

SUMMARY
The adhesion G-protein-coupled receptor GPR133 (ADGRD1) supports growth of the brain malignancy glioblastoma. How the extracellular interactome of GPR133 in glioblastoma modulates signaling remains unknown. Here, we use affinity proteomics to identify the transmembrane protein PTK7 as an extracellular binding partner of GPR133 in glioblastoma. PTK7 binds the autoproteolytically generated N-terminal fragment of GPR133 and its expression in trans increases GPR133 signaling. This effect requires the intramolecular cleavage of GPR133 and PTK7’s anchoring in the plasma membrane. PTK7’s allosteric action on GPR133 signaling is additive with but topographically distinct from orthosteric activation by soluble peptide mimicking the endogenous tethered Stachel agonist. GPR133 and PTK7 are expressed in adjacent cells in glioblastoma, where their knockdown phenocopies each other. We propose that this ligand-receptor interaction is relevant to the pathogenesis of glioblastoma and possibly other physiological processes in healthy tissues.
Graphical Abstract
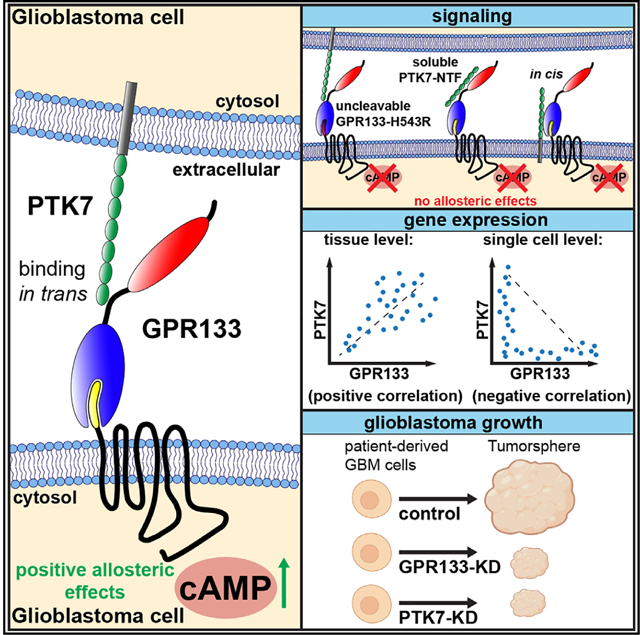
In brief
Frenster et al. identify PTK7 as a GPR133-binding protein in glioblastoma. Interaction of their extracellular domains in trans allosterically increases GPR133 signaling. This effect requires GPR133’s autoproteolytic cleavage and PTK7’s plasma membrane anchorage. PTK7 and GPR133 are expressed in adjacent cells in glioblastoma, where knockdown of either impairs tumor growth.
INTRODUCTION
Adhesion G-protein-coupled receptors (aGPCRs) are thought to serve dual functions as both transmembrane adhesion proteins and transducers of extracellular signals. This protein class is characterized by their long extracellular N termini, which contain receptor-specific adhesive domains that help define the extracellular interactome,1,2 as well as the phylogenetically conserved GPCR autoproteolysis-inducing (GAIN) domain that catalyzes intramolecular cleavage of the N terminus at the GPCR proteolysis site (GPS).3 In C-terminal proximity to the GPS lies an endogenous tethered agonist, the Stachel sequence, which is essential to receptor activation.4–8 A popular model for aGPCR activation invokes ligand binding as a means to convey mechanical forces driving the dissociation of the cleaved extracellular N-terminal fragment (NTF) from the remaining transmembrane-spanning C-terminal fragment (CTF), which contains the Stachel sequence.9,10 This dissociation, in turn, is thought to allow the Stachel sequence to insert itself into the orthosteric binding pocket formed by the transmembrane portion of the receptor to exert agonistic effects.5–8
In an alternative model inspired by the fact that cleavage-deficient mutant aGPCRs maintain signaling competency, ligand binding and mechanical forces allosterically induce conformational changes leading to receptor activation and signaling dependent on orthosteric Stachel agonism but without the requirement for NTF-CTF dissociation. Both these models gained support through the recent publication of cryo-electron microscopy structures of several aGPCRs in their activated state identifying critical residues mediating the interaction of the Stachel sequence with its orthosteric binding pocket within the transmembrane portion of the CTF.5–8
GPR133 (ADGRD1) is a member of the aGPCR class, whose signaling requires the intramolecular agonistic Stachel sequence and leads to Gas-mediated increases in cytosolic cyclic AMP (cAMP).4–8,11 Elucidating GPR133’s mechanisms of action and activation is particularly relevant in the context of glioblastoma (GBM), a primary brain malignancy, in which we found GPR133 to be expressed de novo relative to healthy brain and to be required for tumor growth.12,13 We recently demonstrated that GPR133 undergoes autoproteolytic cleavage at the GPS almost immediately after protein synthesis and that the NTF and CTF remain non-covalently bound in the secretory pathway until they reach the plasma membrane, where dissociation occurs.14 The cleavage and NTF-CTF dissociation of GPR133 correlate with increased receptor signaling, and cleavage is required for positive allosteric modulation of signaling induced by treatment with antibodies targeting the N terminus.14,15 These findings invite a model in which the extracellular interactome of GPR133 and its effects on NTF-CTF dissociation and receptor conformation are predicted to allosterically modulate receptor signaling. However, the identity of extracellular interactors of GPR133 in GBM, as well as whether mechanical forces generated by their binding are involved in receptor signaling, remain unknown. The only previously known GPR133 ligand, the transmembrane cell-surface protein Plexin Domain Containing 2 (PLXDC2), was recently shown to mediate an interaction with GPR133 critical to the female reproductive system in mice.16 However, the underlying mechanism of this interaction, including effects on NTF-CTF dissociation and mechanoactivation requirements, were not assessed.
In this study, we used affinity co-purification and mass spectrometry to discover a number of candidate extracellular binding partners of GPR133 in patient-derived GBM cells. Among them, the single transmembrane-span protein tyrosine kinase 7 (PTK7) binds and activates GPR133 signaling robustly when the receptor and the ligand are expressed in adjacent cells in trans. The allosteric activating effect of PTK7 on receptor signaling requires GPR133 cleavage and PTK7 anchoring to cell membranes or rigid extracellular substrates. This PTK7-GPR133 interaction produces additive effects on receptor activation when combined with soluble Stachel peptide treatment. We show that PTK7 and GPR133 are expressed in complementary patterns in GBM specimens as well as in several healthy human tissues, and that PTK7 knockdown phenocopies GPR133 knockdown in patient-derived GBM cells. These findings support a model in which PTK7 binding in trans allosterically increases GPR133 activation and signaling.
RESULTS
The extracellular interactome of GPR133
To identify the extracellular and plasma-membrane-bound interactome of GPR133 in GBM, we utilized an affinity co-purification/mass spectrometry approach in patient-derived GBM cell cultures (Figure 1A). First, we constructed a GPR133 bait consisting of the full-length receptor with an intracellular (C-terminal) TwinStrep affinity tag and the H543R mutation which prevents autoproteolytic cleavage.3,14,17–21 This mutation was added to prevent the dissociation of the NTF and thus allowed for the co-purification of extracellular interactors via the C-terminal affinity tag. We transduced three separate patient-derived GBM cultures with the lentiviral expression construct for this bait or a control empty vector, let them grow to confluence to increase cell-cell contacts, crosslinked the extracellular interactome with the linker 3,3’-dithiobis(sulfosuccinimidyl propionate) (DTSSP), and purified GPR133 and its interacting proteins using Strep-Tactin in three biological repeats for each condition (Figures 1B and S1A). Mass spectrometric analysis of the eluates detected 2,138 proteins, 569 of which were either extracellular or membrane bound (Figure 1C). Of these, 87 proteins with biologically relevant subcellular compartment annotation showed enrichment within the GPR133 bait condition in all three patient-derived cell cultures, and 23 of them reached statistical significance (p < 0.05) (Figures 1D, 1E, and S1B). We found that, among the significantly enriched (p < 0.05) protein pool prior to filtering for appropriate subcellular localization (n = 55), membrane proteins were over-represented when compared with the total protein pool, which instilled confidence in our approach (Figure S1C).
Figure 1. Co-purification/mass spectrometry-based ligand discovery in patient-derived GBM cells detects GPR133’s extracellular and membrane-bound interactome.
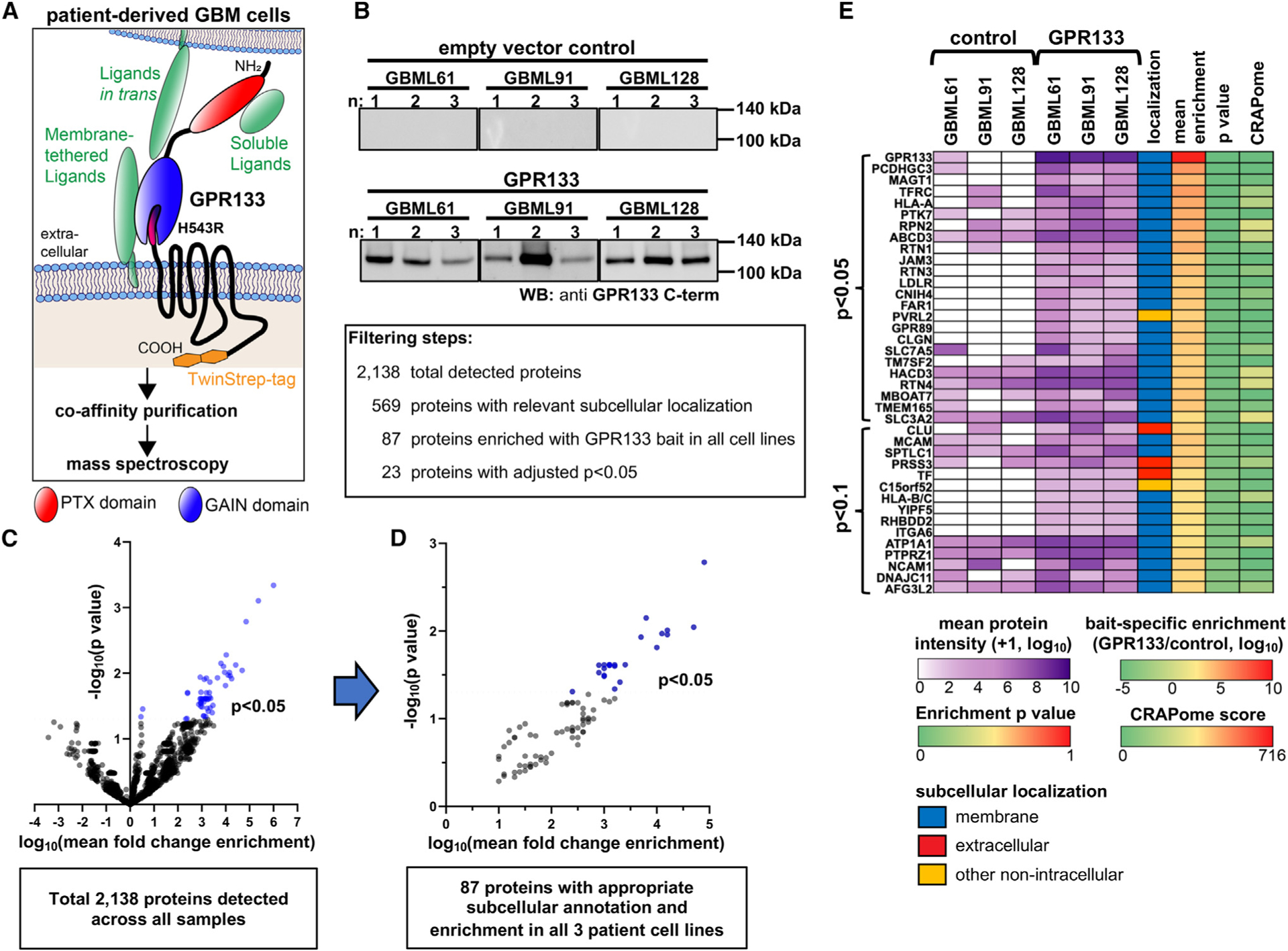
(A) Schematic overview of the affinity co-purification approach. Full-length cleavage-deficient GPR133-H543R with C-terminal intracellular TwinStrep tag was overexpressed in patient-derived GBM cultures. GPR133 and endogenous binding partners were crosslinked with DTSSP, followed by affinity enrichment and analysis by mass spectrometry. GAIN, G-protein-coupled receptor autoproteolysis-inducing domain; PTX, pentraxin-like domain.
