

Abstract
Specificity for a desired enzyme target is an essential property of small-molecule inhibitors. Molecules targeting oncogenic driver mutations in the epidermal growth factor receptor (EGFR) kinase domain have had a considerable clinical impact due to their selective binding to cancer-causing mutants compared to wild type. Despite the availability of clinically approved drugs for cancers driven by EGFR mutants, persistent challenges in drug resistance in the past decades have led to newer generations of drugs with divergent chemical structures. The current clinical challenges are mainly due to acquired resistance to third generation inhibitors, including by the acquisition of the C797S mutation. Several diverse fourth generation candidates and tool compounds that inhibit the C797S mutant have emerged, and their structural characterization has revealed molecular factors that allow for EGFR mutant selective binding. Here, we have reviewed all known structurally-characterized EGFR TKIs targeting clinically-relevant mutations to identify specific features that enable C797S inhibition. Newer generation EGFR inhibitors exhibit consistent and previously underutilized hydrogen bonding interactions with the conserved K745 and D855 residue side chains. We also consider binding modes and hydrogen bonding interactions of inhibitors targeting the classical ATP and the more unique allosteric sites.
Keywords: Kinases, Epidermal growth factor receptor, Lung cancer, Selectivity, Inhibitors, Drugs, Structural biology
Introduction
Tyrosine kinase inhibitors (TKIs) targeting EGFR represent effective therapies in non-small cell lung cancer (NSCLC) due to their ability to selectively inhibit the cancer-causing mutants.(Lynch et al., 2004; Paez et al., 2004) Selective binding for the disease-causing mutation while simultaneously exhibiting limited inhibition of the wild type EGFR is critical for establishing a therapeutic window that maximizes clinical efficacy and limits toxic side effects.(Eck & Yun, 2010) Various structural and pharmacological studies have established that the prevalent activating EGFR mutations, including in-frame deletions in exon 19 (exon19del), insertions in exon 20, and point mutations such as G719S and L858R, all effectively perturb the structure of the kinase domain to ignore ordinary ligand-dependent activation.(Eck & Yun, 2010) The ATP-competitive first-generation 4-anilinoquinazoline-based drugs gefitinib and erlotinib are approved for the treatment of L858R and exon19 del variants and are selective for these activating mutations due to the fortuitously lower ATP affinity for the mutant kinase domain.(Carey et al., 2006; C.-H. Yun et al., 2007) Second-generation inhibitors have emerged based on the 4-anilinoquinazoline scaffold, such as afatinib, that form a covalent bond to the kinase domain through a Michael acceptor that forms an irreversible covalent bond with C797,(Solca et al., 2012) and inactive conformation binding lapatinib.(Moy, Kirkpatrick, Kar, & Goss, 2007) These compounds lack the needed selectivity for EGFR mutants required for clinical efficacy but are approved for other tumors. The emergence of acquired drug resistance to the first-generation 4-anilinoquinazoline drugs through acquisition of the gatekeeper T790M mutation(Kobayashi et al., 2005; C.-H. Yun et al., 2008) led to the development of third-generation anilinopyrimidine-based TKIs with similar C797-targeting Michael acceptors to second-generation compounds, but with marked mutant selectivity.(Oxnard et al., 2016; Zhou, Ercan, Chen, Yun, Li, Capelletti, Cortot, Chirieac, Iacob, Padera, et al., 2009) Of the many third-generation TKIs, AZD9291 (Osimertinib) has received clinical approval for T790M-positive NSCLC and more recently as a first-line therapy.(Soria et al., 2018) Drug resistance as a result of treatment by AZD9291 occurs in part because of the acquisition of a third point mutation (C797S) preventing inhibitors from establishing their potency-conferring covalent bonds.(Niederst et al., 2015; Thress et al., 2015)
To address drug resistance to AZD9291 where no current therapies are available, several drug discovery programs have generated newer fourth-generation inhibitors that target C797S-containing EGFR and are well-positioned for clinical evaluation.(L. Chen, Fu, Zheng, Liu, & Liang, 2017; H.-Y. Zhao, Xi, Xin, & Zhang, 2022) Expectedly, these new inhibitors are structurally divergent from third-generation inhibitors. Additionally, distinct allosteric site targeting inhibitors offer another avenue to target C797S.(D. J. H. De Clercq et al., 2019; Thomas W. Gero et al., 2022; Jia et al., 2016; To et al., 2019) Here, we review the structures and binding modes of these next-generation EGFR inhibitors and show how their structures produce previously underutilized intermolecular interactions from the previous third-generation inhibitors.
EGFR kinase architecture in active versus inactive conformations.
Understanding inhibitor binding modes and their role in enabling drug selectivity first requires a short discussion of the structural elements of the EGFR kinase domain. Generally, protein kinases are phosphoryltransferase enzymes targeting alcohol-containing amino acid side chains (Ser, Thr, and Tyr) of protein targets.(Taylor, Knighton, Zheng, Ten Eyck, & Sowadski, 1992) This activity is enabled by hydrolysis of the γ-phosphate of ATP and requires Mg2+ ions. Specifically, EGFR is a tyrosine kinase (E.C. 2.7.10.1) often involved in autophosphorylation of the long C-terminal tail of other receptor tyrosine kinases (Figure 1).(Hunter, 2009; Lemmon & Schlessinger, 2010) As common in all kinases, the kinase domain in EGFR consists of a conserved ATP-binding site that is located between the N- and C-lobes of the kinase domain. The short peptide linking the lobes is known as the “hinge” (amino acids 790 to 797) and is where the adenosine ring of ATP makes hydrogen bonds (H-bonds) with Q791 and M793. A Thr residue at position 790, known as the “gatekeeper,” is at the N-terminus of the hinge while Cys at 797, a common target of covalent inhibitors, is at the C-terminus. In the inactive conformation, a conserved α-helix (αC-helix, red Figure 2A) is rotated in the “out” position and held in this position by the folded activation loop (A-loop, orange Figure 2A). Activation of EGFR is regulated by extracellular binding of cognate ligands that induce homo-or heterodimerization of kinase domains that induce structural changes within the kinase domain.(Zhang, Gureasko, Shen, Cole, & Kuriyan, 2006) Kinase dimerization leads to inward rotation of the αC-helix and formation of a H-bond between E762 and K745, known as the K-E salt bridge, that is essential for phosphoryl transferase enzymatic activity (Figure 2B). K745 is conserved in all kinases and often referred to as the “catalytic lysine.”(Carrera, Alexandrov, & Roberts, 1993) Another important conserved residue is D855 of the DFG-motif, which is directly involved in H-bonds with ATP and the protein conformation of this element is important in distinguishing certain types of kinase inhibitors.(Müller, Chaikuad, Gray, & Knapp, 2015) While it seems counterintuitive, several new EGFR inhibitors target these active sites conserved residues with noteworthy selectivity for oncogenic mutants compared to wildtype.(See Table1)
Figure 1. The catalytic cycle of a protein kinase.
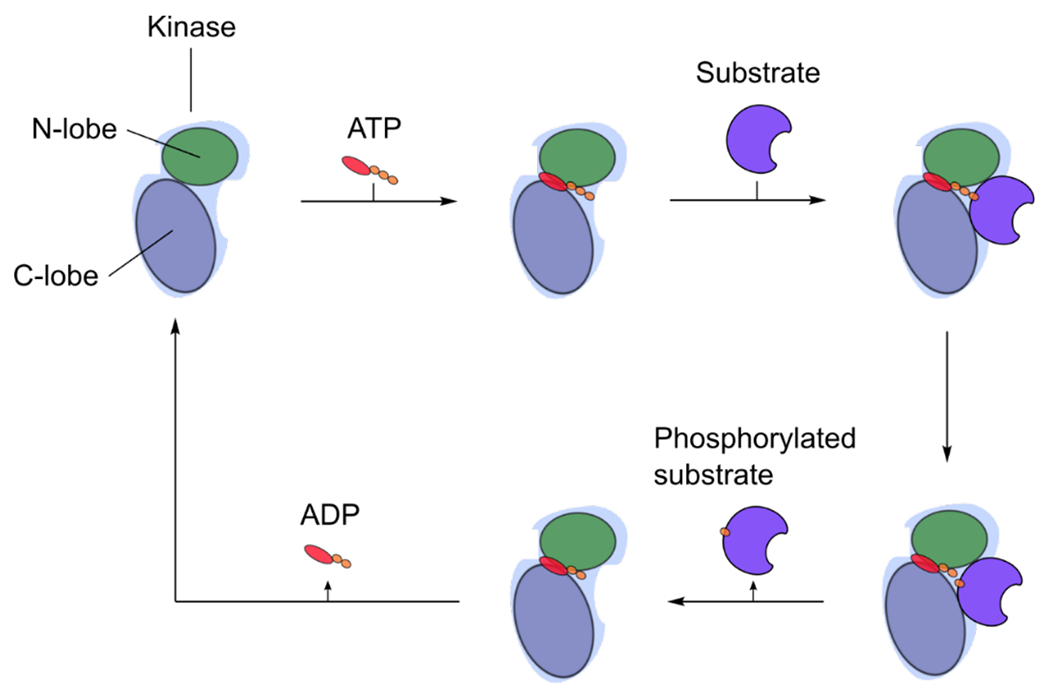
ATP binds to the active site of the kinase, which consists of an N-terminal and C-terminal lobe. The γ-phosphate of ATP is hydrolyzed and transferred to the hydroxyl oxygen of a protein target residue side chain (Ser, Thr or Tyr) and concomitant production of ADP.
Figure 2. The interplay of residues and binding pockets in the inactive and active EGFR kinase domain.
