

Abstract
Conserved residues in protein homolog sequence alignments are structurally or functionally important. For intrinsically disordered proteins or proteins with intrinsically disordered regions (IDRs), however, alignment often fails because they lack a steric structure to constrain evolution. Although sequences vary, the physicochemical features of IDRs may be preserved in maintaining function. Therefore, a method to retrieve common IDR features may help identify functionally important residues. We applied unsupervised contrastive learning to train a model with self‐attention neuronal networks on human IDR orthologs. Parameters in the model were trained to match sequences in ortholog pairs but not in other IDRs. The trained model successfully identifies previously reported critical residues from experimental studies, especially those with an overall pattern (e.g., multiple aromatic residues or charged blocks) rather than short motifs. This predictive model can be used to identify potentially important residues in other proteins, improving our understanding of their functions. The trained model can be run directly from the Jupyter Notebook in the GitHub repository using Binder (mybinder.org). The only required input is the primary sequence. The training scripts are available on GitHub (https://github.com/allmwh/IFF). The training datasets have been deposited in an Open Science Framework repository (https://osf.io/jk29b).
Keywords: intrinsically disordered proteins, liquid–liquid phase separation, unsupervised contrastive machine learning
1. INTRODUCTION
The evolutionary history of DNA/RNA sequences and the proteins they encode can be revealed through multiple sequence alignment methods, enabling the identification of phylogenetic relationships. These methods have been used to identify our extinct Neanderthal and Denisovan cousins through DNA extracted from ancient bones (Green et al., 2010; Meyer et al., 2012), to discover the Archaea domain through prokaryotic ribosomal 16S RNA sequences (Woese and Fox, 1977), and to trace myoglobin and hemoglobin protein sequences back to their globin origins (Hardison, 2012; Suzuki and Imai, 1998). Protein structures also provide insights into protein evolution, as they can be conserved despite changes in the primary sequence. For example, the structural similarity between the motor domains of kinesin and myosin suggests that they have a common ancestor despite low sequence identity (Kull et al., 1996). The shape of a protein also influences its evolution and the conservation of functionally important residues. When conservation levels are mapped onto 3D structures, the most conserved residues are often found in key locations such as the folding core (Echave et al., 2016) or catalytic sites (Craik et al., 1987).
However, intrinsically disordered proteins (IDPs) or proteins with intrinsically disordered regions (IDRs), which are estimated to comprise approximately half of the eukaryotic proteome (Dunker et al., 2000), do not adhere to the structural constraints in evolution. As a result, the sequences of IDPs or IDRs exhibit a broader range of variation compared to their folded counterparts. This phenomenon is exemplified by the example provided in Figure S1. Although some structural evolutionary restraints still apply to some IDRs, especially those that undergo folding‐upon‐binding (Jemth et al., 2018; Karlsson et al., 2022), the evolution of IDRs is mainly constrained by function. One recently recognized function of IDRs is their ability to undergo liquid–liquid phase separation (LLPS) (Alberti et al., 2019; Alberti and Hyman, 2021). This mechanism contributes to the formation of membraneless organelles and explains the spatiotemporal control of many biochemical reactions within a cell (Banani et al., 2017; Shin and Brangwynne, 2017). The proteins within these condensates do not adopt specific conformations (i.e., they still behave like random coils) (Brady et al., 2017; Burke et al., 2015) and thus evolve without structural restraints. Although multiple sequence alignment may work in some instances (e.g., the aromatic residues in the IDRs of TDP‐43 and FUS are conserved, highlighting their potential importance for LLPS [Ho and Huang, 2022]), most IDRs cannot be aligned, especially when there are sequence gaps between orthologs (Light et al., 2013).
The functionally important physicochemical properties of IDPs/IDRs encoded in their primary sequence may be retained during evolution. Aromatic residue patterns (Martin et al., 2020), prion‐like amino‐acids (Patel et al., 2015), charged‐residues blocks (Greig et al., 2020), and coiled‐coil content (Fang et al., 2019) all contribute to LLPS, but these features cannot be revealed by sequence alignment. Multiple sequence alignment methods are, therefore, of limited use in identifying critical residues in IDRs. To overcome this challenge, we propose an unsupervised contrastive machine learning model trained using self‐attention neuronal networks on human IDR orthologs. Our results show that the trained model “pays attention” to crucial residues or features within IDRs. We also provide online access to our model that uses primary sequences as input.