(B) Eluates after Strep-Tactin purification from three different GBM cultures (with three independent biological repeats each) transduced with either TwinStrep-tagged uncleavable GPR133-H543R or an empty vector were analyzed with SDS-PAGE and western blot for GPR133 levels using an anti-GPR133 C-terminal antibody.
(C) Volcano plot of fold enrichment with the GPR133 bait and statistical significance of 2,138 total detected proteins. The 569 candidate proteins with subcellular annotation at the plasma membrane or in the extracellular space were filtered for enrichment with the GPR133 bait versus control vector samples across all cell lines, resulting in 87 consistently enriched proteins.
(D) Volcano plot depicting these 87 proteins, 23 of which were significantly enriched (p < 0.05; blue dots).
(E) Overview heatmap of the 38 most (p < 0.1) significantly enriched GPR133-binding protein candidates with subcellular localizations at the plasma membrane or extracellular space. The purple heatmap depicts detected protein intensities as a log10 mean of three biological replicates per culture per condition. The subcellular localization column was derived using Ensembl database annotations (detailed in STAR Methods). The mean enrichment column depicts the ratio of log10 mean intensities across all cell cultures and replicates from the GPR133 bait samples over the empty vector control samples. The p value column depicts the p value calculated from a mixed-effects model of enrichment with the GPR133 bait across all samples. The CRAPome column depicts the frequency of detection of proteins by mass spectrometry across 716 publicly available affinity purification datasets, and thereby identifies possibly non-specific interactors. See also Figure S1.
PTK7 binds GPR133
To validate binding between GPR133 and top interactors identified in our screen that had been previously implicated in GBM pathogenicity, we generated HEK293T cell lines stably overexpressing TwinStrep-tagged uncleavable GPR133-H543R or an empty vector (Figure S2Ai) and transfected them with C-terminally Myc- and 103His-tagged candidate proteins (PCDHGC3, PTK7, TFRC, NECTIN2/PVRL2, CADM4, and MCAM; Figure S2Aii) (Figure 2Ai). Among these candidates, PTK7 (protein tyrosine kinase 7 [inactive]), and to a lesser extent TFRC (transferrin receptor), demonstrated the most prominent binding to GPR133 in Strep-Tactin-mediated affinity co-purification assays (Figure 2Aii, n = 3 independent experiments). PTK7 is a type I single-pass transmembrane protein consisting of seven extracellular immunoglobulin (Ig)-like C2-type repeats and an intracellular inactive tyrosine kinase domain (Figures 2B and S2B), and has been previously implicated in the pathogenesis of GBM.22 PTK7 not only ranked among the top GPR133 bait-enriched proteins in our screen (Figure 2C) but was also highlighted by the reproducibility of its co-purified protein intensity in eight out of nine biological replicates when normalized to the GPR133 bait (Figures 2D and S2C).
Figure 2. Biochemical validation of candidate binding proteins detects PTK7 as the most robust GPR133 interactor.

(A) Validation affinity co-purification assay testing six of the top interactors identified in the screen with a TwinStrep-tagged GPR133-H543R bait in HEK293T cells. All candidate interactors were C-terminally tagged with the Myc epitope. (Ai) Western blots of input whole-cell lysates stained against Myc tag of ligand candidate proteins and anti-GPR133 C terminus. (Aii) Western blots of eluates after Strep-Tactin purification. Note that PTK7, and to a lesser extent TFRC, co-purify with GPR133. A representative blot of three biological repeats is depicted.
(B) Membrane topology of PTK7. Note that the juxtamembrane portion of PTK7 is cleaved by MT1-MMP (membrane type 1 matrix metalloproteinase) and ADAMs (a disintegrin and metalloproteinases).
(C) PTK7 is identified as one of the top interactors in the enrichment volcano plot as depicted in Figure 1D.
(D) Normalized intensities of peptides corresponding to GPR133 and PTK7 as detected in the mass spectrometry analysis in Figure 1E, detailing all biological replicates.
(E) Comparison of the GPR133-binding interaction of PTK7 vs. the previously reported ligand PLXDC2 using the Strep-Tactin purification paradigm. PTK7 and PLXDC2 (prey) were tagged with the Myc epitope for detection, while GPR133-H543R (bait) was tagged at the C terminus with TwinStrep tag for purification. Note that PTK7 co-purifies with GPR133, while PLXDC2 does not co-purify at detectable amounts under the same experimental conditions. See also Figure S2.
We then compared the PTK7-GPR133 interaction side by side with PLXDC2, the recently described GPR133 ligand expressed in the female reproductive system.16 However, we observed that PLXDC2 was neither detected in our initial screen in the context of patient-derived GBM cells nor did it show detectable co-purification with GPR133 in HEK293T cells in our hands (Figure 2E).
To further understand the nature of the interaction between GPR133 and PTK7, we conducted structure-function assays. First, we deleted the pentraxin (PTX) domain in the N terminus of GPR133 (deleted amino acids [aa] 40–277; Figures S2D and S2E) and found that it is not necessary for the interaction with PTK7 in co-purification experiments (Figures 3A and S2F; n = 2 independent experiments). Second, when we used the His-tagged PTK7 as the bait in co-purification assays using nickel-nitrilotriacetic acid (Ni-NTA) beads, we detected not only the binding of PTK7 to the full-length uncleavable GPR133-H543R but also to a secreted GPR133 NTF (containing aa 1–563) (Figure 3B). These findings suggest that the NTF of GPR133 mediates the interaction with PTK7 and that, within the NTF, the PTX domain is not necessary for PTK7 binding.
Figure 3. Biochemical characterization of the GPR133-PTK7 interaction.

(A) Co-affinity purification of GPR133 variants with co-expressed PTK7. We generated a TwinStrep-tagged GPR133-H543R deletion mutant lacking the PTX domain (GPR133-DPTX-H543R, aa 277–875, contains endogenous signal peptide) and compared its PTK7 binding with that of full-length GPR133-H543R using Strep-Tactin purification. The interaction was not altered by deletion of the PTX domain. Inclusion or omission of DTSSP, an extracellular crosslinker, did not influence the binding. Representative western blots are shown.
(B) Reverse co-purification experiment using PTK7 as bait. Both the full-length GPR133-H543R (aa 1–875) and a secreted TwinStrep-tagged GPR133 NTF (aa 1–563) lacking transmembrane domains co-purified with PTK7 using Ni-NTA beads. Pink, yellow, and red arrowheads denote the monomeric full-length GPR133-H543R, multimers of the full-length GPR133-H543R, or the secreted GPR133 NTF lacking transmembrane domains, respectively. Purple arrowheads denote non-specific antibody off-targets. Multimerization of GPR133 due to its transmembrane domains is reduced but not entirely prevented by the use of 1% ndodecyl b-D-maltoside. GPR133 multimerization is discussed in detail in Frenster et al.14
(C) Affinity co-purification of soluble secreted GPR133 NTF and PTK7 NTF. GPR133 NTF with N-terminal TwinStrep tag and secreted Myc-tagged PTK7 with no transmembrane domains were expressed alone, expressed together in adjacent cells, or co-expressed in the same cells. Secreted PTK7 co-purified with GPR133 in both mixed-culture and co-expression conditions (green arrows). Representative blots are depicted. See also Figure S2.
To test directly whether the extracellular NTFs of GPR133 (aa 1–563) and PTK7 (aa 1–703) bind each other independently of their transmembrane domains, we expressed secreted extracellular fragments of both proteins in either co-expression or mixed-culture systems (Figure 3C). In co-purification assays these soluble NTFs co-eluted reproducibly in both the co-expression and mixed-culture configurations. The co-purification was stronger when these extracellular fragments were expressed in the same cells, likely due to the increased relative abundance and probability of interaction when the two proteins are co-expressed. Nonetheless, the mixed-culture aspect of this assay demonstrated that the extracellular domains of GPR133 and PTK7 can bind each other even when expressed on neighboring cells.
Taken together, our biochemical assays demonstrate that PTK7 and GPR133 robustly bind each other and co-purify independently of the purification method and the choice of bait. The soluble extracellular NTFs of both proteins are sufficient for binding, even when expressed from different cells in a mixed-culture system, but the interaction does not require the N-terminal PTX domain of GPR133.
PTK7 expression in trans activates GPR133 signaling
GPR133 signals through Gas-mediated intracellular cAMP increases.11 To test whether PTK7 influences GPR133 signaling we used HEK293T cells, which provide a good experimental system due to the absence of endogenous GPR133 expression. When co-expressing GPR133 and PTK7 in the same cells (in cis), we observed no increase in intracellular cAMP levels as measured by homogeneous time-resolved fluorescence (HTRF) assays normalized to GPR133 cell-surface expression as measured with ELISAs (Figures S3Ai–Aiii). However, when expressing GPR133 and PTK7 in two separate but co-cultured cell populations, we observed increases in cAMP levels (Figures S3Bi–Biii). To maximize the interaction surface of these co-cultured cells, we designed a three-layer sandwich system in which GPR133-expressing cells were seeded between two layers of PTK7-expressing cells (Figures 4A, S3C, and S3D). We used empty vector controls in both cell populations. In this system, expression of PTK7 in the outer layers increased cAMP levels produced by GPR133 in the middle layer, but not in vector control cell sandwich cultures (Figures 4B and S3Ci–iii). Importantly, this action of PTK7 depended on its anchoring in the membrane of cells in the outer layers because expression of a secreted PTK7 NTF without its transmembrane domain (containing aa 1–703; Figure S2B) did not elicit an effect on GPR133 signaling (Figure 4B). Furthermore, signaling by the cleavage-deficient GPR133-H543R was not influenced by expression of PTK7 in neighboring cells (Figure 4B). Taken together, these data suggest that GPR133 signaling is increased by PTK7 binding in trans and that both GPR133 cleavage and PTK7 membrane anchoring are required for this activation.
Figure 4. PTK7 increases GPR133 signaling through a trans interaction.

(A) Confocal microscopic image from “sandwich cultures” shows layered cells expressing either GPR133 (green) or PTK7 (red). Top view from a central confocal slice (center panel: x and y dimension) and orthogonal views (upper panel: z and x; right panel: z and y) depicted on the same scale. Scale bars, 10 μM.
(B) HTRF assays in “sandwich cultures” demonstrate that PTK7 in neighboring cells increased cAMP levels in cells expressing wild-type (WT) cleaved GPR133, but not the uncleavable H543R mutant. A secreted PTK7 with no transmembrane domain (no TM, aa 1–703) did not influence GPR133 signaling. Two-way ANOVA: middle layer F2, 96 = 4.62, not significant; outer layer F2, 96 = 3.93, not significant; interaction of inner and outer layer F4, 96 = 7.29, p < 0.0001. Tukey’s multiple comparisons: GPR133-WT-expressing cells with empty vector vs. PTK7 co-culture, p < 0.0001; GPR133-WT-expressing cells with full-length PTK7 vs. secreted PTK7 (no TM) co-culture, p < 0.0001. All other comparisons not significant; n = 4–22 independent experiments.
(C) Pre-coating wells with PTK7 NTF (aa 1–703) significantly increased cAMP levels in WT GPR133-expressing, but not H543R uncleavable mutant-expressing cells. COL6 (native purified human collagen VI) had no effect on signaling. Two-way ANOVA: GPR133 variant expressed F2, 80 = 11.65, p < 0.0001; protein coating F2, 80 = 12.90, p < 0.0001; interaction of factors F4, 80 = 9.24, p < 0.0001. Tukey’s multiple comparisons: GPR133-WT-expressing cells on PTK7 NTF-coated vs. uncoated dishes, p < 0.0001; GPR133-WT-expressing cells on PTK7 NTF-coated vs. COL6-coated dishes, p < 0.0001). All other comparisons not significant; n = 5–15 independent experiments.
(D) Combinatorial effect of PTK7 binding and p13 Stachel peptide treatment on GPR133 signaling. HEK293T cells expressing either WT GPR133 or empty vector control were seeded onto PTK7 NTF-coated (1.17 μg/cm2) or control-coated wells. Cells were then treated with synthetic p13 Stachel peptide (500 μM), inactive control peptide (500 μM), or solvent controls, and cAMP levels were measured by HTRF. The combination of PTK7 NTF binding and Stachel peptide elicited an additive response in GPR133 signaling compared with the individual treatments. Two-way ANOVA, GPR133 expression effect F1, 48 = 365.1, p < 0.0001; treatment effect F7, 48 = 13.2, p < 0.0001; interaction of GPR133 expression and treatment F7, 48 = 13.2, p < 0.0001. Tukey’s multiple comparisons: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n = 4 independent experiments.
