03 | Human saliva |
| GCF_020042005.1 | Lc1700 | 2.81896 | 2632 | 98.86 | Human vagina |
| GCF_021278925.1 | lc83 | 2.30843 | 2112 | 98.9 | Human vagina |
| GCF_025194085.1 | CIRM‐BIA 2111 | 2.00737 | 1865 | 99.03 | Human stool |
| GCF_025194045.1 | CIRM‐BIA 2233 | 2.24513 | 2127 | 99.03 | Human vagina |
| This study | LMG11440 | 2.032412 | 2087 | 98.86 | Human vagina |
| This study | LMG12005 | 2.019682 | 2014 | 98.94 | Human vagina |
| This study | LMG18189 | 2.094399 | 2079 | 99.03 | Human saliva |
| This study | LMG11415 | 2.030901 | 2004 | 99.03 | Human saliva |
| This study | LMG18200 | 2.208098 | 2182 | 99.03 | Human stool |
| This study | LB93 | 2.202822 | 2340 | 99.03 | Human vagina |
| This study | LB97 | 2.263389 | 2422 | 99.03 | Human vagina |
The general features of the 22 representative L. crispatus genomes are reported in Table 1 and include an average of 2105 ± 179 predicted Coding Sequences (CDSs) per chromosome (ranging from 2632 to 1865), with an average genome length of 2.20 ± 0.18 Mbp.
Intra‐species genetic variability within the Lactobacillus genus
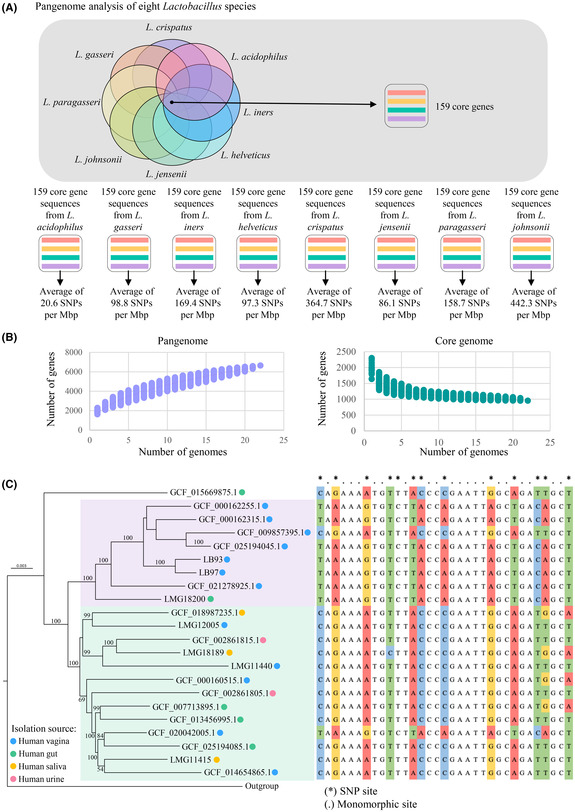
To investigate the level of genomic diversity among L. crispatus strains compared with other species of the Lactobacillus genus, we selected publicly accessible chromosomes belonging to seven different Lactobacillus species known to inhabit various human body sites. Notably, for a robust comparison with the dereplicated 22 representative L. crispatus genomes, we focused on Lactobacillus species for which at least 20 independent conspecific genomes with ANI values between 95% and 98% were retained after accounting for genome completeness >95% (Table S3). Accordingly, 499 Lactobacillus chromosomes were collected and combined with the 22 representative genomes of L. crispatus for pangenome analysis, which led to the identification of 159 core genes, defined as the set of gene families (clusters of orthologous groups [COGs]) shared by each Lactobacillus chromosome tested (Figure 1A).
FIGURE 1.

Comparative analysis between different Lactobacillus species and between 22 non‐identical L. crispatus strains. In panel (A), Venn diagram shows the eight Lactobacillus species sharing the 159 genes used to measure the magnitude of intra‐species genetic diversity. Below, the species‐specific number of SNPs identified within the common protein‐encoding genes is reported for each of the considered Lactobacillus species. Panel (B) depicts the L. crispatus pan‐ and core‐genome size. The number of discovered genes (vertical axe) is plotted as a function of the number of sequentially added genomes (horizontal axe). Panel (C) shows the phylogenomic tree based on the concatenated 959 core genes shared among the 22 non‐identical L. crispatus genomes. The tree was constructed by the neighbour‐joining method. Bootstrap percentages based on 1000 replicates above 50 are shown at node points. For each strain, the isolation source is highlighted with a coloured circle. On the right, an aligned portion of the L. crispatus core genome exemplifies the relationships between phylogenomic clusters and SNP patterns. In the top row, nucleotide positions showing variants are highlighted with an asterisk, while a dot highlights non‐variant sites.
Exploiting this set of 159 core genes, the level of variability at the single nucleotide level was evaluated individually within each Lactobacillus species. In detail, sequences homologous to the 159 core genes (corresponding to an average of 46,760 ± 987 nucleotides) were recovered from each collected genome and compared between strains belonging to the same Lactobacillus species, eventually recording Single Nucleotide Polymorphisms (SNPs) at each nucleotide position.
