

Abstract
Graspetides are a class of RiPPs (ribosomally synthesized and post-translationally modified peptides) defined by the presence of ester or amide side chain-side chain linkages resulting in peptide macrocycles. The graspetide name comes from the ATP-grasp enzymes that install the side chain-side chain linkages. This review covers the early, activity-based isolation of the first graspetides, marinostatins and microviridins, as well as the key genomics-driven experiments that established graspetide as RiPPs. The mechanism and structure of graspetide-associated ATP-grasp enzymes is discussed. Genome mining methods to discover new graspetides as well as the analytical techniques used to determine the linkages in graspetides are described. Extant knowledge on the bioactivity of graspetides as protease inhibitors is reviewed. Further chemical modifications to graspetides as well graspetide engineering studies are also described. We conclude with several suggestions about future directions of graspetide research.
Keywords: Graspetides, RiPPs, natural product
Graspetides: from “grind and find” to genomes
In the 1980s and 1990s, a series of bioactivity-driven natural product isolation campaigns uncovered molecules known as the marinostatins and the microviridins. In the case of the marinostatins, a survey of ocean waters around Japan led to the isolation of a strain of Alteromonas (renamed as Algicola sagamiensis) that secreted a protease inhibitory activity [1]. Biochemical [2] and NMR [3] studies established marinostatin as a 12 aa peptide constrained by two side chain-side chain ester linkages (Figure 1A). In 1990, while studying extracts of the cyanobacterium Microcystis viridis using chromatography, a novel compound, named microviridin, was isolated. Careful NMR analysis of microviridin confirmed that it was a 14 aa peptide with three side chain-side chain linkages (see Glossary), two esters and one amide, as well as acetylation at its N-terminus [4] (Figure 1A). The original microviridin displayed tyrosinase inhibitory activity. Subsequent studies uncovered a wealth of additional related molecules, named microviridins B-J [5–9] produced by a series of different cyanobacteria. These compounds have sequences related to the original microviridin with some compounds named microviridin missing one of the three defining side-chain-side-chain linkages. For example, microviridin D contains only one ester and one amide linkage. The name “microviridin” was retained despite the fact that the producing organisms were no longer M. viridis. These additional microviridins displayed protease inhibitory activity in vitro [5–9], showing a shared bioactivity with the marinostatins. A key breakthrough in the potential ecological role for the microviridins came from studies showing that consumption of a strain of Microcystis cyanobacteria led to a molting disruption in the microcrustacean Daphnia [10]. This molting disruption in Daphnia, known colloquially as water fleas, can prove lethal, demonstrating a potential anti-feeding strategy for Microcystis. Subsequent work by Rohrlach et al. showed that microviridin J was the causative agent of the molting disruption [9, 11].
Figure 1:
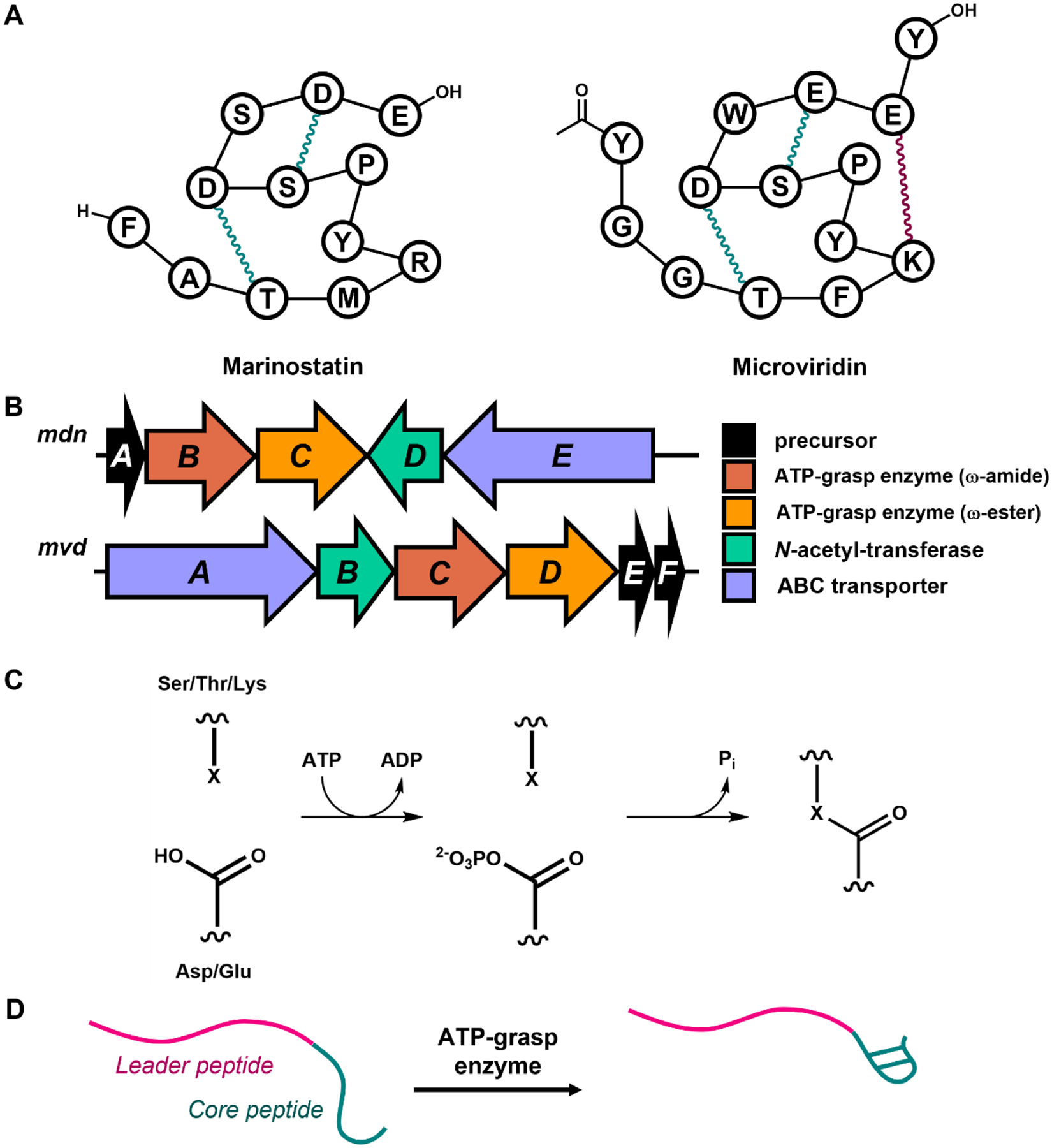
The discovery of graspetides as ribosomally synthesized and post-translationally modified peptides (RiPPs). A: Cartoons depicting the connectivity of (left) marinostatin and (right) microviridin. The ω-ester and ω-amide linkages are colored teal and maroon, respectively. B: The mdn biosynthetic gene cluster (BGC) for microviridin B and the mvd BGC for microviridin K. C: The mechanism of ω-ester or ω-amide linkage formation by a graspetide-associated ATP-grasp enzyme. X represents oxygen or nitrogen from a Ser, Thr, or Lys donor residue. D: Schematics for modifying the C-terminal core peptide (teal) of a graspetide precursor with ATP-grasp enzyme, which requires the leader peptide (pink).
The first suggestion of a biosynthetic route for marinostatins or microviridins came from work by Miyamoto et al. in 1998 [12]. In this study, a genomic library was constructed from the marinostatin producing strain, and Southern blotting experiments using the sequence of marinostatin as a probe ultimately identified an open reading frame (ORF) of 63 aa that includes the 12 aa marinostatin sequence. This was the first indication that peptides from the marinostatin/microviridin class may have been ribosomally derived. This idea was firmly established in 2008 with a pair of reports on microviridin biosynthesis [13–14]. With the aid of then-new cyanobacterial genome sequences, Ziemert et al. were able to define the biosynthetic gene clusters (BGCs) of microviridin B and microviridin J [14]. These BGCs encode a precursor peptide including the microviridin core peptide sequence, two ATP-grasp enzymes, an acetyltransferase, and an ABC transporter (Figure 1B). Expression of the microviridin B and microviridin J BGCs in E. coli led to the production of a series of modified peptides related to the natural products, providing a direct link between the BGCs and the natural products. Philmus et al. examined extracts of the cyanobacterium Planktothrix agardhii and isolated microviridin K, which is the fully dehydrated version of microviridin D. These authors also found the corresponding BGC in the genome sequence of P. agardhii (Figure 1B). To make a direct connection between the product and the BGC, these authors purified the two ATP-grasp enzymes and demonstrated in vitro that the two enzymes installed three dehydrations on the microviridin precursor peptide in an ATP-dependent fashion. With these two seminal works in 2008, it was firmly established that the microviridins belong to the superfamily of ribosomally-derived natural products that are now referred to as RiPPs, or ribosomally synthesized and post-translationally modified peptides. A more recent heterologous expression study also confirmed that marinostatin is a RiPP as well [15].
Given that the key enzymes in the modification of microviridins and marinostatins are ATP-grasp enzymes, it was suggested that these natural products and others for which the biosynthesis follows similar logic be referred to as graspetides [16]. In the past 5+ years there has been a flurry of activity in the discovery and structure of new graspetides, the study of the ATP-grasp enzymes that make graspetides, and in graspetide engineering. This paper will review these breakthroughs, especially in the context of the historical discoveries discussed above. We will close with a brief opinion on future directions for graspetide research.