Data are depicted as mean ± SEM. See also Figures S3 and S4.
To further support the notion that the extracellular N terminus of PTK7 needs to be tethered to the plasma membrane or another rigid substrate for this activation mechanism to occur, we used purified human extracellular domain (NTF, aa 1–704) of PTK7 to pre-coat cell-culture dishes. Indeed, we observed that GPR133-expressing cells seeded on PTK7-coated dishes exhibited significantly higher cAMP levels when compared with cells cultured on untreated dishes using either HTRF assays or a cAMP response element (CRE)-luciferase reporter (Figures 4C, S3E, and S4A). This effect could not be replicated by coating dishes with other proteins, such as collagen VI (COL6, which was detected in a previous unpublished screen for GPR133 interactors by our group, not included in this publication) (Figures 4C and S3E), suggesting that the GPR133-PTK7 interaction is specific rather than due to promiscuous adhesion. Again, cleavage-deficient H543R mutant GPR133 signaling remained unaffected by PTK7 coating, suggesting that the GPR133-PTK7 interaction requires GPR133 cleavage to promote signaling. It is noteworthy that the PTK7 NTF (aa 1–704) does not increase GPR133 signaling when secreted in a soluble form despite binding GPR133, but does activate GPR133 signaling when rigidly immobilized on cell-culture dishes (Figures 4B and 4C).
A recent structural study demonstrated the binding pocket for the tethered endogenous Stachel agonist of GPR133 to be within the 7-transmembrane domain.7 The observation that soluble GPR133 NTF and PTK7 NTF bind each other directly in the absence of transmembrane domains (Figure 3D) suggests that PTK7’s action on GPR133 signaling might be an allosteric effect rather than an interaction with the orthosteric Stachel-binding site. To test directly whether the PTK7 binding allosterically influences GPR133’s response to orthosteric agonism by synthetic p13 peptide mimicking the endogenous Stachel agonist,4 we performed cAMP HTRF assays in GPR133-expressing HEK293T cells cultured on PTK7-coated wells and treated with p13 Stachel peptide (Figure 4D). While both PTK7 binding and administration of Stachel peptide increased GPR133 signaling individually, the combination of both resulted in an additive effect (Figure 4D). To investigate this phenomenon in more detail, we conducted dose-response signaling assays. First, GPR133 signaling was elevated by PTK7 pre-coating in a dose-dependent manner with a maximal response at 586 ng/cm2 of PTK7-NTF coating (Figure S4B). Second, maximal-response PTK7 pre-coating (586 ng/cm2) increased GPR133 signaling comparably in all tested Stachel peptide concentrations without changing the peptide’s dose-response profile (Figure S4C). In contrast, submaximal concentrations of PTK coating (293 ng/cm2) did not manifest this additive effect with Stachel peptide (Figure S4C). These observations support the notion that PTK7 acts allosterically on GPR133 signaling and that this effect is additive with and topographically independent of Stachel orthosteric agonism.
Since PTK7 is a known interactor of the Wnt signaling pathway and has been shown to bind Wnt5a directly, we tested whether Wnt5a administration would have an influence on the interaction between PTK7 and GPR133 (Figure S4D). While we observed a trend indicating that higher levels of Wnt5a (50 ng/mL) might interfere with PTK7’s positive effect on GPR133 signaling, this trend was not statistically significant.
Taken together, our signaling assays point toward a mechanism in which binding of PTK7 to the extracellular domain of GPR133 in trans allosterically increases receptor signaling. This effect requires the autoproteolytic cleavage of GPR133 into an NTF and CTF and the tethering of PTK7’s extracellular domain to the plasma membrane or a rigid support, and is additive with but independent from p13 Stachel peptide orthosteric agonism. A possible mechanism for the positive allosteric effect of PTK7 binding in trans on GPR133 signaling may involve dissociation of the NTF from the CTF of the cleaved receptor.
PTK7 and GPR133 have complementary expression patterns in glioblastoma and healthy human tissues
To interrogate whether these PTK7-GPR133 interactions could occur in human tissues, we first investigated the expression patterns of PTK7 and GPR133 within human GBM biospecimens. Epifluorescent and confocal microscopic analysis of surgically resected GBM biospecimens (n = 5) revealed that both proteins are detectable throughout tumors (Figures 5A, 5B, and S5A), confirming previous reports on PTK722 and GPR13313 being expressed in GBM. We also confirmed that both PTK7 and GPR133 mRNA transcripts are found in GBM using single-cell RNA-sequencing (RNA-seq) data, contrary to HEK293 cells where only PTK7 is found in small amounts (Figures S5B and S5C). In the surgical biospecimens we observed spatial gradients in the expression of both proteins, frequently suggestive of a complementary expression pattern, and confirmed in higher-magnification confocal z stacks that there are indeed adjacent cells predominantly expressing either PTK7 or GPR133 (Figure 5C). We previously demonstrated that GPR133 expression is upregulated in GBM in areas of low oxygen tension through the action of HIF1a.12 However, PTK7 mRNA levels do not seem to be influenced by oxygen levels in our GBM cultures as assessed by qRT-PCR (Figure S5D).
Figure 5. GPR133 and PTK7 expression profiles in GBM and healthy human tissues.
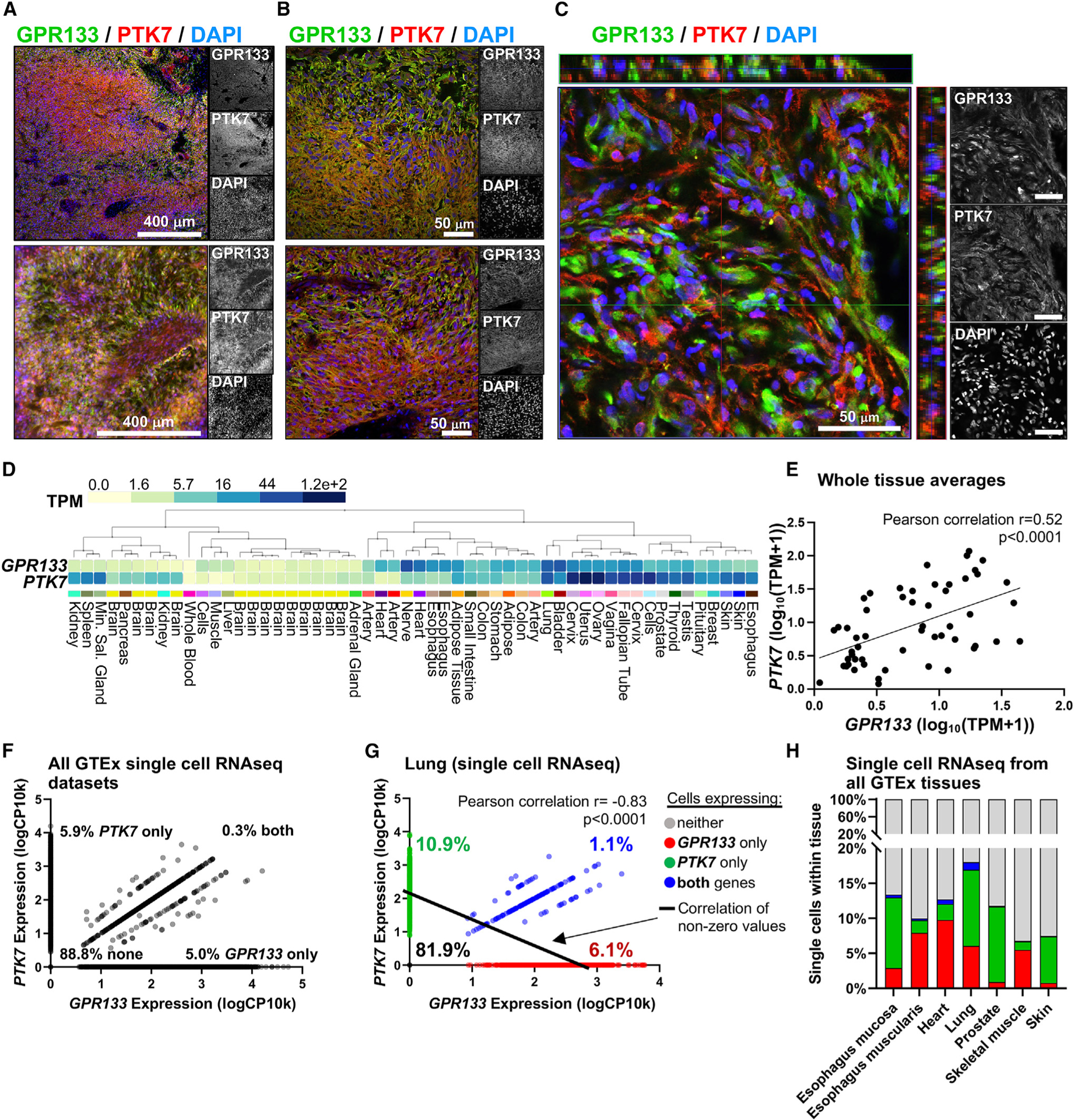
(A) Epifluorescence microscopic images from two GBM surgical specimens show expression of both GPR133 (green) and PTK7 (red) within tumors.
(B) Higher-magnification epifluorescence images demonstrate a complementary expression pattern in GBM specimens.
(C) Confocal microscopic image with orthogonal views of a z stack indicates that cells tend to either predominantly express GPR133 or PTK7.
(D) GTEx portal mRNA data from human tissues shows overlap in PTK7 and GPR133 expression in several tissues.
(E) On a whole-tissue level, the expression of PTK7 mRNA showed a significant positive correlation with GPR133 mRNA (Pearson correlation, p < 0.0001). Each dot represents the whole-tissue average gene expression from the human GTEx tissues listed in (D).
(F) Single-cell gene expression analysis of the seven available GTEx single-cell datasets—esophagus mucosa, esophagus muscularis, heart, lung, prostate, skeletal muscle, and skin—filtered for cells with RNA integrity number (RIN) >8 demonstrates that GPR133 only (5%) and PTK7 only (5.9%) expressing cells are more common than cells co-expressing both genes (0.3%) (130,840 cells included, single-cell expression normalized as log10 of copy number per 10,000 reads).
(G) Within the lung, which exhibits high expression of GPR133 and PTK7 on a whole-tissue level, individual cells preferentially express only GPR133 (6.1%) or only PTK7 (10.9%) rather than co-expressing both genes (1.1%), resulting in a negative correlation (Pearson correlation r = 0.83, p < 0.0001, n = 2,597 cells; cells expressing neither gene were excluded from correlation analysis to avoid zero inflation).
(H) All seven available single-cell datasets from the GTEx portal reflect the same negative correlation between GPR133 and PTK7 expression in human tissues observed in (G), with GPR133 only (red) and PTK7 only (green) expressing cells being more common than cells expressing both genes (blue). Detailed single-cell analyses of these tissues are depicted in Figure S7.
Scale bars in (A) and (B) represent 50 μM or 400 μM as annotated; scale bars in (C) represent 50 μM. See also Figures S5–S7.
Since the increase in GPR133 signaling only occurred when cells were exposed to PTK7 on adjacent cells in trans, we investigated the GPR133 and PTK7 protein expression at the single-cell level by flow cytometry in two independent patient-derived GBM cultures using antibodies targeting their extracellular NTFs (Figure S5E). While we observed that in these cultures only a subset of cells expresses GPR133 or PTK7, the percentage of cells in which we detected only GPR133 or only PTK7 was larger than the percentage of cells co-expressing both proteins in cis. The notion of individual cells predominantly expressing either GPR133 or PTK7, rather than co-expressing both, was confirmed when analyzing publicly available single-cell RNA-seq data from GBM tissues (Figure S5F). Taken together, these observations are aligned with our model that it is the in trans interaction of GPR133 and PTK7 on adjacent cells that allosterically increases GPR133 signaling.
PTK7 contains multiple extracellular metalloprotease recognition sites (MT1-MP/MP14, ADAM12, and ADAM17) as well as an intracellular g-secretase (APH1A, APH1B, NCSTN, PSEN1, and PSENEN) recognition motif (Figures 2B and S2B). We therefore interrogated the single-cell RNA-seq data from GBM tissue for the expression of these proteases. We found that, among the PTK7-expressing and GPR133-expressing cells, a subset express these proteases at detectable levels (Figures S5G and S5H). Given our observation that only membrane-bound PTK7 positively stimulates GPR133 signaling, pro-tease-mediated cleavage of PTK7 might therefore represent a possible level of regulation of this interaction.
