

Abstract
Complete panicle exsertion (CPE) in rice is an important determinant of yield and a desirable trait in breeding. However, the genetic basis of CPE in rice still remains to be completely characterized. An ethyl methane sulfonate (EMS) mutant line of an elite cultivar Samba Mahsuri (BPT 5204), displaying stable and consistent CPE, was identified and named as CPE-110. MutMap and RNA-seq were deployed for unraveling the genomic regions, genes, and markers associated with CPE. Two major genomic intervals, on chromosome 8 (25668481-25750456) and on chromosome 11 (20147154-20190400), were identified to be linked to CPE through MutMap. A non-synonymous SNP (G/A; Chr8:25683828) in the gene LOC_Os08g40570 encoding pyridoxamine 5′-phosphate oxidase with the SNP index 1 was converted to Kompetitive allele-specific PCR (KASP) marker. This SNP (KASP 8-1) exhibited significant association with CPE and further validated through assay in the F2 mapping population, released varieties and CPE exhibiting BPT 5204 mutant lines. RNA-seq of the flag leaves at the booting stage, 1100 genes were upregulated and 1305 downregulated differentially in CPE-110 and BPT 5204. Metabolic pathway analysis indicated an enrichment of genes involved in photosynthesis, glyoxylate, dicarboxylate, porphyrin, pyruvate, chlorophyll, carotenoid, and carbon metabolism. Further molecular and functional studies of the candidate genes could reveal the mechanistic aspects of CPE.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11032-023-01412-1.
Keywords: Rice, CPE, Mutant, MutMap, Causal SNP, RNA-seq
Introduction
Rice (Oryza sativa L.) is an important cereal crop grown worldwide for consumption. Around half of the world’s population consumes rice. Considering the increasing rate of population growth, rice grain production needs to be increased exponentially. Rice productivity is not only hampered by biotic and abiotic stresses but also by the morphology of the crop. Among the morphological trait, panicle exsertion (PE) is an important trait that negatively influence yield. Rice yield is reduced when panicle enclosure occurs during grain-filling stages in cultivated varieties and hybrids (Duan et al. 2012). Panicle enclosure describes that panicles are partly or fully enclosed within the flag leaf sheath leading to undernourished and reduced grain size in varieties whereas in hybrids unfilled grains are dominant. The inability of panicles to exsert fully is commonly considered as a genetic defect (Cruz et al. 2008). Panicle enclosure in the rice genotype is mainly triggered due to the subsiding supply of indigenous hormone, gibberellic acid (GA3) to the panicle, resulting in limited elongation of the upper internodes, subsequently panicle enclosing in the sheath leaf. Spraying GA3 on the rice plants at the initial heading stage can eliminate the panicle enclosure, but the large amount of GA3 increases the grain production cost with environmental pollution. Panicle exsertion trait is quantitatively inherited indicating that multiple genes govern the trait. Quantitative trait loci (QTL) mapping (Bao-Jian et al. 2008; Yang et al. 2009; Herlina and Trijatmiko 2016; Zhao et al. 2016, 2018) and genome-wide association mapping (Dang et al. 2017; Zhan et al. 2019) identified genomic regions/markers for panicle exsertion; however, very few have a significant association and utilized in the breeding program.
Development of mutant lines of elite cultivars provides opportunity for mining several traits of interest and understanding the underlying molecular mechanism. Ethyl methane sulphonate mutants of Samba Mahsuri (BPT 5204), an elite popular cultivar, were developed and evaluated for several traits including, agronomic, yield and, abiotic/biotic stress tolerance (Potupureddi et al. 2021). One of the stabilized mutants, CPE-110, was identified as a completely panicle exerted mutant consistently over years. This mutant was considered for mapping the regions associated with CPE through mutation mapping and RNA-seq (transcriptome analysis).
Currently, next-generation sequencing (NGS) technology has revolutionized the field of biological science and offers extraordinary speed with high sequencing accuracy and cost-effectiveness to study genomic and transcriptome data. Mutation mapping or MutMap is one of the gene mapping approaches based on whole-genome resequencing of pooled DNA from a segregating population (usually F2) that is derived from the cross between mutant and the parent of candidate mutant that allows rapid identification of causal nucleotide changes of mutants (Abe et al. 2012). This NGS-based method has been successfully applied in rice for rapid identification of the QTL as well as candidate genes responsible for important agronomic traits such as pale green leaf (Abe et al. 2012), dwarfism (Abe et al. 2012; Oh et al. 2020), low cadmium accumulation in grain (Cao et al. 2019), tolerance to salt (Takagi et al. 2015), grain size (Yuan et al. 2017), and male sterility (Chen et al. 2014). Rapid pipelines have been developed for the identification of causal region/ SNPs using MutMap (Sugihara et al. 2022).
RNA-seq has emerged as an effective approach for transcriptome profiling with higher coverage and greater resolution which provides precise measurement of gene expression level at a particular condition (Magar et al. 2022). RNA-seq presents the ease of identification and evaluation of thousands of genes in a single analysis as compared to other transcriptome techniques. Beyond quantifying gene expression, RNA-seq enables the discovery of novel transcripts, alternatively spliced genes, and the detection of allele-specific expression (Phule et al. 2019). Recent advances in the RNA-seq workflow, from sample preparation to library construction and data analysis, have facilitated researchers to further elucidate the functional complexity of the transcription (Kukurba and Montgomery 2015). In rice, several genes were identified by transcriptome approach specific to various organs, growth stages, and traits such as developing embryos, cross cells, nucellar epidermis, ovular vascular trace, endosperm and aleurone layer (Wu et al. 2020; Xu et al. 2012), peduncle (Kandpal et al. 2020), pigmented leaf for anthocyanin content (Xu et al. 2021), coleoptile elongation rates under water stress (Hsu and Tung 2017; Phule et al. 2019), tolerance to saline-alkaline stress (Li et al. 2020), response to nitrogen use efficiency (Neeraja et al. 2021), seed dormancy (Xie et al. 2019), resistance to rice blast (Yang et al. 2021), sheath blight (Das et al. 2021), bacterial blight (Wang et al. 2019), bakanae disease (Cheng et al. 2020), and brown planthopper (Satturu et al. 2021). Several authors reported transcriptome analysis for panicle (Zhang et al. 2018) and panicle-related traits at different stages viz panicle initiation and grain filling (Katara et al. 2020) leading to the identification of several candidate genes for the trait of interest.
The complete exsertion of panicle from flag leaf is one of the major factors contributing to grain yield that subsequently enhances productivity; identification of causal SNPs/genes/markers responsible for CPE became the premise of this study. Keeping this in view, the objectives of the present study were to identify the genomic region governing CPE by using MutMap and identification of differentially expressed genes using the RNA-seq.
Materials and methods
Development and phenotyping of MutMap population
To identify genomic regions for CPE, a mapping population was developed using BPT 5204, having incomplete panicle exsertion, and its stabilized EMS mutant, CPE-110, having CPE. During the wet cropping season 2018–2019, CPE-110 was crossed with its parent, BPT 5204, and generated F1 plants (true heterozygotes) were confirmed through genotyping using polymorphic simple sequence repeat (SSR) markers. Furthermore, the seeds of true F1 plants were sown in the field during the wet cropping season 2019–2020 at ICAR-Indian Institute of Rice Research, Hyderabad for F2 seeds. Total 200 F2 plants were grown during the wet cropping season 2020–21 in an augmented block design. The F2 individual plants were phenotyped at the maturity stage for complete panicle exsertion trait. Panicle exsertion was recorded at the maturity stage in terms of panicle enclosure and percent panicle exsertion. We have made three phenotypic classes, completely exserted panicle, intermediate, and incompletely exserted based on panicle exsertion from flag leaf. The panicle exserted completely from the flag leaf was considered as complete panicle exsertion, while a panicle base enclosed 0.1 to 2.0 cm inside the flag leaf was considered as intermediate type; the panicle base enclosed > 2.0 cm in the flag leaf was considered as incompletely exserted panicle. Panicle enclosure was measured in centimeters by observing the extent of coverage of panicles by the flag leaf sheath, and percent panicle exsertion was calculated by the following formula:
MutMap analysis: isolation of DNA, whole genome sequencing, pre-processing, alignment of short reads, and SNP calling
Genomic DNA was extracted from the leaves of BPT 5204, CPE-110 and F2 individuals of BPT 5204 × CPE-110 using NucleoSpin Plant II kit (Macherey-Nagel, Dren, Germany). An equimolar concentration of DNA from 20 F2 plants with completely panicle exserted (CPE), i.e., high phenotypic value (100% panicle exsertion) was pooled together and named as CPE bulk. Thus, BPT 5204, CPE-110, and CPE bulk were prepared for whole genome resequencing (WGRS) separately. About 5 μg DNA was used for the preparation of a sequencing library of average insert size 200–500 bp, according to the protocol for the Paired-End DNA Sample PrepKit (Illumina, USA). The library was sequenced to 40× of genome coverage with the Illumina HiSeq 2500 platform (Illumina, USA). The pipeline of MutMap analysis for panicle exsertion has been depicted in Fig. 1. In brief, 106.98, 108.74, and 121.50 million paired-end short reads from BPT 5204, CPE-110, and CPE bulk were used for the analysis. The raw sequencing data were subjected to quality check to ensure high-quality reads for downstream analyses. The reads with a Phred score < 30, base content biasness, and overrepresented sequences (PCR-over duplication, poly G and poly X tails, and adapter contamination) were filtered out using fastp version 0.20.1 (Chen et al. 2018). Paired-end sequence reads of CPE bulk were aligned to the R498 reference sequence (Du et al. 2017) using BWA (Li and Durbin 2009), SNPs were identified, and SNP indices were calculated in R and scored as homozygous SNPs (SNP index ≥ 0.9) and heterozygous SNPs (SNP index ≥ 0.3 and < 0.9). The SNP indices were plotted against chromosome coordinates in R using ggplot (https://ggplot2.tidyverse.org/). The window size for moving average was set to 1 Mb with an increment of 10 kb (Abe et al. 2012).
