

Abstract
Heterozygous disease-causing variants in BCL11B are the basis of a rare neurodevelopmental syndrome with craniofacial and immunological involvement. Isolated craniosynostosis, without systemic or immunological findings, has been reported in one of the 17 individuals reported with this disorder till date. We report three additional individuals harbouring de novo heterozygous frameshift variants, all lying in the exon 4 of BCL11B. All three individuals presented with the common findings of this disorder i.e. developmental delay, recurrent infections with immunologic abnormalities and facial dysmorphism. Notably, craniosynostosis of variable degree was seen in all three individuals. We, thus add to the evolving genotypes and phenotypes of BCL11B-related BAFopathy and also review the clinical, genomic spectrum along with the underlying disease mechanisms of this disorder.
Keywords: BCL11B, transcription factor, BAFopathy, neurodevelopment, immunodeficiency, craniosynostosis
Introduction
BCL11B (BAF chromatin remodelling complex subunit) encodes a zinc finger transcription factor involved in hematopoietic progenitor cell differentiation as well as development of nervous, cutaneous and craniofacial tissues. Disease-associations for germline BCL11B variants have been reported in 17 individuals (Baxter et al., 2022; Goos et al., 2019; Lessel et al., 2018; Punwani et al., 2016; Qiao et al., 2019). The most penetrant phenotypes for this cohort are immunodeficiency, neurologic deficits and facial anomalies (Table 1). Clinical phenotypes observed less frequently include refractive errors, dental anomalies, feeding difficulties, congenital heart defects and most recently isolated coronal suture craniosynostosis (Goos et al., 2019).
Table 1.
Phenotypes reported with germline variants associated with BCL11B in all the reported individuals including the families in this report
| Phenotype | Number of individuals affected (total number of individuals: 20) |
|---|---|
| Microcephaly | 3/20 (not determined in others) |
| Intellectual disability | 15/20 |
| Speech impairment | 15/20 |
| Motor delay | 16/20 |
| Seizures | 2/20 |
| Autism | 5/20 |
| Tone abnormalities | 2/20 |
| Brain anomalies | 2/20 |
| Heart defects | 1/20 |
| Immunodeficiency | 4/20 |
| Recurrent infections | 12/20 |
| Skin manifestations | 4/20 |
| Craniosynostosis | 3/20 |
| Dental anomalies | 6/20 |
| Feeding difficulties | 5/20 |
| Refractive errors | 6/20 |
| Hirsutism | 2/20 |
| Umbilical hernia | 2/20 |
| Gastrointestinal defects | 1/20 |
We identified three additional individuals with BCL11B-related BAFopathy presenting with neurodevelopmental and immunological features as well as craniosynostosis. This report provides further evidence for craniosynostosis as a significant clinical characteristic in the phenotypic spectrum of this newly described disorder. We also discuss the various disease mechanisms proposed for this gene in the literature.
Clinical history
Proband 1
We ascertained a ten months-old-female born of a consanguineous marriage. There was a history of hypertension during pregnancy in the mother. Proband was born at term via lower segment caesarean section with a birth weight of 3 kg (−0.6SD). She cried immediately at birth and postnatal period was uneventful. She achieved neck holding by three and half months of life, social smile by four months, cooing and babbling by three months, rolling over by six months, sitting with support by seven and half months. At the time of examination at ten months of age, she was able to stand with support and walk with support and she did not have any stranger anxiety. There was history of recurrent lower respiratory tract infections necessitating five hospital admissions in her.
At ten months of age, her length was 65 cm (−2.88 SD), head circumference was 36 cm (−8.52 SD) and weight was 5.87 kg (−4.8 SD). On clinical examination, she had an abnormal head shape with brachycephaly, prominent metopic suture and mild proptosis suggestive of craniosynostosis (Figure 1A.i-ii). She also had microcephaly, mid-face retrusion, high arched palate, hirsutism, hypodontia, umbilical hernia and mild hepatomegaly. Her tone, reflexes and sensory system examination findings were normal with no signs of peripheral neuropathy. Magnetic resonance imaging of the brain showed cephalic index of 86% without any structural abnormalities. Bilateral optic disc edema was noted on ophthalmologic evaluation. At 12 months of age, she was noted to have iron deficiency anemia and another episode of lower respiratory tract infection with high-grade fever. She succumbed during this illness.
