

Abstract
PURPOSE
To describe the clinical and molecular features of metastatic colorectal cancers (mCRCs) bearing uncommon atypical RAS (At-RAS) mutations at codons other than 12, 13, 59, 61, 117, and 146.
MATERIALS AND METHODS
By exploiting five next-generation sequencing sources (Italian collaboration, Memorial Sloan Kettering Cancer Center, Samsung Medical Center, the Biomarker Research for Anti-EGFR Monoclonal Antibodies by Comprehensive Cancer Genomics (BREAC) study, and the Foundation Medicine database), we retrieved 175 At-RAS mutated cases. Molecular data were obtained from 163 samples from Memorial Sloan Kettering Cancer Center and the Foundation Medicine database. Clinical data were available for 27 At-RAS–positive and 467 negative cases from the Italian collaboration, Memorial Sloan Kettering Cancer Center, Samsung Medical Center, and the BREAC study.
RESULTS
At-RAS mutations were identified in 163 (0.9%) of 18,270 mCRCs. Among 133 with evaluable microsatellite instability status, 11 (8%) were microsatellite instability high. POLE exonuclease domain mutations had higher frequency (7%) than expected and were found only in microsatellite-stable tumors with high tumor mutational burden (TMB). Overall, 17% (28 of 163) of At-RAS cases had TMB greater than 20 mutations/Mb. Co-occurring typical RAS/BRAF V600E mutations and NF1 mutations, presumed to cause RAS activation, were found in 30% and 12% of samples, respectively (up to 43% and 50%, respectively, in TMB-high samples). Patients with RAS/BRAF wild-type mCRC achieved a median overall survival (OS) of 42.1 months, whereas those harboring isolated At-RAS, typical RAS, or BRAF V600E mutations showed a median OS of 32.3, 30.0, and 17.9 months, respectively (P < .001). No significant OS difference (P = .240) was found between patients with At-RAS versus typical RAS-mutated mCRC. Only one of six patients evaluable for primary resistance to anti–epidermal growth factor receptors achieved tumor response.
CONCLUSION
At-RAS mutations may be a marker for RAS pathway activation and can be associated with high co-occurrence of POLE exonuclease domain mutations.
INTRODUCTION
RAS proteins, including KRAS and NRAS, are GTPases that cycle from the inactive guanosine diphosphate–bound state to the active guanosine triphosphate (GTP)–bound conformation because of the signaling mediated by tyrosine-kinase receptors.1 Mutations in RAS codons 12, 13, and 61 increase mitogen-activated protein kinase (MAPK) activity by impairing the GTP hydrolysis induced by GTPase-activating proteins; mutations affecting codons 117 and 146 are associated with increased RAS-GTP levels as well.2
Retrospective post hoc analyses of pivotal randomized clinical trials showed lack of clinical benefit or even a detrimental effect when administering anti–epidermal growth factor receptor agents (anti-EGFRs), either as monotherapy or in combination with chemotherapy,3,4 in patients with tumors bearing RAS mutations involving codons 12, 13, 59, 61, 117, and 146 of either KRAS or NRAS. On the basis of these findings, the use of anti-EGFR monoclonal antibodies cetuximab and panitumumab in metastatic colorectal cancer (mCRC) is now restricted to patients with RAS wild-type tumors. Several studies also showed that patients with RAS-mutated mCRC have poorer survival as compared with those with RAS wild-type disease, also when treated with bevacizumab-containing first-line regimens.5
Hotspot mutations affecting codons other than 12, 13, 59, 61, 117, and 146 account for a small subset of RAS mutations. A retrospective, next-generation sequencing (NGS)–based study of 177 patients6 showed that up to 2.8% of mCRCs bear atypical RAS (At-RAS) alterations. Full sequencing of exons 2, 3, and 4 of RAS genes using Sanger sequencing or NGS assays may reveal mutations otherwise missed by hotspot-targeted real-time polymerase chain reaction or multiplex-hotspot MALDI-TOF technologies, which are included in recommendations by current guidelines.7 Because of the widespread use of NGS and other comprehensive genomic profiling techniques that allow sequencing of the entire coding regions of RAS genes, At-RAS mutations are being detected with increasing frequency. However, their functional, prognostic, and predictive role is still largely unknown, leaving a knowledge gap that affects clinical management. Given this, we carried out a multinational effort aimed at unveiling the clinical course and molecular landscape of mCRCs harboring At-RAS mutations as well as exploring their potential role as predictive biomarkers of primary resistance to anti-EGFR therapies.
CONTEXT
Key Objective
To provide molecular and clinical characterization of atypical RAS mutations (ie, those occurring outside major hotspots in codon 12, 13, 59, 61, 117, and 146) in metastatic colorectal cancer.
