

Abstract
As key organelles involved in cellular metabolism, mitochondria frequently undergo adaptive changes in morphology, components and functions in response to various environmental stresses and cellular demands. Previous studies of mitochondria research have gradually evolved, from focusing on morphological change analysis to systematic multiomics, thereby revealing the mitochondrial variation between cells or within the mitochondrial population within a single cell. The phenomenon of mitochondrial variation features is defined as mitochondrial heterogeneity. Moreover, mitochondrial heterogeneity has been reported to influence a variety of physiological processes, including tissue homeostasis, tissue repair, immunoregulation, and tumor progression. Here, we comprehensively review the mitochondrial heterogeneity in different tissues under pathological states, involving variant features of mitochondrial DNA, RNA, protein and lipid components. Then, the mechanisms that contribute to mitochondrial heterogeneity are also summarized, such as the mutation of the mitochondrial genome and the import of mitochondrial proteins that result in the heterogeneity of mitochondrial DNA and protein components. Additionally, multiple perspectives are investigated to better comprehend the mysteries of mitochondrial heterogeneity between cells. Finally, we summarize the prospective mitochondrial heterogeneity-targeting therapies in terms of alleviating mitochondrial oxidative damage, reducing mitochondrial carbon stress and enhancing mitochondrial biogenesis to relieve various pathological conditions. The possibility of recent technological advances in targeted mitochondrial gene editing is also discussed.
Subject terms: Physiology, Target identification, Microarrays
Introduction
A mitochondrion is a double-membrane organelle comprising an outer membrane, inner membrane, and matrix.1 Mitochondria produce most cellular ATP via the tricarboxylic acid cycle (TCA) and the oxidative respiratory (OXPHOS) chain2 and play vital roles in coordinating glucose, lipid, amino acid, and nucleotide metabolism.3 In addition, mitochondria are major sources of the cellular production of reactive oxygen species (ROS) and carry numerous redox pathways.4,5 Mitochondrial proteins catalyze the biosynthesis of Fe-S clusters and one-carbon units6–9 and maintain mitochondrial morphology via, for example, the formation of mitochondrial cristae, networks, and contacts with other organelles.10–22 As semi-independent organelles, mitochondria contain a complete genetic system, which includes the mitochondrial genome (mtDNA) and numerous factors that are crucial for the maintenance and regulation of mtDNA and mitochondrial ribosomes.23–25 The proteins encoded by mtDNA are inserted into the OXPHOS chain located on the inner membrane after translation from ribosomes facilitated by oxidase assembly (OXA),26,27 and numerous cytosolic signaling cascades are connected to mitochondria under physiological and pathophysiological conditions.28,29 The metabolic fitness of mitochondria in response to cellular stress is a measure of mitochondrial quality.30,31 Selective degradation of damaged mitochondria through mitophagy has been identified as the classic mechanism of mitochondrial homeostasis maintenance,32–36 and other autophagy-independent constituents, such as mitochondrion-derived vesicles (MDVs), and pathways, such as the mitocytosis and mitolysosome exocytosis, have been reported.37–39 In contrast to the removal of dysfunctional organelles through mitophagy, the generation of MDVs is a direct outcome of the lateral segregation of cargo into budding vesicles that move along the tubules of functional mitochondria. This process differs from the fission and segregation of mitochondrial fragments.37 The mitocytic pathway is intrinsically associated with cell migration and responses to mild mitochondrial stress to prevent the accumulation of damaged mitochondria.40 The mitolysosome exocytic pathway seems to be a universal process, and different mechanisms mediate mitochondrial exocytosis, such as CD38-mediated transfer, LC3-dependent exopher trafficking and vacuole-mediated cell extrusion. The lysosome-associated mitolysosome exocytic mechanism may also be vital to mitochondrial quality control.39
Mitochondrial heterogeneity has been defined as the variation in mitochondrial features between cells or within the mitochondrial population within a single cell. The key initial step in the field of mitochondrial heterogeneity was the reconstruction of electron micrographs that revealed mitochondrial networks in rat hepatocytes in 1974,41 and this discovery was reproduced in several cell types, including human endothelial cells and astrocytes,42 demonstrating that mitochondrial morphology varies in different cells. In the 1980s, the field was advanced with sequencing of the mouse mitochondrial genome, providing a molecular framework for understanding mtDNA heterogeneity,43 culminating in a summary of the extreme genetic variation within mtDNA in 2021.44 During the 2000s, studies began to focus on the association between the heterogeneity of mitochondrial proteins, noncoding RNAs (ncRNAs), lipids and the cause of mitochondrial-related disease. The mitochondrial proteome of human heart mitochondria was identified in 2003 and included 615 mitochondrion-associated proteins.45 Meanwhile, mitochondrial ncRNAs candidates were systematically identified in mouse mitochondria in 2006,46 and a 2008 study into the nonsynaptic (NS) and synaptic (Syn) mitochondrial lipidomes of the mouse brain revealed that lipidomic heterogeneity influenced energy metabolism.47 In 2020, a mitochondrial circRNA has been reported to alleviate nonalcoholic steatohepatitis (NASH) by reducing mitochondrial ROS (mROS) output.48 More recently, human mitochondrial proteome contains, including 1,134 proteins, was obtained with high confidence (Fig. 1).49
Fig. 1.

Timeline of mitochondrial heterogeneity research. Key discoveries in the field are highlighted. Abbreviations: ncRNA noncoding RNA, mROS mitochondrial reactive oxygen species
In recent decades, mitochondrial purification technology, combined with genomics, transcriptomics, proteomics, and bioinformatics analyses has revealed that mitochondria exhibit a high degree of heterogeneity,45,50,51 and some of this heterogeneity can be explained by the varying proportions of mutant and wild-type mtDNA in eukaryotic cells, a state called heteroplasmy. Other causes of heterogeneity can be cell- and tissue-specific differences in mitochondrial ncRNAs and proteins.52 This review summarizes the heterogeneous characteristics of mitochondria in the pathological conditions of different tissues.
Mitochondrial performance diversity
Historically, fluorescence and electron microscopy have been used to observe mitochondria in mammalian cells.10,13,53 The statistical analysis of mitochondrial morphology hinges on computer image-processing tools, and using these methods, mitochondria have been observed to be ‘tubelike’ structures that are densely packed in the perinuclear space and that emanate outward from the cell periphery. Mitochondrial morphological statistics are a universal part of mitochondrial research. Here, we summarize the methods and software development processes used to quantify mitochondrial morphology (Table 1). The performance of these processes with mitochondria differs significantly in different cells and tissues, and studies have linked different mitochondrial morphologies with molecular mechanisms.
Table 1.
Mitochondrial morphology image analysis
| Basic parameters | Advanced analysis | Platform | Validation | Reference |
|---|---|---|---|---|
| Mitochondrial morphological analysis algorithm | ||||
| Length/width/count/area/ aspect ratio/number/density/form factor/branch length/point/diameters | Colocalization | ImageJ | CV1-4A | 385 |
| Perimeter | Image Pro | Human skin fibroblasts | 386 | |
| Area-weighted form factor | ImageJ | HeLa. PC12 | 387 | |
| 2/3D network-shape integrative analysis | Image Pro | HUVECs | 388 | |
| Mitochondrial movement in neurons | ImageJ | Neurons | 389 | |
| TMRM, calcein | Machine learning | pH values Fs | 390 | |
| Segmentation of mitochondria | Software | Somatosensory cortex | 391 | |
| Mean network size, mitochondrial footprint, | ImageJ | SH-SY5Y cells | 392 | |
| ratio of junctions/ends (J/E), tubules/ends | ImageJ | U2OS cells | 11 | |
| Mitochondrial mass, solidity | ImageJ | Monolayer adherent cells | 393 | |
| Mitochondrial orientation | Software | Pancreatic tumor cell | 394 | |
| Mitochondrial segmentation | ilastik | C. elegans muscle cells | 395 | |
| Mitochondrial motion | Software | PC12, H1299, HFF cells | 396 | |
| Mitochondrial classification algorithm | ||||
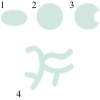 |
Small/swollen globules2, straight/twisted/branched tubules4, loops3 | MicroP | CHO cells | 397 |
| Punctate1. intermediate and filamentous4 | MATLAB | A2780, OVCA-429, A549, Caco-2 cells | 398 | |
| Tubular4. dount2. bolb1 | GemIdent | BEAS-2B, A549, HT108 cells | 399 | |
| Network4, rod-like1, punctate1, and large/round2 | Machine learning | 661w cells | 392 | |
| Punctate1, swollen2, network4 | MATLAB | A549 cells | 400 | |
Mitochondrial morphology
Mitochondrial morphology varies in different tissues54 and depends on the environmental or physiological conditions.55 Changes in mitochondrial morphology are mainly due to the fission and fusion of mitochondria,56–58 the formation and maintenance of mitochondrial cristae,53,59 the contents of the mitochondrial matrix,60,61 and connections with other organelles, such as lysosomes and the endoplasmic reticulum (ER) (Fig. 2).62,63 Mitochondrial fission and fusion occur simultaneously and continuously during metabolic processes in eukaryotic cells.64,65 Fission can produce daughter mitochondria and aids in quality control by allowing damaged mitochondria to be removed and by triggering apoptosis in response to high levels of cellular stress.66,67 The coordination of cytoplasmic, cytoskeletal, and organellar components is required for fission.68 In yeast, the dynamin-related GTPase Dnm1 has been shown to localize to mitochondrial fission sites via recruitment by adaptor proteins (Fis1, Mdv1 and Caf4) to regulate mitochondrial fission.69 The mammalian homolog DRP1 is affected by adaptor proteins (Mff, MiD49, and MiD51) rather than mammalian Fis1, which is involved in mitophagy instead of being implicated as an adaptor protein.67,70 Fusion is a supplementary mitochondrial quality control mechanism that contributes to stress relief by mixing contents of partially damaged mitochondria.71 Fusion can alter mitochondrial function in response to cytosolic signaling. MFN1/2 are mammalian effectors of outer mitochondrial fusion, while OPA1, another key player, is involved in sculpting the inner mitochondrial membrane.72–75 Recent studies have suggested that mitochondrial fission and fusion rates change in response to energetically demanding cellular behaviors76 and extreme conditions (e.g., disease, parasitic infection, and starvation).71 Genetic and environmental factors that affect mitochondrial fission and fusion can also directly impact tissue performance, such as embryonic development,72 organ lesion repair,77–83 and tumorigenesis.84
Fig. 2.

Mitochondrial characteristics under different conditions. Low mitochondrial cristae density is associated with a low cellular energy supply (☹), while a high mitochondrial crista density reflects an adaptation to meet cell energy demands (☺). Mitochondrial contact with many additional organelles, such as the ER, lysosomes and lipid droplets, and the number of contacts mitochondria make with a specific organelle can vary dramatically from only a few contacts to hundreds of contacts per cell. A decrease in the number of mitochondrial connections to other organelles is typically a response to an inefficient metabolic pathway (☹), and in contrast, an increase indicates a response to an active cellular energy metabolic pathway (☺). Mitochondrial fission and fusion are the main pathways of mitochondrial morphology regulation, and under stressful environments, mitochondria are active during mitochondrial fission and produce an increased number of punctate mitochondria (☹), while mitochondrial fusion mediates the formation of mitochondrial networks that adapt to the high energy demands of cells (☺). The content of mitochondria in a cell reflects the intensity of cellular metabolism. Low mitochondrial content in cells is usually associated with low metabolic activity (☹), and high mitochondrial content is associated with high cellular metabolic activity (☺). Intercellular mitochondrial communication is extensive under physiological and pathological conditions, and a low frequency of mitochondrial communication is a response to low cellular adaptation to stressful environments (☹), and in contrast, efficient mitochondrial transfer enhances cellular adaptive capacity (☺)
Mitochondrial cristae remodeling is closely related to mitochondrial respiration, and the electron transport system (ETS) embedded within the mitochondrial cristae directly impacts ATP production.85 Researchers using cryoelectronic tomography have revealed that an increase in energy production is accompanied by an increase in the formation of mitochondrial cristae. In living cells, researchers recently found that OPA1, Yme1L, MICOS, and Sam50, along with the newly identified cristae regulator ATAD3A, controlled mitochondrial cristae dynamics.10,86–88 Genetic and environmental factors that affect mitochondrial cristae formation and maintenance can also impact animal biology, such as the maintenance of constant body temperature,53 lesion repair in multiple organs89,90 and immune responses to the tumor.91 Cold stimulation of brown adipose tissue (BAT) led to enhanced cristae formation, which was attributed to the interorganelle PERK-OGT-TOM70 axis increasing cell respiration through mitochondrial protein import and subsequent cristae formation.92 Notably, PD-1 signaling promotes the exhaustion of activated T cells. A study discovered a reduction in the number and length of mitochondrial cristae in PD-1-stimulated cells.91
Mitochondrial content measured by the number of the mitochondrial genome copies has been shown to be different in the mitochondria of different mammalian organs.93 The contents of intercellular mitochondria is higher in metabolically active cell types than that in less metabolically active cell types.93 For instance, the human myocardium is composed of 23% mitochondria by volume density.94,95 Normal liver cells contain approximately 21% mitochondria.96,97 Mitochondrial content in human skeletal muscle varies from 4% to 15%.98 White adipose tissue (WAT) present with fewer and smaller mitochondria than BAT.99 And the number of mitochondria diminishes during tissue regeneration and cancerogenesis.100–102
Mitochondrial distribution during mitosis
Mitochondrial distribution is usually associated with the cytoskeleton and centrioles.103,104 It depends on the direction of the diffusion currents within cells and is related to the submicroscopic organization of the cytoplasmic matrix and vacuolar system (Fig. 3).105 The ER, which forms a scaffold with mitochondria, is organized into a dense meshwork of subcortical actin cables assembled throughout the cytoplasm of mitotic cells.105 As they are co-oriented with nearby cables, mitochondria are positioned within meshwork pores. Cytochalasin D (CytoD)- or latrunculin A (LatA)-induced meshwork removal caused mitochondria to aggregate, disrupting their symmetrical arrangement and cell division.105
Fig. 3.

Mitochondrial distribution during mitosis. During interphase, the mitochondrial network is evenly distributed in the cytoplasm. During prophase, the mitochondrial network is crumpled and primarily located in the perinuclear area, where punctate mitochondria are apparent. During metaphase, mitochondria move to the equatorial plane in the midline of a cell at right angles to the axis. During anaphase, mitochondria move to the opposite ends of a cell. During telophase, the mitochondrial network is re-formed and grouped at either pole of a cell. a The association of mitochondria with cytoplasmic F-actin may promote mitochondrial distribution during mitosis. b Mitochondrial delivery on microtubules may dock to actin in the cleavage furrow. c Miro-1 is required for transporting mitochondria to the plus ends of microtubules at the cleavage furrow via interaction with KIF5B. d Close association between mitochondria and both ER sheets and actin cables may promote mitochondrial distribution during mitosis. e Myo19 is localized to mitochondria and acted as a novel actin-based motor that controls mitochondrial distribution during mitosis
Mitochondrial components
Mitochondrial composition shows a high degree of heterogeneity. Here, we summarize the differences among mitochondrial genomes, transcriptomes, proteomes, and lipidomes in cells and describe the related underlying molecular mechanisms (Fig. 4).
Fig. 4.

The heterogeneity of mitochondrial components. DNA component: The diversity of the mitochondrial genome arises from base-pair mismatches during the replication of the genome and base mutations after mtDNA short-patch base excision repair (BER). RNA component: Transcripts from the mitochondrial genome include rRNAs, sRNAs, CircRNAs, dsRNAs, IncRNAs, mRNAs and tRNAs. The protein component diversity of the mitochondrial proteome arises from the two main protein resources, the nucleic coding protein import pathway and the mtDNA translation pathway. In addition, mitochondrial proteins are altered via a complex posttranslational modification mechanism. The heterogeneity of mitochondrial lipids is a result of four related lipid transport pathways, including free diffusion, organelle membrane contact, lipid transport proteins and vesicular transport
Mitochondrial DNA
The mitochondrial genome forms a compact, double-stranded circle comprising 16.5 kb with a cytosine-rich light (L) chain and a guanine-rich heavy (H) chain.106 Contrary to the long-held view that most humans harbor only identical mitochondrial genomes, massively parallel deep resequencing has revealed unanticipated extreme genetic variation within mtDNA at multiple levels.44 Multiple mtDNA genotypes are present at both the cellular and organelle levels107 and can be inherited from both parents.108,109 In contrast to the nuclear genome, mtDNA is not supported by histones to protect them against ROS damage.110 Recent studies have shown that the replication sites of mtDNA are physically close to the oxidative respiratory chain, and the production of ROS during oxidative respiration makes mtDNA more susceptible to damage.111 On the other hand, most age-related mtDNA mutations are thought to be due to errors in mtDNA replication,112 which has been confirmed to exert a far greater impact than ROS-mediated mtDNA variation.113 The mechanisms of mtDNA escape from mitochondria to intracellular or extracellular compartments depend on pores formed by Bax and Bak, VDAC oligomers, and mitochondria permeability transition pores (mPTPs).114 The mtDNA continuously and independently replicates during the cell cycle with a half-life that varies from days to weeks depending on the cell type. For example, it is 8–12 days for epithelial cells and 20-25 days for neurons.115,116 Although the polymerase-γ, which is critical for mtDNA replication, shows high fidelity,117 the large number of mtDNA replication cycles that are required over the lifetime of a cell inevitably induces base-pair substitution errors. The origins of human mtDNA variation and its relevance for human diseases, including cancer, neurodegenerative diseases and aging, have been studied. It is commonly assumed that mtDNA is mutated at a faster rate than nuclear DNA (nDNA) in eukaryotes.118
Mitochondrial genome-generated noncoding RNAs
The complexity of transcripts derived from mitochondria is beginning to be understood, and advances in deep-sequencing technology has supported findings of ncRNAs encoded in mitochondria.119 Various ncRNAs are derived from mitochondria, and the mitochondrial genome gives rise to hundreds of circRNAs,48,120,121 at least eight lncRNAs,122 a few dsRNAs123 and various small RNAs.51 Whether moving into the cell membrane or the nucleus or remaining in the mitochondria, these ncRNAs carry out a variety of biological tasks. Global transcriptome profiling revealed that in the human left ventricle, a relatively high abundance (71%) of lncRNAs is encoded by the mitochondrial genome.124 Using a PacBio full-length third-generation sequencing transcriptome dataset, researchers identified two polycistronic transcripts, namely, hsa-MDL1 (mitochondrial D-loop 1) and hsa-MDL1AS (mitochondrial D-loop 1 antisense), generated from a region covering the tRNAPro gene and the full length of the human D-loop region.125 Pan RNA-seq analysis also revealed that the 5’- and 3’-end small RNAs of MDL1 and MDL1AS were ubiquitous.125 Interestingly, highly unstable mitochondrial double-stranded RNA (mt-dsRNA) has been found in HeLa cells.123 Bax-Bak-dependent release of mt-dsRNAs into the cytoplasm triggered the upregulation of interferon-stimulated genes and the activation of innate immune defenses mediated through MDA5-MAVS.123 Second-generation sequencing of mitochondrial RNAs has led to the identification of mammalian mitochondrial genome-encoded circRNAs.120 When investigating the role of mitochondrion-localized circRNAs during metaflammation, researchers showed that one circRNA, named SCAR, bound directly to ATP5B. The interaction of ATP5B and SCAR abrogated mPTP by blocking induced by the cyclophilin D-mPTP interaction and therefore inhibited mROS production.48 Another highly expressed mecciRNA, mc-COX2, a sense RNA encoded by the COX2 locus, was found in the plasma exosomes of chronic lymphocytic leukemia (CLL) patients. The prognosis of CLL was closely correlated with mc-COX2 level, with higher expression levels of mc-COX2 seemingly promoting cell proliferation and protecting cells from apoptosis. Notably, in a comprehensive description of murine and human mitochondrial transcriptomes,51 thousands of small noncoding RNAs (sncRNAs) aligned to the mitochondrial genome at positions corresponding to 16S rRNA, tRNA, and mRNA.
Mitochondrial proteome and protein posttranslational modifications (PTMs)
At least 1100 proteins have been identified with high confidence as members of the mitochondrial proteome.49 However, among these proteins, only 13 proteins are encoded by mtDNA in mammalian cells, included ND1, ND2, ND3, ND4, ND4L, ND5, ND6, CO1, CO2, CO3, ATP6, ATP8 and Cyt b,110 therefore, the remaining 99% must be imported into mitochondria after precursors are synthesized on cytosolic ribosomes.126 With the advancement of mitochondrial isolation technology, the mitochondrial proteome has become a hotspot for organelle proteome research.127,128 The MitoCarta database, a collection of manually annotated mitochondrial proteins with submitochondrial localizations and functions, is a continually updated community resource used for investigating mitochondrial biology.129
The cell-specific mitochondrial proteome composition is associated with the metabolism characterized.130 Large-scale proteomic surveys have provided valuable molecular insights into tissue diversity and indicated that mitochondria obtained from distinct organs share approximately 75% of proteins.131 Furthermore, the use for mitochondrial proteome data has been gradually evolved from identifying differences in mitochondrial content to identifying differences in mitochondrial protein PTMs and characterizing the dynamics of protein interactions. Numerous mitochondrial protein PTMs, such as phosphorylation, acetylation, methylation, ubiquitination, SUMOylation, glycosylation, and nitrosylation, have been reported.132 Pioneering research has revealed that reversible phosphorylation of liver mitochondrial proteins controls ketogenesis.133 Systematic phosphoproteomes of rat liver, heart and skeletal muscle showed that phosphoproteins were involved in amino acid and fatty acid metabolism in liver mitochondria, whereas heart and skeletal muscle were enriched for phosphoproteins involved in energy metabolism.134 Myocardial acetylproteomics demonstrated extensive mitochondrial protein lysine hyperacetylation in the early stages.135 Proteomics techniques have thus revealed that mitochondrial protein PTMs vary between cells.
