

Abstract
The entry of the SARS-CoV-2 virus into a human host cell begins with the interaction between the viral spike protein (S protein) and human angiotensin-converting enzyme 2 (hACE2). Therefore, a possible strategy for the treatment of this infection is based on inhibiting the interaction of the two abovementioned proteins. Compounds that bind to the SARS-CoV-2 S protein at the interface with the alpha-1/alpha-2 helices of ACE2 PD Subdomain I are of particular interest. We present a stepwise optimisation of helical peptide foldamers containing trans-2-aminocylopentanecarboxylic acid residues as the folding-inducing unit. Four rounds of optimisation led to the discovery of an 18-amino-acid peptide with high affinity for the SARS-CoV-2 S protein (Kd = 650 nM) that inhibits this protein–protein interaction with IC50 = 1.3 µM. Circular dichroism and nuclear magnetic resonance studies indicated the helical conformation of this peptide in solution.
Keywords: COVID-19, foldamers, peptides, helix, protein-protein interaction, BLI
Graphical abstract

Introduction
Oligomers with a propensity towards the formation of a well-folded state, called foldamers, have attracted considerable scientific interest due to their ability to mimic the structural behaviour of biomolecules by adopting stable and programmable three-dimensional structures1,2. To move from structure to biological application, foldamers need to exhibit good stability under physiological conditions, allowing their distribution in a living system, and contain appropriately spatially distributed functional groups, which can provide effective interaction with a given molecular target3,4. Since their introduction5,6, numerous studies have been reported on peptide foldamers constructed by modification of the peptide backbone through the incorporation of β-, γ-, δ-amino acid residues in various sequence patterns7–9. One of the most widely studied and best-characterised types of foldamers is chimeric α,β-peptides due to their tendency to form distinctive, residue-controlled secondary structures10. Furthermore, the peptide backbone can be further altered by the introduction of structurally constrained cyclic β-amino acids (containing cyclopropane, cyclobutane, cyclopentane or cyclohexane), which greatly promote self-organisation in α,β-peptides11–15. Due to their synthetic availability and facile insertion into the peptide sequence, β-aminocyclopentanecarboxylic acids (ACPC) are often used in the design of foldameric structures16,17. The ability of a peptide foldamer to form a helix can be indicated by the method of stereochemical patterning: a helical structure is formed if the appropriate ψ and ϕ dihedral angles flanking the amide bond are of the same sign18. It was demonstrated that α,β-peptides containing the backbone patterns ααβ, αααβ, and ααβαααβ can adopt helical conformations16,19,20. Peptide foldamers have had numerous successful applications in medicinal chemistry21–26. This class of compounds can serve as scaffolds for the development of antimicrobial compounds27,28, antifungal agents29, protein–protein interaction (PPI) inhibitors30,31, and antivirals32. Interactions between proteins are of great interest as valuable medicinal targets, and it has been shown that peptide foldamers can inhibit various PPIs, such as the Bcl-2/BH3 domain30, p53/MDM233, and VEGF/VEGFR1 interactions34. One of the possible approaches for inhibiting PPIs in which one side contributes a single α-helix to the interface is based on the mimicry of the critical α-helix by a corresponding foldamer35,36.
The emergence and rapid international spread of the new pathogenic severe acute respiratory syndrome coronavirus (SARS-CoV-2) has caused a global health emergency, leading to the WHO’s declaration of a Public Health Emergency of International Concern (PHEIC). Hence, prompt progress in the design and development of specific antiviral drugs is urgently needed. The SARS-CoV-2 virus invades the host cell through the interaction of the receptor-binding domain (RBD) of its spike (S) protein with the angiotensin-converting enzyme (ACE2) of the host cell37. Therefore, the development of specific inhibitors for the SARS-CoV-2/ACE2 interaction presents an important strategy in the fight against SARS-CoV-2 infection. The crystal structure of the SARS-CoV-2 RBD of the S protein bound to the cell receptor ACE2 revealed the most important residues in the SARS-CoV-2 RBD that are essential for binding38–40. However, the development of S protein/ACE2 interaction inhibitors remains difficult41–45. Screening of 800 million peptides provided S protein binders; however, the identified compounds did not inhibit S protein/ACE2 interactions46. The application of phage display methodology (screening of 109 peptides) also led to finding peptides with high affinity for the RDB of the S protein, but no inhibitory activity of the target PPI has been reported47. Computational studies indicated peptide sequences that should bind to the S protein; however, these candidates either have not been evaluated experimentally or have shown little activity48–53. Stapled and cyclic peptides were found to bind the S protein with moderate micromolar affinity54–56. Only large constructs, including peptide-dendrimer, peptide-antibody, peptide-quantum dots, peptide-DNA constructs, and proteins, were effectively developed to exhibit the desired activity57–62.
In this paper, we present a computer-aided design and synthesis of helical peptide foldamers, as well as an evaluation of their binding to RDB of the S protein and inhibition of the S protein/ACE2 interaction. The incorporation of trans-ACPC residues in the ααβαααβ sequence pattern in the structure of 18-mer peptides rigidifies their helical conformation and thus provides a stable scaffold for the development of biological activity. Several rounds of optimisation led to the discovery of highly active compounds.
Materials and methods
Peptide synthesis
All commercially available reagents and solvents were purchased from Sigma–Aldrich, Merck, Iris Biotech or Bachem and used without further purification. Peptide foldamers were prepared using automated solid-state peptide synthetizers (Biotage® Initiator + Alstra™ and CEM Liberty Blue) on H-Rink amide ChemMatrix resin (loading: 0.59 mmol g−1) with DMF as a solvent. Fmoc deprotection was accomplished using 20% piperidine solution in DMF at 75 °C. The coupling procedures were carried out using 0.5 M DIC and 0.5 M Oxyma solutions (1:1) in DMF. For α-amino acids (5 eq, 0.1 M), single coupling (for Liberty Blue, 4 min at 90 °C) or double coupling (for Biotage, 2 × 15 min at 75 °C) was performed, and for trans-ACPC, single coupling (30 min at 75 °C) was used. Acetylation of the N-terminus was performed using a mixture of NMP/DIPEA/acetic anhydride (80:15:5). The cleavage of the peptides from the resin and deprotection were accomplished using a mixture of TFA/TIS/H2O (95:2.5:2.5) after 3 h of shaking at room temperature. The crude peptide was precipitated with ice-cold diethyl ether and centrifuged (10000 rpm, 15 min, 2 °C). The peptides were purified using preparative HPLC (Knauer Prep, 250 mm × 30 mm preparative column, Thermo Scientific™ Hypersil GOLD™, C18, 12 µm) with a water/acetonitrile (0.05% TFA) eluent system. The purity of the peptides was checked using analytical HPLC (Shimatzu, ReproSil Saphir 100 C18, 5 µ, 150 × 4.6 mm analytical column) with a water/acetonitrile (0.05% TFA) eluent system.
Mass spectrometry
Mass spectra of the synthetised peptides were acquired using a WATERS LCT Premiere XE high-resolution mass spectrometer with an electrospray ionisation (ESI) and time-of-flight (TOF) detector.
Circular dichroism
All CD spectra were recorded using a JASCO J-815 spectropolarimeter between 250 and 190 nm in potassium phosphate buffer (50 mM, pH 7.5) with the following parameters: 25 °C, 0.2 nm resolution, 1.0 nm bandwidth, 20 mdeg sensitivity, 0.25 s response, 50 nm min−1 scanning speed, 5 accumulations, and 0.02 cm cuvette path length. The CD spectra of the buffer alone were recorded and subtracted from the raw data. Samples were obtained by preparing a 0.1 mM solution of purified and lyophilised peptide (as TFA salt) in buffer. The CD intensity is given as the mean residue molar ellipticity (θ [deg × cm2×dmol−1]).
Nuclear magnetic resonance