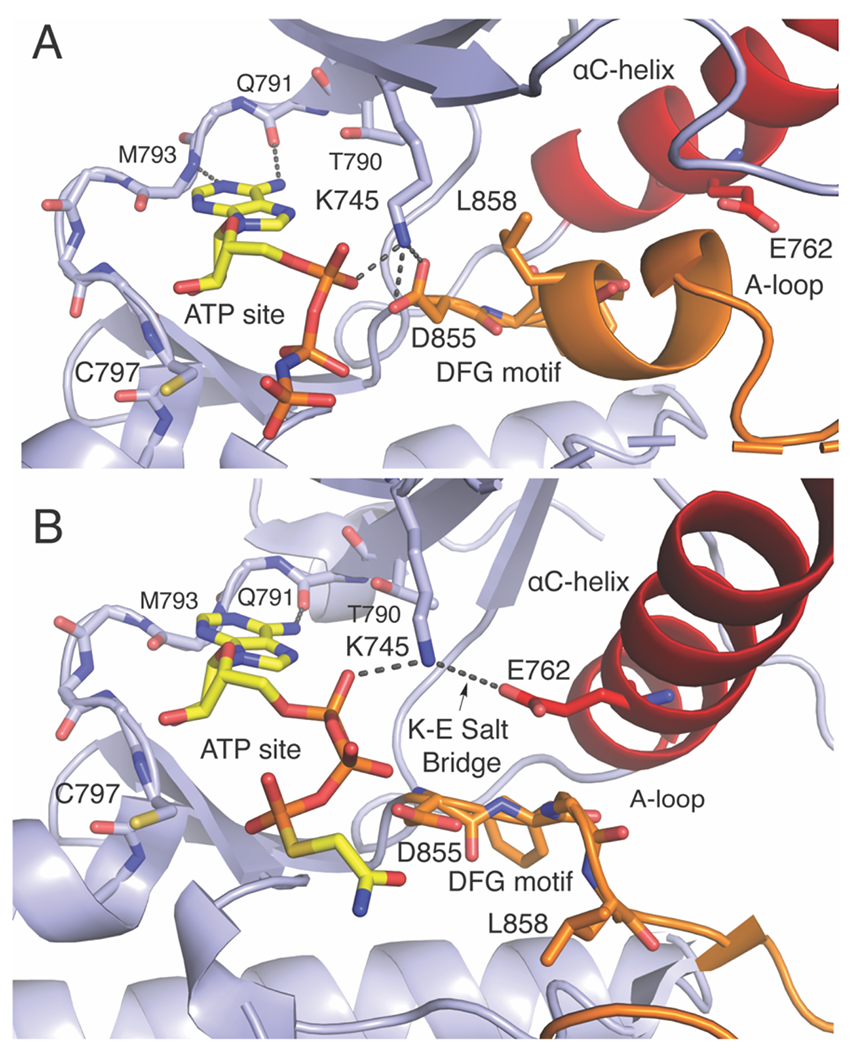
The EGFR kinase domain in the A) inactive (PDB ID 2GS7, ATP site defined by AMP-PNP is colored yellow) and B) active conformations (PDB ID 2GS6, ATP site defined by an ATP analog-peptide conjugate). Activation loop (A-loop) is colored orange and αC-helix colored red. Some hinge region protein side chains are omitted for clarity
Table 1.
Structurally characterized third-generation EGFR inhibitors targeting Ex19del/L858R/T790M mutations.
| Compound Namea | PDB ID | EGFR Kinase Domain αC-helix | Interactions with K745 and D855 | Ref | |
|---|---|---|---|---|---|
| 1 | AZD9291 (Osimertinib) | 6JX4 | in | None | (Xiao-E Yan et al., 2020) |
| 2 | 6JX0 | out | None | (Xiao-E Yan et al., 2020) | |
| 3 | 6JWL | in | None | (Xiao-E Yan et al., 2020) | |
| 4 | 6JXT | out | None | (Xiao-E Yan et al., 2020) | |
| 5 | 4ZAU | in | None | (Yosaatmadja et al., 2015) | |
| 6 | AZD5104 (AZD9291 metabolite) | 7JXL | out | Indole H-bond between D855 | (Tyler S Beyett, To, et al., 2022) |
| 7 | CO-1686 (Rociletinib) | 5XDK | in | None | (Yan, Zhu, Liang, Zhao, Choi, et al., 2017) |
| 8 | 5XDL | in | None | (Yan, Zhu, Liang, Zhao, Choi, et al., 2017) | |
| 9 | 5UWD | out | −CF3 dipole-dipole between K745 | (Niessen et al., 2017) | |
| 10 | WZ4002 | 3IKA | in | None | (Zhou, Ercan, Chen, Yun, Li, Capelletti, Cortot, Chirieac, Iacob, & Padera, 2009) |
| 11 | WZ4003 | 5X2K | in | None | (Zhu et al., 2018) |
| 12 | ASP8273 (Naquotinib) | 5Y9T | in | None | (Hirano et al., 2018) |
| 13 | EGF816 (Nazartinib) | 5FEQ | in | None | (Gerald Lelais et al., 2016) |
| 14 | Compound 43 (Related to EGF816) (1) | 5FEE | in | −CF3 dipole-dipole between K745 | (Gerald Lelais et al., 2016) |
| 15 | 5FED | in | None | (Gerald Lelais et al., 2016) | |
| 16 | PF-06747775 (Mavelertinib) | 7JXI | out | None | (Tyler S. Beyett, To, et al., 2022) |
| 17 | YH25448 (Lazertinib) | 7UKW | out | H-bond between D855 | (D. Heppner et al., 2022) |
| 18 | 7UKV | in | H-bond between D855 | (D. Heppner et al., 2022) | |
| 19 | LDC8201 | 7A6I | in | None | (Lategahn et al., 2022) |
| 20 | TAK-788 | 7B85 | in | None | (Lategahn et al., 2022) |
| 21 | QL-X138 | 4WD5 | in | None | To be published. |
Note that we have listed all compounds according to their generic name or compound code included in the original reference. We have elected to enumerate all of the compounds here that are reported by numeral in the original reference for clarity.
Third Generation Inhibitors (ATP competitive)
To best understand the binding modes of C797S-targeting inhibitors, a brief survey of the binding modes of third-generation inhibitors is necessary. Third-generation EGFR TKIs were developed in response to patents acquiring the gatekeeper T790M resistance mutation as a result of treatment with first-generation inhibitors.(L. Chen et al., 2017; Tan et al., 2018) They are all irreversible inhibitors that form a covalent bond with the thiol of C797, and resistance to these TKIs through the C797S point mutation that renders them essentially inactive.(Niederst et al., 2015; Thress et al., 2015) Generally, their chemical structures contain a hinge binding motif, which is the anilinopyrimidine, and an acrylamide as originally described in the development of WZ4002 (Scheme 1). (Zhou, Ercan, Chen, Yun, Li, Capelletti, Cortot, Chirieac, Iacob, Padera, et al., 2009)
Scheme 1.
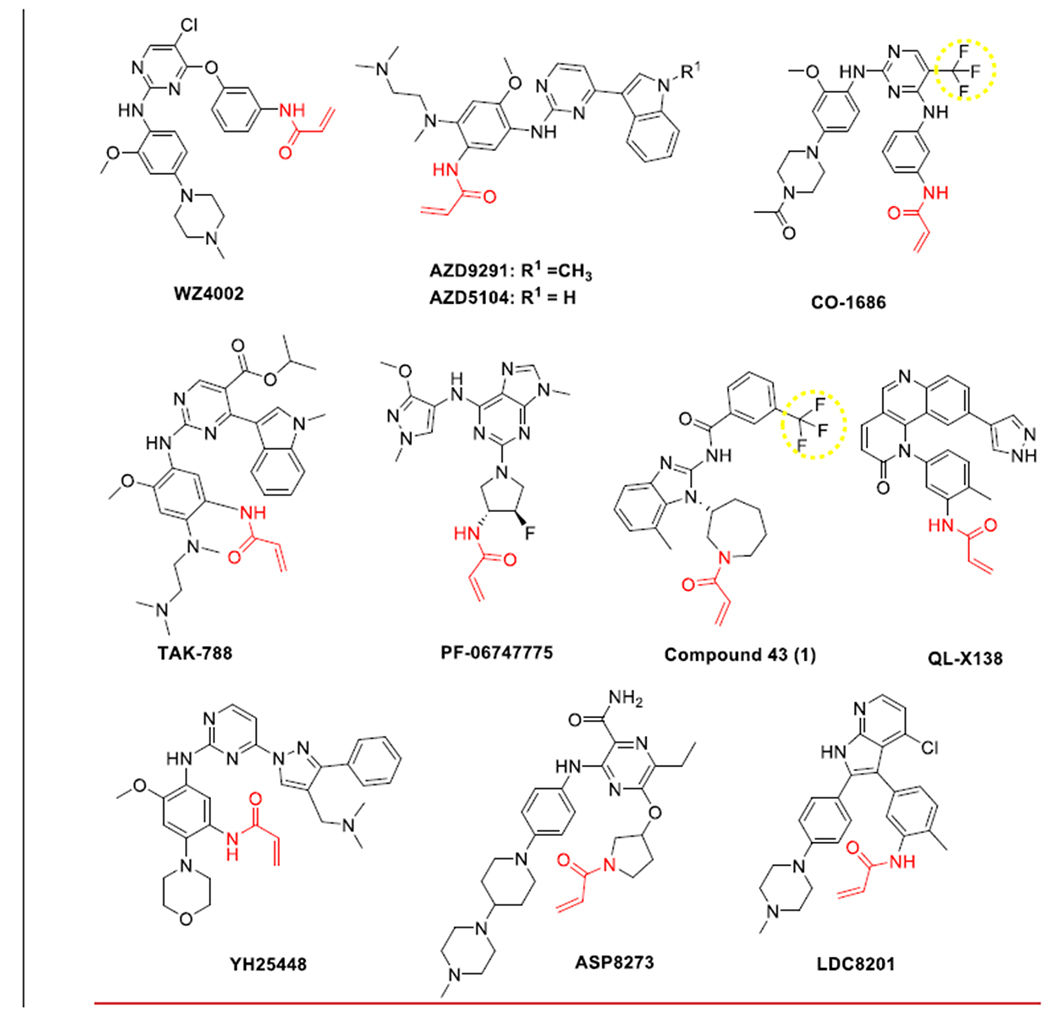
Structurally characterized third-generation EGFR TKIs. Groups that are observed in dipole-dipole interactions with K745 are highlighted with dash yellow circles and the C797-targeting acrylamide warheads are colored red.