The underlying mechanism of mitochondrial heterogeneity is mainly due to mitochondrial protein import and the mitochondrial protein modification pathway. The mitochondrial proteins encoded by nuclear genes are synthesized on cytosolic ribosomes and imported into mitochondria through signal-targeting peptides and pathways through which precursor proteins are imported into mitochondria.3 This presequence pathway is typically recognized by translocase of the outer membrane (TOM) and translocase of the inner membrane (TIM).3 Studies on the TOM complex, consisting of receptor proteins (TOM70, TOM20 and TOM22) and a pore-forming protein (TOM40), revealed that the reversible phosphorylation of TOM complexes contributes to the formation of supercomplexes and controls the activity of distinct import routes.136 Moreover, two of these presequence translocases are differentially distributed across tissues. One form includes the stably expressed housekeeping subunit TIM17B, and the other form includes the stress-regulated subunit TIM17A,137 suggesting that additional regulatory mechanisms contribute to mitochondrial heterogeneity in multicellular organisms. The mechanism of mitochondrial protein PTM heterogeneity may be due to mitochondrion-localized protein modification enzymes. Sirtuin 3, an NAD(+)-dependent protein deacetylase, has been shown to be located in mitochondria and regulates the acetylation levels of mitochondrial proteins.138 A kinase prediction showed important roles for PKA and PKC at the phosphorylation sites of mitochondrial proteins.134
Mitochondrial lipids
Mitochondria are unique organelles for studying membrane biochemistry because their functionality depends on a coordinated supply of proteins and lipids. Most phospholipids, sterols, sphingolipids, and neutral lipids are synthesized within the ER, but mitochondria contribute to the cellular synthesis of phosphatidylethanolamine (PtdEth).139 In recent decades, mitochondrial lipidomic analysis has revealed that mitochondria from various organs carry phosphatidylcholine and phosphatidylethanolamine comprising 5–30% of total phospholipids.140,141 Another subcellular organelle lipidomic study of living cells revealed sophisticated lipid dynamics during mitochondrial cristae dissociation at different stages.142 Despite the gradual recognition of mitochondrial lipid heterogeneity, the molecular mechanisms associated with the regulation of mitochondrial lipids are not clearly understood. Mitochondria play central roles in the catabolic degradation of fatty acids (β-oxidation) and, to some degree, in fatty acid synthesis, which involves frequent communication with other cellular compartments, such as the ER and peroxisomes.59,76 ER-mitochondrion contact sites, which are called mitochondrion-associated membranes (MAMs), allow the exchange of lipids between both organelles,143 and lipid droplets derived from the ER are storage reservoirs for sterols and fatty acids in the form of triacylglycerols (TAGs) and steryl esters.144,145 Hence, intercellular lipid transport from other organelles to mitochondria is clearly important. Similarly, mitochondrial activities depend on lipid exchange between the IMM and outer mitochondrial membrane (OMM).59,146
Intracellular mitochondrial heterogeneity
Studies have revealed intracellular mitochondrial heterogeneity in mitochondrial components, mitochondrial morphology and mitochondrial function in cells that depend on the mitochondrial redox state, membrane potential, respiratory activity and ROS production.147–150 Taking advantage of high-resolution techniques that can be used to identify all types of mtDNA structural variations and single-nucleotide variations (SNVs) in a single cell, intracellular heterogeneity of mtDNA in single neuron mouse cells or human peripheral blood mononuclear cells, hematological cancers, fibroblasts and tumor cell lines has been discovered.106 The most common mtDNA mutation, 8344A>G, has been observed in cells exhibiting a broad range of heteroplasmy (from 0% to 100%).106
Regarding mitochondrial morphology and function, confocal microscopy led to the a distinction being made in mitochondrial subpopulations of specific cell regions based on the immunostaining of mitochondria-specific markers.42 In neurons, distinguishable morphological and compositional variation was found between neuron synaptic mitochondria (sMito) and nonsynaptic mitochondria (nsMito).151–155 Proteomic and enzymatic characteristics of the synaptic mitochondrial subpopulation revealed that the levels of 22 proteins were significantly higher and those of 34 proteins were significantly lower in sMito than in nsMito. These proteins included the mitochondrial ROS clearance-related protein-superoxide dismutase [Mn] (SOD2), TCA-related protein-isocitrate dehydrogenase subunit alpha (IDH3a), aconitate hydratase (ACO2), and ATP-forming β subunit of succinyl-CoA ligase (SuclA2). The OXPHOS-related protein NADH dehydrogenase includes ubiquinone iron-sulfur protein 8 (Ndufs8) and cytochrome c oxidase subunit 5A (Cox5a).156 Further study demonstrated that the 3 most distinct clusters identified between sMito and nsMito by proteomic expression profiling were associated with glycolysis, OXPHOS and inner membrane bioenergetic complexes.157
Intracellular mitochondrial subpopulations may exhibit different responses to substrates and may vary in their sensitivity to deleterious stress.158,159 In neurons, sMito exhibited enhanced respiration activity and increased vulnerability to Ca2+ overload and oxidative damage compared to nsMito.153,156 The enhanced respiration activity of sMito ensures an energy supply for the extension and branching of neuronal axons and dendrites, while increased vulnerability to Ca2+ overload and oxidative damage compared to nsMito makes neuronal axons more vulnerable to oxidative stress. Studies have also revealed that energy shortage and accumulation of Ca2+ and ROS at individual synapses may lead to synaptic loss, which is an early sign of certain neurodegenerative diseases such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Parkinson’s disease (PD).160
Because of the approach established to isolated peridroplet mitochondria (PDM), mitochondrial heterogeneity has also been demonstrated in adipose cells. PDM show unique morphological and enzymatic features compared to cytoplasmic mitochondria.161 The PDM subpopulation of adipose cells has been shown to exhibit enhanced bioenergetic capacity, low fatty acid oxidation capacity and lipid droplet expansion support by providing ATP for triacylglyceride synthesis.161 These intracellular mitochondrial subpopulation characteristics provide an explanation for the capacity of mitochondria within individual cells to be simultaneously involved in different metabolic pathways.
Mitochondrial heterogeneity under pathological conditions
In addition to pathways for energy metabolism, such as the TCA cycle, OXPHOS and fatty acid oxidation (FAO), several other pathways are activated in mitochondria, such as mtDNA transcription and translation; amino acid, lipid, and nucleotide metabolism; calcium homeostasis; apoptosis signaling; and redox process pathways (Fig. 5).3 Annotations of the mitochondrial genome and proteome have allowed for in-depth studies of the biochemical function, evolutionary history, and diversity of mitochondria in cells and tissues (Table 2). More than 150 mtDNA mutations have been associated with maternally inherited syndromes (Table 2).162 Recent research has been aimed to separate pathogenic from benign variants and to create technical tools for the precise editing of the mitochondrial genome.163,164 The majority (31%) of various mitochondrial proteins examined via mitochondrial proteomic analyses exhibit a function related to metabolism.49 The abundance of mitochondrial proteins involved in quality control, signaling, regulatory functions, and membrane dynamics is typically low, and 20% of proteins with low abundance perform unannotated functions and may involve substoichiometric regulatory factors.49 Many other orphan mitochondrial proteins that lack robust functional characterization have been suggested to play biological roles.165 An intensive characterization of the proteome has led to the identification of many mitochondrial proteins that appear to be involved in the reversible PTMs of proteins, such as phosphorylation and acetylation,133,135,166 suggesting a complex signaling network within the organelle. Notably, the current understanding of proteomic signatures in various organs under pathological conditions are important to summarize, as pioneering efforts in recent decades have established the core protein components of mitochondria.167 Genetic and pharmacological mouse models to which mitochondrial proteins can be targeted have provided the basis for a more detailed understanding of mitochondrial heterogeneity in different cellular, tissue and pathological states (Table 3 and Fig. 6), which may be important references for identifying mitochondrial targets for disease therapy.
Fig. 5.

Summary of mitochondrial functions. (1) Fusion and fission. (2) Energy metabolism. (3) Metabolism of amino acids and nucleotides. (4) Mitophagy. (5) mtDNA expression and translation. (6) mtDNA proliferation and mutation. (7) RNA posttranscriptional processing. (8) Protein quality control, degradation and modification. (9) The respiratory chain. (10) ROS clearance. (11) Signaling and the redox process. Abbreviations: MPC mitochondrial pyruvate carrier, CPT carnitine palmitoyltransferase, GLU glucose, FFA free fatty acid, Ser serine, Gly glycine, THF tetrahydrofolate, CH2-THF 5,10-methylene-THF, mtDNA mitochondrial DNA, mRNA messenger RNA, tRNA transfer RNA, ncRNA noncoding RNA, mtUPR mitochondrial unfolded protein response, HSP heat shock protein, ROS reactive oxygen species, CytC cytochrome c, GSH glutathione and GSSG glutathione oxidized
Table 2.
Disease-related mitochondrial omics sequencing data summary
| Organ | Disease | Experimental method | Reference |
|---|---|---|---|
| Nervous system | Mitochondrial encephalomyopathy | mtDNA sequencing | 170,171 |
| Cerebral metabolic abnormalities | mtDNA sequencing | 172 | |
| Parkinson’s disease | mtDNA sequencing. Proteome | 174,175 | |
| Frontotemporal lobar degeneration | mtDNA sequencing | 401 | |
| Neurodegeneration | Proteome | 130 | |
| Alzheimer’s disease | Proteome. Interatomic analysis | 195,402 | |
| Amyotrophic lateral sclerosis | Interatomic analysis | 403 | |
| Spinal muscular atrophy | Proteome | 176 | |
| Neurodegenerative disease | Proteome | 177 | |
| Cardiovascular system | Heart failure | Acetylproteome. Proteome | 135,216,217,404 |
| Myopathy and progressive external ophthalmoplegia | Genome sequencing | 405 | |
| Mitochondrial cardiomyopathy | Genome sequencing | 406 | |
| Leigh syndrome | mtDNA sequencing | 407 | |
| Cardiovascular diseases | mtDNA sequencing | 215 | |
| Cardiac hypertrophy and heart failure | Proteome | 408 | |
| Atrial fibrillation | Proteome | 409 | |
| Proteome comparison of human hearts | Proteome | 410 | |
| Liver | Hepatocellular carcinoma | mtDNA sequencing | 50,230 |
| Nonalcoholic fatty liver disease | Proteome | 232,233,411,412 | |
| Aging and the development of liver diseases | Proteome | 413 | |
| Cholangiocarcinoma | Proteome | 414 | |
| Liver fibrosis | Proteome | 415 | |
| Skeletal muscle | Aging | mtDNA sequencing | 273 |
| Myopathy | mtDNA sequencing | 271 | |
| Obesity and T2D, muscle IR | Proteome | 270,274,277–279,416 | |
| Aging | Proteome | 267,269,280,417 | |
| Myosteatosis | Proteome | 283,418 | |
| Adipose | Aging | Proteome | 291,292,299 |
| Diabetes | Proteome | 289,290,298 | |
| Immune cell | Immune cells | mtDNA sequencing | 315 |
| Innate immune-monocyte-septic shock | Proteome | 316 | |
| Innate immune-macrophage-bacterial resistance | Proteome | 319 | |
| Innate immune-natural killer cells- | Proteome | 419 | |
| adaptive immunity-T-cells | Proteome | 31,320,321 | |
| Cancer | Kidney, colorectal and thyroid cancers | mtDNA sequencing | 420 |
| Nasopharyngeal carcinoma | Proteome | 333 | |
| Human ovarian cancer | Proteome | 334,335 |
Table 3.
Disease-related mitochondrial proteins
| Disease | Regulation protein | Functional distribution | Reference |
|---|---|---|---|
| Neuron system | |||
| Alzheimer’s disease | CPT1P.G, BDH1, SCOT, ACAT1 | FAO | 197,201–203 |
| IDH1, KGDHCP | TCA cycle | 178,180,421 | |
| COX5A, NDUFB8G, OSCPG, UQCRC2G | OXPHOS | 189–194,422–424 | |
| HSPA9, HSP60P, CLPPG | UPRmt | 208,209,425 | |
| Parkinson’s disease | KGDHCG | TCA cycle | 179,426 |
| Miro1G | Mitochondria synaptic transmission, mitophagy | 427–432 | |
| Ndufs2G | OXPHOS | 196 | |
| Cardiovascular system | |||
| Heart failure | NDUFAB1G, NDUFV7G | OXPHOS | 433,434 |
| PDHG, SDHAG, IDH2G | TCA cycle | 135,221,222 | |
| CPT1G, BDH1G | FAO | 223,224,435 | |
| SOD2G, PRX-3G, p66ShcG | ROS reduction | 216,226,436,437 | |
| MPCG | Pyruvate transport | 438 | |
| Liver disease | |||
| Acute liver failure | GSHG | ROS reduction | 236,237 |
| HSP10, HSP60P | UPR | 234,240,241 | |
| GRP75G | Transporters and channels | 234,243 | |
| IDH1G | TCA cycle | 239 | |
| NAFLD | CYP2E1P.G | ROS generation | 256,257 |
| SOD1, SOD2, GclcP, GclmP, Gpx1P.G, GSHG | ROS reduction | 258–262 | |
| NQO1G | Redox process | 439,440 | |
| ACAT, ACAC/ACC CPT1αP, CPT2α, BDH2G | FAO | 253,254,441 | |
| MPC1, MPC2G, SLC25A11P | Transport related to TCA cycle | 255,442,443 | |
| HMGCS2G | Ketogenesis | 444–447 | |
| PHGDHG, PSAT1, PSPH | SSP | 255,448 | |
| Liver fibrosis | p66ShcP.G | ROS generation | 449–451 |
| SOD2P, GSHP | ROS reduction | 420,452–455 | |
| Hmgcs2P.G | Ketone body production | 444,446,456 | |
| IDH1P.G | TCA cycle, AA utilization | 457,458 | |
| Skeletal muscle | |||
| Myopathy | SDHAG | TCA cycle | 459–462 |
| CACT, CPT2P, ACADVL | FAO | 463–465 | |
| COX10P.G, NDUFB8, UQCRC1 | OXPHOS | 462,466–469 | |
| LONP1G, HSP70 | UPRmt | 470,471 | |
| SOD2G | ROS reduction | 472 | |
| SLC25A42G | Coenzyme A import | 473 | |
| Adipose | |||
| Diabetes | UCP1P.G | Thermogenesis | 301–305,474 |
| Ndufv2G, Ndufs4G | OXPHOS | 306,307 | |
| MnSODG | ROS reduction | 308,475 | |
| Immune cell | |||
| Adaptive immunity | CPT1aG, ACATP | FAO | 476–478 |
| SHMT2G | One-carbon | 31,321 | |
| COX10G | OXPHOS | 320 | |
| MPCP.G | Pyruvate import | 323 | |
PIndicates proteins that have been studied pharmacologically; G indicates proteins that have been studied genetically
CPT-1 Carnitine palmitoyltransferase-1, CPT1α Carnitine palmitoyltransferase-1α, BDH1 3OH-Butyrate dehydrogenase, type1, SCOT Succinyl-CoA,3-ketoacid CoA transferase, ACAT1 Acetyl-CoA acetyltransferase 1, IDH1 Isocitrate dehydrogenase (NADP( + )) 1, KGDHC α-Ketoglutarate dehydrogenase complex, COX5A Cytochrome c oxidase subunit 5A, NDUFB8 NADH,ubiquinone oxidoreductase subunit B8, OSCP Oligomycin sensitivity-conferring protein, UQCRC2 Ubiquinol-cytochrome c reductase core protein 2, HSPA9 Mortalin, Hsp60 chaperonin 60, CLPP Caseinolytic protease P, MCAD Medium-chain acyl-CoA dehydrogenase, Miro1 Mitochondrial Rho-GTPase, Ndufs2 NADH dehydrogenase [ubiquinone] iron-sulfur protein 2, NDUFAB1 NADH,ubiquinone oxidoreductase subunit AB1, NDUFV7 NADH dehydrogenase [ubiquinone] flavoprotein 1, PDH Pyruvate dehydrogenase, SDHA Succinate dehydrogenase complex, subunit A, SOD2 Superoxide dismutase 2, PRX-3 Peroxiredoxin 3, p66Shc Src homology 2 domain-containing transforming protein C1, MPC Mitochondrial pyruvate carrier, GSH Glutathione, HSP10 chaperonin 10, GRP75 Glucose-regulated protein 75 kDa, CYP2E1 Cytochrome P450-2E1, SOD1 Superoxide dismutase 1, Gclc Cysteine ligase catalytic subunit, Gclm Glutamate-cysteine ligase modifier subunit, GPX 1 Glutathione peroxidase 1, NQO1 NAD(P)H,Quinone oxidoreductase 1, HMGCS2 3-Hydroxy-3-methylglutaryl CoA synthase 2, ACAT Acetyl-CoA acetyltransferase, ACAC/ACC Acetyl-CoA carboxylase, SLC25A11 Oxoglutarate carrier, PHGDH Phosphoglycerate dehydrogenase, PSAT1 Phosphoserine aminotransferase 1, PSPH Phosphoserine phosphatase, SLC25A42 Mitochondrial coenzyme A (CoA) transporter, ACADVL Acyl-CoA dehydrogenase very long chain, UQCRC1 Cytochrome b-c1 complex subunit 1, LONP1 Lon protease homolog, HSP70 Chaperonin 70, UCP1 Uncoupling protein 1, Ndufv2 NADH dehydrogenase [ubiquinone] Flavoprotein 2, Ndufs4 NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, MnSOD Manganese superoxide dismutase, SHMT2 Serine hydroxymethyl transferase 2, COX10 Cytochrome c oxidase assembly homolog 10, UPRmt mitochondrial unfolding protein response, SSP Serine synthesis pathway, AA utilization Amino acid utilization
Fig. 6.

Comparison of mitochondrial heterogeneity in tissues. Pathways with significant differences in the mitochondrial proteome of tissue in pathological states are marked with cyan colors, and no significant differences in mitochondrial proteomic results or mitochondrial pathways with changes that were not based on mitochondrial proteomic results are marked with pink colors. Based on the research of mitochondrial-targeted transgenic mice model, we summarized the relationship between mitochondrial protein and disease pathological states, all related proteins are marked with magenta. Brain pathology state, Alzheimer’s disease (AD) and Parkinson’s disease (PD), mitochondrial function associated with (2) energy metabolism; (5) mtDNA expression and translation; (6) mtDNA proliferation and mutation; (8) protein quality control, degradation and modification; (9) the respiratory chain; (10) ROS clearance; (11) signaling and the redox process. Skeletal muscle pathology state, hypoxia and myosteatosis, mitochondrial function associated with (2) energy metabolism; (5) mtDNA expression and translation; (6) mtDNA proliferation and mutation; (8) protein quality control, degradation and modification; (9) the respiratory chain; (10), ROS clearance; (11) signaling and the redox process. Cardiovascular pathology states, heart failure (HF) and cardiomyopathy, mitochondrial function associated with (2) energy metabolism; (5) mtDNA expression and translation; (6) mtDNA proliferation and mutation; (8) protein quality control, degradation and modification; (9) the respiratory chain; (10) ROS clearance; (11) signaling and the redox process. Adipose tissue, mitochondrial function associated with (2) energy metabolism; (9) thermogenesis; (10) ROS clearance; (11) signaling and the redox process. The liver, mitochondrial function associated with (2) energy metabolism; (5) mtDNA expression and translation; (6) mtDNA proliferation and mutation; (7) RNA posttranscriptional processing; (8) protein quality control, degradation and modification; (9) the respiratory chain; (10) ROS clearance; (11) signaling and the redox process. Immune system mitochondrial function associated with (2) energy metabolism; (3) metabolism of amino acid and nucleotides; (9) the respiratory chain; (10) ROS clearance; (11) signaling and the redox process. Abbreviations: CPT1c Carnitine Palmitoyltransferase 1C, ACAT acetyl-CoA acetyltransferase 1, IDH1 isocitrate dehydrogenase 1, KGDHC α-ketoglutarate dehydrogenase complex, OSCP oligomycin sensitivity-conferring protein, NDUFB8 NADH:ubiquinone oxidoreductase subunit B8, Ndufs2 NADH dehydrogenase [ubiquinone] iron-sulfur protein 2, HSPA9 Mortalin, Miro1 Mitochondrial Rho-GTPase, SDHA Succinate dehydrogenase complex, subunit A, PDH pyruvate dehydrogenase, BDH1 3OH-Butyrate dehydrogenase, type1, PRX-3 Peroxiredoxin 3, SOD superoxide dismutase, Nnt nicotinamide nucleotide transhydrogenase, GCLC glutamate cysteine ligase catalytic, GRP75 Glucose-regulated protein 75 kDa, HMGCS2 3-Hydroxy-3-methylglutaryl CoA synthase 2, CYP2E cytochrome P450-2E1, COX10 Cytochrome c oxidase assembly homolog 10, LONP Lon protease homolog, SLC25A42 Mitochondrial coenzyme A (CoA) transporter, UCP1 uncoupling protein 1, Ndufv2 NADH dehydrogenase [ubiquinone] Flavoprotein 2, Ndufs4 NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, SHMT2 Serine hydroxymethyl transferase 2 and MPC mitochondrial pyruvate carrier
The nervous system
The brain is a vital organ that consumes massive amounts of energy and depends upon glucose as its main source of energy,168 and a close connection between glucose metabolism and mitochondrial function is critical to brain physiology.169 Due to the disruption of mitochondrial fusion and fission, mitochondrial morphology heterogeneity has emerged in several neurodegenerative diseases (AD and PD).65 Strong evidence has implicated increased mitochondrial component heterogeneity as a central pathological mechanism underpinning neurodegenerative diseases. Multiomics sequencing of neurodegenerative tissues has indicated that abnormalities in the mitochondrial genome and proteome are significantly associated with neuropathological status (Table 2). Major deletions or mutations in the mitochondrial genome of brain tissue have been associated with neurodegenerative diseases, and mtDNA point mutations have been linked to an insufficient energy supply in neurons.170–172 Further research revealed accumulated mtDNA mutations during aging in a mtDNA polymerase (POLG) mutant mouse model and worsened neurodegeneration in an AD mouse model established with mice bred with POLG mutant mice.173 Additionally, mutation accumulation is present in human neurodegenerative diseases. For example, PD patients with primary substantia nigra (SN) neuron mitochondrial defects presented with POLG mutations,174 and substantial loss in the number of SN neurons was observed in patients with POLG mutations, suggesting a correlation between mtDNA mutation accumulation and brain pathology.174 Proteome analysis has revealed the important roles of mitochondria in brain tissue. For example, the proteome of neural stem cells (NSCs) in the subventricular zone (SVZ) from PD patients led to the identification of numerous proteins implicated in mitochondrial activity.175 Spinal muscular atrophy (SMA) is caused by a reduction in survival motor neurons (SMNs) due to disruption in mitochondrion-associated energy-generating functions.176 In addition to mitochondrial energy synthesis in neurons, mitochondrial proteostasis plays an important role in regulating mouse brain autophagic vesicle formation.177 The proteome in multiple neurodegenerative diseases exhibits mitochondrial molecular diversity among three major cerebellar cell types (Purkinje cells, granule cells and astrocytes),130 supporting a correlation between mitochondrial metabolism and the progression of brain diseases, including AD and PD (Fig. 6 and Table 3).