Fig. 1.
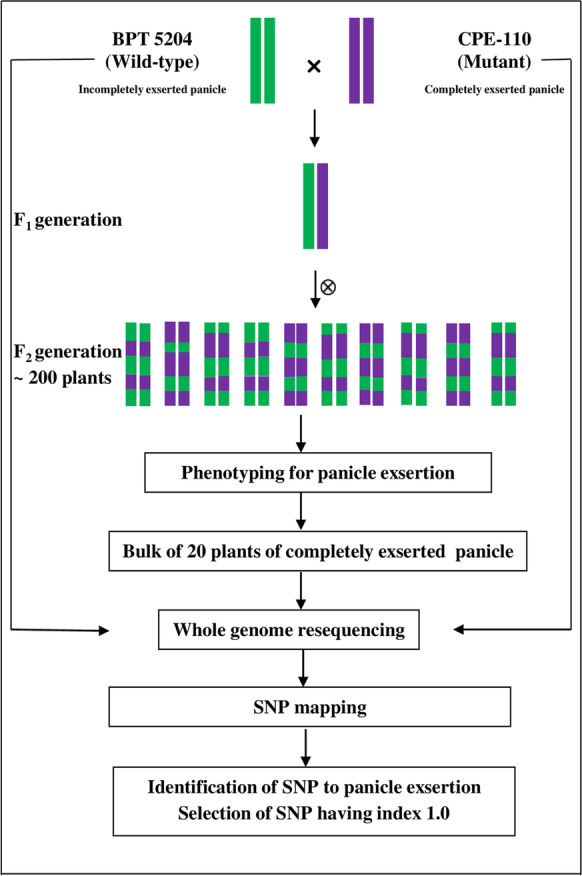
Pipeline of MutMap analysis for identification of genomic region for complete panicle exsertion
Development and assay of kompetitive allele specific PCR (KASP) marker
To validate the causal SNPs for complete panicle exsertion identified using the MutMap approach, a total of 25 genic SNPs (Supplementary Table S1) of eight genes were selected for developing the KASP marker. The 50 bp left and right-flanking sequences of each SNP site were used to design two allele-specific forward and one common reverse primer. The manufacturing of the KASP assays were performed by LGC genomics (London, UK). The chemical profiling for 5 μL reactions was 0.5 μL of template DNA (10 ng), 2.3 μL of 2× KASP master mix, and 0.14 μL of primer mix. The PCR profile of the KASP reaction was followed as pre-read phase at 30 °C for 1 min, hot start at 94 °C for 15 min, touchdown phase of 10 cycles (94 °C for 20 s and 61–51 °C, (dropping 1 °C per cycle) for 60 s) followed by 26 cycles of amplification (94 °C for 20 s; 55 °C for 60 s). The PCR reaction was run in 384-well formatted Applied BiosystemsTM VeritiTM Thermal Cycler. During the pre-read and post-read phases, dyes such as FAMAbs (485 nm), HEXAbs (535 nm), and ROXAbs (575 nm) were utilized to detect fluorescence data.
RNA-seq analysis: RNA isolation, cDNA library preparation, sequencing, pre-processing, and data analysis
The genotypes, BPT 5204 and CPE-110, were utilized for transcriptome analysis. Both the genotypes were grown in the greenhouse (temperature 28 °C, humidity 80%) at ICAR-Indian Institute of Rice Research, Hyderabad during 2021-22. Total RNA was isolated from both the genotypes from flag leaf tissue (middle region) at the panicle initiation stage using the NucleoSpin RNA Plant kit (Macherey-Nagel, Duren, Germany). The quantitative and qualitative assessments of total RNA were conducted using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). The RNA samples with RNA integrity (RIN) value ≥ 8 was pooled in equimolar amounts from three biological replicates prior to the library preparation and subsequent sequencing.
The cDNA libraries were prepared using PrimeScriptTM 1st strand cDNA synthesis kit (Takara, Japan) following the manufacturer’s instructions. Pair-end sequencing was performed using an Illumina HiSeq 2500 sequencing platform (Illumina, San Diego, CA) at Nucleome Informatics Pvt. Ltd., Hyderabad. The raw sequencing data were subjected to quality check to ensure high-quality reads for downstream analyses. The reads with a Phred score < 30, base content biasness, and overrepresented sequences (PCR-over duplication, poly G and poly X tails, and adapter contamination) were filtered out using Fastp version 0.20.1 (Chen et al. 2018). The reads were mapped on the rice reference genome, R498 using a splice-aware alignment algorithm, HISAT2 (v 2.1.0) (Kim et al. 2019). The cleaned reads of RNA-seq data were deposited in NCBI Sequence Read Archive (SRA) database with the BioProject ID PRJNA687517.
Differential gene expression (DEG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) term enrichment
The featureCounts program was used to count the reads at the gene level to check the prevalence of the biotypes of RNA (Liao et al. 2014). Subsequently, NOISeq was used to analyze quality control of count data; normalization and low-count filtering; and differential gene expression analysis (Tarazona et al. 2015). GO term enrichments were carried out by topGO using the parent-child Fisher algorithm, which essentially categorizes the terms by their hierarchical order and performs a hypergeometric test (Alexa and Rahnenfuhrer 2010). These enriched terms were filtered on a p-value cut-off of 0.05, following which the terms are then visualized in the form of a directed acyclic graph (DAG). KEGG term enrichment analysis was performed using the R package clusterProfiler with the terms hosting Benjamini-Hochberg corrected q values (Yu et al. 2012). This was done by selecting the Entrez gene-id values of DEGs (up- and down-regulated genes) separately through the BioMart package.
Validation of differentially expressed genes (DEGs) by quantitative real time-PCR (qRT-PCR)
To validate the RNA-seq DEG data, qRT-PCR was conducted on candidate genes identified by MutMap and RNA-seq. The total RNA of BPT 5204 and CPE-110 was reverse transcribed using PrimeScriptTM 1st strand cDNA synthesis kit (Takara, Japan). The relative expression was analyzed from three biological replicates using the 2−ΔΔCt method with the rice TPH (tumor protein homolog) and Memp (membrane protein) as reference genes (Phule et al. 2019). The locus ids, function, and primer sequences of the genes are listed in Table S2.
Results
Genetic analysis of F2 of BPT 5204 × CPE-110 for CPE
A total of 200 F2 plants along with parents were phenotyped for panicle exsertion during the wet crop season 2019–2020 in the field wet season. The parent, BPT 5204, and its CPE mutant, CPE-110, significantly differed for panicle exsertion (Table 1). The F2 population displayed high phenotypic variability for the trait. The lines in the F2 population for panicle enclosing in flag leaf ranged from 0 to 3.79 cm, whereas percent panicles exserted from flag leaf ranged from 78.8 to 100%. The F2 population exhibited normal distribution for CPE, indicating trait under quantitative control (Fig. 2a, b). Inhibitory gene action was exhibited by the F2 population of BPT 5204 × CPE-110 where the interaction of the homozygous recessive gene with the homozygous/heterozygous dominant gene produced a completely exserted panicle phenotype (Table 1).
Table 1.
Phenotyping of F2 population of CPE-110 × BPT 5204 for CPE
| Trait | Panicle enclosing in flag leaf (cm) | |
| Phenotype | Completely exserted | Incompletely exserted |
| Phenotype class | 0 cm | >0.0 cm |
| Number of F2 genotypes | 29 | 171 |
| Trait | Percent panicle exserted | |
| Phenotype class | 100% | <100% |
| Number of F2 genotypes | 29 | 171 |
| Chi-square (for 13:3) | 2.73 | |
Fig. 2.