To gain insight into whether the GPR133-PTK7 interaction may be relevant to physiological processes, we interrogated the GTEx expression database for GPR133 and PTK7 mRNA levels across healthy human tissues. Indeed, we found co-expression of the two transcripts in several tissues (Figures 5D and S6A). This co-expression was demonstrated by a positive correlation between GPR133 and PTK7 transcript levels across human tissues (Pearson correlation r = 0.52, p < 0.0001) (Figure 5E). The specificity of this correlative expression pattern is further supported by the fact that, when subjecting mRNA levels encoding all significantly enriched GPR133-binding proteins of this study (n = 23) to hierarchical clustering by their similarity of expression across all tissues, PTK7 and GPR133 are paired closest to each other (Figure S6B), with the exception of GPR89. To determine the cell-type-specific expression of PTK7 and GPR133 transcripts within these tissues, we interrogated single-cell RNA-seq data from the GTEx portal. This analysis indicated that despite the positive correlation at the tissue level, individual cells within tissues tend to predominantly express either GPR133 or PTK7 more commonly than co-express both transcripts (Figures 5F–5H). This negative correlation of GPR133 and PTK7 expression at the single-cell level became even more apparent when looking at single cells within individual tissues (Figure S7), especially in the lung, which showed high expression for both genes at the tissue level (Figure 5G; Pearson correlation r = 0.83, p < 0.0001). This often complementary, inversely correlated expression of the two transcripts in different cell types within the same tissue stands in support of our findings that GPR133 signaling is activated by PTK7 only when they are expressed in trans.
PTK7 knockdown in patient-derived GBM cultures phenocopies GPR133 knockdown
PTK7 has been implicated in the pathogenesis of GBM,22 and we have previously demonstrated that GPR133 knockdown prevents growth of GBM cells.12 We theorized that, if PTK7 and GPR133 interact in GBM, knockdown of either protein should produce similar phenotypes. To test this prediction, we used patient-derived GBM cells and lentiviral short hairpin RNA (shRNA) against PTK7 and GPR133. Our previously validated shRNA against GPR13312 and four different shRNA constructs against PTK7 were used to instill confidence in our findings. Western blot analysis confirmed a reduction in endogenous PTK7 protein levels following knockdown with all tested shRNAs (Figures 6Ai and Aii). Tumorsphere formation assays were then used to assess the effect of PTK7 knockdown on clonogenic growth potential. Indeed, sphere formation was impaired with multiple knockdown constructs targeting PTK7, similar to the effects of GPR133 knockdown (Figures 6Bi and Bii; tested in two independent patient-derived GBM cell cultures) and in accordance with our previous observations.12 The fact that knockdown of PTK7 phenocopies the effects of GPR133 knockdown is consistent with our hypothesis that the two proteins interact in GBM.
Figure 6. Knockdown of PTK7 phenocopies that of GPR133 in patient-derived GBM cells.
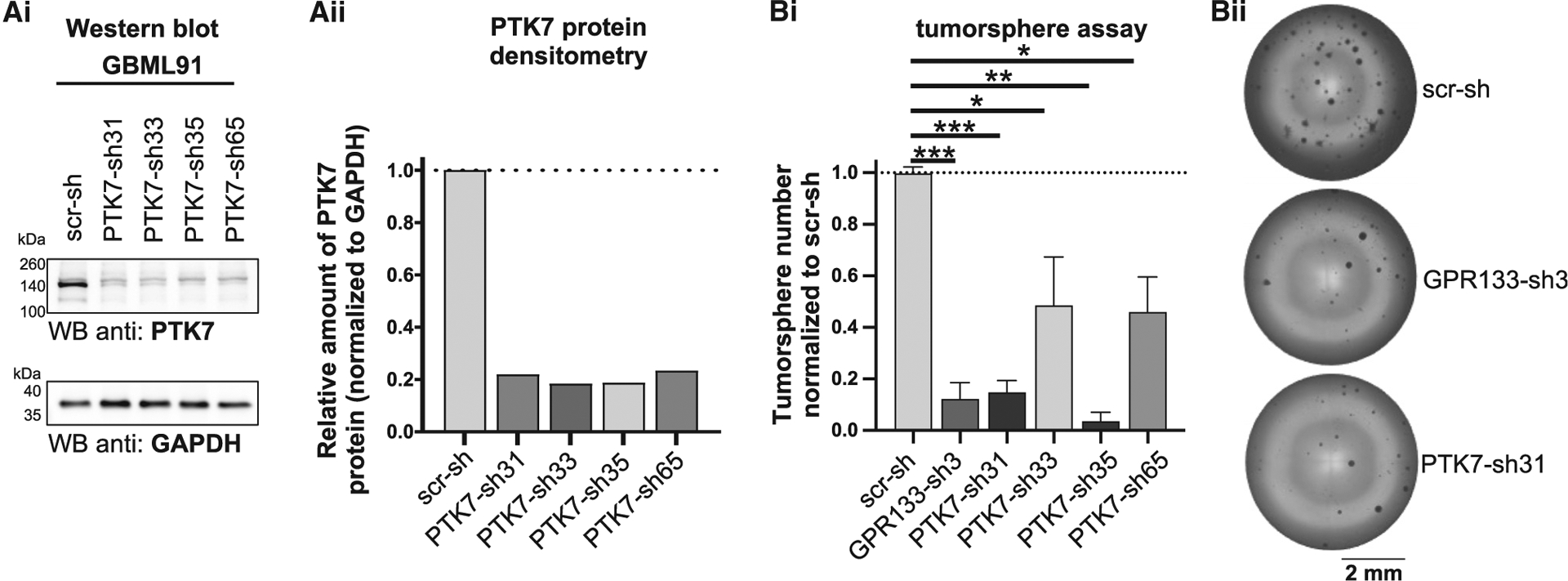
(A) We tested four different shRNAs targeting endogenous PTK7 in patient-derived GBM cells and found significant knockdown for all by western blots (Ai) and densitometric analysis (Aii). A non-targeting scrambled shRNA (scr-sh) was used as control. (Bi) Similar to the effect of our previously described shRNA targeting GPR133 (Bayin et al.12), shRNAs targeting PTK7 reduced tumorsphere formation in vitro (Tukey’s multiple comparisons: *p < 0.05, **p < 0.01, ***p < 0.001; n = 4 independent experiments with six technical replicates per condition each). (Bii) Representative example images of tumorsphere formation assay. Data are depicted as mean ± SEM.
DISCUSSION
The long N termini of adhesion GPCRs are theorized to serve dual roles: first, they adhere to transmembrane proteins or components of the extracellular matrix; and second, they modulate receptor signaling. Here, we demonstrate that PTK7, a type I single-pass transmembrane protein, binds the N terminus of the aGPCR GPR133 to allosterically increase receptor signaling. The effect is observed only when PTK7 and GPR133 interact in trans, i.e., on neighboring cells. In addition, the allosteric activating influence of PTK7 on GPR133 signaling requires the membrane insertion of the former and the autoproteolytic cleavage of the latter, and is additive with soluble Stachel peptide orthosteric agonism.
Both PTK7 and GPR133 are expressed in GBM, a brain malignancy that depends on GPR133 for growth in vitro and in vivo.12,13 The fact that knockdown of PTK7 phenocopies that of GPR133 in patient-derived GBM cells is consistent with our hypothesis that the PTK7-GPR133 interaction is functionally relevant to GBM pathogenesis. Within GBM, flow cytometry and single-cell RNA-seq data indicate that tumor cells tend to not co-express the two proteins (Figure S5). In non-pathological human tissues, PTK7 and GPR133 mRNA expression levels positively correlate across tissue types by bulk RNA-seq analysis but inversely correlate at the single-cell level within these tissues (Figures 5D–5H, S6, and S7). This further supports the notion that the in trans PTK7-GPR133 interaction across cells is the physiologically relevant mode of interaction between these two proteins, and that this interaction may be functionally relevant in multiple tissues.
Our biochemical analyses indicate that the NTF (aa 1–563) of GPR133 is sufficient for the interaction with PTK7 and that its portion extending from the N terminus to the end of the Pentraxin (PTX) domain (aa 31–276) is not required for PTK7 binding. These observations suggest that the interaction depends on sequences distal to the PTX domain within the N terminus of GPR133, possibly including the GAIN domain. Of course, these findings do not exclude the possibility that in vivo the interaction may additionally rely on sequences within the extracellular portion of the CTF of GPR133. The in trans allosteric effect of PTK7 on GPR133 signaling is abolished when GPR133 is rendered uncleavable by the H543R mutation or when PTK7 is not inserted in the plasma membrane but instead secreted as a truncated protein lacking its transmembrane and intracellular portions. Conversely, the positive allosteric effect of this truncated PTK7 is restored when it is anchored on a rigid surface. Collectively, these findings provide insights into mechanistic aspects of GPR133 activation by PTK7 (Figure 7). We propose a model in which the membrane-tethered PTK7 binds GPR133’s NTF in trans and either allosterically induces conformational changes in GPR133, which require the cleaved GAIN domain, or alternatively that the membrane-tethered PTK7 can transduce mechanical forces onto GPR133, facilitating the dissociation of its NTF from the CTF. This new conformation or the absence of its NTF are predicted to allosterically increase GPR133 signaling by facilitating the agonistic interaction of the endogenous Stachel sequence with its orthosteric binding pocket within the transmembrane portion of the receptor, as previously proposed.6,7,14 However, while this model of “mechanical pull and NTF-CTF dissociation” fits all data, we do not have direct evidence of mechanical force transduction. Inherent assumptions in this model are the intramolecular cleavage of GPR133 that allows for a non-rigid receptor conformation amenable to allosteric modulation, including NTF-CTF dissociation, as well as the membrane insertion of PTK7, which ensures that a mechanical force helps induce the aforementioned conformational changes (Figure 7).
Figure 7. Graphical summary of proposed PTK7-GPR133 interaction model.

(A) The extracellular portion of PTK7 binds the GPR133 NTF in trans. This interaction does not depend on either protein’s transmembrane domain nor on GPR133’s pentraxin domain.
(B) The binding may cause conformational changes and/or mechanical dissociation of the GPR133 NTF to increase receptor signaling activity. This interaction is allosteric and does not compete with orthosteric Stachel peptide agonism (in dashed parentheses).
(C) Additional mechanical shear stress may facilitate NTF-CTF dissociation, causing the irreversible activation of GPR133 signaling.
The interaction between PTK7 and GPR133 will require extensive functional characterization in the future. Our data from GBM indicate not only the presence of both proteins in tumors but also a complementary expression pattern, where cells tend to predominantly, but not exclusively, express one or the other. This profile is consistent with our finding that PTK7 activates GPR133 signaling only when expressed in trans, i.e., in adjacent cells, but also raises other important questions. First, our HEK293T data indicate that co-expression of the two proteins in the same cell has no effect on GPR133 signaling. In GBM, tumor cells tend to have a predilection for predominantly expressing one or the other protein, but a subpopulation of cells co-expresses both proteins together. A logical future question will be how the relative weights of cis and trans interactions influence GPR133 signaling. Second, the juxtamembrane extracellular region of PTK7 is subject to proteolysis by metalloproteases, with such cleavages predicted to untether the N terminus of PTK7 from the membrane. Our experiments have indicated that eliminating the membrane insertion of PTK7 abolishes effects on GPR133 signaling, suggesting that in vivo proteolytic cleavage of PTK7 by metalloproteases may also prevent any agonistic effects on GPR133 signaling. Given that metalloproteases are abundantly expressed in GBM, juxtamembrane cleavage of PTK7 represents a potentially crucial regulatory mechanism of the PTK7-GPR133 interaction. Third, we have so far analyzed the agonistic influence of the PTK7-GPR133 interaction on GPR133 signaling, but its effects on PTK7 function are unknown. PTK7 is a known component of Wnt signaling complexes,23–25 and the Wnt system, in turn, is an important tumorigenic mechanism in GBM.26 Our signaling data suggested a trend of Wnt5a administration interfering with PTK7’s positive allosteric effect on GPR133 signaling, which could suggest that Wnt5a and GPR133 are competing for PTK7 binding. However, this observation did not reach statistical significance and will need to be investigated further in the future. While our present study has interrogated the effects of this interaction from a GPR133-centric view, we intend to dissect additional effects of this interaction on PTK7 function and Wnt signaling in the future.