The TCA cycle, OXPHOS, and FAO are predominant pathways linked to metabolic changes in mitochondria under pathological circumstances. As the key enzyme in the TCA cycle, the alpha-ketoglutarate dehydrogenase complex (KGDHC) showed a 44% reduction in the activity in familial AD patient brain samples,178 and KGDHC immunostaining in PD patient tissues showed a reduction in the number of melanized neurons.179 Subsequent research showed that the loss of KGDHC-enriched cells was proportional to the total loss of neurons.180 Other TCA core enzymes, such as pyruvate dehydrogenase (PDH) and isocitrate dehydrogenase-1 (IDH1), have been analyzed in subsequent clinical studies,181,182 and the results suggested TCA enzymes were functional in the pathological brain. In addition, the OXPHOS complex was suppressed under neurodegenerative conditions,183–185 indicating lower mitochondrial oxidative respiration efficiency and increased ROS production, both of which impair neurons.186–188 A transgenic AD mouse model showed decreased expression of NADH:ubiquinone oxidoreductase subunit B8 (NDUFB8), which is a nuclear DNA-encoded subunit that is integral to the assembly of Complex I.189 Further research revealed that NDUFB8 protein O-GlcNAcylation impairment was involved in the high-fat diet (HFD)-induced neurodegenerative process.190 Similarly, other respiratory chain proteins, such as oligomycin sensitivity-conferring protein (OSCP), physically interacts with amyloid beta (Aβ) in the brains of AD individuals and AD mouse models.191 Moreover, restored mitochondrial bioenergetics enhanced cognition in an AD (5xFAD) mouse model by blocking the deleterious impact of CypD on OSCP or by overexpressing OSCP.192,193 Increased binding among Aβ, the apoE4 fragment, Tau and respiratory chain proteins leads to mitochondrial dysfunction.194,195 Complex I activity was disrupted during OXPHOS by Ndufs2 deletion and caused human-like PD in mice.196 These studies suggest a correlation between mitochondrial respiratory chain proteins and the progression of brain disease. Regarding mitochondrial FAO adaptation in brain pathology, both AD and PD models showed a systemic shift from glycolysis to lipid metabolism,197,198 and the expression of mitochondrial lipid metabolism-related proteins was elevated in AD and PD models.197,199 Fatty acid metabolism was also upregulated in both pathologies to compensate for neuronal glucose hypometabolism. The carnitine palmitoyl transferase (CPT) system is crucial for mitochondrial β-oxidation of long-chain fatty acids (Table 3).200 Overexpression of CPT1c in the mouse brain caused microencephaly, and CPT1c-KO mice demonstrated a marked reduction in spatial learning ability201,202 and increased sensitivity to oxidative stress.203 In addition, genetic or pharmacological inhibition of acetyl-CoA acetyltransferase 1 (ACAT), an enzyme that catalyzes the final step in the mitochondrial beta-oxidation pathway, is thought to exert an inhibitory effect on the brain lesion process.204–207
In addition to the removal of damaged mitochondria in neuronal cells via fusion and fission and mitophagy to maintain mitochondrial homeostasis, the mitochondrial protein unfolded response (UPRmt), which is involved in the maintenance of mitochondrial function, is becoming better understood. A transcriptome analysis of the prefrontal cortex of AD patients revealed that UPRmt-related genes (HSPA9, HSP60, and YMEL1L) were upregulated, while genetic or chemical inhibition of HSPA9 strongly induced mitochondrial fragmentation and synergistically increased Aβ-mediated cytotoxicity as well as mitochondrial dysfunction.208 UPRmt is a conserved mitochondrial stress response signature in diseases involving Aβ proteotoxicity in both humans and mice.209
The cardiovascular system
The cardiovascular system is a vital organ system that delivers essential substances to all cells to support basic functions. This network is composed of the heart, the centralized pump; blood vessels that distribute blood throughout the body; and blood, which transports different substances. Cardiovascular homeostasis relies heavily on mitochondrial fatty acid-driven oxidative phosphorylation for ATP production.210 Cardiovascular disease, including myocardial infarction, and cardiomyopathies of different etiologies, including forms of arrhythmia, hypertension, atherosclerosis, and other vascular conditions, is the main “killer” in humans,211 and mitochondrial dysfunction is a central etiological determinant of cardiovascular disease.212 Strong evidence has shown mitochondrial morphological heterogeneity in several cardiovascular diseases,213 and the interchange of the mitochondrial morphology between elongated interconnected mitochondrial networks and a fragmented disconnected arrangement has been found to be relevant in various aspects of cardiovascular diseases. Recently, multiomics studies of cardiovascular disease have demonstrated a role for mitochondrial component heterogeneity in the cardiovascular system, specifically suggesting that increased mitochondrial heterogeneity plays a role in cardiovascular pathology (Table 2). Genome sequencing has led to the identification of mitochondrial gene mutations, including mitochondrial genomic tRNA and respiratory chain coding gene mutants that are linked to cardiovascular diseases.214,215 Systematic studies have described the pleiotropic effects of different mtDNA variants and identified novel associations between these variants and previously uncharacterized complex and quantitative traits.44 Moreover, high-throughput proteomic and metabolomic analysis of a 2-week ventricular-tachypaced congestive heart failure dog model revealed significant myocardial mitochondrial alterations, particularly the downregulation of oxidant proteins (superoxide dismutase (SOD) and peroxiredoxin (PRX-3)) and the upregulation of TCA core enzymes (malate dehydrogenase (DH), α-/β-enolase (ENO1 and ENO3) and pyruvate dehydrogenase (PDHA1)).216 Global proteomic surveys of cardiac ventricles isolated from failing human hearts led to the identification of 25 proteins with significantly changed expression, with 7 proteins located in mitochondria and associated with metabolism, antioxidant activity and the UPRmt 217 Therefore, cardiovascular metabolic derangements, oxidant clearance and proteostasis contribute to cardiovascular pathogenesis.
Myocardial metabolic disorders are largely affected by the TCA cycle, OXPHOS and FAO. During pathological heart remodeling, cardiac metabolism is reprogrammed to increase reliance on glucose and significantly increase glycolysis, whereas OXPHOS and FAO are downregulated.28 Increased glycolysis is associated with the uncoupling of OXPHOS, resulting in increased lactate production and inhibition of the branched chain amino acid (BCAA) degradation pathway via downregulated KLF15, which promotes a hypertrophic response in cardiomyocytes.218,219 All these factors reduce the efficiency of ATP synthesis and exacerbate pathological remodeling.219,220 Mitochondrial metabolism adaptation associated with early cardiac pathology contributes to the pathogenesis of heart failure (HF). During the progression from compensated cardiac hypertrophy to HF, net mitochondrial protein acetylation increases, and the acetylation of some of these proteins, such as succinate dehydrogenase complex. subunit A (SDHA), decreases their catalytic function, suggesting that mitochondrial protein acetylation homeostasis is a potential driver of the development of the energy metabolism dysregulation that contributes to heart failure.135 One-half of the acetylation sites have been identified previously as potential targets of sirtuin 3 (SIRT3) deacetylase activity in the mouse heart.166 Other TCA cycle enzymes (ACO2, IDH2, and MDH2) have also been shown to be acetylated, and heart-specific knockout or overexpression of TCA-associated enzymes in mice demonstrated exacerbated or attenuated the acquisition of cardiac pathological phenotypes (Table 3). Cardiac PDH E1a deficiency caused a large myocardial infarct area and increased macrophage infiltration in the heart, while PDH activated by dichloroacetate in WT hearts during ischemia/reperfusion increased glucose oxidation and reduced myocardial infarct size.221 Moreover, in an IDH2-deficient mouse model, mitochondrial dysfunction and cardiac hypertrophy were promoted by PDH activation.222 Mitochondrial long-chain fatty acid (LCFA) oxidation, the main FAO pathway involved in myocardial energy supply, was also inhibited due to the reduced activity of the rate-limiting enzyme CPT1. Heterozygous CPT1-knockout mice subjected to the transverse aorta constriction exhibited exacerbated cardiac hypertrophy and remodeling.223 Heart-specific (3OH-Butyrate dehydrogenase, type1) BDH1-overexpressing transgenic mice were resistant to fibrosis, contractile dysfunction, and oxidative damage, suggesting that increased ketone body utilization decreased oxidative stress and protected against HF.224 Hence, targeting key enzymes of mitochondrial FAO is important for the treatment of heart disease.
ROS accumulation can damage cellular lipids, proteins, and DNA, and a pioneering study revealed that mitochondrial antioxidant proteins (SOD and PRX-3) were downregulated in an HF animal model. A mitochondrion-located redox enzyme (PRX-3) tissue-specific knockout mouse model showed impaired antioxidant capacity and exacerbated cardiac dysfunction and oxidative stress during HF.225 Cardiomyocyte-specific SOD2-deficient mice die at ~4 months due to HF and showed mitochondrial architecture alterations, with prominent disruption of cristae and increased vacuole formation.226 Another novel mechanism related to the ROS clearance pathway has also been reported. Research has suggested that nicotinamide nucleotide transhydrogenase (Nnt) mediates a reverse reaction in which NADPH is consumed to support NADH and ATP production under pathological heart conditions, which results in a reduction in NADPH-linked antioxidative capacity,227 Inhibition of Nnt led to the reversal of its catalytic function in a mouse model and protected against oxidative stress, HF, and death.227
The liver
The liver is the hub of intermediary metabolism supporting key anabolic pathways that synthesize glucose, lipids, and ketones to carefully meet the energy requirements of peripheral tissues.29 A study revealed that the primary hepatic mitochondrial structure showed mostly discrete globular or short tubular mitochondria,228 while the mitochondrial morphology of primary hepatocytes isolated from rats continuously fed ethanol showed increased heterogeneity.228 In a mouse model carrying mtDNA point mutations, elongated mitochondrial networks with an artificial loop structure, depressed autophagy, high mitochondrial respiration and an upregulated antioxidative response were found in liver tissue sections and isolated hepatocytes, which indicated that mtDNA mutations accelerated liver ballooning and degeneration.229 Next-generation sequencing (NGS) applied to hepatitis B virus (HBV)-related hepatocellular carcinoma patients revealed that patients with D-loop mutations in mtDNA were more likely to undergo relapse.230 In addition, mitochondrial genome nucleotide polymorphism sites were identified in genes related to nonalcoholic fatty liver disease (NAFLD) development.231 Multiomics has suggested that metabolic remodeling plays key role in liver pathogenesis.232,233 Mitochondrial proteome sequencing of mouse acute liver injury, NAFLD or liver fibroblasts showed the high plasticity of mitochondrial proteins (Table 2).
Acetaminophen (APAP) is a widely used analgesic and antipyretic drug, the overdose of which causes severe centrilobular hepatic necrosis in humans and experimental animals. Recent studies showed that the expression of chaperone proteins HSP10 and HSP60 and glutathione (GSH) was reduced in mitochondria due to treatment of APAP at toxic doses in mice that had been fasted overnight.234 The potential therapeutic benefits of GSH, HSP10 and HSP60 have been described in liver disease.235 A mechanism explaining the decrease in GSH content during liver disease first involves a decrease in the GSH biosynthesis rate. Then, hepatocyte-specific knockout of glutamate cysteine ligase catalytic (GCLC) protein, the catalytic subunit of the rate-limiting and regulatory enzyme glutamate cysteine ligase (GCL) in the GSH biosynthetic pathway, showed marked mitochondrial morphology changes and a profound decrease in ATP generation in conjunction with histological features of hepatic steatosis.236 Livers from GCLC-specific-knockout mice developed spontaneous liver pathologies characteristic of various clinical stages of liver injury.236,237 In addition to the de novo GSH synthesis pathway, GSH is found in both the reduced (GSH) and oxidized states (GSSG), and the reduction in GSSG with a commensurate increase in GSH mediated by the enzyme GSH reductase aids in maintaining the GSH level.238 Both the reduction and oxidation pathways require NADPH to provide reducing equivalents, and high IDH1 expression in the liver is an important source of cytosolic NADPH. Studies have identified that IDH1-knockout mice were more sensitive to LPS-induced sepsis, which was attributed to a large increase in the hepatocyte apoptosis rate.239 Although liver-specific HSP10- and HSP60- knockout mouse models have not yet been reported, research has revealed that chlorogenic acid, liquiritigenin and liquiritin alleviated hepatic inflammatory injury by inhibiting HSP60 release.240,241 Interestingly, 75-kDA glucose-regulated protein (GRP75) is a major component of both the mitochondrial quality control system and mitochondria-associated membrane,242 and overexpression of GRP75 in the liver decreased cytochrome c expression in CCL4-induced liver injury.243
Further studies revealed that the regulation of hepatic mitochondrial proteins at the molecular level during liver injury and disease is associated with mitochondrial metabolism (TCA cycle, OXPHOS, FAO, and ketogenesis), ROS and protein stabilization (Table 3). Studies on liver mitochondria isolated from a rat model of spontaneous diabetes revealed that the levels of FAO and OXPHOS proteins were increased after the rats were rendered diabetic, while the levels of ROS-detoxifying enzymes were decreased.244,245 Further study revealed reversible phosphorylation that was widespread in mitochondrial proteins related to OXPHOS, the TCA cycle, FAO, the urea cycle, hormone metabolism, and glycolipid biosynthesis,133 with the enzymes involved in ketogenesis (Hmgcs2), lipogenesis (Gpam), and retinol metabolism (Dhrs4) the most significantly changed via modification.133 A key mitochondrial enzyme in ketogenesis, 3-hydroxy-3-methylglutaryl CoA synthase 2 (HMGCS2), showed the greatest change in phosphorylation,133 indicating that phosphorylation of HMGCS2 plays a key role in the regulation of liver mitochondrial ketogenesis metabolism. Patients with NASH presented with significant increases in hepatic mitochondrial FAO,246 the TCA cycle,247 OXPHOS245 and ketogenesis,232 and both mice and humans under chronic nutritional overload showed the impaired mitochondrial function of the TCA cycle,232 OXPHOS232,248 and ketogenesis.249,250 Thus, mitochondrial metabolic function is upregulated in NASH to accommodate the rapid accumulation of hepatic triglycerides. In an NAFLD model, fat extracted by the liver coupled with mismatched mitochondrial fat disposal capacity led to fat accumulation in hepatocytes. A study suggested that the accumulation of plasma triglycerides derived from the liver and dietary free fatty acids (FFAs) and de novo lipogenesis in simple steatosis are associated with mitochondrial metabolism adaptation.246,247,251 This adaptation of mitochondrial metabolism is the central feature of NAFLD.248,252
In addition, mice fed an HFD showed normal mitochondrial energetics but accumulated lipotoxic byproducts, including ceramides and diacylglycerols. This finding highlights the complex interaction between early compensatory oxidative mechanisms and the inefficient storage/disposal of FFAs.233 The mitochondrial FAO pathway regulation mechanism plays an important role in liver pathology progression, and studies have shown that CPT1α, CPT2α, ACAT and ACAC/ACC are core regulatory sites.253 Permanently active CPT1 mutants enhanced hepatic FAO and autophagy, reduced liver steatosis, and improved glucose homeostasis in HFD mice, suggesting that CPT1 gene therapy reduced HFD-induced dysregulation.254 To further illustrate the interactions between lipid metabolism and other cellular metabolic functions, a systematic analysis was performed with hepatocytes to distinguish NASH-specific metabolic features. This study identified PHGDH, SHMT1 and SHMT2 as potential mitochondrion-targeted therapeutic options for NASH and showed that downregulation of these proteins resulted in serine synthesis pathway blockade.255 In addition to the fat-processing burden placed on the hepatic mitochondria, a high ROS burden has been associated with liver pathology. In high hepatic cholesterol-induced NASH and fibrosis, mitochondrial proteins related to ROS generation, such as cytochrome P450-2E1 (CYP2E), were upregulated,256,257 and Cyp2e1-null mice showed resistance to high cholesterol-induced NASH and fibrosis,256 while ROS clearance proteins, including SOD1, SOD2, Gclc, Gclm and Gpx1, were downregulated.258–262 Silibinin is used for the clinical treatment of NASH because it significantly activates antioxidase activity (CAT, GSH-Px and HO-1) and inhibits pro-oxidase activity (CYP2E1 and CYP4A) to reduce ROS generation.257
The skeletal muscle
The influence of skeletal muscle, which accounts for as much as 40% of body mass, has multiple implications for mobility, injury, and metabolic diseases and thus exerts a major impact on overall quality of life263. Skeletal muscle plays a prominent role in metabolic homeostasis and is closely associated with mitochondrial oxidative metabolism263. Mitochondria form a reticulum within muscle cells and are classified into subsarcolemmal (SS) mitochondria and intermyofibrillar (IMF) mitochondrial types.264 SS mitochondria exhibit a circular morphology, while IMF mitochondria exhibit a long and branched morphology.265 Mitochondrial morphology heterogeneity has been observed in a mouse skeletal muscle aging model, suggesting that mitochondrial morphology heterogeneity is associated with skeletal muscle pathology.265
Muscle mitochondrial metabolism diversity depends on different types of muscle fibers266–268 and different muscle pathology conditions (obesity, type 2 diabetes (T2D), and aging)267,269,270 (Table 2). In 1991, a male patient with myopathy and neuropathy presented with large-scale deletion of the mitochondrial genome at nucleotides 6570-14150.271 Mitochondrial genome mutations and large-scale deletions were also found in Leigh syndrome and myopathy.272 Further study identified two new point mutations, A189G and T408A, which had accumulation in muscle tissue, but not in other tissues, of several older individuals.273 This study revealed a close association between mtDNA mutations and specific mutagenic machinery.
The SS and IMF mitochondrial subpopulations indicate different susceptibility to obesity, with IMF mitochondria showing upregulated TCA cycle enzyme activity but downregulated OXPHOS protein activity, in contrast to SS mitochondria.274 Furthermore, skeletal muscle consists of three major fiber types: slow oxidative Type 1 fibers, fast oxidative Type 2a fibers, and fast glycolytic Type 2x fibers.275 Characterization of the proteome of isolated single fibers from the extensor digitorum longus muscle in mice revealed that the abundance of proteins involved in OXPHOS, FAO, and the TCA cycle varied between the individual muscle fiber types, with Type 2a and Type 1 fibers showing a greater abundance of OXPHOS and FAO proteins.276 A proteome analysis of the skeletal muscle of patients with T2D taken by biopsy led to the identification of ATP synthase β-subunit (ATP5F1B) downregulation. Moreover, ATP5F1B was phosphorylated in vivo, and the levels of a downregulated ATP5F1B phospho-isoform in diabetic muscle correlated inversely with fasting plasma glucose levels.277 A subsequent study on mitochondrial OXPHOS protein phosphorylation reported that abnormal site-specific phosphorylation of ATP5F1B, together with reduced OXPHOS protein content, contributed to mitochondrial dysfunction during muscle insulin resistance.278 Recent studies have even described an accurate mitochondrial protein atlas of T2D pathological states. A comparison of the mitochondrial proteomes between T2D and nondiabetic skeletal muscle samples identified mitochondrial functions (OXPHOS, TCA, FAO, and the ROS response) related to T2D.279 Thus, muscle mitochondrial proteomic studies provide guidance and direction for studying the regulatory mechanisms of mitochondrial protein content and modifications in T2D.