Figure 1.
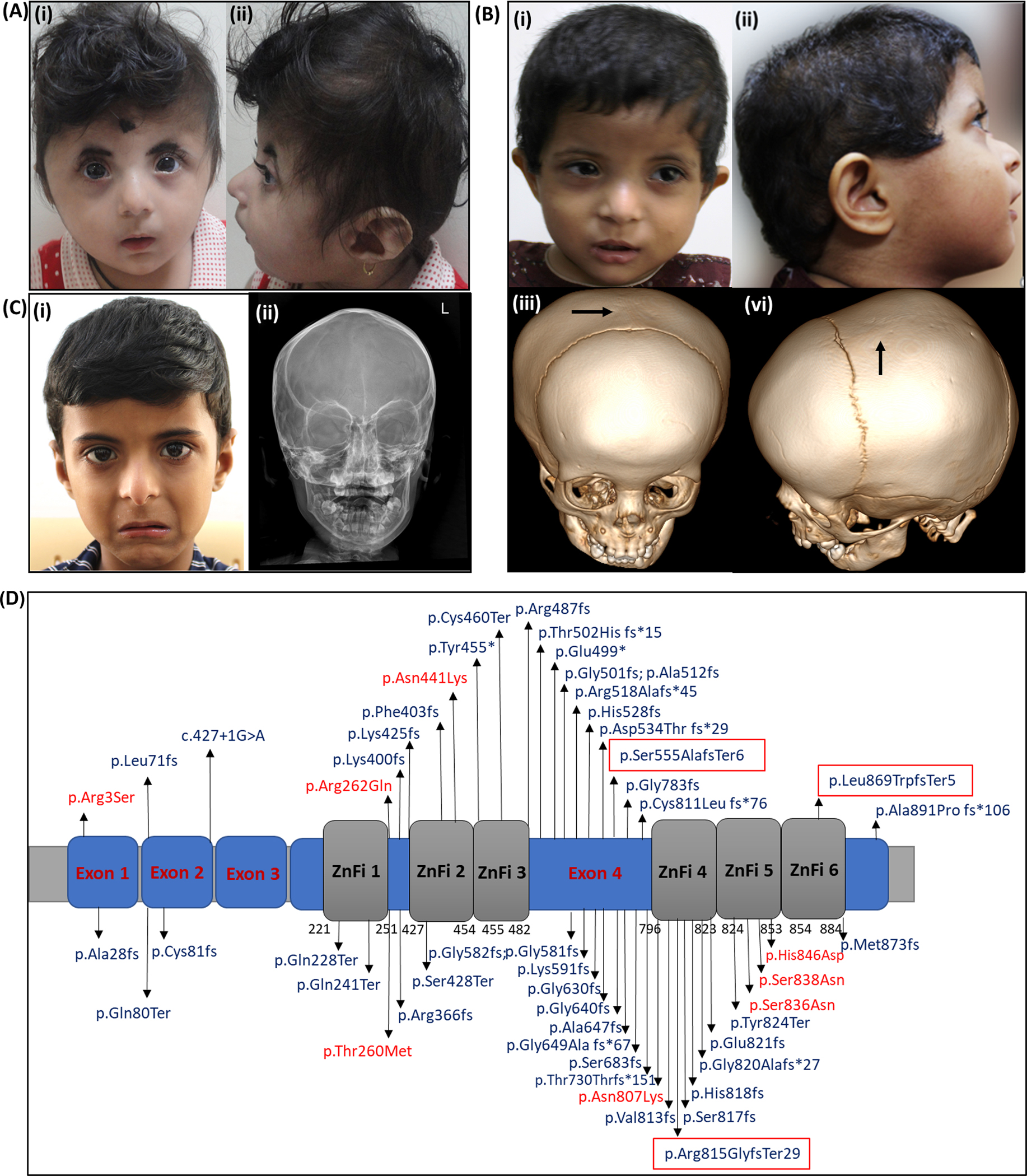
(A) Proband 1 with (i) facial dysmorphism and (ii) brachycephaly (B) Proband 2 with (i) facial dysmorphism (ii) scaphocephaly and dysmorphism (iii) and (iv) showing sagittal suture craniosynostosis on computed tomography of head (C) Proband 3 depicting (i) facial dysmorphic features and (ii) X ray skull showing sagittal suture craniosynostosis. (D) Schematic representation of structure of BCL11B highlighting current and reported exonic variants. Missense variants are highlighted in red, truncating variants in blue and the variants within the red box are present in families from the current study.
