

Abstract
Background:
Two recent randomized clinical trials of escalating doses of allopurinol for the progression of chronic kidney disease (CKD) reported no benefits but potentially increased risk for death. Whether the risk could occur in patients with gout and concurrent CKD remains unknown.
Objective:
To examine the relation of allopurinol initiation, allopurinol dose escalation, and achieving target serum urate (SU) level after allopurinol initiation to all-cause mortality in patients with both gout and CKD.
Design:
Cohort study.
Setting:
The Health Improvement Network U.K. primary care database (2000 to 2019).
Participants:
Patients aged 40 years or older who had gout and concurrent moderate-to-severe CKD.
Measurements:
The association between allopurinol initiation and all-cause mortality over 5-year follow-up in propensity score (PS)–matched cohorts was examined. Analysis of hypothetical trials were emulated: achieving target SU level (<0.36 mmol/L) versus not achieving target SU level and dose escalation versus no dose escalation for mortality over 5-year follow-up in allopurinol initiators.
Results:
Mortality was 4.9 and 5.8 per 100 person-years in 5277 allopurinol initiators and 5277 PS-matched noninitiators, respectively (hazard ratio [HR], 0.85 [95% CI, 0.77 to 0.93]). In the target trial emulation analysis, the HR of mortality for the achieving target SU level group compared with the not achieving target SU level group was 0.87 (CI, 0.75 to 1.01); the HR of mortality for allopurinol in the dose escalation group versus the no dose escalation group was 0.88 (CI, 0.73 to 1.07).
Limitation:
Residual confounding cannot be ruled out.
Conclusion:
In this population-based data, neither allopurinol initiation, nor achieving target SU level with allopurinol, nor allopurinol dose escalation was associated with increased mortality in patients with gout and concurrent CKD.
Primary Funding Source:
Project Program of National Clinical Research Center for Geriatric Disorders.
Gout is the most common form of inflammatory arthritis (1), and its incidence and prevalence have increased during the past few decades (2–8). The cornerstone of long-term management of gout is urate-lowering therapy. Rheumatology treatment guidelines for gout recommend a treat-to-target approach of lowering serum urate (SU) level to below 0.36 mmol/L for all patients with recurrent gout flares, tophi, or radiographic joint damage due to gout (9–12). The most widely used urate-lowering medication is allopurinol (13), which is started at a low dose, increased over weeks to months to achieve the SU level target, and continued indefinitely (9, 11, 14). Besides its urate lowering effect, several observational studies also examined the relation of allopurinol use to the risk for death in patients with gout; the results, however, are inconclusive (15–23).
Chronic kidney disease (CKD) is a comorbidity present in 20% or more of patients with gout (24, 25). Moreover, many studies have found that hyperuricemia is associated with an increased risk for incident CKD and its sequalae (26–28); thus, SU has been considered a potential therapeutic target for halting the progression of CKD. Recently, results from 2 randomized controlled trials (RCTs) showed no renal function–preserving benefits of allopurinol in patients with renal disease but without gout (29, 30). Neither trial set SU level–based inclusion criterion; participants’ mean SU levels were 0.49 mmol/L and 0.36 mmol/L at enrollment, respectively. Unexpectedly, both trials and pooled analyses indicated that allopurinol was associated with a 2-fold increased risk for death in patients with renal disease (31, 32).
Whether allopurinol use will increase mortality in patients with both gout and CKD remains unknown. We did a population-based cohort study to assess the relation of allopurinol initiation to mortality in patients with gout and concurrent moderate-to-severe CKD. In addition, we conducted 2 cohort studies emulating RCTs to examine the effects of achieving target SU level with allopurinol and allopurinol dose escalation on mortality.
METHODS
Data Source
We used data from The Health Improvement Network (THIN), an electronic health records database from general practitioners (GPs) in the United Kingdom. It consists of approximately 17 million persons in the United Kingdom. The computerized information includes sociodemographic characteristics, anthropometric characteristics, lifestyle factors, and details from visits to GPs (that is, prescriptions, diagnoses from specialist referrals, hospital admissions, and results of laboratory tests). The Read classification system is used to code specific diagnoses (33), whereas a dictionary based on the Multilex classification system is used to code drugs (34). The validity of THIN for use in clinical and epidemiologic research studies has been shown in a previous study (35). The scientific review committee for THIN (21SRC003) and the institutional review board at Xiangya Hospital approved this study, with waiver of informed consent. This study followed the recommendations of the RECORD (REporting of studies Conducted using Observational Routinely-collected Data) statement and the extension RECORD-PE for pharmacoepidemiology studies (36).
Study Design and Cohort Definition
We included patients who were 40 to 89 years old, had gout and concurrent moderate-to-severe CKD from 1 January 2000 to 1 January 2018, and had at least 1 year of continuous enrollment with GPs before entering the study. The diagnosis of gout was based on the presence of at least 1 Read code for gout (37–39). Moderate-to-severe CKD (≥stage 3) was identified by either estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2 on at least 2 occasions more than 90 days apart within 1 year, with no intervening eGFR of 75 mL/min/1.73 m2 or greater, or at least 1 Read code for CKD stage 3 to 5, hemodialysis, or peritoneal dialysis (39). The date of the first allopurinol prescription was assigned as the index date for allopurinol initiators, hereafter called the initiators, and a random date within that time block was assigned as the index date for noninitiators. Persons were excluded if they had cancer (including cancer of the cervix in situ and nonmelanoma skin cancer) or kidney transplant before the index date, or had no SU measure before the index date, or were prescribed other urate-lowering medication (for example, febuxostat, probenecid, benzbromarone, and sulphinpyrazone) during the 1 year before the index date.
First, we did a propensity score (PS)–matched cohort study to compare mortality in initiators versus noninitiators. We calculated PS for initial prescription of allopurinol using logistic regression within each 1-year time block, and persons with missing covariate data for PS calculation were excluded from this analysis. For each initiator, we identified a nonallopurinol initiator within the same time block using a PS greedy matching algorithm (40) (Figure 1). Persons who were PS matched in the early accrual time blocks were ineligible to be included in the later accrual time blocks (see Supplement section 1.1 for details, available at Annals.org).
Figure 1.
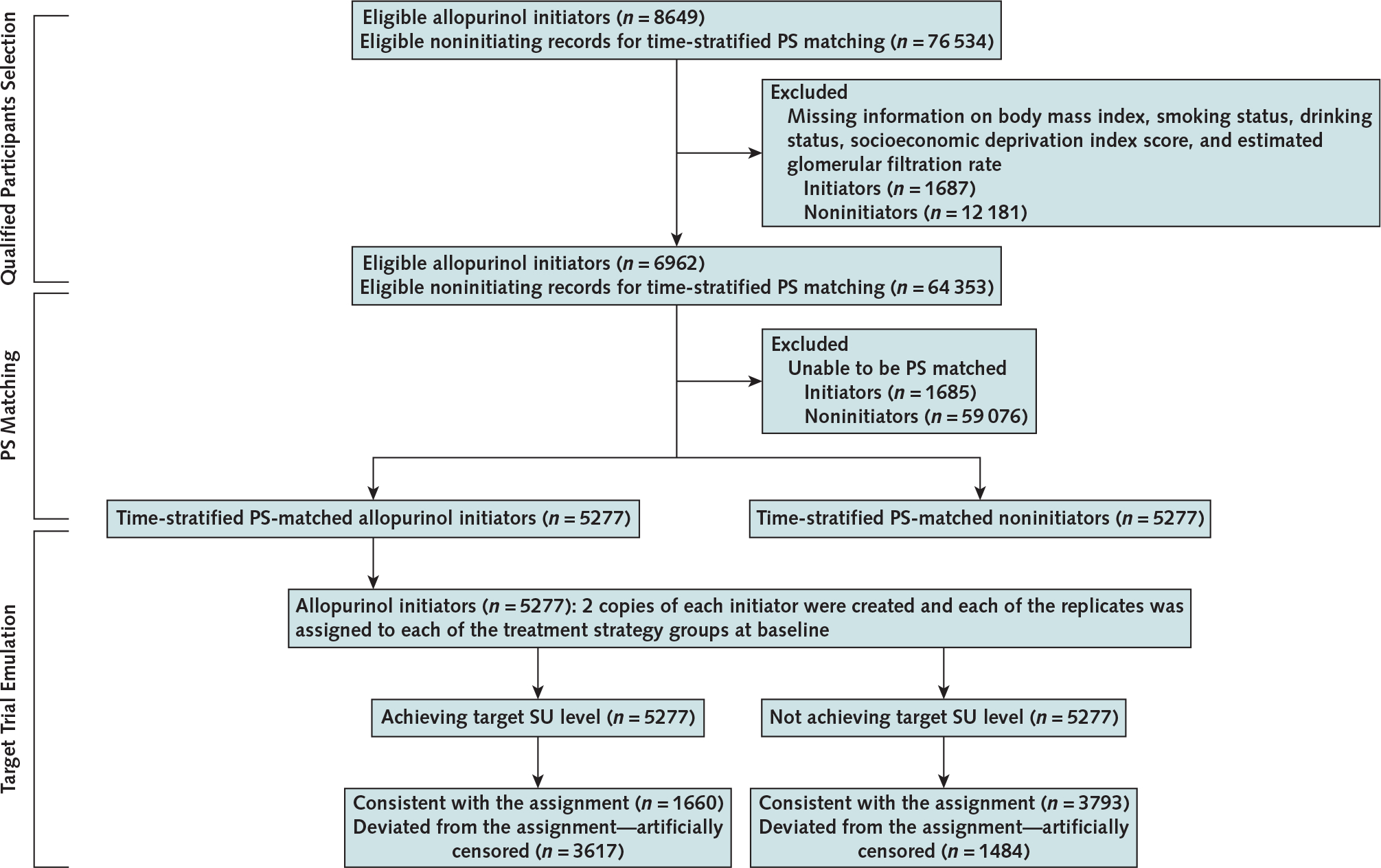
Flow chart of eligible persons for examining the relation of allopurinol initiation and achieving target SU level with allopurinol to all-cause mortality, THIN 2000 to 2019.