Knowledge Generated
We provide the first evidence, to our knowledge, that the prognostic role of atypical RAS mutations is superimposable to that of typical RAS and that tumors bearing these alterations are characterized by a high prevalence of POLE exonuclease domain mutations and high tumor mutational burden.
Relevance
Our results suggest the clinical meaning of atypical RAS mutations as markers of activation of RAS pathway and highlight the potential clinical relevance of testing At-RAS–mutated tumors for the presence of POLE exonuclease domain mutations to identify a niche of patients who may be candidates for immunotherapy approaches.
MATERIAL AND METHODS
Patient Population
As shown in Figure 1, mCRC cases harboring At-RAS mutations were retrieved from five screening sources: patients screened at three Italian institutions (Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Azienda Ospedaliera Universitaria Pisana, and Azienda Ospedaliera Universitaria Integrata di Verona), the Memorial Sloan Kettering Cancer Center database, the Samsung Medical Center database, the Biomarker Research for Anti-EGFR Monoclonal Antibodies by Comprehensive Cancer Genomics (BREAC) Japanese study,8 and the Foundation Medicine database. Extended RAS sequencing data for each screening platform are detailed in the Data Supplement.
FIG 1.

Study flow diagram. At-RAS, atypical RAS; BREAC, Biomarker Research for Anti-EGFR Monoclonal Antibodies by Comprehensive Cancer Genomics (BREAC); mCRC, metastatic colorectal cancer; NGS, next-generation sequencing.
We were able to retrieve complete clinical data for 33 patients with At-RAS–mutated mCRC, and we compared them with a cohort of 467 At-RAS–negative cases retrieved by the Italian institutions and the BREAC study in terms of baseline characteristics and overall survival (OS). Six patients were excluded because of the co-occurrence of typical RAS or BRAF V600E mutations in their specimens. Extended molecular data were available for patients from the Memorial Sloan Kettering Cancer Center database and the Foundation Medicine database. Functional At-RAS mutation characterization, namely MAPK pathway activation compared with RAS wild-type status, was reported according to Loree et al.9 Mutational frequency of recurrently mutated genes was compared with historical data reported by Yaeger et al10 in the setting of mCRC. The study was approved by the Fondazione IRCCS Istituto Nazionale dei Tumori di Milano Institutional Review Board (study ID: INT 117/15) and conducted according to the ethical principles for medical research involving human subjects adopted in the Declaration of Helsinki. All patients who were alive signed a written informed consent.
Clinical Characteristics
Clinical, pathologic, and molecular characteristics at the time of diagnosis of mCRC were collected. These included sex, age, Eastern Cooperative Oncology Group performance status, primary tumor location (right- v left-sided), mucinous histology, primary tumor resection, time to metastases (synchronous v metachronous), number of metastatic sites (one v more than one), and microsatellite instability (MSI) status.
Assessment of Primary Resistance to Anti-EGFR Treatment
To assess primary resistance to anti-EGFR antibody therapies, we evaluated the response to anti-EGFRs for patients with tumors bearing any At-RAS mutation but no co-occurring typical RAS or BRAF mutations. Only patients receiving cetuximab or panitumumab monotherapy, or in combination with irinotecan if previous irinotecan refractoriness was clearly demonstrated, were included, as previously described.11
Molecular Analyses
The type of At-RAS mutations, the presence of nonsilent mutations, and comutated genes were analyzed for each case. A VCF file was annotated with COSMIC (https://cancer.sanger.ac.uk/cosmic) and TransVar12 databases at the protein change level. Analysis and visualization of mutations were then performed using R environment (http://www.Bioconductor.org), with MAFtools13 and COSMIC.67 libraries. OncoKB (https://oncokb.org/#/) was used to define the oncogenic annotation. Tumor mutational burden (TMB) was calculated as the total number of nonsilent mutations divided by the territory targeted (Mb) by each NGS panel assay (MSK-IMPACT 341, MSK-IMPACT 410, or FoundationOne), and MSI was determined on 95 or 114 loci using validated methods, as previously described.14,15
Cancer Driver Mutation Evaluation
To obtain a whole panorama of At-RAS mutations, we first focused on identifying driver point mutations including single-nucleotide polymorphisms and short indels in each individual tumor by computing multiple tools: cancer driver mutations through cancer genome interpreter database (https://www.cancergenomeinterpreter.org/home) and OncodriveMUT algorithm16; Provean tool17 (http://provean.jcvi.org/genome_submit_2.php?species=human); and transFIC (https://bbglab.irbbarcelona.org/fannsdb/help/transfic.html) assessing SIFT,18 Polyphen,17 and MutationAssessor19 tools. This comprehensive analysis can overcome numerous biases that confound the analyses to find driver mutations from the passenger ones.