Mechanism of graspetide formation
As discussed above, the side-chain-side-chain crosslinks that define graspetide natural products are installed by one or more ATP-grasp enzymes. These enzymes also catalyze important reactions in primary metabolism [17]. Briefly, the ATP-grasp activates the side-chain of Glu or Asp acceptor residue as an acyl phosphate, consuming one molecule of ATP. The linkages are formed with subsequent attack by a nucleophilic sidechain of a donor residue, which can be Ser, Thr, or Lys (Figure 1C). A series of in vitro biochemical and structural studies have led to an understanding of how these ATP-grasp enzymes engage and modify their peptide substrates. The in vitro studies on microviridin K by Philmus et al. established that the side-chain-side-chain linkages in microviridin K can only be installed when the leader peptide, the N-terminal portion of the 48 aa microviridin K precursor MvdE, is present [13] (Figure 1D). In other words, while the 48 aa MvdE precursor peptide is cyclized by the ATP-grasp enzymes MvdC and MvdD, a linear 14 aa core peptide corresponding to the final microviridin product is unable to be cyclized [13]. This leader peptide-directed biosynthesis has subsequently been appreciated as a hallmark of RiPP biosynthesis [18–19] wherein the leader peptide positions the core peptide for modification within RiPP maturation enzymes. Mutagenesis studies on the leader peptide of microviridin L carried out heterolgously in E. coli demonstrated the importance of a conserved heptapeptide motif, PFFARFL, in the leader peptide of microviridins [20]. Mutagenesis of this motif led to either incomplete or no processing of the microviridin core. It was suggested that this motif served to dock the microviridin precursor within the ATP-grasp enzymes. Later work showed that leader peptide of MdnA, precursor to microviridin J, could be truncated N-terminal to the PFFARFL while still being correctly cyclized by the cognate ATP grasp enzymes [21].
The ordering of crosslink introduction into microviridins and other graspetides has been studied using both mutagenesis methods and by analyzing partially cyclized substrates. In the case of microviridin K, it was established that its two ester linkages must be installed before the amide linkage can be formed [13]. In a followup by this same group, mutagenesis studies demonstrated that the two ester linkages are installed in a specific order [22]. This precise order of crosslink installation was also observed in the biosynthesis of the graspetide thuringinin [23]. This peptide forms a stem-loop structure with two ester linkages (see Figure 3), and it has been demonstrated that the innermost crosslink, forming the loop, is installed first, followed by formation of the stem crosslink. We have observed similar ordering in the stem-loop graspetide amycolimiditide, a peptide with four ester crosslinks in its stem. Like thuringinin, the first ester to form in amycolimiditide establishes the loop of the peptide with subsequent esters installed in order along the stem [24].
Figure 3:
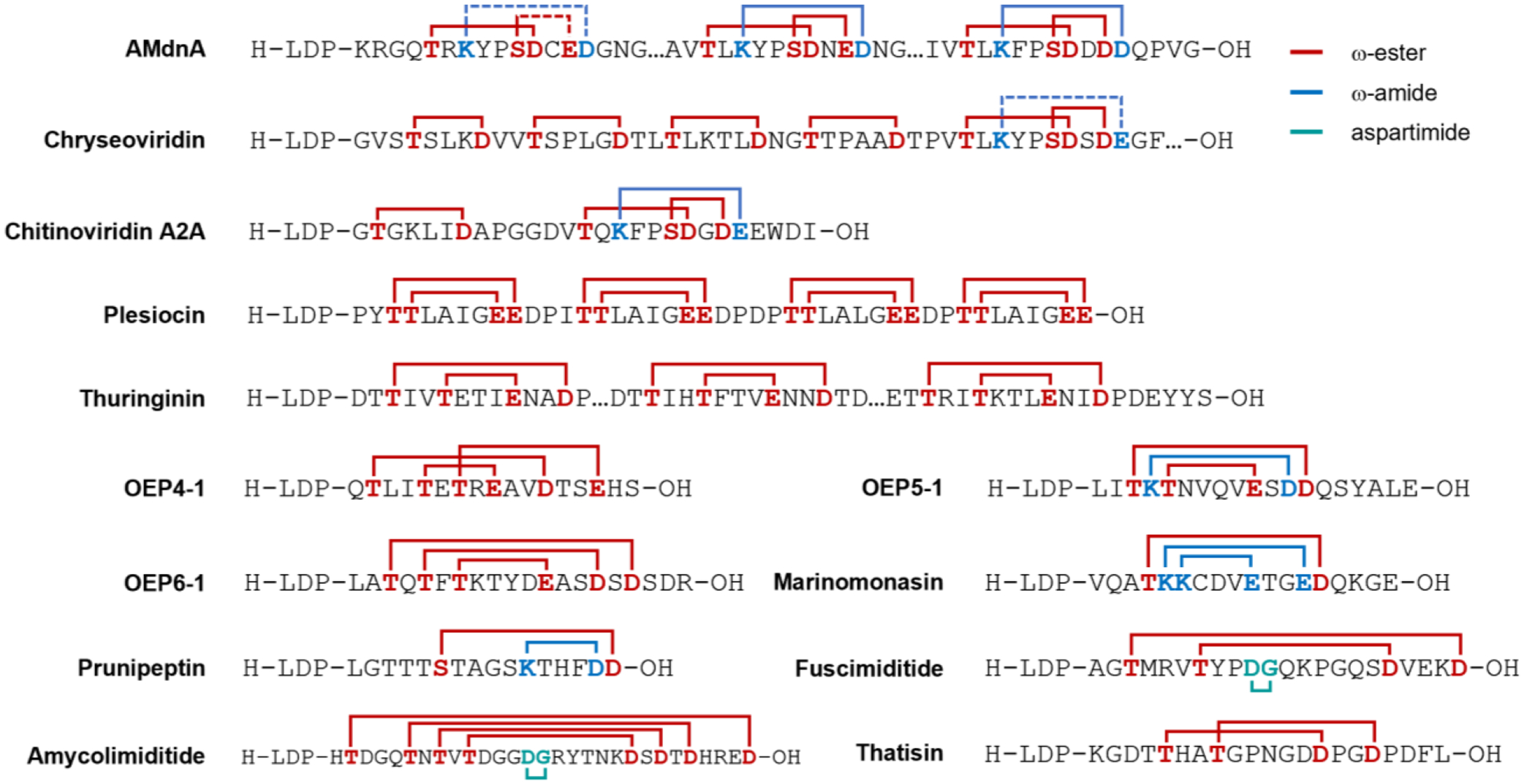
Connectivity of selected graspetides with novel patterns of crosslinks. Red line indicates an ω-ester linkage between Ser/Thr and Asp/Glu. Blue line indicates an ω-amide linkage between Lys and Asp/Glu. Cyan line indicates an aspartimide moiety. Dashed line indicates an expected linkage based on the typical microviridin crosslinking pattern that is not proven experimentally yet. LDP at the N-terminus of these sequences stands for leader peptide.
The ATP-grasp enzymes involved in microviridin and other graspetide biosynthesis have been the topic of several different structural studies. Bruner and colleagues solved the first crystal structures of microviridin-associated ATP-grasp enzymes, MdnB and MdnC, encoded in the microviridin J BGC [21]. As expected from other ATP-grasp enzymes as well as size exclusion chromatography, both MdnB and MdnC crystallized as dimers. A structure of the complex between MdnC and the leader peptide of MdnA was also solved. This structure showed that the conserved PFFARFL motif in the leader peptide folds into an α-helix and the Arg residue of this motif docks with a pair of well-conserved acidic sidechains in MdnC. Two additional structures of graspetide ATP-grasp enzymes were published in 2021 providing further details about substrate recognition in these enzymes (Figure 2). The enzyme CdnC, an ATP-grasp enzyme found in a graspetide BGC in Chryseobacterium gregarium, installs ester linkages in a microviridin-like core as well as in 4 additional 5–6 aa macrocycles with a single ester (see Figure 3). Bewley and colleagues solved a structure of CdnC that includes a leader peptide segment, a core peptide (not attached to the leader), and ADP [25]. Of note is that the two molecules of the CdnC dimer in the crystals are asymmetric; one CdnC molecule is a quaternary complex with all components present (leader, core, and ADP), while the other molecule lacks the core peptide substrate (Figure 2). This asymmetry highlights rearrangements that occur upon core peptide binding. Kim and colleagues solved a series of structures of PsnB, the ATP-grasp enzyme responsible for the cyclization of plesiocin (Figure 3). This group also solved the structure of a quaternary complex with separate leader and core peptides and an ADP nucleotide (Figure 2). Like the chryseoviridin case, the crystallized dimers were asymmetric. In the most asymmetric dimer, one molecule of PsnB was in its apo state while the other molecule was fully liganded with leader and core peptides and the nucleotide [26]. From these structures emerges a model in which the binding of each component (leader peptide, nucleotide, and core peptide) induces conformational changes to the enzyme resulting in a catalytically competent active site. Most recently, a structure of the ATP-grasp enzyme AMdnB, which processes multiple microviridin-like core peptides [27] (discussed in more detail below), was solved in its apo form [28]. Comparison of this structure to the MdnB structure revealed differences in the homodimer interface, which may be relevant to the iterative processing of multicore graspetides.