PS= propensity score; SU= serum urate; THIN= The Health Improvement Network.
Second, we emulated analyses of hypothetical target trial using a cloning, censoring, and weighting approach to assess the effect of either achieving target SU levels or allopurinol dose escalation on mortality in initiators using observational data (41–43) (Figure 1). We created a data set with 2 copies of each initiator at baseline and assigned each of the replicates to either 1 of the intervention groups (that is, achieving target SU level group vs. not achieving target SU level group, allopurinol dose-escalation group vs. no allopurinol dose escalation group). We allowed for a grace period of 1 year after allopurinol initiation for persons to achieve the target SU level or escalate the allopurinol dose (14, 44) (see Supplement section 1.2 for details, available at Annals.org).
Assessment of Outcome
The outcome was all-cause mortality over the 5 years after the index date. The death date recorded in THIN is linked to the National Health Service; thus, a change in vital status to “dead” is immediately updated in the person’s electronic health record.
Assessment of Covariates
We obtained covariate information before the index date on sociodemographic characteristics (age, sex, socioeconomic deprivation index score, and region), anthropometric characteristics (body mass index), lifestyle factors (smoking status and alcohol consumption), CKD severity (grades 3 to 5), SU level, and comorbidities (congestive heart failure, myocardial infarction, stroke, hypertension, angina, diabetes mellitus, hyperlipidemia, chronic obstructive pulmonary disease, ischemic heart disease, pneumonia or infection, varicose veins, depression, and lupus). Medication use (that is, antihypertensive drug, statin, antidiabetic drug, diuretics, aspirin, systemic corticosteroid, topical corticosteroid, nonsteroidal anti-inflammatory drugs, nitrates, and colchicine) was identified by at least 1 prescription during the 1 year before the index date. Serum creatinine level was obtained from the database before the index date. The eGFR was calculated from serum creatinine levels using the Modification of Diet in Renal Disease formula (45). We identified the presence of comorbidities using Read codes, as recorded by GPs. Finally, we calculated the number of visits to a GP and hospital admissions during the 1 year before the index date.
Statistical Analysis
The baseline characteristics of initiators were compared with those of PS-matched noninitiators using standardized differences. Person-years of follow-up for each participant were calculated as the amount of time from the index date to the first of the following events: death, disenrollment from a GP practice participating in THIN, 5 years of follow-up, or the end of the study (30 April 2019). We computed mortality rates and plotted cumulative incidence curves of death for initiators and noninitiators, respectively. We estimated the absolute rate difference in mortality between the 2 comparison groups. We obtained the hazard ratio (HR) of mortality for the allopurinol initiation using Cox proportional hazards models and the SE of the HR using “sandwich estimation” (46). We tested the proportional hazards assumption using the Kolmogorov supremum test (47). When the proportional hazards assumption was violated, we conducted a weighted Cox regression to obtain a weighted average HR (48). We did 7 sensitivity analyses to assess the robustness of the study findings. First, we did an as-treated analysis to account for nonadherence to treatment under investigation by censoring the follow-up when either initiators discontinued allopurinol treatment (that is, no prescription refill for allopurinol after a period of more than 60 days) or noninitiators started treatment with either allopurinol or febuxostat. Second, we did an analysis among persons who were enrolled in THIN for at least 1 year and developed gout during the follow-up. Third, we multiplied imputed missing data values of covariates (that is, body mass index, smoking status, alcohol drinking, socioeconomic deprivation index score, and eGFR) using a sequential regression method. To minimize random error, we imputed 5 data sets, did the PS matching, calculated the HRs and their 95% CIs from each imputed data set, and averaged these measures using the Rubin rule (49). Fourth, we did an analysis in participants whose gout diagnosis was defined by Read code plus receiving medication for gout (that is, colchicine or nonsteroidal anti-inflammatory drugs). This definition had a positive predictive value of 90% in the General Practice Research Database (50), in which 60% of participants overlap with THIN. Fifth, we introduced a 60-day lag time to exclude participants who died within the 60 days after the index date. Sixth, we calculated the E-value to quantitatively evaluate the minimum residual confounding effect that would nullify an association observed in the primary analyses (51). Finally, we assessed the robustness of our findings using other PS-matching methods (that is, optimal matching, 1: up to 2 matching, 1:up to 3 matching, and 1:up to 4 matching).
We emulated a hypothetical target trial to assess the effect of achieving target SU level with allopurinol, defined as any updated SU measurement less than 0.36 mmol/L within 1 year after allopurinol initiation, on mortality using observational data. Each initiator was assigned to both the achieving target SU level group and the not achieving target SU level group. We divided the follow-up time into 5 one-year time blocks starting from allopurinol initiation. Replicates assigned to the achieving target SU level group were censored at 1 year after allopurinol initiation if they did not achieve the target SU level. Replicates assigned to the not achieving target SU level group were censored if they achieved the target SU level at any time within 1 year after allopurinol initiation. Because censoring may lead to potential selection bias, we used inverse probability weights to account for censoring (42). The denominator of the inverse probability weight was the probability that a replicate adhered to his or her assigned group using the logistic regression, which consisted of the baseline covariates described earlier (see the Assessment of Covariates section). We fitted a pooled logistic regression model for mortality, including an indicator for achieving target SU level, year of follow-up (linear and quadratic term), and baseline confounders in the weighted population (52, 53). The odds ratio generated from this model approximated the HR because the outcome is rare. We used a robust SE to compute 95% CI for HR estimates. We estimated absolute 5-year mortality by fitting the pooled logistic models with product terms between the achieving target SU level indicator and the year of follow-up variables. The models’ predicted values were then used to estimate mortality from baseline (52). The mortality curves were standardized to the baseline variables (54). We used a nonparametric bootstrap with 100 samples to compute the 95% CI for absolute estimates. We took the same approach to assess the effect of allopurinol dose escalation versus no dose escalation within 1 year after initiation of allopurinol treatment on mortality (Supplement sections 1.1 and 1.2; Appendix Figure 1, available at Annals.org).
All analyses were done using SAS software, version 9.4 (SAS Institute), and a 2-sided P value of 0.05 or less was considered statistically significant for all tests.
Role of the Funding Source
The funding source had no role in the design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication.
RESULTS
Time-Stratified PS–Matched Cohorts
As shown in the Appendix Table (available at Annals. org), before PS matching, initiators had a higher SU level; a lower eGFR; a higher prevalence of prescriptions of diuretics, systemic corticosteroids, nonsteroidal anti-inflammatory drugs, and colchicine; and more GP visits than noninitiators (noninitiators could be included in more than 1 recruital time block before PS matching). After PS matching, the characteristics between the 2 comparison cohorts were well balanced, with all standardized differences less than 0.10 (Table 1). Among 5277 PS-matched initiators and noninitiators, the mean age was 74 years, and 40% were women. The mean values of body mass index, SU, and eGFR were 30.2 kg/m2, 0.52 mmol/L, and 47.5 mL/min/1.73 m2, respectively. The final mean levels of SU during 5-year follow-up were 0.42 mmol/L (SD, 0.13) and 0.48 mmol/L (SD, 0.12) in initiators and noninitiators, respectively. The final mean levels of eGFR during 5-year follow-up were 47.2 mL/min/1.73 m2 (SD, 16.8) and 46.1 mL/min/1.73 m2 (SD, 16.5) in initiators and noninitiators, respectively.
Table 1.