Statistical Analysis
The Fisher’s exact test, v2 test, or Mann-Whitney test were used when appropriate to assess the associations of At-RAS mutations with investigated characteristics. We investigated the impact of At-RAS mutations on OS, defined as the time from diagnosis of metastatic disease to death or last follow-up for patients who were alive. OS analysis was determined according to the Kaplan-Meier method, and survival curves were compared by means of the log-rank test. The association of At-RAS mutations and clinicopathological characteristics with OS was assessed in univariate analyses, and a multivariable Cox proportional hazard model was built, including as covariates variables associated with survival with a P value < .10 in the univariate analyses. Hazards proportionality was assumed and verified using the goodness-of-fit v2 test.
RESULTS
Clinicopathological and Molecular Features of Patients Bearing At-RAS–Mutated mCRC
As listed in Table 1, when focusing on the 27 patients with mCRC with clinical information available and isolated At-RAS mutations, this population was not significantly associated with any specific clinical or pathologic feature, as compared with BRAF, typical RAS, or RAS/BRAF wild-type subgroups. The list of specific At-RAS mutations with functional characterization, co-occurring typical RAS/BRAF V600 mutations, and MSI status is detailed in the Data Supplement.
TABLE 1.
Patient Characteristics According to the Presence of At-RAS, BRAF, and Typical RAS Mutations and RAS/BRAF Wild-Type Status

In the Foundation Medicine (from August 2012 to January 2018) and Memorial Sloan Kettering Cancer Center (from January 2014 to January 2018) data sets, 142 and 21 cases with At-RAS mutations were retrieved out of a total of 16,324 and 1,946 mCRC samples analyzed from unique patients, respectively, with an overall incidence of 0.9%. A total of 56 unique At-RAS mutations were detected (Fig 2A), of which 30 KRAS and three NRAS were known oncogenic missense mutations or indels as defined by the OncoKb database. Overall, At-KRAS and -NRAS mutations occur primarily in the GTPase domain and consequently may affect the ability of RAS GTPase-activating proteins, such as NF1, to bind RAS and regulate its signaling (Data Supplement). Co-occurring mutations involving typical RAS and/or BRAF V600E were reported in 30% of cases with At-RAS mutations. In addition, multiple At-RAS mutations occurred in five (3%) samples from 163 patients. At-RAS mutations found to be hotspots were KRAS pV14I, KRAS pL19F, and KRAS pQ22K (each present in > 10% of the samples), which may be endowed with a higher MAPK activity than RAS wild-type protein.9 In addition, from 52 At-RAS annotated mutations, 71% (37 of 52) may be recognized as driver mutations (Data Supplement). In our data set, a specific At-RAS mutation could be possibly recognized as driver in 76% of cases, suggesting a functional effect of a subset of At-RAS alterations in mCRC tumorigenesis. In 24% of cases with At-RAS mutations possibly recognized as passengers, we then explored the co-occurrence with other mutational events to define possible cooperative mechanisms involved in tumor growth. Some unique frequently comutated genes with At-RAS passenger mutations were involved in critical tumorigenic pathways such as MAPK, ATM, and RAS positive regulation of phosphorylation signaling (Data Supplement).