Considering the nucleotide variations with an occurrence rate above 20% to exclude sequencing errors, the eight inspected Lactobacillus lineages exhibited a total number of intra‐species SNPs per Mbp ranging from 20.6 to 442.3, with L. johnsonii and L. acidophilus showing the lowest and the highest number of SNPs, respectively (Figure 1A). Thus, these data highlighted how, on average, the various species of the Lactobacillus genus display different levels of intra‐species genetic diversity, which has the potential to translate into intra‐species phenotypic variability.
Remarkably, the L. crispatus core gene set returned an average of 364.7 SNPs per Mbp, emerging among the species with the higher genetic variation, even compared with other notorious Lactobacillus species inhabiting the human vaginal tract, such as L. gasseri, L. iners and L. jensenii (Figure 1A). Specifically, among L. crispatus members, most SNPs were found in gene sequences coding for transmembrane transport mechanisms (26%), followed by genes predicted to be involved in the biosynthesis of extracellular protein components (14.5%) and carbohydrate metabolism (10.4%). In contrast, within more niche‐specialised Lactobacillus species, for example L. gasseri, the genes with higher number of SNPs were predicted to be involved in DNA‐related processes (21.4%) and protein‐protein interaction (12%).
Pan‐ and core‐genome analysis of the L. crispatus species
Chromosome sequences of the 22 non‐identical L. crispatus strains were submitted to gene re‐annotation and subsequently analysed from a pangenome perspective, providing information on the ubiquitous genetic backbone of conspecific chromosomes and the intra‐species genetic diversity (Tettelin et al., 2005). In total, the pangenome of L. crispatus includes 6512 COGs, whose accumulation curve, depicting the expansion of the pangenome as a function of the number of genomes included, is still far from being saturated. Thus, indicating that L. crispatus species is characterised by an open pangenome where the total gene pool obtainable for this species has not yet been fully disclosed (Figure 1B). Moreover, we determined the current L. crispatus core genome to be comprising 959 COGs that were conserved across all the 22 analysed strains (15% of the pangenome) while an average of 157.3 ± 54.3 genes per genome were associated with only one strain.
Based on the core gene sequences obtained from the 22 non‐redundant L. crispatus genomes, a phylogenetic tree was constructed to evaluate the evolution of the species (Figure 1C). According to the clustering relationship, the 22 strains assessed were divided into two main clusters, one of which was intriguingly composed only of L. crispatus strains isolated from the female reproductive tract (Figure 1C, violet shadows). Moreover, this phylogenetic tree also displayed a second phylogenetic cluster of strains isolated from different human body districts, encompassing vagina, gut, saliva and urine (Figure 1C, green shadow). Notably, this mixed group may include a few strains that can survive/colonise in closely related niches, like different human body districts.
To investigate the intra‐species genomic variability of L. crispatus taxon, we measured the genetic diversity at the single‐nucleotide level by comparing the whole genome sequences (wgSNPs) and the corresponding core genome (cgSNPs). Specifically, from the core genome‐based phylogenomic tree (Figure 1), we selected the L. crispatus strain placed at the deepest split, that is the most divergent chromosome (RefSeq genome assembly GCF_015669875), which was used as a reference sequence to compute pairwise alignments and SNPs extraction. Overall, the 22 L. crispatus strains showed an average of 28,811 wgSNPs (representing about 2% of the genome sequence), most of which (about 90%) resided within CDSs.
For cgSNPs evaluation, each of the 22 homologous nucleotide sequences obtained by concatenating the 959 COGs shared among all the non‐identical L. crispatus strains (corresponding to an average of 904,903 bases) was examined for sequence variations against the concatenated (core) gene set of the reference L. crispatus assembly. This procedure resulted in the identification of an average of 15,007 cgSNPs (representing a variation rate of 1 for every 56 nucleotides) that was lowered when compared with previous analyses of polymorphic sites within clinically relevant microorganisms such as Pseudomonas aeruginosa and Escherichia coli (showing 159,609 SNPs within the concatenated core genes of 3,629,979 bp and 266,969 SNPs within a core genome of 2,159,296 bp, respectively) (Muthukumarasamy et al., 2020; Shakya et al., 2020). Albeit the core genome resulted rather conserved within these 22 L. crispatus strains, it might be worth mentioning that the observed micro‐diversity lies within DNA sequences that code for proteins. Therefore, it could have evolutionary importance contributing to intra‐species diversity by affecting protein domains. Indeed, the nucleotide sequence variation among the L. crispatus core genome was not randomly distributed, but phylogenetically co‐clustered strains showed common patterns of SNP profiles (Figure 1C), thus indicating that the observed SNPs are representing evolutionary trajectories and not mere random mutations or sequencing errors.
Exploration of the micro‐diversity in the ubiquitous features of L. crispatus genome and identification of fast evolving genes
With the aim of defining whether and which categories of genes are more concerned by a rapid sequence evolution, we calculated the level of polymorphisms for each protein‐coding gene constituting the L. crispatus core genome. Like what has been performed above, the homologous gene sequences from the L. crispatus GCF_015669875 were used as reference in pairwise comparisons of each individual core gene. Accordingly, the number of SNPs resulting from the average of all alignment pairs ranged from zero to 347.45 ± 150.30 per gene (Table S4).
Among the genes with the lower average number of SNP sites (lower than 5.8, corresponding to the data below the 25th percentile, Figure 2A), we identified protein‐coding sequences involved in putative housekeeping functions, including ribosome assembly and function and central glycolysis regulation, as well as DNA replication and cell division (Table S4).