Baseline Characteristics of Patients With Gout and CKD by PS–Matched Allopurinol Initiator and Noninitiator Status
| Characteristic | Allopurinol Initiators | Noninitiators | Standardized Difference |
|---|---|---|---|
|
| |||
| Patients, n | 5277 | 5277 | - |
| Demographic characteristic | |||
| Mean age (SD), y | 73.9 (9.0) | 74.0 (9.6) | 0.008 |
| Mean socioeconomic deprivation index score (SD)* | 2.7 (1.3) | 2.7 (1.3) | 0.013 |
| Female, % | 38.8 | 39.8 | 0.021 |
| Mean body mass index (SD), kg/m2 | 30.2 (5.7) | 30.1 (5.8) | 0.015 |
| Mean SU level (SD), mmol/L | 0.52 (0.10) | 0.52 (0.10) | 0.041 |
| Mean estimated glomerular filtration rate (SD), mL/min/1.73 m2 | 47.5 (14.0) | 47.5 (14.1) | 0.001 |
| Lifestyle factors, % | |||
| Drinking | 0.009 | ||
| None | 20.4 | 20.8 | |
| Past | 3.7 | 3.8 | |
| Current | 75.9 | 75.5 | |
| Smoking | 0.013 | ||
| None | 49.3 | 48.7 | |
| Past | 44.6 | 44.9 | |
| Current | 6.1 | 6.3 | |
| Region, % | <0.001 | ||
| England | 78.2 | 78.2 | |
| Northern Ireland | 5.2 | 5.2 | |
| Scotland | 8.0 | 8.0 | |
| Wales | 8.6 | 8.6 | |
| Stage of CKD, % | 0.036 | ||
| Stage 3 | 96.0 | 95.7 | |
| Stage 4 | 3.0 | 3.5 | |
| Stage 5 | 0.9 | 0.7 | |
| Comorbidity, % | |||
| Congestive heart failure | 19.3 | 19.3 | <0.001 |
| Hypertension | 81.6 | 81.0 | 0.014 |
| Chronic obstructive pulmonary disease | 9.7 | 10.1 | 0.011 |
| Myocardial infarction | 15.3 | 15.2 | 0.005 |
| Angina | 20.0 | 19.9 | 0.001 |
| Diabetes | 29.1 | 28.5 | 0.012 |
| Hyperlipidemia | 25.6 | 24.3 | 0.029 |
| Ischemic heart disease | 34.3 | 33.5 | 0.017 |
| Pneumonia or infection | 9.3 | 9.8 | 0.017 |
| Stroke | 7.6 | 7.5 | 0.003 |
| Transient ischemic attack | 6.7 | 6.5 | 0.005 |
| Varicose veins | 9.8 | 11.1 | 0.044 |
| Depression | 10.2 | 10.4 | 0.007 |
| Lupus | 0.3 | 0.3 | 0.007 |
| Medication, % † | |||
| Angiotensin-converting enzyme inhibitors | 53.6 | 53.8 | 0.004 |
| β-receptor inhibitor | 47.7 | 47.1 | 0.013 |
| Calcium-channel blockers | 36.1 | 36.3 | 0.003 |
| Statin | 62.0 | 60.7 | 0.027 |
| Antidiabetic | 20.0 | 19.8 | 0.005 |
| Anticoagulants | 19.0 | 20.1 | 0.026 |
| Aspirin | 41.2 | 40.1 | 0.022 |
| Thiazide diuretics | 31.5 | 31.8 | 0.006 |
| Loop diuretics | 42.3 | 42.7 | 0.008 |
| Potassium-sparing diuretics | 13.3 | 13.6 | 0.007 |
| Systemic corticosteroid | 21.5 | 21.6 | 0.005 |
| Topical corticosteroid | 14.5 | 14.1 | 0.014 |
| Nitrates | 13.9 | 13.9 | 0.001 |
| Nonsteroidal anti-inflammatory drugs | 68.8 | 70.5 | 0.037 |
| Colchicine | 43.8 | 44.4 | 0.012 |
| Mean Health care use (SD) † | |||
| Hospitalizations | 0.6 (1.3) | 0.6 (1.4) | 0.011 |
| General practice visits | 9.8 (7.4) | 9.9 (9.7) | 0.019 |
| Specialist referrals | 0.8 (1.2) | 0.8 (1.3) | 0.003 |
CKD = chronic kidney disease; PS = propensity score; SU = serum urate.
The socioeconomic deprivation index was measured by the Townsend deprivation index, which was grouped into quintiles from 1 (least deprived) to 5 (most deprived).
Frequency during the past year.
Mortality was lower in initiators than in noninitiators (Figure 2). As shown in Table 2, 811 deaths (incidence rate, 4.9 per 100 person-years) occurred during 5 years of follow-up in initiators and 922 deaths (incidence rate, 5.8 per 100 person-years) occurred in their comparators. In initiators, compared with noninitiators, the rate difference of mortality was −0.9 (95% CI, −1.4 to −0.4) per 100 person-years and the HR of mortality was 0.85 (CI, 0.77 to 0.93). The proportional hazard assumption was violated, and the weighted average HR of mortality for initiators was 0.88 (CI, 0.80 to 0.97). Results from all sensitivity analyses also showed that initiators had a modestly lower mortality than noninitiators (Table 2). The E-value was 1.63 (CI, 1.36 to 1.92), indicating that the relation of potential residual confounders to both allopurinol initiation and death must be 1.63 or greater to nullify the modestly protective association between allopurinol initiation and mortality seen in the primary analyses. Similar results were also seen when other PS-matching methods were used to control for confounding (data not shown).
Figure 2.
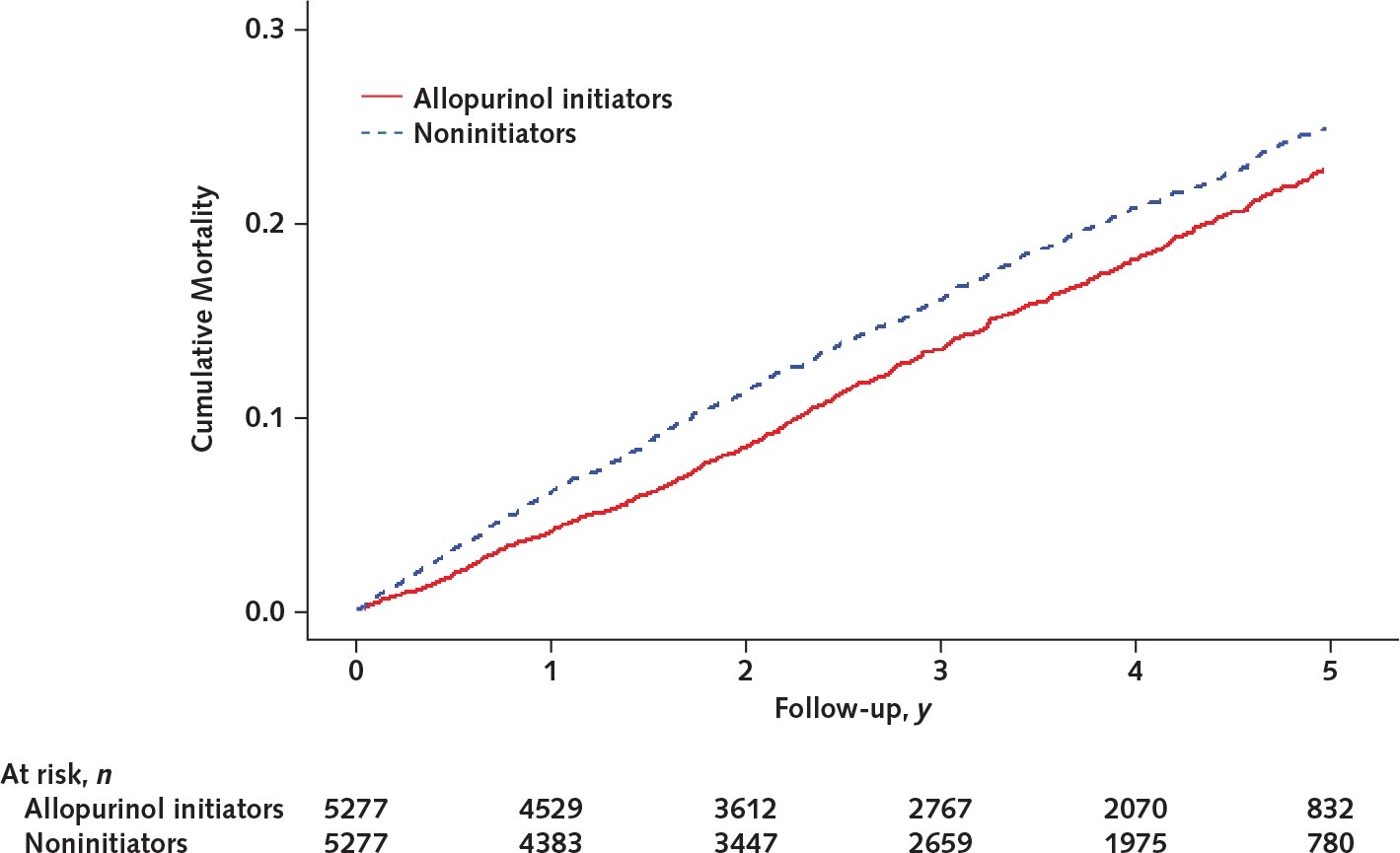
Five-year cumulative mortality between allopurinol initiators and noninitiators in the propensity score–matched study.
Table 2.
Relation of Allopurinol Initiation to All-Cause Mortality in Patients With Gout and CKD in the PS–Matched Cohort Study
| Analysis | Allopurinol Initiators/Noninitiators |
Rate Difference* per 100 Person-Years (95% CI) | Hazard Ratio† (95% CI) | |||
|---|---|---|---|---|---|---|
| Patients, n | Deaths, n | Mean Follow-up, y | Rate per 100 Person-Years | |||
|
| ||||||
| Primary analysis | 5277/5277 | 811/922 | 3.1/3.0 | 4.9/5.8 | −0.9 (−1.4 to −0.4) | 0.85 (0.77 to 0.93) |
| As-treated analysis‡ | 5277/5277 | 318/599 | 1.5/2.0 | 4.1/5.7 | −1.6 (−2.3 to−1.0) | 0.72 (0.63 to 0.82) |
| Incident gout cases | 4313/4313 | 643/698 | 3.1/3.0 | 4.8/5.4 | −0.6 (−1.1 to 0.0) | 0.89 (0.80 to 0.99) |
| Missing data imputation§ | 5980/5980 | 926/1067 | 3.1/3.0 | 4.9/5.9 | −1.0 (−1.5 to −0.5) | 0.83 (0.76 to 0.91) |
| Redefined gout cases | 4714/4714 | 704/783 | 3.1/3.0 | 4.8/5.6 | −0.8 (−1.3 to −0.2) | 0.86 (0.78 to 0.96) |
| 60-d lag analysis | 5226/5226 | 771/882 | 3.0/2.9 | 4.9/5.8 | −0.9 (−1.4 to −0.4) | 0.84 (0.76 to 0.93) |
CKD = chronic kidney disease; PS = propensity score.