FIG 2.
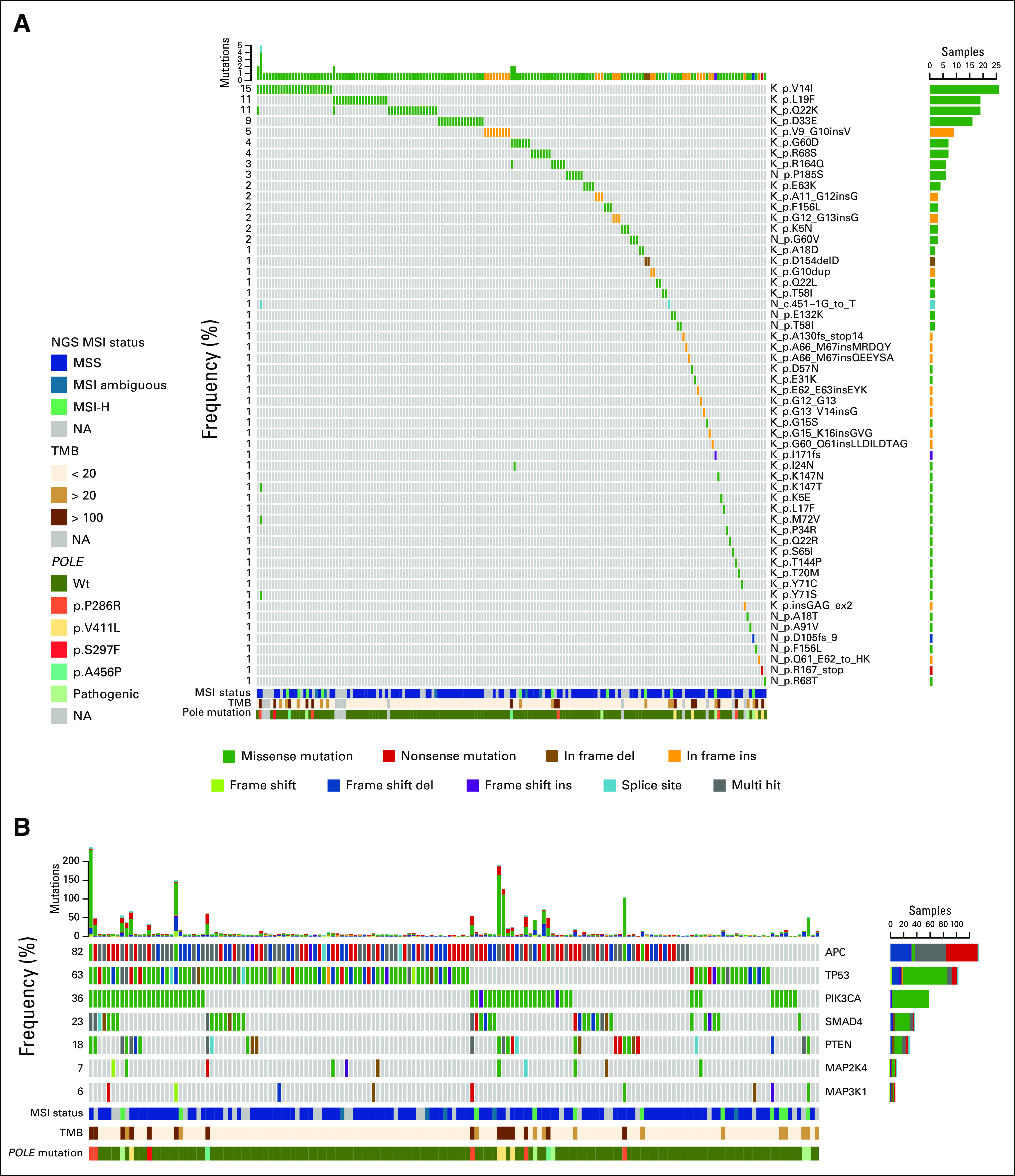
Summary of mutagenesis and molecular features associated with atypical RAS mutations in patients with extensive next-generation sequencing (NGS) data from screening sources of Memorial Sloan Kettering Cancer Center database and Foundation Medicine database including 163 patients. (A) At-RAS mutations oncoplot. Each column represents one patient and each row one specific atypical RAS mutation or gene. The upper bar plot indicates the number of mutations per sample, and the right bar plot describes the mutational frequency among the overall population, indicating the variant classification by colors. Multi-hit variants are genes that present different mutations in the same sample. Lower molecular feature heat maps indicate clinical microsatellite instability (MSI) status, tumor mutational burden (TMB), and POLE exonuclease domain (ED) mutations. (B) Target sequencing oncoplots of most frequently comutated genes (> 18%) with At-RAS mutations and MAPK-mutated genes. Lower molecular feature heat maps indicate clinical MSI status, TMB, and POLE ED mutations. MSI-H, MSI-high; NA, not available; Wt, wild type.
Recurrently mutated genes identified by NGS for Memorial Sloan Kettering Cancer Center database and Foundations Medicine database patient screening sources (N = 163) are outlined in the Data Supplement. Sixty-seven recurrently (≥ 4%) mutated genes were identified to be comutated with At-RAS, including most frequently APC (82%), TP53 (63%), PIK3CA (36%), SMAD4 (23%), and PTEN (18%; Fig 2B). Regarding MSI status from 133 At-RAS tumors with available data, 8% of tumors were classified as MSI-high (MSI-H) and 92% as MSS, in agreement with the frequency observed in typical RAS-mutated tumors10 (n = 354; MSI-H, 11%; MSS, 89%). There was a broad range of recurrently comutated genes in our cohort and typical RAS-mutated tumors from Yaeger et al10 (Data Supplement). For instance, NF1 mutations occurred in 12% of cases with At-RAS. POLE exonuclease domain (ED) mutations (P286R, S459F, V411L, A456P, and S297F) were reported in 7% of samples with At-RAS mutations (12 of 163), compared with 0.71% of all-comers and 0.77% of samples with typical RAS mutations10 (P < .001).
Cases with At-RAS and POLE ED mutations were exclusively MSS but had a median TMB of 219 mutations/Mb (range, 107 to 364 mutations/Mb). Despite the enrichment in POLE mutations, median TMB in samples harboring At-RAS mutations (range, 0 to 363.65 mutations/Mb; median, 5.21 mutations/Mb) was similar compared with cases with typical RAS mutations10 (range, 0.97 to 337.86 mutations/Mb; median, 6.79 mutation/Mb).