Due to its clinical success, AZD9291 (osimertinib) has been the subject of intense study with the goal of understanding its selectivity for T790M mutant EGFR.(Yosaatmadja et al., 2015) Recent X-ray crystallography and molecular modeling studies have shown that AZD9291 exhibits T790M-selective binding through a previously underappreciated rotation of the N-methylindole group producing favorable van der Waals interactions with the T790M mutant methionine residue (Figure 3A).(Xiao- E. Yan et al., 2020) More recently, a structurally-related inhibitor YH25448 (lazertinib) was shown to possess improved medicinal chemistry properties over AZD9291 due to the presence similar van der Waals interactions with T790M and a unique intramolecular H-bond between amine and amide group in addition to an H-bond with the D855 of the DFG motif (Figure 3B).(D.E. Heppner et al., 2022; J. Yun et al., 2019).
Figure 3. Structures and binding modes of selected third-generation EGFR TKIs.
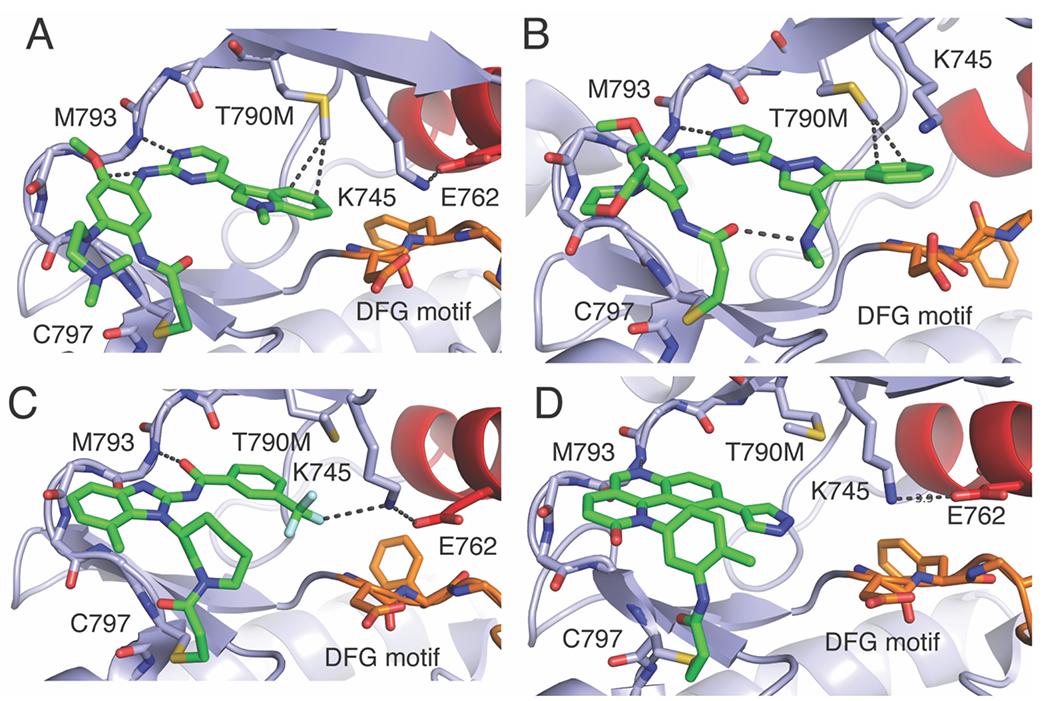
A) AZD9291 in complex with EGFR(T790M) in the active conformation PDB 6JX0. B) YH25448 in complex with T790M/V948R in the inactive conformation PDB ID 7UKW. C) Compound 43 (1) (Gérald Lelais et al., 2016)(related to EGF816) in complex with EGFR(T790M) active conformation with −CF3 in a dipole-dipole interaction with K745 H-bond PDB ID 5FEE. D) QL-X138 in complex with EGFR(T790M) in active conformation with pyrazole directed toward K745 PDB ID 7A6I.
One important point of difference that will be made in the following section is that nearly all the reversible binding fourth-generation inhibitors make H-bonding interactions with K745, which are seen in a minority of third-generation inhibitors (Table 1). One example is the phase 1 metabolite of AZD9291 (AZD5104), which is known to have similar potency against WT EGFR and the demethylation of the indole nitrogen leads to a H-bond with D855 (PDB ID 7JXL).(Tyler S. Beyett, To, et al., 2022) Other inhibitors that show H-bonding interactions with K745 include compound 43 (1) (related to EGF816)(Gérald Lelais et al., 2016) through a −CF3 group (Figure 3C). Moreover, CO-1686(Yan, Zhu, Liang, Zhao, Geun Choi, et al., 2017) shows a dipole-dipole interaction through a −CF3 group with K745. (Table 1) While not seen in this active conformation structure, a potential H-bond is possible via the pyrazole of QL-X138 and could be relevant in the inactive (αC-helix out) conformation where the K-E salt bridge is not formed (Figure 3D).
Fourth Generation Inhibitors (ATP competitive)
The emergence of acquired drug resistance to AZD9291, and other third-generation TKIs, through the C797S mutation presented a variety of opportunities for developing novel inhibitors and the earliest examples of reversible-binding ATP-competitive inhibitors emerged serendipitously.(Niederst et al., 2015; Park et al., 2020; Thress et al., 2015) (Table 2) The first series of ATP-competitive inhibitors that exhibited C797S activity were based on a trisubstituted imidazole core.(Günther, Juchum, Kelter, Fiebig, & Laufer, 2016) Optimization yielded imidazole inhibitors that bound reversibly to C797S-containing EGFR kinases with low nanomolar biochemical potencies.(Günther et al., 2016; Günther et al., 2017; Juchum et al., 2017) A series of reversible and irreversible imidazole inhibitors showed comparable H-bond to the catalytic K745 residue only in αC-helix outward inactive crystal structures (Scheme 2, Figure 4A).(David E. Heppner, Günther, Wittlinger, Laufer, & Eck, 2020) These imidazole-K745 H-bonds are observed in addition to H-bonds between K745 and D855.(David E. Heppner et al., 2020) Selective N-methylation of the imidazole nitrogens showed that blocking the K745 H-bond was responsible for the C797S-dependent reversible binding properties. This highlights the functional importance of this H-bonding interaction for enabling stronger reversible binding and implies that additional interactions not ordinately seen in the majority of third-generation TKIs may be the key to enhancing the potency of small-molecule inhibitors against C797S. However, the imidazole-based inhibitors are most potent against WT EGFR implying that H-bonds with K745 are necessary for tight reversible binding but not mutant selectivity.
Table 2.
Structurally characterized fourth-generation EGFR inhibitors targeting C797S mutants.
Note that we have listed all compounds according to their generic name or compound code included in the original reference. We have elected to enumerate all compounds here that are reported by numeral in the original reference for clarity.
Scheme 2.
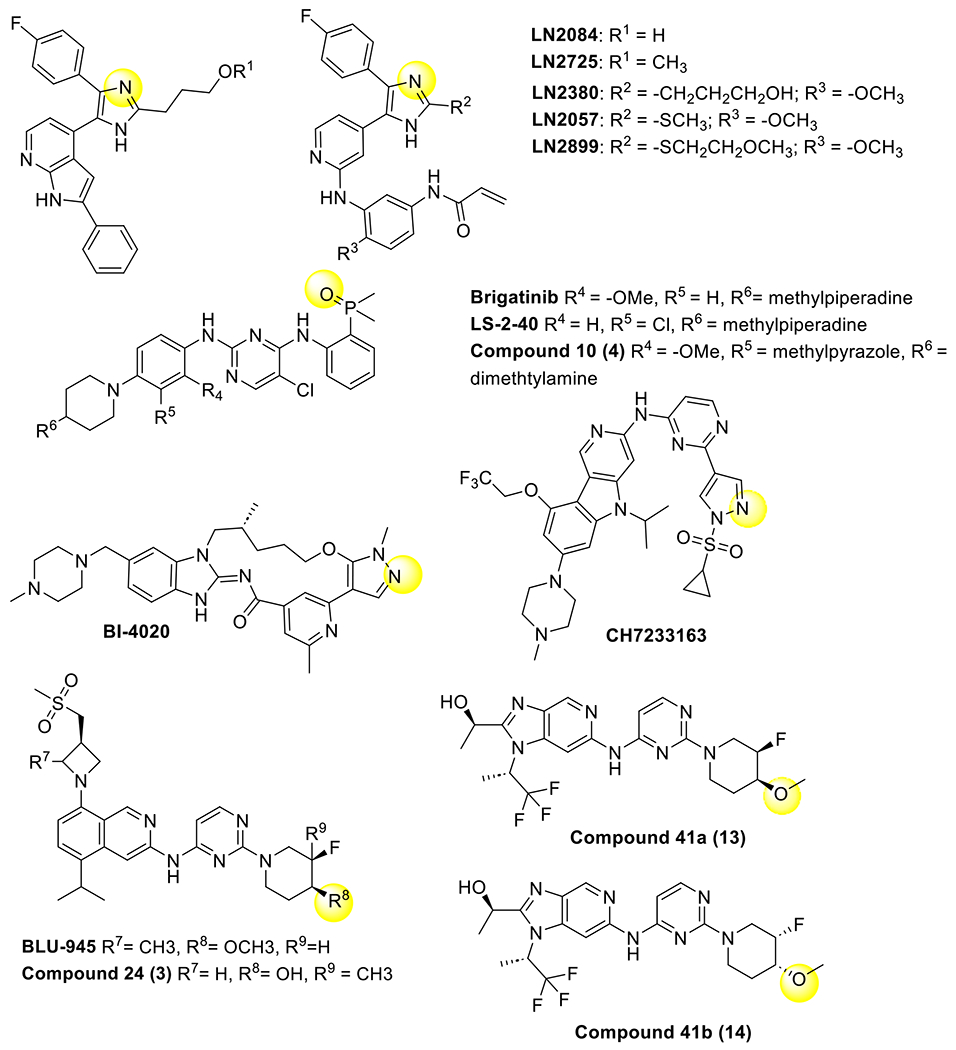
Structurally characterized ATP-competitive fourth-generation EGFR TKIs and related analogues (Table 2). Groups that are engaged in H-bonds with K745 are highlighted with yellow circles.