2. METHODS
2.1. Training dataset preprocessing
Human protein sequences were retrieved from UniProt (UniProt, 2019) and the corresponding orthologs were obtained from the Orthologous Matrix (OMA) database (Altenhoff et al., 2021). Chordate orthologs were aligned using Clustal Omega (Sievers et al., 2011). The PONDR (Romero et al., 1997) VSL2 algorithm was used to predict the IDR of the human proteins and to define the boundaries of the aligned sequences (Figure 1a). Aligned regions were defined as subgroups. N‐terminal methionines were removed to assist learning (methionine is coded by the start codon in protein synthesis). After removing gaps within the aligned sequences, all sequences were padded to a length of 512 amino acids (repeating from the N‐terminus; Figure 1a). The few sequences longer than 512 amino acids (56,086 out of 2,402,346, 2.3%) were truncated from the C‐terminus. To pad or to truncate the sequence to 512 is for unifying the dimension of our training dataset, keeping the consistency of the model input size. The repeated sequences preserve the relative ordering of the original sequence. This can be beneficial for tasks where the order is important in sequence classification. The training dataset thus consisted of 28,955 ortholog subgroups from 13,476 human protein families with IDRs longer than 40 amino acids.
FIGURE 1.
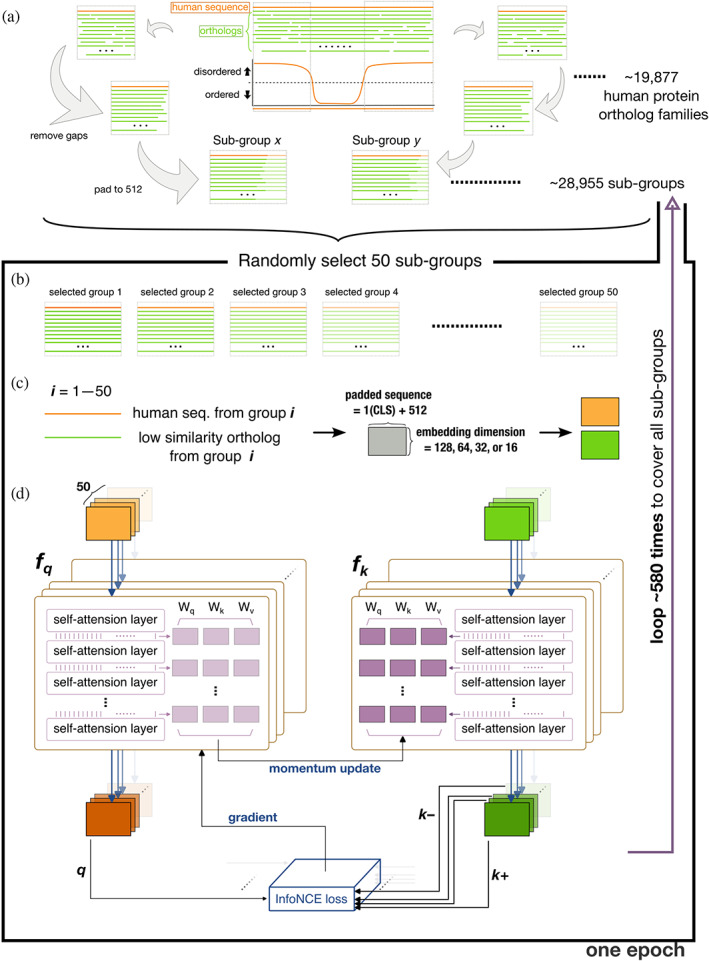
Flowchart of the training scheme. (a) Schematic representation of how the training datasets were constructed from human sequences (orange lines) and orthologs (green lines). (b) A training batch made up of 50 randomly selected subgroups. (c) Embedding of the human sequence and one of its orthologs from the same subgroup (selection probability weighted by dissimilarity) to different dimensions (as a tensor for each sequence). (d) The architecture of the training model. The steps in panels (b)–(d) were repeated 580 times to cover all subgroups in the training set, and the whole process (a training epoch) was repeated 400 times.