To assess the impact of TMB on the molecular landscape, we divided our cohort into two groups on the basis of TMB as follows: TMB low (≤ 20 mutations/Mb; n = 135 [83%]) and TMB high (> 20 mutations/Mb; n = 28 [17%]). Cases classified as TMB high were enriched in POLE ED mutations (TMB high, 42%; TMB low, 0%) and POLE pathogenic mutations (TMB high, 57%; TMB low, 0.7%) and were associated with specific recurrently comutated genes (Data Supplement). Furthermore, typical RAS and/or BRAF V600E mutations were more common in the TMB-high group (43%) than in TMB low (28%). We also found a significant increase in NF1 mutation frequency in TMB-high tumors (50%) compared with TMB low (4.4%).
Prognostic Role of At-RAS Mutations
At a median follow-up of 73.9 months (95% CI, 64.1 to 85.3 months), although patients with RAS/BRAF wild-type mCRC achieved a median OS of 42.1 months (95% CI, 37.9 to 45.6 months), those harboring At-RAS mutations, typical RAS mutations, or BRAF V600E mutations achieved median OS of 32.3 (95% CI, 24 to non-assessable), 30.0 (95% CI, 26.2 to 34.7), or 17.9 (95% CI, 11.2 to 30.4) months, respectively (P < .001; Fig 3). In this latter group, patients with tumors bearing At-RAS mutations had no significant difference in terms of OS when compared with those with typical RAS mutations (hazard ratio [HR], 0.73; 95% CI, 0.46 to 1.16; P = .240) but significantly longer OS than those bearing BRAF V600E mutations (HR, 0.39; 95% CI, 0.18 to 0.82; P = .005).
FIG 3.

Kaplan-Meier curves for overall survival (OS) according to RAS, atypical RAS (At-RAS), and BRAF V600E mutational (mut) status. HR, hazard ratio; NA, non-assessable; Ref, reference.
In the multivariable Cox model (Table 2), including other prognostic covariates, At-RAS–, RAS-, and BRAF V600E–based classification was independently associated with OS (P = .03), with an HR versus RAS/BRAF wild type of 0.74 (95% CI, 0.40 to 1.35) for At-RAS mutations, 1.17 (95% CI, 0.90 to 1.53) for typical RAS mutations, and 2.53 (95% CI, 1.28 to 5.00) for BRAF V600E mutations. Also, primary tumor resection (P < .001) and number of metastatic sites (P = .006) were significantly associated with OS in the multivariable analysis.
TABLE 2.
Association of Atypical RAS, RAS, and BRAF Mutational Status and Known Prognostic Baseline Characteristics With Overall Survival

Predictive Role of At-RAS Mutations Regarding Anti-EGFR Monoclonal Antibodies
Among 27 patients with At-RAS–mutated mCRC and available clinical data, six fulfilled the clinical criteria for the primary resistance cohort (Data Supplement; Table 3). One patient experienced a partial response to panitumumab monotherapy, two patients had stable disease lasting less than 6 months, and three patients had progressive disease. All samples were assessed using the Primary Resistance in RAS and BRAF Wild-Type Metastatic Colorectal Cancer Patients Treated With Anti-EGFR Monoclonal Antibodies (PRESSING) panel,11 which groups together several genomic alterations associated with anti-EGFR primary resistance beyond RAS and BRAF mutations. None of the resistant patients with At-RAS mutations and primary resistance showed a concomitant genomic alteration known to be associated with anti-EGFR primary resistance.
TABLE 3.
Response to Anti-EGFR–Based Treatment in Patients With mCRC With At-RAS Mutations

DISCUSSION
Recent advances in the depiction of the molecular landscape of mCRC provides evidence for the potential impact of several biomarkers other than RAS and BRAF mutational status and MSI status for improving the clinical management of affected patients. For this reason, NGS techniques able to provide feasible comprehensive molecular characterization of mCRC from routinely obtained clinical specimens in relevant timeframes have become frequent in clinical practice. As a consequence, clinicians need to understand how to manage and properly interpret a wide amount of molecular information of unknown or uncertain clinical significance. Among them, characterization of rare alterations is extremely challenging. Here we focus on At-RAS mutations to identify the similarities and differences of cases with At-RAS versus those with typical RAS mutations and their independent weight on patients’ outcomes.