FIGURE 2.

Identification of the 52 HVGs. In panel (A), Box‐Whisker plot was used to represent the gene distribution based on the number of SNP sites obtained by comparing the nucleotide sequence of every 959 protein‐coding genes shared among all the 22 Lactobacillus crispatus chromosomes. For each gene, the number of SNP sites was expressed as the average of all the pairwise comparisons against the reference sequence (homologous gene sequence of L. crispatus GCF_015669875). The Q3 + 1.5IQR was used as a cut‐off to select the 52 HVGs. Panel (B) reports the functional annotation and the number of SNP sites of each HVG.
In contrast, by considering the data above the third quartile (Q3 + 1.5·Inter‐Quartile Range, Figure 2A), we identified 52 genes with the highest average number of SNP sites (ranging from 347.45 ± 150.30 to 90.25 ± 28.76), that is the most highly variable genes (HVGs), which therefore represented the set of genes that have been likely under the strongest selection pressure (Figure 2B, Table S4). Interestingly, among the HVGs, we identified mainly genes involved in the biosynthesis and rearrangement of cell wall components, such as lipoteichoic acids and peptidoglycan, as well as transmembrane transport of a variety of substrates, including carbohydrates and micronutrients (Figure 2B, Table S4). The presence of such micro‐diversity in proteins directly mediating interactions with the environment likely reflects adaptive mechanisms to the changing biotic and abiotic components, thereby leading to possible different competitive abilities and (sub‐) niche specialisation. Indeed, the intra‐species heterogeneity observed in the L. crispatus core genes emerged less marked when the genomes of 22 L. crispatus strains were compared based on their ecological niche, thus showing greater gene sequence homogeneity among genomes sharing the same environment (Figure 2A). However, vaginal‐derived L. crispatus strains showed a significantly higher number of HVGs than those isolated from the human gut (Mann–Whitney test, p‐value = 0.007), indicating that the vaginal environment exerts crucial ecological forces driving the L. crispatus genome evolution.
A new phylogenomic tree, representing the evolutionary outcomes determined by mutational hotspots within the L. crispatus species, was generated employing the nucleotide sequences of the 52 HVGs (Figure 3). Specifically, this tree was composed of three main clusters, where not all the strains maintained the same distribution compared to the original phylogenomic tree based on the whole core genome (Figure 3). Indeed, the HVG‐based tree better distinguished among strains from closely related niches, highlighting that the evolution of the HVGs does not strictly follow the overall strain speciation, probably reflecting a relatively recent adaptation to specific environmental stimuli, such as multiple human body site colonisation or inter‐strain niche competition. Accordingly, based on the picture emerging from the HVG‐derived phylogenetic distribution, we selected four highly divergent L. crispatus strains, that is LB97, LMG11440, LMG18200 and LMG11415, which were used for in vitro phenotypical assays aimed at investigating the link between evolutionary trajectories, grow performances and competitive abilities.
FIGURE 3.

Phylogenetic analysis based on the 52 HVG sequences. Proteomic tree based on concatenating the 52 protein encoding core genes identified as highly variable across the 22 non‐identical Lactobacillus crispatus genomes. Phylogenetic groups are highlighted in different colours. For comparison, the phylogenomic tree resulting from the whole core genome (presented in Figure 1) is visualised using the redial layout. For each strain, the coloured circle represents the isolation source, while the diameter of the black circle is proportional to the number of SNPs identified within the core genome using the GCF_015669875.1 genome sequence as reference.
In vitro evaluation of L. crispatus growth performances on selected carbohydrate sources in an in vitro human vaginal model
To investigate how the high level of genetic heterogeneity identified within L. crispatus could influence the respective growth abilities, the four selected L. crispatus strains (LB97, LMG11440, LMG18200 and LMG11415, whose genome diversity corresponded to ANI pairwise values <98%, Table S2) were cultivated on six different carbohydrate sources including glycogen, which is the primary bacterial nutritional source in the vaginal lumen (Tester & Al‐Ghazzewi, 2018; Zhang et al., 2022), along with other glycogen‐like α‐glucans, which may also represent a substrate for the bacterial enzymatic arsenal involved in carbohydrates breakdown of the vaginal environment (Figure 4A, Table S5). The optical density (OD) was registered after 48 h of anaerobic cultivation, and the growth on MRS was used as control condition (Table S5). Upon one‐way ANOVA test with Bonferroni correction (cut‐off p‐value < 0.05), the comparative growth assay showed widespread statistically significant differences across the four L. crispatus strains. In particular, L. crispatus LB97, isolated from the human vagina, showed greater growth performances on most of the carbohydrates tested, including glycogen (final OD >1.2; all Bonferroni‐corrected p‐values < 0.05), thus demonstrating a metabolic specialisation consistent with its isolation niche. Conversely, the gut‐derived L. crispatus LMG18200 exhibited the lowest growth when glycogen, starch and pullulan were used as the unique carbon source (all final OD measures ~0.3; all Bonferroni‐corrected p‐values < 0.05), suggesting the incapability of this strain to metabolise long‐chain α‐glucans (Figure 4A, Tables S5 and S6). Moreover, all strains appeared nearly equally limited in mucin utilisation (all final OD <0.3; Bonferroni‐corrected p‐values > 0.05; Figure 4A, Tables S5 and S6).