The rate in allopurinol initiators minus the rate in noninitiators.
The rate in the allopurinol initiators divided by the rate in noninitiators.
This analysis censored the follow-up at the time when allopurinol initiators discontinued the allopurinol treatment (i.e., no prescription refill for allopurinol with a period of more than 60 d) or noninitiators started treatment (initiated with allopurinol or febuxostat).
We imputed 5 data sets and did the PS matching for each imputed data set. Patients, deaths, mean follow-up, rate, rate difference, and hazard ratio refer to the average value of the 5 imputed data sets.
Target Trial Emulation in Allopurinol Initiators
In 5277 initiators, 1484 achieved the target SU level within 1 year after the index date. The median value of initial allopurinol dose was 100 mg/d (range, 100 to 300 mg/d), the median value of final allopurinol dose was 300 mg/d (range, 100 to 900 mg/d), and the final mean level of SU during the 5-year follow-up was 0.30 mmol/L (SD, 0.08) in participants who achieved the target SU level. In 3793 participants who did not achieve the target SU level within 1 year after the index date, about 33% discontinued treatment after the first prescription. The median value of either initial or final allopurinol dose was 100 mg/d (range, 100 to 300 mg/d for initial dose; range, 100 to 900 mg/d for final dose), and the final mean level of SU during the 5-year follow-up was 0.47 mmol/L (SD, 0.11). For achieving target SU level, compared with not achieving target SU level, the difference in 5-year mortality was −1.6 percentage points (CI, −3.6 to −0.5 percentage points) and the HR was 0.87 (CI, 0.75 to 1.01) (Table 3; Appendix Figure 2, available at Annals.org).
Table 3.
Relations of Achieving Target SU Level (<0.36 mmol/L) and Allopurinol Dose Escalation to All-Cause Mortality in Allopurinol Initiators With Gout and CKD in the Target Trial Emulation Studies
| Variable | Achieving Target SU Level /Not Achieving Target SU Level | Dose Escalation/ No Dose Escalation |
|---|---|---|
|
| ||
| Patients, n | 5277/5277 | 3696/3696 |
| Weighted deaths, n | 660/754 | 483/518 |
| Weighted risk over 5 y, % | 13.2/14.8 | 13.9/15.2 |
| Inverse probability weighting risk difference, % (95% CI) | −1.6 (−3.6 to −0.5) | −1.4 (−3.7 to 0.4) |
| Inverse probability weighting hazard ratio (95% CI) | 0.87 (0.75 to 1.01) | 0.88 (0.73 to 1.07) |
CKD = chronic kidney disease; SU = serum urate.
In 5277 initiators, we excluded 1581 (30%) participants who did not have information on initial dose of allopurinol. In the remaining 3696 initiators, 773 increased their allopurinol dose within 1 year after the index date. In the allopurinol dose escalators, the median value of initial allopurinol dose before the dose escalation was 100 mg/d (range, 100 to 300 mg/d), and the corresponding value of final allopurinol dose after the dose escalation was 300 mg/d (range, 150 to 600 mg/d). The final mean SU level during the 5-year follow-up was 0.35 mmol/L (SD, 0.10). In the allopurinol nondose escalators, the final mean SU level was 0.39 mmol/L (SD, 0.11) during the 5-year follow-up. Compared with no dose escalation, the difference in 5-year mortality for dose escalation was −1.4 percentage points (CI, −3.7 to 0.4 percentage points), and the corresponding HR was 0.88 (CI, 0.73 to 1.07) (Table 3; Appendix Figure 3, available at Annals.org).
DISCUSSION
In this large database of GP electronic health records from the United Kingdom, allopurinol initiation was associated with a modestly lower mortality compared with nonallopurinol use in participants with both gout and moderate-to-severe CKD. In addition, emulating a target RCT in allopurinol initiators, we showed that a treat-to-target approach of lowering SU level with allopurinol does not seem to increase mortality in participants with both gout and CKD.
Many observational studies have found that persons with hyperuricemia are at an increased risk for incident and progressive renal disease compared with the general population (26, 55, 56). However, findings from Mendelian randomization studies do not support a causal relationship between SU levels and renal function (57–59). In addition, despite potential renoprotective benefits suggested in previous observational studies (39, 60–64) and earlier RCTs (65–69), 2 large-scale, recent RCTs (the CKD-FIX [Controlled trial of slowing of Kidney Disease progression From the Inhibition of Xanthine oxidase] [29] and the PERL [Preventing Early Renal Loss in Diabetes] trials [30]) reported no protective effect of allopurinol on renal function deterioration in patients with either stage 3 or 4 CKD without gout or in those with both type 1 diabetes and early-to-moderate diabetic kidney disease. Furthermore, both trials reported that mortality was numerically higher in the allopurinol treatment group than in the placebo group, with pooled analysis showing a relative risk of 2.07 (CI, 0.98 to 4.34; P= 0.06) (31, 32).
Although some previous studies found that allopurinol use was associated with lower mortality in patients with gout or persons with hyperuricemia (15–18), others have not confirmed this (19, 20). A recent meta-analysis found that several studies that reported a protective effect of allopurinol use on mortality may have immortal time bias or immeasurable time bias (21). The meta-analysis of studies that did not have such biases found a null association (21). Nevertheless, no previous study has assessed the effect of allopurinol initiation on mortality, specifically in patients with both gout and CKD. Our study found that neither allopurinol use, nor achieving target SU level, nor allopurinol dose escalation with allopurinol seem to increase mortality in participants with gout and concurrent CKD. These findings provide empirical evidence that adopting current gout treatment guidelines does not seem to have a detrimental effect on mortality in patients with both gout and CKD.
Two strengths of our study merit comment. First, owing to ethical and logistic difficulties, it is likely infeasible to conduct an RCT to assess whether allopurinol use increases mortality in participants with both gout and CKD. Using a real-world, population-based electronic database and a study design emulating a RCT, we showed that the strategy of achieving target SU level with allopurinol did not show a detrimental effect on mortality in patients with both gout and CKD. These findings are pertinent to the management of gout in the context of CKD. Second, we assessed both absolute and relative effects of allopurinol initiation, achieving target SU level via allopurinol, and allopurinol dose escalation on mortality and evaluated the effect using both intention-to-treat and as-treated approaches. All results were consistent, indicating the robustness of our study findings.
Our study has some limitations. First, although we used rigorous approaches to control for confounding, some covariates, such as disease severity; causes of CKD (for example, IgA nephropathy); and finer geographic areas across England, Northern Ireland, Scotland, or Wales, may not be well captured in THIN; thus, residual confounding cannot be ruled out. For example, we were unable to adjust for severity of various comorbidities. More frail and sicker patients may be less likely to be prescribed and continue their “preventive medication” (for example, allopurinol), particularly for nonimmediately fatal conditions, or physicians may be reluctant to escalate the allopurinol dose for those patients. Consequently, residual confounding due to severity of comorbidities could lead to a potentially biased protective effect of allopurinol on mortality. Second, allopurinol initiators and those who achieved target SU levels may have received better health care for their overall health needs than their comparators, which could also lead to lower mortality. Third, previous studies have reported that allopurinol use was associated with a decreased risk for cardiovascular mortality compared with nonuse (70) but a modestly increased risk for cardiovascular events compared with probenecid initiators (71). However, owing to a lack of recent data on cause-specific mortality in THIN, we were unable to assess the effect of allopurinol use on the risk for cardiovascular mortality in participants with both gout and CKD.
Our findings are clinically relevant in gout care because CKD is a common comorbidity of gout, and allopurinol is most commonly used with escalating doses to achieve and maintain a SU target below a subsaturation point of urate crystals, which will eventually decrease gout flare frequency and tissue urate crystal burden (72–74). However, the findings from 2 recent RCTs that allopurinol use may increase mortality in participants without gout but with CKD have raised concerns about whether a treat-to-target approach of lowering SU level would be safe for patients with gout and concurrent CKD. To that end, our findings provide reassurance that such a strategy does not have an apparent detrimental effect on mortality in patients with both gout and CKD (9–12).
In conclusion, in this population-based data, neither allopurinol initiation, nor achieving target SU level with allopurinol, nor allopurinol dose escalation was associated with an increased risk for death in patients with gout and concurrent CKD.
Supplementary Material
Financial Support:
By the Project Program of National Clinical Research Center for Geriatric Disorders (Dr. Zeng: Xiangya Hospital 2020LNJJ03), the National Natural Science Foundation of China (Dr. Lei: 81930071 and 81772413, Dr. Zeng: 82072502), and the National Institutes of Health (Dr. Neogi: K24 AR070892 and P30 AR072571).
Appendix Table.