The NMR experiments were performed on a Bruker AvanceTM spectrometer operating at 600.58 MHz for 1H, equipped with a 5.0 mm PA BBO probe. The NMR spectra of peptide 27 at 298 K were recorded in methanol d3. The temperature was controlled to ± 0.1 K. TOCSY and NOESY experiments were performed for chemical shift and structure assignment. All NMR spectra were acquired with suppression of the solvent OH signal using a 3–9-19 pulse sequence with gradients. Typical TOCSY - homonuclear Hartman (Hahn transfer using the mlev17 sequence for mixing using two power levels for excitation and spinlock) and 2D NOESY were recorded in phase-sensitive mode with a spectral width of 8620 Hz in both dimensions using 4k data points and a relaxation delay of 2 s. These spectra were acquired with 1024 increments of 18 scans for TOCSY and 80 scans for 2D NOESY. Mixing times were set at 80 ms and 200 ms for the TOCSY and 2D NOESY experiments, respectively. The data were acquired and processed using Topspin 3.1 (BrukerBioSpin, Rheinstetten, Germany). The processed spectra were assigned with the help of the SPARKY program63.
NMR structure calculation
NMR structure generation for peptide 27 was performed in Xplor-NIH v. 2.41.164. Initially, 100 random conformations were generated from the sequence using the seqTOpsf protocol. NMR-derived interproton contacts were classified by the standard method with upper distance limits of 2.5 Å for strong contacts, 3.5 Å for medium and 5 Å for weak, and the lower distance limit was set to 1.8 Å. For the β-amino acid, three backbone torsions were also restrained to match the previous experimental data65 with a tolerance of 30 degree deviation. Standard simulated annealing protocols implemented in Xplor-NIH were used and composed of the following steps: (1) high temperature dynamics (3500 K, 800 ps or 8000 steps), (2) simulated annealing performed from 3500 K to 25 K with 12.5 K step, at each temperature, short dynamics was done (100 steps or 0.2 ps); (3) gradient minimisation of the final structure. Finally, the top ten lowest energy structures were superimposed and averaged.
Biolayer interferometry
Biolayer interferometry (BLI) binding data were obtained using an Octet RED96 (ForteBio) and processed using the instrument’s integrated software. All steps were performed at 30 °C with shaking at 400 rpm in a black 96-well plate, with a working volume of 200 µL in each well. Fc-tagged RBD (AcroBiosystems) was loaded onto protein A biosensors (ForteBio) at a concentration of 3 µg mL−1 in binding buffer (10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, 0.05% surfactant P20 Tween, 0.5% BSA) for 600 s. After baseline measurement in binding buffer alone, the binding kinetics were monitored by dipping the biosensors in wells containing the tested peptide at the indicated concentration (association step, 300 s) and then dipping the sensors back into the binding buffer solution (dissociation step, 300 s). The initial screening of the tested peptides was performed at 50 µM, while for the accurate determination of Kd value, BLI screening was executed for a range of different peptide concentrations depending on their binding affinities. Tested peptides were diluted from concentrated stock solution using binding buffer. Collected raw data were background subtracted using the reference biosensors with the buffer alone.
Homogeneous time-resolved fluorescence (HTRF) measurements
The HTRF assay was performed using an HTRF 96-well low volume white plate (CisBio #66PL96025), antibodies: PAb Anti Human IgG-Eu cryptate (CisBio #61HFCKLA) and MAb Anti 6HIS-XL665 (CisBio #61HISXLA), proteins: Human ACE2/ACEH Protein, Fc Tag (MALS verified) (AcroBioSystem #AC2-H5257) and SARS-CoV-2 (COVID-19) S protein RBD, His Tag (MALSverified) (AcroBioSystem #SPD-C52H3) and PPI Europium Detection Buffer (CisBio # 61DB9RDF). For initial screening of peptide activity, stock solutions were prepared at 1 mM and 50 µM in H2OmilliQ and then diluted to 500 µM and 5 µM concentrations with PPI Detection Buffer. The concentrations of peptides in the wells were 50 µM and 0.5 µM. To determine the half-maximal inhibitory concentrations (IC50) of the tested compounds, measurements were performed on individual dilution series. The peptide 27 concentrations in the wells were as follows: 50 µM, 25 µM, 5 µM, 0.5 µM, 250 nM, 50 nM, and 5 nM. Concentrations of proteins and antibodies in wells were as follows: PAb Anti Human IgG-Eu cryptate − 1 nM, MAb Anti 6HIS-XL665–10 nM, Human ACE2/ACEH Protein, Fc Tag − 20 nM, SARS-CoV-2 (COVID-19) S protein RBD, His Tag − 10 nM. Proteins and antibodies were diluted from stock solutions using PPI Europium Detection Buffer. The assay was performed using the CisBio standard protocol. First, 2 µl of peptide was added to the well, followed by 4 µl of Fc-ACE2 and 4 µl of His-RBD. After this step, the plate was incubated at room temperature for 15 min, and then 10 µl of antibody mixture (1:1, v:v) was added. After mixing all components, the plate was incubated for 2 h at room temperature followed by TR-FRET measurements on a CLARIOstar® fluorimeter, with an excitation filter at 337 nm and fluorescence wavelength measurements at 620 and 665 nm. Collected data were background subtracted using the negative control, normalised to the positive control, averaged, and fitted with the DoseResp Function to determine the IC50 value using Origin2019.
Molecular modelling
The first series of peptides (peptides A1-A3 and analogs) was designed on the basis of the complex of native ACE2 and SARS-CoV-2 RBD (pdb: 6m0j)66. The next series was designed on the basis of previously reported ACE2/SARS-CoV-2 RBD miniprotein inhibitors: LCB1 (pdb: 7jzu) and LCB3 (pdb: 7jzm)62. Full-length proteins were truncated to single helices responsible for most of the interaction with RBD (Ile21-Ser43 in ACE2, Ala22-Lys38 in LCB1 and Asn1-His18 in LCB3) and foldamerized by introduction of transACPC residues, giving different realisations of the previously experimentally characterised βααβαααβ motif19. The minimisation of the obtained structures was performed in Discovery Studio (BIOVIA) using the Smart Minimiser algorithm and the CHARMm force field, up to an energy change of 0.0 or RMS gradient of 0.1. Complexes with RBD were designed by superimposition of inhibitors onto full-length proteins and optimised using the Local Docking protocol (Rosetta), and different analogs of these peptides were designed by the FastDesign protocol (Rosetta).
Results and discussion
The available structural and biochemical data derived on the basis of the crystal structures of the SARS-CoV and SARS-CoV-2 S protein complexes with ACE267,68, cryoEM studies69, and biochemical binding data of ACE2 with SARS-CoV-2 S68 serve as a good starting point for the design of small peptide-based inhibitors of the SARS-CoV-2 S/ACE2 interaction. Studies have shown that among all secondary structures that form the ACE2 protein, most contacts with the SARS-CoV-2 RBD originate from the N-terminal α1-helix (ACEα1, Table S2)38. Therefore, an approach to designing new SARS-CoV-2 S inhibitors was to mimic the indicated α1-helix motif. A short peptide built up of natural amino acids, such as the ACEα1 helix, when synthesised alone, may not be conformationally stable; therefore, additional stabilisation of the helical conformation is essential. With this in mind, the following points should be addressed: (a) retaining as many of the original binding residues as possible to preserve the biological activity; (b) incorporating structurally constrained trans-ACPC residues to promote the formation of stable helical structures; and (c) further modifications with different natural amino acids to furnish the peptide sequence with additional contacts. Therefore, our strategy can be described as a three-step procedure (Figure 1). Initially, a helical fragment of a known protein that interacts with the RBD of the S protein is selected. Here, we used either human ACE2 or de novo designed LCB1 or LCB3 proteins. Subsequently, we incorporate constrained β-amino acid residues (i.e. trans-ACPC) to rigidify the three-dimensional structure of the chosen fragment. Because introduced β-amino acid residues should not disturb peptide interaction with the target, sequence patterns placing trans-ACPC on one side of the helix are applied – namely, ααβαααβ or ααααααβ. Finally, the obtained constructs are optimised iteratively towards optimal binding to the target using computer-aided methods.
Figure 1.