FIGURE 4.

Differential growth and competitive abilities between Lactobacillus crispatus strains. Panel (A) shows the optical density (OD) registered after 48 h of anaerobic growth in different nutritive substrates. Panel (B) illustrates the design of the bioreactor‐based experiment simulating the vaginal environment. In panel (C), the bar chart reports the quantification of the metagenomic reads (using average RPKM measures) mapping marker genes unique to each L. crispatus strain throughout the 48 h of growth in the bioreactor. The standard deviations are plotted as error bars. Different lowercase letters indicate significant differences at p‐value < 0.05 according to the Bonferroni test. In panel (D), alignment of partial amino acid sequences corresponding to the type II pullulanase genes of L. crispatus LB97 and LMG11440 strains genes highlights two amino acid substitutions Gln (Q) to Lys (K) and Arg (R) to Thr (T).
Furthermore, to determine the reciprocal competitive ability of the different L. crispatus strains, the growth performances of the four selected strains were evaluated through a co‐cultivation experiment involving a bioreactor model simulating the nutritional and chemical–physical conditions of the vaginal environment (Pan et al., 2020) (Figure 4B). The proliferation trend of each strain was followed for 48 h by mapping the sequenced metagenomic reads at multiple time points against a set of strain‐specific marker genes (Tables S7 and S8). In accordance with what was observed in the carbohydrate grow assay, the vaginal isolate LB97 showed a notable proliferation ability, over dominating the four‐strain L. crispatus community at every co‐cultivation time‐point (Table S8, all Bonferroni‐corrected p‐values < 0.05; Figure 4C).
In contrast, the strains LMG11415 (isolated from the human saliva) and LMG18200 (isolated from the human intestine) were clearly overwhelmed (Figure 4C). Consistently, close examination of the genomes of these four L. crispatus strains revealed that these latter were lacking in any glycogen‐degrading encoding gene while the proliferating vaginal‐derived LB97 and LMG11440 strains carried a type II pullulanase acting on both α‐1,6‐ and α‐1,4‐glycosidic bonds, which therefore achieves complete glycogen degradation, as reported in recent studies (van der Veer et al., 2019; Zhang et al., 2022). Nevertheless, LB97 and LMG11440 substantially differ in their proliferation capabilities (Table S8, Bonferroni‐corrected p‐values < 0.05; Figure 4C).
Accordingly, the metabolic potential of the gene sequences identified in the comparative genome analysis as associated uniquely with the L. crispatus LB97 was investigated using the MetaCyc database (Caspi et al., 2018). The results revealed that the predicted proteome of this strain is characterised by the presence of protein‐encoding genes involved in the uptake and metabolism of galactitol and polyamines, as well as a locus‐encoding proteins dedicated to the ascorbate degradation, which can contribute to the maintenance of the host's vaginal health (Linares et al., 2011) (Table S9). Moreover, the unique gene repertoire of the LB97 strain also contained a gene encoding for a mucin‐binding protein (MucBP domain), thus corroborating for this strain the hypothesis of a host mucin role as adhesion site rather than a carbon source, as previously reported in the literature (Muthukumarasamy et al., 2020; Shakya et al., 2020; Tester & Al‐Ghazzewi, 2018) and confirmed by the growth assay described above (Figure 4A). However, our functional investigation did not detect genes possibly involved in the metabolism of the nutritional sources constituting the used glycogen‐based culture medium (Table S9).
Interestingly, when SNPs were calculated between these latter two L. crispatus genome sequences, a total of 27,906 nucleotide positions showed differences of which 8238 (29% of the total SNPs) corresponded to amino acid replacements, thus resulting in strain‐specific intragenic variants, which can contribute to generating phenotypic differences (Table S10). Moreover, alignment of their type II pullulanase gene revealed four variations at single nucleotide level (Table S11, Figure S1), two of which resulted in amino acid substitutions (Figures 4D and S1). In more detail, these non‐synonymous SNPs lie within the protein Carbohydrate Binding Module (CBM) (Figure 4D), with possible repercussions on the efficiency of the protein binding to its substrate, as also evidenced by the 3D protein structure prediction (Figure S2).
Overall, the finding of isolate‐specific intragenic SNPs, and particularly those within the pullulanase‐encoding gene, possibly explains the growth and competitiveness differences observed between the vaginal‐isolated L. crispatus LB97 and LMG11440 strains cultivated on the simulated vaginal medium.
CONCLUSION
In this study, a comparative genome analysis involving 41 newly decoded human L. crispatus genomes coupled with 200 publicly available genome sequences from this species allowed us to deeply investigate the L. crispatus core gene evolution by connecting data from single nucleotide variations, phylogenomic reconstructions and in vitro experiments.
Compared with other Lactobacillus species, including those inhabiting the human vaginal tract, that is L. iners, L. gasseri and L. jensenii, a higher level of sequence variation at the single nucleotide level was observed within the gene pool shared among the inspected L. crispatus strains, thus highlighting a within‐species diversity driven by conserved genes evolution.