It is becoming clear that several adhesion GPCRs exhibit promiscuity in interactions through their N termini. As an example, CD97 (ADGRE5) is known to bind several ligands: CD55, CD90, chondroitin sulfate, and integrin.27–31 Similarly, GPR133 was recently shown to interact with PLXDC2, a plexin-domain-containing protein, in the female reproductive system. Here, we demonstrate that GPR133 binding to PTK7 in the context of GBM increases receptor signaling. This suggests that the interactome of the N terminus of GPR133 is context dependent and that additional ligands may exist in other tissues where GPR133 is expressed. In our ligand discovery assay, we identified several GPR133 interactors containing Ig domains, such as PTK7, JAM3, NECTIN2, MCAM, NCAM1, and CADM4, although only PTK7 among them showed reproducible binding and modulation of signaling. However, our screen indicated additional interactors lacking Ig domains, for example, TFRC. Detailed structure-function studies and functional assays will be needed to elucidate the biochemical basis and phenotypic consequences of such interactions.
In summary, we describe a novel interaction of GPR133 with PTK7, which results in positive allosteric modulation of GPR133 signaling when the two proteins are expressed on adjacent cells. This effect on receptor signaling depends on both the intramolecular cleavage of the N terminus of GPR133 at the GPS and the membrane insertion of PTK7. Potential mechanisms include PTK7-mediated mechanical forces that allosterically generate conformational changes in GPR133, with or without NTF dissociation, that lead to increased Gas signaling. Additional work will be required to dissect the significance of this interaction in GBM pathogenesis, as well as in physiological processes in tissues where both GPR133 and PTK7 are expressed.
Limitations of the study
Our affinity proteomics screen identified PTK7 as a GPR133-binding protein and a positive allosteric modulator of GPR133 signaling. However, we did not test every significantly enriched protein during our validation screen and may have missed other true interactors. Furthermore, the binding validation assays were conducted with intracellular C-terminal affinity tags, which could potentially interfere with GPR133’s interaction with the candidate proteins and thereby result in false-negative results. Thus, while we validated PTK7 as a GPR133 ligand, we cannot exclude the possibility of false-negative results when other candidates were tested. By the same token, our observed absence of binding between GPR133 and PLXDC2 is not meant to disprove or discourage any follow-up studies toward their interaction in the female reproductive system.
Our data demonstrate that both GPR133 cleavage and PTK7 anchoring to cell membranes in trans or to rigid substrates are required for increased GPR133 signaling. Secreted soluble NTF of PTK7 is insufficient to increase GPR133 signaling. These observations, together with our previous work on receptor activation following GPR133 NTF dissociation, suggests a model in which PTK7 binding in trans facilitates the removal of the GPR133 NTF. However, while we propose this model, we do not have direct evidence of any force transduction between PTK7 and GPR133.
Our signaling assays in HEK293T cells demonstrate an increase in GPR133 signaling when adjacent cells overexpress PTK7. However, HEK293T cells express low baseline levels of PTK7 even without overexpression (Figure S5C). This raises the possibility that our results underestimate the positive allosteric effect of PTK7 on GPR133 signaling, since our assays compare overexpression of PTK7 with baseline expression levels rather than to absence of PTK7.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dimitris Placantonakis (dimitris.placantonakis@nyulangone.org).
Materials availability
Expression plasmids generated in this study (detailed in the key resource table) will be available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER/CAT# |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GPR133-CTF | Sigma-Aldrich | HPA042395; RRID:AB_10796906 |
| Mouse monoclonal anti-GPR133-NTF | Not commercially available (Bayin et al.12; Frenster et al.13) | Clone “8E3E8” |
| Goat polyclonal anti-GAPDH | Thermo Fisher | PA1-9046; RRID:AB_1074703 |
| Mouse monoclonal anti-Myc-tag (9B11) | Cell Signaling | 2276S; RRID:AB_331783 |
| Rabbit polyclonal anti-PTK7 (CTF) | Proteintech Group Inc. | 17799-1-AP; RRID:AB_2878442 |
| Rabbit polyclonal anti-PTK7 (NTF) | Thermo Fisher | PA5-82070; RRID:AB_2789231 |
| Mouse monoclonal anti-StrepTag (StrepMAB-Classic-HRP conj.) | IBA | 2-1509-001 |
| Secondary Donkey anti-Mouse Alexa Fluor Plus 488 | Thermo Fisher | A32766; RRID:AB_2762823 |
| Secondary Donkey anti-Rabbit Alexa Fluor Plus 555 | Thermo Fisher | A32794; RRID:AB_2762834 |
| Secondary Donkey anti-Goat Alexa Fluor 647 | Thermo Fisher | A21447; RRID:AB_141844 |
| Secondary Donkey anti-Mouse Alexa Fluor 647 | Thermo Fisher | A31571; RRID:AB_162542 |
| Secondary Chicken anti-Mouse HRP-conjugated | Thermo Fisher | A15975; RRID:AB_2534649 |
| Secondary Chicken anti-Rabbit HRP-conjugated | Thermo Fisher | A15987; RRID:AB_2534661 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human PTK7/CCK4 Fc chimera protein | R&D | 9799-TK-050 |
| Human Collagen Type VI | Rockland | 009-001-108 |
| Recombinant human/mouse Wnt-5a protein | R&D | 645-WN-010/CF |
| 3-Isobutyl-1-methylxanthine (IBMX) | Sigma-Aldrich | I7018–100MG |
| DDM (n-dodecyl b-D-maltoside) (10%) | Thermo Scientific | BN2005 |
| Neurobasal Medium | Gibco | 21103049 |
| EGF | R&D | 236-EG-01M |
| bFGF | R&D | 233-FB-01M |
| GlutaMAX Supplement | Gibco | 35050061 |
| MEM Non-Essential Amino Acids | Gibco | 11140050 |
| N2-Supplement | Gibco | 17-502-049 |
| B27-Supplement | Gibco | 12587010 |
| Poly-L-Ornithine solution | Sigma-Aldrich | P4957–50mL |
| Lipofectamine 2000 | Invitrogen | 11668–019 |
| Accutase | Innovative Cell Technologies | AT104 |
| Gravity flow Strep-Tactin Sepharose column | IBA | 2-1202-001 |
| 10x Buffer E; Strep-Tactin Elution Buffer with Desthiobiotin | IBA | 2-1000-025 |
| 10x Buffer R; Strep-Tactin Regeneration Buffer with HABA | IBA | 2-1002-100 |
| 10x Buffer W; Strep-Tactin Wash Buffer | IBA | 2-1003-100 |
| MagStrep “type3” XT magnetic beads | IBA | 2-4090-002 |
| 10x Buffer BXT | IBA | 2-1042-025 |
| BioLock Biotin blocking solution | IBA | 2-0205-250 |
| Desthiobiotin | IBA | 2-1000-001 |
| Critical commercial assays | ||
| cAMP Gs Dynamic kit (HTRF) | CisBio | 62AM4PEC |
| ELISA: TMB Stabilized Chromogen | Thermo Fisher | SB02 |
| ELISA: Stop Solution | Thermo Fisher | SS04 |
| Cells-to-CT kit | Thermo Fisher | AM1729 |
| HPRT TaqMan probes | Thermo Fisher | Hs02800695_m1 |
| GPR133 TaqMan probes | Thermo Fisher | Hs00914797_m1 |
| PTK7 TaqMan probes | Thermo Fisher | Hs00897151_m1 |
| Deposited data | ||
| Mass spectrometry raw data from affinity purifications | This paper | MassIVE database: MSV000090520 |
| Experimental models: Cell lines | ||
| Lenti-X 293T cell Line | Takara | 632180 |
| Patient-derived GBM cultures | Derived in our lab, not commercially available | GBML61/91/109/128 |
| Recombinant DNA | ||
| pLVX-EF1α-mCherry-N1 Vector | Takara | 631986 |
| pLVX_EF1α-mCherry_PGK-GPR133-WT | Not commercially available (Frenster et al.14) | N/A |
| pLVX_EF1α-mCherry_PGK-GPR133-H543R | Not commercially available (Frenster et al.14) | N/A |
| pLVX_EF1α-mCherry_PGK-GPR133-H543R-C-terminal_TwinStrep | Not commercially available (Frenster et al.14) | N/A |
| pLVX_EF1α-mCherry_PGK-GPR133-DPTX-H543R-C-terminal_TwinStrep | Not commercially available (Frenster et al.14) | N/A |
| pLVX_EF1α-mCherry_PGK-GPR133(NTF)-H543R-Stachel-TwinStrep (secreted, noTM) | This paper | N/A |
| pLVX_EF1α-PTK7-Myc-10xHis-t2A-mCherry | This paper | N/A |
| pLVX_EF1α-PTK7-NTF-Myc-10xHis-t2A-mCherry (secreted, no TM) | This paper | N/A |
| pLVX_EF1α-PCDHG3-Myc-10xHis-t2A-mCherry | This paper | N/A |
| pLVX_EF1α-TFRC-Myc-10xHis-t2A-mCherry | This paper | N/A |
| pLVX_EF1α-NECTIN2-Myc-10xHis-t2A-mCherry | This paper | N/A |
| pLVX_EF1α-CADM4-Myc-10xHis-t2A-mCherry | This paper | N/A |
| pLVX_EF1α-MCAM-Myc-10xHis-t2A-mCherry | This paper | N/A |
| pLVX_EF1α-PLXDC2-Myc-10xHis-t2A-mCherry | This paper | N/A |
| pLKO-scramble-shRNA | Bayin et al.12 | N/A |
| pLKO-GPR133-sh3 | (Bayin et al.12 | N/A |
| pLKO-PTK7-sh31 (TRCN0000006431) | Millipore Sigma | SHCLNG-NM_002821 |
| pLKO-PTK7-sh33 (TRCN0000006433) | Millipore Sigma | SHCLNG-NM 002821 |
| pLKO-PTK7-sh35 (TRCN0000006435) | Millipore Sigma | SHCLNG-NM_002821 |
| pLKO-PTK7-sh65 (TRCN0000199565) | Millipore Sigma | SHCLNG-NM_002821 |
| Software and algorithms | ||
| ImageJ | Schneider et al.32 | https://imagej.nih.gov/ij |
| GTEx database | N/A | https://gtexportal.orghome |
| Ensembl database | Howe et al.33 | https://www.ensembl.org/index.html |
| CRAPome | Mellacheruvu et al.34 | https://reprint-apms.org |
Data and code availability
Mass spectrometry raw data generated by this study have been deposited in the MassIVE (Mass Spectrometry Interactive Virtual Environment) database of UCSD and are publicly available under the identifier MassIVE: MSV000090520.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture
Patient-derived glioblastoma (GBM) cultures were established and maintained as previously described.12,14,35,36 In brief, fresh bio-specimens from patients undergoing surgical resection of GBM were obtained after informed consent (NYU IRB study 12–01130). These tumor-specimens were mechanically minced with surgical blades and further enzymatically dissociated to single cells using Accutase (Innovative Cell Technologies, Cat# AT104). Patient-derived GBM cells were maintained in spheroid suspension cultures, or if experimentally required, grown as attached cultures on dishes pretreated with poly-L-ornithine (Sigma, Cat# P4957) and laminin (Thermo Fisher, Cat# 23017015). GBM growth medium was formulated as 500 mL Neurobasal medium (Gibco, Cat# 21103049) supplemented with 5 mL N2 (Gibco, Cat# 17-502-049), 500 mL B27 (Gibco, Cat# 12587010), 5 mL non-essential amino acids (Gibco, Cat# 11140050), 5 mL GlutaMax (Gibco, Cat# 35050061), and was freshly supplemented with 20 ng/mL recombinant basic Fibroblast Growth Factor (bFGF; R&D, Cat# 233-FB-01M) and 20 ng/mL Epidermal Growth Factor (EGF; R&D, Cat# 236-EG-01M) every 48 h. These fresh patient-derived cultures were profiled for mutations and copy number variations (CNV) (Table S1) on an Ion Torrent S5 instrument using a focused next-generation sequencing panel of 50 genes (NYU Oncomine focus assay; Table S2).37,38 All parental tumors were isocitrate dehydrogenase (IDH) wild-type GBM.
Lenti-X HEK293T cells (Takara, Cat# 632180) were cultured according to the manufacturer’s protocols in DMEM (Gibco, Cat# 11965–118) supplemented with 10% fetal bovine serum (Peak Serum, Cat# PS-FB2) and sodium pyruvate (Gibco, Cat# 11360070).