Aging-associated mitochondrial function decline contributes to insulin resistance in elderly individuals,280 suggesting that increases in intramyocellular fatty acid metabolites may be results of an age-associated reduction in mitochondrial oxidative and phosphorylation activity.280 This result was confirmed via a quantitative proteomic analysis of skeletal muscle collected from young and elderly individuals.269 Of the mitochondrial proteins identified, the levels of 173 mitochondrial proteins were changed with age, and these proteins were related to OXPHOS, the TCA cycle and mitochondrial homeostasis. Interestingly, this change in skeletal muscle mitochondrial protein level was partially reversed by physical activity.281,282 Myosteatosis is the pathological accumulation of lipids that can occur in conjunction with atrophy and fibrosis following skeletal muscle injury. A pioneering study determined that mitochondrial dysfunction leads to the accumulation of lipids in myosteatosis.283 Research evaluating changes during muscle fiber force production showed that mitochondrial FAO was reduced in the early injury process and that the levels of glycolytic metabolites in muscles generally increased and that these metabolites showed a greater capacity to oxidize pyruvate at later points.283
The adipose tissue
Whole-body adipose tissue content and type are controlled in response to various internal and external cues (e.g., nutritional status and temperature).284,285 The regulatory processes involved in fat storage and oxidation in white adipocytes and thermogenic adipocytes (brown and beige adipocytes) play central roles in body energy homeostasis,286,287 and adipose tissue mitochondrial dysfunction in pathological states such as obesity, insulin resistance, and chronic inflammation is closely associated with adipose malfunction.288–292 Indeed, BAT and beige adipocytes exhibit fragmented round-shaped mitochondria, while white adipocytes exhibit elongated organelles with high levels of ATP synthesis.293 This mitochondrial morphology heterogeneity can determine uncoupling protein 1 (UCP1) content, suggesting that mitochondrial morphology is associated with thermogenesis.293 Increased somatic mtDNA mutations resulting from POLG mutation is associated with a reduced lifespan and premature onset of aging-related phenotypes, such as weight loss and reduced subcutaneous fat.294 Mitochondrial thymidine kinase 2 (Tk2) is vital to maintaining appropriate levels of mtDNA. A mouse model with null mutation of Tk2 showed mtDNA depletion, moderate hypotrophy in adipose tissues, and reduced fat accumulation.295 Moreover, overexpression of mitochondrial targeted 8-oxoguanine DNA glycosylase, which has been associated with the base-excision repair pathway, protected against diet-induced obesity, insulin resistance, and adipose tissue inflammation.296
Mitochondrial proteomics has revealed the relevance of mitochondria in adipose tissue and the systemic implications of their impaired function. WAT mitochondria not only selectively express proteins that support anabolic functions but also degrade xenobiotics, while BAT mitochondrial proteins are particularly suited to catabolic functioning.297 Adipose tissue plays important pathophysiological roles in metabolic abnormalities, such as obesity, T2DM and aging. A proteomic analysis of visceral adipose tissue (VAT) in early T2DM patients and control individuals showed downregulation of the TCA cycle, FAO and OXPHOS, while the mitochondrion-related ROS response was upregulated.289,298 Interestingly, aging led to a change similar to that of T2D pathology with mitochondrial remodeling,291,299 indicating that the mitochondrial proteome plays an important role in the aging-related pathology of T2D.300 BAT mitochondria dissipate chemical energy as heat through thermogenic respiration, which requires UCP1.301–305 Studies have revealed that UCP1 C253 sulfenylation302 and K56/K151 hypersuccinylation304 play important roles in UCP1-dependent thermogenesis and whole-body energy expenditure. Further study found significant inhibition of thermogenic responses in UCP1-C253A-mutant mice.305 UCP1-dependent thermogenesis in adipose tissue plays an important role in obesity. Furthermore, quantitative mitochondrial proteomics of BAT and beige adipose tissue has led to the identification of arginine/creatine metabolism as a beige adipose signature induced in response to cold exposure.301
Although a recent study identified other UCP1-independent thermogenic mechanisms,303 mitochondrial TCA metabolism and pyruvate dehydrogenase activity are associated with ATP-dependent thermogenesis, suggesting that mitochondrial thermogenesis function plays an important role in BAT and beige adipose function. An increasing number of mitochondrial proteins have been identified in adipose pathologies, such as OXPHOS proteins (Ndufv2 and Ndufs4)306,307 and ROS-response proteins (MnSOD).308 A study identified that overexpression of Ndufv2 in adipose tissue mediated increases in mitochondrial biogenesis by regulating supercomplex assembly and elevating mitochondrial ROS production.306 Genetic deletion of Ndufs4 in adipose tissue resulted in an increased propensity to develop diet-induced weight gain, glucose intolerance, and elevated levels of fat-related inflammatory genes, specifically in young male mice. Both studies linked mouse adipose phenotypes to the mitochondrial respiratory chain and identified sex differences at the genetic level.306,307
The immune system
The immune system is important in protecting against infection and cancer. Studies have focused on mitochondrial functions in immune cells.309 The immune system consists of the innate immune system and the adaptive immune system. Innate immune response cells include monocytes, macrophages/dendritic cells, granulocytes (neutrophils, eosinophils, and basophils), and innate lymphocytic cells, including natural killer (NK) cells. The adaptive immune system consists of T and B lymphocytes.309 These cells recognize a foreign agent and mount an inflammatory response, and previous works have revealed that mitochondria are rapidly reprogrammed to meet the demands of effective immune responses.310 Notably, NK cell mitochondria exhibited a small spherical mitochondrial shape, while NK cells infected by human immunodeficiency virus exhibited a long and tubular mitochondrial morphology. The discovery of mitochondrial heterogeneity in immune cells has also been reproduced in macrophages, monocytes and T lymphocytes.311,312 Mitochondrial genome sequencing has been applied to immune cells. A pioneering study showed that the mtAtp8 polymorphism increased the adaptive potential of CD4+ T cells when OXPHOS was impaired.313 Further study of the mtAtp8 (m.7778G>T) polymorphism in CD4+ T cells showed a differential cellular respiration profile that led to modified cytokine production in the CD4+ T cells.314 These observations showed that mtDNA mutations affected the immune system, but immune cells still maintained proper functioning despite their high mtDNA mutation load. One possible mechanism may involve mtDNA replication that lags cell proliferation, which is evident in both pro-B and pre-B progenitor cells, because it reduces the number of mtDNA copies per cell and causes a genetic bottleneck.315
Mitochondrial proteomics fuels the study of mitochondrial adaptive changes during the immune response. Mitochondrial proteomics has highlighted the close association between mitochondrial energy metabolism and the innate immune response. Monocytes are key inflammation coordinators and act as direct effectors of innate immunity. A previous study explored the proteome of monocytes in sepsis and revealed that glycolytic proteins showed consistent positive regulation, while TCA and OXPHOS were negatively regulated in the sepsis group, and these differences were largely reversed in the recovery group.316 Macrophages play an important role in pathogen elimination via phagocytosis, in which pathogens are deactivated by the gradual acidification of the phagosome and exposure to mitochondrion-derived ROS.317,318 A recent study on macrophage mitochondria in bacterial infections suggested that OXPHOS was negatively regulated, consistent with increased mitochondrial ROS generation.319 The mitochondrial proteome has also been assessed in adaptive immune cells, such as in T cells during activation.31,320,321 A pioneering study showed that naïve CD4+ T-cell activation induced a unique program of mitochondrial one-carbon metabolism.31 Supplementing cell cultures with exogenous serine and inhibition of the mitochondrial serine catabolic enzyme SHMT2 illustrated the critical role of the mitochondrial one-carbon metabolism pathway in T-cell activation and survival.31 In particular, the activation of aged naïve T cells was enhanced by the addition of products of one-carbon metabolism (formate and glycine).321 Other proteomic research on T cells has also suggested that mitochondria are critical for immune function. Systemic reconstruction of regulatory networks underlying T-cell activation led to the identification of mitochondrial pathways, including mitoribosomes and Complex IV-mediated OXPHOS. T-cell COX10-specific-knockout mice showed greatly elevated cell death rates and impaired cell proliferation.320 In addition to the mitochondrial proteomic study of immune cells, pharmacological and genetic inhibition of other mitochondrial proteins has yielded a broad overview of immune responses.322,323 Acsbg1, a member of the ACSL family, was selectively expressed in Treg cells, and genetic deletion of Acsbg1 not only caused mitochondrial dysfunction but also dampened other metabolic pathways.322 Furthermore, genetic deletion of mitochondrial pyruvate carrier (MPC) drove CD8+ T-cell differentiation toward the acquisition of a memory T-cell phenotype due to increasing glutamine levels and FAO. In contrast, short-term inhibition of MPC in activated T cells enhanced antitumor activity.323
Mitochondrial heterogeneity in cancer
Cancer involves the rapid proliferation of abnormal cells that accumulate into tumors that grow beyond their usual boundaries and can spread to other organs via metastasis.324 Cancer heterogeneity clearly contributes to the plasticity of cancer cells.325 A corollary to this view is that cancer plasticity is mainly affected by the tumor microenvironment via epigenetic modification,326,327 while increasing evidence tends to support the importance of mitochondrial heterogeneity in cancer heterogeneity.328,329 Accumulation of mtDNA mutations during aging has been reported in various tumor types, such as kidney, colorectal and thyroid cancers.330,331 More than 85% of mtDNA obtained from 21 cancer tissues and 38 cancer types carried mutations, with the ND5 gene located in mtDNA the most frequently mutated in most cancer cells and with ND4 frequently mutated in prostate and lung cancers.330 Sequencing of the mtDNA mutation in tumor epithelial cells demonstrated that 44% of mtDNA mutations in adenomas and 85% of mtDNA mutations in adenocarcinomas were tumor specific. Moreover, these mutations have also been observed in the mtDNA of normal crypts, suggesting that mtDNA heterogeneity in normal crypts may provide a selective metabolic advantage during tumorigenesis.331
Studies targeting the mitochondrial proteome have provided solid evidence for cancer mitochondrial heterogeneity (Fig. 7).332 Different mitochondrial proteins have been identified between nasopharyngeal carcinoma (NPC) metastatic and nonmetastatic cell sublines, and these proteins, such as PRDX3 and SOD2, have been associated with ROS and redox pathways.333 Suppression of mitochondrial PRDX3 in the ROS pathway enhanced the mobility potential of NPC metastatic cancer cells.333 Furthermore, as determined via the mitochondrial proteomic analysis of human ovarian cancer (OC) cells compared to drug-resistant cell sublines334,335 or human OC tissues, a series of enzyme profiles associated with the TCA and OXPHOS pathways was significantly different in OC cells compared to platinum-resistant cell sublines.336,337 Some of these enzymes are crucial in tumor progression and maintenance. For example, IDH is overexpressed in OC tissues and plays roles in growth and proliferation, suggesting that mitochondrial heterogeneity contributes to cancer cell drug resistance.338 Intriguingly, therapeutics targeting mitochondrial IDH have been implemented in clinical practice.338 Research on the human OC mitochondrial phosphoproteome led to the identification of 48 differentially phosphorylated mitochondrial proteins between OC tissue and paracarcinoma tissue.339 The mitochondrial proteome profiling of human OC and NPCs has provided ideal models to study therapeutic targets in various cancers.332
Fig. 7.

Mitochondrial heterogeneity in cancer. Glycolysis, a potential protein target of key TCA cycle enzymes, is in purple. Fatty acid metabolism, a potential protein target of FAO key enzymes, is in red. Glutamate metabolism, a potential protein target of glutamine metabolism, is in orange. One-carbon metabolism, potential protein targets of one-carbon metabolism are in orange. Abbreviations: PDH pyruvate dehydrogenase, IDH isocitrate dehydrogenase, CPT1 carnitine palmitoyltransferase 1, CPT2 carnitine palmitoyltransferase 2, GLS glutaminase enzyme, GDH glutamate dehydrogenase, SHMT2 serine hydroxymethyltransferase, 2, GCS glycine cleavage system, MTHFD2 methylenetetrahydrofolate dehydrogenase, and MTHFD1L methylenetetrahydrofolate dehydrogenase 1-like
A critical metabolic phenotype observed in cancer cells is based on the Warburg effect, in which ATP generation shifts from oxidative phosphorylation to glycolysis, even under normal oxygen concentrations.340 It is a long-held view that this cancer cell metabolic reprogramming arises from mitochondrial defects that inhibit the ability to oxidize glucose carbon effectively into carbon dioxide. However, recent research has revealed that most mitochondria in cancer cells are reprogrammed, not subject to functional defects, to function as biosynthetic organelles,341 which play critical roles in both tumorigenesis and metastasis.328,342,343 Regarding tumorigenesis, heterogeneity of the SDH subunit has been observed in hereditary paragangliomas and pheochromocytomas, and SDH defects account for 10–70% of inherited paragangliomas and 10–30% of pheochromocytomas.344 Inhibition of SDH causes the loss of mitochondrial Complex II activity and increased production of mitochondrial ROS that result in the high expression of angiogenic factor in paraganglioma.345 In multiple cutaneous and uterine leiomyomata and in aggressive forms of renal cell cancer, fumarate hydratase (FH) heterogeneity is critical to tumor formation.346 Because FH is a vital enzyme of the TCA cycle, the heterogeneity of FH in renal cancer cells causes a metabolic shift from the TCA cycle to a linear metabolic pathway beginning with glutamine intake and ending with bilirubin excretion in mitochondria.346
In addition, increased mitochondrial redox activities in cancer have been correlated with cancer metastasis.347 Higher mitochondrial membrane potential has been associated with cancer cell survival and invasiveness.348–350 Recent research has revealed that mitochondrial heterogeneity potentiates cancer cell dissemination and metastasis.329,342 In the same host with breast cancer, circulating cancer cells exhibited enhanced mitochondrial biogenesis and respiration compared to cancer cells in the primary tumors,351 and heterogeneity in mitochondrial PHGDH protein promoted breast cancer metastasis.329 In other models of cancer metastasis, such as oral squamous cell carcinoma and melanoma, invading leader cells have been shown to be associated with an increase in mitochondrial membrane potential and translation rate.342
Therapy targeting mitochondrial heterogeneity
Mitochondrial heterogeneity has been under-diagnosed in clinical practice because of insufficient understanding about the mechanism of the mitochondrial abnormalities underlying various disease pathologic condition. The situation makes it a challenge to develop effective therapies. At yet, there is no clinical treatment for the loss-of-function mutation in the mitochondrial genes, but clinically-approved therapies are available to alleviate the symptoms.352,353 Based on the rationale that mitochondrial dysfunction from sustained damage to the organelle’s DNA, proteins and lipids, treatment strategies aimed at oxidative damage, carbon stress and mutation of mitochondrial genes are discussed.
Therapies targeting mitochondrial oxidative damage
Tissues and organs that have high metabolic rate have higher possibility to suffer from oxidative stress, for example cardiovascular system and nervous system. Several therapies aimed at mitochondrial oxidative stress have been implemented. The use of antioxidants to prevent ROS-induced damage has emerged as the prime therapy. MitoQ is a mitochondria-targeted antioxidant, and preclinical studies demonstrate that MitoQ reduces myocardium damage and improves cardiac function in different animal models of cardiomyopathy.354,355 According to preclinical investigations, CoQ10, a component of ETC, attenuates oxidative stress and cardiomyocyte remodeling in rodents with diabetic cardiomyopathy. Treatment of heart failure patients with CoQ10 reduces the number of adverse cardiovascular events and rates of hospitalization and mortality.356 Meanwhile, MitoQ and CoQ10 have been tested in metabolic disease and neurodegeneration disease. MitoQ attenuated weight gain and ameliorates hepatic dysfunction in obese rodents and oxidative stress, synaptic loss and amyloid beta peptide accumulation which is associated with preserved cognitive function in a rodent model of AD. Clinical data suggest that MitoQ, as antioxidant treatment, increase insulin sensitivity in patients with diabetes.357 Other attractive compounds such as Bendavia and Cyclosporine A, that reduce ROS release, may have demonstrated clinical referential value.358,359 Several studies have revealed that inhibited ROS generation shows different aspects in cardiovascular system, immune system and liver alleviated pathologic conditions. Dimethyl malonate (DMM), inhibitor of SDH, has been decreased succinate accumulation and oxidation during heart ischemia and reperfusion (IR).360 Further study has observed DMM to inhibit LPS-induced mtROS generation in inflammatory macrophage and to promote an anti-inflammatory outcome.361 While inhibition of SDH in activated T cells has impaired T cell activation and function.362 These studies revealed that appropriate clinic treatment of mtROS inhibition should be based on the tissue mitochondrial heterogeneity.
Therapies targeting mitochondrial carbon stress
Mitochondrial carbon stress exhibits high level of activated acyl-CoA and the depletion of the mitochondrial NAD+ pool, which is also manifested in pathogenic conditions, such as liver disease, AD, IR and diabetes. Mitochondrial carbon stress may result in protein acylation, the accumulation of misfolded and damaged proteins, and the disruption of protein function and proteostasis.352,353 Clinical therapies targeting mitochondrial carbon stress have also been implemented. A related common pathway of mitochondrial carbon stress is the depletion of NAD+. Therefore, many protective effects have been demonstrated in fatty liver disease, AD and diabetes by administrating compounds such as nicotinamide (NAM), NR, and NMN, whose function by replenishing NAD+ levels and activating sirtuins is to counteract carbon stress.209,363,364 Furthermore, clinical treatments that protect the mitochondria from carbon stress directly by affecting TCA cycle and OXPHOS in pathologic states. As mentioned, SDH, as a target to alleviate pathologic process has been reported in IR and immune response. More TCA cycle enzymes emerged as a target to release carbon stress in pathological conditions have been reported, including PDH and IDH. PDH activated by dichloroacetate in WT heart during IR has been observed to reduce myocardial infarct size. As for the IDH, previous studies have found that IDH-deficient mouse model has shown mitochondrial dysfunction and cardiac hypertrophy.222 Moreover, inhibition of complex I by mitochondria-targeted S-nitrosating agent MitoSNO has been used to decrease ROS production and cardiac IR injury.365 It is likely that many other agents that act in a similar way against type 2 diabetes. Metformin is widely used for the treatment of type 2 diabetes that inhibits complex I, elevates the ADP:ATP ratio and thereby activates liver AMPK to slow liver gluconeogenesis.366 Recent study has revealed that metformin ameliorates acute respiratory distress syndrome by inhibiting complex I through disruption of ATP and mtDNA synthesis.367
In addition, compounds that target mitochondria, such as doxycycline (DOX),368,369 nicotinamide riboside (NR) and olaparib (AZD2281 or AZD), exhibit the ability to enhance UPRmt.370 DOX markedly induces the transcription of UPRmt, increases mitophagy and induces respiration gene expression in GMC101 worms and reduces intracellular Aβ deposits in a human neuroblastoma cell line (SH-SY5Y).370 The treatment of NR and AZD to GMC101 worms also induces the UPRmt and improves lifespan, but the treatment of NR and AZD to CL2122 improves the UPRmt only in aging.370 These studies provide clinical prospect in targeting unfold protein accumulation.
Therapies targeting mitochondrial biogenesis and dynamic
Intercellular and intracellular imbalances of mitochondrial biogenesis and distribution cause pathological states in various tissues and organs.371,372 This kind of heterogeneity has become a potential target of clinical interventions. Instead of directly affecting mitochondria, more alternative therapeutic strategy is to alter mitochondrial amount by enhancing mitochondrial biogenesis and modulating mitochondrial dynamic by inhibiting the protein machinery. In targeting mitochondrial biogenesis, previous studies have demonstrated many drugs interact with mitochondrial biogenesis pathway by altering the activity of transcription factors. These drugs include anti-diabetic drugs pioglitazone and rosiglitazone as well as by the lipid metabolism modifiers thiazolidinediones which increase PGC1α expression levels and enhance mitochondrial biogenesis.373–375 Meanwhile, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), agonist directly activating PGC1α, has been demonstrated to enhance mitochondrial biogenesis by activating PGC1α, resulting in a partial repair of mitochondrial myopathy mice model.376
Small compounds have been developed to target mitochondrial dynamic. Such as Mdivi1, mitochondrial division inhibitor 1, which decreases DRP1 activity can slows mitochondrial fission.377 Previous studies have revealed that Mdivi1 is neuroprotective in neurodegeneration model by inhibiting Drp1-dependent mitochondrial fission.378 But in many cases it still remains unclear of the beneficial effect of modulating mitochondrial dynamic. One of the important aspects of modulating mitochondrial dynamic is that they are intimately linked to the mitochondrial quality control. As for compounds that induce mitophagy to maintain mitochondrial quality, the urolithin A (UA) prevents the accumulation of dysfunctional mitochondria with aging, extends lifespan in C. elegans and increases muscle function in rodents.379 Mitochondrial transfer has been shown to be an attractive prospect that could be a potential therapeutic approach in targeting mitochondrial biogenesis. Previous studies have reported that healthy mitochondria were released from astrocytes into the extracellular space and then were transferred into energetically stressed neurons to maintain neural tissue hemostasis.380
Therapies targeting mitochondrial gene mutation
Correction of mitochondria mutation gene is emerging as a critical intervention approach targeting mitochondrial heterogeneity. In previous studies, mitochondrial-targeted restriction endonucleases or transcription activator-like effector nucleases (TALENs) have been applied to selectively reduced human mutated mtDNA levels responsible for Leber’s hereditary optic neuropathy.381 Recently, mitochondrion-targeted zinc-finger nucleases (mtZEN) have been applied to specifically eliminate the deleterious mtDNA m.5024C>T mutation in vivo throughout the mouse heart.164 In combination with programmable nucleases and tissue-specific adeno-associated viruses, this mtDNA gene-editing tool may offer a potentially universal route for treating mtDNA heterogeneity-related diseases.164 Meanwhile, the mitochondria-targeted meganucleases (mitoARCUS) is equally capable of eliminating mtDNA m.5024C>T mutation throughout the mouse liver and skeleton muscle in vivo, with a relatively small size and the ability to recognize one-base mutation.382 Current studies have achieved to catalyze C•G-to-T•A conversions and A•T -to- G•C conversions in human mtDNA with high target specificity and product purity.163,383 Research have engineered a interbacterial toxin variant, named DddA variant, that catalyzes the deamination of cytidines within dsDNA until brought together on target DNA.163 Fusion of the DddA variant, TALENs and uracil glycosylase inhibitor resulted in RNA-free DddA-derived cytosine base editors (DdCBEs), which catalyzes C•G-to-T•A conversions in human mtDNA, and the application of DdCBEs to disease-associated mtDNA mutations has been shown to induce the recovery of respiration rates and OXPHOS in human cells.163 Further studies have made an fusion of DddA variant, TALENs and an engineered deoxyadenosine deaminase to resulting in catalyzing A•T -to- G•C conversion in human mt DNA.383 These studies have provided prospect in clinical treatment of disease-causing mtDNA mutation. However, mitochondrial genome editing efficiency is not yet sufficient for application in clinical therapies.383 On the other hand, studies have revealed that mitochondrion-targeted genetic editing may induce extensive off-target changes in the nuclear genome,384 which might restrict the application of mitochondrial DNA base editors.