Interestingly, the genetic heterogeneity observed within the L. crispatus species appears to be reflected at the phenotypic level. In fact, when different L. crispatus strains were co‐cultivated in a bioreactor‐based model simulating the vaginal environment, substantial differences were noted in the colonisation and competition efficiency. Although members of the L. crispatus species were previously thought to utilise the glycogen hydrolysis products generated in the vaginal environment by the human α‐amylase (Mirmonsef et al., 2014; Tester & Al‐Ghazzewi, 2018), recent evidences showed that members of this taxon produce the enzyme to independently degrade glycogen, annotated as a type II pullulanase (van der Veer et al., 2019; Zhang et al., 2022). In this context, while the absence of this gene was noted for those L. crispatus strains unable to stably proliferate on glycogen under in vitro conditions, we identified two amino acid substitutions within the type II pullulanase carbohydrate‐binding module arising from non‐synonymous SNPs, which could explain the different proliferation and dominance abilities observed in vitro for the L. crispatus strains investigated in this study.
Remarkably, while the strain‐specific accessory genetic content has been historically pointed out as one of the main sources of variability resulting from intra‐species evolution, data collected in the framework of this study revealed that the evolution of the core genome could contribute to generate marked strain‐specific phenotypic traits. Thus, understanding this evolutionary driving force could be relevant for unravelling strain‐specific capabilities to successfully dominate the female reproductive tract and, ultimately, selecting suitable L. crispatus strains that could be applied for novel bacterial therapy strategies.
AUTHOR CONTRIBUTIONS
Chiara Tarracchini: Data curation (lead); formal analysis (lead); investigation (lead); methodology (equal); software (equal); writing – original draft (lead). Chiara Argentini: Data curation (equal); investigation (equal); validation (equal). Giulia Alessandri: Data curation (supporting); methodology (supporting). Gabriele Andrea Lugli: Data curation (supporting); software (equal). Leonardo Mancabelli: Data curation (supporting); supervision (supporting). Federico Fontana: Data curation (supporting); software (supporting). Rosaria Anzalone: Investigation (supporting). Alice Viappiani: Investigation (supporting). Francesca Turroni: Conceptualization (equal); project administration (supporting); supervision (supporting); writing – review and editing (supporting). Marco Ventura: Conceptualization (lead); project administration (lead); writing – review and editing (lead). Christian Milani: Conceptualization (supporting); data curation (supporting); methodology (supporting); project administration (supporting); supervision (lead); writing – review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Figure S1.
{kind=link}
Figure S2.
{kind=link}
Table S1.
Table S2.
Table S3.
Table S4.
Table S5.
Table S6.
Table S7.
Table S8.
Table S9.
Table S10.
Table S11.
ACKNOWLEDGEMENTS
We thank GenProbio Srl for the financial support to the Laboratory of Probiogenomics. The cost of the equipment (Sirius One MALDI‐TOF Bioanalyzer) used for this experimental investigation was partly supported by the University of Parma through the Scientific Instrumentation Upgrade Programme 2020. Part of this research is conducted using the High‐Performance Computing (HPC) facility of the University of Parma. C.A. is supported by Fondazione Cariparma, Parma, Italy.
Tarracchini, C. , Argentini, C. , Alessandri, G. , Lugli, G.A. , Mancabelli, L. , Fontana, F. et al. (2023) The core genome evolution of Lactobacillus crispatus as a driving force for niche competition in the human vaginal tract. Microbial Biotechnology, 16, 1774–1789. Available from: 10.1111/1751-7915.14305
Contributor Information
Marco Ventura, Email: marco.ventura@unipr.it.
Christian Milani, Email: christian.milani@unipr.it.
DATA AVAILABILITY STATEMENT
Genome sequences of the 41 newly sequenced L. crispatus were deposited in NCBI‐SRA (Short Read Archive) repository with accession number PRJNA947599.
REFERENCES
- Abdelmaksoud, A.A. , Koparde, V.N. , Sheth, N.U. , Serrano, M.G. , Glascock, A.L. , Fettweis, J.M. et al. (2016) Comparison of Lactobacillus crispatus isolates from Lactobacillus‐dominated vaginal microbiomes with isolates from microbiomes containing bacterial vaginosis‐associated bacteria. Microbiology (Reading), 162(3), 466–475. Available from: https://pubmed.ncbi.nlm.nih.gov/26747455/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argentini, C. , Fontana, F. , Alessandri, G. , Lugli, G.A. , Mancabelli, L. , Ossiprandi, M.C. et al. (2022) Evaluation of modulatory activities of Lactobacillus crispatus strains in the context of the vaginal microbiota. Microbiology Spectrum, 10(2), e0273321. Available from: https://pubmed.ncbi.nlm.nih.gov/35266820/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A.A. , Dvorkin, M. , Kulikov, A.S. et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19(5), 455–477. Available from: https://pubmed.ncbi.nlm.nih.gov/22506599/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum, H.E. (2017) The human microbiome. Advances in Medical Sciences, 62(2), 414–420. Available from: https://pubmed.ncbi.nlm.nih.gov/28711782/ [DOI] [PubMed] [Google Scholar]