Distribution pattern of CPE in F2 population of CPE-110 × BPT 5204 for CPE. a Length of panicle choked in flag leaf. b Percent panicle exserted
Whole-genome resequencing and alignment of short reads of CPE bulk of MutMap population
The CPE bulk and its parents, BPT 5204 and CPE-110, were sequenced to ∼40x genome coverage. The statistical details on the number of raw reads, mapped reads, depth of genome coverage, average coverage, and average quality are presented in Table 2. On aligning, 121.13, 106.5, and 107.5 million paired-end reads of CPE bulk, BPT 5204, and CPE-110, respectively, were mapped to the R498 reference genome (Table 2) with genome coverage 97.53, 95.02, and 94.98%, respectively. We obtained 17.85, 15.74, and 15.69 cleaned gigabases for CPE bulk, BPT 5204, and CPE-110, respectively.
Table 2.
Summary of QC data BPT 5204, CPE-110, and CPE bulk of F2 population of BPT 5204 × CPE-110
| Genotype/QC description | BPT 5204 | CPE-110 | Bulk of complete panicle exsertion |
|---|---|---|---|
| Depth of genome coverage (X) | 40.25 | 40.13 | 45.64 |
| Genome Coverage with R498 reference genome (%) | 95.02 | 94.98 | 97.53 |
| No. of raw PE reads (million) | 106.98 | 108.74 | 121.50 |
| number of mapped reads | 106.50 | 107.50 | 121.13 |
| Total Bases (Gb) | 15.74 | 15.69 | 17.85 |
| Homozygous SNPs | 630123 | 667155 | 353547 |
Identification of CPE-associated genes and SNPs using MutMap
The Illumina short reads obtained for CPE bulk, BPT 5204, and CPE-110 were separately aligned to the reference sequence of R498 and identified 353547, 630123, and 667155 homozygous SNPs respectively (Table 2). Upon comparison of CPE bulk with wild-type BPT 5204, SNP index plots were generated. Two major peaks, one on chromosome 8 (25668481-25750456) and the other on chromosome 11 (20147154-20190400) with SNP index = 1, were recorded (Fig. 3; Supplementary Fig. 1). We mined the MutMap candidate regions of chromosomes 8 and 11 using the RAP-DB database (http://rapdb.dna.affrc.go.jp/) and identified a total of 15 and ten genes, respectively (Table 3).
Fig. 3.

Genomic region (shown in rectangular shape) on a chromosome 8 and b chromosome 11 exhibiting for complete panicle exsertion. X-axis indicates the physical position of the chromosome, and the Y-axis indicates the average SNP-index. SNP index plot regression lines were obtained by averaging SNP indices from a moving window of five consecutive SNPs and shifting the window of one SNP at a time. The X-axis value of each averaged SNP index was set at a midpoint between the first and the fifth SNP
Table 3.
List of genes identified from the MutMap region of chromosome 8 and 11 for complete panicle exsertion using F2 MutMap population of CPE-110 × BPT 5204
| SN | Gene | Chromosome | Description | CDS coordinates (5′-3′) |
|---|---|---|---|---|
| 1 | LOC_Os08g40550 | 8 | Retrotransposon protein, putative, unclassified, expressed | 25673540–25668739 |
| 2 | LOC_Os08g40555 | 8 | ATPase, E1-E2 type, putative, expressed | 25675948–25674428 |
| 3 | LOC_Os08g40560 | 8 | ZOS8-11 - C2H2 zinc finger protein, expressedX | 25682411–25676686 |
| 4 | LOC_Os08g40570 | 8 | Pyridoxamine 5′-phosphate oxidase family protein, putative, expressed | 25686522–25683429 |
| 5 | LOC_Os08g40580 | 8 | Methyltransferase domain containing protein, expressed | 25690627–25687620 |
| 6 | LOC_Os08g40590 | 8 | Oxysterol-binding protein, putative, expressed | 25694808–25699639 |
| 7 | LOC_Os08g40600 | 8 | Thaumatin, putative, expressed | 25701623–25700147 |
| 8 | LOC_Os08g40610 | 8 | 30S ribosomal protein S16, putative, expressed | 25704363–25702617 |
| 9 | LOC_Os08g40615 | 8 | Expressed protein | 25707694–25708488 |
| 10 | LOC_Os08g40620 | 8 | rabGAP/TBC domain-containing protein, putative, expressed | 25712814–25717641 |
| 11 | LOC_Os08g40630 | 8 | mTERF domain containing protein, expressed | 25721973–25719257 |
| 12 | LOC_Os08g40640 | 8 | Retrotransposon protein, putative, Ty1-copia subclass, expressed | 25733446 - 25723118 |
| 13 | LOC_Os08g40650 | 8 | Senescence-induced receptor-like serine/threonine-protein kinase precursor, putative, expressed | 25735566–25743180 |
| 14 | LOC_Os08g40660 | 8 | Retrotransposon protein, putative, unclassified, expressed | 25750374–25745440 |
| 15 | LOC_Os11g34370 | 11 | Phospholipase, patatin family, putative, expressed | 20147936–20144591 |
| 16 | LOC_Os11g34380 | 11 | Retrotransposon protein, putative, unclassified | 20148368–20149117 |
| 17 | LOC_Os11g34390 | 11 | Glycosyltransferase, putative, expressed | 20154022–20152496 |
| 18 | LOC_Os11g34400 | 11 | Retrotransposon protein, putative, unclassified | 20157516–20157190 |
| 19 | LOC_Os11g34410 | 11 | Retrotransposon protein, putative, unclassified, expressed | 20158507–20164146 |
| 20 | LOC_Os11g34420 | 11 | Retrotransposon protein, putative, Ty3-gypsy subclass, expressed | 20167991–20164768 |
| 21 | LOC_Os11g34430 | 11 | Retrotransposon protein, putative, unclassified | 20169257–20168931 |
| 22 | LOC_Os11g34440 | 11 | Phospholipase A2, putative, expressed | 20176341–20174651 |
| 23 | LOC_Os11g34450 | 11 | 14-3-3 protein, putative, expressed | 20178405–20181583 |
| 24 | LOC_Os11g34460 | 11 | OsFBO10 - F-box and other domain containing protein, expressed | 20187327–20182477 |
Furthermore, upon comparison with BPT 5204, we identified 20 (11 genic and 7 inter-genic) and 37 (14 genic and 23 inter-genic) homozygous SNPs in the MutMap region of chromosome 8 (81.9 kb) and 11 (43.2 kb), respectively. Of the 11 genic SNPs identified in the MutMap region of chromosome 8, three SNPs each were observed in the exon, intron, and promoter region respectively while two SNPs were observed in the 3' UTR region of genes. Out of the three exonic SNPs, each SNP was located in LOC_Os08g40560, LOC_Os08g40640, and LOC_Os08g40660 genes, respectively (Table 4). The genes viz, LOC_Os08g40560, LOC_Os08g40570, LOC_Os08g40610, and LOC_Os08g40615 encoded ZOS8-11-C2H2 zinc finger protein, pyridoxamine 5′-phosphate oxidase family protein, 30S ribosomal protein S16, and expressed protein respectively while two genes viz, LOC_Os08g40640 and LOC_Os08g40660 encoded retrotransposon. Likewise, of 14 genic SNPs identified in the MutMap region of chromosome 11, nine and five SNPs were located in the exon and intron region of two genes respectively. Of nine exonic SNPs, six and three SNPs were observed in LOC_Os11g34370 and LOC_Os11g34460, respectively (Table 4). The gene, LOC_Os11g34370 encoded for phospholipase, patatin family, is involved in the lipid metabolic process, whereas the gene, F-box, and other domain-containing protein (LOC_Os11g34460) have catalytic and protein-binding activity, involved in the signal transduction and biosynthetic processes including nucleobase, nucleoside, nucleotide, and nucleic acid metabolic process as well as in the flower development.
Table 4.