Figure 4. Structures and binding modes of reversible-binding fourth generation EGFR TKIs that inhibit C797S.
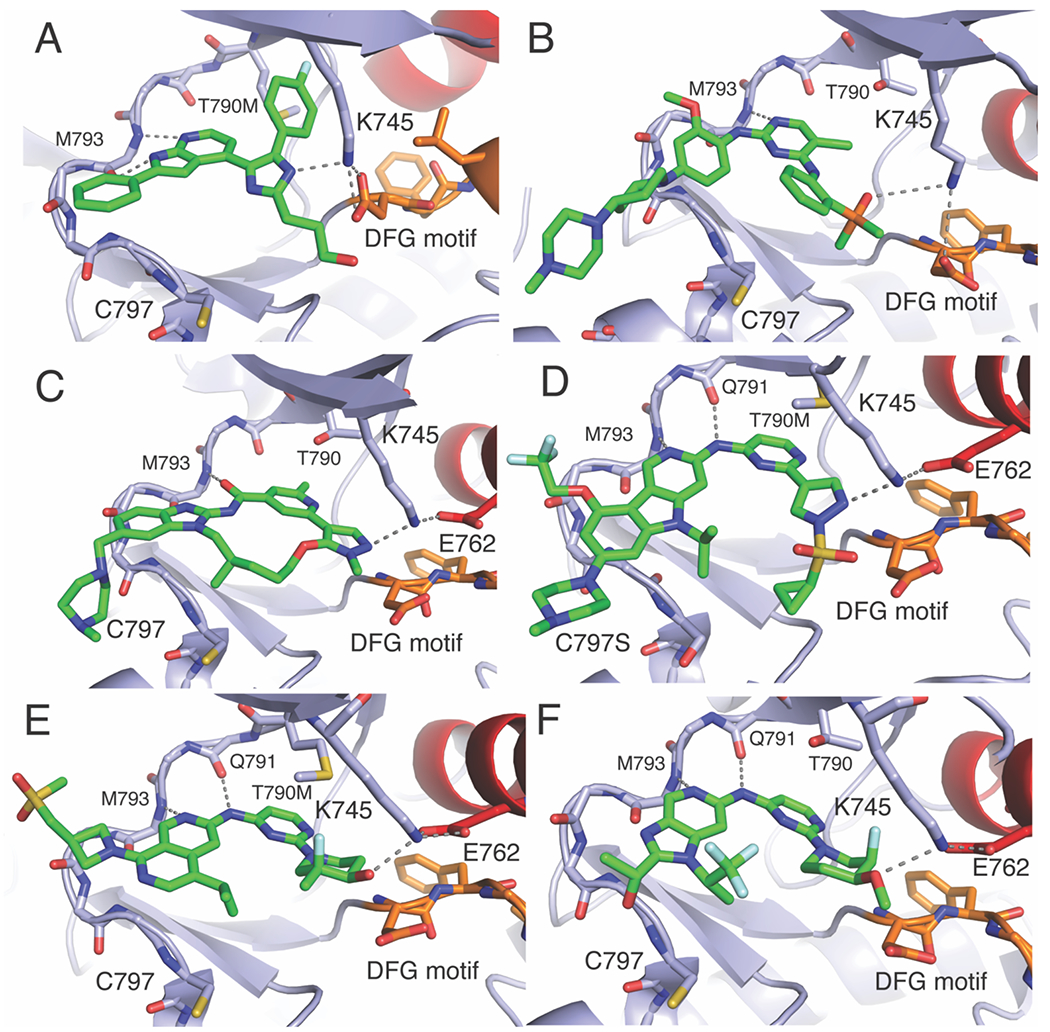
A) The reversible-binding trisubstituted imidazole LN2084 in complex with EGFR(T790M/V948R) PDB ID 6V5N B) Brigatinib bound to WT EGFR 7AEM. C) BI-4020 in complex with WT EGFR PDB ID 7KXZ. D) CH7233163 in complex with T790M/C797S PDB ID 6LUB. E) Compound 24 (3) in complex with T790M EGFR (Note that this analogue contains an alcohol where this group is methylated in BLU-945) PDB ID 8D76). F) Compound 41a (13) in complex with WT EGFR PDB ID 5CAV.
Another compound that unexpectedly showed potency against C797S was brigatinib, which was originally described as an ALK inhibitor (Scheme 2, Figure 4B). This compound as well as chemical derivatives showed off-target inhibition of EGFR(del19/T790M/C797S) that was enhanced if combined with the anti-EGFR antibody cetuximab.(S. Li, Zhang, Zhu, Lei, Lai, Peng, Tong, Pang, Lu, Ding, et al., 2022; Uchibori et al., 2017b) Recent structures of brigatinib and variants bound to EGFR revealed an H-bond from the phosphene oxide to K745, which is analogous to the binding mode as seen to K1150 in ALK (Figure 4B).(Floch, Finlay, Bianco, Bickerton, Colclough, Cross, Cuomo, Guerot, Hargreaves, Martin, et al., 2019) Alternative structures based on optimization of brigatinib have been produced and have been shown to be selective for EGFR C797S.(S. Li, Zhang, Zhu, Lei, Lai, Peng, Tong, Pang, Lu, Ding, et al., 2022)
More recent activity in pharmaceutical companies have generated distinct ATP-competitive reversible inhibitors with activity against C797S. At Boehringer Ingelheim, structure-guided design of a macrocyclic benzimidazole inhibitor (BI-4020), related to the third-generation EGF816, shows tight binding to C797S-containing EGFR and interacts with K745 in both active and inactive conformations in through a novel pyrazole moiety (Figure 4C).(Tyler S. Beyett, Jaimin K. Rana, Ilse K. Schaeffner, David E. Heppner, & Michael J. Eck, 2022; Engelhardt et al., 2019) In another case, an extensive drug development campaign by Blueprint Therapeutics led to the development of Compound 24 (3) the analogue of BLU-945 based on an isoquinoline scaffold, which exhibits selectivity for C797S (Figure 4E)(Eno et al., 2022). Like other inhibitors for C797S variants, the crystal structures of analogs of Compound 24 (3) show that a critical alcohol is positioned toward the K745 residues in the inactive conformation. Additionally, the Chugai Pharmaceutical Company developed a molecule CH7233163 that overcomes AZD9291 resistance and exhibits H-bonds with K745 through a pyrazole similar to BI-4020 in the active kinase conformation (Figure 4D). These inhibitors, as well as those based on brigatinib, are more selective for the mutant EGFR as compared to WT. Furthermore, molecules published prior to the emergence of C797S that have structural resemblance to BLU-945 and analogues, show binding modes that also show H-bonds to K745 but to our knowledge have yet to be assayed for C797S activity (Scheme 2 compounds 41a-b).(Heald, Bowman, Bryan, Burdick, Chan, Chan, Chen, Clausen, Dominguez-Fernandez, Eigenbrot, et al., 2015) Since the imidazole-based molecules are not selective while these scaffolds are, it is difficult to generalize that H-bonds to K745 are needed for mutant selectivity but appear to drive stronger reversible binding. A distinguishing feature of the imidazoles (Figure 4A) versus the other mutant-selective scaffolds (Figure 4B–F) could be variations in interactions near the gatekeeper T790 residue, but further studies are needed to confirm such a structure-activity relationship. Overall, reversible-binding ATP-competitive inhibitors with promise against C797S-containing EGFR are distinct from earlier third-generation TKIs and suggest new mutant-selective molecules can gain needed potency through binding conserved residues.
Fourth-Generation Allosteric Inhibitors (Type 3)
An alternative approach to target C797S and T790M EGFR mutations is to bind a drug molecule within a pocket distinct and distant from the ATP binding site. Such allosteric inhibitors have recently been discovered through a targeted high-throughput screen that selected for molecules with selectivity for the L858R/T790M mutant.(D. J. H. De Clercq et al., 2019; Jia et al., 2016) Two scaffolds identified, a phenyl glycine (triblade)(Jia et al., 2016; To, Beyett, Jang, Feng, Bahcall, Haikala, Shin, Heppner, Rana, Leeper, et al., 2022; To et al., 2019) and dibenzodiazepinone,(D. J. H. De Clercq et al., 2019) have led to drug development efforts including quinazolinones (Thomas W. Gero et al., 2022) and related molecules (Table 3).(Obst-Sander, Ricci, Kuhn, Friess, Koldewey, Kuglstatter, Hewings, Goergler, Steiner, Rueher, et al., 2022) Generally from a variety of X-ray cocrystal structures (Scheme 3, Figure 5A–B), these inhibitors are ATP non-competitive and bind to a site that is made accessible due to the outward αC-helix conformation that is adjacent to the ATP site.(Jia et al., 2016) Inhibitors that bind this pocket are intrinsically mutant selective as the L858R point mutation allows for access to this site via permanent unfolding of the A-loop.(Tyler S. Beyett, To, et al., 2022; Jia et al., 2016) Cocrystal structures containing the L858R mutation, which are underrepresented due to being relatively difficult to crystallize, show H-bonding interactions between the mutant Arg and the 2-isoindolinone of JBJ-09-063 potentially also contributing to L858R selectivity (Figure 5C).(Tyler S. Beyett, To, et al., 2022) Additionally, selectivity for T790M is enabled by thioether-aromatic ring interactions and otherwise insensitive to the C797S mutation due to the relatively long distance from the allosteric pocket to this amino acid side chain.
Table 3.