Figure 2:
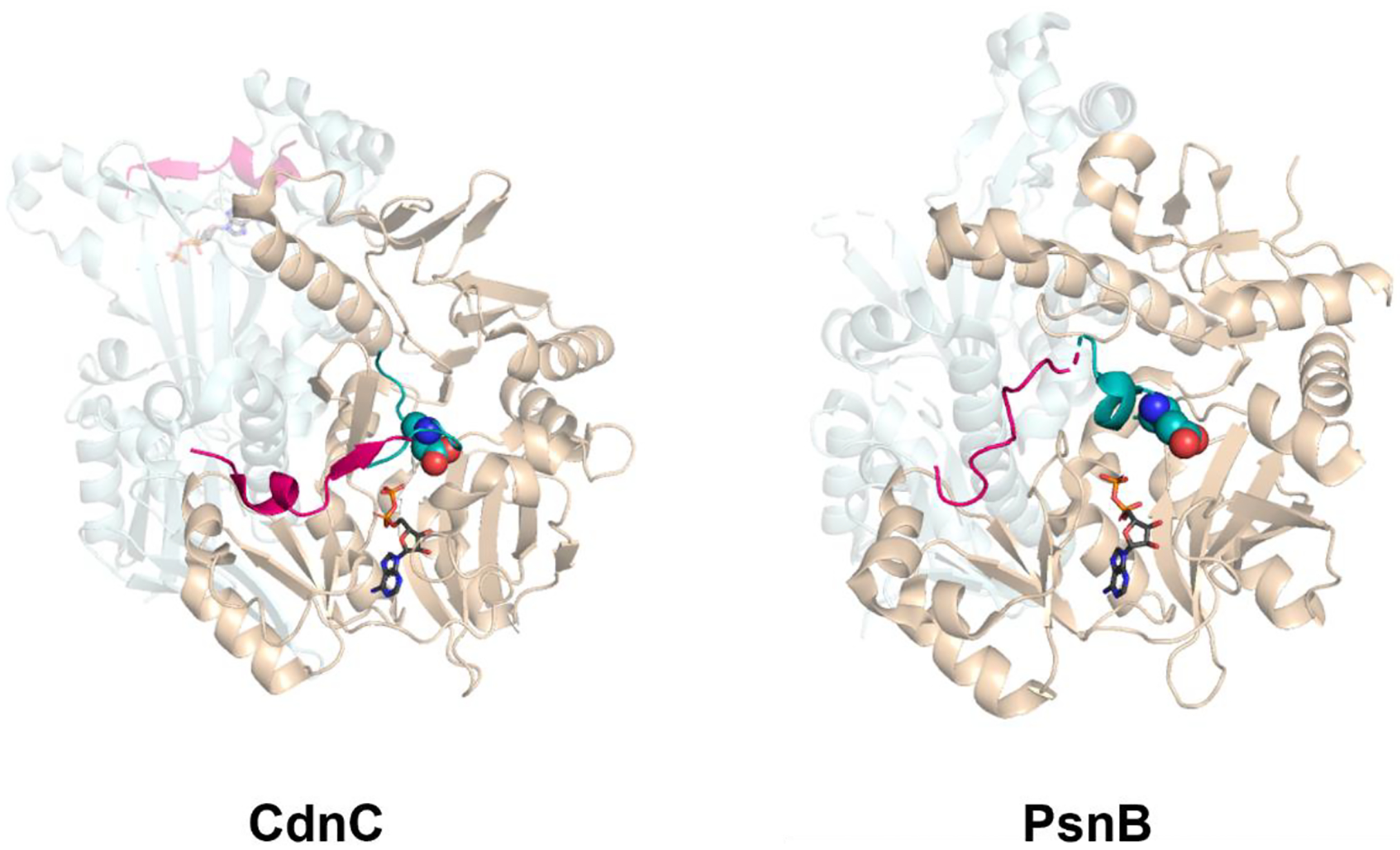
The crystal structures of ATP-grasp enzyme dimers with ADP and graspetide precursor. Left: CdnC, the ester-forming ATP-grasp enzyme for chryseoviridin (PDB ID: 7MGV); right: PsnB, the ATP-grasp enzyme for plesiocin (PDB ID: 7DRM). The ATP-grasp enzyme complexed with the precursor (leader peptide in pink and core peptide in teal) and ADP (grey) is colored wheat. The other monomer without the core peptide (CdnC) or in apo state (PsnB) is colored silver. The side chain of the first acceptor residue in the core peptide is represented as spheres.
Discovery of new graspetides with multiple cores and different crosslink patterns
Next-generation sequencing has led to the explosively rapid expansion of bacterial genome databases, tremendously aiding recent genome mining efforts for discovering new graspetides [16]. Recent phylogenomic studies continued to reveal new microviridin-type graspetides across Cyanobacteria, Proteobacteria, and Bacteroidetes [29–32], including grimoviridin [33] and microviridin 1777 [34]. Some of them contain unprecedented multiple cores additionally installed by their cognate ATP-grasp enzymes. The first type of multicore microviridins to be experimentally characterized was AMdnA, produced by the filamentous cyanobacterium Anabaena sp. PCC7120 [14, 27–28]. AMdnA contains three microviridin-like core repeats containing the TxKxPSDx(D/E)(D/E) consensus motif. As demonstrated by the in vitro reconstitution experiments, the last two repeats are fully tri-cyclized by the ATP-grasp enzymes AMdnC and AMdnB, whereas the first repeat is only singly esterified. Another type of multicore microviridins displays tandem macrolactone rings installed by an ATP-grasp enzyme preceding the microviridin-type core peptide at the C-terminal end. This is the case for chryseoviridin [25] and chitinoviridin A2A [35], displaying four 5–6 residue macrolactone rings and one 6 residue macrolactone ring, respectively, before the C-terminal true microviridin-like core.
Plesiocin [36] was the first graspetide characterized to have a different structure than microviridins and marinostatins. Its BGC is comprised of three genes encoding a precursor (psnA1), an ATP-grasp enzyme (psnB), and an unknown membrane protein (psnC), with an additional precursor gene (psnA2) located distal to the BGC. The graspetide precursor encoded by psnA2 was selected for further experimental characterization. Plesiocin has four core repeats, each comprising 9–13 residues with the conserved sequence motif TTxxxxEE. Interstitial Asp-Pro dipeptides located between the four core repeats were used as sites for cleavage using 70% (v/v) formic acid. Each core was determined to be doubly dehydrated, indicating the presence of two crosslinks for each repeat. The connectivity was determined by analyzing the two singly dehydrated partial hydrolysates of each core repeat. Each core repeat was revealed to have a hairpin structure in which the first Thr formed the outermost ω-ester linkage with the second Glu and the second Thr formed the innermost ω-ester linkage with the first Glu. The overall structure of plesiocin was determined as a four-leafed bicyclic hairpin peptide with four-residue loops. Thuringinin [23] is another graspetide that does not resemble microviridin or marinostatin and has a similar overall structure as plesiocin. While close homologs have up to seven core repeats, the graspetide with three repeats was chosen for experimental characterization. Two crosslinks were determined to be localized in each core repeat with the consensus sequence TxxTxxxExxDxD. The ω-esters were located between the second Thr and Glu residue and the first Thr and the first Asp residue. Hence, thuringinin has a similar stem-loop hairpin repeat structure like plesiocin, but with three-residue loop and two interstitial residues between the crosslinks.
Genome mining efforts after the discovery of plesiocin and thuringinin revealed about four thousand predicted graspetides in publicly available genomes [30, 37–38]. The graspetides were classified initially into 12 groups by Kim and co-workers [37]. The classification was expanded to 24 groups by Mitchell and co-workers [38], based on each group’s conserved consensus sequence or putative presence of additional PTM enzymes. Makarova et al. also carried out genome mining for graspetides, classifying them into 174 separate families based on predicted connectivity and putative modification genes [30]. Of the original 12 groups proposed by Kim and co-workers, microviridin and marinostatin-type peptides were categorized as Group 1 graspetides. Plesiocin-like peptides formed Group 2, and thuringinin-like graspetides formed Group 3. Group 4, 5, and 6 graspetides were experimentally characterized by similar techniques for elucidating plesiocin and thuringinin connectivity [37]. Group 4 graspetides exhibit the TxxTxTxExxDxxE consensus core sequence with connectivity of three ω-ester linkages shown in Figure 3. The presence of an ester crosslink overarching the two remaining macrocyclic linkages suggest a caged-like structure similar to that of microviridins. Group 5 and 6 graspetides have TKTxxxxExDD and TxTxTxxxxExxDxD motifs with connectivity for stem-loop hairpin as shown in Figure 3. Unlike other graspetides with stem-loop hairpin structure, Group 5 and 6 graspetides have unequal and inconsistent number of interstitial residues between the crosslinks. Another unique observation is that both ω-amide and ω-ester linkages in Group 5 graspetide are installed by the same ATP-grasp enzyme, demonstrating an unprecedented dual functionality. Bioinformatics analysis of Group 4–6 graspetides also revealed examples with multiple core repeats [37].
Marinomonasin is a Group 7 graspetide with one ω-ester and two ω-amide linkages [39]. The structure is another example of stem-loop hairpin connectivity with highly constrained stem with no interstitial residue between the crosslinks except two residues between innermost and middle ω-amide linkages at the C-terminal side. Prunipeptin is a Group 11 graspetide with one ω-ester and one ω-amide linkages forming a bicyclic hairpin structure with unequal number of interstitial residues between the crosslinks [40]. Fuscimiditide [41] and amycolimiditide [24] are Group 13 graspetides, most of which putatively contain an aspartimide moiety installed by enzyme homologs of protein L-isoaspartyl (D-aspartyl) O-methyltransferase (PIMT) [42]. The aspartimidylation of these graspetides will be discussed later. Thatisin is a Group 16 graspetide that is named after the Thr-His-Ala-Thr tetrapeptide within the N-terminal crosslinked region [38]. In this study, the connectivity was determined as two interlocking ω-ester crosslinks. The methods used to determine the positions of crosslinks within these graspetides is discussed in the next section.
Methods for determining crosslinks in graspetides
Determining the location of crosslinks within graspetide is key to understand their overall structure. A variety of methods exist using NMR and mass spectrometry to determine crosslinking positions. The gold standard of these methods is 2D NMR experiments. Proton-carbon HMBC experiments can be used to unequivocally assign either ester or amide crosslinks while the NOESY experiment can be used for amide crosslinks (Figure 4) [4, 24, 41]. Ester and amide crosslinks can also be disrupted in the mass analyzer in MALDI-MS/MS experiments [36–37]. In the case of amides, the acceptor residue (Glu or Asp) is transformed into an acylium ion, marking this residue as the site of modification. For esters, a McLafferty rearrangement can occur, resulting in a dehydrated donor residue (Figure 4). Another approach to determining the identity of acceptor residues in graspetide crosslinks is treat the peptide with an appropriate nucleophile (methoxide, hydrazine) or reducing agent (LiBH4), both of which will cleave the ester bond and mark the acceptor sidechain for detection by mass spectrometry (Figure 4) [4, 23–24, 36–38, 43–44].