Identification of putative candidate genes for complete panicle exsertion using F2 MutMap population of CPE-110 × BPT 5204
| Gene | Chromosome | Position | Location in gene | Ref allele | Alt allele | SNP index | Gene function |
|---|---|---|---|---|---|---|---|
| LOC_Os08g40560 | 8 | 25681951 | Intron | G | A | 1.0 | ZOS8-11-C2H2 zinc finger protein, expressed |
| 25682026 | Exon | G | A | 1.0 | |||
| LOC_Os08g40570 | 8 | 25683828 | Promoter | G | A | 1.0 | Pyridoxamine 5′-phosphate oxidase family protein, putative, expressed |
| 25683858 | Promoter | C | T | 1.0 | |||
| 25683907 | Promoter | C | T | 1.0 | |||
| LOC_Os08g40610 | 8 | 25702666 | 3′ UTR | C | T | 1.0 | 30S ribosomal protein S16, putative, expressed |
| LOC_Os08g40615 | 8 | 25708269 | 3′ UTR | G | A | 1.0 | Expressed protein |
| LOC_Os08g40640 | 8 | 25723394 | Intron | C | T | 1.0 | Retrotransposon protein, putative, Ty1-copia subclass, expressed |
| 25725623 | Exon | C | T | 1.0 | |||
| LOC_Os08g40660 | 8 | 25748406 | Intron | G | A | 1.0 | Retrotransposon protein, putative, unclassified, expressed |
| 25748564 | Exon | G | A | 1.0 | |||
| LOC_Os11g34370 | 11 | 20147154 | Intron | C | T | 1.0 | Phospholipase, patatin family, putative, expressed |
| 20147420 | Intron | G | A | 1.0 | |||
| 20147484 | Exon | C | T | 1.0 | |||
| 20147595 | Exon | C | T | 1.0 | |||
| 20147655 | Exon | G | A | 1.0 | |||
| 20147736 | Exon | C | T | 1.0 | |||
| 20147770 | Exon | G | A | 1.0 | |||
| 20147800 | Exon | C | T | 1.0 | |||
| 20147929 | 3′ UTR | G | A | 1.0 | |||
| LOC_Os11g34460 | 11 | 20182992 | Exon | G | A | 1.0 | OsFBO10-F-box and other domain containing protein, expressed |
| 20183408 | Exon | C | T | 1.0 | |||
| 20186295 | Intron | C | T | 1.0 | |||
| 20186389 | Intron | G | A | 1.0 | |||
| 20186415 | Intron | C | T | 1.0 |
Validation of causal SNP for complete panicle exsertion by kompetitive allele specific PCR (KASP)
To validate the SNP identified for complete panicle exsertion through the MutMap approach, KASP assays were developed for 25 SNPs. The list of developed KASP markers is represented in Supplementary Table S1. Furthermore, 25 KASP markers used for genotyping in the same population, that is, F2 of BPT 5204 × CPE-110. After genotyping, only one KASP marker, KASP 8-1 (Chr8:25683828; G/A), displayed a strong association with the CPE trait in the F2 population (Fig. 4). Furthermore, to know the association of KASP 8-1 for CPE, this marker was screened in completely panicle exserted lines such as stabilized mutants (n = 12) and released varieties (n = 15). The results indicated that KASP 8-1 marker exhibited co-segregation with panicle exsertion (Fig. 4). The KASP 8-1 marker was located in LOC_Os08g40570 which encodes a Pyridoxamine 5′-phosphate oxidase family protein.
Fig. 4.

Kompetitive allele specific primer assay of KASP 8-1 marker and its co-segregation with complete panicle exsertion
RNA-seq statistics
Using the Illumina sequencing platform, a total of 35.89 million paired-end reads consisting of 5.1 Giga base (Gb) were generated for BPT 5204; of this, a total of 4.8 Gb (95.6%) passed in quality control. Like-wise, for CPE-110, 39.72 million reads comprising 5.6 Gb were generated; of this, a total of 5.4 Gb (95.7%) passed in quality control and retained for further analysis (Table 5). Using HISAT2, a total of 16.64 (92.7%) and 18.20 million paired-end reads (91.7%) were aligned with reference genome for BPT 5204 and CPE-110, respectively. Of these, 13.43 (74.8%) and 11.61 (58.5%) million paired-end reads from BPT 5204 and CPE-110, respectively, were uniquely mapped (Table 6).
Table 5.
Summary of Quality Control (QC) data of CPE-110 and BPT 5204 through RNA-seq
| Genotype/QC description | BPT 5204 | CPE-110 |
|---|---|---|
| Number of paired-end reads (million) | 35.89 | 39.72 |
| Total bases (million) | 5109.31 | 5663.27 |
| % duplication | 18.45 | 37.63 |
| Q30 fraction | 0.95 | 0.95 |
| Q30 bases (million) | 4884.05 | 5417.90 |
| GC content fraction | 0.56 | 0.53 |
Table 6.
Overview of mapping status of RNA-seq data of CPE-110 and BPT 5204
| Genotype/mapping description | BPT 5204 | CPE-110 |
|---|---|---|
| Paired-end reads mapped uniquely (million) | 13.43 | 11.61 |
| Paired-end reads mapped discordantly uniquely (million) | 0.10 | 0.07 |
| Paired-end reads one mate mapped uniquely (million) | 0.59 | 0.49 |
| Paired-end reads multimapped (million) | 2.44 | 5.91 |
| Paired-end reads one mate multimapped (million) | 0.07 | 0.11 |
| Paired-end reads neither mate aligned | 1.30 | 1.65 |
| % mapped to Reference genome | 92.72 | 91.68 |
Identification of differentially expressed genes between CPE-110 and BPT 5204
Using NOISeq, genes with counts per million (CPM) > 1 were retained for analyses, leading to the retention of 18,248 genes from a total of 38,978 genes. NOISeq matrix with the 18,248 genes was used for analyzing the level of gene expression using Trimmed Mean of M-values (TMM) normalization, with a |log2FC| ≥1 and with a probability value of > 0.95. In total, 2469 differentially expressed genes (DEGs) were observed between CPE-110 and BPT 5204 in flag leaf tissue during the panicle initiation stage for complete panicle exsertion (Fig. 5a). Of these, 1100 genes were upregulated while 1305 were downregulated in CPE-110 (Fig. 5b; Supplementary Table S3 and S4). The transcriptome analysis between the completely exserted panicle line (CPE-110) and incompletely exserted panicle line (BPT 5204) gave us a deeper insight into the differentially expressed genes (DEGs). In this investigation, several subsets of DEGs were identified which are potentially related to panicle exsertion (Table 7; Fig. 6).
Fig. 5.

Study of differential gene expression for complete panicle exsertion by scatter plots a volcano plot, created by taking the log2FC on the X-axis and the –log (p-value) on the Y-axis. b MA plots created by taking log scaled average expression on the X-axis and log2FC on the Y-axis
Table 7.