- Bohbot, J.M. , Daraï, E. , Bretelle, F. , Brami, G. , Daniel, C. & Cardot, J.M. (2018) Efficacy and safety of vaginally administered lyophilized Lactobacillus crispatus IP 174178 in the prevention of bacterial vaginosis recurrence. Journal of Gynecology Obstetrics and Human Reproduction, 47(2), 81–86. Available from: https://pubmed.ncbi.nlm.nih.gov/29196153/ [DOI] [PubMed] [Google Scholar]
- Caspi, R. , Billington, R. , Fulcher, C.A. , Keseler, I.M. , Kothari, A. , Krummenacker, M. et al. (2018) The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Research, 46(D1), D633–D639. Available from: https://pubmed.ncbi.nlm.nih.gov/29059334/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, C.R. , Wierzbicki, M.R. , French, A.L. , Morris, S. , Newmann, S. , Reno, H. et al. (2020) Randomized trial of lactin‐V to prevent recurrence of bacterial vaginosis. The New England Journal of Medicine, 382(20), 1906–1915. Available from: https://pubmed.ncbi.nlm.nih.gov/32402161/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- France, M.T. , Mendes‐Soares, H. & Forney, L.J. (2016) Genomic comparisons of Lactobacillus crispatus and Lactobacillus iners reveal potential ecological drivers of community composition in the vagina. Applied and Environmental Microbiology, 82(24), 7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.L. , LoCascio, P.F. , Land, M.L. , Larimer, F.W. & Hauser, L.J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. Available from: https://pubmed.ncbi.nlm.nih.gov/20211023/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhas, M. , Eberl, L. & Glass, J.I. (2011) Essence of life: essential genes of minimal genomes. Trends in Cell Biology, 21(10), 562–568. Available from: https://pubmed.ncbi.nlm.nih.gov/21889892/ [DOI] [PubMed] [Google Scholar]
- Jumper, J. , Evans, R. , Pritzel, A. , Green, T. , Figurnov, M. , Ronneberger, O. et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. Available from: https://www.nature.com/articles/s41586‐021‐03819‐2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K.I. & Miyata, T. (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066. Available from: https://pubmed.ncbi.nlm.nih.gov/12136088/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen, K. , Hallin, P. , Rødland, E.A. , Stærfeldt, H.H. , Rognes, T. & Ussery, D.W. (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research, 35(9), 3100–3108. Available from: https://pubmed.ncbi.nlm.nih.gov/17452365/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. & Salzberg, S.L. (2012) Fast gapped‐read alignment with bowtie 2. Nature Methods, 9(4), 357–359. Available from: https://pubmed.ncbi.nlm.nih.gov/22388286/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepargneur, J.P. (2016) Lactobacillus crispatus as biomarker of the healthy vaginal tract. Annales de Biologie Clinique, 74(4), 421–427. Available from: https://pubmed.ncbi.nlm.nih.gov/27492695/ [DOI] [PubMed] [Google Scholar]
- Linares, D. , Michaud, P. , Delort, A.M. , Traïkia, M. & Warrand, J. (2011) Catabolism of L‐ascorbate by Lactobacillus rhamnosus GG. Journal of Agricultural and Food Chemistry, 59(8), 4140–4147. Available from: https://pubmed.ncbi.nlm.nih.gov/21401096/ [DOI] [PubMed] [Google Scholar]
- Lowe, T.M. & Eddy, S.R. (1997) tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research, 25(5), 955–964. Available from: https://pubmed.ncbi.nlm.nih.gov/9023104/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A. , Duranti, S. , Albert, K. , Mancabelli, L. , Napoli, S. , Viappiani, A. et al. (2019) Unveiling genomic diversity among members of the species bifidobacterium pseudolongum, a widely distributed gut commensal of the animal kingdom. Applied and Environmental Microbiology, 85(8), e03065‐18. Available from: https://pubmed.ncbi.nlm.nih.gov/30737347/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A. , Milani, C. , Mancabelli, L. , Van Sinderen, D. & Ventura, M. (2016) MEGAnnotator: a user‐friendly pipeline for microbial genomes assembly and annotation. FEMS Microbiology Letters, 363(7), 49. Available from: https://academic.oup.com/femsle/article/363/7/fnw049/2197823 [DOI] [PubMed] [Google Scholar]
- Mancabelli, L. , Mancino, W. , Lugli, G.A. , Milani, C. , Viappiani, A. , Anzalone, R. et al. (2021) Comparative genome analyses of Lactobacillus crispatus isolated from different ecological niches reveal an environmental adaptation of this species to the human vaginal environment. Applied and Environmental Microbiology, 87(8), 1–21. Available from: https://pubmed.ncbi.nlm.nih.gov/33579685/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mändar, R. , Sõerunurk, G. , Štšepetova, J. , Smidt, I. , Rööp, T. , Kõljalg, S. et al. (2023) Impact of Lactobacillus crispatus‐containing oral and vaginal probiotics on vaginal health: a randomised double‐blind placebo controlled clinical trial. Beneficial Microbes, 14, 143–152. Available from: https://pubmed.ncbi.nlm.nih.gov/36856121/ [DOI] [PubMed] [Google Scholar]