Baseline Characteristics of Patients With Gout and CKD by Allopurinol Initiator and Noninitiator Status Before PS Matching
| Characteristic | Allopurinol Initiators | Noninitiators* | Standardized Difference† |
|---|---|---|---|
|
| |||
| Patients, n | 6962 | 64 353 | – |
| Demographic characteristic | |||
| Mean age (SD), y | 73.9 (9.1) | 74.9 (9.0) | 0.050 |
| Mean socioeconomic deprivation index (SD)‡ | 2.7 (1.3) | 2.6 (1.3) | 0.017 |
| Female, % | 40.0 | 32.3 | 0.074 |
| Mean body mass index (SD), kg/m2 | 30.2 (5.8) | 29.4 (5.4) | 0.071 |
| Mean SU level (SD), mmol/L | 0.54 (0.11) | 0.42 (0.12) | 0.471 |
| Mean estimated glomerular filtration rate (SD), mL/min/1.73 m2 | 46.6 (15.1) | 50.2 (14.8) | 0.114 |
| Lifestyle factors, % | |||
| Drinking | 0.018 | ||
| None | 20.8 | 19.2 | |
| Past | 3.7 | 3.9 | |
| Current | 75.5 | 76.9 | |
| Smoking | 0.001 | ||
| None | 49.0 | 49.3 | |
| Past | 45.0 | 44.5 | |
| Current | 6.1 | 6.2 | |
| Region, % | 0.061 | ||
| England | 77.0 | 82.5 | |
| Northern Ireland | 5.6 | 5.5 | |
| Scotland | 8.3 | 4.3 | |
| Wales | 9.1 | 7.7 | |
| Stage of CKD, % | 0.005 | ||
| Stage 3 | 95.6 | 95.6 | |
| Stage 4 | 3.4 | 3.2 | |
| Stage 5 | 1.0 | 1.2 | |
| Comorbidity, % | |||
| Congestive heart failure | 21.2 | 14.1 | 0.087 |
| Hypertension | 81.9 | 81.5 | 0.005 |
| Chronic obstructive pulmonary disease | 10.1 | 9.0 | 0.017 |
| Myocardial infarction | 15.6 | 14.6 | 0.013 |
| Angina | 20.5 | 20.7 | 0.002 |
| Diabetes | 29.1 | 30.4 | 0.014 |
| Hyperlipidemia | 25.5 | 25.2 | 0.004 |
| Ischemic heart disease | 35.2 | 33.3 | 0.018 |
| Pneumonia or infection | 9.3 | 9.7 | 0.007 |
| Stroke | 7.6 | 8.4 | 0.015 |
| Transient ischemic attack | 6.4 | 7.4 | 0.018 |
| Varicose veins | 10.5 | 10.7 | 0.002 |
| Depression | 10.2 | 9.6 | 0.010 |
| Lupus | 0.3 | 0.4 | 0.007 |
| Medication, % § | |||
| Angiotensin-converting enzyme inhibitors | 54.7 | 48.7 | 0.058 |
| β-receptor inhibitor | 49.1 | 41.9 | 0.069 |
| Calcium-channel blockers | 35.8 | 39.7 | 0.039 |
| Statin | 62.4 | 61.1 | 0.013 |
| Antidiabetic | 20.2 | 19.9 | 0.003 |
| Anticoagulants | 20.0 | 16.1 | 0.048 |
| Aspirin | 41.2 | 41.5 | 0.003 |
| Thiazide diuretics | 31.8 | 21.3 | 0.114 |
| Loop diuretics | 46.0 | 29.7 | 0.163 |
| Potassium-sparing diuretics | 14.9 | 8.8 | 0.089 |
| Systemic corticosteroid | 23.3 | 14.0 | 0.116 |
| Topical corticosteroid | 14.7 | 14.1 | 0.007 |
| Nitrates | 14.5 | 12.4 | 0.031 |
| Nonsteroidal anti-inflammatory drugs | 70.4 | 51.4 | 0.203 |
| Colchicine | 49.5 | 12.2 | 0.411 |
| Mean Health care use (SD) § | |||
| Hospitalizations | 0.6 (1.4) | 0.6 (1.4) | 0.018 |
| General practice visits | 10.2 (7.9) | 7.9 (7.7) | 0.143 |
| Specialist referrals | 0.8 (1.3) | 0.7 (1.2) | 0.039 |
CKD = chronic kidney disease; PS = propensity score; SU = serum urate.
The number for noninitiators represents the number of records (i.e., participants could be included in more than 1 recruitment time block).
We used generalized estimating equations to calculate the standardized differences accounting for correlation among the duplicated participants.
The socioeconomic deprivation index was measured by the Townsend deprivation index, which was grouped into quintiles from 1 (least deprived) to 5 (most deprived).
Frequency during the past year.
Appendix Figure 1.
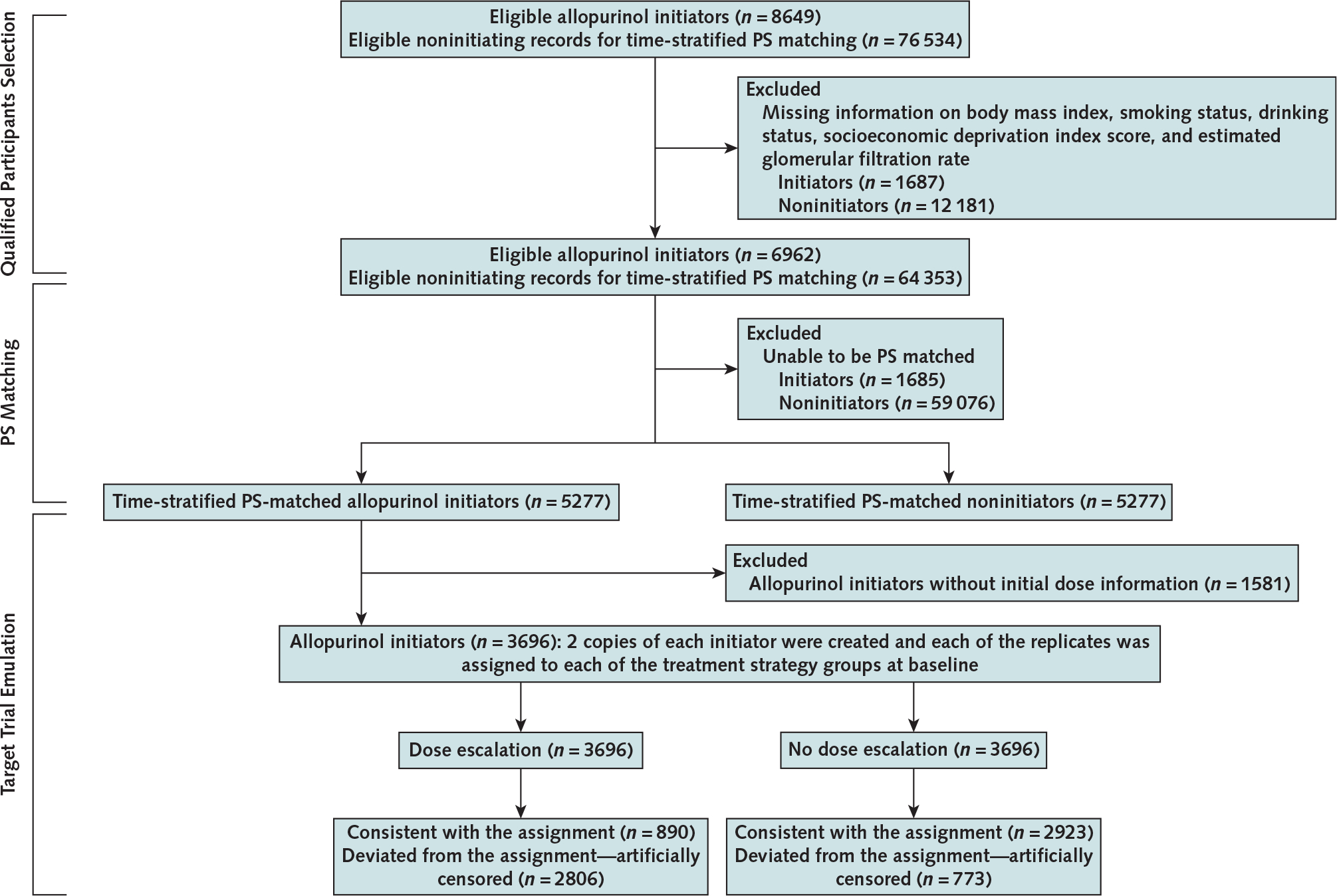
Flow chart of eligible persons for examining the relation of allopurinol initiation and allopurinol dose escalation to all-cause mortality, THIN 2000 to 2019.
PS = propensity score; THIN = The Health Improvement Network.
Appendix Figure 2.