The strategy for the development of SARS-CoV-2 S protein-human ACE2 interaction inhibitors based on peptide foldamers.
In the first step, trans-ACPC residues were incorporated into the α1 helix of the ACE2 protein following two patterns: ααβαααβ (A1 and A2) and ααααααβ (A3) (Table S2). Cyclopentane-containing β-amino acid residues were introduced in place of seven predominantly nonpolar amino acid residues. In the case of applied sequence patterning, the heptad residue repeat gives helices in which β residues are aligned along one side of the helix, forming a “β-stripe” oriented towards the solvent (Figure S1)70. This one-sided orientation enables the binding residues to be closely allocated and therefore oriented towards the binding surface of the RBD protein. The circular dichroism (CD) spectrum of ACEα1 showed a Cotton effect at 200 nm, which is characteristic for unstructured peptides (Figure S2). However, peptides with ααβαααβ patterning (A1 and A2) as well as with ααααααβ patterning (A3) exhibited a different CD spectrum with a strong minimum at 204 nm, which indicates the formation of a helical structure.30
The binding affinity of the designed peptides to the RBD of the S protein was evaluated using biolayer interferometry (BLI) experiments. The BLI technique is based on the measurement of the interference pattern of white light reflected from a biolayer surface. In this case, biosensors coated with a protein A biolayer were used for the immobilisation of Fc-tagged RBD. Subsequently, the interaction of the corresponding peptides (analytes) with the RBD was monitored in real time by recording changes in light interference due to the association of the analyte. The initial screening was performed for 50 µM solutions of foldameric peptides (BLI sensograms for all tested peptides are given in the Supporting Information). First, the native ACEα1 helix was synthesised, and its binding affinity measurement revealed a very low response (small shift in nanometers) towards binding to the RBD protein. In continuation, all designed and synthesised foldamers derived from the ACEα1 helix (peptides A1, A2, and A3) as well as their fragments (decapeptides A1a-c, A2a-c, and A3a-c) and mutants (A1_v1-A1_v4 and A2_v1-A2_v4, Table S2) were tested using the BLI assay. The sensograms of several foldameric helices (A1v3, A2v2, A2v3) gave a noticeably higher response signal in comparison to the native helix (Figure S3). However, the signal corresponding to the reference sensor revealed the occurrence of non-specific binding of these peptides to the empty protein A sensor. Unfortunately, because of the very low response of the BLI signal, it was not possible to determine the dissociation constant values (Kd) for this group of foldameric helices.
Although it was possible to foldamerize the fragment of ACE2 that interacts with RBD by incorporation of trans-ACPC residues, and peptides with stable helical conformation were obtained, they did not provide a satisfactory improvement in the binding affinity towards RBD. Therefore, we implemented a different strategy based on the modification of the in silico-designed proteins LCB1 and LCB3, which are reported to exert high binding to the SARS-CoV-2 S protein.40 The LCB1 and LCB3 proteins are built from three helical fragments, and similar to the ACE2 protein, most of their binding interactions with the SARS-CoV-2 RBD originate from one of these helices. In the case of the LCB1 protein, most of the binding residues are located in the middle helix (sequence 1, Table 1), while LCB3 interacts with the RBD predominantly through the N-terminal helix (sequence 6, Table 1). Therefore, these fragments were selected for the further optimisation and design of potential foldameric SARS-CoV-2 inhibitors (peptides 1 and 6, Table 1). The first set of designed helical foldamers (peptides 2–5 and 7–10, Table 1) was obtained by incorporating four trans-ACPC residues into the ααβαααβ pattern by replacing nonpolar amino acid residues. Furthermore, tryptophan residues were introduced in sequences to provide hydrophobic interacting side chains, which could fit into the cavities formed on the surface of the binding domain (Figure 2). CD measurements revealed that peptides 1 and 6 were partially folded into α-helices, while a substantial increase in the conformational stability of the foldameric peptides 2–5 and 7–10 was observed in comparison to the original sequences (Figure 3), with a pronounced contribution of the helical structure.
Table 1.
Amino acid sequences of the first set of designed helical foldamers (peptides 2–5 and 7–10) and their affinities to the RBD of the S protein.

|
*Positions highlighted in yellow were mutated in comparison to peptides 1 and 6, respectively. Trans-(1S,2S)-2-aminocyclopentane-carboxylic acid residues are shown as black pentagons. n. a. – not active.
Figure 2.