List of candidate genes identified through RNA-seq for complete panicle exsertion
| SN | RAP gene ID | MSU gene ID | Description |
|---|---|---|---|
| 1 | Os11g0189600 | LOC_Os11g08569 | 2,3-Oxidosqualene cyclase, triterpene synthase, parkeol synthase |
| 2 | Os11g0311100 | None | Similar to pantothenate kinase family protein |
| 3 | Os07g0454200 | LOC_Os07g27120 | Similar to hydroxyproline-rich glycoprotein gas29p precursor |
| 4 | Os02g0522100 | LOC_Os02g32230 | Similar to cDNA clone:J013126H19, full insert sequence |
| 5 | Os07g0450000 | LOC_Os07g26740 | Similar to 60S ribosomal protein L44 |
| 6 | Os07g0472200 | LOC_Os07g28910 | Similar to zinc-finger protein Lsd1 |
| 7 | Os04g0511200 | LOC_Os04g43200 | EFA27 for EF hand, abscisic acid, 27kD |
| 8 | Os02g0541500 | LOC_Os02g33720 | Similar to predicted protein |
| 9 | Os07g0663800 | LOC_Os07g46852 | Similar to oxidoreductase, short chain dehydrogenase/reductase family protein, expressed |
| 10 | Os01g0124200 | LOC_Os01g03340 | Similar to Bowman-Birk trypsin inhibitor |
| 11 | Os01g0603800 | LOC_Os01g41930 | Similar to Triticum sp. (pAWJL3) leucine rich repeat region mRNA (fragment) |
| 12 | Os11g0286800 | LOC_Os11g18366 | Squalene cyclase domain containing protein |
| 13 | Os03g0689400 | LOC_Os03g48320 | Similar to NB-ARC domain containing protein, expressed |
| 14 | Os03g0654700 | LOC_Os03g45210 | Protein of unknown function DUF1637 family protein |
| 15 | Os10g0468500 | LOC_Os10g33040 | Serine/threonine protein kinase-related domain containing protein |
| 16 | Os01g0778900 | None | Similar to serine-rich protein-related |
| 17 | Os10g0446800 | LOC_Os10g30970 | Similar to predicted protein |
| 18 | Os07g0501700 | LOC_Os07g31830 | C2 calcium-dependent membrane targeting domain containing protein |
| 19 | Os09g0466400 | LOC_Os09g29130 | Zinc finger homeodomain (ZF-HD) class homeobox transcription factor, rice morphogenesis, modulation of leaf rolling |
| 20 | Os02g0137700 | LOC_Os02g04510 | NAD(P)-binding domain containing protein |
| 21 | Os04g0578600 | LOC_Os04g48930 | Similar to H0404F02.15 protein |
| 22 | Os07g0249800 | LOC_Os07g14600 | Similar to IAA-amino acid hydrolase ILR1-like 8 |
| 23 | Os03g0817100 | LOC_Os03g60250 | Uncharacterised protein family UPF0497, trans-membrane plant domain containing protein |
| 24 | Os02g0136800 | LOC_Os02g04420 | Protein of unknown function DUF1677, Oryza sativa family protein |
| 25 | Os11g0492300 | LOC_Os11g29990 | Similar to NB-ARC domain containing protein |
| 26 | Os01g0764800 | LOC_Os01g55940 | Indole-3-acetic acid (IAA)-amido synthetase, disease resistance, abiotic stress tolerance |
| 27 | Os03g0111300 | LOC_Os03g02050 | Nonspecific lipid-transfer protein 2 (nsLTP2) (7 kDa lipid transfer protein) |
| 28 | Os04g0531750 | LOC_Os04g44924 | Similar to OSIGBa0125M19.13 protein |
| 29 | Os01g0870400 | LOC_Os01g65010 | Similar to S-domain class receptor-like kinase3 |
| 30 | Os11g0485200 | LOC_Os11g29490 | ATPase, P-type, K/Mg/Cd/Cu/Zn/Na/Ca/Na/H-transporter family protein |
| 31 | Os09g0558100 | LOC_Os09g38560 | Similar to low-temperature induced protein lt101.2 |
| 32 | Os07g0228400 | LOC_Os07g12560 | Cyclin-like F-box domain containing protein |
| 33 | Os02g0147200 | LOC_Os02g05400 | Similar to cDNA clone:J013125B21, full insert sequence |
| 34 | Os10g0469600 | LOC_Os10g33130 | Similar to leucine-rich repeat family protein, expressed |
| 35 | Os05g0177500 | LOC_Os05g08480 | UDP-glucuronosyl/UDP-glucosyltransferase family protein |
| 36 | Os11g0444000 | LOC_Os11g25720 | Similar to UDP-glucosyltransferase BX8 |
| 37 | Os07g0689600 | LOC_Os07g48980 | Nicotianamine synthase 3 (EC 2.5.1.43) (S-adenosyl-L-methionine:S-adenosyl-L-methionine:S-adenosyl-methionine 3-amino-3-carboxypropyltransferase 3 |
| 38 | Os03g0342100 | LOC_Os03g22230 | Pollen Ole e 1 allergen/extensin domain containing protein |
| 39 | Os03g0197900 | LOC_Os03g10150 | Protein of unknown function DUF623, plant domain containing protein |
| 40 | Os07g0235700 | LOC_Os07g13160 | Similar to cDNA clone:J023006M12, full insert sequence |
| 41 | Os07g0477250 | LOC_Os07g29440 | Similar to S-adenosylmethionine synthetase 2 |
| 42 | Os07g0199350 | None | Similar to HAT family dimerisation domain-containing protein |
| 43 | Os07g0561800 | LOC_Os07g37454 | Similar to carbohydrate transporter/sugar porter |
| 44 | Os10g0100700 | LOC_Os10g01080 | Vitamin B6 biosynthesis protein family protein |
Fig. 6.

Heat map of expression profiles of highly significant DEGs related to complete panicle exsertion. Color from red to green indicates that the FPKM values were small to large, red color indicates low level of gene expression, and green color indicates high level of gene expression
Genes underlying MutMap identified QTLs
In the current study, two genes, namely, LOC_Os11g34390 and LOC_Os11g34370, were identified to be downregulated in CPE-110 at the panicle initiation stage. The genes are located in the region (chrom 11:20147154-20190400) identified on chromosome 11 through MutMap. The gene LOC_Os11g34390 encodes glycosyltransferase 6, a galactosyl transferase family protein that is involved in the biosynthesis of xyloglucan. Likewise, LOC_Os11g34370 is encoded by patatin family protein which is involved in the lipid metabolic process (GO:0006629), mainly in triacylglycerol degradation. The downregulation of these genes along with the observed mutations are compelling to speculate a possible role of these genes in complete panicle exsertion in CPE-110.
To validate RNA-seq DEG data, qRT-PCR was conducted on twenty-four functionally relevant genes which were highly up and down-regulated in both the genotypes. The expression of eighteen genes was found to be according to RNA-seq result, while the other six genes, namely, Os01g0764800, Os01g0124200, Os10g0100700, Os11g0286800, Os11g0189600, and Os07g0454200, revealed the opposite expression to RNA-seq (Supplementary Fig. 2; Supplementary Table S3 and S4).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment
Gene Ontology (GO) enrichment was performed for DEGs to gain insights into the enrichment of functional categories that could be associated with CPE. The annotated DEGs were categorized into three major groups viz. biological process (BP), molecular function (MF), and cellular component (CC). The identified GO terms were further classified into down-regulated and up-regulated groups. In downregulated DEGs, a total of 130 GO terms were assigned, including 75, 20, and 35 in BP, CC and MF, respectively, whereas, in up-regulated DEGs, a total of 47 GO terms were assigned, including 26, 8, and 13 in BP, CC, and MF, respectively (Supplementary Table S5-S10). Overall, among the BP category, the significantly upregulated GO terms were “regulation of nucleobase-containing compound metabolic process” followed by “dephosphorylation,” “nucleosome organization,” and “chromatin assembly or disassembly.” In CC category, nucleus,” “plasma membrane,” “Sm-like protein family complex,” “cell periphery,” and “small nuclear ribonucleoprotein complex” were the most significant GO terms, whereas, in molecular functions category, phosphatase inhibitor activity, phosphatase regulator activity, and oxidoreductase activity revealed most significant GO terms (Table 8; Fig. 7). Interestingly, in BP category, two GO terms, namely, GO:0009765 and GO:0019684, were specific to down-regulated DEGs and were involved in the “photosynthesis, light harvesting (11 DEGs)” and “photosynthesis, light reaction (11 DEGs)” processes, respectively, suggesting the role of these genes for complete panicle exsertion. Likewise, 11 GO terms were significantly downregulated in a cellular component, mainly in the plastid (58 DEGs) and its components like photosystem (15 DEGs), plastid stroma (2 DEGs), chloroplast thylakoid (83 DEGs), photosystem I reaction center (4 DEGs), photosystem II oxygen-evolving complex (10 DEGs), plastoglobule (2 DEGs), thylakoid lumen (2 DEGs), and photosynthetic membrane (45 DEGs) (Table 9). In the molecular function category, a total of 22 GO terms were downregulated and mostly involved in the oxidoreductase activity (9 DEGs), metallopeptidase activity (9 DEGs), peptidyl-prolyl cis-trans isomerase activity (7 DEGs), protein heterodimerization activity (14 DEGs), etc. (Table 9; Fig. 7).
Table 8.
List of GO terms significantly up regulated in CPE-110 related to cellular component, biological process, and molecular function
| GO.ID | Term | DEGs | p value |
|---|---|---|---|
| Biological process | |||
| GO:0006413 | Translational initiation | 3 | 0.04 |
| GO:0006979 | Response to oxidative stress | 2 | 0.02 |
| GO:0009072 | Aromatic amino acid family metabolic process | 3 | 0.05 |
| GO:0009741 | Response to brassinosteroid | 1 | 0.05 |
| GO:0010119 | Regulation of stomatal movement | 1 | 0.03 |
| GO:0014070 | Response to organic cyclic compound | 1 | 0.04 |
| GO:0006333 | Chromatin assembly or disassembly | 3 | 0.00 |
| GO:0019219 | Regulation of nucleobase-containing compound metabolic process | 23 | 0.00 |
| GO:0043446 | Cellular alkane metabolic process | 1 | 0.03 |
| GO:0043447 | Alkane biosynthetic process | 1 | 0.04 |
| GO:0044786 | Cell cycle DNA replication | 2 | 0.01 |
| GO:0065004 | Protein-DNA complex assembly | 3 | 0.01 |
| GO:0071248 | Cellular response to metal ion | 1 | 0.04 |
| GO:0071824 | Protein-DNA complex subunit organization | 3 | 0.02 |
| Cellular component | |||
| GO:0030008 | TRAPP complex | 2 | 0.02 |
| GO:0030532 | Small nuclear ribonucleoprotein complex | 3 | 0.02 |
| GO:0071944 | Cell periphery | 22 | 0.02 |
| GO:0099023 | Vesicle tethering complex | 2 | 0.02 |
| GO:0120114 | Sm-like protein family complex | 3 | 0.01 |
| GO:1990072 | TRAPPIII protein complex | 1 | 0.05 |
| Molecular function | |||
| GO:0003725 | Double-stranded RNA binding | 1 | 0.01 |
| GO:0016208 | AMP binding | 1 | 0.03 |
| GO:0016624 | Oxidoreductase activity, acting on the aldehyde or oxo group of donors, disulfide as acceptor | 1 | 0.04 |
| GO:0019208 | Phosphatase regulator activity | 5 | 0.00 |
| GO:0019212 | Phosphatase inhibitor activity | 5 | 0.00 |
Fig. 7.