Structurally characterized allosteric and spanning ATP-allosteric hybrid inhibitors effective against L858R, T790M, and C797S mutants
| Compound Namea | PDB ID | EGFR Kinase Domain αC-helix | Interactions with K745 and D855 | Ref | |
|---|---|---|---|---|---|
| 1 | EAI001 | 5D41 | out | H-bond between aminothiazole and D855, H-bond between AMP-PNP and K745 | (Jia et al., 2016) |
| 2 | EAI002 | 6P8Q | out | H-bond between carbonyl and K745. | (D. J. H. De Clercq et al., 2019) |
| 3 | EAI045 | 5ZWJ | out | H-bond between aminothiazole and D855, H-bond between isoindolinone and K745 | (P. Zhao, Yao, Zhu, Chen, & Yun, 2018) |
| 4 | 6P1L | out | H-bond between aminothiazole and D855, H-bond between AMP-PNP and K745 | (D. J. De Clercq et al., 2019) | |
| 5 | JBJ-09-063 | 7JXQ | out | H-bond between aminothiazole and D855, H-bond between AMP-PNP and K745 | (To, Beyett, Jang, Feng, Bahcall, Haikala, Shin, Heppner, Rana, Leeper, et al., 2022) |
| 6 | JBJ-09-063 | 7K1I | out | H-bond between aminothiazole and D855, H-bond between AMP-PNP and K745. H-bond between L858R and K745 | (Tyler S Beyett, To, et al., 2022; To, Beyett, Jang, Feng, Bahcall, Haikala, Shin, Heppner, Rana, & Leeper, 2022) |
| 7 | JBJ-04-125-02 | 6DUK | out | H-bond between aminothiazole and D855, H-bond between AMP-PNP and K745 | (To et al., 2019) |
| 8 | DDC4002 | 6P1D | out | H-bond between aminothiazole and D855, H-bond between AMP-PNP and K745 | (D. J. H. De Clercq et al., 2019) |
| 9 | Compound 34 (15) | 7LTX | out | H-bond between NH of amide and D855. | (Thomas W Gero et al., 2022) |
| 10 | Compound 38 (16) | 8A2B | out | H-bond between aminothiazole and D855. H-bond between inhibitor ketone and K745 | (Obst-Sander, Ricci, Kuhn, Friess, Koldewey, Kuglstatter, Hewings, Goergler, Steiner, & Rueher, 2022) |
| 11 | Compound 57 (17) | 8A2D | out | H-bond between aminothiazole and D855 | (Obst-Sander, Ricci, Kuhn, Friess, Koldewey, Kuglstatter, Hewings, Goergler, Steiner, & Rueher, 2022) |
| Simultaneously Bound ATP Site TKIs and allosteric inhibitors | |||||
| 12 | AZD9291 and EAI045 | 7JXM | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Tyler S Beyett, To, et al., 2022) |
| 13 | AZD9291 and DDC4002 | 6XL4 | out | H-bond between benzodiazepinone ketone and K745. | (Tyler S Beyett, To, et al., 2022) |
| 14 | AZD9291 and JBJ-04-125-02 | 7JXP | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Tyler S Beyett, To, et al., 2022) |
| 15 | PF-06747775 and JBJ-04-125-02 | 7JXK | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Tyler S Beyett, To, et al., 2022) |
| 16 | AZD9291 and JBJ-09-063 | 7JXW | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Tyler S Beyett, To, et al., 2022) |
| 17 | ASP8273 and JBJ-09-063 | 7LG8 | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Tyler S. Beyett, To, et al., 2022) |
| 18 | AZD9291 and JBJ-09-063 | 7K1H | out | H-bond between aminothiazole and D855. H-bond between isoindolinone ketone and K745. H-bond between L858R and K745 | (Tyler S Beyett, To, et al., 2022) |
| 19 | PF-06747775 and JBJ-04-125-02 | 7JXK | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Tyler S Beyett, To, et al., 2022) |
| 20 | PF-06747775 and EAI001 | 6Z4D | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Niggenaber et al., 2020) |
| 21 | AZD9291 and EAI045 | 6Z4B | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Niggenaber et al., 2020) |
| 22 | Spebrutinib and EAI001 | 7A2A | out | H-bond with aminothiazole and D855. H-bond between isoindolinone ketone and K745. | (Niggenaber et al., 2020) |
Note that we have listed all compounds according to their generic name or compound code included in the original reference. We have elected to enumerate all of the compounds here that are reported by numeral in the original reference for clarity.
Scheme 3.
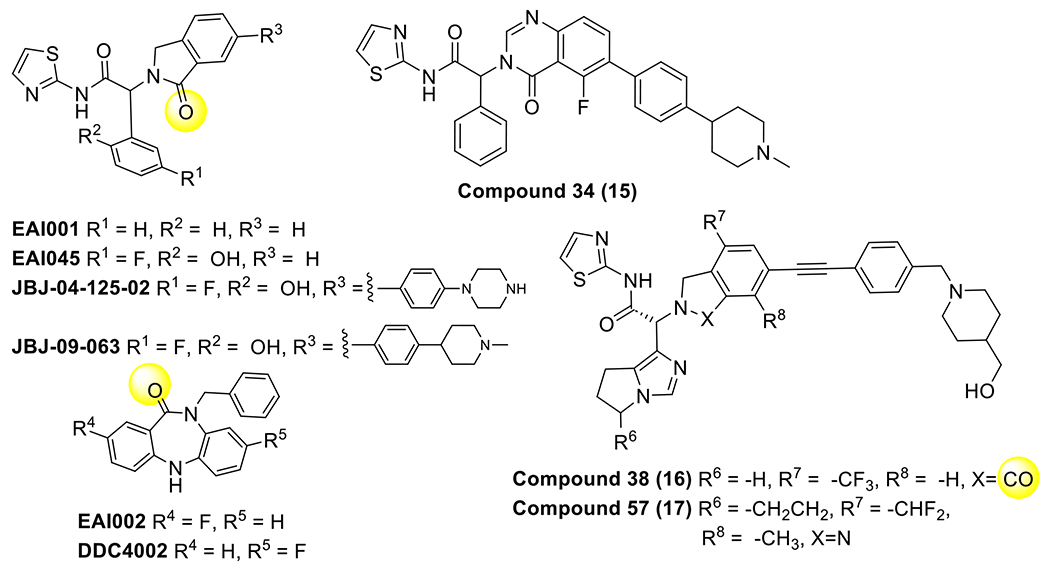
All structurally characterized allosteric EGFR TKIs and related analogues (Table 3). Groups that form H-bonds with K745 are highlighted with yellow circles.
Figure 5. Binding modes of mutant-selective allosteric inhibitors.
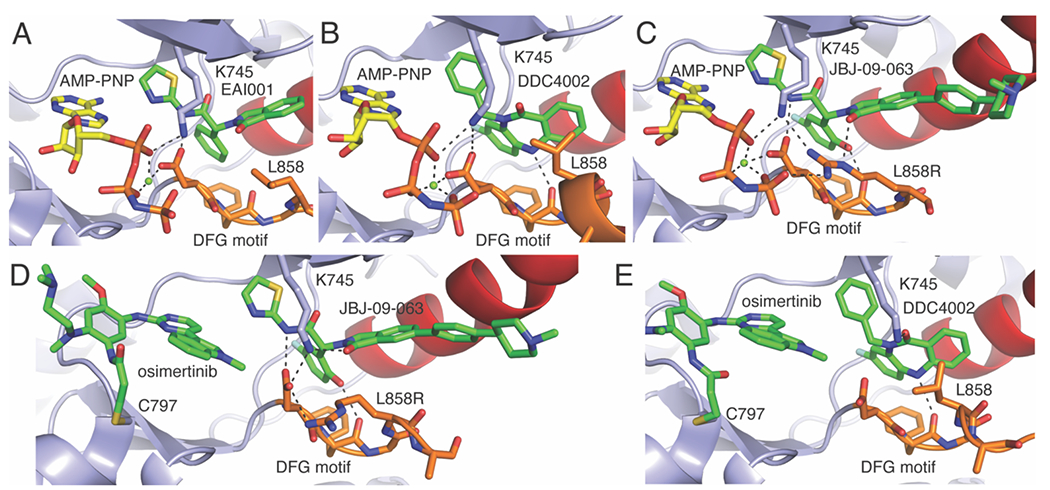
A) EAI001 bound in the allosteric pocket adjacent to the ATP site (AMP-PNP is colored yellow) PDB ID 5D41. B) DDC4002 bound in the allosteric pocket adjacent to ATP (AMP-PNP) PDB ID 6P1D. C) JBJ-09-063 bound in the allosteric pocket showing L858R H-bonding interactions with allosteric inhibitor PDB ID 7K1I. D) Cocrystal structure of JBJ-09-063 bound simultaneously with AZD9291 PDB ID 7JXW. E) Cocrystal structure of DDC4002 bound simultaneously with AZD9291 PDB ID 6XL4.
With respect to interactions with conserved residue side chains, the aminothiazole-containing variants (e.g., EAI001 and related derivatives) all form H-bonds with D855 that bridges the allosteric pocket and H-bonds with ATP (Figure 5A,C). Such an H-bond is not possible with the benzodiazepinone inhibitors (Figure 5B).(D. J. H. De Clercq et al., 2019) Notably, the catalytic K745 does not interact with the allosteric inhibitors but is involved in H-bonds with ATP (AMP-PNP in the crystal structures, Figure 5A–B) in all scaffolds crystallized as well as interacting with L858R that include this mutation (Figure 5C).(To, Beyett, Jang, Feng, Bahcall, Haikala, Shin, Heppner, Rana, Leeper, et al., 2022) Recent cocrystal structures of allosteric inhibitors simultaneously bound with ATP-competitive third generation inhibitors, such as AZD9291 (Figure 5D), show a “swing” of K745 from the ATP side and over to the allosteric inhibitor now H-bonding with the 2-isoindolinone ketone (Figure 5D–E).(Tyler S. Beyett, To, et al., 2022; Niggenaber et al., 2020) Additionally, structural rearrangement of the P-loop F723 into a pi-stack with certain phenyl-containing allosteric inhibitors (e.g., JBJ-04-125-02, JBJ-09-063) are the source of positive cooperativity of ATP-competitive and allosteric inhibitors,(Tyler S. Beyett, To, et al., 2022) which is an important insight given that these inhibitors synergize with TKIs in vivo.(To, Beyett, Jang, Feng, Bahcall, Haikala, Shin, Heppner, Rana, Leeper, et al., 2022; To et al., 2019) Both the structural rearrangements of F723 and K745 toward the allosteric pocket indicate that displacement of ATP can lead to binding differences of amino acid side chains toward the allosteric site, which likely impact pharmacological efficacy of these next-generation inhibitors.