Each training batch consisted of 50 randomly selected subgroups (Figure 1b). The human sequence from each subgroup was paired with one of its orthologs (one of the nonhuman sequences in the same subgroup, Figure 1c). The selection probability was weighted by the Levenshtein distance (Levenshtein, 1966) from the human sequence to favor low similarity pairings. Figure S2 shows how different the sequences typically were in these ortholog subgroups, along with the corresponding selection probabilities. The most dissimilar sequences (high probability of being selected for training) in each ortholog group were also deposited in Open Science Framework. A classifier token (CLS) was added to the start of the selected sequences, and these were mapped to a matrix with an embedding dimension of 128 (embed_dim; Figure 1c). Each residue in the protein sequence was embedded in a numerical vector, as another dimension alongside the padding (512). We have tested different embedding sizes from 16, 32, 64, and 128. The embedding size of 128 is sufficient for converged training performance.
2.2. Training architecture
The training architecture was a self‐supervised contrastive learning model, Momentum Contrast version 3 (MoCo v3) (Chen et al., 2021). In the computer vision field, this model was designed to learn meaningful representations from pictures without relying on explicit labels or annotations. By applying this architecture to our task, the model learned to distinguish between similar and dissimilar protein sequences and capture their underlying patterns and features. This approach allowed us to use large amounts of unlabeled protein data to train our model effectively. The base encoder in MoCo v3 was replaced with a classical self‐attention network (Vaswani et al., 2017) for fitting our amino acid letters input. We used eight‐head attention and tested six attention layers. The attention layers help our model to focus on different parts of the protein sequence when making predictions and allow the model to give more importance to specific regions. Fifty human sequences from the same batch and their corresponding orthologs (the ones with the lowest similarity to each human sequence, as mentioned above) were sent to the momentum encoders (f q , f k respectively, following the original nomenclature (Chen et al., 2021)), and calculated in parallel (Figure 1d). The outputs from each human sequence and its ortholog were a query (q) and key (k+; the positive sample for each query). The output of the other 49 orthologs were the negative samples (k–). All 50 combinations of q, k+, and k– were formulated to minimize a contrastive loss using the adopted InfoNCE (van den Oord et al., 2018):
| (1) |
where τ is a temperature hyper‐parameter (set to 0.02). The loss was computed in a symmetrized manner (Chen et al., 2021), that is, the human sequences (q) were also sent to f k , and the orthologs (k) were sent to the f q with correspondent outputs for calculating the InfoNCE loss. The parameters between the attention layers of f q (light purple blocks in Figure 1d) were updated according to a gradient to minimize the cross‐entropy loss (Equation 1). The parameters in f k (dark purple blocks) were updated by the momentum encoder: (1 – m) • query_encoder + m • key_encoder, with m set to 0.999 by default (Chen et al., 2021).
This scheme (Figure 1b–d) was repeated ~580 times to include all 28,955 subgroups in each training epoch. The training consists of 400 epochs, and the InfoNCE loss is sufficiently converged (Figure S3). After the training, the attention scores for each residue in an input sequence are predicted by the trained model. The attention score represents the measure of importance or relevance assigned to each amino acid position within a protein sequence. It indicates how much attention or focus the model attributes to that specific position when making predictions. A higher attention score suggests that the model considers that position to be more influential within the protein. The attention score is a representation of the model's internal weighting and should be interpreted in the context of its impact on the model's predictions rather than directly linked to specific physical properties of the amino acids.
The model was built on PyTorch and the training was performed on a Nvidia Telsa P100 16G GPU.