- Martínez‐Carranza, E. , Barajas, H. , Alcaraz, L.D. , Servín‐González, L. , Ponce‐Soto, G.Y. & Soberón‐Chávez, G. (2018) Variability of bacterial essential genes among closely related bacteria: the case of Escherichia coli . Frontiers in Microbiology, 9(MAY), 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes‐Soares, H. , Suzuki, H. , Hickey, R.J. & Forneya, L.J. (2014) Comparative functional genomics of Lactobacillus spp. reveals possible mechanisms for specialization of vaginal lactobacilli to their environment. Journal of Bacteriology, 196(7), 1458–1470. Available from: https://pubmed.ncbi.nlm.nih.gov/24488312/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirmonsef, P. , Hotton, A.L. , Gilbert, D. , Burgad, D. , Landay, A. , Weber, K.M. et al. (2014) Free glycogen in vaginal fluids is associated with Lactobacillus colonization and low vaginal pH. PLoS One, 9(7), e102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukumarasamy, U. , Preusse, M. , Kordes, A. , Koska, M. , Schniederjans, M. , Khaledi, A. et al. (2020) Single‐nucleotide polymorphism‐based genetic diversity analysis of clinical Pseudomonas aeruginosa isolates. Genome Biology and Evolution, 12(4), 396–406. Available from: https://pubmed.ncbi.nlm.nih.gov/32196089/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala, T. , Kankainen, M. , Castro, J. , Cerca, N. , Edelman, S. , Westerlund‐Wikström, B. et al. (2014) Comparative genomics of Lactobacillus crispatus suggests novel mechanisms for the competitive exclusion of Gardnerella vaginalis. BMC Genomics, 15(1), 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olm, M.R. , Crits‐Christoph, A. , Bouma‐Gregson, K. , Firek, B.A. , Morowitz, M.J. & Banfield, J.F. (2021) inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nature Biotechnology, 39(6), 727–736. Available from: https://www.nature.com/articles/s41587‐020‐00797‐0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, A.J. , Taylor, B. , Delaney, A.J. , Soares, J. , Seemann, T. , Keane, J.A. et al. (2016) SNP‐sites: rapid efficient extraction of SNPs from multi‐FASTA alignments. Microbial Genomics, 2(4), e000056. Available from: https://pubmed.ncbi.nlm.nih.gov/28348851/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, M. , Hidalgo‐Cantabrana, C. , Goh, Y.J. , Sanozky‐Dawes, R. & Barrangou, R. (2020) Comparative analysis of Lactobacillus gasseri and Lactobacillus crispatus isolated from human urogenital and gastrointestinal tracts. Frontiers in Microbiology, 22(10), 3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puebla‐Barragan, S. , Watson, E. , van der Veer, C. , Chmiel, J.A. , Carr, C. , Burton, J.P. et al. (2021) Interstrain variability of human vaginal Lactobacillus crispatus for metabolism of biogenic amines and antimicrobial activity against urogenital pathogens. Molecules, 26(15), 4538. Available from: https://pubmed.ncbi.nlm.nih.gov/34361691/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset, F. , Cabezas‐Caballero, J. , Piastra‐Facon, F. , Fernández‐Rodríguez, J. , Clermont, O. , Denamur, E. et al. (2021) The impact of genetic diversity on gene essentiality within the Escherichia coli species. Nature Microbiology, 6(3), 301–312. Available from: https://www.nature.com/articles/s41564‐020‐00839‐y [DOI] [PubMed] [Google Scholar]
- Sanozky‐Dawes, R. & Barrangou, R. (2022) Lactobacillus, glycans and drivers of health in the vaginal microbiome. Microbiome Research Reports, 1(3), 18. Available from: https://www.oaepublish.com/mrr/article/view/4861, https://oaepublish.com/mrr/article/view/4860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulthess, B. , Bloemberg, G.V. , Zbinden, R. , Böttger, E.C. & Hombach, M. (2014) Evaluation of the Bruker MALDI Biotyper for identification of gram‐positive rods: development of a diagnostic algorithm for the clinical laboratory. Journal of Clinical Microbiology, 52(4), 1089–1097. Available from: https://pubmed.ncbi.nlm.nih.gov/24452159/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya, M. , Ahmed, S.A. , Davenport, K.W. , Flynn, M.C. , Lo, C.C. & Chain, P.S.G. (2020) Standardized phylogenetic and molecular evolutionary analysis applied to species across the microbial tree of life. Scientific Reports, 10(1), 1–15. Available from: https://www.nature.com/articles/s41598‐020‐58356‐1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer, F. & Bäckhed, F. (2013) The gut microbiota — masters of host development and physiology. Nature Reviews Microbiology, 11(4), 227–238. Available from: https://www.nature.com/articles/nrmicro2974 [DOI] [PubMed] [Google Scholar]
- Stapleton, A.E. , Au‐Yeung, M. , Hooton, T.M. , Fredricks, D.N. , Roberts, P.L. , Czaja, C.A. et al. (2011) Randomized, placebo‐controlled phase 2 trial of a Lactobacillus crispatus probiotic given intravaginally for prevention of recurrent urinary tract infection. Clinical Infectious Diseases, 52(10), 1212–1217. Available from: https://pubmed.ncbi.nlm.nih.gov/21498386/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strati, F. & Facciotti, F. (2022) Gut microbiota‐derived metabolites in host physiology. In: Troisi, J. (Ed.) Metabolomics Perspectives: From Theory to Practical Application [Internet]. Cambridge, MA: Academic Press, pp. 515–534. Available from: https://www.researchgate.net/publication/367747520_Gut_microbiota‐derived_metabolites_in_host_physiology [Google Scholar]