All cells were maintained in humidified cell culture incubators at 37°C and 5% CO2. HEK293T cells were cultured at 21% O2, while patient-derived GBM cells were cultured at 4% O2 to more closely resemble physiological conditions.
METHOD DETAILS
Ligand discovery
To detect membrane-bound or extracellular proteins interacting with the full-length GPR133 receptor, three separate patient-derived GBM cultures (GBML61, GBML91, GBML128) were lentivirally transduced with either a C-terminally TwinStrep-tagged and H543R mutated uncleavable GPR133 overexpression cassette, or an empty vector control. Both lentiviral vectors express mCherry for easy identification of transduced cells. Transduced cells were isolated by fluorescence-activated cell sorting (FACS) and expanded under adherent culture conditions. For each experiment, ten confluent 10 cm plates for each GBM cell line and construct were used as input and the entire experiment was repeated in three independent experiments. On the day of the experiment, adherent cell cultures were washed once with HBSS (+Ca2+, +Mg2+) and subsequently fixed using the cell membrane impermeable 3,3’-dithiobis(sulfosuccinimidyl propionate) (DTSSP, Thermo Fisher Scientific, Cat#21578) crosslinking agent at a concentration of 1 mM for 2 h at 4°C. The crosslinking reaction was quenched by supplementing it with 20 mM Tris. Cells were washed once with ice-cold PBS, scraped off the culture dishes, and pelleted by centrifugation. Cell pellets were lysed in RIPA buffer supplemented with 1% n-dodecyl b-D-maltoside (DDM) (Thermo, Cat# BN2005) and protease inhibitor cocktail (Thermo, Cat# 78429), and with 1/10 volume of 10X Buffer W (IBA, Cat# 2-1003-100) and BioLock (IBA, Cat# 2-0205-250) to block free biotin. After 20 min of mixing and incubation on ice, solutions were precleared by centrifugation at 5,000 × g for 15 min. The precleared lysates were slowly run through Strep-Tactin Sepharose columns (IBA, Cat#2-1202-001) by gravity flow, followed by multiple washing steps, as recommended in the manufacturer’s protocols. All wash buffers were supplemented with 0.1% DDM to remain above the critical micellar concentration of the detergent. The GPR133 bait and all co-immunoprecipitated proteins were eluted using the manufacturer’s elution buffer containing 2.5 mM desthiobiotin (IBA, Cat#2-1000-025). Elutions were analyzed by Western blot and mass spectrometry.
Protein identification: Nanoflow liquid chromatography-coupled to tandem mass spectrometry (LC-MS/MS)
Sample processing
Affinity purified samples were subjected to SDS/PAGE to fractionate proteins and remove contaminants. The resulting gels were washed 3 times in distilled deionized H2O for 15 min each and visualized by staining overnight with EZ-Run Protein Gel Staining Solution (Thermo Fisher Scientific, Cat# BP36201). Stained protein gel regions were typically excised into 4 gel sections per gel lane, and destained using standard published protocols.39 In-gel digestion was performed overnight with mass spectrometry grade trypsin (Trypsin Gold, Promega, Cat# V5280) at 5 ng/mL in 50 mM NH4HCO3 digest buffer. After acidification with 10% formic acid (final concentration of 0.5–1% formic acid), resulting peptides were desalted using hand-packed, reversed phase Empore C18 Extraction Disks (3M, Cat#3M2215), following an established method.40
Mass spectrometry data acquisition
Desalted peptides were concentrated to a very small droplet by vacuum centrifugation and reconstituted in 10 mL 0.1% formic acid in H2O. For two technical replicate analyses, 90% of the peptide material was used for liquid chromatography, followed by data-dependent acquisition mode (DDA) tandem mass spectrometry (LC-MS/MS). A Q Exactive HF mass spectrometer was coupled directly to an EASY-nLC 1000 (Thermo Fisher Scientific, Cat#LC120) equipped with a self-packed 75 μM x 20-cm reverse phase column (ReproSil-Pur C18, 3M, Dr. Maisch GmbH, Germany) for peptide separation. Analytical column temperature was maintained at 50°C by a column oven (Sonation GmbH, Germany). Peptides were eluted with a 3–40% acetonitrile gradient over 110 min at a flow rate of 250 nL/min. The mass spectrometer was operated in DDA mode with survey scans acquired at a resolution of 120,000 (at m/z 200) over a scan range of 300–1750 m/z. Up to 15 of the most abundant precursors from the survey scan were selected with an isolation window of 1.6 Th for fragmentation by higher-energy collisional dissociation with normalized collision energy (NCE) of 27. The maximum injection time for the survey and MS/MS scans was 60 ms and the ion target value (AGC) for both scan modes was set to 3e6.
Mass spectrometry data processing
The mass spectra files were processed using the MaxQuant proteomics data analysis workflow (version 1.6.0.1) with the Andromeda search engine.41,42 Raw mass spectrometry files were used to extract peak lists which were searched with the Andromeda search engine against the human proteome and a file containing contaminants, such as human keratins. Trypsin digestion was specified allowing up to 2 missed cleavages with the minimum required peptide length set to be seven amino acids. N-acetylation of protein N-termini, oxidation of methionines and deamidation of asparagines and glutamines were set as variable modifications. For the initial main search, parent peptide masses were allowed mass deviation of up to 20 ppm. Peptide spectral matches and protein identifications were filtered using a target-decoy approach at a false discovery rate of 1%. We used the raw MS1 intensity for protein quantitation. All mass spectrometry raw data were deposited in the MassIVE (Mass Spectrometry Interactive Virtual Environment) database of UCSD and are publicly available under the identifier MSV000090520.
Computational analysis of ligand candidates
The resulting list of detected proteins was annotated with subcellular localizations using the “cellular_component” entry of the Ensemble database as ref.33. Of the 2,138 total detected proteins, 569 with either membrane-, extracellular-, or other non-intracellular annotations were kept. The category “membrane” localization was a combination of the annotation terms: membrane, plasma membrane, integral component of membrane, integral component of plasma membrane, voltage-gated calcium channel complex, apical part of cell, cell junction. The category “extracellular” was a combination of the annotation terms: extracellular exosome, extracellular region, extracellular space. The category “other non-intracellular” was a combination of the annotation terms: cell, cellular_component, endoplasmic reticulum, Golgi apparatus, Golgi membrane, intracellular membrane-bounded organelle, NA. To assess differential protein levels in samples containing the GPR133 bait or the control empty vector, a linear mixed-effects model was built for each protein, with log transformed intensity (log10(value + 1)) as the response, group membership (GPR133 vs. control) as the fixed effect and GBM culture as the random effect. p values for the fixed effect from all models were then ranked and subjected to multiple testing correction with Benjamini-Hochberg procedure. Mixed effects models were fit using R software and the ‘nlme’ package. Of the correctly localized candidate proteins, only those enriched with the GPR133 bait in all three patient-derived cell lines were kept, resulting in a final list of 87 candidate binding proteins. These 87 candidates were screened for non-specific binding by interrogating the CRAPome database (https://reprint-apms.org/)34 and were assigned a value from 0 to 716 as a measure of frequency of detection by mass spectrometry in other affinity coimmunoprecipitation experiments.
Western blot analysis
Whole cell lysates were generated by lysing cells in RIPA buffer (Thermo, Cat#89900) supplemented with Halt complete protease inhibitor cocktail (Thermo, Cat# 78429) and 1% n-dodecyl b-D-maltoside (DDM) (Thermo, Cat# BN2005). After 15 min on ice, lysates were sonicated using a Qsonica rod sonicator at 25% power for 3 s. Whole cell lysates were precleared by centrifugation at 15,000 × g for 10 min at 4°C. Protein concentrations were determined using the DC protein assay kit II (BioRad, Cat# 5000112) and standardized across all samples within an experiment. Protein lysates were reduced in Laemmli buffer (BioRad, Cat# 1610747) containing b-mercaptoethanol at 37°C for 30 min, but were not boiled to prevent aggregation of hydrophobic transmembrane regions. Equal amounts of protein were separated by SDS-PAGE and transferred to 0.2 μM nitrocellulose membranes (BioRad, Cat# 1620112). After blocking the membranes in 2% BSA in Tris-buffered saline (TBS)-Tween for 1 h at room temperature, they were incubated with primary antibodies (listed in the key resources table) at 4°C overnight and visualized either with HRP-conjugated secondary antibodies for chemiluminescence, or with up to 3 simultaneous fluorescent Alexa Fluor Plus-conjugated secondary antibodies. Images were acquired using the iBrightFL1000 system (Invitrogen).
Affinity purification with the Strep-Tactin system
TwinStrep-tagged proteins were affinity purified from whole cell lysates or cell culture supernatants using MagStrep “type3” XT beads (IBA, Cat# 2-4090-002) according to the manufacturer’s protocols. All steps were performed on ice or at 4°C. In brief, whole cell lysates containing 1% n-dodecyl b-D-maltoside (DDM) (Thermo, Cat# BN2005) and protease inhibitor cocktail (Thermo, Cat# 78429) or cell culture medium were pretreated with 1/10 volume of 10X Buffer W (IBA, Cat# 2-1003-100) and BioLock (IBA, Cat# 2-0205-250) for 15 min on ice to block free biotin. Solutions were then precleared by centrifugation at 15,000 × g for 10 min. After input aliquots were obtained from the precleared protein solutions, MagStrep beads were added and samples were incubated rotating at 4°C overnight. The following day, MagStrep beads were separated using a magnetic rack and washed 5 times in a large excess of 1X Buffer W containing 0.1% DDM and protease inhibitors. Proteins were eluted from MagStrep beads in 3 consecutive steps of 20 mL 2X Buffer BXT (IBA, Cat# 2-1042-025) containing 1% DDM. Elutions were pooled and analyzed by Western blot.
Affinity purification with His-tag Ni-NTA system
10xHistidine tagged proteins were purified from whole cell lysates using HisPur Ni-NTA beads (Thermo, Cat# 88221), according to the manufacturer’s protocols. All steps were performed on ice or at 4°C. In brief, whole cell lysates containing 1% DDM and protease inhibitor cocktail were precleared by centrifugation at 15,000 × g for 10 min and supplemented with 10 mM imidazole (Sigma-Aldrich, Cat# I202). After obtaining input aliquots, pre-washed Ni-NTA beads were added, and samples were rotated at 4°C overnight. The following day, the Ni-NTA beads were pelleted by centrifugation at 700 × g for 3 min and were subsequently washed 4 times in PBS containing 50 mM imidazole and 0.1% DDM. Proteins were eluted from Ni-NTA beads in 3 consecutive steps of 20 mL 1 M imidazole in PBS containing 1% DDM. Elutions were pooled and analyzed by Western blot.
Immunofluorescent staining and microscopy
Fresh surgical GBM biospecimens were obtained after informed consent (NYU IRB study 12–01130) from patients undergoing surgical resection and were immediately fixed in 4% paraformaldehyde (PFA, Sigma-Aldrich, Cat# P6148) solution at 4°C overnight. Specimens were then transferred to 30% sucrose solution and incubated at 4°C for 48 h, before embedding and freezing in Tissue-Plus OCT (Thermo, Cat#23-730-571). Specimens were cryosectioned into 30 μM-thick slices, recovered on Superfrost glass slides (Thermo, Cat#12-550-15) and stored at−20°C. On the day of staining, slides were brought to room temperature, fixed again in 4% PFA for 10 min, washed 3 times with PBS containing 0.1% Triton X-100 (PBS-T) and blocked with 10% bovine serum albumin (BSA) in PBS-T for 1 h at room temperature. Primary antibodies were mixed in 1% BSA in PBS-T solution at the concentrations indicated in the key resources table and incubated at 4°C overnight. After 3 washes with PBS-T, secondary Alexa fluorophore-conjugated antibodies were added at a concentration of 1:1,000 in 1% BSA in PBS-T and incubated for 1 h at room temperature. After 3 additional washes in PBS-T, nuclei were counterstained with 500 ng/mL of 4’,6-diamidino-2-phenylindole (DAPI, Sigma, CAT#D8417) for 10 min at room temperature. Slides were washed again and mounted under a cover glass. Epifluorescent microscopy was conducted on a Zeiss AxioObserver and confocal laser scanning microscopy was conducted on a Zeiss LSM700. Images were analyzed using ImageJ software.