Bivalent ATP-allosteric Inhibitors
The proximity of the ATP and allosteric sites has motivated the design of spanning molecules that bind extensively within these sites (Table 3).(Q. Li et al., 2019; Wittlinger et al., 2022) Such spanning inhibitors based on 4-anilinoquinazoline and trisubstituted imidazole ATP-site scaffolds hybridized with groups resembling EAI045 have offered proof-of-concept examples of such molecules, however structurally and functionally much remains unexplored. X-ray cocrystal structures of the imidazole bivalent molecules indicate that these molecules bind to both allosteric and ATP sites with H-bonding interactions with K745 and D855 mostly resembling the parent ATP-site imidazole inhibitor (Figure 6A, cf. Figure 4A). Interestingly, inhibitors based on pyrrolopyrimidine ATP-site scaffolds were designed prior to the discovery of C797S and anchor a urea-cyclohexane moiety within the EGFR allosteric pocket.(Sogabe et al., 2013) Crystal structures show that urea group in the allosteric pocket directs the K745 “swing” toward the allosteric pocket since the ATP site part of this molecule cannot accept H-bonds from K745 (Figure 6B). While few examples have been studied, spanning ATP-allosteric inhibitors do not appear to guarantee mutant-selective. Distinctly the 4-anilinoquinazolines are similarly potent between WT and mutants (Q. Li et al., 2019) and the trisubstituted imidazoles exhibit modest selectivity.(Wittlinger et al., 2022)
Figure 6. Structurally-characterized spanning inhibitors covalently linking the ATP and allosteric pockets.
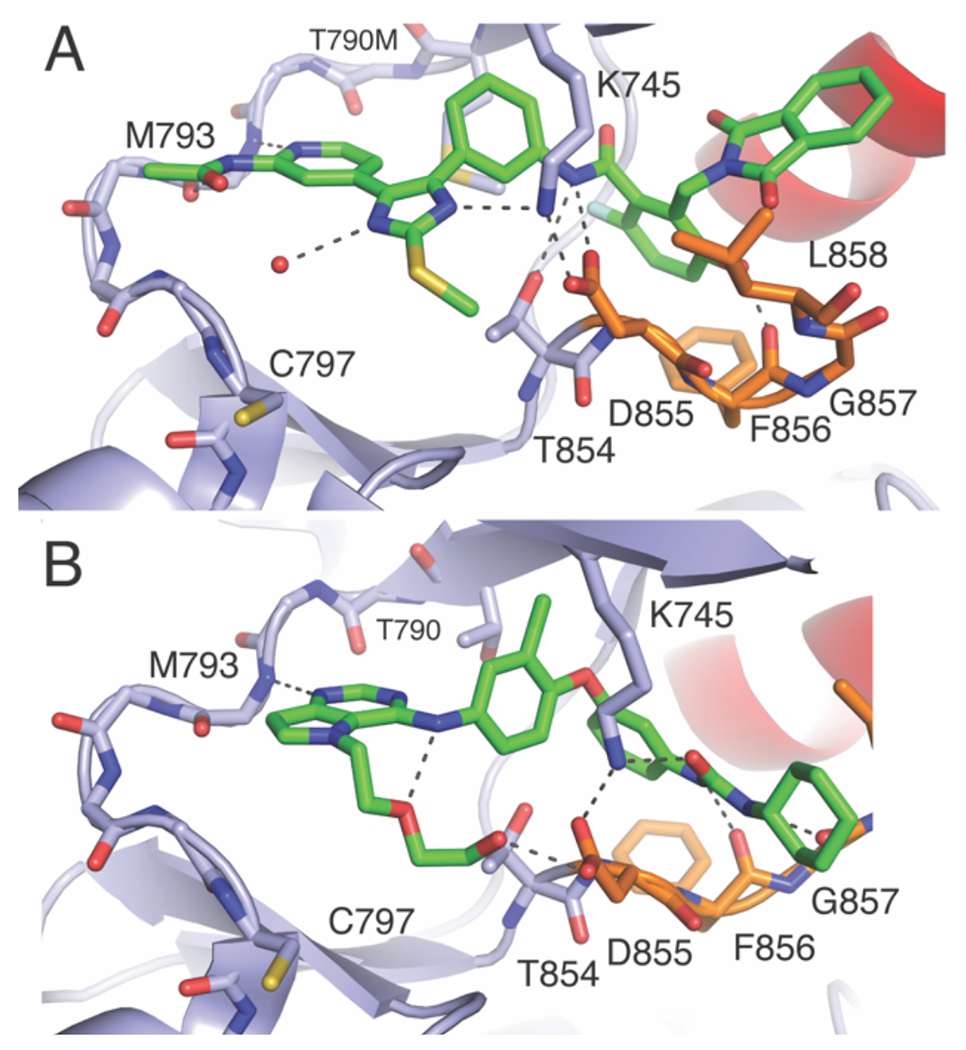
A) Trisubstituted imidazole (PDB ID 6WXN) and B) pyrrolopyrimidine (PBD ID 3W2S) ATP-site inhibitors.
Concluding Thoughts
Over the past decades, designing drugs with sufficient specificity against activating mutations in EGFR has been a persistently “moving target”. Throughout, structural biology has remained a critical asset in directing new inhibitor mechanisms (covalent versus reversible binding) as well as the exploitation of unique intermolecular interactions. As the newer generation of ATP-competitive C797S-targeting EGFR inhibitors have been reported, distinct H-bonding interactions with conserved catalytic residues K745 and D855 drive needed potency, although not guaranteeing mutant selectivity. Allosteric inhibitors, by virtue of their distinct binding pocket, are robustly mutant selective and make H-bonding interactions with K745 in circumstances where ATP is replaced with an ATP site TKI. Moreover, simultaneous binding of both allosteric inhibitors and ATP-competitive inhibitors have shown synergize effect against mutant EGFR. Lessons from these examples are valuable given the need for C797S-selective therapies as well as the recent emergence of interest in targeting Lys residues with covalent warheads.(Arafet et al., 2023; P. Chen et al., 2022; Liu, Yue, Tsai, & Shen, 2019; Pettinger, Jones, & Cheeseman, 2017; Reja, Wang, Lyu, Haeffner, & Gao, 2022) Future studies and drug design of spanning ATP-allosteric inhibitors as well as Lys-targeting (covalent and non-covalent) in kinases may offer valuable insights to how producing strong interactions to conserved residues may aid in producing inhibitors with selective binding to activating mutations.
Scheme 4.
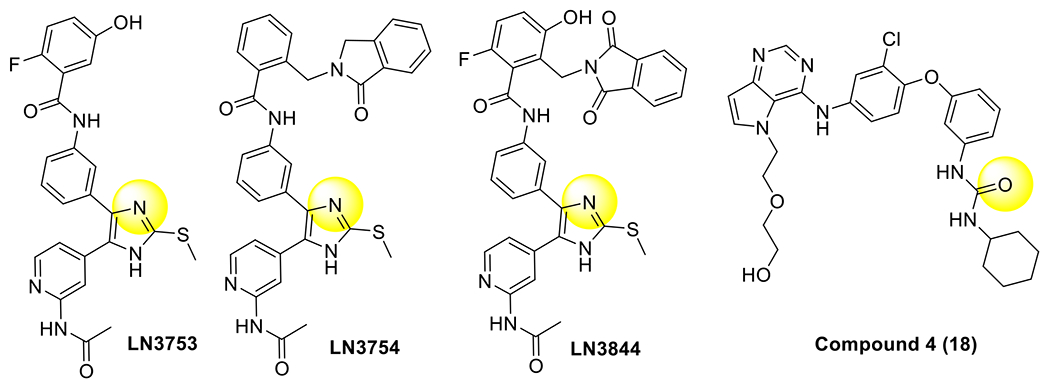
All structurally characterized bivalent EGFR TKIs.
Table 4.
Structurally characterized of bivalent ATP-allosteric EGFR inhibitors
| Name a | PDB ID | EGFR Kinase Domain αC-helix | Interactions with K745 and D855 | Ref | |
|---|---|---|---|---|---|
| 1 | LN3753 (2a) | 6WA2 | out | H-bond between imidazole and K745. H-bond between D855 and amide and K745. | (Wittlinger et al., 2022) |
| 2 | LN3754 | 6WAK | out | H-bond between imidazole and K745. H-bond between D855 and amide and K745. | (Wittlinger et al., 2022) |
| 3 | LN3844 (2c) | 6WXN | out | H-bond between imidazole and K745. H-bond between D855 and amide and K745. | (Wittlinger et al., 2022) |
| 4 | Compound 4 (18) | 3W2S | out | H-bond between urea and K745. | (Sogabe et al., 2013) |
Note that we have listed all compounds according to their generic name or compound code included in the original reference. We have elected to enumerate all of the compounds here that are reported by numeral in the original reference for clarity.
Acknowledgments.
We acknowledge support from the State University of New York at Buffalo for startup funding (DEH) and the National Center for Advancing Translational Sciences of the NIH UL1TR001412-07 (K Scholar Award to DEH). T.S.B is supported by a Ruth L. Kirschstein National Research Service Award (F32CA247198-01). SAL. and iFIT are funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (EXC 2180-390900677). TüCAD2 is funded by the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments. National Institutes of Health Grants R01CA116020 and R35CA242461 (to M.J.E).