3. RESULTS
3.1. The trained model attributes a high attention score to experimentally confirmed critical residues
Studies have shown that the aromatic residues (phenylalanine, tyrosine, and tryptophan) in the IDRs of TDP‐43 (Li et al., 2018b), FUS (Lin et al., 2017a) and hnRNP‐A1 (Molliex et al., 2015) are critical for LLPS‐related functions. These residues obtain a high attention score in our model (Figure 2a). The aromatic residues (two tryptophans and 10 tyrosines) in galectin‐3 (Lin et al., 2017b) also score highly (Figure 2b, left panel). Interestingly, although a zebrafish's galectin, which has an IDR, differs substantially in primary sequence from human galectin‐3 (Supplementary Figure S4), the aromatic residues (mostly tryptophan instead of tyrosine) also have high attention scores (Figure 2b, right panel). Note that this zebrafish's galectin was not in the OMA ortholog database used for training (OMA number: 854142). Charged residues (purple arrows in Figure 2c) reported to be associated with condensation in NPM1 (Mitrea et al., 2018), FMRP (Tsang et al., 2019), and Caprin1 (Wong et al., 2020) also obtain high attention scores (Figure 2c). Our model also assigns high attention scores to the methionines in Pbp‐1 (labeled in Figure 2d; Pbp‐1 is the yeast ortholog of human Ataxin‐2), which have been shown to be critical for redox‐sensitive regulation (Kato et al., 2019). Altogether, these results indicate that the trained model correctly identifies known key IDR residues.
FIGURE 2.
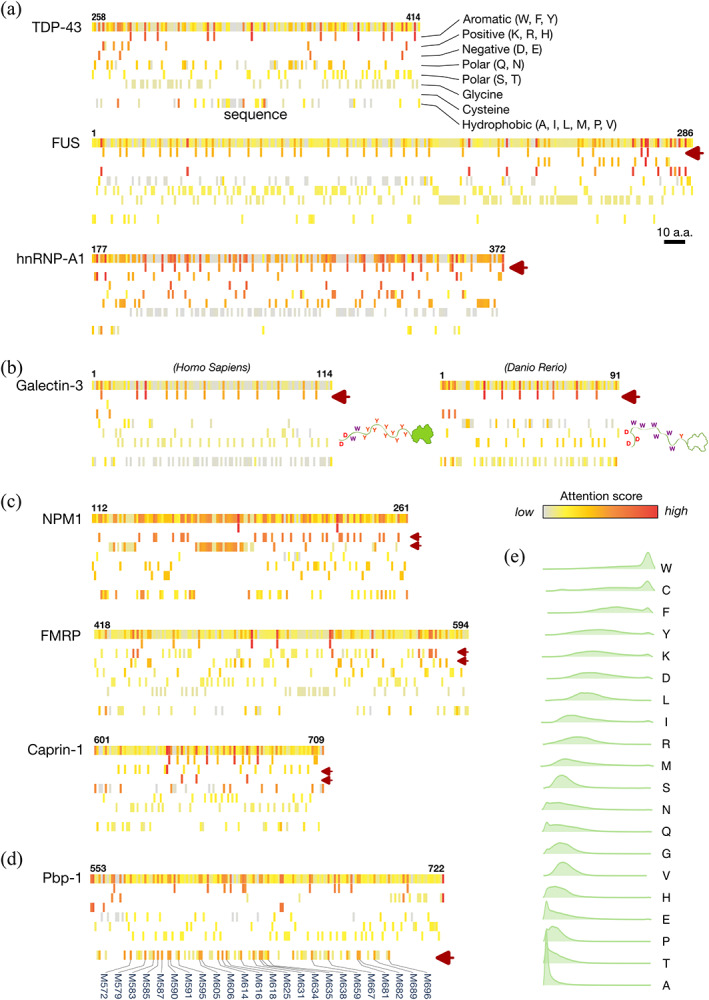
Results of the trained model for reference proteins and attention score distributions for individual amino acids. (a–d) Sequences and attention scores for the intrinsically disordered regions of (a) the RNA‐binding proteins TDP‐43, FUS, and hnRNP‐A1, (b) human and zebrafish galectin‐3, (c) NPM1, FMRP, and Caprin‐1, and (d) Pbp‐1. The attention scores appear as heatmaps from high (red) to low (gray) in the top row of each protein along with residue numbers. Amino acids with different physical properties are shown on separate rows as indicated in panel (a). Purple arrows indicate amino acids of known functional importance. (e) Half‐violin plots of the distribution of attention scores in human IDRs for each amino acid, sorted by median value from high (tryptophan, W) to low (alanine, A). IDRs, intrinsically disordered regions.