Ligand candidate expression plasmids for GPR133-binding validation experiments
All expression plasmids are based on the lentiviral vector pLVX-EF1a-mCherry-N1 (Takara, Cat# 631986). Codon-optimized variants of GPR133 were previously published and described.14 To narrow down the list of candidates for binding validation experiments, six candidate proteins from the primary screen were chosen either due to their high enrichment and/or previously published evidence for a role in GBM pathogenicity. Ligand candidate cDNAs were obtained from the following commercially available plasmids: PTK7 (Addgene, Cat#65250), PCDHGC3 (SinoBiological, Cat#HG22064-UT), TFRC (Addgene, Cat#69610), NECTIN2 (SinoBiological, Cat#HG10005-G), CADM4 (SinoBiological, Cat#HG16033-G), MCAM (SinoBiological, Cat#HG10115-M), PLXDC2 (SinoBiological, Cat#HG23536-U). All ligand cDNAs were C-terminally tagged with 10xHistidine and Myc tags and subcloned into the same pLVX backbone to generate bicistronic expression cassettes through a T2A cleavage site (ligand-T2A-mCherry). The C-terminal 10xHis and Myc tags are on the intracellular side of the transmembrane proteins for all candidates except TFRC.
Lentiviral production and infection
Lentiviruses were generated and used as previously described.43 In short, Lenti-X HEK293T cells (Takara, Cat# 632180) were grown in 10 cm dishes to 80% confluency and co-transfected with expression plasmids of interest and second-generation packaging plasmids (psPax2 + pMD2.G) using Lipofectamine 2000. Lentiviral particles were collected from the cell culture supernatant at 24, 48, and 72 h after transfection and concentrated using the Lenti-X concentrator (Contech Takara, Cat# 631231) according to the manufacturer’s instructions. Stably transduced cell lines were generated by lentiviral transduction in the presence of 4 μg/mL protamine sulfate at a multiplicity of infection of three (MOI = 3), followed by purification of mCherry-expressing cells via FACS.
HTRF signaling assays
HTRF assays were performed using the cAMP Gs dynamic kit (CisBio, Cat# 62AM4PEC) following the manufacturer’s protocols with slight modifications (Frenster et al. 2021). In brief, HEK293T were either stably transduced with lentiviruses or acutely transfected with expression plasmids using Lipofectamine 2000 (Invitrogen, Cat# 11668–019), according to the manufacturer’s protocol. Twenty-four hours after transfection, cells were reseeded onto 96-well plates pretreated with poly-L-lysine (Sigma, Cat# P4707) at a density of 75,000 cells per well.
In experiments where culture vessels were pre-coated with proteins, 96-well plates were pre-treated with poly-L-lysine (PLL), rinsed with distilled deionized H2O, and coated with 30 mL of: varying concentrations of PTK7 (R&D, Cat# 9799-TK-050), as indicated; or 12.5 μg/mL COL6 (Rockland, Cat# 009-001-108) resulting in a total of 375 ng of protein per well (1.17 μg/cm2); or varying concentrations of Wnt5a (R&D, Cat#645-WN-010/CF), as indicated. Plates were left to air-dry overnight and rinsed with distilled deionized H20 once before plating cells.
Alternatively, cells were plated as multi-layer sandwich cultures by seeding layers of alternating transgenic cells on top of each other with 12 h’ time intervals between each layer to allow for attachment.
Twenty-four hours after the final seeding of cells, the medium was exchanged with 50 mL of fresh medium with 1 mM 3-isobutyl-1-methylxanthine (IBMX) (Sigma-Aldrich, Cat# I7018–100MG). In some experiments, synthetic Stachel p13 peptide (TNFAILMQVVPLEOH) or its inactive control (TNAAIAAQVVPLE-OH) were added at the indicated final concentrations. Peptides were dissolved as described previously.4,44 In short, purified peptides were dissolved at 100 mM in 100% DMSO and further diluted into 10 mM stocks (10% DMSO) using a 50 mM, pH 8, Tris buffer. These 10 mM stocks were used for cell treatment after dilution to the indicated concentration in IBMX-containing medium. Additionally, in some experiments, recombinant Wnt5a was added to IBMX-containing medium, as indicated. Cells were then incubated at 37°C for an additional 30 min. Following cell lysis, cAMP levels were measured using the cAMP Gs dynamic kit on the FlexStation 3 (Molecular Devices), according to the manufacturer’s protocol.
CRE-luciferase cAMP assay
HEK293T cells stably overexpressing GPR133 or vector control, were transfected with a cAMP Response Element (CRE)-Luciferase reporter plasmid (Progema, Cat# E8471). Twenty-four hours later cells were transferred onto PTK7 (1.17 μg/cm2) or PLL-only pre-coated culture wells as indicated above. Twenty-four hours after re-seeding and 48 h after transfection, cells were lysed and luciferase activity was determined using the Bright-Glo Luciferase Assay System (Promega, Cat#E2650).
Enzyme-linked immunosorbent assay (ELISA)
Cells were seeded on 96-well plates simultaneously with each corresponding HTRF signaling assay, as described above. Twenty-four hours after seeding, cells were washed with HBSS (+Ca2+/+Mg2+) and fixed with 4% PFA for 20 min at room temperature. Whole cell ELISAs were performed under permeabilizing conditions in the presence of 0.1% Triton X-100 throughout, while surface ELISAs were performed under non-permeabilizing conditions in the absence of any detergent. The fixed cells were first blocked in HEK293T medium containing 10% FBS for 1 h at room temperature, and then incubated with primary antibodies diluted in HEK293T medium at concentrations indicated in the key resources table for 1 h at room temperature. After 3 thorough washes with PBS, cells were incubated with horseradish peroxidase-conjugated secondary antibodies diluted 1:500 in HEK293T medium for 1 h at room temperature. After 3 additional washes with PBS, cells were overlayed with TMB-stabilized chromogen for 10 min (Thermo Fisher, Cat# SB02) followed by an equal volume of acidic stop solution (Thermo Fisher, Cat# SB04). Absorbance was read at 450 nm using a Synergy H1 multi-mode plate reader (Biotek). Non-specific background was determined using “secondary-antibody only” control wells and subtracted from sample wells.
Flow cytometric analysis of GPR133 and PTK7 surface protein expression
For flow cytometric analysis, patient-derived GBM cultures were enzymatically dissociated to single cells by incubating GBM spheroids in Accutase (Innovative Cell Technologies, Cat# AT104) for 5 min at 37°C. After washing, cells were incubated with primary antibodies against the extracellular domains of GPR133 (mouse monoclonal antibody clone “8E3E8”) and PTK7 (rabbit polyclonal, Thermo Fisher, Cat#PA5–82070) diluted at 1:200 in flow cytometry buffer (PBS -Ca2+/Mg2+, 2 mM EDTA, 0.5% BSA) for 1 h rotating at 4°C. After 3 washes in flow cytometry buffer, cells were incubated with appropriate secondary antibodies for 20 min at room temperature in the dark. After additional 3 washes, cells were analyzed on a BD LSRFortessa Cell Analyzer. Secondary antibody-only controls were used as negative controls for gating of true positive cells in each channel. Single-color controls were used for bleed-through compensation to distinguish single from double-positive cells. Data were analyzed with FlowJo software.
In silico analysis of tissue and single cell expression profiles
Expression of GPR133 and ligand candidates in healthy tissues was conducted using publicly available data from the GTEx portal. Hierarchical clustering of mRNA levels of ligand candidates against GPR133 by human tissue was conducted using the Multi-Gene Query of the Genotype-Tissue Expression (GTEx) web portal (https://gtexportal.org/home/multiGeneQueryPage), without exclusion of any available tissues. Single cell transcriptome analysis was performed using the GTEx single cell dataset as released by Eraslan et al. (https://www.biorxiv.org/content/10.1101/2021.07.19.452954v1.full; doi: https://doi.org/10.1101/2021.07.19.452954). The raw data file GTEx_8_tissues_snRNAseq_atlas_071421.public_obs.h5ad was extracted using python 2.7.16. Expression values in log(CP10K + 1) were combined with the annotation of all cells in R 4.2.0. Single cell expression data for GPR133 and PTK7 from 209,126 cells was extracted from the cited dataset and only cells with RIN>8 were used in the analysis.
Single cell RNAseq analysis in GBM tissue was conducted using publicly available data with the GEO accession number GSM3828672. This dataset was created using Smartseq2 and was pre-processed to TPM by the authors of the dataset as detailed in the GEO entry.
HEK293 bulk RNAseq analysis was conducted using publicly available data with the GEO accession number GSE158834. Specifically, only the dataset’s wildtype control conditions (GSM4811760, GSM4811761, GSM4811762) were used for analysis of unperturbed baseline gene expression.
Tumorsphere formation assays
Patient-derived GBM cultures were grown as described above. Cells were dissociated to a single cell suspension and infected at a multiplicity of infection of 3 (MOI = 3) with lentiviruses expressing shRNAs in the backbone vector pLKO. Cells received either a non-targeting control shRNA (CCTAAGGTTAAGTCGCCCTCG), a previously described shRNA against GPR133 (CCTGCAGGGACT GTTCATATT) (Bayin, et al. 2016), or PTK7-targeting shRNAs from the Sigma MISSION library (Millipore Sigma, Cat# SHCLNGNM_002821): sh31, CACAGGGTTAATGAGTCTCTT; sh33, CCACAGCACAAGTGATAAGAT; sh34, CCTCATGTTCTACTGCAAGAA; sh35, CCTGAGGATTTCCAAGAGCAA; sh65, GCCACTCATCTGCCAACTTTG). Two days after infection, cells were dissociated to single cell suspensions and selected in 5 μg/mL puromycin for 5 days. Seven days post infection, cells were dissociated to single cells, counted, and plated at 500 cells per well (96-well plate) in 100 mL medium. Cells were allowed to grow and initiate tumorspheres in 96-well plates for 2 weeks, while growth factors and fresh media were supplemented every other day. After 2 weeks of growth, the 96-well plates were imaged using automated tile scanning on an EVOS imaging system (Thermo Fisher Scientific). Tile scans of each well were exported, and sphere number was analyzed in ImageJ. An area of 4,000 μM2 (sphere diameter 70 μM) was used as a minimum size cutoff during analysis. Each experimental condition was quantified from 6 technical replicate wells, which was counted as a single data point in statistical analyses.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical comparisons were conducted in GraphPad Prism (v9). Replicates were represented as mean ± SEM (standard error of the mean), unless otherwise noted. Statistical significance was calculated using Students t-test; and one-way or two-way analysis of variance (ANOVA) with post hoc Tukey’s or Sidak’s multiple comparisons tests. GTEx expression correlation was calculated as Pearson r correlation with two-tailed testing. The threshold of significance was set at p < 0.05 throughout.
Supplementary Material
Highlights.
PTK7 is a GPR133-binding protein in glioblastoma
PTK7-GPR133 binding takes place between their extracellular N-terminal domains
PTK7 binding in trans increases GPR133 signaling
Knockdown of either GPR133 or PTK7 impairs glioblastoma tumorsphere formation
ACKNOWLEDGMENTS
J.D.F. was supported by an NYSTEM Stem Cell Biology training grant to NYU Grossman School of Medicine (C32560GG). G.S. was supported by a DFG postdoctoral fellowship (STE 2843/1–1). N.R.-B. was supported by a T32 Cell Biology training grant (T32GM136542) to NYU Grossman School of Medicine and an NYSTEM Stem Cell Biology training grant to NYU Grossman School of Medicine (C32560GG). D.B. was supported by F30 CA247418. S.H. was supported by the DFG HO 6389/2–2 ‘KFO 337’. N.S. was supported by DFG CRC 1423 (project number A6) and FOR 2149 (project number P4). I.L. and T.S. were supported by DFG FOR2149 (project numbers 266022790 P4 to T.S. and P5 to I.L.), CRC1052 (project number 209933838 B6), and CRC1423 (project number 421152132). T.N. was supported by S10 RR027990 and P30 NS050276. D.G.P. was supported by NIH/NINDS R01 NS102665, NYSTEM (NY State Stem Cell Science) IIRP C32595GG, NIH/NINDS R01 NS124920, NIH/NINDS R21 NS126806, NIH/NIBIB R01 EB028774 (to Dr. Steven Baete at NYU Grossman School of Medicine), NIH/NCI R21CA263402 (to Dr. David Zagzag at NYU Grossman School of Medicine), NIH/NCATS 2UL1TR001445, NYU Grossman School of Medicine, and DFG FOR2149 as Mercator fellow. The NYU Microscopy Core facility was supported by the NYU Perlmutter Cancer Center support grant P30CA016087. The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank the Microscopy core facility at NYU Grossman School of Medicine for assistance with confocal microscopy imaging. We thank Dr. Andreas Kloetgen for helping with the annotation of subcellular localizations of candidate proteins detected during the ligand discovery affinity screen.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112679.