Literature Cited
- Arafet K, Scalvini L, Galvani F, Martí S, Moliner V, Mor M, & Lodola A (2023). Mechanistic Modeling of Lys745 Sulfonylation in EGFR C797S Reveals Chemical Determinants for Inhibitor Activity and Discriminates Reversible from Irreversible Agents. Journal of Chemical Information and Modeling. doi: 10.1021/acs.jcim.2c01586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyett TS, Rana JK, Schaeffner IK, Heppner DE, & Eck MJ (2022). Structural analysis of the macrocyclic inhibitor BI-4020 binding to EGFR kinase. bioRxiv, 2022.2008.2027.505540. doi: 10.1101/2022.08.27.505540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyett TS, To C, Heppner DE, Rana JK, Schmoker AM, Jang J, … Eck MJ (2022). Molecular basis for cooperative binding and synergy of ATP-site and allosteric EGFR inhibitors. Nature Communications, 13(1), 2530. doi: 10.1038/s41467-022-30258-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, … Sliwkowski MX (2006). Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer research, 66(16), 8163–8171. [DOI] [PubMed] [Google Scholar]
- Carrera AC, Alexandrov K, & Roberts TM (1993). The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proceedings of the National Academy of Sciences, 90(2), 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Fu W, Zheng L, Liu Z, & Liang G (2017). Recent progress of small-molecule epidermal growth factor receptor (EGFR) inhibitors against C797S resistance in non-small-cell lung cancer: miniperspective. Journal of medicinal chemistry, 61(10), 4290–4300. [DOI] [PubMed] [Google Scholar]
- Chen P, Sun J, Zhu C, Tang G, Wang W, Xu M, … Gao L (2022). Cell-Active, Reversible, and Irreversible Covalent Inhibitors That Selectively Target the Catalytic Lysine of BCR-ABL Kinase. Angewandte Chemie International Edition, e202203878. [DOI] [PubMed] [Google Scholar]
- De Clercq DJ, Heppner DE, To C, Jang J, Park E, Yun C-H, … Scott DA (2019). Discovery and optimization of dibenzodiazepinones as allosteric mutant-selective EGFR inhibitors. ACS Medicinal Chemistry Letters, 10(11), 1549–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq DJH, Heppner DE, To C, Jang J, Park E, Yun C-H, … Gray NS (2019). Discovery and Optimization of Dibenzodiazepinones as Allosteric Mutant-Selective EGFR Inhibitors. ACS Medicinal Chemistry Letters, 10(11), 1549–1553. doi: 10.1021/acsmedchemlett.9b00381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eck MJ, & Yun C-H (2010). Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, 1804(3), 559–566. doi: 10.1016/j.bbapap.2009.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt H, Böse D, Petronczki M, Scharn D, Bader G, Baum A, … McConnell DB (2019). Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. Journal of medicinal chemistry, 62(22), 10272–10293. doi: 10.1021/acs.jmedchem.9b01169 [DOI] [PubMed] [Google Scholar]
- Eno MS, Brubaker JD, Campbell JE, De Savi C, Guzi TJ, Williams BD, … Kim J (2022). Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer. Journal of medicinal chemistry, 65(14), 9662–9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay MRV, Barton P, Bickerton S, Bista M, Colclough N, Cross DA, … Guerot CM (2021). Potent and selective inhibitors of the epidermal growth factor receptor to overcome C797S-mediated resistance. Journal of medicinal chemistry, 64(18), 13704–13718. [DOI] [PubMed] [Google Scholar]
- Floch N, Finlay MRV, Bianco A, Bickerton S, Colclough N, Cross DA, … Martin MJ (2019). Evaluation of the therapeutic potential of phosphine oxide pyrazole inhibitors in tumors harboring EGFR C797S mutation. Cancer Research, 79(13_Supplement), 4451–4451. [Google Scholar]
- Floch N, Finlay MRV, Bianco A, Bickerton S, Colclough N, Cross DA, … Ward RA (2019). Abstract 4451: Evaluation of the therapeutic potential of phosphine oxide pyrazole inhibitors in tumors harboring EGFR C797S mutation. Cancer research, 79(13_Supplement), 4451–4451. doi: 10.1158/1538-7445.Am2019-4451 [DOI] [Google Scholar]
- Gero TW, Heppner DE, Beyett TS, To C, Azevedo SC, Jang J, … Shin BH (2022). Quinazolinones as allosteric fourth-generation EGFR inhibitors for the treatment of NSCLC. Bioorganic & Medicinal Chemistry Letters, 68, 128718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gero TW, Heppner DE, Beyett TS, To C, Azevedo SC, Jang J, … Scott DA (2022). Quinazolinones as allosteric fourth-generation EGFR inhibitors for the treatment of NSCLC. Bioorganic & medicinal chemistry letters, 68, 128718. doi: 10.1016/j.bmcl.2022.128718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther M, Juchum M, Kelter G, Fiebig H, & Laufer S (2016). Lung Cancer: EGFR Inhibitors with Low Nanomolar Activity against a Therapy-Resistant L858R/T790M/C797S Mutant. Angewandte Chemie International Edition, 55(36), 10890–10894. doi: 10.1002/anie.201603736 [DOI] [PubMed] [Google Scholar]
- Günther M, Lategahn J, Juchum M, Döring E, Keul M, Engel J, … Laufer S (2017). Trisubstituted pyridinylimidazoles as potent inhibitors of the clinically resistant L858R/T790M/C797S EGFR mutant: targeting of both hydrophobic regions and the phosphate binding site. Journal of medicinal chemistry, 60(13), 5613–5637. [DOI] [PubMed] [Google Scholar]
- Heald R, Bowman KK, Bryan MC, Burdick D, Chan B, Chan E, … Eigenbrot C (2015). Noncovalent mutant selective epidermal growth factor receptor inhibitors: a lead optimization case study. Journal of medicinal chemistry, 58(22), 8877–8895. [DOI] [PubMed] [Google Scholar]
- Heald R, Bowman KK, Bryan MC, Burdick D, Chan B, Chan E, … Heffron TP (2015). Noncovalent Mutant Selective Epidermal Growth Factor Receptor Inhibitors: A Lead Optimization Case Study. Journal of medicinal chemistry, 58(22), 8877–8895. doi: 10.1021/acs.jmedchem.5b01412 [DOI] [PubMed] [Google Scholar]
- Heppner D, Beyett T, Shaurova T, Pham C, Wittlinger F, Schaeffner I, … Eck M (2022). Structural Basis for Inhibition of Mutant EGFR with Lazertinib (YH25448). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner DE, Günther M, Wittlinger F, Laufer SA, & Eck MJ (2020). Structural Basis for EGFR Mutant Inhibition by Trisubstituted Imidazole Inhibitors. Journal of medicinal chemistry, 63(8), 4293–4305. doi: 10.1021/acs.jmedchem.0c00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner DE, Günther M, Wittlinger F, Laufer SA, & Eck MJ (2020). Structural basis for EGFR mutant inhibition by trisubstituted imidazole inhibitors. Journal of medicinal chemistry, 63(8), 4293–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner DE, Wittlinger F, Beyett TS, Shaurova T, Urul DA, Buckley B, … Hershberger PA (2022). Structural Basis for Inhibition of Mutant EGFR with Lazertinib (YH25448). ACS Medicinal Chemistry Letters, 13(12), 1856–1863. doi: 10.1021/acsmedchemlett.2c00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Yasuda H, Hamamoto J, Nukaga S, Masuzawa K, Kawada I, … Sakagami H (2018). Pharmacological and Structural Characterizations of Naquotinib, a Novel Third-Generation EGFR Tyrosine Kinase Inhibitor, in EGFR-Mutated Non–Small Cell Lung CancerCharacterization of Naquotinib in EGFR-Mutated Lung Cancer. Molecular cancer therapeutics, 17(4), 740–750. [DOI] [PubMed] [Google Scholar]
- Hunter T. (2009). Tyrosine phosphorylation: thirty years and counting. Current opinion in cell biology, 21(2), 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Yun C-H, Park E, Ercan D, Manuia M, Juarez J, … Eck MJ (2016). Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature, 534(7605), 129–132. doi: 10.1038/nature17960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juchum M, Günther M, Döring E, Sievers-Engler A, Lämmerhofer M, & Laufer S (2017). Trisubstituted imidazoles with a rigidized hinge binding motif act as single digit nM inhibitors of clinically relevant EGFR L858R/T790M and L858R/T790M/C797S mutants: an example of target hopping. Journal of medicinal chemistry, 60(11), 4636–4656. [DOI] [PubMed] [Google Scholar]
- Kashima K, Kawauchi H, Tanimura H, Tachibana Y, Chiba T, Torizawa T, & Sakamoto H (2020). CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Molecular Cancer Therapeutics, 19(11), 2288–2297. doi: 10.1158/1535-7163.Mct-20-0229 [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, … Halmos B (2005). EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. New England Journal of Medicine, 352(8), 786–792. doi: 10.1056/NEJMoa044238 [DOI] [PubMed] [Google Scholar]
- Lategahn J, Tumbrink HL, Schultz-Fademrecht C, Heimsoeth A, Werr L, Niggenaber J, … Menninger S (2022). Insight into Targeting Exon20 Insertion Mutations of the Epidermal Growth Factor Receptor with Wild Type-Sparing Inhibitors. Journal of medicinal chemistry. [DOI] [PubMed] [Google Scholar]
- Lelais G, Epple R, Marsilje TH, Long YO, McNeill M, Chen B, … Bursulaya B (2016). Discovery of (R, E)-N-(7-Chloro-1-(1-[4-(dimethylamino) but-2-enoyl] azepan-3-yl)-1 H-benzo [d] imidazol-2-yl)-2-methylisonicotinamide (EGF816), a Novel, Potent, and WT Sparing Covalent Inhibitor of Oncogenic (L858R, ex19del) and Resistant (T790M) EGFR Mutants for the Treatment of EGFR Mutant Non-Small-Cell Lung Cancers. Journal of medicinal chemistry, 59(14), 6671–6689. [DOI] [PubMed] [Google Scholar]
- Lelais G, Epple R, Marsilje TH, Long YO, McNeill M, Chen B, … Michellys P-Y (2016). Discovery of (R,E)-N-(7-Chloro-1-(1-[4-(dimethylamino)but-2-enoyl]azepan-3-yl)-1H-benzo[d]imidazol-2-yl)-2-methylisonicotinamide (EGF816), a Novel, Potent, and WT Sparing Covalent Inhibitor of Oncogenic (L858R, ex19del) and Resistant (T790M) EGFR Mutants for the Treatment of EGFR Mutant Non-Small-Cell Lung Cancers. Journal of medicinal chemistry, 59(14), 6671–6689. doi: 10.1021/acs.jmedchem.5b01985 [DOI] [PubMed] [Google Scholar]