Crystal structure of LCB3-RBD complex (A), PDB id 7JZM (residues 1–18 of LCB3 are shown), and the modelled foldameric peptide 10 bound to the RBD of S protein (B). Peptides are shown in stick representation; trans-ACPC residues’ carbon atoms are coloured green, tryptophan residues’ carbon atoms are coloured pink. The surface of the RBD is coloured according to interpolated charge: blue – positive, grey – neutral, red – negative. Key intermolecular interactions are shown as dashed lines: green – hydrogen bonds, orange – charge assisted hydrogen bonds, pink – hydrophobic interactions, white – hydrogen bond donor/π interactions.
Figure 3.

CD spectra of (A) peptides (2–5) derived from LCB1 helix (1) and (B) peptides (6–10) derived from LCB3 helix (6), dissolved in potassium phosphate buffer, 50 mM, pH 7.5.
The affinity of these peptides to bound RBD was also screened using the BLI method. Within the first group of foldamers, derived from LCB1 (peptides 2–5, Table 1), none of the peptides showed any improvement in the intensity of the response on the BLI sensogram, and it was not possible to perform a reliable calculation of the Kd value. Among the second set of foldamers, obtained by altering the LCB3 protein fragment (peptides 7–10, Table 1), derivative 10 gave a significantly stronger response than its native variant. To determine the Kd value with high accuracy, we performed BLI screening of different concentrations of peptide 10 (Figure 4), and a value of 82.5 µM was obtained after global fitting. Peptide foldamer 10, compared to the original sequence 6, possesses two tryptophan residues oriented towards two cavities on the binding RBD surface, contributing significantly to the binding affinity (Figure 2(B)).
Figure 4.

BLI-based screening of the binding affinity properties of peptide 10 towards RBD. Association and dissociation steps are given for different concentrations of peptide 10, ranging from 1 to 100 µM.
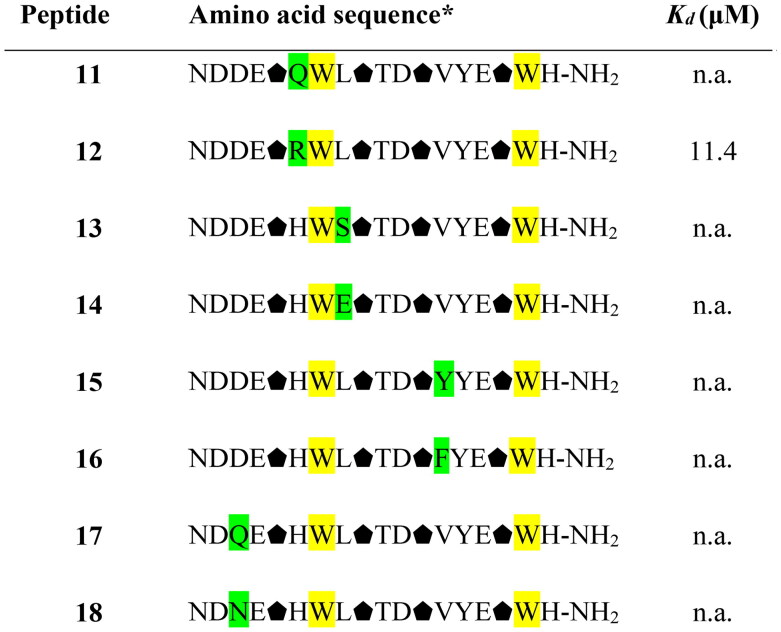
The consecutive optimisation of the foldameric structure was focused on further alteration of the peptide 10 sequence. Therefore, eight new variants (peptides 11–18, Table 2) were designed, synthesised, and tested for their binding affinity. Each of the new peptides contained one mutation in comparison to the parent peptide 10. The alterations for the first four peptides were made regarding the amino acids adjacent to the Trp7 residue by introducing polar and charged amino acids, which could contribute to the binding affinity of the foldamers (Figure 5). Mutations were also made by the introduction of voluminous hydrophobic residues (Tyr, Phe) or polar amino acids (Gln, Asn). The highest affinity with a Kd value of 11.4 µM (Figure 6) was obtained for peptide 12, which differs from the original sequence in one arginine residue located next to Trp7. The Arg6 side chain forms additional hydrogen bonds with the hydroxyl groups of Tyr453 and Ser494 located on the surface of the RBD protein (Figure 5).
Table 2.
Amino acid sequences of the second set of designed helical foldamers (11–18) derived from peptide 10 and their binding affinities to the RBD of the S protein.
|
*Positions highlighted in yellow and green indicate original mutations introduced to design peptide 10 and newly introduced mutations, respectively. Trans-(1S,2S)-2-aminocyclopentane-carboxylic acid residues are shown as black pentagons. n. a. – not active.
Figure 5.

The modelled complex of peptide 12 with the RDB of the S protein. Peptide 12 is shown in stick representation; trans-ACPC residues’ carbon atoms are coloured green, mutated residues’ carbon atoms are coloured pink. The surface of the RBD is coloured according to interpolated charge: blue – positive, grey – neutral, red – negative. Key intermolecular interactions are shown as dashed lines: green – hydrogen bonds, orange – charge assisted hydrogen bonds, pink – hydrophobic interactions, white – hydrogen bond donor/π interactions.
Figure 6.

BLI-based screening of the binding affinity properties of peptide 12 towards RBD. Association and dissociation steps are given for the different concentrations of peptide 12, ranging from 1 to 100 µM.
The third round of optimisation was based on mutations of peptide 12 (Table 3). The attempt to increase activity by introducing hydrophobic residues (peptides 19–21, Table 3) instead of the polar threonine residue resulted in a loss of binding affinity. A similar effect was obtained for foldamers in which Trp7 was replaced by different hydrophobic residues (peptides 22–24, Table 3), as well as for peptide with one additional Trp residue in the sequence (peptide 26, Table 3). Next, we examined the effect of introducing an additional arginine residue into the sequence by placing it at the C- or N-terminus (peptides 25 and 27, Table 3). Mutation of the N-terminus with Arg resulted in a significant increase in the binding activity of peptide 27, with a Kd value of 1.1 µM (Figure 7). Modelling studies revealed that N-terminal arginine forms additional interactions with the RBD through Gln498 and Thr500 residues (Figure 8). Among the other tested foldamers, peptides 29 and 32 containing homoglutamic acid also showed pronounced binding affinities with Kd values of 6.2 and 6.5 µM, respectively.
Table 3.
Amino acid sequences of the third set of designed helical foldamers (19–32) derived from peptide 12 and their binding affinities to the RBD of the S protein.

|
* Positions highlighted in yellow, green and cyan indicate mutations of peptide 10, peptide 12 and newly introduced mutations, respectively. Trans-(1S,2S)-2-aminocyclopentane-carboxylic acid residues are shown as black pentagons. n. a. – not active.
Figure 7.

BLI-based screening of the binding affinity properties of peptide 27 towards RBD. Association and dissociation steps are given for different concentrations of peptide 27, ranging from 0.25 to 10 µM.
Figure 8.