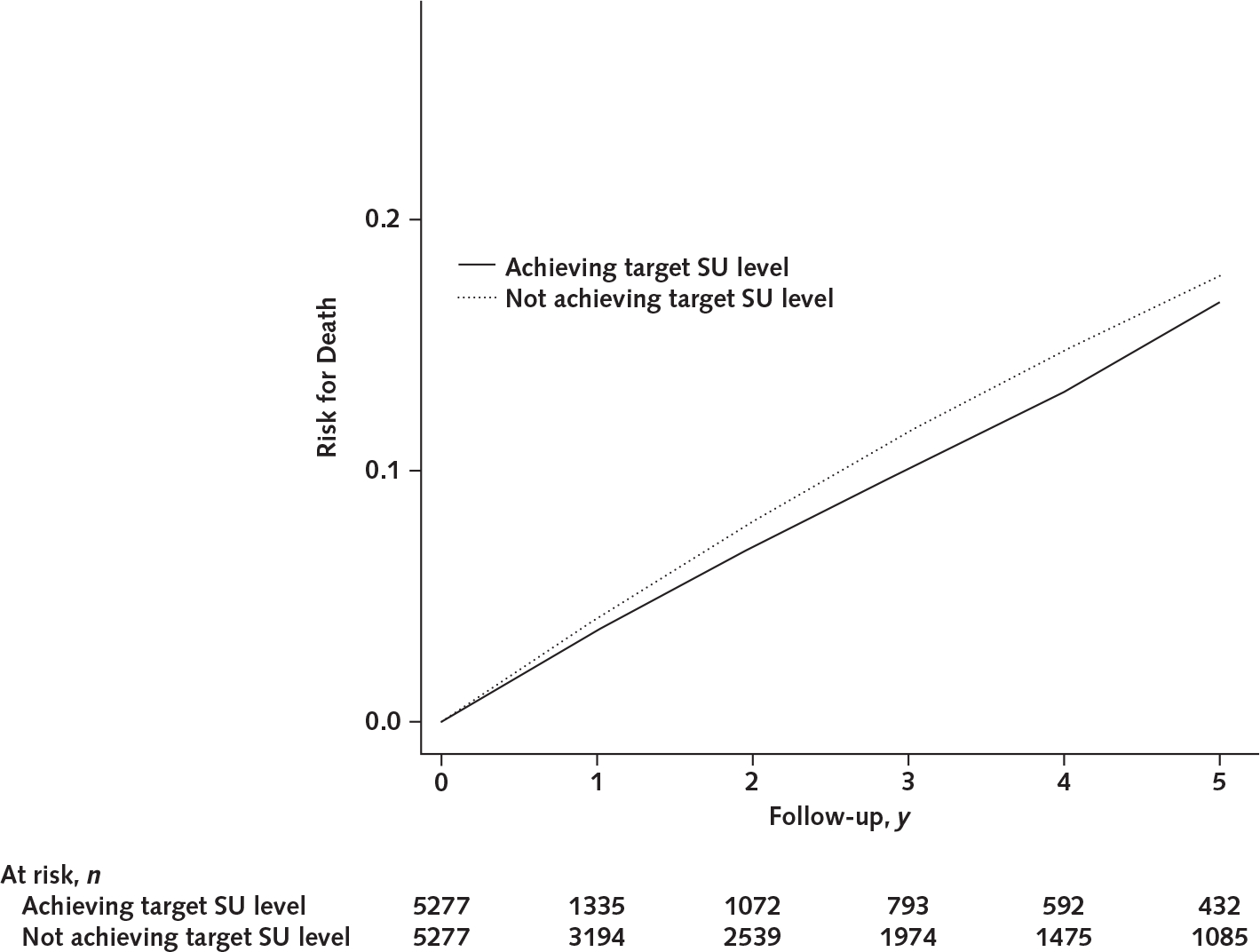
Five-year risk for death between patients achieving target SU level and those not achieving target SU level with allopurinol in the target trial emulation study.
SU = serum urate.
Appendix Figure 3.
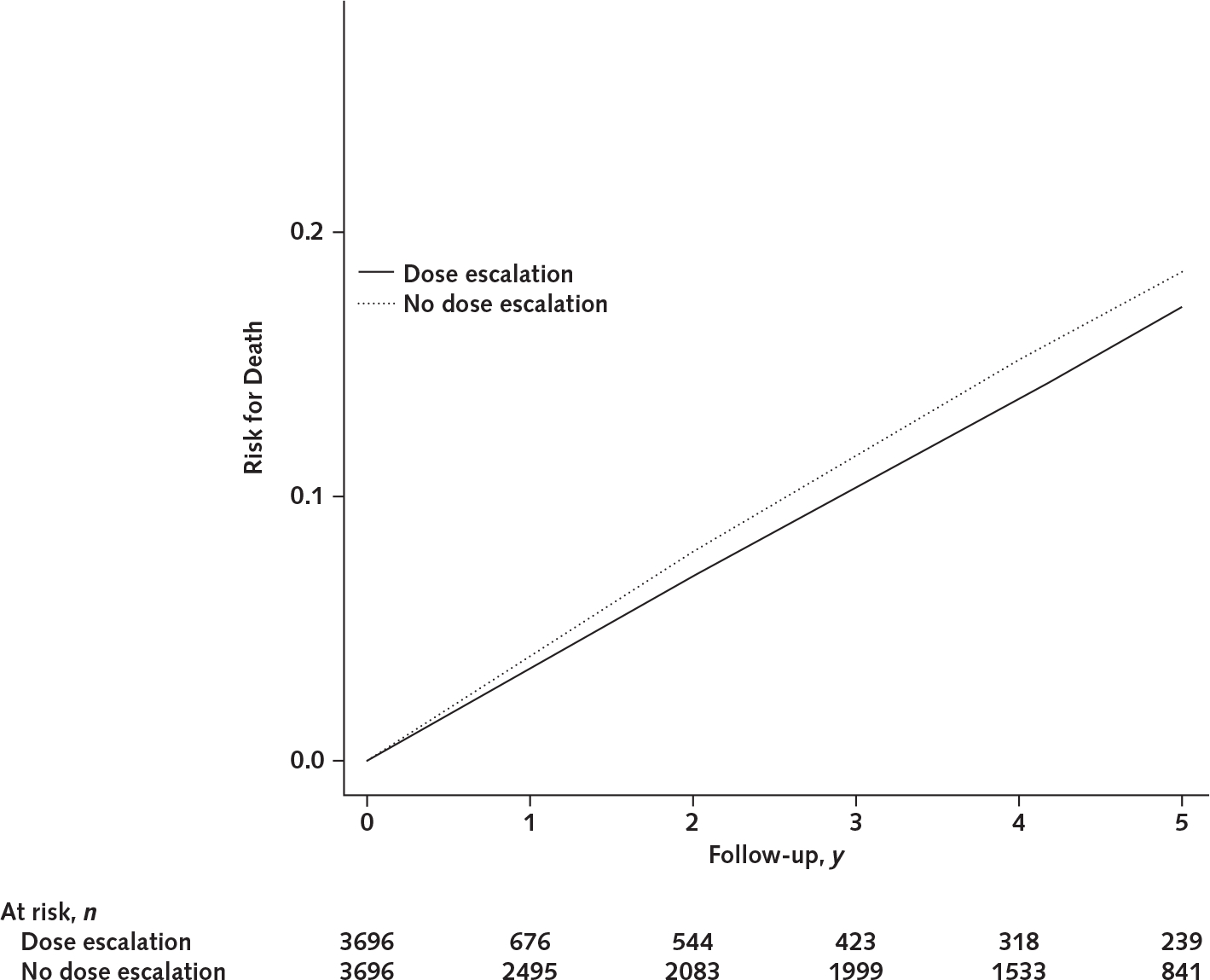
Five-year risk for death between allopurinol dose escalation and no allopurinol dose escalation in the target trial emulation study.
Footnotes
Disclaimer: The interpretation of these data is the sole responsibility of the authors. THIN is a registered trademark of Cegedim SA in the United Kingdom and other countries. Reference made to the THIN database is intended to be descriptive of the data asset licensed by IQVIA. This work uses deidentified data provided by patients as a part of their routine primary care.
Disclosures: Disclosures can be viewed at www.acponline.org/authors/icmje/ConflictOfInterestForms.do?msNum=M21-2347.
Reproducible Research Statement: Study protocol: Available from Dr. Lei (e-mail, lei_guanghua@csu.edu.cn). Statistical code: See Supplement section 2 (available at Annals.org). Data set: Available for purchase from info@the-health-improvement-network.co.uk.
See also: Web-Only Supplement
Contributor Information
Jie Wei, Health Management Center, Xiangya Hospital, Central South University, Changsha, China.
Hyon K. Choi, Division of Rheumatology, Allergy, and Immunology, Department of Medicine, and the Mongan Institute, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts.
Tuhina Neogi, Section of Rheumatology, Boston University School of Medicine, Boston, Massachusetts.
Nicola Dalbeth, Department of Medicine, University of Auckland, Auckland, New Zealand.
Robert Terkeltaub, Rheumatology, Allergy-Immunology Section, San Diego VA Medical Center, San Diego, California.
Lisa K. Stamp, Department of Medicine, University of Otago, Christchurch, New Zealand.
Houchen Lyu, Department of Orthopedics, General Hospital of Chinese PLA, Beijing, and Department of Orthopaedics, Xiangya Hospital, Central South University, Changsha, China.
Natalie McCormick, Division of Rheumatology, Allergy, and Immunology, Department of Medicine, and the Mongan Institute, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts, and Arthritis Research Canada, Richmond, British Columbia, Canada.
Jingbo Niu, Selzman Institute for Kidney Health, Section of Nephrology, Department of Medicine, Baylor College of Medicine, Houston, Texas.
Chao Zeng, Department of Orthopaedics, Xiangya Hospital, Central South University, and Hunan Key Laboratory of Joint Degeneration and Injury, and National Clinical Research Center of Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China.
Guanghua Lei, Department of Orthopaedics, Xiangya Hospital, Central South University, and National Clinical Research Center of Geriatric Disorders, Xiangya Hospital, Central South University, and Hunan Key Laboratory of Joint Degeneration and Injury, Changsha, China.