DECLARATION OF INTERESTS
D.G.P. and NYU Grossman School of Medicine own a patent in the European Union and Hong Kong titled “Method for treating high grade glioma” on the use of GPR133 as a treatment target in glioma. D.G.P. has received consultant fees from Tocagen, Synaptive Medical, Monteris, Advantis, and Robeaute in the past.
REFERENCES
- 1.Vizurraga A, Adhikari R, Yeung J, Yu M, and Tall GG (2020). Mechanisms of adhesion G protein-coupled receptor activation. J. Biol. Chem 295, 14065–14083. 10.1074/jbc.REV120.007423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langenhan T, Aust G, and Hamann J (2013). Sticky signaling–adhesion class G protein-coupled receptors take the stage. Sci. Signal 6, re3. 10.1126/scisignal.2003825. [DOI] [PubMed] [Google Scholar]
- 3.Araç D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Südhof TC, and Brunger AT (2012). A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J 31, 1364–1378. 10.1038/emboj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liebscher I, Schön J, Petersen SC, Fischer L, Auerbach N, Demberg LM, Mogha A, Cöster M, Simon KU, Rothemund S, et al. (2014). A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep. 9, 2018–2026. 10.1016/j.celrep.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barros-Álvarez X, Nwokonko RM, Vizurraga A, Matzov D, He F, Papasergi-Scott MM, Robertson MJ, Panova O, Yardeni EH, Seven AB, et al. (2022). The tethered peptide activation mechanism of adhesion GPCRs. Nature 604, 757–762. 10.1038/s41586-022-04575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ping YQ, Xiao P, Yang F, Zhao RJ, Guo SC, Yan X, Wu X, Zhang C, Lu Y, Zhao F, et al. (2022). Structural basis for the tethered peptide activation of adhesion GPCRs. Nature 604, 763–770. 10.1038/s41586-022-04619-y. [DOI] [PubMed] [Google Scholar]
- 7.Qu X, Qiu N, Wang M, Zhang B, Du J, Zhong Z, Xu W, Chu X, Ma L, Yi C, et al. (2022). Structural basis of tethered agonism of the adhesion GPCRs ADGRD1 and ADGRF1. Nature 604, 779–785. 10.1038/s41586-022-04580-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao P, Guo S, Wen X, He QT, Lin H, Huang SM, Gou L, Zhang C, Yang Z, Zhong YN, et al. (2022). Tethered peptide activation mechanism of the adhesion GPCRs ADGRG2 and ADGRG4. Nature 604, 771–778. 10.1038/s41586-022-04590-8. [DOI] [PubMed] [Google Scholar]
- 9.Wilde C, Mitgau J, Suchý T, Schöneberg T, and Liebscher I (2022). Translating the force - mechano-sensing GPCRs. Am. J. Physiol. Cell Physiol 322, C1047–C1060. 10.1152/ajpcell.00465.2021. [DOI] [PubMed] [Google Scholar]
- 10.Lin HH, Ng KF, Chen TC, and Tseng WY (2022). Ligands and beyond: mechanosensitive adhesion GPCRs. Pharmaceuticals 15, 219. 10.3390/ph15020219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohnekamp J, and Schöneberg T (2011). Cell adhesion receptor GPR133 couples to Gs protein. J. Biol. Chem 286, 41912–41916. 10.1074/jbc.C111.265934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bayin NS, Frenster JD, Kane JR, Rubenstein J, Modrek AS, Baitalmal R, Dolgalev I, Rudzenski K, Scarabottolo L, Crespi D, et al. (2016). GPR133 (ADGRD1), an adhesion G-protein-coupled receptor, is necessary for glioblastoma growth. Oncogenesis 5, e263. 10.1038/oncsis.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frenster JD, Kader M, Kamen S, Sun J, Chiriboga L, Serrano J, Bready D, Golub D, Ravn-Boess N, Stephan G, et al. (2020). Expression profiling of the adhesion G protein-coupled receptor GPR133 (ADGRD1) in glioma subtypes. Neurooncol. Adv 2, vdaa053. 10.1093/noajnl/vdaa053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frenster JD, Stephan G, Ravn-Boess N, Bready D, Wilcox J, Kieslich B, Wilde C, Sträter N, Wiggin GR, Liebscher I, et al. (2021). Functional impact of intramolecular cleavage and dissociation of adhesion G protein-coupled receptor GPR133 (ADGRD1) on canonical signaling. J. Biol. Chem 296, 100798. 10.1016/j.jbc.2021.100798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephan G, Frenster JD, Liebscher I, and Placantonakis DG (2022). Activation of the adhesion G protein-coupled receptor GPR133 (ADGRD1) by antibodies targeting its N-terminus. J. Biol. Chem 298, 101949. 10.1016/j.jbc.2022.101949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bianchi E, Sun Y, Almansa-Ordonez A, Woods M, Goulding D, Martinez-Martin N, and Wright GJ (2021). Control of oviductal fluid flow by the G-protein coupled receptor Adgrd1 is essential for murine embryo transit. Nat. Commun 12, 1251. 10.1038/s41467-021-21512-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin HH, Stacey M, Yona S, and Chang GW (2010). GPS proteolytic cleavage of adhesion-GPCRs. Adv. Exp. Med. Biol 706, 49–58. 10.1007/978-1-4419-7913-1_4. [DOI] [PubMed] [Google Scholar]
- 18.Krasnoperov V, Lu Y, Buryanovsky L, Neubert TA, Ichtchenko K, and Petrenko AG (2002). Post-translational proteolytic processing of the calcium-independent receptor of alpha-latrotoxin (CIRL), a natural chimera of the cell adhesion protein and the G protein-coupled receptor. Role of the G protein-coupled receptor proteolysis site (GPS) motif. J. Biol. Chem 277, 46518–46526. 10.1074/jbc. M206415200. [DOI] [PubMed] [Google Scholar]
- 19.Volynski KE, Silva JP, Lelianova VG, Atiqur Rahman M, Hopkins C, and Ushkaryov YA (2004). Latrophilin fragments behave as independent proteins that associate and signal on binding of LTX(N4C). The EMBO journal 23, 4423–4433. 10.1038/sj.emboj.7600443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moriguchi T, Haraguchi K, Ueda N, Okada M, Furuya T, and Akiyama T (2004). DREG, a developmentally regulated G protein-coupled receptor containing two conserved proteolytic cleavage sites. Gene Cell. 9, 549–560. 10.1111/j.1356-9597.2004.00743.x. [DOI] [PubMed] [Google Scholar]
- 21.Okajima D, Kudo G, and Yokota H (2010). Brain-specific angiogenesis inhibitor 2 (Bai2) may be activated by proteolytic processing. J. Recept. Signal Transduct. Res 30, 143–153. 10.3109/10799891003671139. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q, Zhang C, Yuan J, Fu J, Wu M, Su J, Wang X, Yuan X, and Jiang W (2015). PTK7 regulates Id1 expression in CD44-high glioma cells. Neuro Oncol. 17, 505–515. 10.1093/neuonc/nou227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peradziryi H, Tolwinski NS, and Borchers A (2012). The many roles of PTK7: a versatile regulator of cell-cell communication. Arch. Biochem. Biophys 524, 71–76. 10.1016/j.abb.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Dunn NR, and Tolwinski NS (2016). Ptk7 and mcc, unfancied components in non-canonical Wnt signaling and cancer. Cancers 8, 68. 10.3390/cancers8070068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganier L, Betzi S, Derviaux C, Roche P, Dessaux C, Muller C, Hoffer L, Morelli X, and Borg JP (2022). Discovery of small-molecule inhibitors of the PTK7/beta-catenin interaction targeting the Wnt signaling pathway in colorectal cancer. ACS Chem. Biol 17, 1061–1072. 10.1021/acschembio.1c00826. [DOI] [PubMed] [Google Scholar]
- 26.Barzegar Behrooz A, Talaie Z, Jusheghani F, Łos MJ, Klonisch T, and Ghavami S (2022). Wnt and PI3K/Akt/mTOR survival pathways as therapeutic targets in glioblastoma. Int. J. Mol. Sci 23, 1353. 10.3390/ijms23031353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamann J, Vogel B, van Schijndel GM, and van Lier RA (1996). The seven-span transmembrane receptor CD97 has a cellular ligand (CD55, DAF). J. Exp. Med 184, 1185–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wandel E, Saalbach A, Sittig D, Gebhardt C, and Aust G (2012). Thy-1 (CD90) is an interacting partner for CD97 on activated endothelial cells. J. Immunol 188, 1442–1450. 10.4049/jimmunol.1003944. [DOI] [PubMed] [Google Scholar]
- 29.Stacey M, Chang GW, Davies JQ, Kwakkenbos MJ, Sanderson RD, Hamann J, Gordon S, and Lin HH (2003). The epidermal growth factor-like domains of the human EMR2 receptor mediate cell attachment through chondroitin sulfate glycosaminoglycans. Blood 102, 2916–2924. 10.1182/blood-2002-11-3540. [DOI] [PubMed] [Google Scholar]
- 30.Niu M, Xu S, Yang J, Yao D, Li N, Yan J, Zhong G, and Song G (2021). Structural basis for CD97 recognition of the decay-accelerating factor CD55 suggests mechanosensitive activation of adhesion GPCRs. J. Biol. Chem 296, 100776. 10.1016/j.jbc.2021.100776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu D, Duan L, Rodda LB, Lu E, Xu Y, An J, Qiu L, Liu F, Looney MR, Yang Z, et al. (2022). CD97 promotes spleen dendritic cell homeostasis through the mechanosensing of red blood cells. Science 375, eabi5965. 10.1126/science.abi5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider C, Rasband W, and Eliceiri K (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howe KL, Achuthan P, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, Bhai J, et al. (2021). Ensembl 2021. Nucleic Acids Res. 49, D884–D891. 10.1093/nar/gkaa942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736. 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frenster JD, and Placantonakis DG (2018). Establishing primary human glioblastoma tumorsphere cultures from operative specimens. Methods Mol. Biol 1741, 63–69. 10.1007/978-1-4939-7659-1_4. [DOI] [PubMed] [Google Scholar]
- 36.Bayin NS, Frenster JD, Sen R, Si S, Modrek AS, Galifianakis N, Dolgalev I, Ortenzi V, Illa-Bochaca I, Khahera A, et al. (2017). Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells. Oncotarget 8, 64932–64953. 10.18632/oncotarget.18117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehrotra M, Duose DY, Singh RR, Barkoh BA, Manekia J, Harmon MA, Patel KP, Routbort MJ, Medeiros LJ, Wistuba II, and Luthra R (2017). Versatile ion S5XL sequencer for targeted next generation sequencing of solid tumors in a clinical laboratory. PLoS One 12, e0181968. 10.1371/journal.pone.0181968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hovelson DH, McDaniel AS, Cani AK, Johnson B, Rhodes K, Williams PD, Bandla S, Bien G, Choppa P, Hyland F, et al. (2015). Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia 17, 385–399. 10.1016/j.neo.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kishinevsky S, Wang T, Rodina A, Chung SY, Xu C, Philip J, Tal-done T, Joshi S, Alpaugh ML, Bolaender A, et al. (2018). HSP90-incorporating chaperome networks as biosensor for disease-related pathways in patient-specific midbrain dopamine neurons. Nat. Commun 9, 4345. 10.1038/s41467-018-06486-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 41.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, and Mann M (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res 10, 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 42.Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 43.Frenster JD, Inocencio J, and Placantonakis DG (2018). Lentiviral transduction of primary human glioblastoma cultures. Methods Mol. Biol 1741, 81–89. 10.1007/978-1-4939-7659-1_6. [DOI] [PubMed] [Google Scholar]
- 44.Demberg LM, Winkler J, Wilde C, Simon KU, Schön J, Rothemund S, Schöneberg T, Prömel S, and Liebscher I (2017). Activation of adhesion G protein-coupled receptors: agonist specificity of STACHEL sequence-derived peptides. J. Biol. Chem 292, 4383–4394. 10.1074/jbc.M116.763656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry raw data generated by this study have been deposited in the MassIVE (Mass Spectrometry Interactive Virtual Environment) database of UCSD and are publicly available under the identifier MassIVE: MSV000090520.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.