Although we found that At-RAS mutations are not associated with specific clinicopathological characteristics in mCRC, the prognostic role of isolated At-RAS mutations seems highly superimposable with that of typical RAS mutations, with a worse prognosis when compared with RAS and BRAF V600E wild type and a clearly better outcome than the BRAF V600E mutant subgroup. Therefore, At-RAS mutations may confer biologic aggressiveness to the disease through MAPK hyperactivation, similar to typical RAS mutations. Supporting this, in a preliminary report Loree et al9 functionally characterized a wide set of At-RAS mutations by means of an ectopic expression assay, and although the median MAPK activity of At-RAS mutations was lower than that of typical RAS mutations, it was higher than RAS wild type. Here we provide additional evidence that even though not all RAS mutations are created equal,20 and their additional characterization may be useful, their overall clinical impact is quite clear.
Regarding the molecular landscape of At-RAS–mutated tumors, we showed that MSI-H status had a frequency of 8% (similar to previous reports in the metastatic setting). As expected, POLE ED mutations were found only in MSS tumors21 with a significantly higher frequency (7%) as compared with the literature.10 Accordingly, an enrichment of TMB-high tumors was observed among those harboring At-RAS mutations as compared with historical data.10
Intriguingly, a high proportion of cases with At-RAS mutations (30%) displayed co-occurring typical RAS and/or BRAF V600E mutations. This enrichment was not only due to MSI-H or TMB-high tumors. Given that 92% of cases with At-RAS mutations were MSS and 83% were TMB low, the occurrence of At-RAS mutations was not limited to hyper- or ultramutated tumors enriched with passenger mutations. It is arguable that the mild hyperactivation of the oncogenic MAPK pathway driven by some At-RAS mutations may require additional hits (eg, a co-occurring RAS or NF1 mutation) to efficiently drive tumor progression. In contrast, the effect of typical RAS or BRAF V600E mutations may be able to promote cancer progression by itself, thus explaining the mutual exclusivity of these alterations. Because of co-occurring typical RAS mutations in up to 30% of cases, At-RAS mutations could therefore closely resemble the history of non-V600E BRAF mutations, particularly those with low-activity BRAF mutations.22,23 Actually, oncogenic RAS and non-V600E BRAF mutations cooperate for malignant transformation in preclinical models.24 Similarly, here we show an enriched frequency of coalterations in NF1, a negative regulator of RAS proteins.25 Moreover, by means of complex computational algorithms, the majority (76%) of At-RAS mutations here could be predicted as driver mutations, even if additional demonstration of the transforming capability of specific At-RAS mutations should be performed thanks to preclinical validation studies.
A crucial but still unanswered issue is the magnitude of benefit that patients with At-RAS mutations may derive from anti-EGFR agents, because patients with RAS wild-type tumors, especially if left-sided, gain substantial survival benefit from these agents when compared to bevacizumab in first-line treatment.7 To explore the predictive weight of At-RAS mutations, we focused on patients with clearly assessable activity of anti-EGFRs. Only one of six patients fulfilling these criteria achieved a response to single-agent panitumumab. A KRAS p.K5N missense mutation, whose functional behavior has not been elucidated so far, was found in that case. In all the other cases, no concomitant genomic alterations potentially related to intrinsic resistance to anti-EGFRs were reported. Among patients who achieved stable disease as best response, disease stabilization was short-lasting (ie, < 6 months). Because our results on the negative predictive role of At-RAS mutations on the efficacy of anti-EGFRs are descriptive and could be biased by several confounding factors, they cannot be used to drive decision making in clinical practice. However, even if validation studies of our preliminary findings are challenging, retrospective or pooled analyses of randomized clinical trials should be ideally carried out, possibly by means of targeted genomic panels including At-RAS mutations combined with other multiple, albeit uncommon, primary resistance alterations, as recently performed by our group.26 We highly emphasize that a prospective international data set and preclinical collaborations should be established to better assess the individual role of specific At-RAS mutations as predictive biomarkers.
The main limitations of our study include the small subset of patients with At-RAS–mutated tumors with available data about clinical outcome, especially about response to anti-EGFR agents; the lack of comprehensive genomic sequencing data for all patients included in the study; and the heterogeneity of NGS platforms adopted. Our experience underlines the need to collect information from multinational collaborations to shed a light on rare or very rare genomic alterations. Because of the small sample size of our clinical cohort, we are not able to draw definitive conclusions. However, our data suggest a close phenotypic similarity between isolated At-RAS and typical RAS mutations that is reasonably driven by At-RAS mutations with established increased MAPK activity or by the cooperation of multiple activating signals in the MAPK pathway. Finally, the presence of At-RAS mutations was significantly associated with the co-occurrence of POLE ED mutations. Therefore, because At-RAS mutations may be easily found, thanks to simple assays such as Sanger sequencing or small NGS panels, clinicians could consider POLE sequencing for patients with ascertained At-RAS–mutated mCRC, because of potential benefit from immunotherapy in the ultramutated/POLE ED–mutated subgroup.27
ACKNOWLEDGMENT
S.R.-C. received the “Cátedra Salvador Zubirán” UNAM/INCMNSZ and is part of the national system of researchers, CONACYT (CVU 363453).