Figure 4:
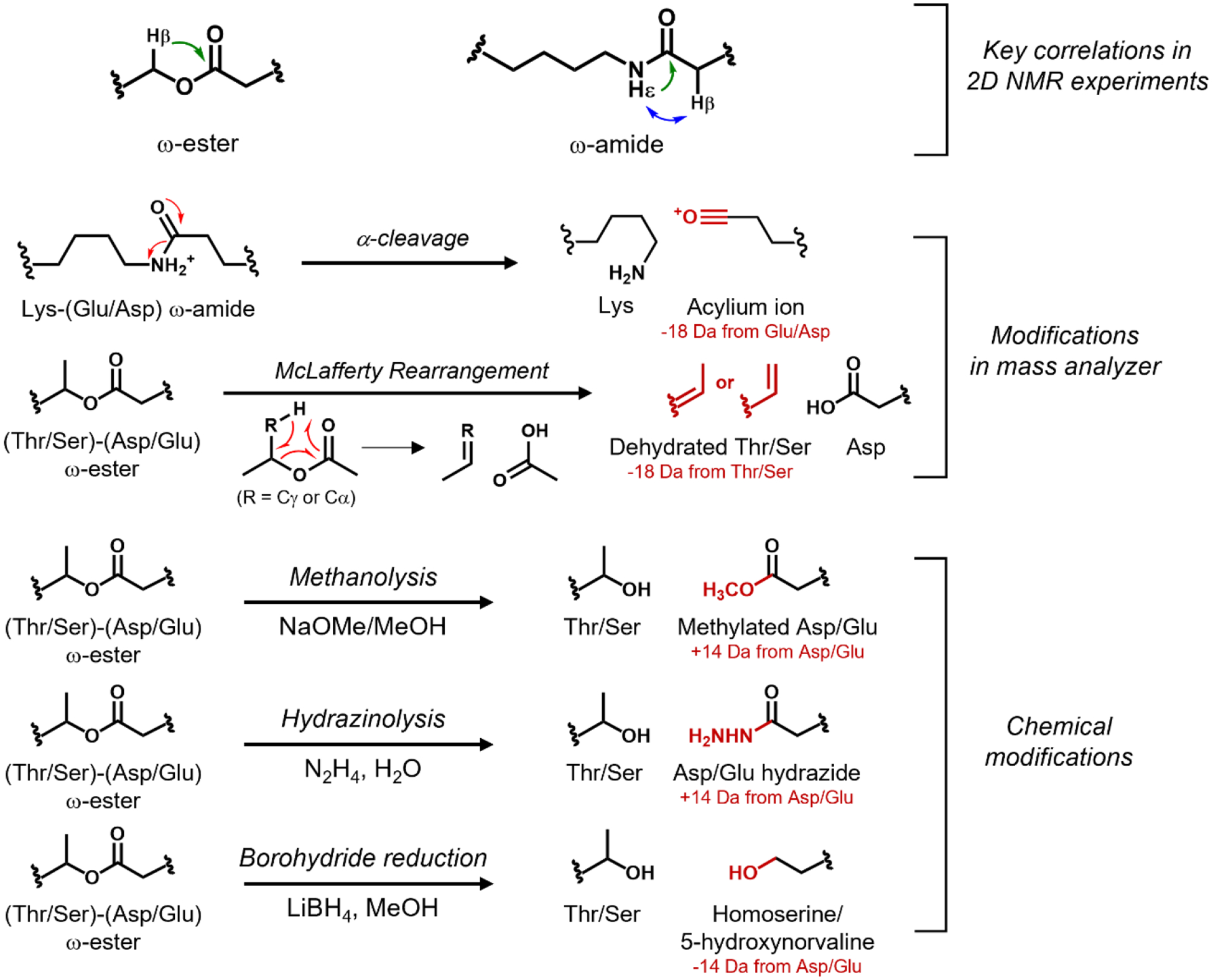
NMR and MS/MS-based methodology for locating ω-ester or ω-amide crosslinks. An HMBC correlation (green) between a ω-proton (Hβ) of Ser or Thr and the side chain carbonyl carbon of Asp or Glu indicates an ω-ester linkage. An ω-amide linkage is indicated by the HMBC correlation between the ε-proton of Lys (Hε) and the side chain carbonyl carbon of Asp or Glu, or a NOESY correlation between the Hε of Lys and a Hβ of Asp or Hγ of Glu; Asp is shown in the figure. For MS/MS-based methods, red color indicates the resulting chemical modification and mass difference that serve as a footprint for crosslinking. The α-cleavage and McLafferty rearrangement occur during MS/MS experiments. Methanolysis, hydrazinolysis, and borohydride reduction are chemical reactions performed before MS(/MS) analysis.
Bioactivities of graspetides
Graspetides with known bioactivities are mostly microviridins that inhibit various serine proteases [45–46], with a few potently inhibiting elastase [5, 7, 36, 47], chymotrypsin [34, 36], trypsin [9], and subtilisin [3, 48] at low nanomolar ranges (≤100 nM) and others weakly inhibiting tyrosinase [4], thrombin [49], and the aforementioned proteases [6, 8, 50–52]. As protease inhibitors, graspetides have received attention as potential therapeutic agents that down-regulate malicious proteolytic activities [45]. For example, potent elastase inhibitors microviridins have been called for testing against neutrophil elastase [45–46], whose excessive activity in the lungs may cause pulmonary emphysema and cystic fibrosis [53]. Thrombin, which proteolytically converts fibrinogen into an insoluble fibrin clot as part of a procoagulant activity [54], is another therapeutic target suggested for graspetides as an anticoagulant against excessive blood clotting [45]. For this purpose, microviridin B was shown to inhibit thrombin with an EC50 value of 4.58 μM [49]. So far, however, no graspetides have advanced to clinical utility.
Microviridin J is a potent trypsin inhibitor, and its mechanism and potential ecological role were extensively studied. Besides inhibiting laboratory-grade porcine trypsin, microviridin J was also found to inhibit trypsin-like proteases produced from the water flea Daphnia magna [9]. Lethal molting disruption was observed in a similar microcrustacean Daphnia pulcaria when co-incubated with purified microviridin J or its producer Microcystis UWOCC MRC [10] or UWOCC CBS [11]. Hence, it was hypothesized that the microviridin could inhibit trypsin-like proteases in Daphnia that are essential for developing new integuments [9]. Similarly, microviridin C and seven other putative microviridins from Microcystis strains HUB08B03, HUB11G02 and HUB19B05 were found to moderately inhibit the trypsin-like activity of Daphnia [55]. Recently, another graspetide, microviridin 1777 from Microcystis aeruginosa strain EAWAG 127, was discovered to kill the fairy shrimp Thamnocephalus platyurus at LD50 value of 95 μM [34], underscoring the trend that microviridins produced from Microcystis cyanobacteria serve as lethal agents against microcrustaceans by a mechanism of proteolysis inhibition.
The mechanism of protease inhibition performed by a graspetide was revealed by Weiz et al. from the co-crystallization of microviridin J with trypsin [56]. The structure revealed that the ω-ester-linked Thr4, Arg5, and ω-amide-linked Lys6 acted as P2, P1, and P1’ residues of a trypsin substrate, respectively. Thr4 (P2) γ-methyl group was oriented towards Leu99 residue of the trypsin S2 binding pocket; Arg-5 (P1) was shown to interact with Asp189 carboxyl group of the trypsin S1 binding pocket; the aliphatic stretch of Lys6 (P1’) side chain was shown to form van der Waals contacts with Cys42-Cys58 disulfide linkage at the S1’ site of trypsin. In addition to these stabilizing factors for microviridin-trypsin interaction, the C-terminal part of microviridin J spanning from Ser9 to Trp14 adopted a helical conformation stabilized by intramolecular covalent linkages of the graspetide, whose ester and amide groups form additional polar contacts within the peptide. This makes microviridin J unable to be processed by trypsin while strongly binding it. In addition to confirming the fifth position of microviridin J, which is variable across different microviridins, as the specificity-determining P1 residue, the authors were able to shift the target protease specificity of microviridin J by substituting the fifth residue with different residues. For example, the F5L variant of microviridin L transformed the subtilisin inhibitor into an elastase inhibitor (IC50 = 0.13 μM) [56]. Similarly, Taichi et al. modified marinostatin to be a trypsin inhibitor with Ki of 5.3 nM by swapping its P1 and P1’ residues (Met and Arg, respectively) [48], and Lee et al. substituted the Leu3 P1 residue of the last core repeat of plesiocin with arginine, creating a variant capable of simultaneously inhibiting chymotrypsin and trypsin with Ki of 5.4 and 130 nM, respectively [57]. Additionally, for plesiocin, the variant containing only one of the four repeats showed weaker inhibition against elastase and chymotrypsin, indicating that the number of core repeats positively influences the inhibitory activity.
Recently, Saha et al. demonstrated that microviridins might additionally play a role as quorum-sensing modulators within a cyanobacterial community [58]. A bioluminescence assay was performed with E. coli containing lasI-lasR or luxI-luxR receptors controlling the expression of luxCDABE. The tested microviridins, microviridin-1688, microviridin-1739, and microviridin-1748, inhibited bioluminescence against only the lasI-lasR receptor by 32–55%, demonstrating their capability of influencing intercellular communication in a discriminatory fashion. As further evidence for relevance to quorum-sensing, the yield of microviridin production from native organisms was shown to be affected by the presence of autoinducers [58] and cell density [59–60].