Conclusion
Mitochondrial biology has been extensively studied using traditional biochemical and molecular methods for decades. Technological advances have driven a steep change in our understanding of mitochondrial biology. Systematic studies on mitochondrial multi-omics have led to the discovery of mitochondrial heterogeneity in different cells and tissues (Fig. 8). However, we still know very little about the molecular mechanisms involved, especially the complexity of the mitochondrial genome. A mtDNA phenotype that is neutral in one context may be deleterious in others. Thus, understanding mtDNA-specific heteroplasmic variants’ behavior is extremely important for clinical studies designed to “clear up” pathogenic mtDNA mutations. In the rapidly evolving field of mitochondrial proteomics, the comprehensive characterization of the mitochondrial protein inventory offers exciting opportunities for systematic analysis of this organelle underneath physiological and pathological conditions. Hundreds of mitochondrial proteins that do not appear in the current catalog will likely be identified via a combination of high-throughput and traditional biochemical strategies, resulting in a complete mitochondrial proteome profiling. It will then be important to understand the extent of splicing variants and PTMs of all these proteins, as well as their specific localization in mitochondria. An advanced challenge regarding the mitochondrial proteome is then to understand how mitochondrial proteins function together in pathways and complexes. Using high-throughput methods such as RNAi, protein‒protein interaction mapping and computational prediction, most of the uncharacterized proteins will be annotated.
Fig. 8.

Heterogeneity of mitochondrial proteins in tissues. The mitochondrial protein expression data were obtained from MitoCarta 3.0 (http://www.broadinstitute.org/pubs/MitoCarta). a Mitochondrial protein expression in 14 human tissues as determined with MitoPathways. Columns represent different tissues, and rows represent the relative expression levels of different proteins. The R package (pheatmap) was used to draw the heatmap. b Mitochondrial protein expression of MitoPathways in 14 mouse tissues. Columns represent different tissues, and rows represent the relative expression levels of different proteins. The R package (pheatmap) was used to draw the heatmap. c Number of distinct proteins detected and not detected in 14 human tissues. d Number of distinct proteins detected and not detected in 14 mouse tissues. e Heatmap showing mitochondrial protein heterogeneity in 6 tissues. Maroon represents upregulated proteins in tissues under pathological conditions, and blue represents downregulated proteins in tissues under pathological conditions
As the protein inventory and complexes of mitochondria are refined, it will become important to describe their heterogeneity in different tissues, developmental states and diseases. The first step in the current characterization of mitochondrial heterogeneity is the genetic study of rare and common variants of the mitochondrial proteome. Because of next-generation sequencing technologies, large projects, such as the 1000 Genomes Project, will soon catalog the range of normal mitochondrial genomic variants. Additionally, resequencing of individuals with extreme mitochondrial phenotypes may reveal an additional set of high-propensity variants. However, establishing genetic links between specific extreme phenotypes and mitochondrial gene mutations remains very challenging. To study the molecular mechanisms of mitochondrial function adaptation and related alterations in different tissues, developmental states and diseases, it is important to obtain high-resolution mitochondrial proteome data and a method to target mitochondria. However, multi-omics technique also presents some limitations. First, due to the limitation of MS-based sequencing, the mitochondrial proteome might miss some bona fide mitochondrial proteins, especially proteins less than 10 kDa or with unfavorable proteolytic cleavage sites.49 Second, it is still a challenge to identify proteins that are in mitochondria under specific conditions.49
Supplementary information
Dataset 1,Dataset 2,Dataset 3, Dataset 4
Acknowledgements
This study was performed with the support of the National Natural Science Foundation of China (82002339, 81820108020), Shanghai Frontiers Science Center of Degeneration and Regeneration in Skeletal System (BJ1-9000-22-4002), Shanghai Municipal Hospital Orthopedic Specialist Alliance, and Shanghai Municipal Health Commission key priority discipline project; Shanghai Spinal Disease and Trauma Orthopedics Research Center.
Author contributions
L.C. drafted and conceived the initial manuscript. M.Z. performed the bioinformatics analysis. H.L., D.L., P.L., Y.Z., provided the essential assistant for our final manuscript. J.G., W.Z., C.Z., and L.C. provided the essential ideas for this work. J.G., and L.C. drew the figures and arranged the tables. J.G., Z.W., and C.Z. revised the manuscript. All authors have read and approved the article.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Long Chen, Mengnan Zhou, Hao Li
Contributor Information
Changqing Zhang, Email: zhangcq@sjtu.edu.cn.
Weiguo Zou, Email: zouwg94@sibcb.ac.cn.
Junjie Gao, Email: colingjj@163.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41392-023-01546-w.
References
- 1.Friedman JR, et al. Mitochondrial form and function. Nature. 2014;505:335–343. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sierra MF, et al. Assembly of the mitochondrial system. Purification of a mitochondrial product of the ATPase. PNAS. 1973;70:3155–3159. doi: 10.1073/pnas.70.11.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfanner N, et al. Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019;20:267–284. doi: 10.1038/s41580-018-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zorov DB, et al. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sena LA, et al. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HE, et al. Lipid biosynthesis coordinates a mitochondrial-to-cytosolic stress response. Cell. 2016;166:1539–1552.e1516. doi: 10.1016/j.cell.2016.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martensson CU, et al. Acylglycerol kinase: mitochondrial protein transport meets lipid biosynthesis. Trends Cell Biol. 2017;27:700–702. doi: 10.1016/j.tcb.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Zheng Y, et al. Mitochondrial one-carbon pathway supports cytosolic folate integrity in cancer cells. Cell. 2018;175:1546–1560.e1517. doi: 10.1016/j.cell.2018.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Reyes I, et al. Mitochondrial one-carbon metabolism maintains redox balance during hypoxia. Cancer Discov. 2014;4:1371–1373. doi: 10.1158/2159-8290.CD-14-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu C, et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 2020;11:940. doi: 10.1038/s41419-020-03152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ouellet M, et al. A novel algorithm identifies stress-induced alterations in mitochondrial connectivity and inner membrane structure from confocal images. PLoS Comput. Biol. 2017;13:e1005612. doi: 10.1371/journal.pcbi.1005612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ban T, et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017;19:856–863. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- 13.Modi S, et al. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun. 2019;10:4399. doi: 10.1038/s41467-019-12382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaurov I, et al. The diverged trypanosome MICOS complex as a hub for mitochondrial cristae shaping and protein import. Curr. Biol. 2018;28:3393–3407.e3395. doi: 10.1016/j.cub.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Galloway CA, et al. Mitochondrial morphology in metabolic diseases. Antioxid. Redox Signal. 2013;19:415–430. doi: 10.1089/ars.2012.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah SI, et al. Mitochondrial fragmentation and network architecture in degenerative diseases. PLoS One. 2019;14:e0223014. doi: 10.1371/journal.pone.0223014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rafelski SM, et al. Mitochondrial network size scaling in budding yeast. Science. 2012;338:822–824. doi: 10.1126/science.1225720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng W, et al. Mitochondria-lysosome contacts regulate mitochondrial Ca dynamics via lysosomal TRPML1. PNAS. 2020;117:19266–19275. doi: 10.1073/pnas.2003236117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim D, et al. Ca(2+) handling at the mitochondria-ER contact sites in neurodegeneration. Cell Calcium. 2021;98:102453. doi: 10.1016/j.ceca.2021.102453. [DOI] [PubMed] [Google Scholar]
- 20.Parakh S, et al. The mitochondrial-associated ER membrane (MAM) compartment and its dysregulation in Amyotrophic Lateral Sclerosis (ALS) Semin. Cell Dev. Biol. 2021;112:105–113. doi: 10.1016/j.semcdb.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Giacomello M, et al. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 2016;23:1417–1427. doi: 10.1038/cdd.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng W, et al. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. PNAS. 2020;117:19266–19275. doi: 10.1073/pnas.2003236117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crimi M, et al. The mitochondrial genome, a growing interest inside an organelle. Int. J. Nanomed. 2008;3:51–57. doi: 10.2147/ijn.s2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Souza AR, et al. Mitochondrial transcription and translation: overview. Essays Biochem. 2018;62:309–320. doi: 10.1042/EBC20170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kummer E, et al. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol. 2021;22:307–325. doi: 10.1038/s41580-021-00332-2. [DOI] [PubMed] [Google Scholar]
- 26.Wiedemann N, et al. Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 2017;86:685–714. doi: 10.1146/annurev-biochem-060815-014352. [DOI] [PubMed] [Google Scholar]
- 27.Neupert W. A perspective on transport of proteins into mitochondria: a myriad of open questions. J. Mol. Biol. 2015;427:1135–1158. doi: 10.1016/j.jmb.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Zhou B, et al. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018;128:3716–3726. doi: 10.1172/JCI120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sunny NE, et al. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 2017;28:250–260. doi: 10.1016/j.tem.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Field CS, et al. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for treg suppressive function. Cell Metab. 2020;31:422–437.e425. doi: 10.1016/j.cmet.2019.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ron-Harel N, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 2016;24:104–117. doi: 10.1016/j.cmet.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malpartida AB, et al. Mitochondrial dysfunction and mitophagy in parkinson’s disease: from mechanism to therapy. Trends Biochem. Sci. 2021;46:329–343. doi: 10.1016/j.tibs.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Pickles S, et al. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018;28:R170–R185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu NN, et al. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid. Med. Cell. Longev. 2019;2019:9825061. doi: 10.1155/2019/9825061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neuspiel M, et al. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008;18:102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 36.Towers CG, et al. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev. Cell. 2021;56:2029–2042.e2025. doi: 10.1016/j.devcel.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soubannier V, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 38.Jiao H, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021;184:2896–2910 e2813. doi: 10.1016/j.cell.2021.04.027. [DOI] [PubMed] [Google Scholar]
- 39.Bao F, et al. Mitolysosome exocytosis, a mitophagy-independent mitochondrial quality control in flunarizine-induced parkinsonism-like symptoms. Sci. Adv. 2022;8:eabk2376. doi: 10.1126/sciadv.abk2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng Y, et al. Mdivi-1, a mitochondrial fission inhibitor, reduces angiotensin-II- induced hypertension by mediating VSMC phenotypic switch. Biomed. Pharmacother. 2021;140:111689. doi: 10.1016/j.biopha.2021.111689. [DOI] [PubMed] [Google Scholar]
- 41.Brandt JT, et al. The structure of rat liver mitochondria: a reevaluation. Biochem. Biophys. Res. Commun. 1974;59:1097–1104. doi: 10.1016/s0006-291x(74)80091-4. [DOI] [PubMed] [Google Scholar]
- 42.Collins TJ, et al. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bibb MJ, et al. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26:167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- 44.Stewart JB, et al. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 2021;22:106–118. doi: 10.1038/s41576-020-00284-x. [DOI] [PubMed] [Google Scholar]
- 45.Taylor SW, et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 46.Lung B, et al. Identification of small non-coding RNAs from mitochondria and chloroplasts. Nucleic Acids Res. 2006;34:3842–3852. doi: 10.1093/nar/gkl448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiebish MA, et al. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J. Neurochem. 2008;106:299–312. doi: 10.1111/j.1471-4159.2008.05383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao Q, et al. Targeting mitochondria-located circRNA SCAR alleviates NASH via reducing mROS output. Cell. 2020;183:76–93.e22. doi: 10.1016/j.cell.2020.08.009. [DOI] [PubMed] [Google Scholar]
- 49.Morgenstern M, et al. Quantitative high-confidence human mitochondrial proteome and its dynamics in cellular context. Cell Metab. 2021;33:2464–2483.e2418. doi: 10.1016/j.cmet.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X, et al. Multi-regional sequencing reveals intratumor heterogeneity and positive selection of somatic mtDNA mutations in hepatocellular carcinoma and colorectal cancer. Int. J. Cancer. 2018;143:1143–1152. doi: 10.1002/ijc.31395. [DOI] [PubMed] [Google Scholar]
- 51.Mercer TR, et al. The human mitochondrial transcriptome. Cell. 2011;146:645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He Q, et al. Tissue-specific expression atlas of murine mitochondrial tRNAs. J. Biol. Chem. 2021;297:100960. doi: 10.1016/j.jbc.2021.100960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gomez-Valades AG, et al. Mitochondrial cristae-remodeling protein OPA1 in POMC neurons couples Ca(2+) homeostasis with adipose tissue lipolysis. Cell Metab. 2021;33:1820–1835 e1829. doi: 10.1016/j.cmet.2021.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heine KB, et al. Mitochondrial behaviour, morphology, and animal performance. Biol. Rev. Camb. Philos. Soc. 2020;95:730–737. doi: 10.1111/brv.12584. [DOI] [PubMed] [Google Scholar]
- 55.Choi J, et al. Comparative analysis of the mitochondrial morphology, energy metabolism, and gene expression signatures in three types of blastocyst-derived stem cells. Redox Biol. 2020;30:101437. doi: 10.1016/j.redox.2020.101437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Z, et al. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 57.Giedt RJ, et al. Computational imaging reveals mitochondrial morphology as a biomarker of cancer phenotype and drug response. Sci. Rep. 2016;6:32985. doi: 10.1038/srep32985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kleele T, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. 2021;593:435–439. doi: 10.1038/s41586-021-03510-6. [DOI] [PubMed] [Google Scholar]
- 59.Kojima R, et al. Maintenance of cardiolipin and crista structure requires cooperative functions of mitochondrial dynamics and phospholipid transport. Cell Rep. 2019;26:518–528.e516. doi: 10.1016/j.celrep.2018.12.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacob KJ, et al. Mitochondrial content, but not function, is altered with a multimodal resistance training protocol and adequate protein intake in leucine-supplemented pre/frail women. Front. Nutr. 2020;7:619216. doi: 10.3389/fnut.2020.619216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bulthuis EP, et al. Mitochondrial morphofunction in mammalian cells. Antioxid. Redox Signal. 2019;30:2066–2109. doi: 10.1089/ars.2018.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedman JR, et al. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wong YC, et al. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 2018;554:382–386. doi: 10.1038/nature25486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nasrallah CM, et al. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014;10:650–658. doi: 10.1038/nrendo.2014.160. [DOI] [PubMed] [Google Scholar]
- 65.Burte F, et al. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurosci. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- 66.Ni HM, et al. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi: 10.1016/j.redox.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kraus F, et al. Function and regulation of the divisome for mitochondrial fission. Nature. 2021;590:57–66. doi: 10.1038/s41586-021-03214-x. [DOI] [PubMed] [Google Scholar]
- 68.Korobova F, et al. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–467. doi: 10.1126/science.1228360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mears JA, et al. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2011;18:20–26. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee JE, et al. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. 2016;540:139–143. doi: 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Youle RJ, et al. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen H, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chandhok G, et al. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 2018;93:933–949. doi: 10.1111/brv.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li M, et al. Mitochondrial fusion via OPA1 and MFN1 supports liver tumor cell metabolism and growth. Cells. 2020;9:121. doi: 10.3390/cells9010121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li C, et al. STING1 promotes ferroptosis through MFN1/2-dependent mitochondrial fusion. Front. Cell Dev. Biol. 2021;9:698679. doi: 10.3389/fcell.2021.698679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park H, et al. Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J. Clin. Investig. 2019;129:694–711. doi: 10.1172/JCI120606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boutant M, et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017;36:1543–1558. doi: 10.15252/embj.201694914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Song M, et al. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 2017;26:872–883.e875. doi: 10.1016/j.cmet.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhatia D, et al. Conditional deletion of myeloid-specific mitofusin 2 but not mitofusin 1 promotes kidney fibrosis. Kidney Int. 2022;101:963–986. doi: 10.1016/j.kint.2022.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shirakabe A, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload–induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ding M, et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1alpha pathway. J. Pineal Res. 2018;65:e12491. doi: 10.1111/jpi.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Favaro G, et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019;10:2576. doi: 10.1038/s41467-019-10226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang J, et al. Inhibition of Drp1 SUMOylation by ALR protects the liver from ischemia-reperfusion injury. Cell Death Differ. 2021;28:1174–1192. doi: 10.1038/s41418-020-00641-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deng L, et al. MFN2 knockdown promotes osteogenic differentiation of iPSC-MSCs through aerobic glycolysis mediated by the Wnt/beta-catenin signaling pathway. Stem Cell Res. Ther. 2022;13:162. doi: 10.1186/s13287-022-02836-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cogliati S, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu W, et al. Chchd2 regulates mitochondrial morphology by modulating the levels of Opa1. Cell Death Differ. 2020;27:2014–2029. doi: 10.1038/s41418-019-0482-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peralta S, et al. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell. Sci. 2018;131:jcs217075. doi: 10.1242/jcs.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arguello T, et al. ATAD3A has a scaffolding role regulating mitochondria inner membrane structure and protein assembly. Cell Rep. 2021;37:110139. doi: 10.1016/j.celrep.2021.110139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Genin EC, et al. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 2016;8:58–72. doi: 10.15252/emmm.201505496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dietz JV, et al. Mitochondrial contact site and cristae organizing system (MICOS) machinery supports heme biosynthesis by enabling optimal performance of ferrochelatase. Redox Biol. 2021;46:102125. doi: 10.1016/j.redox.2021.102125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ogando J, et al. PD-1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8(+) T lymphocytes. J. Cell. Physiol. 2019;7:151. doi: 10.1186/s40425-019-0628-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Latorre-Muro P, et al. A cold-stress-inducible PERK/OGT axis controls TOM70-assisted mitochondrial protein import and cristae formation. Cell Metab. 2021;33:598–614 e597. doi: 10.1016/j.cmet.2021.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Veltri KL, et al. Distinct genomic copy number in mitochondria of different mammalian organs. J. Cell. Physiol. 1990;143:160–164. doi: 10.1002/jcp.1041430122. [DOI] [PubMed] [Google Scholar]
- 94.Barth E, et al. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992;24:669–681. doi: 10.1016/0022-2828(92)93381-s. [DOI] [PubMed] [Google Scholar]
- 95.Schaper J, et al. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985;56:377–391. doi: 10.1161/01.res.56.3.377. [DOI] [PubMed] [Google Scholar]
- 96.Szeto HH, et al. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016;90:997–1011. doi: 10.1016/j.kint.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 97.Shami GJ, et al. Three-dimensional ultrastructure of giant mitochondria in human non-alcoholic fatty liver disease. Sci. Rep. 2021;11:3319. doi: 10.1038/s41598-021-82884-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Larsen S, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012;590:3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kopecky J, et al. Mitochondrial uncoupling and lipid metabolism in adipocytes. Biochem. Soc. Trans. 2001;29:791–797. doi: 10.1042/bst0290791. [DOI] [PubMed] [Google Scholar]
- 100.Burke PJ. Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer. 2017;3:857–870. doi: 10.1016/j.trecan.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kalyanaraman B, et al. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018;14:316–327. doi: 10.1016/j.redox.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tan YQ, et al. Mitochondria: the metabolic switch of cellular oncogenic transformation. Biochim Biophys. Acta Rev. Cancer. 2021;1876:188534. doi: 10.1016/j.bbcan.2021.188534. [DOI] [PubMed] [Google Scholar]
- 103.Pollister, A. W. Mitochondrial orientations and molecular patterns. Physiol. Zool.14, 268–280 (1941).