Yuqing Zhang, Division of Rheumatology, Allergy, and Immunology, Department of Medicine, and the Mongan Institute, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts.
Reference
- 1.Dehlin M, Jacobsson L, Roddy E. Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat Rev Rheumatol. 2020;16:380–390. doi: 10.1038/s41584-020-0441-1 [DOI] [PubMed] [Google Scholar]
- 2.Kuo CF, Grainge MJ, Mallen C, et al. Rising burden of gout in the UK but continuing suboptimal management: a nationwide population study. Ann Rheum Dis. 2015;74:661–7. doi: 10.1136/annrheumdis-2013-204463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rai SK, Aviña-Zubieta JA, McCormick N, et al. The rising prevalence and incidence of gout in British Columbia, Canada: population-based trends from 2000 to 2012. Semin Arthritis Rheum. 2017;46:451–456. doi: 10.1016/j.semarthrit.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zobbe K, Prieto-Alhambra D, Cordtz R, et al. Secular trends in the incidence and prevalence of gout in Denmark from 1995 to 2015: a nationwide register-based study. Rheumatology (Oxford). 2019;58:836–839. doi: 10.1093/rheumatology/key390 [DOI] [PubMed] [Google Scholar]
- 5.Kim JW, Kwak SG, Lee H, et al. Prevalence and incidence of gout in Korea: data from the national health claims database 2007–2015. Rheumatol Int. 2017;37:1499–1506. doi: 10.1007/s00296-017-3768-4 [DOI] [PubMed] [Google Scholar]
- 6.Dehlin M, Drivelegka P, Sigurdardottir V, et al. Incidence and prevalence of gout in Western Sweden. Arthritis Res Ther. 2016;18:164. doi: 10.1186/s13075-016-1062-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Safiri S, Kolahi AA, Cross M, et al. Prevalence, incidence, and years lived with disability due to gout and its attributable risk factors for 195 countries and territories 1990–2017: a systematic analysis of the global burden of disease study 2017. Arthritis Rheumatol. 2020;72:1916–1927. doi: 10.1002/art.41404 [DOI] [PubMed] [Google Scholar]
- 8.Xia Y, Wu Q, Wang H, et al. Global, regional and national burden of gout, 1990–2017: a systematic analysis of the Global Burden of Disease Study. Rheumatology (Oxford). 2020;59:1529–1538. doi: 10.1093/rheumatology/kez476 [DOI] [PubMed] [Google Scholar]
- 9.FitzGerald JD, Dalbeth N, Mikuls T, et al. 2020 American College of Rheumatology guideline for the management of gout. Arthritis Care Res (Hoboken). 2020;72:744–760. doi: 10.1002/acr.24180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hui M, Carr A, Cameron S, et al. ; British Society for Rheumatology Standards, Audit and Guidelines Working Group. The British Society for Rheumatology guideline for the management of gout. Rheumatology (Oxford). 2017;56:e1–e20. doi: 10.1093/rheumatology/kex156 [DOI] [PubMed] [Google Scholar]
- 11.Richette P, Doherty M, Pascual E, et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. 2017;76:29–42. doi: 10.1136/annrheumdis-2016-209707 [DOI] [PubMed] [Google Scholar]
- 12.Sivera F, Andrés M, Carmona L, et al. Multinational evidence-based recommendations for the diagnosis and management of gout: integrating systematic literature review and expert opinion of a broad panel of rheumatologists in the 3e initiative. Ann Rheum Dis. 2014;73:328–35. doi: 10.1136/annrheumdis-2013-203325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SC, Neogi T, Kim E, et al. Trends in utilization of urate-lowering therapies following the US Food and Drug Administration’s boxed warning on febuxostat. Arthritis Rheumatol. 2021;73:542–543. doi: 10.1002/art.41550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stamp LK, Chapman PT, Barclay ML, et al. A randomised controlled trial of the efficacy and safety of allopurinol dose escalation to achieve target serum urate in people with gout. Ann Rheum Dis. 2017;76:1522–1528. doi: 10.1136/annrheumdis-2016-210872 [DOI] [PubMed] [Google Scholar]
- 15.Luk AJ, Levin GP, Moore EE, et al. Allopurinol and mortality in hyperuricaemic patients. Rheumatology (Oxford). 2009;48:804–6. doi: 10.1093/rheumatology/kep069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen KS, Pottegård A, Lindegaard HM, et al. Effect of allopurinol on cardiovascular outcomes in hyperuricemic patients: a cohort study. Am J Med. 2016;129:299–306.e2. doi: 10.1016/j.amjmed.2015.11.003 [DOI] [PubMed] [Google Scholar]
- 17.Dubreuil M, Zhu Y, Zhang Y, et al. Allopurinol initiation and all-cause mortality in the general population. Ann Rheum Dis. 2015;74:1368–72. doi: 10.1136/annrheumdis-2014-205269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen JH, Lan JL, Cheng CF, et al. Effect of urate-lowering therapy on the risk of cardiovascular disease and all-cause mortality in patients with gout: a case-matched cohort study. J Rheumatol. 2015;42:1694–701. doi: 10.3899/jrheum.141542 [DOI] [PubMed] [Google Scholar]
- 19.Kuo CF, Grainge MJ, Mallen C, et al. Effect of allopurinol on all-cause mortality in adults with incident gout: propensity score-matched landmark analysis. Rheumatology (Oxford). 2015;54:2145–50. doi: 10.1093/rheumatology/kev246 [DOI] [PubMed] [Google Scholar]
- 20.Ju C, Lai RWC, Li KHC, et al. Comparative cardiovascular risk in users versus non-users of xanthine oxidase inhibitors and febuxostat versus allopurinol users. Rheumatology (Oxford). 2020;59:2340–2349. doi: 10.1093/rheumatology/kez576 [DOI] [PubMed] [Google Scholar]
- 21.Suissa S, Suissa K, Hudson M. Effectiveness of allopurinol in reducing mortality: time-related biases in observational studies. Arthritis Rheumatol. 2021;73:1749–1757. doi: 10.1002/art.41710 [DOI] [PubMed] [Google Scholar]
- 22.Hay CA, Prior JA, Belcher J, et al. Mortality in patients with gout treated with allopurinol: a systematic review and meta-analysis. Arthritis Care Res (Hoboken). 2021;73:1049–1054. doi: 10.1002/acr.24205 [DOI] [PubMed] [Google Scholar]
- 23.Coburn BW, Michaud K, Bergman DA, et al. Allopurinol dose escalation and mortality among patients with gout: a national propensity-matched cohort study. Arthritis Rheumatol. 2018;70:1298–1307. doi: 10.1002/art.40486 [DOI] [PubMed] [Google Scholar]
- 24.Bevis M, Blagojevic-Bucknall M, Mallen C, et al. Comorbidity clusters in people with gout: an observational cohort study with linked medical record review. Rheumatology (Oxford). 2018;57:1358–1363. doi: 10.1093/rheumatology/key096 [DOI] [PubMed] [Google Scholar]
- 25.Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007–2008. Am J Med. 2012;125:679–687.e1. doi: 10.1016/j.amjmed.2011.09.033 [DOI] [PubMed] [Google Scholar]
- 26.Weiner DE, Tighiouart H, Elsayed EF, et al. Uric acid and incident kidney disease in the community. J Am Soc Nephrol. 2008;19:1204–11. doi: 10.1681/ASN.2007101075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obermayr RP, Temml C, Gutjahr G, et al. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol. 2008;19: 2407–13. doi: 10.1681/ASN.2008010080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu CY, Iribarren C, McCulloch CE, et al. Risk factors for end-stage renal disease: 25-year follow-up. Arch Intern Med. 2009;169:342–50. doi: 10.1001/archinternmed.2008.605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Badve SV, Pascoe EM, Tiku A, et al. ; CKD-FIX Study Investigators. Effects of allopurinol on the progression of chronic kidney disease. N Engl J Med. 2020;382:2504–2513. doi: 10.1056/NEJMoa1915833 [DOI] [PubMed] [Google Scholar]
- 30.Doria A, Galecki AT, Spino C, et al. ; PERL Study Group. Serum urate lowering with allopurinol and kidney function in type 1 diabetes. N Engl J Med. 2020;382:2493–2503. doi: 10.1056/NEJMoa1916624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allopurinol Conway R. and chronic kidney disease [Letter]. N Engl J Med. 2020;383:1689. doi: 10.1056/NEJMc2026125 [DOI] [PubMed] [Google Scholar]
- 32.McCormick N, Zhang Y, Choi HK. Allopurinol and chronic kidney disease [Letter]. N Engl J Med. 2020;383:1689–1690. doi: 10.1056/NEJMc2026125 [DOI] [PubMed] [Google Scholar]
- 33.Chisholm J The read clinical classification [Editorial]. BMJ. 1990;300:1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Databank First. FDB Multilex. Accessed at www.fdbhealth.co.uk/solutions/multilex-clinical-decision-support on 8 March 2021.