Graspetide modifications beyond the signature ω-ester/amide crosslinks
An on-going interest in the graspetide field is a further diversification of graspetide structure from post-translational modifications other than the signature ester/amide crosslinking performed by ATP-grasp enzymes. As discussed earlier, most of Group 13 graspetides are synthesized from BGCs that additionally encode a gene for an enzyme homologous to protein L-isoaspartyl/D-aspartyl O-methyltransferase (PIMT) [38, 41]. This enzyme is known as a catalyst for repairing aged proteins with accumulated isoaspartate (isoAsp) β-amino acid, which is a hydrolysis product of aspartimide formed by asparagine deamidation or aspartate dehydration [61]. PIMT selectively transforms L-isoAsp and D-Asp into L-aspartimide, which hydrolyzes back to L-isoAsp or L-Asp, creating an iterative loop that overall reverts L-isoAsp into L-Asp [62]. On the other hand, the O-methyltransferase that modifies graspetides were shown to transform both Asp and isoAsp into aspartimide (Figure 5) [24, 41]. Aspartimide on graspetides can withstand hydrolysis for days, depending on the ionic strength of the solvent, at a neutral pH [24, 41]. On the other hand, an aspartimide on a synthetic linear peptide was shown to hydrolyze within few hours under similar conditions [61, 63], showing that aspartimides on graspetides are more stable. Aspartimide on a linear peptide typically hydrolyzes at about 3–4:1 ratio of isoAsp and Asp [61, 63]. This ratio differs widely across aspartimide-containing graspetides (~95% isoAsp for fuscimiditide, ~70% isoAsp for amycolimiditide) [24, 41], as well as lanthipeptide (~70% isoAsp) [64] and lasso peptides (exclusively Asp) [65].
Figure 5:
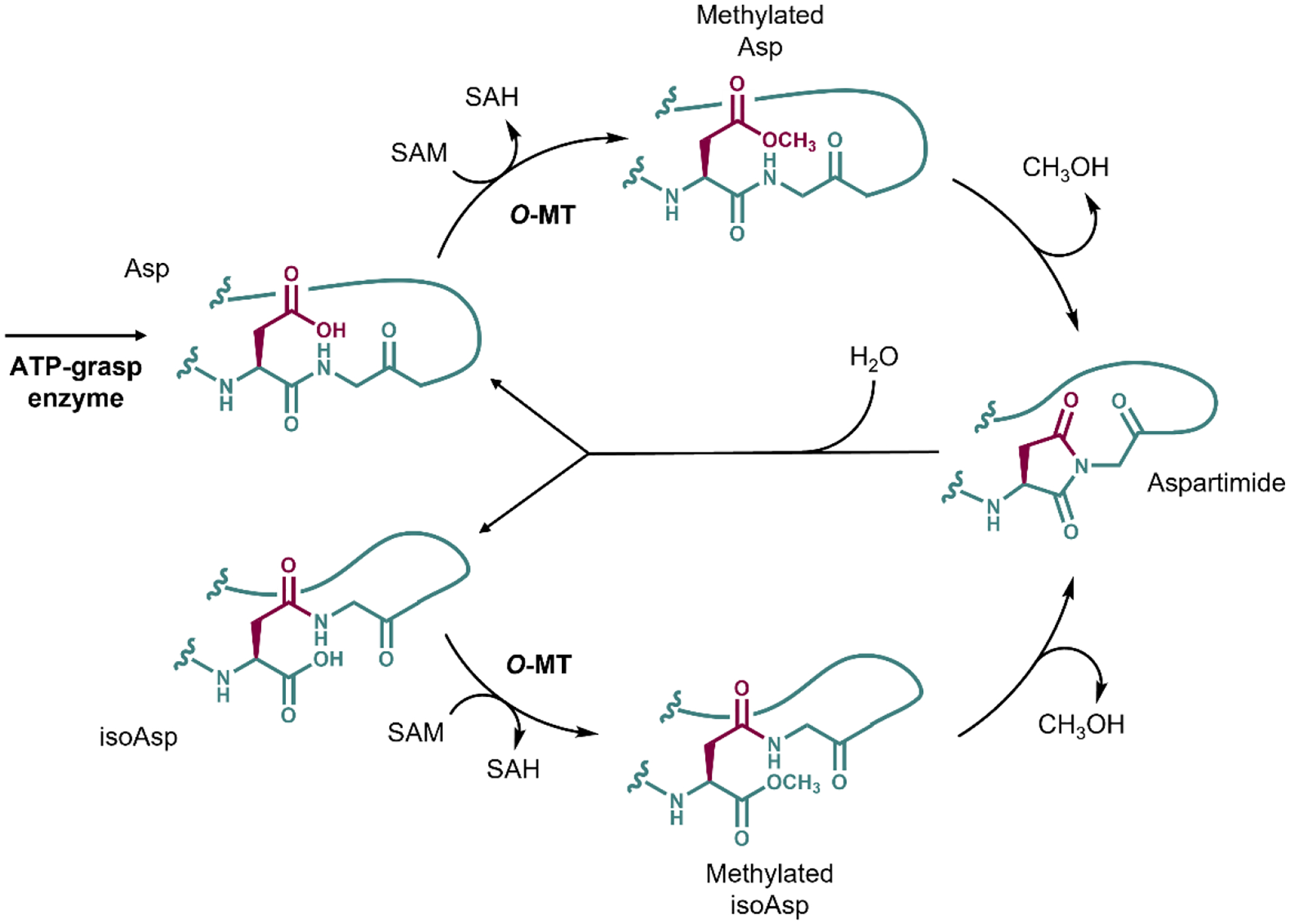
The aspartimidylation pathway of Group 13 graspetides catalyzed by O-MT. The original side chain of Asp is colored maroon, while the rest of the peptide is colored teal. The final product of the reaction catalyzed by ATP-grasp enzyme is the graspetide precursor with all ω-ester linkages with Asp in the aspartimidylation site. The O-MT methylates the Asp using SAM as a methyl group donor. The aspartimide forms spontaneously, resulting in CH3OH leaving the peptide. The aspartimide can be hydrolyzed at the side chain or the backbone, generating Asp (top) or isoAsp (bottom) at the aspartimidylation site. The O-MT recognizes both species, able to re-aspartimidylate the graspetide.
So far, the experimentally verified Group 13 graspetides, fuscimiditide and amycolimiditide, have a stem-loop hairpin-like structure, with ω-ester crosslinks forming the stem of the peptide. In our amycolimiditide study, we demonstrated that all ester crosslinks are required for efficient aspartimidylation by comparative kinetics experiment across amycolimiditide variants lacking ester crosslinks. We also established that the follower residue of the aspartimide must be Gly for successful aspartimidylation, unlike the aspartimidylated lasso peptides. Most of the fuscimiditide and amycolimiditide homologs contain the conserved Asp-Gly site between the N-terminal nucleophile-enriched region and the C-terminal acid-enriched region in the core peptide sequences. Similarly, Stephenson and Clarke in 1989 demonstrated that spontaneous aspartimidylation occurs fastest with Gly as the follower residue in linear peptides [66].
The bioactivity of aspartimidylated graspetides and other aspartimidylated RiPPs has not been established yet. It is unclear whether these RiPP natural products are bioactive in the pre-aspartimidylated, aspartimidylated, or isoAsp form. Possibly, the isoAsp-containing graspetides are bioactive weapons against other organisms, initially made inactive as the aspartimidylated form within the host organism by the coexpressed O-methyltransferase. Alternatively, the aspartimide serves as an electrophilic handle for binding an endogenous protein, making the aspartimidylated graspetide an internally bioactive molecule.
The recent genome mining efforts by Ramesh et al. and Makarova et al. led to the discovery of novel graspetides whose BGCs encode an additional PTM enzyme, such as radical SAM (rSAM) methyltransferases with SPASM domain, SagB-like dehydrogenase, and YcaO-like cyclodehydratases [30, 38]. The rSAM methyltransferases containing the SPASM domain are commonly RiPP-modifying enzymes that catalyze PTMs such as Trp-Lys C-C bond formation in streptide [67], Trp-Trp ether linkage formation in darobactin [68], and Cα-S thioether crosslinking in ruminococcin C [69–70]. On the other hand, SagB-like dehydrogenase, and YcaO-like cyclodehydratases are biosynthetic enzymes involved in thiazole and oxazole heterocyclization widely observed across different RiPP classes like linear azol(in)e-containing peptides (LAPs) and thiopeptides [16, 30]. So far, there is no experimental validation confirming whether these enzymes can install additional modifications on graspetides. This raises questions regarding what modifications are performed by these enzymes on graspetides, whether the chemical pathway would involve a pre-formed ω-ester/amide linkages, and the extent of structural diversification or bioactivity modulation due to such modification.
Engineering of graspetide scaffolds
Besides searching for graspetides with novel linkage patterns and additional PTMs, researchers have attempted to artificially introduce nonnative modifications to graspetides. Kanaori et al.’s work on marinostatin was the first to examine the properties of an artificially modified graspetide, featuring a marinostatin variant whose ω-ester linkages are replaced with disulfide bonds [3]. This variant, named MST-2SS, was determined to have a more flexible, less constrained structure, based on fewer NOEs and higher proton exchange rates in NMR experiments. MST-2SS exhibited a similar inhibitory activity against subtilisin BPN’, with Ki of 3.4 nM, similar to 1.5 nM from the original marinostatin. The disulfide substitution was also applied recently in the amycolimiditide study to examine the importance of ω-ester linkages for the methyltransferase-catalyzed aspartimidylation [24]. When the outermost crosslink of amycolimiditide was replaced with a disulfide bond, methylation was slowed down at a rate similar to the methylation rate of the amycolimiditide variant with missing outermost linkage. Reduction of the disulfide bond with dithiothreitol did not affect the methylation rate, indicating that the ester functional groups on the graspetide stem are important for recognition by the methyltransferase. Overall, the disulfide substitution study probes whether an ester crosslink of a graspetide serves solely as a conformational constraint.