- 104.Christiansen EG. Orientation of the mitochondria during mitosis. Nature. 1949;163:361. doi: 10.1038/163361a0. [DOI] [PubMed] [Google Scholar]
- 105.Moore AS, et al. Actin cables and comet tails organize mitochondrial networks in mitosis. Nature. 2021;591:659–664. doi: 10.1038/s41586-021-03309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jaberi E, et al. Identification of unique and shared mitochondrial DNA mutations in neurodegeneration and cancer by single-cell mitochondrial DNA structural variation sequencing (MitoSV-seq) EBioMedicine. 2020;57:102868. doi: 10.1016/j.ebiom.2020.102868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gorman GS, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015;77:753–759. doi: 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McWilliams TG, et al. Mitochondrial DNA can be inherited from fathers, not just mothers. Nature. 2019;565:296–297. doi: 10.1038/d41586-019-00093-1. [DOI] [PubMed] [Google Scholar]
- 109.Luo S, et al. Biparental inheritance of mitochondrial DNA in humans. PNAS. 2018;115:13039–13044. doi: 10.1073/pnas.1810946115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rackham O, et al. Organization and expression of the mammalian mitochondrial genome. Nat. Rev. Genet. 2022;23:606–623. doi: 10.1038/s41576-022-00480-x. [DOI] [PubMed] [Google Scholar]
- 111.Bogenhagen DF, Mitochondrial DNA. nucleoid structure. Biochim. Biophys. Acta. 2012;1819:914–920. doi: 10.1016/j.bbagrm.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 112.Fontana GA, et al. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020;48:11244–11258. doi: 10.1093/nar/gkaa804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ju YS, et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife. 2014;3:e02935. doi: 10.7554/eLife.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Perez-Trevino P, et al. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165761. doi: 10.1016/j.bbadis.2020.165761. [DOI] [PubMed] [Google Scholar]
- 115.Korr H, et al. Mitochondrial DNA synthesis studied autoradiographically in various cell types in vivo. BJMBR. 1998;31:289–298. doi: 10.1590/s0100-879x1998000200012. [DOI] [PubMed] [Google Scholar]
- 116.Gross NJ, et al. Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J. Biol. Chem. 1969;244:1552–1562. [PubMed] [Google Scholar]
- 117.Graziewicz MA, et al. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem. Rev. 2006;106:383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 118.Allio R, et al. Large variation in the ratio of mitochondrial to nuclear mutation rate across animals: implications for genetic diversity and the use of mitochondrial DNA as a molecular marker. Mol. Biol. Evol. 2017;34:2762–2772. doi: 10.1093/molbev/msx197. [DOI] [PubMed] [Google Scholar]
- 119.Liu X, et al. Mitochondria encoded non-coding RNAs in cell physiology. Front. Cell Dev. Biol. 2021;9:713729. doi: 10.3389/fcell.2021.713729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu X, et al. Identification of mecciRNAs and their roles in the mitochondrial entry of proteins. Sci. China Life Sci. 2020;63:1429–1449. doi: 10.1007/s11427-020-1631-9. [DOI] [PubMed] [Google Scholar]
- 121.Wu Z, et al. Mitochondrial Genome-Derived circRNA mc-COX2 Functions as an Oncogene in Chronic Lymphocytic Leukemia. Mol. Ther. Nucleic Acids. 2020;20:801–811. doi: 10.1016/j.omtn.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jusic A, et al. Mitochondrial noncoding RNA-regulatory network in cardiovascular disease. Basic Res. Cardiol. 2020;115:23. doi: 10.1007/s00395-020-0783-5. [DOI] [PubMed] [Google Scholar]
- 123.Dhir A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018;560:238–242. doi: 10.1038/s41586-018-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang KC, et al. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 2014;129:1009–1021. doi: 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gao S, et al. Two novel lncRNAs discovered in human mitochondrial DNA using PacBio full-length transcriptome data. Mitochondrion. 2018;38:41–47. doi: 10.1016/j.mito.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 126.Schmidt O, et al. Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 127.Merkley ED, et al. The succinated proteome. J. Mass Spectrom. 2014;33:98–109. doi: 10.1002/mas.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Baeza J, et al. Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem. Sci. 2016;41:231–244. doi: 10.1016/j.tibs.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rath S, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021;49:D1541–D1547. doi: 10.1093/nar/gkaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fecher C, et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019;22:1731–1742. doi: 10.1038/s41593-019-0479-z. [DOI] [PubMed] [Google Scholar]
- 131.Mootha VK, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 132.Peng Y, et al. Post-translational modifications on mitochondrial metabolic enzymes in cancer. Free Radic. Biol. Med. 2022;179:11–23. doi: 10.1016/j.freeradbiomed.2021.12.264. [DOI] [PubMed] [Google Scholar]
- 133.Grimsrud PA, et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 2012;16:672–683. doi: 10.1016/j.cmet.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bak S, et al. Tissue specific phosphorylation of mitochondrial proteins isolated from rat liver, heart muscle, and skeletal muscle. J. Proteome Res. 2013;12:4327–4339. doi: 10.1021/pr400281r. [DOI] [PubMed] [Google Scholar]
- 135.Horton JL, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016;2:e84897. doi: 10.1172/jci.insight.84897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schmidt O, et al. Regulation of mitochondrial protein import by cytosolic kinases. Cell. 2011;144:227–239. doi: 10.1016/j.cell.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 137.Sinha D, et al. Unraveling the intricate organization of mammalian mitochondrial presequence translocases: existence of multiple translocases for maintenance of mitochondrial function. Mol. Biol. Cell. 2014;34:1757–1775. doi: 10.1128/MCB.01527-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hirschey MD, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Scharwey M, et al. Mitochondrial lipid transport at a glance. J. Cell. Sci. 2013;126:5317–5323. doi: 10.1242/jcs.134130. [DOI] [PubMed] [Google Scholar]
- 140.Mitchell TW, et al. Membrane phospholipid composition may contribute to exceptional longevity of the naked mole-rat (Heterocephalus glaber): a comparative study using shotgun lipidomics. Exp. Gerontol. 2007;42:1053–1062. doi: 10.1016/j.exger.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 141.Albert CJ, et al. Myocardial lipidomics. Developments in myocardial nuclear lipidomics. Front Biosci. 2007;12:2750–2760. doi: 10.2741/2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhanghao K, et al. High-dimensional super-resolution imaging reveals heterogeneity and dynamics of subcellular lipid membranes. Nat. Commun. 2020;11:5890. doi: 10.1038/s41467-020-19747-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rieusset J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death Dis. 2018;9:388. doi: 10.1038/s41419-018-0416-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Luo J, et al. Intracellular cholesterol transport by sterol transfer proteins at membrane contact sites. Trends Biochem. Sci. 2019;44:273–292. doi: 10.1016/j.tibs.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 145.Encinar Del Dedo J, et al. Coupled sterol synthesis and transport machineries at ER-endocytic contact sites. J. Cell Biol. 2021;220:e202010016. doi: 10.1083/jcb.202010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Miliara X, et al. Structural insight into the TRIAP1/PRELI-like domain family of mitochondrial phospholipid transfer complexes. EMBO Rep. 2015;16:824–835. doi: 10.15252/embr.201540229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Park MK, et al. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001;20:1863–1874. doi: 10.1093/emboj/20.8.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hollenbeck PJ, et al. The axonal transport of mitochondria. J. Cell. Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kuznetsov AV, et al. Subcellular heterogeneity of mitochondrial function and dysfunction: evidence obtained by confocal imaging. Mol. Cell. Biochem. 2004;256-257:359–365. doi: 10.1023/b:mcbi.0000009881.01943.68. [DOI] [PubMed] [Google Scholar]
- 150.Kuznetsov AV, et al. Functional imaging of mitochondria in saponin-permeabilized mice muscle fibers. J. Cell Biol. 1998;140:1091–1099. doi: 10.1083/jcb.140.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lai JC, et al. Synaptic and non-synaptic mitochondria from rat brain: isolation and characterization. J. Neurochem. 1977;28:625–631. doi: 10.1111/j.1471-4159.1977.tb10434.x. [DOI] [PubMed] [Google Scholar]
- 152.Brown MR, et al. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J. Biol. Chem. 2006;281:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- 153.Stauch KL, et al. Quantitative proteomics of synaptic and nonsynaptic mitochondria: insights for synaptic mitochondrial vulnerability. J. Proteome Res. 2014;13:2620–2636. doi: 10.1021/pr500295n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Maeda R, et al. High throughput single cell analysis of mitochondrial heteroplasmy in mitochondrial diseases. Sci. Rep. 2020;10:10821. doi: 10.1038/s41598-020-67686-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Faitg J, et al. 3D neuronal mitochondrial morphology in axons, dendrites, and somata of the aging mouse hippocampus. Cell Rep. 2021;36:109509. doi: 10.1016/j.celrep.2021.109509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Völgyi K, et al. Synaptic mitochondria: a brain mitochondria cluster with a specific proteome. J. Proteom. 2015;120:142–157. doi: 10.1016/j.jprot.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 157.Graham LC, et al. Proteomic profiling of neuronal mitochondria reveals modulators of synaptic architecture. Mol. Neurodegener. 2017;12:77. doi: 10.1186/s13024-017-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kristian T, et al. Heterogeneity of the calcium-induced permeability transition in isolated non-synaptic brain mitochondria. J. Neurochem. 2002;83:1297–1308. doi: 10.1046/j.1471-4159.2002.01238.x. [DOI] [PubMed] [Google Scholar]
- 159.Zorov DB, et al. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pekkurnaz G, et al. Mitochondrial heterogeneity and homeostasis through the lens of a neuron. Nat. Metab. 2022;4:802–812. doi: 10.1038/s42255-022-00594-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Benador IY, et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 2018;27:869–885 e866. doi: 10.1016/j.cmet.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ruiz-Pesini E, et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35:D823–D828. doi: 10.1093/nar/gkl927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mok BY, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583:631–637. doi: 10.1038/s41586-020-2477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gammage PA, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018;24:1691–1695. doi: 10.1038/s41591-018-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rensvold JW, et al. Defining mitochondrial protein functions through deep multiomic profiling. Nature. 2022;606:382–388. doi: 10.1038/s41586-022-04765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dittenhafer-Reed KE, et al. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 2015;21:637–646. doi: 10.1016/j.cmet.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Calvo SE, et al. The mitochondrial proteome and human disease. Annu Rev. Genomics Hum. Genet. 2010;11:25–44. doi: 10.1146/annurev-genom-082509-141720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mergenthaler P, et al. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–597. doi: 10.1016/j.tins.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang S, et al. Glucose metabolic crosstalk and regulation in brain function and diseases. Prog. Neurobiol. 2021;204:102089. doi: 10.1016/j.pneurobio.2021.102089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hamalainen RH, et al. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243 A > G mutation in human induced pluripotent stem cell-derived disease model. PNAS. 2013;110:E3622–E3630. doi: 10.1073/pnas.1311660110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lorenz C, et al. Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell. 2017;20:659–674 e659. doi: 10.1016/j.stem.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 172.Weiduschat N, et al. Cerebral metabolic abnormalities in A3243G mitochondrial DNA mutation carriers. Neurology. 2014;82:798–805. doi: 10.1212/WNL.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kukreja L, et al. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2014;9:16. doi: 10.1186/1750-1326-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Reeve A, et al. The impact of pathogenic mitochondrial DNA mutations on substantia nigra neurons. J. Neurosci. Res. 2013;33:10790–10801. doi: 10.1523/JNEUROSCI.3525-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Donega V, et al. Transcriptome and proteome profiling of neural stem cells from the human subventricular zone in Parkinson’s disease. Acta Neuropathol. Commun. 2019;7:84. doi: 10.1186/s40478-019-0736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Thelen MP, et al. Mitochondrial defects in the respiratory complex I contribute to impaired translational initiation via ROS and energy homeostasis in SMA motor neurons. Acta Neuropathol. Commun. 2020;8:223. doi: 10.1186/s40478-020-01101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Goldsmith J, et al. Proteomic profiling shows mitochondrial nucleoids are autophagy cargo in neurons: implications for neuron maintenance and neurodegenerative disease. Autophagy. 2022;18:2003–2005. doi: 10.1080/15548627.2022.2056865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sheu KF, et al. Abnormality of the alpha-ketoglutarate dehydrogenase complex in fibroblasts from familial Alzheimer’s disease. Ann. Neurol. 1994;35:312–318. doi: 10.1002/ana.410350311. [DOI] [PubMed] [Google Scholar]
- 179.Mizuno Y, et al. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann. Neurol. 1994;35:204–210. doi: 10.1002/ana.410350212. [DOI] [PubMed] [Google Scholar]
- 180.Ko LW, et al. Selective loss of KGDHC-enriched neurons in Alzheimer temporal cortex: does mitochondrial variation contribute to selective vulnerability? J. Mol. Neurosci. 2001;17:361–369. doi: 10.1385/JMN:17:3:361. [DOI] [PubMed] [Google Scholar]
- 181.Holmquist L, et al. Lipoic acid as a novel treatment for Alzheimer’s disease and related dementias. Pharmacol. Ther. 2007;113:154–164. doi: 10.1016/j.pharmthera.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 182.Walker JM, et al. Differential protein expression in the hippocampi of resilient individuals identified by digital spatial profiling. Acta Neuropathol. Commun. 2022;10:23. doi: 10.1186/s40478-022-01324-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Area-Gomez E, et al. Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J. Clin. Investig. 2019;129:34–45. doi: 10.1172/JCI120848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Koopman WJ, et al. OXPHOS mutations and neurodegeneration. EMBO J. 2013;32:9–29. doi: 10.1038/emboj.2012.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kawamata H, et al. Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J. Cell Biol. 2017;216:3917–3929. doi: 10.1083/jcb.201709172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dias V, et al. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013;3:461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nissanka N, et al. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018;592:728–742. doi: 10.1002/1873-3468.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Simpson DSA, et al. ROS generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants. 2020;9:743. doi: 10.3390/antiox9080743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Francis BM, et al. Reduced levels of mitochondrial complex I subunit NDUFB8 and linked complex I + III oxidoreductase activity in the TgCRND8 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2014;39:347–355. doi: 10.3233/JAD-131499. [DOI] [PubMed] [Google Scholar]
- 190.Zuliani I, et al. High-fat diet leads to reduced protein O-GlcNAcylation and mitochondrial defects promoting the development of Alzheimer’s disease signatures. Int. J. Mol. Sci. 2021;22:3746. doi: 10.3390/ijms22073746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Beck SJ, et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016;7:11483. doi: 10.1038/ncomms11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Gauba E, et al. Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis. 2019;121:138–147. doi: 10.1016/j.nbd.2018.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Gauba E, et al. Modulation of OSCP mitigates mitochondrial and synaptic deficits in a mouse model of Alzheimer’s pathology. Neurobiol. Aging. 2021;98:63–77. doi: 10.1016/j.neurobiolaging.2020.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nakamura T, et al. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener. 2009;4:35. doi: 10.1186/1750-1326-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tracy TE, et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell. 2022;185:712–728 e714. doi: 10.1016/j.cell.2021.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gonzalez-Rodriguez P, et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature. 2021;599:650–656. doi: 10.1038/s41586-021-04059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Demarest TG, et al. Biological sex and DNA repair deficiency drive Alzheimer’s disease via systemic metabolic remodeling and brain mitochondrial dysfunction. Acta Neuropathol. 2020;140:25–47. doi: 10.1007/s00401-020-02152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fais M, et al. Parkinson’s disease-related genes and lipid alteration. Int. J. Mol. Sci. 2021;22:7630. doi: 10.3390/ijms22147630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ding F, et al. Ovariectomy induces a shift in fuel availability and metabolism in the hippocampus of the female transgenic model of familial Alzheimer’s. PLoS One. 2013;8:e59825. doi: 10.1371/journal.pone.0059825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Virmani A, et al. The carnitine palmitoyl transferase (CPT) system and possible relevance for neuropsychiatric and neurological conditions. Mol. Neurobiol. 2015;52:826–836. doi: 10.1007/s12035-015-9238-7. [DOI] [PubMed] [Google Scholar]
- 201.Reamy AA, et al. Carnitine palmitoyltransferase-1c gain-of-function in the brain results in postnatal microencephaly. J. Neurochem. 2011;118:388–398. doi: 10.1111/j.1471-4159.2011.07312.x. [DOI] [PubMed] [Google Scholar]
- 202.Carrasco P, et al. Ceramide levels regulated by carnitine palmitoyltransferase 1 C control dendritic spine maturation and cognition. J. Biol. Chem. 2012;287:21224–21232. doi: 10.1074/jbc.M111.337493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ding Y, et al. Carnitine palmitoyltransferase 1 (CPT1) alleviates oxidative stress and apoptosis of hippocampal neuron in response to beta-Amyloid peptide fragment Abeta(25-35) Bioengineered. 2021;12:5440–5449. doi: 10.1080/21655979.2021.1967032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Giovannoni MP, et al. Selective ACAT inhibitors as promising antihyperlipidemic, antiathero-sclerotic and anti-Alzheimer drugs. Mini Rev. Med Chem. 2003;3:576–584. doi: 10.2174/1389557033487890. [DOI] [PubMed] [Google Scholar]
- 205.Huttunen HJ, et al. Knockdown of ACAT-1 reduces amyloidogenic processing of APP. FEBS Lett. 2007;581:1688–1692. doi: 10.1016/j.febslet.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bryleva EY, et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. PNAS. 2010;107:3081–3086. doi: 10.1073/pnas.0913828107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Murphy SR, et al. Acat1 knockdown gene therapy decreases amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Ther. 2013;21:1497–1506. doi: 10.1038/mt.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Park SJ, et al. Down-regulation of mortalin exacerbates Abeta-mediated mitochondrial fragmentation and dysfunction. J. Biol. Chem. 2014;289:2195–2204. doi: 10.1074/jbc.M113.492587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sorrentino V, et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bonora M, et al. Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019;16:33–55. doi: 10.1038/s41569-018-0074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Murphy E, et al. Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ. Res. 2016;118:1960–1991. doi: 10.1161/RES.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Brown DA, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017;14:238–250. doi: 10.1038/nrcardio.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ong SB, et al. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010;88:16–29. doi: 10.1093/cvr/cvq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hahn A, et al. Mitochondrial genome (mtDNA) mutations that generate reactive oxygen species. Antioxidants. 2019;8:392. doi: 10.3390/antiox8090392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Dabravolski SA, et al. The role of mitochondrial DNA mutations in cardiovascular diseases. Int. J. Mol. Sci. 2022;23:952. doi: 10.3390/ijms23020952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.De Souza AI, et al. Proteomic and metabolomic analysis of atrial profibrillatory remodelling in congestive heart failure. J. Mol. Cell. Cardiol. 2010;49:851–863. doi: 10.1016/j.yjmcc.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 217.Li W, et al. Proteomic analysis of metabolic, cytoskeletal and stress response proteins in human heart failure. J. Cell. Mol. Med. 2012;16:59–71. doi: 10.1111/j.1582-4934.2011.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Pound KM, et al. Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ. Res. 2009;104:805–812. doi: 10.1161/CIRCRESAHA.108.189951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Shao D, et al. Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nat. Commun. 2018;9:2935. doi: 10.1038/s41467-018-05362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kolwicz SC, Jr, et al. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013;113:603–616. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sun W, et al. Cardiac-specific deletion of the Pdha1 gene sensitizes heart to toxicological actions of ischemic stress. Toxicol. Sci. 2016;151:193–203. doi: 10.1093/toxsci/kfw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ku HJ, et al. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic. Biol. Med. 2015;80:84–92. doi: 10.1016/j.freeradbiomed.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 223.He L, et al. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation. 2012;126:1705–1716. doi: 10.1161/CIRCULATIONAHA.111.075978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Uchihashi M, et al. Cardiac-specific Bdh1 overexpression ameliorates oxidative stress and cardiac remodeling in pressure overload-induced heart failure. Circ. Heart Fail. 2017;10:e004417. doi: 10.1161/CIRCHEARTFAILURE.117.004417. [DOI] [PubMed] [Google Scholar]
- 225.Sonn SK, et al. Peroxiredoxin 3 deficiency induces cardiac hypertrophy and dysfunction by impaired mitochondrial quality control. Redox Biol. 2022;51:102275. doi: 10.1016/j.redox.2022.102275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sharma S, et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol. 2020;37:101740. doi: 10.1016/j.redox.2020.101740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Nickel AG, et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–484. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 228.Das S, et al. Mitochondrial morphology and dynamics in hepatocytes from normal and ethanol-fed rats. Pharmacol. Ther. 2012;464:101–109. doi: 10.1007/s00424-012-1100-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Niemann J, et al. An mtDNA mutation accelerates liver aging by interfering with the ROS response and mitochondrial life cycle. Free Radic. Biol. Med. 2017;102:174–187. doi: 10.1016/j.freeradbiomed.2016.11.035. [DOI] [PubMed] [Google Scholar]
- 230.Yin C, et al. NGS-based profiling reveals a critical contributing role of somatic D-loop mtDNA mutations in HBV-related hepatocarcinogenesis. Ann. Oncol. 2019;30:953–962. doi: 10.1093/annonc/mdz105. [DOI] [PubMed] [Google Scholar]