SUPPORT
Supported by MSK Cancer Center core grant P30 CA 008748.
AUTHOR CONTRIBUTIONS
Conception and design: Filippo Pietrantonio, Giovanni Randon, Daniele Rossini, Jeffrey S. Ross, Daisuke Kotani, Vincent A. Miller, Filippo de Braud, Chiara Cremolini
Provision of study material or patients: Seung Tae Kim, Beatrice Borelli, Alfredo Falcone
Collection and assembly of data: Filippo Pietrantonio, Rona Yaeger, Alexa B. Schrock, Giovanni Randon, Daniele Rossini, Giovanni Fucà, Jeffrey S. Ross, Daisuke Kotani, Russell Madison, Seung Tae Kim, Lisa Salvatore, Filippo Pagani, Beatrice Borelli, Federica Perrone, Jeeyun Lee, Takayuki Yoshino, Filippo de Braud, Alfredo Falcone, Jaclyn F. Hechtman, Chiara Cremolini
Data analysis and interpretation: Filippo Pietrantonio, Rona Yaeger, Alexa B. Schrock, Giovanni Randon, Sandra Romero-Cordoba, Daniele Rossini, Giovanni Fucà, Jeffrey S. Ross, Daisuke Kotani, Alessandra Raimondi, Federica Perrone, Maria Di Bartolomeo, Vincent A. Miller, Siraj M. Ali, Takayuki Yoshino, Chiara Cremolini
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Filippo Pietrantonio
Consulting or Advisory Role: Amgen, Merck Serono, Bayer, Eli Lilly, Sanofi, Roche, SERVIER
Rona Yaeger
Research Funding: Array BioPharma, GlaxoSmithKline, Novartis, Boehringer Ingelheim
Travel, Accommodations, Expenses: Array BioPharma
Alexa B. Schrock
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Giovanni Randon
Travel, Accommodations, Expenses: Sanofi/Aventis
Jeffrey S. Ross
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Research Funding: Foundation Medicine
Daisuke Kotani
Honoraria: Merck Serono, Chugai Pharmaceutical, Takeda, Eli Lilly Japan
Consulting or Advisory Role: Merck Serono
Russell Madison
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Lisa Salvatore
Honoraria: Roche, Merck Serono, SERVIER, Bayer
Consulting or Advisory Role: Merck Serono, SERVIER, Bayer
Travel, Accommodations, Expenses: Sanofi, Merck Serono, Bayer
Maria Di Bartolomeo
Honoraria: Eli Lilly, MSD Oncology, SERVIER
Consulting or Advisory Role: Eli Lilly, MSD Oncology
Research Funding: Eli Lilly (Inst)
Travel, Accommodations, Expenses: Roche, Sanofi
Vincent A. Miller
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine, Mirati Therapeutics
Consulting or Advisory Role: Revolution Medicines
Patents, Royalties, Other Intellectual Property: Receive periodic royalties related to T790M patent awarded to Memorial Sloan Kettering Cancer Center
Siraj M. Ali
Employment: Foundation Medicine
Leadership: Incysus
Stock and Other Ownership Interests: Exelixis, Blueprint Medicines, Agios Pharmaceuticals, Genocea Biosciences
Consulting or Advisory Role: Revolution Medicines, Azitra (I), Princepx Tx (I)
Patents, Royalties, Other Intellectual Property: Patents via Foundation Medicine; Patents via Seres Health on microbiome stuff in non-neoplastic disease (I)
Takayuki Yoshino
Research Funding: Chugai Pharmaceutical (Inst), Sanofi (Inst), Sumitomo Dainippon Pharma (Inst), GlaxoSmithKline (Inst)
Filippo De Braud
Consulting or Advisory Role: Ignyta, Pfizer, Amgen, Novartis, Daiichi Sankyo, Bristol-Myers Squibb, Dompè, Pierre Fabre, Roche, Octimet Oncology, Incyte, Teofarma, EMD Serono, Pharm Research Associates
Speakers' Bureau: MSD, Novartis, Bristol-Myers Squibb, Roche, Pfizer, Menarini
Research Funding: Novartis (Inst), Roche (Inst), MSD (Inst), Ignyta (Inst), MedImmune (Inst), Nektar (Inst), Bristol-Myers Squibb (Inst), Merck Serono (Inst), Bayer (Inst), Celgene (Inst), GlaxoSmithKline (Inst), Boehringer Ingelheim (Inst), Eli Lilly (Inst), Pfizer (Inst), SERVIER (Inst)
Alfredo Falcone
Honoraria: Eli Lilly, Roche, Merck, SERVIER, Amgen
Consulting or Advisory Role: Amgen, Bayer, Bristol-Myers Squibb, Eli Lilly, Merck, Roche, SERVIER
Research Funding: Amgen (Inst), Bayer (Inst), Merck (Inst), Roche (Inst), Sanofi (Inst), MSD (Inst), SERVIER (Inst)
Travel, Accommodations, Expenses: Amgen, Bayer, Roche, Merck, SERVIER
Jaclyn F. Hechtman
Honoraria: Medscape
Consulting or Advisory Role: Navigant Consulting, Axiom Biotechnologies
Research Funding: Bayer
Chiara Cremolini
Honoraria: Roche, Amgen, Bayer, SERVIER
Consulting or Advisory Role: Roche, Bayer, Amgen
Speakers' Bureau: SERVIER
Research Funding: Merck
Travel, Accommodations, Expenses: Roche, SERVIER
No other potential conflicts of interest were reported.