Franz and Koehnke engineered microviridin J as a hybrid RiPP that contains thiazole moieties [71], which is widely observed across different classes of RiPPs, including LAPs, cyanobactins, thiopeptides, and bottromycins [72]. The strategy for nonnative thiazole backbone heterocyclization entailed leader peptide exchange. The microviridin core variant with Cys substitutions was C-terminally fused to the cyanobactin heterocyclase LynD leader peptide and sortase SrtA recognition sequence. After the free Cys residues were modified into thiazoles by LynD, SrtA was used to perform transpeptidation, exchanging the LynD leader peptide with the microviridin J leader peptide. The ATP-grasp enzyme MdnC successfully modified the peptide, yielding a hybrid microviridin J containing thiazole moieties. This opens up possible avenues for engineering graspetides, as thiazole incorporation on protein loops and linkers has been demonstrated to improve stability or confer an additional function [73].
Scholz et al. demonstrated that various functional groups can be incorporated at the N-terminus of microviridins B and K [74]. The functional tags studied were Nε-biotinyl-L-lysine for biotin-avidin pulldown experiments, a dansyl group as a fluorophore, or O-propargyl-L-tyrosine for click chemistry. These functional groups were incorporated during solid phase peptide synthesis, and the resulting linear peptide was then macrocyclized by the constitutively active ATP-grasp enzymes LP-MvdD and LP-MvdC that do not require the leader peptide to be intact on the precursor. This technique was developed to facilitate bioactivity screening of graspetides of unknown function. However, this requires the cognate ATP-grasp enzymes to be constitutively activated like LP-MvdD and LP-MvdC, which may not be successful. So far, the only ATP-grasp enzymes enabled for constitutive activation are those for microviridins and plesiocin [57, 75].
To engineer microviridins for an improved or modified bioactivity, Weiz et al. generated a microviridin library obtained by random mutagenesis on the highly variable positions 1, 2, 3, 5, and 14 [56]. Most variants were only partially processed by the ATP-grasp enzymes, with the most successfully processed variant containing Phe at position 1 and Ala at position 2. Another screening attempt was made with position 5 variants, and several active inhibitors against different proteases were obtained, demonstrating that position 5 controls the target specificity of microviridin. Similarly, for plesiocin, the residue that modulates the peptide’s specificity of target protease was shown to be the first residue of the loop of a plesiocin hairpin-like core peptide repeat [57]. While the wildtype plesiocin has leucine at this position, substituting it to arginine at the last core peptide yielded the variant Psn-LLLR that exhibits bifunctional protease inhibitor activity against elastase and trypsin. A different type of graspetide diversification was centered on manipulating the sizes and positions of macrocycles [22, 56]. For microviridins and plesiocin, the attempts were unsuccessful and resulted in incomplete processing by the ATP-grasp enzymes. So far, the only successful attempt was observed in amycolimiditide, which was fully esterified with one or two Gly insertions in the loop. Further attempts across multiple graspetides are necessary to examine the full potential of using graspetide and ATP-grasp enzyme for generating synthetic peptides with diversified structures. Few results from studies on plesiocin [57] and thatisin [38] indicate that mutagenesis on the core peptide may yield ester linkages placed at nonnative locations, exhibiting greater flexibility in macrocyclic peptide design than using a microviridin-type graspetide as the starting template.
Concluding Remarks and Future Perspectives
In this paper we have covered how the study of graspetides has grown expanded from a bioactivity-driven enterprise to a primarily genome-driven effort. Much has been learned about the diversity of different graspetide structures in nature as well as ATP-grasp enzymes that make them. Genome mining efforts show that the field has just scratched the surface in terms of the diversity of sequences and structures that make up graspetides. Much still remains to be learned about graspetides, however (see Outstanding Questions). Connecting these molecules to biological functions [76] will aid in understanding why so many different microorganisms from different ecological niches produce graspetides of different shapes and sizes. The ATP-grasp enzymes that make graspetides may be a fertile testing ground to answer questions about how these enzymes can install multiple modifications in a single precursor peptide and in a specific order to boot. This catalytic challenge is faced by many different RiPP biosynthetic enzymes that install specific sets of modifications [77–78]. Finally, with many new graspetide scaffolds appearing in the past ~3 years, we anticipate that protein and peptide engineers will turn their attention to these scaffolds as a new way to generate macrocyclized peptides either by rational design or library approaches [79].
Outstanding Questions.
What are the bioactivities exhibited by novel graspetides beyond microviridin and plesiocin? How do these graspetides interact with their targets, and how do their interactions differ from microviridins’ binding interactions with serine proteases?
What are the underlying factors responsible for the extensive structural diversity in crosslinking patterns and multivalency amongst graspetides?
What purposes do the graspetides other than microviridins serve? How do their protease-inhibiting or other activities impact the native producer, neighboring organisms, and the ecological niche?
How does the quaternary structure of ATP-grasp enzymes and their graspetide precursors transition after each crosslinking step?
What roles do additional post-translational modifications play, and how do they modulate the bioactivities of graspetides?
How are the biosynthetic gene clusters for graspetides transcriptionally regulated?
The protease-inhibiting activities of graspetides have been proposed for various biotechnological applications, including therapeutics, but their feasibility remains uncertain. What are the limiting factors affecting the applicability of graspetides in biotechnology?
Highlights.
Genome mining revealed novel graspetides with unique patterns of ω-ester or ω-amide crosslinks.
The crystal structures of ATP-grasp enzymes complexed with the precursors and cofactors were elucidated, deepening our understand of how these RiPP peptides undergo enzymatic maturation.
Experimental and bioinformatic studies have identified graspetides with additional post-translational modifications beyond the ω-ester and ω-amide crosslinks.
Researchers have explored various ways of engineering graspetides, screening variants for desired bioactivities or incorporating nonnative chemical moieties.
Acknowledgements
RiPPs research in the Link Lab is supported by the National Institutes of Health Grant GM107036 and a grant from Princeton University School of Engineering and Applied Sciences (Focused Research Team on Precision Antibiotics and Innovation Funds).
Glossary
- Acceptor
The acidic residue providing the carbonyl carbon (Asp/Glu) for the ω-ester/amide linkage.
- ATP-grasp enzyme
A family of enzymes containing an ATP-grasp fold that uses ATP to catalyze a covalent linkage formation. For graspetides, this enzyme is responsible for the formation of an ω-ester linkage, ω-amide linkage, or both.
- Biosynthetic gene cluster (BGC)
A group of adjacently located genes involved in RiPP maturation, tailoring, export, or the regulation of the BGC.
- Core (peptide)
A portion of graspetide that is macrocyclized by its outermost ω-ester/amide linkage.
- Donor
The residue containing the nucleophilic side chain (Ser/Thr/Lys) making the ω-ester/amide linkage.
- Graspetides
A family of RiPPs containing ester or amide crosslinks installed by ATP-grasp enzymes.
- RiPPs
A group of peptide natural products that are first ribosomally synthesized as a linear precursor protein and then post-translationally modified and shortened N-terminally (and C-terminally in some cases) by the associated biosynthetic and tailoring enzymes.
- Side chain-side chain linkage
a covalent linkage formed by the side chain of one amino acid residue and that of another residue. A common example in proteins and peptides is the disulfide linkage between two cysteine residues.
- ω-ester/amide linkage
A side chain-side chain linkage between the hydroxyl/amine group of a Ser/Thr/Lys residue (i.e. donor) and the carbonyl carbon of an Asp/Glu residue (i.e. acceptor).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare that they have no competing interests.