Gene Ontology enrichment analysis of the DEGs. X-axis represents the sub-categories; Y-axis represents number of genes in each sub-category
Table 9.
List of GO terms significantly down regulated in CPE-110 related to biological process, cellular component, and molecular function
| GO.ID | Term | DEGs | p_value |
|---|---|---|---|
| Biological process | |||
| GO:0009765 | Photosynthesis, light harvesting | 11 | 2E-06 |
| GO:0019684 | Photosynthesis, light reaction | 12 | 5E-05 |
| Cellular component | |||
| GO:0009521 | Photosystem | 15 | 0.01 |
| GO:0009532 | Plastid stroma | 2 | 0.04 |
| GO:0009534 | Chloroplast thylakoid | 34 | 0.03 |
| GO:0009536 | Plastid | 58 | 0.00 |
| GO:0009538 | Photosystem I reaction center | 4 | 0.00 |
| GO:0009579 | Thylakoid | 49 | 0.00 |
| GO:0009654 | Photosystem II oxygen evolving complex | 10 | 0.03 |
| GO:0010287 | Plastoglobule | 2 | 0.01 |
| GO:0031977 | Thylakoid lumen | 2 | 0.01 |
| GO:0031984 | Organelle subcompartment | 34 | 0.00 |
| GO:0034357 | Photosynthetic membrane | 45 | 0.00 |
| Molecular function | |||
| GO:0003755 | Peptidyl-prolyl cis-trans isomerase activity | 7 | 0.00 |
| GO:0004176 | ATP-dependent peptidase activity | 5 | 0.01 |
| GO:0004222 | Metalloendopeptidase activity | 5 | 0.04 |
| GO:0004417 | Hydroxyethylthiazole kinase activity | 1 | 0.04 |
| GO:0008124 | 4-Alpha-hydroxytetrahydrobiopterin dehydratase activity | 2 | 0.01 |
| GO:0008237 | Metallopeptidase activity | 9 | 0.00 |
| GO:0008887 | Glycerate kinase activity | 1 | 0.04 |
| GO:0016168 | Chlorophyll binding | 1 | 0.05 |
| GO:0016642 | Oxidoreductase activity, acting on the CH-NH2 group of donors, disulfide as acceptor | 2 | 0.01 |
| GO:0016667 | Oxidoreductase activity, acting on a sulfur group of donors | 4 | 0.02 |
| GO:0016703 | Oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of one atom of oxygen (internal monooxygenases or internal mixed function oxidases) | 3 | 0.04 |
| GO:0016709 | Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, NAD(P)H as one donor, and incorporation of one atom of oxygen | 2 | 0.03 |
| GO:0016765 | Transferase activity, transferring alkyl or aryl (other than methyl) groups | 4 | 0.04 |
| GO:0016846 | Carbon-sulfur lyase activity | 3 | 0.02 |
| GO:0019156 | Isoamylase activity | 1 | 0.03 |
| GO:0030170 | Pyridoxal phosphate binding | 2 | 0.02 |
| GO:0030410 | Nicotianamine synthase activity | 3 | 0.00 |
| GO:0035251 | UDP-glucosyltransferase activity | 4 | 0.01 |
| GO:0043743 | LPPG:FO 2-phospho-L-lactate transferase activity | 1 | 0.04 |
| GO:0046982 | Protein heterodimerization activity | 14 | 0.00 |
| GO:0070279 | Vitamin B6 binding | 2 | 0.02 |
| GO:0102193 | Protein-ribulosamine 3-kinase activity | 1 | 0.05 |
The KEGG pathway enrichment analysis was performed to determine the metabolic pathways in which the DEGs were involved. In total, 11 pathways were significantly enriched in both down and up-regulated DEGs where 9 and 2 pathways were specific to down and up-regulated DEGs in CPE-110 as compared to BPT 5204, respectively. The down-regulated DEGs were significantly over-represented in “photosynthesis” (20 genes), photosynthesis-antenna proteins (12 genes), glyoxylate and dicarboxylate metabolism (17 genes), carbon metabolism (30 genes), carbon fixation in photosynthetic organisms (14 genes), porphyrin and chlorophyll metabolism (10 genes), glycine, serine and threonine metabolism (10 genes), pyruvate metabolism (12 genes), and carotenoid biosynthesis (6 genes) (Table 10). In the case of up-regulated DEGs, two pathways were identified, namely, phenylalanine, tyrosine and tryptophan biosynthesis (7 genes), and peroxisome (9 genes) (Table 10).
Table 10.
Involvement of up and down regulated DEGs in different metabolic pathways for CPE
| KEGG ID | Pathway | Gene ratio | p value | Gene ID |
|---|---|---|---|---|
| Involvement of up regulated DEGs in different metabolic pathways in BPT 5204 for CPE | ||||
| osa00400 | Phenylalanine, tyrosine and tryptophan biosynthesis | 7/152 | 0.00 | 4328828/4333918/4335756/4341584/4343946/4345872/4349157 |
| osa04146 | Peroxisome | 9/152 | 0.00 | 4324062/4327232/4332347/4337714/4337904/4342124/4345945/4349764/4350881 |
| Involvement of down regulated DEGs in different metabolic pathways in CPE-110 for CPE | ||||
| osa00196 | Photosynthesis - antenna proteins | 12/223 | 0.00 | 4324599/4328623/4330828/4333359/4340892/4343583/4343604/4343709/4345663/4346803/4347166/4350176 |
| osa00195 | Photosynthesis | 20/223 | 0.00 | 4324479/4324933/4326537/4327150/4332431/4332745/4334300/4335799/4337500/4339593/4339833/4342192/4342370/4343366/4343515/4343570/4344899/4346326/4351694/4352085 |
| osa00630 | Glyoxylate and dicarboxylate metabolism | 17/223 | 0.00 | 4324401/4326849/4326980/4327981/4329690/4331509/4332108/4334274/4336245/4337051/4337272/4337447/4339682/4343993/4345962/4349114/4350456 |
| osa01200 | Carbon metabolism | 30/223 | 0.00 | 107276220/4324401/4325531/4326849/4326980/4327981/4329690/4330413/4330673/4331130/4331495/4331509/4331761/4332364/4334274/4335227/4336044/4336245/4337051/4338750/4339204/4339682/4341496/4342543/4343993/4345962/4347022/4347204/4349114/4350456 |
| osa00710 | Carbon fixation in photosynthetic organisms | 14/223 | 0.00 | 4325531/4330413/4331495/4331761/4332364/4334274/4335227/4336044/4338750/4339204/4339682/4341496/4342543/4343993 |
| osa00860 | Porphyrin and chlorophyll metabolism | 10/223 | 0.00 | 4326901/4330711/4332843/4333259/4341462/4346136/4348519/4348648/4349004/4349433 |
| osa00260 | Glycine, serine and threonine metabolism | 10/223 | 0.00 | 107276062/4324401/4326849/4326980/4327981/4336624/4337051/4345962/4349114/4350456 |
| osa00620 | Pyruvate metabolism | 12/223 | 0.00 | 4325531/4326849/4326980/4330673/4331130/4334274/4337361/4338750/4339682/4343993/4344858/4347022 |
| osa00906 | Carotenoid biosynthesis | 6/223 | 0.00 | 4328572/4330451/4335984/4336753/4345810/4352846 |
Discussion
Panicle exsertion is an important agronomic trait that influences grain yield in the rice crop. Shrunken and unfilled grains are generally observed in the incompletely exserted panicle rice genotypes. In this study, we utilized BPT 5204 and its mutant CPE-110 for the identification of genes governing CPE. BPT 5204 is a high yielding medium slender, highly adapted cultivar having incomplete panicle exsertion trait, grown in mostly central and southern regions of India, while CPE-110 is a completely exserted panicle, stabilized mutant, morphologically like BPT 5204. We observed inhibitory genetic inheritance pattern in F2 generation (n = 200) for panicle exsertion indicating the possibility of interaction between one homozygous recessive gene and another dominant homozygous/heterozygous gene leading to CPE. Our result differs with other studies where panicle exsertion was inherited as a dominant (Pandey and Gupta 1993) or recessive trait (Cruz et al. 2008; Zhao et al. 2018). For rapid identification of the gene controlling the panicle exsertion, MutMap, a method based on whole-genome sequencing of bulked DNA of F2 segregating population derived from BPT 5204 × CPE-110, was used in the present study.