Modelled complex of peptide 27 with the RBD of the S protein. Peptide 27 is shown in stick representation; trans-ACPC residues’ carbon atoms are coloured green, mutated residues’ carbon atoms are coloured pink. The surface of the RBD is coloured according to interpolated charge: blue – positive, grey – neutral, red – negative. Key intermolecular interactions are shown as dashed lines: green – hydrogen bonds, orange – charge assisted hydrogen bonds, pink – hydrophobic interactions, white – hydrogen bond donor/π interactions.
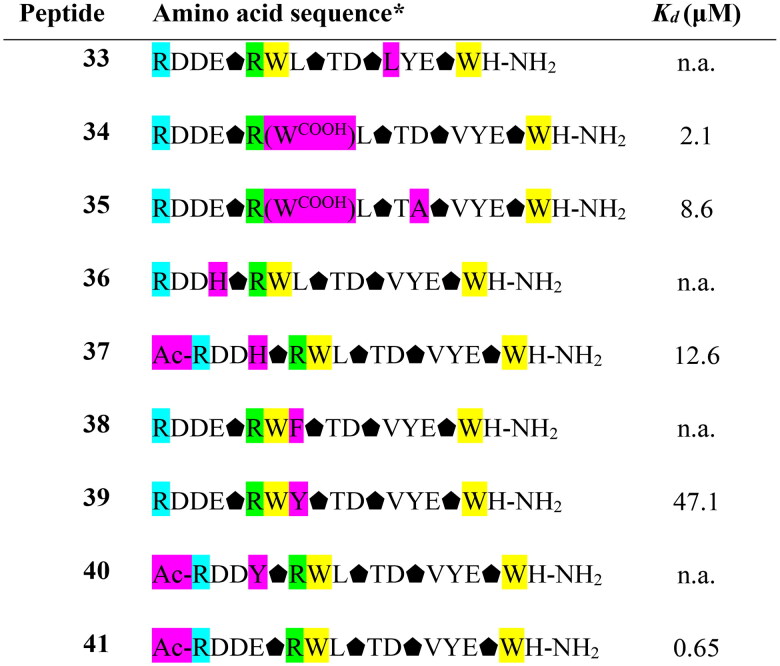
The last cycle of optimizations was focused on alterations of the sequence of peptide 27, which is the most active one. These alterations resulted in the synthesis of nine new foldamers (peptides 33–41, Table 4), which, according to their CD spectra (Figure 9), retained the helical structure of the initial sequences. Among these derivatives, peptide 34 containing a nonnatural amino acid, εN-carboxymethyltryptophan (WCOOH), instead of Trp showed good binding affinity with Kd = 2.1 µM. One of the peptides in this group, peptide 41, showed the highest activity thus far, with a Kd value of 0.65 µM (Figure 10).
Table 4.
Amino acid sequences of the fourth set of designed helical foldamers (33–41) derived from peptide 27 and their binding affinities to the RBD of the S protein.
|
*Positions highlighted in yellow, green, cyan and purple indicate mutations in peptide 10, peptide 12, and peptide 27 and newly introduced mutations, respectively. Trans-(1S,2S)-2-aminocyclopentane-carboxylic acid residues are shown as black pentagons. n. a. – not active. WCOOH – εN-carboxymethyltryptophan.
Figure 9.

CD spectra of peptides 33–41 dissolved in 50 mM potassium phosphate buffer, pH 7.5.
Figure 10.

BLI-based screening of the binding affinity properties of peptide 41 towards RBD. Association and dissociation steps are given for different concentrations of peptide 41, ranging from 0.25 to 10 µM.
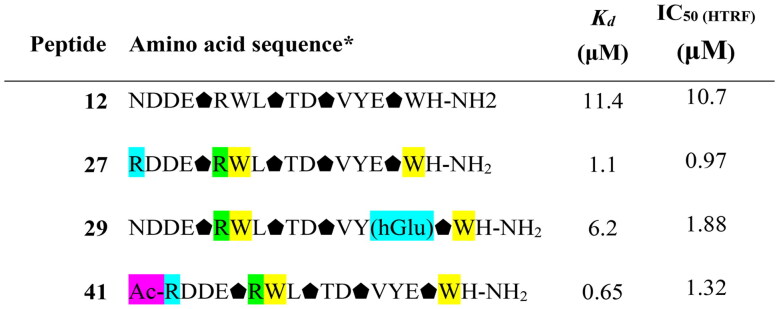
The high binding affinity of peptide 27 to the RBD protein is not explicit proof of its ability to inhibit the interaction of the SARS-CoV-2 S protein with human ACE2. To prove that the binding of peptide 27 is specific and inhibits the RBD/ACE2 interaction, we also performed an additional screening using a homogeneous time-resolved fluorescence (HTRF) assay. This technology was introduced more than two decades ago in the field of drug target studies and has been widely used ever since for competitive inhibition and specific binding investigations71. To our knowledge, the application of this method to inhibition studies of the RBD/ACE2 interaction has not yet been presented in the published literature. The assay is based on the interaction between human ACE2/ACEH protein, Fc Tag, and SARS-CoV-2 S protein RBD, His Tag. Each of the interacting partners is coupled with a fluorescent label, namely, PAb Anti Human IgG-Eu cryptate and MAb Anti 6HIS-XL665, respectively. The change in the level of energy transfer between the fluorophores involved is monitored at different concentrations of the examined foldameric helices (Figure 11). The IC50 values obtained from the HTRF assay (Table 5) are in good agreement with the Kd values. The best value was obtained for peptide 27, with a calculated IC50 value of 0.97 µM.
Figure 11.

HTRF assay evaluation of the S RBD/ACE2 inhibitory activity of selected peptides.
Table 5.
Comparison of Kd values obtained via BLI screening and IC50 values obtained from the HTRF assay for selected peptides with the most pronounced activity.
|
* amino acid residue coding and colouring is the same as in Table 4
Peptide 27 was further studied in detail using NMR spectroscopy. Based on the TOCSY and NOESY spectra, all resonances were unambiguously identified (Table S4), and numerous nonsequential interproton contacts were identified (Table S5). In particular, 24 medium range i-i + 3 contacts accompanied by i-i + 2 and i-i + 4 interactions were present, indicating that the peptide adopts a helical conformation (Figure 12A). The simulated annealing protocol with NMR-derived restraints provided a well-folded helical structure of peptide 27 (Figure 12B-C). In particular, the fragment between ACPC5 and ACPC16 is fully defined. Moreover, the observed conformation is consistent with the conformations resulting from inhibitor–protein molecular modelling studies, as presented above.
Figure 12.