3.2. Most amino acids have broadly distributed attention scores except tryptophan and cysteine, whose presence in IDRs hints at potential importance
Figure 2e compares the attention score distributions of the amino acids in human IDRs. The differences are striking, but the attention scores are not correlated with other physical properties, such as disorder/order propensity (Radivojac et al., 2007; Vihinen et al., 1994), prion‐likeness (Lancaster et al., 2014), or prevalence in human IDRs (Figure S5). The attention scores of alanine are always low. Although poly‐alanine promotes α‐helix formation (Polling et al., 2015), which is known to contribute to IDR functions (Chiu et al., 2022; Conicella et al., 2020; Li et al., 2018a), our model ignores this amino acid. This is probably because α‐helices are also promoted by other amino‐acid types, such as leucine or methionine (Levitt, 1978; Pace and Scholtz, 1998), in different combinations not involving alanine. The training process did not include structure information, and thus structure‐related sequence motifs could not be learned by our model. At the other end of the distribution, tryptophan and cysteine systematically obtain high attention scores. These structure‐promoting amino acids rarely appear in unstructured regions (Radivojac et al., 2007; Uversky and Dunker, 2010; Vihinen et al., 1994); therefore, their appearance in IDRs hints at their potential importance. Although little is known about the role of cysteine in IDRs, its involvement in tuning structural flexibility and stability has been recently discussed (Bhopatkar et al., 2020), and our results may have also predicted its currently ignored importance in IDRs. Tryptophan, in contrast, is well‐known to act as LLPS‐driving “stickers” in IDRs (Li et al., 2018b; Sheu‐Gruttadauria and MacRae, 2018; Wang et al., 2018), and bioinformatic analysis shows that they may have evolved in the IDRs of specific proteins to assist LLPS (Ho and Huang, 2022).
The fact that most amino acids, including those highlighted in Figure 2a–d, have broad attention score distributions (Figure 2e), excludes the possibility that our model is biased toward particular amino‐acid types rather than sequence content as a whole. Moreover, in the machine learning procedure, the protein sequences were embedded into higher dimension matrices (as sequences of digits; Figure 1c), and amino‐acid type information was lost when the matrices were transformed into tensors along with the self‐attention layers (Figure 1d). These results support the predictive ability of the trained model.
4. DISCUSSION
Genetic information, in the form of a linear combination of nucleic or amino acids, becomes more diverse over time. Comparing levels of diversity between different species reveals how closely related they are. In terms of amino acids, multiple sequence alignment not only highlights phylogenetic relationships between proteins but also facilitates homology modeling for structure prediction (Balakrishnan et al., 2011; Morcos et al., 2011; Weigt et al., 2009). Machine learning approaches have recently been used to incorporate information from evolution to train structure prediction models (AlQuraishi, 2019; Xu, 2019), and the highly accurate predictions from AlphaFold (Jumper et al., 2021) and RoseTTAFold (Baek et al., 2021) have revolutionized structural biology. In contrast, the structural conformations of IDRs lack a one‐to‐one correspondence with the primary sequence, and multiple sequence alignment often fails (Ho and Huang, 2022; Lindorff‐Larsen and Kragelund, 2021). These limitations make IDR structural ensembles challenging to predict. A few attempts have been reported, such as using generative autoencoders to learn from short molecular dynamics simulations (Bhopatkar et al., 2020). The potential and challenges of machine learning in IDR ensemble prediction are also discussed (Lindorff‐Larsen and Kragelund, 2021).
Sequence pattern prediction faces similar challenges, including the lack of a sufficient stock of “ground‐truth” training data for validating the model performance, such as image databases or the Protein Data Bank. Nevertheless, unsupervised learning architectures have been developed to train models without labeled datasets (Hinton and Sejnowski, 1999), and this type of approach is particularly well‐suited for IDRs. For instance, Saar et al. (2021) used a language‐model‐based classifier to predict whether IDRs undergo LLPS. Moses and coworkers pioneered the use of unsupervised contrastive learning, using protein orthologs as augmentation (Lu et al., 2020), to train their model to identify IDR characteristics (Lu et al., 2022). While we also used ortholog sequences as training data, our approach differs in several key ways. We used self‐attention networks, rather than convolutional neural networks, to capture the distal features in the entire protein sequence. Additionally, we trained our model using the latest contrastive learning architecture (MoCo v3), which greatly reduces memory usage for larger batches and enhances efficiency. In contrast to other masked language models (Brandes et al., 2022; Elnaggar et al., 2022; Rives et al., 2021), our approach is the first, to the best of our knowledge, to combine contrastive learning and self‐attention in extracting features using natural language processing for protein sequence analysis. Furthermore, our trained model directly “pays attention” to potentially critical residues in the entire sequence, rather than mapping the primary sequence to learned motifs (Lu et al., 2022).