References
- 1.Imada C; Simidu U; Taga N, Isolation and characterization of marine bacteria producing alkaline protease inhibitor. Nippon Suisan Gakkaishi 1985, 51 (5), 799–803. [Google Scholar]
- 2.Imada C; Hara S; Maeda M; Simidu U, Amino acid sequences of marinostatins C-1 and C-2 from marine Alteromonas sp. Nippon Suisan Gakkaishi 1986, 52 (8), 1455–1459. [Google Scholar]
- 3.Kanaori K; Kamei K; Taniguchi M; Koyama T; Yasui T; Takano R; Imada C; Tajima K; Hara S, Solution structure of marinostatin, a natural ester-linked protein protease inhibitor. Biochemistry 2005, 44 (7), 2462–8. [DOI] [PubMed] [Google Scholar]
- 4.Ishitsuka MO; Kusumi T; Kakisawa H; Kaya K; Watanabe MM, Microviridin. A novel tricyclic depsipeptide from the toxic cyanobacterium Microcystis viridis. Journal of the American Chemical Society 1990, 112 (22), 8180–8182. [Google Scholar]
- 5.Okino T; Matsuda H; Murakami M; Yamaguchi K, New microviridins, elastase inhibitors from the blue-green alga Microcystis aeruginosa. Tetrahedron 1995, 51 (39), 10679–10686. [Google Scholar]
- 6.Shin HJ; Murakami M; Matsuda H; Yamaguchi K, Microviridins D-F, serine protease inhibitors from the cyanobacterium Oscillatoria agardhii (NIES-204). Tetrahedron 1996, 52 (24), 8159–8168. [Google Scholar]
- 7.Murakami M; Sun Q; Ishida K; Matsuda H; Okino T; Yamaguchi K, Microviridins, elastase inhibitors from the cyanobacterium Nostoc minutum (NIES-26). Phytochemistry 1997, 45 (6), 1197–1202. [Google Scholar]
- 8.Fujii K; Sivonen K; Naganawa E; Harada K.-i., Non-Toxic Peptides from Toxic Cyanobacteria, Oscillatoria agardhii. Tetrahedron 2000, 56 (5), 725–733. [Google Scholar]
- 9.Rohrlack T; Christoffersen K; Hansen PE; Zhang W; Czarnecki O; Henning M; Fastner J; Erhard M; Neilan BA; Kaebernick M, Isolation, characterization, and quantitative analysis of Microviridin J, a new Microcystis metabolite toxic to Daphnia. J Chem Ecol 2003, 29 (8), 1757–70. [DOI] [PubMed] [Google Scholar]
- 10.Kaebernick M; Rohrlack T; Christoffersen K; Neilan BA, A spontaneous mutant of microcystin biosynthesis: genetic characterization and effect on Daphnia. Environ Microbiol 2001, 3 (11), 669–79. [DOI] [PubMed] [Google Scholar]
- 11.Rohrlack T; Christoffersen K; Kaebernick M; Neilan BA, Cyanobacterial protease inhibitor microviridin J causes a lethal molting disruption in Daphnia pulicaria. Appl Environ Microbiol 2004, 70 (8), 5047–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyamoto K; Tsujibo H; Hikita Y; Tanaka K; Miyamoto S; Hishimoto M; Imada C; Kamei K; Hara S; Inamori Y, Cloning and nucleotide sequence of the gene encoding a serine proteinase inhibitor named marinostatin from a marine bacterium, Alteromonas sp. strain B-10–31. Biosci Biotechnol Biochem 1998, 62 (12), 2446–9. [DOI] [PubMed] [Google Scholar]
- 13.Philmus B; Christiansen G; Yoshida WY; Hemscheidt TK, Post-translational modification in microviridin biosynthesis. Chembiochem 2008, 9 (18), 3066–73. [DOI] [PubMed] [Google Scholar]
- 14.Ziemert N; Ishida K; Liaimer A; Hertweck C; Dittmann E, Ribosomal synthesis of tricyclic depsipeptides in bloom-forming cyanobacteria. Angew Chem Int Ed Engl 2008, 47 (40), 7756–9. [DOI] [PubMed] [Google Scholar]
- 15.Unno K; Nakagawa H; Kodani S, Heterologous production of new protease inhibitory peptide marinostatin E. Biosci Biotechnol Biochem 2021, 85 (1), 97–102. [DOI] [PubMed] [Google Scholar]
- 16.Montalban-Lopez M; Scott TA; Ramesh S; Rahman IR; van Heel AJ; Viel JH; Bandarian V; Dittmann E; Genilloud O; Goto Y; Grande Burgos MJ; Hill C; Kim S; Koehnke J; Latham JA; Link AJ; Martinez B; Nair SK; Nicolet Y; Rebuffat S; Sahl HG; Sareen D; Schmidt EW; Schmitt L; Severinov K; Sussmuth RD; Truman AW; Wang H; Weng JK; van Wezel GP; Zhang Q; Zhong J; Piel J; Mitchell DA; Kuipers OP; van der Donk WA, New developments in RiPP discovery, enzymology and engineering. Nat Prod Rep 2021, 38 (1), 130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer LM; Abhiman S; Maxwell Burroughs A; Aravind L, Amidoligases with ATP-grasp, glutamine synthetase-like and acetyltransferase-like domains: synthesis of novel metabolites and peptide modifications of proteins. Mol Biosyst 2009, 5 (12), 1636–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oman TJ; van der Donk WA, Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat Chem Biol 2010, 6 (1), 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Link AJ, Biosynthesis: Leading the way to RiPPs. Nat Chem Biol 2015, 11 (8), 551–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiz AR; Ishida K; Makower K; Ziemert N; Hertweck C; Dittmann E, Leader peptide and a membrane protein scaffold guide the biosynthesis of the tricyclic peptide microviridin. Chem Biol 2011, 18 (11), 1413–21. [DOI] [PubMed] [Google Scholar]
- 21.Li K; Condurso HL; Li G; Ding Y; Bruner SD, Structural basis for precursor protein-directed ribosomal peptide macrocyclization. Nat Chem Biol 2016, 12 (11), 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Philmus B; Guerrette JP; Hemscheidt TK, Substrate specificity and scope of MvdD, a GRASP-like ligase from the microviridin biosynthetic gene cluster. ACS Chem Biol 2009, 4 (6), 429–34. [DOI] [PubMed] [Google Scholar]
- 23.Roh H; Han Y; Lee H; Kim S, A Topologically Distinct Modified Peptide with Multiple Bicyclic Core Motifs Expands the Diversity of Microviridin-Like Peptides. Chembiochem 2019, 20 (8), 1051–1059. [DOI] [PubMed] [Google Scholar]
- 24.Choi B; Elashal HE; Cao L; Link AJ, Mechanistic Analysis of the Biosynthesis of the Aspartimidylated Graspetide Amycolimiditide. J Am Chem Soc 2022, 144 (47), 21628–21639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao G; Kosek D; Liu HB; Ohlemacher SI; Blackburne B; Nikolskaya A; Makarova KS; Sun J; Barry CE Iii; Koonin EV; Dyda F; Bewley CA, Structural Basis for a Dual Function ATP Grasp Ligase That Installs Single and Bicyclic omega-Ester Macrocycles in a New Multicore RiPP Natural Product. J Am Chem Soc 2021, 143 (21), 8056–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song I; Kim Y; Yu J; Go SY; Lee HG; Song WJ; Kim S, Molecular mechanism underlying substrate recognition of the peptide macrocyclase PsnB. Nat Chem Biol 2021, 17 (11), 1123–1131. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y; Li K; Yang G; McBride JL; Bruner SD; Ding Y, A distributive peptide cyclase processes multiple microviridin core peptides within a single polypeptide substrate. Nat Commun 2018, 9 (1), 1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li G; Patel K; Zhang Y; Pugmire JK; Ding Y; Bruner SD, Structural and biochemical studies of an iterative ribosomal peptide macrocyclase. Proteins 2022, 90 (3), 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmed MN; Reyna-Gonzalez E; Schmid B; Wiebach V; Sussmuth RD; Dittmann E; Fewer DP, Phylogenomic Analysis of the Microviridin Biosynthetic Pathway Coupled with Targeted Chemo-Enzymatic Synthesis Yields Potent Protease Inhibitors. ACS Chem Biol 2017, 12 (6), 1538–1546. [DOI] [PubMed] [Google Scholar]
- 30.Makarova KS; Blackburne B; Wolf YI; Nikolskaya A; Karamycheva S; Espinoza M; Barry CE 3rd; Bewley CA; Koonin EV, Phylogenomic analysis of the diversity of graspetides and proteins involved in their biosynthesis. Biol Direct 2022, 17 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osterholm J; Popin RV; Fewer DP; Sivonen K, Phylogenomic Analysis of Secondary Metabolism in the Toxic Cyanobacterial Genera Anabaena, Dolichospermum and Aphanizomenon. Toxins (Basel) 2020, 12 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearson LA; Crosbie ND; Neilan BA, Distribution and conservation of known secondary metabolite biosynthesis gene clusters in the genomes of geographically diverse Microcystis aeruginosa strains. Marine and Freshwater Research 2020, 71 (5), 701–716. [Google Scholar]
- 33.Unno K; Kaweewan I; Nakagawa H; Kodani S, Heterologous expression of a cryptic gene cluster from Grimontia marina affords a novel tricyclic peptide grimoviridin. Appl Microbiol Biotechnol 2020, 104 (12), 5293–5302. [DOI] [PubMed] [Google Scholar]
- 34.Sieber S; Grendelmeier SM; Harris LA; Mitchell DA; Gademann K, Microviridin 1777: A Toxic Chymotrypsin Inhibitor Discovered by a Metabologenomic Approach. J Nat Prod 2020, 83 (2), 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang T; Wang X; Zhao H; Huo L; Fu C, Uncovering a Subtype of Microviridins via the Biosynthesis Study of FR901451. ACS Chem Biol 2022. [DOI] [PubMed] [Google Scholar]
- 36.Lee H; Park Y; Kim S, Enzymatic Cross-Linking of Side Chains Generates a Modified Peptide with Four Hairpin-like Bicyclic Repeats. Biochemistry 2017, 56 (37), 4927–4930. [DOI] [PubMed] [Google Scholar]
- 37.Lee H; Choi M; Park JU; Roh H; Kim S, Genome Mining Reveals High Topological Diversity of omega-Ester-Containing Peptides and Divergent Evolution of ATP-Grasp Macrocyclases. J Am Chem Soc 2020, 142 (6), 3013–3023. [DOI] [PubMed] [Google Scholar]
- 38.Ramesh S; Guo X; DiCaprio AJ; De Lio AM; Harris LA; Kille BL; Pogorelov TV; Mitchell DA, Bioinformatics-Guided Expansion and Discovery of Graspetides. ACS Chem Biol 2021, 16 (12), 2787–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaweewan I; Nakagawa H; Kodani S, Heterologous expression of a cryptic gene cluster from Marinomonas fungiae affords a novel tricyclic peptide marinomonasin. Appl Microbiol Biotechnol 2021, 105 (19), 7241–7250. [DOI] [PubMed] [Google Scholar]
- 40.Unno K; Kodani S, Heterologous expression of cryptic biosynthetic gene cluster from Streptomyces prunicolor yields novel bicyclic peptide prunipeptin. Microbiol Res 2021, 244, 126669. [DOI] [PubMed] [Google Scholar]