- Tachedjian, G. , Aldunate, M. , Bradshaw, C.S. & Cone, R.A. (2017) The role of lactic acid production by probiotic lactobacillus species in vaginal health. Research in Microbiology, 168(9–10), 782–792. Available from: https://pubmed.ncbi.nlm.nih.gov/28435139/ [DOI] [PubMed] [Google Scholar]
- Tarracchini, C. , Fontana, F. , Mancabelli, L. , Lugli, G.A. , Alessandri, G. , Turroni, F. et al. (2022) Gut microbe metabolism of small molecules supports human development across the early stages of life. Frontiers in Microbiology, 13, 1006721. Available from: https://pubmed.ncbi.nlm.nih.gov/36177457/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarracchini, C. , Lugli, G.A. , Mancabelli, L. , Milani, C. , Turroni, F. & Ventura, M. (2020) Assessing the genomic variability of Gardnerella vaginalis through comparative genomic analyses: evolutionary and ecological implications. Applied and Environmental Microbiology, 87(1), 1–16. Available from: https://pubmed.ncbi.nlm.nih.gov/33097505/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester, R. & Al‐Ghazzewi, F.H. (2018) Intrinsic and extrinsic carbohydrates in the vagina: a short review on vaginal glycogen. International Journal of Biological Macromolecules, 112, 203–206. [DOI] [PubMed] [Google Scholar]
- Tettelin, H. , Masignani, V. , Cieslewicz, M.J. , Donati, C. , Medini, D. , Ward, N.L. et al. (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan‐genome”. Proceedings of the National Academy of Sciences of the United States of America, 102(39), 13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni, F. , Rizzo, S.M. , Ventura, M. & Bernasconi, S. (2022) Cross‐talk between the infant/maternal gut microbiota and the endocrine system: a promising topic of research. Microbiome Research Reports, 1(2), 14. Available from: https://www.oaepublish.com/mrr/article/view/4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni, F. , van Sinderen, D. & Ventura, M. (2022) Bifidobacteria: insights into the biology of a key microbial group of early life gut microbiota. Microbiome Research Reports, 1, 2. Available from: https://www.bing.com/search?q=Bifidobacteria%3A+insights+into+the+biology+of+a+key+microbial+group+of+early+life+gut+microbiota&cvid=b6d628128deb4e00a9ea38a06b32a4b0&aqs=edge.69i57j69i58.294j0j9&FORM=ANAB01&PC=U531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veer, C. , Hertzberger, R.Y. , Bruisten, S.M. , Tytgat, H.L.P. , Swanenburg, J. , de Kat, A.‐B.A. et al. (2019) Comparative genomics of human Lactobacillus crispatus isolates reveals genes for glycosylation and glycogen degradation: implications for in vivo dominance of the vaginal microbiota. Microbiome [Internet], 7(1), 1–14. Available from: https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168‐019‐0667‐9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner, B.B. (2019) The contribution of the gut microbiome to neurodevelopment and neuropsychiatric disorders. Pediatric Research, 85(2), 216–224. [DOI] [PubMed] [Google Scholar]
- Werner, G. , Fleige, C. , Feßler, A.T. , Timke, M. , Kostrzewa, M. , Zischka, M. et al. (2012) Improved identification including MALDI‐TOF mass spectrometry analysis of group D streptococci from bovine mastitis and subsequent molecular characterization of corresponding Enterococcus faecalis and Enterococcus faecium isolates. Veterinary Microbiology, 160(1–2), 162–169. Available from: https://pubmed.ncbi.nlm.nih.gov/22677481/ [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Li, L. , Zhang, T. & Zhong, J. (2022) Characterization of a novel type of glycogen‐degrading amylopullulanase from Lactobacillus crispatus . Applied Microbiology and Biotechnology, 106(11), 4053–4064. Available from: https://pubmed.ncbi.nlm.nih.gov/35612627/ [DOI] [PubMed] [Google Scholar]
- Zhang, Q. , Zhang, L. , Ross, P. , Zhao, J. , Zhang, H. & Chen, W. (2020) Comparative genomics of Lactobacillus crispatus from the gut and vagina reveals genetic diversity and lifestyle adaptation. Genes (Basel), 11(4), 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. (2022) KaKs_Calculator 3.0: calculating selective pressure on coding and non‐coding sequences. Genomics, Proteomics & Bioinformatics, 20(3), 536–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Xiao, J. , Wu, J. , Zhang, H. , Liu, G. , Wang, X. et al. (2012) ParaAT: a parallel tool for constructing multiple protein‐coding DNA alignments. Biochemical and Biophysical Research Communications, 419(4), 779–781. Available from: https://pubmed.ncbi.nlm.nih.gov/22390928/ [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Tang, H. & Ye, Y. (2012) RAPSearch2: a fast and memory‐efficient protein similarity search tool for next‐generation sequencing data. Bioinformatics, 28(1), 125–126. Available from: https://pubmed.ncbi.nlm.nih.gov/22039206/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Wu, J. , Yang, J. , Sun, S. , Xiao, J. & Yu, J. (2012) PGAP: pan‐genomes analysis pipeline. Bioinformatics, 28(3), 416–418. Available from: https://pubmed.ncbi.nlm.nih.gov/22130594/ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Table S1.
Table S2.
Table S3.
Table S4.
Table S5.
Table S6.
Table S7.
Table S8.
Table S9.
Table S10.
Table S11.
Data Availability Statement
Genome sequences of the 41 newly sequenced L. crispatus were deposited in NCBI‐SRA (Short Read Archive) repository with accession number PRJNA947599.