- 35.Lewis JD, Schinnar R, Bilker WB, et al. Validation studies of the Health Improvement Network (THIN) database for pharmacoepidemiology research. Pharmacoepidemiol Drug Saf. 2007;16:393–401. [DOI] [PubMed] [Google Scholar]
- 36.Langan SM, Schmidt SA, Wing K, et al. The reporting of studies conducted using observational routinely collected health data statement for pharmacoepidemiology (RECORD-PE). BMJ. 2018; 363:k3532. doi: 10.1136/bmj.k3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Peloquin CE, Dubreuil M, et al. Sleep apnea and the risk of incident gout: a population-based, body mass index-matched cohort study. Arthritis Rheumatol. 2015;67:3298–302. doi: 10.1002/art.39330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlesinger N, Lu N, Choi HK. Gout and the risk of incident erectile dysfunction: a body mass index-matched population-based study. J Rheumatol. 2018;45:1192–1197. doi: 10.3899/jrheum.170444 [DOI] [PubMed] [Google Scholar]
- 39.Vargas-Santos AB, Peloquin CE, Zhang Y, et al. Association of chronic kidney disease with allopurinol use in gout treatment. JAMA Intern Med. 2018;178:1526–1533. doi: 10.1001/jamainternmed.2018.4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seeger JD, Williams PL, Walker AM. An application of propensity score matching using claims data. Pharmacoepidemiol Drug Saf. 2005;14:465–76. [DOI] [PubMed] [Google Scholar]
- 41.Lyu H, Yoshida K, Zhao SS, et al. Delayed denosumab injections and fracture risk among patients with osteoporosis: a population-based cohort study. Ann Intern Med. 2020;173:516–526. doi: 10.7326/M20-0882 [DOI] [PubMed] [Google Scholar]
- 42.Hernán MA. How to estimate the effect of treatment duration on survival outcomes using observational data. BMJ. 2018;360: k182. doi: 10.1136/bmj.k182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hernán MA, Robins JM. Using big data to emulate a target trial when a randomized trial is not available. Am J Epidemiol. 2016;183:758–64. doi: 10.1093/aje/kwv254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mikuls TR, Cheetham TC, Levy GD, et al. Adherence and outcomes with urate-lowering therapy: a site-randomized trial. Am J Med. 2019;132:354–361. doi: 10.1016/j.amjmed.2018.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–70. [DOI] [PubMed] [Google Scholar]
- 46.Webster-Clark M, Stürmer T, Wang T, et al. Using propensity scores to estimate effects of treatment initiation decisions: state of the science. Stat Med. 2021;40:1718–1735. doi: 10.1002/sim.8866 [DOI] [PubMed] [Google Scholar]
- 47.Lin DY, Wei LJ, Ying Z. Checking the Cox model with cumulative sums of Martingale-based residuals. Biometrika. 1993;80:557–72. doi: 10.2307/2337177 [DOI] [Google Scholar]
- 48.Dunkler D, Ploner M, Schemper M, et al. Weighted Cox regression using the R package coxphw. J Stat Softw. 2018;84:1–26. doi: 10.18637/jss.v084.i0230450020 [DOI] [Google Scholar]
- 49.Rubin DB. Multiple Imputation for Nonresponse in Surveys. J Wiley; 2004. [Google Scholar]
- 50.Meier CR, Jick H. Omeprazole, other antiulcer drugs and newly diagnosed gout. Br J Clin Pharmacol. 1997;44:175–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.VanderWeele TJ, Ding P. Sensitivity analysis in observational research: introducing the E-value. Ann Intern Med. 2017;167:268–274. doi: 10.7326/M16-2607 [DOI] [PubMed] [Google Scholar]
- 52.Emilsson L, García-Albeniz X, Logan RW, et al. Examining bias in studies of statin treatment and survival in patients with cancer. JAMA Oncol. 2018;4:63–70. doi: 10.1001/jamaoncol.2017.2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hernán MA, Brumback B, Robins JM. Marginal structural models to estimate the causal effect of zidovudine on the survival of HIV-positive men. Epidemiology. 2000;11:561–70. [DOI] [PubMed] [Google Scholar]
- 54.Danaei G, García Rodríguez LA, Cantero OF, et al. Electronic medical records can be used to emulate target trials of sustained treatment strategies. J Clin Epidemiol. 2018;96:12–22. doi: 10.1016/j.jclinepi.2017.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Domrongkitchaiporn S, Sritara P, Kitiyakara C, et al. Risk factors for development of decreased kidney function in a southeast Asian population: a 12-year cohort study. J Am Soc Nephrol. 2005;16:791–9. [DOI] [PubMed] [Google Scholar]
- 56.Kuo CF, Luo SF, See LC, et al. Hyperuricaemia and accelerated reduction in renal function. Scand J Rheumatol. 2011;40:116–21. doi: 10.3109/03009742.2010.507218 [DOI] [PubMed] [Google Scholar]
- 57.Hughes K, Flynn T, de Zoysa J, et al. Mendelian randomization analysis associates increased serum urate, due to genetic variation in uric acid transporters, with improved renal function. Kidney Int. 2014;85:344–51. doi: 10.1038/ki.2013.353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahola AJ, Sandholm N, Forsblom C, et al. ; FinnDiane Study Group. The serum uric acid concentration is not causally linked to diabetic nephropathy in type 1 diabetes. Kidney Int. 2017;91:1178–1185. doi: 10.1016/j.kint.2016.11.025 [DOI] [PubMed] [Google Scholar]
- 59.Jordan DM, Choi HK, Verbanck M, et al. No causal effects of serum urate levels on the risk of chronic kidney disease: a Mendelian randomization study. PLoS Med. 2019;16:e1002725. doi: 10.1371/journal.pmed.1002725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanbay M, Ozkara A, Selcoki Y, et al. Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearance, and proteinuria in patients with normal renal functions. Int Urol Nephrol. 2007;39:1227–33. [DOI] [PubMed] [Google Scholar]
- 61.Pai BH, Swarnalatha G, Ram R, et al. Allopurinol for prevention of progression of kidney disease with hyperuricemia. Indian J Nephrol. 2013;23:280–6. doi: 10.4103/0971-4065.114499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levy GD, Rashid N, Niu F, et al. Effect of urate-lowering therapies on renal disease progression in patients with hyperuricemia. J Rheumatol. 2014;41:955–62. doi: 10.3899/jrheum.131159 [DOI] [PubMed] [Google Scholar]
- 63.Krishnamurthy A, Lazaro D, Stefanov DG, et al. The effect of allopurinol on renal function. J Clin Rheumatol. 2017;23:1–5. doi: 10.1097/RHU.0000000000000480 [DOI] [PubMed] [Google Scholar]
- 64.Singh JA, Yu S. Are allopurinol dose and duration of use nephroprotective in the elderly? A Medicare claims study of allopurinol use and incident renal failure. Ann Rheum Dis. 2017;76:133–139. doi: 10.1136/annrheumdis-2015-209046 [DOI] [PubMed] [Google Scholar]
- 65.Goicoechea M, Garcia de Vinuesa S, Verdalles U, et al. Allopurinol and progression of CKD and cardiovascular events: long-term follow-up of a randomized clinical trial. Am J Kidney Dis. 2015;65:543–9. doi: 10.1053/j.ajkd.2014.11.016 [DOI] [PubMed] [Google Scholar]
- 66.Siu YP, Leung KT, Tong MK, et al. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51–9. [DOI] [PubMed] [Google Scholar]
- 67.Liu P, Chen Y, Wang B, et al. Allopurinol treatment improves renal function in patients with type 2 diabetes and asymptomatic hyperuricemia: 3-year randomized parallel-controlled study. Clin Endocrinol (Oxf). 2015;83:475–82. doi: 10.1111/cen.12673 [DOI] [PubMed] [Google Scholar]
- 68.Goicoechea M, de Vinuesa SG, Verdalles U, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5:1388–93. doi: 10.2215/CJN.01580210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kao MP, Ang DS, Gandy SJ, et al. Allopurinol benefits left ventricular mass and endothelial dysfunction in chronic kidney disease. J Am Soc Nephrol. 2011;22:1382–9. doi: 10.1681/ASN.2010111185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.MacIsaac RL, Salatzki J, Higgins P, et al. Allopurinol and cardiovascular outcomes in adults with hypertension. Hypertension. 2016;67:535–40. doi: 10.1161/HYPERTENSIONAHA.115.06344 [DOI] [PubMed] [Google Scholar]
- 71.Kim SC, Neogi T, Kang EH, et al. Cardiovascular risks of Probenecid versus allopurinol in older patients with gout. J Am Coll Cardiol. 2018;71:994–1004. doi: 10.1016/j.jacc.2017.12.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shoji A, Yamanaka H, Kamatani N. A retrospective study of the relationship between serum urate level and recurrent attacks of gouty arthritis: evidence for reduction of recurrent gouty arthritis with antihyperuricemic therapy. Arthritis Rheum. 2004;51:321–5. [DOI] [PubMed] [Google Scholar]
- 73.Dalbeth N, Billington K, Doyle A, et al. Effects of allopurinol dose escalation on bone erosion and urate volume in gout: a dual-energy computed tomography imaging study within a randomized, controlled trial. Arthritis Rheumatol. 2019;71:1739–1746. doi: 10.1002/art.40929 [DOI] [PubMed] [Google Scholar]
- 74.Doherty M, Jenkins W, Richardson H, et al. Efficacy and cost-effectiveness of nurse-led care involving education and engagement of patients and a treat-to-target urate-lowering strategy versus usual care for gout: a randomised controlled trial. Lancet. 2018;392:1403–1412. doi: 10.1016/S0140-6736(18)32158-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.