- 41.Elashal HE; Koos JD; Cheung-Lee WL; Choi B; Cao L; Richardson MA; White HL; Link AJ, Biosynthesis and characterization of fuscimiditide, an aspartimidylated graspetide. Nat Chem 2022, 14 (11), 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mishra PKK; Mahawar M, PIMT-Mediated Protein Repair: Mechanism and Implications. Biochemistry (Mosc) 2019, 84 (5), 453–463. [DOI] [PubMed] [Google Scholar]
- 43.Smith RF; Bates AC; Battisti AJ; Byrnes PG; Mroz CT; Smearing TJ; Albrecht FX, Reactions of Hydrazines with Esters and Carboxylic Acids. Journal of Organic Chemistry 1968, 33 (2), 851–&. [Google Scholar]
- 44.Soai K; Ookawa A; Hayashi H, Novel Functional-Group Selectivity in Reductions with Lithium Borohydride in Mixed-Solvents Containing Methanol. J Chem Soc Chem Comm 1983, (12), 668–669. [Google Scholar]
- 45.do Amaral SC; Monteiro PR; Neto J; Serra GM; Goncalves EC; Xavier LP; Santos AV, Current Knowledge on Microviridin from Cyanobacteria. Mar Drugs 2021, 19 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ongpipattanakul C; Desormeaux EK; DiCaprio A; van der Donk WA; Mitchell DA; Nair SK, Mechanism of Action of Ribosomally Synthesized and Post-Translationally Modified Peptides. Chem Rev 2022, 122 (18), 14722–14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vegman M; Carmeli S, Three aeruginosins and a microviridin from a bloom assembly of Microcystis spp. collected from a fishpond near Kibbutz Lehavot HaBashan, Israel. Tetrahedron 2014, 70 (38), 6817–6824. [Google Scholar]
- 48.Taichi M; Yamazaki T; Kawahara K; Motooka D; Nakamura S; Harada S; Teshima T; Ohkubo T; Kobayashi Y; Nishiuchi Y, Structure-activity relationship of marinostatin, a serine protease inhibitor isolated from a marine organism. J Pept Sci 2010, 16 (7), 329–36. [DOI] [PubMed] [Google Scholar]
- 49.Anas ARJ; Mori A; Tone M; Naruse C; Nakajima A; Asukabe H; Takaya Y; Imanishi SY; Nishizawa T; Shirai M; Harada KI, FVIIa-sTF and Thrombin Inhibitory Activities of Compounds Isolated from Microcystis aeruginosa K-139. Mar Drugs 2017, 15 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ziemert N; Ishida K; Weiz A; Hertweck C; Dittmann E, Exploiting the natural diversity of microviridin gene clusters for discovery of novel tricyclic depsipeptides. Appl Environ Microbiol 2010, 76 (11), 3568–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gatte-Picchi D; Weiz A; Ishida K; Hertweck C; Dittmann E, Functional analysis of environmental DNA-derived microviridins provides new insights into the diversity of the tricyclic peptide family. Appl Environ Microbiol 2014, 80 (4), 1380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reshef V; Carmeli S, New microviridins from a water bloom of the cyanobacterium Microcystis aeruginosa. Tetrahedron 2006, 62 (31), 7361–7369. [Google Scholar]
- 53.Polverino E; Rosales-Mayor E; Dale GE; Dembowsky K; Torres A, The Role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest 2017, 152 (2), 249–262. [DOI] [PubMed] [Google Scholar]
- 54.Al-Amer OM, The role of thrombin in haemostasis. Blood Coagul Fibrinolysis 2022, 33 (3), 145–148. [DOI] [PubMed] [Google Scholar]
- 55.Czarnecki O; Henning M; Lippert I; Welker M, Identification of peptide metabolites of Microcystis (Cyanobacteria) that inhibit trypsin-like activity in planktonic herbivorous Daphnia (Cladocera). Environ Microbiol 2006, 8 (1), 77–87. [DOI] [PubMed] [Google Scholar]
- 56.Weiz AR; Ishida K; Quitterer F; Meyer S; Kehr JC; Muller KM; Groll M; Hertweck C; Dittmann E, Harnessing the evolvability of tricyclic microviridins to dissect protease-inhibitor interactions. Angew Chem Int Ed Engl 2014, 53 (14), 3735–8. [DOI] [PubMed] [Google Scholar]
- 57.Lee C; Lee H; Park JU; Kim S, Introduction of Bifunctionality into the Multidomain Architecture of the omega-Ester-Containing Peptide Plesiocin. Biochemistry 2020, 59 (3), 285–289. [DOI] [PubMed] [Google Scholar]
- 58.Saha S; Bulzu PA; Urajova P; Mares J; Konert G; Camara Manoel J; Macho M; Ewe D; Hrouzek P; Masojidek J; Ghai R; Saurav K, Quorum-Sensing Signals from Epibiont Mediate the Induction of Novel Microviridins in the Mat-Forming Cyanobacterial Genus Nostoc. mSphere 2021, 6 (4), e0056221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pereira DA; Pimenta AM; Giani A, Profiles of toxic and non-toxic oligopeptides of Radiocystis fernandoii (Cyanobacteria) exposed to three different light intensities. Microbiol Res 2012, 167 (7), 413–21. [DOI] [PubMed] [Google Scholar]
- 60.Dehm D; Krumbholz J; Baunach M; Wiebach V; Hinrichs K; Guljamow A; Tabuchi T; Jenke-Kodama H; Sussmuth RD; Dittmann E, Unlocking the Spatial Control of Secondary Metabolism Uncovers Hidden Natural Product Diversity in Nostoc punctiforme. ACS Chem Biol 2019, 14 (6), 1271–1279. [DOI] [PubMed] [Google Scholar]
- 61.Geiger T; Clarke S, Deamidation, Isomerization, and Racemization at Asparaginyl and Aspartyl Residues in Peptides - Succinimide-Linked Reactions That Contribute to Protein-Degradation. Journal of Biological Chemistry 1987, 262 (2), 785–794. [PubMed] [Google Scholar]
- 62.Clarke S, A Protein Carboxyl Methyltransferase that Recognizes Age-Damaged Peptides and Proteins and Participates in their Repair. In S-Adenosylmethionine-dependent Methyltransferases: Structures and Function, Cheng X; Blumenthal RM, Eds. World Scientific Publishing Company: Singapore, 1999; pp 123–148. [Google Scholar]
- 63.Murray ED; Clarke S, Metabolism of a Synthetic L-Isoaspartyl-Containing Hexapeptide in Erythrocyte Extracts - Enzymatic Methyl Esterification Is Followed by Nonenzymatic Succinimide Formation. Journal of Biological Chemistry 1986, 261 (1), 306–312. [PubMed] [Google Scholar]
- 64.Acedo JZ; Bothwell IR; An L; Trouth A; Frazier C; van der Donk WA, O-Methyltransferase-Mediated Incorporation of a beta-Amino Acid in Lanthipeptides. J Am Chem Soc 2019, 141 (42), 16790–16801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao L; Beiser M; Koos JD; Orlova M; Elashal HE; Schroder HV; Link AJ, Cellulonodin-2 and Lihuanodin: Lasso Peptides with an Aspartimide Post-Translational Modification. J Am Chem Soc 2021, 143 (30), 11690–11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stephenson RC; Clarke S, Succinimide Formation from Aspartyl and Asparaginyl Peptides as a Model for the Spontaneous Degradation of Proteins. Journal of Biological Chemistry 1989, 264 (11), 6164–6170. [PubMed] [Google Scholar]
- 67.Schramma KR; Bushin LB; Seyedsayamdost MR, Structure and biosynthesis of a macrocyclic peptide containing an unprecedented lysine-to-tryptophan crosslink. Nat Chem 2015, 7 (5), 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Imai Y; Meyer KJ; Iinishi A; Favre-Godal Q; Green R; Manuse S; Caboni M; Mori M; Niles S; Ghiglieri M; Honrao C; Ma X; Guo JJ; Makriyannis A; Linares-Otoya L; Bohringer N; Wuisan ZG; Kaur H; Wu R; Mateus A; Typas A; Savitski MM; Espinoza JL; O’Rourke A; Nelson KE; Hiller S; Noinaj N; Schaberle TF; D’Onofrio A; Lewis K, A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576 (7787), 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balty C; Guillot A; Fradale L; Brewee C; Boulay M; Kubiak X; Benjdia A; Berteau O, Ruminococcin C, an anti-clostridial sactipeptide produced by a prominent member of the human microbiota Ruminococcus gnavus. J Biol Chem 2019, 294 (40), 14512–14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balty C; Guillot A; Fradale L; Brewee C; Lefranc B; Herrero C; Sandstrom C; Leprince J; Berteau O; Benjdia A, Biosynthesis of the sactipeptide Ruminococcin C by the human microbiome: Mechanistic insights into thioether bond formation by radical SAM enzymes. J Biol Chem 2020, 295 (49), 16665–16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Franz L; Koehnke J, Leader peptide exchange to produce hybrid, new-to-nature ribosomal natural products. Chem Commun (Camb) 2021, 57 (52), 6372–6375. [DOI] [PubMed] [Google Scholar]
- 72.Cox CL; Doroghazi JR; Mitchell DA, The genomic landscape of ribosomal peptides containing thiazole and oxazole heterocycles. BMC Genomics 2015, 16, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walker JA; Hamlish N; Tytla A; Brauer DD; Francis MB; Schepartz A, Redirecting RiPP Biosynthetic Enzymes to Proteins and Backbone-Modified Substrates. ACS Cent Sci 2022, 8 (4), 473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scholz S; Kerestetzopoulou S; Wiebach V; Schnegotzki R; Schmid B; Reyna-Gonzalez E; Ding L; Sussmuth RD; Dittmann E; Baunach M, One-Pot Chemoenzymatic Synthesis of Microviridin Analogs Containing Functional Tags. Chembiochem 2022, e202200345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reyna-Gonzalez E; Schmid B; Petras D; Sussmuth RD; Dittmann E, Leader Peptide-Free In Vitro Reconstitution of Microviridin Biosynthesis Enables Design of Synthetic Protease-Targeted Libraries. Angew Chem Int Ed Engl 2016, 55 (32), 9398–401. [DOI] [PubMed] [Google Scholar]
- 76.Cao L; Do T; Link AJ, Mechanisms of action of ribosomally synthesized and posttranslationally modified peptides (RiPPs). J Ind Microbiol Biotechnol 2021, 48 (3–4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Habibi Y; Weerasinghe NW; Uggowitzer KA; Thibodeaux CJ, Partially Modified Peptide Intermediates in Lanthipeptide Biosynthesis Alter the Structure and Dynamics of a Lanthipeptide Synthetase. J Am Chem Soc 2022, 144 (23), 10230–10240. [DOI] [PubMed] [Google Scholar]
- 78.Freeman MF; Gurgui C; Helf MJ; Morinaka BI; Uria AR; Oldham NJ; Sahl HG; Matsunaga S; Piel J, Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides. Science 2012, 338 (6105), 387–90. [DOI] [PubMed] [Google Scholar]
- 79.Do T; Link AJ, Protein Engineering in Ribosomally Synthesized and Post-translationally Modified Peptides (RiPPs). Biochemistry 2023, 62 (2), 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]