Our research investigates the viability and potential of using contrastive learning and self‐attention networks to identify features within proteins' IDRs. Our tool offers a convenient resource for biochemists and cell biologists to identify overall features in an IDR sequence, such as a predominance of aromatic residues or blocks of charged residues (Figure 2). Moreover, our model provides intuitive results highlighting potentially important residues for researchers to target in mutagenesis or truncation experiments. Nevertheless, we are aware that the predictive capacity of our approach could be enhanced through the use of larger training datasets, including nonhuman orthologues, or by using increased computational resources, such as additional GPUs, to enable training with larger batch sizes.
We have created online access to our model, IFF (IDP Feature Finder), which only requires a primary sequence or UniProt ID as input. We expect our program to be useful in various research fields, notably cell biology, to efficiently identify critical residues in proteins with IDRs, such as those that undergo LLPS.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Data S1. Supporting Information.
ACKNOWLEDGMENTS
The authors thank the IT Service Center at NYCU for GPU access. This work was supported by the National Science and Technology Council of Taiwan (110‐2113‐M‐A49A‐504‐MY3). The authors are also grateful to Prof. Wen‐Shyong Tzou (National Taiwan Ocean University) for his help.
Ho W‐L, Huang H‐C, Huang J. IFF: Identifying key residues in intrinsically disordered regions of proteins using machine learning. Protein Science. 2023;32(9):e4739. 10.1002/pro.4739
Review Editor: Nir Ben‐Tal
REFERENCES
- Alberti S, Gladfelter A, Mittag T. Considerations and challenges in studying liquid‐liquid phase separation and biomolecular condensates. Cell. 2019;176(3):419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Hyman AA. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat Rev Mol Cell Biol. 2021;22(3):196–213. [DOI] [PubMed] [Google Scholar]
- AlQuraishi M. End‐to‐end differentiable learning of protein structure. Cell Syst. 2019;8(4):292–301.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenhoff AM, Train CM, Gilbert KJ, Mediratta I, Mendes de Farias T, Moi D, et al. OMA orthology in 2021: website overhaul, conserved isoforms, ancestral gene order and more. Nucleic Acids Res. 2021;49(D1):D373–D379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three‐track neural network. Science. 2021;373(6557):871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79(4):1061–1078. [DOI] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18(5):285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhopatkar AA, Uversky VN, Rangachari V. Disorder and cysteines in proteins: a design for orchestration of conformational see‐saw and modulatory functions. Prog Mol Biol Transl Sci. 2020;174:331–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady JP, Farber PJ, Sekhar A, Lin YH, Huang R, Bah A, et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell‐specific protein on phase separation. Proc Natl Acad Sci U S A. 2017;114(39):E8194–E8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes N, Ofer D, Peleg Y, Rappoport N, Linial M. ProteinBERT: a universal deep‐learning model of protein sequence and function. Bioinformatics. 2022;38(8):2102–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke KA, Janke AM, Rhine CL, Fawzi NL. Residue‐by‐residue view of in vitro FUS granules that bind the C‐terminal domain of RNA polymerase II. Mol Cell. 2015;60(2):231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XL, Xie SN, He KM. An empirical study of training self‐supervised vision transformers. 2021 IEEE/CVF International Conference on Computer Vision (ICCV 2021). 2021:9620–9.