REFERENCES
- 1.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D: RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer 11:761-774, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janakiraman M, Vakiani E, Zeng Z, et al. : Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res 70:5901-5911, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Douillard JY, Oliner KS, Siena S, et al. : Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369:1023-1034, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Pietrantonio F, Cremolini C, Petrelli F, et al. : First-line anti-EGFR monoclonal antibodies in panRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Crit Rev Oncol Hematol 96:156-166, 2015 [DOI] [PubMed] [Google Scholar]
- 5.Cremolini C, Loupakis F, Antoniotti C, et al. : FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol 16:1306-1315, 2015 [DOI] [PubMed] [Google Scholar]
- 6.Harlé A, Filhine-Tresarrieu P, Husson M, et al. : Rare RAS mutations in metastatic colorectal cancer detected during routine RAS genotyping using next generation sequencing. Target Oncol 11:363-370, 2016 [DOI] [PubMed] [Google Scholar]
- 7.Yoshino T, Arnold D, Taniguchi H, et al. : Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: A JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann Oncol 29:44-70, 2018 [DOI] [PubMed] [Google Scholar]
- 8.Shinozaki E, Yoshino T, Yamazaki K, et al. : Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: The Biomarker Research for anti-EGFR monoclonal Antibodies by Comprehensive Cancer genomics (BREAC) study. Br J Cancer 117:1450-1458, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loree JM, Miron B, Holla V, et al: Not all RAS mutations created equal: Functional and clinical characterization of 80 different KRAS and NRAS mutations. J Clin Oncol 35, 2017 (suppl 15; abstr 3589)
- 10.Yaeger R, Chatila WK, Lipsyc MD, et al. : Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33:125-136.e3, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cremolini C, Morano F, Moretto R, et al. : Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: The PRESSING case-control study. Ann Oncol 28:3009-3014, 2017 [DOI] [PubMed] [Google Scholar]
- 12.Zhou W, Chen T, Chong Z, et al. : TransVar: A multilevel variant annotator for precision genomics. Nat Methods 12:1002-1003, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayakonda A, Lin DC, Assenov Y, et al. : Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28:1747-1756, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalmers ZR, Connelly CF, Fabrizio D, et al. : Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9:34, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fabrizio DA, George TJ Jr, Dunne RF, et al. : Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointest Oncol 9:610-617, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Perez A, Lopez-Bigas N: Functional impact bias reveals cancer drivers. Nucleic Acids Res 40:e169, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi Y, Sims GE, Murphy S, et al. : Predicting the functional effect of amino acid substitutions and indels. PLoS One 7:e46688, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar P, Henikoff S, Ng PC: Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073-1081, 2009 [DOI] [PubMed] [Google Scholar]
- 19.Reva B, Antipin Y, Sander C: Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res 39:e118, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller MS, Miller LD: RAS mutations and oncogenesis: Not all RAS mutations are created equally. Front Genet 2:100, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Domingo E, Freeman-Mills L, Rayner E, et al. : Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol 1:207-216, 2016 [DOI] [PubMed] [Google Scholar]
- 22.Cremolini C, Di Bartolomeo M, Amatu A, et al. : BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol 26:2092-2097, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. : Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548:234-238, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heidorn SJ, Milagre C, Whittaker S, et al. : Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140:209-221, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philpott C, Tovell H, Frayling IM, et al. : The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics 11:13, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morano F, Corallo S, Lonardi S, et al: Negative hyper-selection of RAS and BRAF wild-type metastatic colorectal cancer patients receiving panitumumab-based maintenance therapy: A pre-specified exploratory analysis of the Valentino study. J Clin Oncol (in press) [DOI] [PMC free article] [PubMed]
- 27. Silberman R, Steiner DF, Lo AA, et al: Complete and prolonged response to immune checkpoint blockade in POLE-mutated colorectal cancer. JCO Precis Oncol 10.1200/PO.18.00214. [DOI] [PubMed]