Regular NOESY contacts between HA and HN atoms of nonadjacent residues of peptide 27 (A). Contacts between i - i + 2, i - i + 3 and i - i + 4 are shown in green, blue and pink, respectively. Averaged structure of peptide 27 calculated on the basis of NMR-derived restraints shown in stick representation (B) and superimposition of 10 lowest energy structures (C). Green dotted lines represent hydrogen bonds.
In summary, four rounds of optimisation of the peptide sequence were performed starting from the foldamerized version of the helical fragment of the LCB3 protein, peptide 7. In the first group, peptide 10 was found to be most active due to the presence of two Trp residues at positions 7 and 17, which anchored the peptide in two small clefts of the RDB of the S protein. Subsequently, it was indicated that replacing position 7 with an Arg residue can significantly improve the binding to the target by the formation of hydrogen bonds with Ser494 and Tyr453 (peptide 12). Further analogs were obtained screening positions 1, 4, 7, 10, 14, 15 and 18, but only the replacement of position 1 with Arg (interaction with Gln498) and the extension of Glu15 to hGlu15 provided peptides with binding properties, with the most active peptide 27 described by Kd = 1.1 µM. The fourth round of the trial did not provide significant improvement of the studied activity, although several peptides active in the micromolar range were found. The comparison of interaction energy calculated using the MMGBSA method (Table S8) for starting peptide 7 and subsequent optimizations (peptides 10, 12 and 27) clearly shows the increasing trend that was also observed experimentally. Moreover, molecular dynamics simulation confirmed the stability of the structure of the peptide 27–RBD complex (Figure S190). Interestingly, radius of gyration for 27-RBD complex was significantly lower than that observed for the complex of the starting peptide 1, what indicated its higher stability and is in agreement with obtained experimental results.
Conclusions
Analysis of the binding affinity of several dozen foldameric peptides in four rounds of optimisation led to the discovery of highly active molecules that bind to the RBD of the SARS-CoV-2 S protein and inhibit its interaction with human ACE2. The incorporation of conformationally restricted trans-ACPC units into the ααβαααβ pattern significantly rigidified the helical structure of the obtained peptides regardless of the introduced mutations. The results of CD and NMR measurements were consistent with the previously reported conformation of such peptides that was assumed in the molecular modelling of inhibitor-protein complexes. Due to rational control of conformational behaviour, peptide foldamers were found to be excellent scaffolds for the construction of inhibitors of S protein/ACE2 interactions, although the development of efficient peptide-based inhibitors of this PPI has previously proven challenging.
Supplementary Material
Funding Statement
The work was financially supported by the National Science Centre, Poland, Grant No. 2020/01/0/ST4/00064 (to Ł.B.). The authors would like to thank Dassault Systems for providing a free 6-month license for the BIOVIA Discovery Studio software package.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data will be made available on request.
References
- 1.Cheng RP, Gellman SH, DeGrado WF.. β-peptides: from structure to function. Chem Rev. 2001;101(10):3219–3232. [DOI] [PubMed] [Google Scholar]
- 2.Gellman SH. Foldamers: a manifesto. Acc Chem Res. 1998;31(4):173–180. [Google Scholar]
- 3.Goodman CM, Choi S, Shandler S, DeGrado WF.. Foldamers as versatile frameworks for the design and evolution of function. Nat Chem Biol. 2007;3(5):252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinek TA, Fülöp F.. Peptidic foldamers: ramping up diversity. Chem. Soc. Rev. 2012;41(2):687–702. [DOI] [PubMed] [Google Scholar]
- 5.Seebach D, Overhand M, Kühnle FNM, Martinoni B, Oberer L, Hommel U, Widmer H. β Peptides: synthesis by Arndt-Eistert homologation with concomitant peptide coupling. Structure determination by NMR and CD spectroscopy and by X-ray crystallography. Helical secondary structure of a β-hexapeptide in solution and its stability towards Pe. HCA. 1996;79(4):913–941. [Google Scholar]
- 6.Appella DH, Christianson LA, Klein DA, Powell DR, Huang X, Barchi JJ, Gellman SH.. Residue-based control of helix shape in β-peptide oligomers. Nature. 1997;387(6631):381–384. [DOI] [PubMed] [Google Scholar]
- 7.Appella DH, Christianson LA, Klein DA, Richards MR, Powell DR, Gellman SH.. Synthesis and structural characterization of helix-forming β-peptides: trans-2-aminocyclopentanecarboxylic acid oligomers. J Am Chem Soc. 1999;121(33):7574–7581. [Google Scholar]
- 8.Sharma GVM, Jayaprakash P, Narsimulu K, Ravi Sankar A, Ravinder Reddy K, Radha Krishna P, Kunwar AC.. A left-handed 9-helix in γ-peptides: synthesis and conformational studies of oligomers with dipeptide repeats of c-linked carbo-γ4-amino acids and γ-aminobutyric acid. Angew Chem Int Ed. 2006;45(18):2944–2947. [DOI] [PubMed] [Google Scholar]
- 9.Arndt H-D, Ziemer B, Koert U.. Folding propensity of cyclohexylether-δ-peptides. Org Lett. 2004;6(19):3269–3272. [DOI] [PubMed] [Google Scholar]
- 10.Pilsl LKA, Reiser O.. α/β-peptide foldamers: state of the art. Amino Acids. 2011;41(3):709–718. [DOI] [PubMed] [Google Scholar]
- 11.Fülöp F, Martinek TA, Tóth GK.. Application of alicyclic β-amino acids in peptide chemistry. Chem Soc Rev. 2006;35(4):323. [DOI] [PubMed] [Google Scholar]
- 12.De Pol S, Zorn C, Klein CD, Zerbe O, Reiser O.. Surprisingly stable helical conformations inα/β-peptides by incorporation ofcis-β-aminocyclopropane carboxylic acids. Angew Chem Int Ed. 2004;43(4):511–514. [DOI] [PubMed] [Google Scholar]
- 13.Torres E, Gorrea E, Burusco KK, Da Silva E, Nolis P, Rúa F, Boussert S, Díez-Pérez I, Dannenberg S, Izquierdo S, et al. Folding and self-assembling with β-oligomers based on (1R,2S)-2-aminocyclobutane-1-carboxylic acid. Org Biomol Chem. 2010;8(3):564–575. [DOI] [PubMed] [Google Scholar]
- 14.Shin S, Lee M, Guzei IA, Kang YK, Choi SH.. 12/10-helical β-peptide with dynamic folding propensity: coexistence of right- and left-handed helices in an enantiomeric foldamer. J Am Chem Soc. 2016;138(40):13390–13395. [DOI] [PubMed] [Google Scholar]