- 231.Dabravolski SA, et al. Mitochondrial mutations and genetic factors determining NAFLD risk. Int. J. Mol. Sci. 2021;22:4459. doi: 10.3390/ijms22094459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Satapati S, et al. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012;53:1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Patterson RE, et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 2016;310:E484–E494. doi: 10.1152/ajpendo.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ruepp SU, et al. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol. Sci. 2002;65:135–150. doi: 10.1093/toxsci/65.1.135. [DOI] [PubMed] [Google Scholar]
- 235.Vairetti M, et al. Changes in glutathione content in liver diseases: an update. Antioxidants. 2021;10:364. doi: 10.3390/antiox10030364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Chen Y, et al. Hepatocyte-specific Gclc deletion leads to rapid onset of steatosis with mitochondrial injury and liver failure. Hepatology. 2007;45:1118–1128. doi: 10.1002/hep.21635. [DOI] [PubMed] [Google Scholar]
- 237.Chen Y, et al. Glutathione defense mechanism in liver injury: insights from animal models. Food Chem. Toxicol. 2013;60:38–44. doi: 10.1016/j.fct.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Couto N, et al. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016;95:27–42. doi: 10.1016/j.freeradbiomed.2016.02.028. [DOI] [PubMed] [Google Scholar]
- 239.Itsumi M, et al. Idh1 protects murine hepatocytes from endotoxin-induced oxidative stress by regulating the intracellular NADP(+)/NADPH ratio. Cell Death Differ. 2015;22:1837–1845. doi: 10.1038/cdd.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Huang Z, et al. Liquiritigenin and liquiritin alleviated monocrotaline-induced hepatic sinusoidal obstruction syndrome via inhibiting HSP60-induced inflammatory injury. Toxicology. 2019;428:152307. doi: 10.1016/j.tox.2019.152307. [DOI] [PubMed] [Google Scholar]
- 241.Hu F, et al. Chlorogenic acid alleviates acetaminophen-induced liver injury in mice via regulating Nrf2-mediated HSP60-initiated liver inflammation. Eur. J. Pharm. 2020;883:173286. doi: 10.1016/j.ejphar.2020.173286. [DOI] [PubMed] [Google Scholar]
- 242.Zhao Q, et al. GRP75 regulates mitochondrial-supercomplex turnover to modulate insulin sensitivity. Diabetes. 2022;71:233–248. doi: 10.2337/db21-0173. [DOI] [PubMed] [Google Scholar]
- 243.E Q, et al. Over-expression of GRP75 inhibits liver injury induced by oxidative damage. Acta Biochim. Biophys. Sin. 2013;45:129–134. doi: 10.1093/abbs/gms098. [DOI] [PubMed] [Google Scholar]
- 244.Deng WJ, et al. Proteome, phosphoproteome, and hydroxyproteome of liver mitochondria in diabetic rats at early pathogenic stages. Mol. Cell Proteom. 2010;9:100–116. doi: 10.1074/mcp.M900020-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Buchner DA, et al. Increased mitochondrial oxidative phosphorylation in the liver is associated with obesity and insulin resistance. Obesity. 2011;19:917–924. doi: 10.1038/oby.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Miele L, et al. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am. J. Gastroenterol. 2003;98:2335–2336. doi: 10.1111/j.1572-0241.2003.07725.x. [DOI] [PubMed] [Google Scholar]
- 247.Sunny NE, et al. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Koliaki C, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 249.Schmid AI, et al. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care. 2011;34:448–453. doi: 10.2337/dc10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Fritsch M, et al. Time course of postprandial hepatic phosphorus metabolites in lean, obese, and type 2 diabetes patients. Am. J. Clin. Nutr. 2015;102:1051–1058. doi: 10.3945/ajcn.115.107599. [DOI] [PubMed] [Google Scholar]
- 251.Iozzo P, et al. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology. 2010;139:846–856. doi: 10.1053/j.gastro.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 252.Donnelly KL, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Filali-Mouncef Y, et al. The menage a trois of autophagy, lipid droplets and liver disease. Autophagy. 2022;18:50–72. doi: 10.1080/15548627.2021.1895658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Weber M, et al. Liver CPT1A gene therapy reduces diet-induced hepatic steatosis in mice and highlights potential lipid biomarkers for human NAFLD. FASEB J. 2020;34:11816–11837. doi: 10.1096/fj.202000678R. [DOI] [PubMed] [Google Scholar]
- 255.Mardinoglu A, et al. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014;5:3083. doi: 10.1038/ncomms4083. [DOI] [PubMed] [Google Scholar]
- 256.Abdelmegeed MA, et al. Cytochrome P450-2E1 promotes fast food-mediated hepatic fibrosis. Sci. Rep. 2017;7:39764. doi: 10.1038/srep39764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Liu Y, et al. Silibinin ameliorates hepatic lipid accumulation and oxidative stress in mice with non-alcoholic steatohepatitis by regulating CFLAR-JNK pathway. Acta Pharmaceutica Sin. B. 2019;9:745–757. doi: 10.1016/j.apsb.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Miyata M, et al. Selenoneine ameliorates hepatocellular injury and hepatic steatosis in a mouse model of NAFLD. Nutrients. 2020;12:1898. doi: 10.3390/nu12061898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Merry TL, et al. Hepatocyte glutathione peroxidase-1 deficiency improves hepatic glucose metabolism and decreases steatohepatitis in mice. Diabetologia. 2016;59:2632–2644. doi: 10.1007/s00125-016-4084-3. [DOI] [PubMed] [Google Scholar]
- 260.Chen X, et al. Adropin protects against liver injury in nonalcoholic steatohepatitis via the Nrf2 mediated antioxidant capacity. Redox Biol. 2019;21:101068. doi: 10.1016/j.redox.2018.101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Barbosa PO, et al. Açaí (Euterpe oleracea Martius) supplementation improves oxidative stress biomarkers in liver tissue of dams fed a high-fat diet and increases antioxidant enzymes’ gene expression in offspring. Biomed. Pharmacother. 2021;139:111627. doi: 10.1016/j.biopha.2021.111627. [DOI] [PubMed] [Google Scholar]
- 262.Huang YS, et al. Genetic variations of superoxide dismutase 2 and cytochrome P450 2E1 in non-alcoholic steatohepatitis. Liver Int. 2014;34:931–936. doi: 10.1111/liv.12533. [DOI] [PubMed] [Google Scholar]
- 263.Kruse R, et al. The mitochondrial proteomic signatures of human skeletal muscle linked to insulin resistance. Int. J. Mol. Sci. 2020;21:5374. doi: 10.3390/ijms21155374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ferreira R, et al. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics. 2010;10:3142–3154. doi: 10.1002/pmic.201000173. [DOI] [PubMed] [Google Scholar]
- 265.Leduc-Gaudet J-P, et al. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget. 2015;6:17923–17937. doi: 10.18632/oncotarget.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Koves TR, et al. Subsarcolemmal and intermyofibrillar mitochondria play distinct roles in regulating skeletal muscle fatty acid metabolism. Am. J. Physiol. Cell Physiol. 2005;288:C1074–C1082. doi: 10.1152/ajpcell.00391.2004. [DOI] [PubMed] [Google Scholar]
- 267.Murgia M, et al. Single muscle fiber proteomics reveals fiber-type-specific features of human muscle aging. Cell Rep. 2017;19:2396–2409. doi: 10.1016/j.celrep.2017.05.054. [DOI] [PubMed] [Google Scholar]
- 268.Deshmukh AS, et al. Deep muscle-proteomic analysis of freeze-dried human muscle biopsies reveals fiber type-specific adaptations to exercise training. Nat. Commun. 2021;12:304. doi: 10.1038/s41467-020-20556-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Ubaida-Mohien C, et al. Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. Elife. 2019;8:e49874. doi: 10.7554/eLife.49874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Giebelstein J, et al. The proteomic signature of insulin-resistant human skeletal muscle reveals increased glycolytic and decreased mitochondrial enzymes. Diabetologia. 2012;55:1114–1127. doi: 10.1007/s00125-012-2456-x. [DOI] [PubMed] [Google Scholar]
- 271.Molnar M, et al. A large-scale deletion of mitochondrial DNA in a case with pure mitochondrial myopathy and neuropathy. Acta Neuropathol. 1996;91:654–658. doi: 10.1007/s004010050480. [DOI] [PubMed] [Google Scholar]
- 272.Brautbar A, et al. The mitochondrial 13513G>A mutation is associated with Leigh disease phenotypes independent of complex I deficiency in muscle. Mol. Genet Metab. 2008;94:485–490. doi: 10.1016/j.ymgme.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 273.Wang Y, et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. PNAS. 2001;98:4022–4027. doi: 10.1073/pnas.061013598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Kras KA, et al. Obesity modifies the stoichiometry of mitochondrial proteins in a way that is distinct to the subcellular localization of the mitochondria in skeletal muscle. Metabolism. 2018;89:18–26. doi: 10.1016/j.metabol.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Schiaffino S, et al. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011;91:1447–1531. doi: 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- 276.Murgia M, et al. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015;16:387–395. doi: 10.15252/embr.201439757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Hojlund K, et al. Proteome analysis reveals phosphorylation of ATP synthase beta -subunit in human skeletal muscle and proteins with potential roles in type 2 diabetes. J. Biol. Chem. 2003;278:10436–10442. doi: 10.1074/jbc.M212881200. [DOI] [PubMed] [Google Scholar]
- 278.Hojlund K, et al. Human ATP synthase beta is phosphorylated at multiple sites and shows abnormal phosphorylation at specific sites in insulin-resistant muscle. Diabetologia. 2010;53:541–551. doi: 10.1007/s00125-009-1624-0. [DOI] [PubMed] [Google Scholar]
- 279.Chae S, et al. A mitochondrial proteome profile indicative of type 2 diabetes mellitus in skeletal muscles. Exp. Mol. Med. 2018;50:1–14. doi: 10.1038/s12276-018-0154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Petersen KF, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Hood DA, et al. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu. Rev. Physiol. 2019;81:19–41. doi: 10.1146/annurev-physiol-020518-114310. [DOI] [PubMed] [Google Scholar]
- 282.Ubaida-Mohien C, et al. Physical activity associated proteomics of skeletal muscle: being physically active in daily life may protect skeletal muscle from aging. Front. Physiol. 2019;10:312. doi: 10.3389/fphys.2019.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Gumucio JP, et al. Reduced mitochondrial lipid oxidation leads to fat accumulation in myosteatosis. FASEB J. 2019;33:7863–7881. doi: 10.1096/fj.201802457RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Aldiss P, et al. Exercise-induced ‘browning’ of adipose tissues. Metabolism. 2018;81:63–70. doi: 10.1016/j.metabol.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Koenen M, et al. Obesity, adipose tissue and vascular dysfunction. Circ. Res. 2021;128:951–968. doi: 10.1161/CIRCRESAHA.121.318093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wang QA, et al. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013;19:1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Abdullahi A, et al. Taming the flames: targeting white adipose tissue browning in hypermetabolic conditions. Endocr. Rev. 2017;38:538–549. doi: 10.1210/er.2017-00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Perez-Perez R, et al. Attenuated metabolism is a hallmark of obesity as revealed by comparative proteomic analysis of human omental adipose tissue. J. Proteom. 2012;75:783–795. doi: 10.1016/j.jprot.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 289.Kim SJ, et al. A protein profile of visceral adipose tissues linked to early pathogenesis of type 2 diabetes mellitus. Mol. Cell Proteom. 2014;13:811–822. doi: 10.1074/mcp.M113.035501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Lindinger PW, et al. Important mitochondrial proteins in human omental adipose tissue show reduced expression in obesity. J. Proteom. 2015;124:79–87. doi: 10.1016/j.jprot.2015.03.037. [DOI] [PubMed] [Google Scholar]
- 291.Gomez-Serrano M, et al. Proteome-wide alterations on adipose tissue from obese patients as age-, diabetes- and gender-specific hallmarks. Sci. Rep. 2016;6:25756. doi: 10.1038/srep25756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Gomez-Serrano M, et al. Differential proteomic and oxidative profiles unveil dysfunctional protein import to adipocyte mitochondria in obesity-associated aging and diabetes. Redox Biol. 2017;11:415–428. doi: 10.1016/j.redox.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Michurina SS, et al. Mitochondrial dynamics keep balance of nutrient combustion in thermogenic adipocytes. Mitochondrion. 2021;59:157–168. doi: 10.1016/j.mito.2021.05.001. [DOI] [PubMed] [Google Scholar]
- 294.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 295.Villarroya J, et al. Thymidine kinase 2 deficiency-induced mitochondrial DNA depletion causes abnormal development of adipose tissues and adipokine levels in mice. PLoS One. 2011;6:e29691. doi: 10.1371/journal.pone.0029691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Komakula SSB, et al. The DNA repair protein OGG1 protects against obesity by altering mitochondrial energetics in white adipose tissue. Sci. Rep. 2018;8:14886. doi: 10.1038/s41598-018-33151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Forner F, et al. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metab. 2009;10:324–335. doi: 10.1016/j.cmet.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 298.Carruthers NJ, et al. The human type 2 diabetes-specific visceral adipose tissue proteome and transcriptome in obesity. Sci. Rep. 2021;11:17394. doi: 10.1038/s41598-021-96995-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Plubell DL, et al. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell Proteom. 2017;16:873–890. doi: 10.1074/mcp.M116.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Rangel-Azevedo C, et al. Progressive brown adipocyte dysfunction: Whitening and impaired nonshivering thermogenesis as long-term obesity complications. J. Nutr. Biochem. 2022;105:109002. doi: 10.1016/j.jnutbio.2022.109002. [DOI] [PubMed] [Google Scholar]
- 301.Kazak L, et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell. 2015;163:643–655. doi: 10.1016/j.cell.2015.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Chouchani ET, et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. 2016;532:112–116. doi: 10.1038/nature17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Ikeda K, et al. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat. Med. 2017;23:1454–1465. doi: 10.1038/nm.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Wang G, et al. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol. Cell. 2019;74:844–857 e847. doi: 10.1016/j.molcel.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Mills EL, et al. Cysteine 253 of UCP1 regulates energy expenditure and sex-dependent adipose tissue inflammation. Cell Metab. 2022;34:140–157 e148. doi: 10.1016/j.cmet.2021.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Chella Krishnan K, et al. Sex-specific genetic regulation of adipose mitochondria and metabolic syndrome by Ndufv2. Nat. Metab. 2021;3:1552–1568. doi: 10.1038/s42255-021-00481-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Choi KM, et al. Adipose mitochondrial complex i deficiency modulates inflammation and glucose homeostasis in a sex-dependent manner. Endocrinology. 2022;163:bqac018. doi: 10.1210/endocr/bqac018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wang PW, et al. Biphasic response of mitochondrial biogenesis to oxidative stress in visceral fat of diet-induced obesity mice. Antioxid. Redox Signal. 2014;20:2572–2588. doi: 10.1089/ars.2013.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Faas MM, et al. Mitochondrial function in immune cells in health and disease. Biochim Biophys. Acta Mol. Basis Dis. 2020;1866:165845. doi: 10.1016/j.bbadis.2020.165845. [DOI] [PubMed] [Google Scholar]
- 310.Cervantes-Silva MP, et al. Alterations in mitochondrial morphology as a key driver of immunity and host defence. EMBO Rep. 2021;22:e53086. doi: 10.15252/embr.202153086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Gao Z, et al. Mitochondrial dynamics controls anti-tumour innate immunity by regulating CHIP-IRF1 axis stability. Nat. Commun. 2017;8:1805. doi: 10.1038/s41467-017-01919-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Li Y, et al. Imaging of macrophage mitochondria dynamics in vivo reveals cellular activation phenotype for diagnosis. Theranostics. 2020;10:2897–2917. doi: 10.7150/thno.40495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Yu X, et al. The mtDNA nt7778 G/T polymorphism affects autoimmune diseases and reproductive performance in the mouse. Hum. Mol. Genet. 2009;18:4689–4698. doi: 10.1093/hmg/ddp432. [DOI] [PubMed] [Google Scholar]
- 314.Schilf P, et al. A mitochondrial polymorphism alters immune cell metabolism and protects mice from skin inflammation. Int. J. Mol. Sci. 2021;22:1006. doi: 10.3390/ijms22031006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Tang Z, et al. A genetic bottleneck of mitochondrial DNA during human lymphocyte development. Mol. Biol. Evol. 2022;39:msac090. doi: 10.1093/molbev/msac090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.de Azambuja Rodrigues PM, et al. Proteomics reveals disturbances in the immune response and energy metabolism of monocytes from patients with septic shock. Sci. Rep. 2021;11:15149. doi: 10.1038/s41598-021-94474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Wong CO, et al. Lysosomal degradation is required for sustained phagocytosis of bacteria by macrophages. Cell Host Microbe. 2017;21:719–730 e716. doi: 10.1016/j.chom.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Abuaita BH, et al. Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host Microbe. 2018;24:625–636 e625. doi: 10.1016/j.chom.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Fu Q, et al. Proteomic analysis of murine macrophages mitochondria and lysosomes reveal Cathepsin D as a potential broad-spectrum antimicrobial protein. J. Proteom. 2020;223:103821. doi: 10.1016/j.jprot.2020.103821. [DOI] [PubMed] [Google Scholar]
- 320.Tan H, et al. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity. 2017;46:488–503. doi: 10.1016/j.immuni.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ron-Harel N, et al. Defective respiration and one-carbon metabolism contribute to impaired naive T cell activation in aged mice. PNAS. 2018;115:13347–13352. doi: 10.1073/pnas.1804149115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kanno T, et al. Acsbg1-dependent mitochondrial fitness is a metabolic checkpoint for tissue T(reg) cell homeostasis. Cell Rep. 2021;37:109921. doi: 10.1016/j.celrep.2021.109921. [DOI] [PubMed] [Google Scholar]
- 323.Wenes M, et al. The mitochondrial pyruvate carrier regulates memory T cell differentiation and antitumor function. Cell Metab. 2022;34:731–746 e739. doi: 10.1016/j.cmet.2022.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Martin GS. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001;2:467–475. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 325.Prasetyanti PR, et al. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer. 2017;16:41. doi: 10.1186/s12943-017-0600-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Feinberg AP, et al. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 327.Bissell MJ, et al. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Begum HM, et al. Spatial regulation of mitochondrial heterogeneity by stromal confinement in micropatterned tumor models. Sci. Rep. 2019;9:11187. doi: 10.1038/s41598-019-47593-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Rossi M, et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature. 2022;605:747–753. doi: 10.1038/s41586-022-04758-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Yuan Y, et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020;52:342–352. doi: 10.1038/s41588-019-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Smith AL, et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer. 2020;1:976–989. doi: 10.1038/s43018-020-00112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Li N, et al. Mass spectrometry-based mitochondrial proteomics in human ovarian cancers. J. Mass Spectrom. 2020;39:471–498. doi: 10.1002/mas.21618. [DOI] [PubMed] [Google Scholar]
- 333.Liu J, et al. Mitochondrial proteomics of nasopharyngeal carcinoma metastasis. BMC Med. Genom. 2012;5:62. doi: 10.1186/1755-8794-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Chen M, et al. Quantitative proteomic analysis of mitochondria from human ovarian cancer cells and their paclitaxel-resistant sublines. Cancer Sci. 2015;106:1075–1083. doi: 10.1111/cas.12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Dai Z, et al. Mitochondrial comparative proteomics of human ovarian cancer cells and their platinum-resistant sublines. Proteomics. 2010;10:3789–3799. doi: 10.1002/pmic.200900685. [DOI] [PubMed] [Google Scholar]
- 336.Li N, et al. The lncRNA SNHG3 regulates energy metabolism of ovarian cancer by an analysis of mitochondrial proteomes. J. Gynecol. Oncol. 2018;150:343–354. doi: 10.1016/j.ygyno.2018.06.013. [DOI] [PubMed] [Google Scholar]
- 337.Chen X, et al. Quantitative proteomics analysis identifies mitochondria as therapeutic targets of multidrug-resistance in ovarian cancer. Theranostics. 2014;4:1164–1175. doi: 10.7150/thno.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Dahl ES, et al. Targeting IDH1 as a prosenescent therapy in high-grade serous ovarian cancer. Mol. Cancer Res. 2019;17:1710–1720. doi: 10.1158/1541-7786.MCR-18-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Li N, et al. Quantitative analysis of the human ovarian carcinoma mitochondrial phosphoproteome. Aging. 2019;11:6449–6468. doi: 10.18632/aging.102199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 341.Ahn CS, et al. Mitochondria as biosynthetic factories for cancer proliferation. Cell Metab. 2015;3:1. doi: 10.1186/s40170-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Delaunay S, et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature. 2022;607:593–603. doi: 10.1038/s41586-022-04898-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Yang J, et al. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 344.Baysal BE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 345.Gimenez-Roqueplo AP, et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am. J. Hum. Genet. 2001;69:1186–1197. doi: 10.1086/324413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Frezza C, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225–228. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 347.Porporato PE, et al. Mitochondrial metabolism and cancer. Cell Res. 2018;28:265–280. doi: 10.1038/cr.2017.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Heerdt BG, et al. Mitochondrial membrane potential (delta psi(mt)) in the coordination of p53-independent proliferation and apoptosis pathways in human colonic carcinoma cells. Cancer Res. 1998;58:2869–2875. [PubMed] [Google Scholar]
- 349.Heerdt BG, et al. The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res. 2005;65:9861–9867. doi: 10.1158/0008-5472.CAN-05-2444. [DOI] [PubMed] [Google Scholar]
- 350.Kuwahara Y, et al. The involvement of mitochondrial membrane potential in cross-resistance between radiation and docetaxel. Int J. Radiat. Oncol. Biol. Phys. 2016;96:556–565. doi: 10.1016/j.ijrobp.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 351.LeBleu VS, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014;16:1001–1015. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Murphy MP, et al. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018;17:865–886. doi: 10.1038/nrd.2018.174. [DOI] [PubMed] [Google Scholar]
- 353.Bozi LHM, et al. Mitochondrially-targeted treatment strategies. Mol. Asp. Med. 2020;71:100836. doi: 10.1016/j.mam.2019.100836. [DOI] [PubMed] [Google Scholar]
- 354.Goh KY, et al. Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biol. 2019;21:101100. doi: 10.1016/j.redox.2019.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Ribeiro Junior RF, et al. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018;117:18–29. doi: 10.1016/j.freeradbiomed.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.De Blasio MJ, et al. Therapeutic targeting of oxidative stress with coenzyme Q10 counteracts exaggerated diabetic cardiomyopathy in a mouse model of diabetes with diminished PI3K(p110α) signaling. Free Radic. Biol. Med. 2015;87:137–147. doi: 10.1016/j.freeradbiomed.2015.04.028. [DOI] [PubMed] [Google Scholar]