In recent years, wild type and its mutant have emerged as an ideal material for detecting candidate genes because of high genetic similarity which eliminates most genetic background noise. MutMap identifies the SNPs introduced by mutagenesis that can be deployed further for trait improvement. The markers are used to identify the regions harboring the mutation responsible for a given phenotype, and the causal SNPs are readily identified due to sufficient sequence coverage in that region (Abe et al. 2012). In rice, using the MutMap approach, genes have been identified for important agronomic traits. For instance, 08SG2/OsBAK1 for small grain size (Yuan et al. 2017), OsRR22 for salinity tolerance (Takagi et al. 2015), WB1 for endosperm development (Wang et al. 2018), OsNRAMP5 for low Cd uptake and accumulation (Cao et al. 2019), MER3 for male sterility (Chen et al. 2014), OsCAO1 for pale green leaf (Abe et al. 2012), and dwf1 for dwarfism (Oh et al. 2020).
In the present study, we have identified 25 SNPs of eight putative candidate genes of novel genomic region on chromosome 8 (25668481-25750456) and chromosome 11 (20147154-20190400) for CPE using MutMap. After designing the KASP assay for 25 SNPs, one KASP marker, KASP 8-1, exhibited strong co-segregation with the trait. The KASP 8-1 (chrom8: 25683828) is located in the promoter region of LOC_Os08g40570, encoded for pyridoxamine 5′-phosphate oxidase family protein. Here, we found an SNP in the cis-acting regulatory DNA element (promoter) of LOC_Os08g40570 of CPE-110 that could be associated with CPE. The three signal sequences were recognized at SNP of promoter, namely, TATTAG (Fusada et al. 2005) (TACTAG in BPT 5204; TATTAG in CPE-110), GTAC (Kropat et al. 2005) (GTAC in BPT 5204; GTAT in CPE-110), and YACT (Gowik et al. 2004) (YACT in BPT 5204; YATT in CPE-110), regulating the expression of pyridoxamine 5′-phosphate oxidase. In wild type parent, BPT 5204, pyridoxamine 5′-phosphate oxidase oxidizes pyridoxamine-5-PO4 (PMP) and pyridoxine-5- PO4 (PNP) to pyridoxal-5-PO4. The pyridoxal-5-PO4 acts as a cofactor for ACC deaminase which catalyzes 1-aminocyclopropane-1-carboxylic acid (ACC) to ammonia and alpha-ketoglutaric acid resulting in lowering the synthesis of ethylene in wild type, BPT 5204. It has been reported that ethylene induces gibberellic acid synthesis by inducing kaurene synthase and down-regulating AP2-ERF (Qi et al. 2011). Also, in RNA-seq data, we found that LOC_Os10g01080 (Os10g0100700), a vitamin B6 biosynthesis protein family protein, was highly downregulated in mutant (11 fold change) CPE-110. Thus, it can be predicted that in mutant CPE-110, mutation in pyridoxamine 5′-phosphate oxidase resulted in enhanced ethylene and gibberellic acid that finally result in complete panicle exsertion from flag leaf (Fig. 8). We found the reduced expression of LOC_Os08g40570 and LOC_Os10g01080 in mutant CPE-110 via quantitative real time PCR (Fig. 9).
Fig. 8.

Role of LOC_Os08g40570 (pyridoxamine 5′-phosphate oxidase) in panicle exsertion
Fig. 9.

Reduced expressions of LOC_Os08g40570 and LOC_Os10g01080 demonstrated by quantitative real time PCR
Overall, this study revealed potential genomic regions in rice that could be associated with complete panicle exsertion. The transcriptome profiling points towards a possible involvement of gibberellic acid metabolism and other pathways in determining the extent of panicle exsertion. The study has resulted in the development of an SNP marker (KASP 8-1, pyridoxamine 5′-phosphate oxidase family protein) that could be of potential use in introgressing CPE trait into rice cultivars displaying panicle enclosure and hybrid rice programs. Earlier genomic regions, namely, qPEL10.2 on chromosome 10 (Dang et al. 2017) and qPE12 on chromosome 12 (Zhao et al. 2018) related with panicle exsertion, were identified; however, we identified novel genomic region as well as potential candidate gene for complete panicle exsertion in rice. Additionally, candidate genes have been identified, that might be of prime importance in governing CPE in rice, thus providing scope for functional characterization of these genes. The results from this study are expected to accelerate the genetic improvement of rice, especially in hybrid seed production.
Supplementary information
(DOCX 1.47 mb)
(DOCX 74 kb)
(XLSX 13 kb)
(XLSX 11 kb)
(XLSX 478 kb)
(XLSX 614 kb)
(XLSX 13 kb)
(XLSX 10 kb)
(XLSX 11 kb)
(XLSX 10 kb)
(XLSX 9 kb)
(XLSX 9 kb)
Author contribution
MSM conceived the study; MSM and KMB planned and designed the experiments; AH, SB, GCG, NM, AP, EPV, PGA, and PV performed phenotyping, validation experiments, and genotyping. MSM and HKP contributed to the genetic material. SD, GCG, and KA executed scripts for MutMap data. AH, KMB, SB, and NM analyzed the data. AH and KMB drafted the manuscript. MSM, RMS, KMB, and HKP critically revised the manuscript. All authors reviewed and approved the submission.
Funding
Authors are thankful to the directors, ICAR-IIRR and CSIR-CCMB and Council of Scientific and Industrial Research, Government of India, for the financial support. This work was supported by the Council of Scientific and Industrial Research, Government of India, Grant No. 34/1/TD-AgriNutriBiotech/NCP-FBR 2019-RPPBDD/TMD-SeMI to Dr. Maganti S Madhav and Dr. Hitendra K Patel.
Data availability
All the experiments and data analyses were conducted in ICAR-Indian Institute of Rice Research Hyderabad, India. MutMap analysis was done at CSIR-Centre for Cellular and Molecular Biology, Hyderabad, India.
Declarations
Ethics approval
We declare that these experiments complied with the ethical standards in India.
Consent to participate
Not applicable.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Anil A Hake and Suneel Ballichatla contributed equally to this paper.
Contributor Information
Kalyani M. Barbadikar, Email: kalyaniaau@gmail.com
Sheshu Madhav Maganti, Email: sheshu24@gmail.com.