- 15.Berlicki Ł, Pilsl L, Wéber E, Mándity IM, Cabrele C, Martinek TA, Fülöp F, Reiser O.. Unique α,β- and α,α,β,β-peptide foldamers based on cis-β-aminocyclopentanecarboxylic acid. Angew Chem Int Ed. 2012;51(9):2208–2212. [DOI] [PubMed] [Google Scholar]
- 16.Choi SH, Guzei IA, Spencer LC, Gellman SH.. Crystallographic characterization of helical secondary structures in α/β-peptides with 1:1 residue alternation. J Am Chem Soc. 2008;130(20):6544–6550. [DOI] [PubMed] [Google Scholar]
- 17.Choi SH, Guzei IA, Spencer LC, Gellman SH.. Crystallographic characterization of helical secondary structures in 2:1 and 1:2 α/β-peptides. J Am Chem Soc. 2009;131(8):2917–2924. [DOI] [PubMed] [Google Scholar]
- 18.Mándity IM, Wéber E, Martinek TA, Olajos G, Tóth GK, Vass E, Fülöp F.. Design of peptidic foldamer helices: a stereochemical patterning approach. Angew Chem Int Ed. 2009;48(12):2171–2175. [DOI] [PubMed] [Google Scholar]
- 19.Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH.. Structural and Biological mimicry of protein surface recognition by α/β-peptide foldamers. Proc Natl Acad Sci USA. 2009;106(35):14751–14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szefczyk M, Węglarz-Tomczak E, Fortuna P, Krzysztoń A, Rudzińska-Szostak E, Berlicki Ł.. Controlling the helix handedness of Ααβ-peptide foldamers through sequence shifting. Angew Chem. 2017;129(8):2119–2123. [DOI] [PubMed] [Google Scholar]
- 21.Cabrele C, Martinek TA, Reiser O, Berlicki Ł.. Peptides containing β-amino acid patterns: challenges and successes in medicinal chemistry. J Med Chem. 2014;57(23):9718–9739. [DOI] [PubMed] [Google Scholar]
- 22.Johnson LM, Gellman SH.. α-helix mimicry with α/β-peptides. Methods Enzymol. 2013;523:407–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girvin ZC, Gellman SH.. Foldamer catalysis. J Am Chem Soc. 2020;142(41):17211–17223. [DOI] [PubMed] [Google Scholar]
- 24.Klein M. Stabilized helical peptides: overview of the technologies and its impact on drug discovery. Expert Opin Drug Discov. 2017;12(11):1117–1125. [DOI] [PubMed] [Google Scholar]
- 25.Gopalakrishnan R, Frolov AI, Knerr L, Drury WJ, Valeur E.. Therapeutic potential of foldamers: from chemical biology tools to drug candidates? J Med Chem. 2016;59(21):9599–9621. [DOI] [PubMed] [Google Scholar]
- 26.Oba M. Cell-penetrating peptide foldamers: drug-delivery tools. ChemBioChem. 2019;20(16):2041–2045. [DOI] [PubMed] [Google Scholar]
- 27.Arvidsson PI, Ryder NS, Weiss HM, Gross G, Kretz O, Woessner R, Seebach D.. Antibiotic and hemolytic activity of a B2/B3 peptide capable of folding into a 12/10-helical secondary structure. Chem Bio Chem. 2003;4(12):1345–1347. [DOI] [PubMed] [Google Scholar]
- 28.Schmitt MA, Weisblum B, Gellman SH.. Interplay among folding, sequence, and lipophilicity in the antibacterial and hemolytic activities of α/β-peptides. J Am Chem Soc. 2007;129 (2):417–428. [DOI] [PubMed] [Google Scholar]
- 29.Lee M-R, Raman N, Gellman SH, Lynn DM, Palecek SP.. Hydrophobicity and helicity regulate the antifungal activity of 14-helical β-peptides. ACS Chem Biol. 2014;9(7):1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boersma MD, Haase HS, Peterson-Kaufman KJ, Lee EF, Clarke OB, Colman PM, Smith BJ, Horne WS, Fairlie WD, Gellman SH.. Evaluation of diverse α/β-backbone patterns for functional α-helix mimicry: analogues of the Bim BH3 domain. J Am Chem Soc. 2012;134(1):315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wójcik P, Berlicki Ł.. Peptide-based inhibitors of protein-protein interactions. Bioorg Med Chem Lett. 2015;26(3):707–713. [DOI] [PubMed] [Google Scholar]
- 32.Stephens OM, Kim S, Welch BD, Hodsdon ME, Kay MS, Schepartz A.. Inhibiting HIV fusion with a beta-peptide foldamer. J Am Che. Soc. 2005;127(38):13126–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fortuna P, Twarda-Clapa A, Skalniak L, Ożga K, Holak TA, Berlicki Ł.. Systematic ‘Foldamerization’ of peptide inhibiting P53-MDM2/X interactions by the incorporation of trans- or cis-2-aminocyclopentanecarboxylic acid residues. Eur J Med Chem. 2020;208:112814. [DOI] [PubMed] [Google Scholar]
- 34.Checco JW, Gellman SH.. Iterative nonproteinogenic residue incorporation yields α/β-Peptides with a helix-loop-helix tertiary structure and high affinity for VEGF. Chem Bio Chem. 2017;18(3):291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horne WS. Peptide and peptoid foldamers in medicinal chemistry. Expert Opin Drug Discov. 2011;6(12):1247–1262. [DOI] [PubMed] [Google Scholar]
- 36.Bullock BN, Jochim AL, Arora PS.. Assessing helical protein interfaces for inhibitor design. J Am Chem Soc. 2011;133(36):14220–14223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q.. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu C, Wang Y, Liu C, Zhang C, Han W, Hong X, Wang Y, Hong Q, Wang S, Zhao Q, et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci Adv. 2021;7(1):eabe5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu L, Chen Q, Liu K, Wang J, Han P, Zhang Y, Hu Y, Meng Y, Pan X, Qiao C, et al. Broad Host range of SARS-CoV-2 and the molecular basis for SARS-CoV-2 binding to cat ACE2. Cell Discov. 2020;6(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panchal D, Kataria J, Patel K, Crowe K, Pai V, Azizogli A, Kadian N, Sanyal S, Roy A, Dodd‐O J, et al. Peptide‐based inhibitors for SARS‐CoV‐2 and SARS‐CoV. Adv Therap. 2021;4(10):2100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schütz D, Ruiz-Blanco YB, Münch J, Kirchhoff F, Sanchez-Garcia E, Müller JA.. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv Drug Deliv Rev. 2020;167:47–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bojadzic D, Alcazar O, Chen J, Chuang ST, Condor Capcha JM, Shehadeh LA, Buchwald P.. Small-molecule inhibitors of the coronavirus spike: ACE2 protein-protein interaction as blockers of viral attachment and entry for SARS-CoV-2. ACS Infect Dis. 2021;7(6):1519–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang D, Hamdoun S, Chen R, Yang L, Ip CK, Qu Y, Li R, Jiang H, Yang Z, Chung SK, et al. Identification of natural compounds as SARS-CoV-2 entry inhibitors by molecular docking-based virtual screening with bio-layer interferometry. Pharmacol Res. 2021;172:105820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carino A, Moraca F, Fiorillo B, Marchianò S, Sepe V, Biagioli M, Finamore C, Bozza S, Francisci D, Distrutti E, et al. Hijacking SARS-CoV-2/ACE2 receptor interaction by natural and semi-synthetic steroidal agents acting on functional pockets on the receptor binding domain. Front Chem. 2020;8:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pomplun S, Jbara M, Quartararo AJ, Zhang G, Brown JS, Lee YC, Ye X, Hanna S, Pentelute BL.. De novo discovery of high-affinity peptide binders for the SARS-CoV-2 spike protein. ACS Cent Sci. 2021;7(1):156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu L, Wang R, Wen T, Liu L, Wang T, Liu S, Xu H, Wang C.. Peptide binder with high-affinity for the SARS-CoV-2 spike receptor-binding domain. ACS Appl Mater Interfaces. 2022;14(25):28527–28536. [DOI] [PubMed] [Google Scholar]