- Chiu SH, Ho WL, Sun YC, Kuo JC, Huang JR. Phase separation driven by interchangeable properties in the intrinsically disordered regions of protein paralogs. Commun Biol. 2022;5(1):400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conicella AE, Dignon GL, Zerze GH, Schmidt HB, D'Ordine AM, Kim YC, et al. TDP‐43 alpha‐helical structure tunes liquid‐liquid phase separation and function. Proc Natl Acad Sci U S A. 2020;117(11):5883–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik CS, Roczniak S, Largman C, Rutter WJ. The catalytic role of the active site aspartic acid in serine proteases. Science. 1987;237(4817):909–913. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- Echave J, Spielman SJ, Wilke CO. Causes of evolutionary rate variation among protein sites. Nat Rev Genet. 2016;17(2):109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnaggar A, Heinzinger M, Dallago C, Rehawi G, Wang Y, Jones L, et al. ProtTrans: toward understanding the language of life through self‐supervised learning. IEEE Trans Pattern Anal Mach Intell. 2022;44(10):7112–7127. [DOI] [PubMed] [Google Scholar]
- Fang X, Wang L, Ishikawa R, Li Y, Fiedler M, Liu F, et al. Arabidopsis FLL2 promotes liquid‐liquid phase separation of polyadenylation complexes. Nature. 2019;569(7755):265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, et al. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig JA, Nguyen TA, Lee M, Holehouse AS, Posey AE, Pappu RV, et al. Arginine‐enriched mixed‐charge domains provide cohesion for nuclear speckle condensation. Mol Cell. 2020;77(6):1237–1250 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison RC. Evolution of hemoglobin and its genes. Cold Spring Harb Perspect Med. 2012;2(12):a011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton G, Sejnowski TJ, editors. Unsupervised learning: foundations of neural computation. US: MIT Press; 1999. [Google Scholar]
- Ho WL, Huang JR. The return of the rings: evolutionary convergence of aromatic residues in the intrinsically disordered regions of RNA‐binding proteins for liquid‐liquid phase separation. Protein Sci. 2022;31(5):e4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemth P, Karlsson E, Vögeli B, Guzovsky B, Andersson E, Hultqvist G, et al. Structure and dynamics conspire in the evolution of affinity between intrinsically disordered proteins. Sci Adv. 2018;4(10):eaau4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson E, Sorgenfrei FA, Andersson E, Dogan J, Jemth P, Chi CN. The dynamic properties of a nuclear coactivator binding domain are evolutionarily conserved. Commun Biol. 2022;5(1):286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Yang YS, Sutter BM, Wang Y, McKnight SL, Tu BP. Redox state controls phase separation of the yeast Ataxin‐2 protein via reversible oxidation of its methionine‐rich low‐complexity domain. Cell. 2019;177(3):711–721 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380(6574):550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster AK, Nutter‐Upham A, Lindquist S, King OD. PLAAC: a web and command‐line application to identify proteins with prion‐like amino acid composition. Bioinformatics. 2014;30(17):2501–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenshtein VI. Binary codes capable of correcting deletions, insertions, and reversals. Soviet physics doklady; 1966. Soviet Union. [Google Scholar]
- Levitt M. Conformational preferences of amino acids in globular proteins. Biochemistry. 1978;17(20):4277–4285. [DOI] [PubMed] [Google Scholar]
- Li HR, Chen TC, Hsiao CL, Shi L, Chou CY, Huang JR. The physical forces mediating self‐association and phase‐separation in the C‐terminal domain of TDP‐43. Biochim Biophys Acta. 2018a;1866(2):214–223. [DOI] [PubMed] [Google Scholar]
- Li HR, Chiang WC, Chou PC, Wang WJ, Huang JR. TAR DNA‐binding protein 43 (TDP‐43) liquid‐liquid phase separation is mediated by just a few aromatic residues. J Biol Chem. 2018b;293(16):6090–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light S, Sagit R, Sachenkova O, Ekman D, Elofsson A. Protein expansion is primarily due to indels in intrinsically disordered regions. Mol Biol Evol. 2013;30(12):2645–2653. [DOI] [PubMed] [Google Scholar]
- Lin Y, Currie SL, Rosen MK. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J Biol Chem. 2017a;292:19110–19120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Qiu DC, Chang WH, Yeh YQ, Jeng US, Liu FT, et al. The intrinsically disordered N‐terminal domain of galectin‐3 dynamically mediates multisite self‐association of the protein through fuzzy interactions. J Biol Chem. 2017b;292(43):17845–17856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff‐Larsen K, Kragelund BB. On the potential of machine learning to examine the relationship between sequence, structure, dynamics and function of intrinsically disordered proteins. J Mol Biol. 2021;433(20):167196. [DOI] [PubMed] [Google Scholar]