References
- Abe A, Kosugi S, Yoshida K, et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol. 2012;30:174–178. doi: 10.1038/nbt.2095. [DOI] [PubMed] [Google Scholar]
- Alexa A, Rahnenfuhrer J. topGO: topGO: Enrichment analysis for Gene Ontology. R Package Version. 2010;2(18):0. [Google Scholar]
- Bao-Jian Q, Xiao-Biao ZHU, Ying-Ying W, De-Lin H. Mapping of QTL for three panicle exsertion-related traits in rice under different growing environments. Acta Agron Sin. 2008;34:389–396. [Google Scholar]
- Cao ZZ, Lin XY, Yang YJ, et al. Gene identification and transcriptome analysis of low cadmium accumulation rice mutant (lcd1) in response to cadmium stress using MutMap and RNA-seq. BMC Plant Biol. 2019;19:1–13. doi: 10.1186/s12870-019-1867-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Yan W, Wang N, et al. Cloning of a rice male sterility gene by a modified MutMap method. Yi Chuan. 2014;36:85–93. doi: 10.3724/SP.J.1005.2014.00085. [DOI] [PubMed] [Google Scholar]
- Cheng A-P, Chen S-Y, Lai M-H, et al. Transcriptome analysis of early defenses in rice against Fusarium fujikuroi. Rice. 2020;13:1–15. doi: 10.1186/s12284-020-00426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz RP, Milach SCK, Federizzi LC. Inheritance of pinacle exsertion in rice. Sci Agric. 2008;65:502–507. doi: 10.1590/S0103-90162008000500009. [DOI] [Google Scholar]
- Dang X, Fang B, Chen X et al (2017) Favorable marker alleles for panicle exsertion length in rice (Oryza sativa L.) mined by association mapping and the RSTEP-LRT method. Front. Plant Sci 8. 10.3389/fpls.2017.02112 [DOI] [PMC free article] [PubMed]
- Das A, Mazahar M, Sahu A et al (2021) Differential regulation of rice transcriptome to Rhizoctonia solani infection. bioRxiv. 10.1101/2021.05.05.442799
- Du H, Yu Y, Ma Y, et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat Commun. 2017;8:15324. doi: 10.1038/ncomms15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan YL, Guan HZ, Ming ZHUO, et al. Genetic analysis and mapping of an enclosed panicle mutant locus esp1 in rice (Oryza sativa L.) J Integr Agric. 2012;1:1933–1939. doi: 10.1016/S2095-3119(12)60449-3. [DOI] [Google Scholar]
- Fusada N, Masuda T, Kuroda H, et al. Identification of a novel cis-element exhibiting cytokinin-dependent protein binding in vitro in the 5′-region of NADPH-protochlorophyllide oxidoreductase gene in cucumber. Plant Mol Biol. 2005;59:631–645. doi: 10.1007/s11103-005-0579-x. [DOI] [PubMed] [Google Scholar]
- Gowik U, Burscheidt J, Akyildiz M, et al. Cis-regulatory elements for mesophyll-specific gene expression in the C4 plant Flaveria trinervia, the promoter of the C4 phosphoenolpyruvate carboxylase gene. Plant Cell. 2004;16:1077–1090. doi: 10.1105/tpc.019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlina L, Trijatmiko KR. Identification of quantitative trait loci (QTL) for awn, incomplete panicle exertion and total spikelet number in an F2 population derived from a backcross inbred line, bio-148, and the recurrent parent, IR64. Makara J Sci. 2016;20:3. doi: 10.7454/mss.v20i1.5657. [DOI] [Google Scholar]
- Hsu S-K, Tung C-W (2017) RNA-seq analysis of diverse rice genotypes to identify the genes controlling coleoptile growth during submerged germination. Front Plant Sci 762. 10.3389/fpls.2017.00762 [DOI] [PMC free article] [PubMed]
- Kandpal M, Vishwakarma C, Krishnan K, et al. Gene expression dynamics in rice peduncles at the heading stage. Front Genet. 2020;11:584678. doi: 10.3389/fgene.2020.584678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katara JL, Verma RL, Parida M, et al. Differential expression of genes at panicle initiation and grain filling stages implied in heterosis of rice hybrids. Int J Mol Sci. 2020;21:1080. doi: 10.3390/ijms21031080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropat J, Tottey S, Birkenbihl RP, et al. A regulator of nutritional copper signaling in Chlamydomonas is an SBP domain protein that recognizes the GTAC core of copper response element. Proc Natl Acad Sci. 2005;102:18730–18735. doi: 10.1073/pnas.0507693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukurba KR, Montgomery SB (2015) RNA sequencing and analysis. Cold Spring Harb Protoc 11:951–969. 10.1101/pdb.top084970 [DOI] [PMC free article] [PubMed]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Ma C, Tai H, et al. Comparative transcriptome analysis of two rice genotypes differing in their tolerance to saline-alkaline stress. PLoS One. 2020;15:e0243112. doi: 10.1371/journal.pone.0243112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Magar ND, Shah P, Harish K, et al. Gene expression and transcriptome sequencing: basics, analysis, advances. IntechOpen: InGene Expression; 2022. [Google Scholar]
- Neeraja CN, Barbadikar KM, Krishnakanth T, et al. Down regulation of transcripts involved in selective metabolic pathways as an acclimation strategy in nitrogen use efficient genotypes of rice under low nitrogen. 3 Biotech. 2021;11:80. doi: 10.1007/s13205-020-02631-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Cheon K-S, Kang D-Y, et al. MutMap analysis of a rice dwarf mutant line. Korean Soc Breed Sci. 2020;52:9–19. doi: 10.9787/KJBS.2020.52.1.9. [DOI] [Google Scholar]
- Pandey DK, Gupta HS. Genetics of panicle exsertion in cold-tolerant rice (Oryza sativa) Plant Breed. 1993;111:82–85. doi: 10.1111/j.1439-0523.1993.tb00610.x. [DOI] [Google Scholar]
- Phule AS, Barbadikar KM, Maganti SM, et al. RNA-seq reveals the involvement of key genes for aerobic adaptation in rice. Sci Rep. 2019;9:1–10. doi: 10.1038/s41598-019-41703-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potupureddi G, Balija V, Ballichatla S, et al. Mutation resource of Samba Mahsuri revealed the presence of high extent of variations among key traits for rice improvement. PLoS One. 2021;16:e0258816. doi: 10.1371/journal.pone.0258816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Sun F, Wang Q, et al. Rice ethylene-response AP2/ERF factor OsEATB restricts internode elongation by down-regulating a gibberellin biosynthetic gene. Plant Physiol. 2011;157:216–228. doi: 10.1104/pp.111.179945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satturu V, Kudapa HB, Muthuramalingam P, et al. RNA-seq based global transcriptome analysis of rice unravels the key players associated with brown planthopper resistance. Int J Biol Macromol. 2021;191:118–128. doi: 10.1016/j.ijbiomac.2021.09.058. [DOI] [PubMed] [Google Scholar]
- Sugihara Y, Young L, Yaegashi H, et al. High-performance pipeline for MutMap and QTL-seq. PeerJ. 2022;10:e13170. doi: 10.7717/peerj.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H, Tamiru M, Abe A, et al. MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat Biotechnol. 2015;33:445–449. doi: 10.1038/nbt.3188. [DOI] [PubMed] [Google Scholar]
- Tarazona S, Furió-Tarí P, Turrà D, et al. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43:e140–e140. doi: 10.1093/nar/gkv711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Tariq R, Ji Z, et al. Transcriptome analysis of a rice cultivar reveals the differentially expressed genes in response to wild and mutant strains of Xanthomonas oryzae pv. oryzae. Sci Rep. 2019;9:1–13. doi: 10.1038/s41598-019-39928-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang Y, Sun L, et al. WB1, a regulator of endosperm development in rice, is identified by a modified MutMap method. Int J Mol Sci. 2018;19:2159. doi: 10.3390/ijms19082159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T-Y, Müller M, Gruissem W, et al. Genome wide analysis of the transcriptional profiles in different regions of the developing rice grains. Rice. 2020;13:1–19. doi: 10.1186/s12284-020-00421-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Bai J, Yang YY, et al. The RNA-seq transcriptome analysis identified genes related to rice seed dormancy. Biol Plant. 2019;63:308–313. doi: 10.32615/bp.2019.035. [DOI] [Google Scholar]
- Xu H, Gao Y, Wang J. Transcriptomic analysis of rice (Oryza sativa) developing embryos using the RNA-seq technique. PLoS One. 2012;7:e30646. doi: 10.1371/journal.pone.0030646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Pan R, Zhang Y, et al. RNA-seq-based profiling of pl mutant reveals transcriptional regulation of anthocyanin biosynthesis in rice (Oryza sativa L.) Int J Mol Sci. 2021;22:9787. doi: 10.3390/ijms22189787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Li S, Xiao Y, et al. Transcriptome analysis of rice response to blast fungus identified core genes involved in immunity. Plant Cell Environ. 2021;44:3103–3121. doi: 10.1111/pce.14098. [DOI] [PubMed] [Google Scholar]
- Yang D-W, Zhu Z, Zhang Y-D, et al. Substitution mapping of QTL for panicle exertion using CSSL in rice (Oryza sativa L.) Yi Chuan. 2009;31:741–747. doi: 10.3724/sp.j.1005.2009.00741. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. Omi a J Integr Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Fan S, Huang J, et al. 08SG2/OsBAK1 regulates grain size and number, and functions differently in Indica and Japonica backgrounds in rice. Rice. 2017;10:1–12. doi: 10.1186/s12284-017-0165-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan C, Hu J, Pang Q, et al. Genome-wide association analysis of panicle exsertion and uppermost internode in rice (Oryza sativa L.) Rice. 2019;12:1–13. doi: 10.1186/s12284-019-0330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Sun P, He Q, et al. Transcriptome analysis of near-isogenic line provides novel insights into genes associated with panicle traits regulation in rice. PLoS One. 2018;13:e0199077. doi: 10.1371/journal.pone.0199077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Zhao Q, Zhao L, et al. Characterization and fine mapping of qPE12, a new locus controlling rice panicle exsertion. Euphytica. 2018;214:1–10. doi: 10.1007/s10681-017-2104-0. [DOI] [Google Scholar]
- Zhao CF, Chen T, Zhao QY, et al. Analysis of QTLs for panicle exsertion and its relationship with yield and yield-related traits in rice (Oryza sativa L.) Genet Mol Res. 2016;15:gmr.15027423. doi: 10.4238/gmr.15027423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 1.47 mb)
(DOCX 74 kb)
(XLSX 13 kb)
(XLSX 11 kb)
(XLSX 478 kb)
(XLSX 614 kb)
(XLSX 13 kb)
(XLSX 10 kb)
(XLSX 11 kb)
(XLSX 10 kb)
(XLSX 9 kb)
(XLSX 9 kb)
Data Availability Statement
All the experiments and data analyses were conducted in ICAR-Indian Institute of Rice Research Hyderabad, India. MutMap analysis was done at CSIR-Centre for Cellular and Molecular Biology, Hyderabad, India.