- 357.Bond ST, et al. The antioxidant moiety of MitoQ Imparts minimal metabolic effects in adipose tissue of high fat fed mice. Front. Physiol. 2019;10:543. doi: 10.3389/fphys.2019.00543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Mewton N, et al. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J. Am. Coll. Cardiol. 2010;55:1200–1205. doi: 10.1016/j.jacc.2009.10.052. [DOI] [PubMed] [Google Scholar]
- 359.Shi J, et al. Bendavia restores mitochondrial energy metabolism gene expression and suppresses cardiac fibrosis in the border zone of the infarcted heart. Life Sci. 2015;141:170–178. doi: 10.1016/j.lfs.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Chouchani ET, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Mills EL, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–470.e413. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Nastasi C, et al. Inhibition of succinate dehydrogenase activity impairs human T cell activation and function. Sci. Rep. 2021;11:1458. doi: 10.1038/s41598-020-80933-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Yoshino J, et al. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Gariani K, et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology. 2016;63:1190–1204. doi: 10.1002/hep.28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Chouchani ET, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Owen MR, et al. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochemical J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 367.Xian H, et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity. 2021;54:1463–1477.e1411. doi: 10.1016/j.immuni.2021.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Moullan N, et al. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 2015;10:1681–1691. doi: 10.1016/j.celrep.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Houtkooper RH, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Sorrentino V, et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Aung LHH, et al. Therapeutic potential and recent advances on targeting mitochondrial dynamics in cardiac hypertrophy: a concise review. Mol. Ther. Nucleic Acids. 2021;25:416–443. doi: 10.1016/j.omtn.2021.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Longo M, et al. Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): new perspectives for a fairy-tale ending? Metabolism. 2021;117:154708. doi: 10.1016/j.metabol.2021.154708. [DOI] [PubMed] [Google Scholar]
- 373.Whitaker RM, et al. Mitochondrial biogenesis as a pharmacological target: a new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol. 2016;56:229–249. doi: 10.1146/annurev-pharmtox-010715-103155. [DOI] [PubMed] [Google Scholar]
- 374.Hondares E, et al. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: an autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology. 2006;147:2829–2838. doi: 10.1210/en.2006-0070. [DOI] [PubMed] [Google Scholar]
- 375.Butterick TA, et al. Pioglitazone increases PGC1-α signaling within chronically ischemic myocardium. Basic Res. Cardiol. 2016;111:37. doi: 10.1007/s00395-016-0555-4. [DOI] [PubMed] [Google Scholar]
- 376.Viscomi C, et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Cassidy-Stone A, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Ruiz A, et al. Mitochondrial division inhibitor 1 (mdivi-1) protects neurons against excitotoxicity through the modulation of mitochondrial function and intracellular Ca2+ signaling. Front Mol. Neurosci. 2018;11:3. doi: 10.3389/fnmol.2018.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Ryu D, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016;22:879–888. doi: 10.1038/nm.4132. [DOI] [PubMed] [Google Scholar]
- 380.Hayakawa K, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–555. doi: 10.1038/nature18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Reddy P, et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell. 2015;161:459–469. doi: 10.1016/j.cell.2015.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Zekonyte U, et al. Mitochondrial targeted meganuclease as a platform to eliminate mutant mtDNA in vivo. Nat. Commun. 2021;12:3210. doi: 10.1038/s41467-021-23561-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Cho SI, et al. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell. 2022;185:1764–1776.e1712. doi: 10.1016/j.cell.2022.03.039. [DOI] [PubMed] [Google Scholar]
- 384.Lei Z, et al. Mitochondrial base editor induces substantial nuclear off-target mutations. Nature. 2022;606:804–811. doi: 10.1038/s41586-022-04836-5. [DOI] [PubMed] [Google Scholar]
- 385.De Vos KJ, et al. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr. Biol. 2005;15:678–683. doi: 10.1016/j.cub.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 386.Koopman WJ, et al. Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytom. A. 2006;69:1–12. doi: 10.1002/cyto.a.20198. [DOI] [PubMed] [Google Scholar]
- 387.Cribbs JT, et al. Functional characterization of phosphorylation sites in dynamin-related protein 1. Meth. Enzymol. 2009;457:231–253. doi: 10.1016/S0076-6879(09)05013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Nikolaisen J, et al. Automated quantification and integrative analysis of 2D and 3D mitochondrial shape and network properties. PLoS One. 2014;9:e101365. doi: 10.1371/journal.pone.0101365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Miller KE, et al. Automated measurement of fast mitochondrial transport in neurons. Front. Cell. Neurosci. 2015;9:435. doi: 10.3389/fncel.2015.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Iannetti EF, et al. Multiplexed high-content analysis of mitochondrial morphofunction using live-cell microscopy. Nat. Protoc. 2016;11:1693–1710. doi: 10.1038/nprot.2016.094. [DOI] [PubMed] [Google Scholar]
- 391.Marquez Neila P, et al. A fast method for the segmentation of synaptic junctions and mitochondria in serial electron microscopic images of the brain. Neuroinformatics. 2016;14:235–250. doi: 10.1007/s12021-015-9288-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Valente AJ, et al. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017;119:315–326. doi: 10.1016/j.acthis.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 393.Bosch A, et al. Automated quantitative analysis of mitochondrial morphology. Methods Mol. Biol. 2019;2040:99–115. doi: 10.1007/978-1-4939-9686-5_6. [DOI] [PubMed] [Google Scholar]
- 394.Rohani A, et al. Mito Hacker: a set of tools to enable high-throughput analysis of mitochondrial network morphology. Sci. Rep. 2020;10:18941. doi: 10.1038/s41598-020-75899-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Fischer CA, et al. MitoSegNet: easy-to-use deep learning segmentation for analyzing mitochondrial morphology. iScience. 2020;23:101601. doi: 10.1016/j.isci.2020.101601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Lefebvre A, et al. Automated segmentation and tracking of mitochondria in live-cell time-lapse images. Nat. Methods. 2021;18:1091–1102. doi: 10.1038/s41592-021-01234-z. [DOI] [PubMed] [Google Scholar]
- 397.Peng JY, et al. Automatic morphological subtyping reveals new roles of caspases in mitochondrial dynamics. PLoS Comput. Biol. 2011;7:e1002212. doi: 10.1371/journal.pcbi.1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Lihavainen E, et al. Mytoe: automatic analysis of mitochondrial dynamics. Bioinformatics. 2012;28:1050–1051. doi: 10.1093/bioinformatics/bts073. [DOI] [PubMed] [Google Scholar]
- 399.Ahmad T, et al. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013;4:e461. doi: 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Zahedi A, et al. Deep analysis of mitochondria and cell health using machine learning. Sci. Rep. 2018;8:16354. doi: 10.1038/s41598-018-34455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Nie Y, et al. Heteroplasmic mitochondrial DNA mutations in frontotemporal lobar degeneration. Acta Neuropathol. 2022;143:687–695. doi: 10.1007/s00401-022-02423-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Hesse R, et al. Comparative profiling of the synaptic proteome from Alzheimer’s disease patients with focus on the APOE genotype. Acta Neuropathol. Commun. 2019;7:214. doi: 10.1186/s40478-019-0847-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Blokhuis AM, et al. Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta Neuropathol. 2016;132:175–196. doi: 10.1007/s00401-016-1575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Brea-Calvo G, et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 2015;96:309–317. doi: 10.1016/j.ajhg.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Feichtinger RG, et al. Biallelic C1QBP mutations cause severe neonatal-, childhood-, or later-onset cardiomyopathy associated with combined respiratory-chain deficiencies. Am. J. Hum. Genet. 2017;101:525–538. doi: 10.1016/j.ajhg.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Willcox JAL, et al. Neither cardiac mitochondrial DNA variation nor copy number contribute to congenital heart disease risk. Am. J. Hum. Genet. 2022;109:961–966. doi: 10.1016/j.ajhg.2022.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Galera-Monge T, et al. The mutation m.13513G>A impairs cardiac function, favoring a neuroectoderm commitment, in a mutant-load dependent way. J. Cell. Physiol. 2019;234:19511–19522. doi: 10.1002/jcp.28549. [DOI] [PubMed] [Google Scholar]
- 408.Mohamed BA, et al. Proteomic analysis of short-term preload-induced eccentric cardiac hypertrophy. J. Transl. Med. 2016;14:149. doi: 10.1186/s12967-016-0898-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Doll S, et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 2017;8:1469. doi: 10.1038/s41467-017-01747-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Linscheid N, et al. Quantitative proteome comparison of human hearts with those of model organisms. PLoS Biol. 2021;19:e3001144. doi: 10.1371/journal.pbio.3001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Eccleston HB, et al. Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid. Redox Signal. 2011;15:447–459. doi: 10.1089/ars.2010.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Simões ICM, et al. The alterations of mitochondrial function during NAFLD progression—an independent effect of mitochondrial ROS production. Int. J. Mol. Sci. 2021;22:6848. doi: 10.3390/ijms22136848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Bárcena B, et al. Aging induces hepatic oxidative stress and nuclear proteomic remodeling in liver from wistar rats. Antioxidants. 2021;10:1535. doi: 10.3390/antiox10101535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Zhang X, et al. Proteomic analysis of liver proteins of mice exposed to 1,2-dichloropropane. Arch. Toxicol. 2020;94:2691–2705. doi: 10.1007/s00204-020-02785-4. [DOI] [PubMed] [Google Scholar]
- 415.Cao W, et al. Txn1, Ctsd and Cdk4 are key proteins of combination therapy with taurine, epigallocatechin gallate and genistein against liver fibrosis in rats. Biomed. Pharmacother. 2017;85:611–619. doi: 10.1016/j.biopha.2016.11.071. [DOI] [PubMed] [Google Scholar]
- 416.Hwang H, et al. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes. 2010;59:33–42. doi: 10.2337/db09-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Robinson MM, et al. Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans. Cell Metab. 2017;25:581–592. doi: 10.1016/j.cmet.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Migliavacca E, et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019;10:5808. doi: 10.1038/s41467-019-13694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Grabowski P, et al. Proteome analysis of human neutrophil granulocytes from patients with monogenic disease using data-independent acquisition. Mol. Cell Proteom. 2019;18:760–772. doi: 10.1074/mcp.RA118.001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Wang G, et al. Liver fibrosis can be induced by high salt intake through excess reactive oxygen species (ROS) production. J. Agric. Food Chem. 2016;64:1610–1617. doi: 10.1021/acs.jafc.5b05897. [DOI] [PubMed] [Google Scholar]
- 421.Shi Q, et al. Inactivation and reactivation of the mitochondrial alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2011;286:17640–17648. doi: 10.1074/jbc.M110.203018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Vaillant-Beuchot L, et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021;141:39–65. doi: 10.1007/s00401-020-02234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Zhao W, et al. Estrogen deficiency induces mitochondrial damage prior to emergence of cognitive deficits in a postmenopausal mouse model. Front. Aging Neurosci. 2021;13:713819. doi: 10.3389/fnagi.2021.713819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Chen F, et al. Molecular signatures of mitochondrial complexes involved in alzheimer’s disease via oxidative phosphorylation and retrograde endocannabinoid signaling pathways. Oxid. Med. Cell. Longev. 2022;2022:9565545. doi: 10.1155/2022/9565545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Chung SJ, et al. Lack of association of mortalin (HSPA9) and other mitochondria-related genes with risk of Parkinson’s and Alzheimer’s diseases. Neurobiol. Aging. 2017;49:215 e219–215 e210. doi: 10.1016/j.neurobiolaging.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 426.Yang L, et al. Mice deficient in dihydrolipoyl succinyl transferase show increased vulnerability to mitochondrial toxins. Neurobiol. Dis. 2009;36:320–330. doi: 10.1016/j.nbd.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Shaltouki A, et al. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 2018;136:607–620. doi: 10.1007/s00401-018-1873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Lahiri V, et al. Functional impairment in RHOT1/Miro1 degradation and mitophagy is a shared feature in familial and sporadic Parkinson disease. Autophagy. 2017;13:1259–1261. doi: 10.1080/15548627.2017.1327512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Hsieh CH, et al. Miro1 Marks Parkinson’s disease subset and miro1 reducer rescues neuron loss in Parkinson’s Models. Cell Metab. 2019;30:1131–1140 e1137. doi: 10.1016/j.cmet.2019.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Safiulina D, et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 2019;38:e99384. doi: 10.15252/embj.201899384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Krüger R, et al. Impaired mitochondrial–endoplasmic reticulum interaction and mitophagy in Miro1-mutant neurons in Parkinson’s disease. Hum. Mol. Genet. 2020;29:1353–1364. doi: 10.1093/hmg/ddaa066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Li L, et al. A mitochondrial membrane-bridging machinery mediates signal transduction of intramitochondrial oxidation. Nat. Metab. 2021;3:1242–1258. doi: 10.1038/s42255-021-00443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Hou T, et al. NDUFAB1 confers cardio-protection by enhancing mitochondrial bioenergetics through coordination of respiratory complex and supercomplex assembly. Cell Res. 2019;29:754–766. doi: 10.1038/s41422-019-0208-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Shi X, et al. ndufa7 plays a critical role in cardiac hypertrophy. J. Cell. Mol. Med. 2020;24:13151–13162. doi: 10.1111/jcmm.15921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Carley AN, et al. Short-chain fatty acids outpace ketone oxidation in the failing heart. Circulation. 2021;143:1797–1808. doi: 10.1161/CIRCULATIONAHA.120.052671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Matsushima S, et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006;113:1779–1786. doi: 10.1161/CIRCULATIONAHA.105.582239. [DOI] [PubMed] [Google Scholar]
- 437.Wang Y, et al. P66Shc deletion ameliorates oxidative stress and cardiac dysfunction in pressure overload-induced heart failure. J. Card. Fail. 2020;26:243–253. doi: 10.1016/j.cardfail.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 438.Cluntun AA, et al. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. 2021;33:629–648.e610. doi: 10.1016/j.cmet.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Mohs A, et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 2021;74:638–648. doi: 10.1016/j.jhep.2020.09.037. [DOI] [PubMed] [Google Scholar]
- 440.Di Francesco A, et al. NQO1 protects obese mice through improvements in glucose and lipid metabolism. NPJ Aging Mech. Dis. 2020;6:13. doi: 10.1038/s41514-020-00051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Otsuka H, et al. Deficiency of 3-hydroxybutyrate dehydrogenase (BDH1) in mice causes low ketone body levels and fatty liver during fasting. J. Inherit. Metab. Dis. 2020;43:960–968. doi: 10.1002/jimd.12243. [DOI] [PubMed] [Google Scholar]
- 442.McCommis KS, et al. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 2015;22:682–694. doi: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Tan M, et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. 2020;27:2143–2157. doi: 10.1038/s41418-020-0491-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Cotter DG, et al. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014;124:5175–5190. doi: 10.1172/JCI76388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.d’Avignon DA, et al. Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. JCI Insight. 2018;3:e99762. doi: 10.1172/jci.insight.99762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Qian X, et al. Exercise in mice ameliorates high-fat diet-induced nonalcoholic fatty liver disease by lowering HMGCS2. Aging. 2021;13:8960–8974. doi: 10.18632/aging.202717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Asif S, et al. Hmgcs2-mediated ketogenesis modulates high-fat diet-induced hepatosteatosis. Mol. Metab. 2022;61:101494. doi: 10.1016/j.molmet.2022.101494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Sim WC, et al. Downregulation of PHGDH expression and hepatic serine level contribute to the development of fatty liver disease. Metabolism. 2020;102:154000. doi: 10.1016/j.metabol.2019.154000. [DOI] [PubMed] [Google Scholar]
- 449.Fu R, et al. Protocatechuic acid-mediated miR-219a-5p activation inhibits the p66shc oxidant pathway to alleviate alcoholic liver injury. Oxid. Med. Cell. Longev. 2019;2019:3527809. doi: 10.1155/2019/3527809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Zhao Y, et al. p66Shc contributes to liver fibrosis through the regulation of mitochondrial reactive oxygen species. Theranostics. 2019;9:1510–1522. doi: 10.7150/thno.29620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Wang Z, et al. Inhibition of p66Shc oxidative signaling via CA-induced upregulation of miR-203a-3p alleviates liver fibrosis progression. Mol. Ther. Nucleic Acids. 2020;21:751–763. doi: 10.1016/j.omtn.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Nahon P, et al. Myeloperoxidase and superoxide dismutase 2 polymorphisms comodulate the risk of hepatocellular carcinoma and death in alcoholic cirrhosis. Hepatology. 2009;50:1484–1493. doi: 10.1002/hep.23187. [DOI] [PubMed] [Google Scholar]
- 453.Al-Serri A, et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012;56:448–454. doi: 10.1016/j.jhep.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 454.von Montfort C, et al. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J. Hepatol. 2012;57:852–859. doi: 10.1016/j.jhep.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Zou Y, et al. Rac2 deficiency attenuates CCl(4)-induced liver injury through suppressing inflammation and oxidative stress. Biomed. Pharmacother. 2017;94:140–149. doi: 10.1016/j.biopha.2017.07.074. [DOI] [PubMed] [Google Scholar]
- 456.Wang YH, et al. Loss of HMGCS2 enhances lipogenesis and attenuates the protective effect of the ketogenic diet in liver cancer. Cancers. 2020;12:1797. doi: 10.3390/cancers12071797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Xu L, et al. Bmal1 inhibits phenotypic transformation of hepatic stellate cells in liver fibrosis via IDH1/alpha-KG-mediated glycolysis. Acta Pharmacol. Sin. 2022;43:316–329. doi: 10.1038/s41401-021-00658-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Ye J, et al. IDH1 deficiency attenuates gluconeogenesis in mouse liver by impairing amino acid utilization. PNAS. 2017;114:292–297. doi: 10.1073/pnas.1618605114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Renkema GH, et al. SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors. Eur. J. Hum. Genet. 2015;23:202–209. doi: 10.1038/ejhg.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Siebers EM, et al. Sdha+/- rats display minimal muscle pathology without significant behavioral or biochemical abnormalities. J. Neuropathol. Exp. Neurol. 2018;77:665–672. doi: 10.1093/jnen/nly042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Brown AD, et al. Mitochondrial fragmentation and dysfunction in type IIx/IIb diaphragm muscle fibers in 24-month old fischer 344 rats. Front. Physiol. 2021;12:727585. doi: 10.3389/fphys.2021.727585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Fogarty MJ, et al. Aging reduces succinate dehydrogenase activity in rat type IIx/IIb diaphragm muscle fibers. J. Appl Physiol. 2020;128:70–77. doi: 10.1152/japplphysiol.00644.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Boufroura FZ, et al. A new AMPK activator, GSK773, corrects fatty acid oxidation and differentiation defect in CPT2-deficient myotubes. Hum. Mol. Genet. 2018;27:3417–3433. doi: 10.1093/hmg/ddy254. [DOI] [PubMed] [Google Scholar]
- 464.Fatehi F, et al. Adult-onset very-long-chain acyl-CoA dehydrogenase deficiency (VLCADD) Eur. J. Neurol. 2020;27:2257–2266. doi: 10.1111/ene.14402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Rubio-Gozalbo ME, et al. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol. Asp. Med. 2004;25:521–532. doi: 10.1016/j.mam.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 466.Weinheimer EM, et al. Resistance exercise and cyclooxygenase (COX) expression in human skeletal muscle: implications for COX-inhibiting drugs and protein synthesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R2241–R2248. doi: 10.1152/ajpregu.00718.2006. [DOI] [PubMed] [Google Scholar]
- 467.Pataky MW, et al. High-fat diet-induced insulin resistance in single skeletal muscle fibers is fiber type selective. Sci. Rep. 2017;7:13642. doi: 10.1038/s41598-017-12682-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Wang X, et al. Scinderin promotes fusion of electron transport chain dysfunctional muscle stem cells with myofibers. Nat. Aging. 2022;2:155–169. doi: 10.1038/s43587-021-00164-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Unni S, et al. Tryptophan oxidation in the UQCRC1 subunit of mitochondrial complex III (ubiquinol-cytochrome C reductase) in a mouse model of myodegeneration causes large structural changes in the complex: a molecular dynamics simulation study. Sci. Rep. 2019;9:10694. doi: 10.1038/s41598-019-47018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Xu Z, et al. Disuse-associated loss of the protease LONP1 in muscle impairs mitochondrial function and causes reduced skeletal muscle mass and strength. Nat. Commun. 2022;13:894. doi: 10.1038/s41467-022-28557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Shin CS, et al. LONP1 and mtHSP70 cooperate to promote mitochondrial protein folding. Nat. Commun. 2021;12:265. doi: 10.1038/s41467-020-20597-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Bruton JD, et al. Reactive oxygen species and fatigue-induced prolonged low-frequency force depression in skeletal muscle fibres of rats, mice and SOD2 overexpressing mice. J. Physiol. 2008;586:175–184. doi: 10.1113/jphysiol.2007.147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Shamseldin HE, et al. Mutation of the mitochondrial carrier SLC25A42 causes a novel form of mitochondrial myopathy in humans. J. Hum. Genet. 2016;135:21–30. doi: 10.1007/s00439-015-1608-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Tews D, et al. Elevated UCP1 levels are sufficient to improve glucose uptake in human white adipocytes. Redox Biol. 2019;26:101286. doi: 10.1016/j.redox.2019.101286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Han YH, et al. Adipocyte-specific deletion of manganese superoxide dismutase protects from diet-induced obesity through increased mitochondrial uncoupling and biogenesis. Diabetes. 2016;65:2639–2651. doi: 10.2337/db16-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.van der Windt GJ, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Klein Geltink RI, et al. Metabolic conditioning of CD8(+) effector T cells for adoptive cell therapy. Nat. Metab. 2020;2:703–716. doi: 10.1038/s42255-020-0256-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Schmidt NM, et al. Targeting human Acyl-CoA:cholesterol acyltransferase as a dual viral and T cell metabolic checkpoint. Nat. Commun. 2021;12:2814. doi: 10.1038/s41467-021-22967-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset 1,Dataset 2,Dataset 3, Dataset 4