- Lu AX, Lu AX, Moses A. Evolution is all you need: phylogenetic augmentation for contrastive learning. arXiv. 2020. [Google Scholar]
- Lu AX, Lu AX, Pritišanac I, Zarin T, Forman‐Kay JD, Moses AM. Discovering molecular features of intrinsically disordered regions by using evolution for contrastive learning. PLoS Comput Biol. 2022;18(6):e1010238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EW, Holehouse AS, Peran I, Farag M, Incicco JJ, Bremer A, et al. Valence and patterning of aromatic residues determine the phase behavior of prion‐like domains. Science. 2020;367(6478):694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, et al. A high‐coverage genome sequence from an archaic Denisovan individual. Science. 2012;338(6104):222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrea DM, Cika JA, Stanley CB, Nourse A, Onuchic PL, Banerjee PR, et al. Self‐interaction of NPM1 modulates multiple mechanisms of liquid‐liquid phase separation. Nat Commun. 2018;9(1):842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163(1):123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, et al. Direct‐coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci U S A. 2011;108(49):E1293–E1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J. 1998;75(1):422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162(5):1066–1077. [DOI] [PubMed] [Google Scholar]
- Polling S, Ormsby AR, Wood RJ, Lee K, Shoubridge C, Hughes JN, et al. Polyalanine expansions drive a shift into alpha‐helical clusters without amyloid‐fibril formation. Nat Struct Mol Biol. 2015;22(12):1008–1015. [DOI] [PubMed] [Google Scholar]
- Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92(5):1439–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rives A, Meier J, Sercu T, Goyal S, Lin Z, Liu J, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci U S A. 2021;118(15):e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P, Obradovic Z, Dunker AK. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Inform Ser Workshop Genome Inform. 1997;8:110–124. [PubMed] [Google Scholar]
- Saar KL, Morgunov AS, Qi R, Arter WE, Krainer G, Lee AA, et al. Learning the molecular grammar of protein condensates from sequence determinants and embeddings. Proc Natl Acad Sci U S A. 2021;118(15):e2019053118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu‐Gruttadauria J, MacRae IJ. Phase transitions in the assembly and function of human miRISC. Cell. 2018;173(4):946–957 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science. 2017;357(6357):eaaf4382. [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol. 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Imai K. Evolution of myoglobin. Cell Mol Life Sci. 1998;54(9):979–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang B, Arsenault J, Vernon RM, Lin H, Sonenberg N, Wang LY, et al. Phosphoregulated FMRP phase separation models activity‐dependent translation through bidirectional control of mRNA granule formation. Proc Natl Acad Sci U S A. 2019;116(10):4218–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Dunker AK. Understanding protein non‐folding. Biochim Biophys Acta. 2010;1804(6):1231–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oord A, Li Y, Vinyals O. Representation learning with contrastive predictive coding. arXiv. 2018; abs/1807.03748. [Google Scholar]
- Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, et al. Attention is all you need. Advances in Neural Information Processing Systems 30 (NIPS 2017). 2017:6000–10.
- Vihinen M, Torkkila E, Riikonen P. Accuracy of protein flexibility predictions. Proteins. 1994;19(2):141–149. [DOI] [PubMed] [Google Scholar]
- Wang J, Choi JM, Holehouse AS, Lee HO, Zhang X, Jahnel M, et al. A molecular grammar governing the driving forces for phase separation of prion‐like RNA binding proteins. Cell. 2018;174(3):688–699 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigt M, White RA, Szurmant H, Hoch JA, Hwa T. Identification of direct residue contacts in protein‐protein interaction by message passing. Proc Natl Acad Sci U S A. 2009;106(1):67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A. 1977;74(11):5088–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LE, Kim TH, Muhandiram DR, Forman‐Kay JD, Kay LE. NMR experiments for studies of dilute and condensed protein phases: application to the phase‐separating protein CAPRIN1. J Am Chem Soc. 2020;142(5):2471–2489. [DOI] [PubMed] [Google Scholar]
- Xu J. Distance‐based protein folding powered by deep learning. Proc Natl Acad Sci U S A. 2019;116(34):16856–16865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.