- 48.Pei P, Qin H, Chen J, Wang F, He C, He S, Hong B, Liu K, Qiao R, Fan H, et al. Computational Design of ultrashort peptide inhibitors of the receptor-binding domain of the SARS-CoV-2 s protein. Brief Bioinform. 2021;22(6):bbab243. [DOI] [PubMed] [Google Scholar]
- 49.Rajpoot S, Solanki K, Kumar A, Zhang KYJ, Pullamsetti SS, Savai R, Faisal SM, Pan Q, Baig MS.. In-silico design of a novel tridecapeptide targeting spike protein of SARS-CoV-2 variants of concern. Int J Pept Res Ther. 2022;28(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bibilashvili RS, Sidorova MV, Dudkina US, Palkeeva ME, Molokoedov AS, Kozlovskaya LI, Egorov AM, Ishmukhametov AA, Parfyonova EV.. Peptide inhibitors of the interaction of the SARS-CoV-2 receptor-binding domain with the ACE2 cell receptor. Biochem Moscow Suppl Ser B. 2021;15(4):274–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kalita P, Tripathi T, Padhi AK.. Computational protein design for COVID-19 research and emerging therapeutics. ACS Cent Sci. 2023;9(4):602–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan DC, Morris C, Mahindra A, Blair CM, Tejeda G, Herbert I, Turnbull ML, Lieber G, Willett BJ, Logan N, et al. Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 spike protein do not prevent virus internalization. Pept Sci. 2021;113(4):e24217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Padhi AK, Seal A, Khan JM, Ahamed M, Tripathi T.. Unraveling the mechanism of arbidol binding and inhibition of SARS-CoV-2: insights from atomistic simulations. Eur J Pharmacol. 2021;894:173836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miki T, Namii K, Seko K, Kakehi S, Moro G, Mihara H.. Pattern enrichment analysis for phage selection of stapled peptide ligands. Chem Sci. 2022;13(43):12634–12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Norman A, Franck C, Christie M, Hawkins PME, Patel K, Ashhurst AS, Aggarwal A, Low JKK, Siddiquee R, Ashley CL, et al. Discovery of cyclic peptide ligands to the SARS-COV-2 spike protein using mrna display. ACS Cent Sci. 2021;7(6):1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hampton JT, Lalonde TJ, Tharp JM, Kurra Y, Alugubelli YR, Roundy CM, Hamer GL, Xu S, Liu WR.. Novel regioselective approach to cyclize phage-displayed peptides in combination with epitope-directed selection to identify a potent neutralizing macrocyclic peptide for SARS-CoV-2. ACS Chem Biol. 2022;17(10):2911–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kruse M, Altattan B, Laux EM, Grasse N, Heinig L, Möser C, Smith DM, Hölzel R.. Characterization of binding interactions of SARS-CoV-2 spike protein and DNA-peptide nanostructures. Sci Rep. 2022;12(1):12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pramanik A, Mayer J, Sinha SS, Sharma PC, Patibandla S, Gao Y, Corby LR, Bates JT, Bierdeman MA, Tandon R, et al. Human ACE2 peptide-attached plasmonic-magnetic heterostructure for magnetic separation, surface enhanced raman spectroscopy identification, and inhibition of different variants of SARS-CoV-2 infections. ACS Appl Bio Mater. 2022;5(9):4454–4464. [DOI] [PubMed] [Google Scholar]
- 59.Pramanik A, Sharma PC, Patibandla S, Gao Y, Ruppa-Kasani V, Goli J, Kumar A, Chatterjee A, Sinha SS, Bates JT, et al. Blocking SARS-CoV-2 delta variant (B.1.617.2) spike protein receptor-binding domain binding with the ACE2 receptor of the host cell and inhibiting virus infections using human host defense peptide-conjugated graphene quantum dots. ACS Omega. 2022;7(9):8150–8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labriola JM, Miersch S, Chen G, Chen C, Pavlenco A, Saberianfar R, Caccuri F, Zani A, Sharma N, Feng A, et al. Peptide–antibody fusions engineered by phage display exhibit an ultrapotent and broad neutralization of SARS-CoV-2 variants. ACS Chem Biol. 2022;17(7):1978–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mesias V, St D, Zhu H, Tang X, Dai X, Guo Y, Liu W, Huang J.. Effective ACE2 peptide–nanoparticle conjugation and its binding with the SARS-Cov-2 RBD quantified by dynamic light scattering. Chem Commun. 2021;57(57):6979–6982. [DOI] [PubMed] [Google Scholar]
- 62.Cao L, Goreshnik I, Coventry B, Case JB, Miller L, Kozodoy L, Chen RE, Carter L, Walls AC, Park Y-J, et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370(6515):426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goddard TD, K DG.. Sparky, 3rd ed.; University of California: San Francisco, CA, USA, 2001. [Google Scholar]
- 64.Schwieters CD, Kuszewski JJ, Marius Clore G.. Using Xplor-NIH for NMR molecular structure determination. Prog Nucl Magn Reson Spectrosc. 2006;48(1):47–62. [Google Scholar]
- 65.Martinek TA, Tóth GK, Vass E, Hollósi M, Fülöp F.. Cis-2-aminocyclopentanecarboxylic acid oligomers adopt a sheetlike structure: switch from helix to nonpolar strand. Angew Chem Int Ed. 2002;41(10):1718–1721. [DOI] [PubMed] [Google Scholar]
- 66.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q.. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li F, Li W, Farzan M, Harrison SC.. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. [DOI] [PubMed] [Google Scholar]
- 68.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. [DOI] [PubMed] [Google Scholar]
- 69.Benton DJ, Wrobel AG, Xu P, Roustan C, Martin SR, Rosenthal PB, Skehel JJ, Gamblin SJ.. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature. 2020;588(7837):327–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horne WS, Price JL, Gellman SH.. Interplay among side chain sequence, backbone composition, and residue rigidification in polypeptide folding and assembly. Proc Natl Acad Sci USA. 2008;105(27):9151–9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Degorce F. HTRF: a technology tailored for drug discovery – a review of theoretical aspects and recent applications. TOCHGENJ. 2009;3(1):22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.