- Lemmon MA, & Schlessinger J (2010). Cell signaling by receptor tyrosine kinases. Cell, 141(7), 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhang T, Li S, Tong L, Li J, Su Z, … Xu Y (2019). Discovery of Potent and Noncovalent Reversible EGFR Kinase Inhibitors of EGFRL858R/T790M/C797S. ACS Medicinal Chemistry Letters, 10(6), 869–873. doi: 10.1021/acsmedchemlett.8b00564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhang T, Zhu S-J, Lei C, Lai M, Peng L, … Ding J (2022). Optimization of Brigatinib as New Wild-Type Sparing Inhibitors of EGFRT790M/C797S Mutants. ACS Medicinal Chemistry Letters, 13(2), 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhang T, Zhu S-J, Lei C, Lai M, Peng L, … Ding K (2022). Optimization of Brigatinib as New Wild-Type Sparing Inhibitors of EGFRT790M/C797S Mutants. ACS Medicinal Chemistry Letters, 13(2), 196–202. doi: 10.1021/acsmedchemlett.1c00555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Yue Z, Tsai C-C, & Shen J (2019). Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. Journal of the American Chemical Society, 141(16), 6553–6560. doi: 10.1021/jacs.8b13248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, … Haluska FG (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. New England Journal of Medicine, 350(21), 2129–2139. [DOI] [PubMed] [Google Scholar]
- Moy B, Kirkpatrick P, Kar S, & Goss P (2007). Lapatinib. Nature Reviews Drug Discovery, 6(6), 431–432. doi: 10.1038/nrd2332 [DOI] [PubMed] [Google Scholar]
- Müller S, Chaikuad A, Gray NS, & Knapp S (2015). The ins and outs of selective kinase inhibitor development. Nature Chemical Biology, 11(11), 818–821. doi: 10.1038/nchembio.1938 [DOI] [PubMed] [Google Scholar]
- Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, … Engelman JA (2015). The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clinical Cancer Research, 21(17), 3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen S, Dix MM, Barbas S, Potter ZE, Lu S, Brodsky O, … Gajiwala KS (2017). Proteome-wide map of targets of T790M-EGFR-directed covalent inhibitors. Cell chemical biology, 24(11), 1388–1400. e1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggenaber J, Heyden L, Grabe T, Müller MP, Lategahn J, & Rauh D (2020). Complex Crystal Structures of EGFR with Third-Generation Kinase Inhibitors and Simultaneously Bound Allosteric Ligands. ACS Medicinal Chemistry Letters, 11(12), 2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obst-Sander U, Ricci A, Kuhn B, Friess T, Koldewey P, Kuglstatter A, … Rueher D (2022). Discovery of Novel Allosteric EGFR L858R Inhibitors for the Treatment of Non-Small-Cell Lung Cancer as a Single Agent or in Combination with Osimertinib. Journal of medicinal chemistry. [DOI] [PubMed] [Google Scholar]
- Obst-Sander U, Ricci A, Kuhn B, Friess T, Koldewey P, Kuglstatter A, … Jaeschke G (2022). Discovery of Novel Allosteric EGFR L858R Inhibitors for the Treatment of Non-Small-Cell Lung Cancer as a Single Agent or in Combination with Osimertinib. Journal of medicinal chemistry, 65(19), 13052–13073. doi: 10.1021/acs.jmedchem.2c00893 [DOI] [PubMed] [Google Scholar]
- Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, … Jänne PA (2016). Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small-cell lung cancer. Journal of clinical oncology, 34(28), 3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, … Boggon TJ (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science, 304(5676), 1497–1500. [DOI] [PubMed] [Google Scholar]
- Park S, Ku BM, Jung HA, Sun J-M, Ahn JS, Lee S-H, … Ahn M-J (2020). EGFR C797S as a Resistance Mechanism of Lazertinib in Non-small Cell Lung Cancer with EGFR T790M Mutation. Cancer Research and Treatment: Official Journal of Korean Cancer Association, 52(4), 1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinger J, Jones K, & Cheeseman MD (2017). Lysine-Targeting Covalent Inhibitors. Angewandte Chemie International Edition, 56(48), 15200–15209. doi: 10.1002/anie.201707630 [DOI] [PubMed] [Google Scholar]
- Reja RM, Wang W, Lyu Y, Haeffner F, & Gao J (2022). Lysine-Targeting Reversible Covalent Inhibitors with Long Residence Time. Journal of the American Chemical Society, 144(3), 1152–1157. doi: 10.1021/jacs.1c12702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogabe S, Kawakita Y, Igaki S, Iwata H, Miki H, Cary DR, … Ishikawa T (2013). Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Medicinal Chemistry Letters, 4(2), 201–205. doi: 10.1021/ml300327z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, … Adolf GR (2012). Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. Journal of Pharmacology and Experimental Therapeutics, 343(2), 342–350. doi: 10.1124/jpet.112.197756 [DOI] [PubMed] [Google Scholar]
- Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, … Kurata T (2018). Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. New England Journal of Medicine, 378(2), 113–125. [DOI] [PubMed] [Google Scholar]
- Tan C-S, Kumarakulasinghe NB, Huang Y-Q, Ang YLE, Choo JR-E, Goh B-C, & Soo RA (2018). Third generation EGFR TKIs: current data and future directions. Molecular Cancer, 17(1), 29. doi: 10.1186/s12943-018-0778-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Knighton DR, Zheng J, Ten Eyck LF, & Sowadski JM (1992). Structural framework for the protein kinase family. Annual review of cell biology, 8(1), 429–462. [DOI] [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, … Oxnard GR (2015). Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nature medicine, 21(6), 560–562. doi: 10.1038/nm.3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- To C, Beyett TS, Jang J, Feng WW, Bahcall M, Haikala HM, … Leeper BA (2022). An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nature Cancer, 3(4), 402–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To C, Beyett TS, Jang J, Feng WW, Bahcall M, Haikala HM, … Jänne PA (2022). An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nature Cancer, 3(4), 402–417. doi: 10.1038/s43018-022-00351-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJ, … Heppner DE (2019). Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer discovery, 9(7), 926–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchibori K, Inase N, Araki M, Kamada M, Sato S, Okuno Y, … Katayama R (2017a). Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nature communications, 8(1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchibori K, Inase N, Araki M, Kamada M, Sato S, Okuno Y, … Katayama R (2017b). Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nature Communications, 8(1), 14768. doi: 10.1038/ncomms14768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittlinger F, Heppner DE, To C, Günther M, Shin BH, Rana JK, … Laufer SA (2022). Design of a “Two-in-One” Mutant-Selective Epidermal Growth Factor Receptor Inhibitor That Spans the Orthosteric and Allosteric Sites. Journal of medicinal chemistry, 65(2), 1370–1383. doi: 10.1021/acs.jmedchem.1c00848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X-E, Ayaz P, Zhu S-J, Zhao P, Liang L, Zhang CH, … Huang X (2020). Structural basis of AZD9291 selectivity for EGFR T790M. Journal of medicinal chemistry, 63(15), 8502–8511. [DOI] [PubMed] [Google Scholar]
- Yan X-E, Ayaz P, Zhu S-J, Zhao P, Liang L, Zhang CH, … Yun C-H (2020). Structural Basis of AZD9291 Selectivity for EGFR T790M. Journal of medicinal chemistry, 63(15), 8502–8511. doi: 10.1021/acs.jmedchem.0c00891 [DOI] [PubMed] [Google Scholar]
- Yan X-E, Zhu S-J, Liang L, Zhao P, Choi HG, & Yun C-H (2017). Structural basis of mutant-selectivity and drug-resistance related to CO-1686. Oncotarget, 8(32), 53508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X-E, Zhu S-J, Liang L, Zhao P, Geun Choi H, & Yun C-H (2017). Structural basis of mutant-selectivity and drug-resistance related to CO-1686. Oncotarget, 8(32). Retrieved from https://www.oncotarget.com/article/18588/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosaatmadja Y, Silva S, Dickson JM, Patterson AV, Smaill JB, Flanagan JU, … Squire CJ (2015). Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. Journal of structural biology, 192(3), 539–544. [DOI] [PubMed] [Google Scholar]
- Yun C-H, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, & Eck MJ (2007). Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell, 11(3), 217–227. doi: 10.1016/j.ccr.2006.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, … Eck MJ (2008). The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proceedings of the National Academy of Sciences, 105(6), 2070–2075. doi: 10.1073/pnas.0709662105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Hong MH, Kim S-Y, Park C-W, Kim S, Yun MR, … Koh JS (2019). YH25448, an irreversible EGFR-TKI with potent intracranial activity in EGFR mutant non–small cell lung cancer. Clinical Cancer Research, 25(8), 2575–2587. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, & Kuriyan J (2006). An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell, 125(6), 1137–1149. [DOI] [PubMed] [Google Scholar]
- Zhao H-Y, Xi X-X, Xin M, & Zhang S-Q (2022). Overcoming C797S mutation: The challenges and prospects of the fourth-generation EGFR-TKIs. Bioorganic Chemistry, 128, 106057. doi: 10.1016/j.bioorg.2022.106057 [DOI] [PubMed] [Google Scholar]
- Zhao P, Yao M-Y, Zhu S-J, Chen J-Y, & Yun C-H (2018). Crystal structure of EGFR T790M/C797S/V948R in complex with EAI045. Biochemical and biophysical research communications, 502(3), 332–337. doi: 10.1016/j.bbrc.2018.05.154 [DOI] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Chen L, Yun C-H, Li D, Capelletti M, … Padera R (2009). Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature, 462(7276), 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Chen L, Yun C-H, Li D, Capelletti M, … Jänne PA (2009). Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature, 462(7276), 1070–1074. doi: 10.1038/nature08622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S-J, Zhao P, Yang J, Ma R, Yan X-E, Yang S-Y, … Yun C-H (2018). Structural insights into drug development strategy targeting EGFR T790M/C797S. Oncotarget, 9(17), 13652. [DOI] [PMC free article] [PubMed] [Google Scholar]