Antenatal scan in her mother at 22 weeks of gestation during second pregnancy (III.1) showed nuchal edema, echogenic intra-cardiac foci and echogenic bowel. Earlier, carrier testing was performed in parents in view of three pregnancy losses. Both are carriers for a known stopgain variant, c.1867C>T p.(Arg623Ter) in exon 12 of GUSB (NM_000181.4). The proband also carried this variant in heterozygous state.
The detailed methodology is given in supplementary information. On trio exome sequencing in the family, a de novo heterozygous variant, c.1662_1668del p.(Ser555AlafsTer6) in exon 4 of BCL11B (NM_138576.4) was identified in her (ClinVar ID: VCV001342021.2). The variant was validated using Sanger sequencing in the family (Supplementary figure 1).
In addition to this, two other de novo heterozygous likely pathogenic variants were identified in her i.e. c.881G>A p.(Arg294Gln) in exon 10 of KRIT1 (NM_194454.3) and c.328C>Tp.(Arg110Cys) in exon 6 of MORC2 (NM_001303256.3). We validated the de novo status of the MORC2 variant by Sanger sequencing (Supplementary figure 1).
Proband 2
Proband 2 is a one-year-one-month old female born to a non-consanguineously married couple. She was born at term via normal vaginal delivery with a birth weight of 3.35 kg (+0.42 SD). There were no significant postnatal events. She attained social smile by four months, neck holding by five months of age, roll over by eight months, sitting with support by nine months and independent sitting by ten months. She could babble by four months and could speak monosyllables by eight months. She developed stranger anxiety by ten months of age. There is history of multiple hospital admissions in view of cough, fever and breathing problems. At three months of age, she had an episode of intestinal intussusception.
At one-year-one-month of age, her weight was 7.7 kg (−1.3 SD), length was 76 cm (0.19 SD) and head circumference was 43 cm (−1.94 SD). She had low posterior hairline, scaphocephaly, tall forehead, small and down-slanting palpebral fissures, telecanthus, depressed nasal root with convex nasal bridge, low set and anteverted external ears, small earlobes, long and smooth philtrum, thin upper vermilion and a small mouth (Figure 1B. i-ii). On central nervous system examination, her tone and reflexes were normal. Rest of the systemic examination was unremarkable.
Her haemoglobin levels were 8.6 g/dL (reference levels: 11.1–14.2g/dL) and peripheral smear showed microcytic hypochromic anemia. Her total leukocyte count was 14.7×103/μl (Reference range: 9.0–16.0 ×103)/ μl. Her neutrophil count was 28% (42–74%) and lymphocyte count was 54.5% (18–44%) while absolute lymphocyte count was 12.10 × 103 μl (3.5–11) and absolute eosinophil count was 1.31 ×103 (01.−1.0). On computed tomography of head, sagittal suture craniosynostosis was noted (Figure 1B. iii-vi). Magnetic resonance imaging of the brain was unremarkable.
Trio exome sequencing in the family revealed a de novo heterozygous pathogenic variant, c.2605del p.(Leu869TrpfsTer5) in exon 4 of BCL11B (NM_138576.4) (ClinVar ID: SCV002757784.1). The variant was validated by Sanger sequencing in the family (Supplementary figure 2).
Proband 3
Proband 3 is a five-years-old male born to a non-consanguineous married couple. Antenatal scans at fifth month of gestation showed a ventricular cyst in the brain, which resolved with subsequent scans. At seven months of gestation, his mother had papular rashes over her body. The proband was born at term via lower segment caesarean section in view of non-progression of labour and cord around the neck with a birth weight of 3.2 kg (mean). He cried immediately after birth and postnatal period was uneventful. He achieved social smile by five months, sitting with support by eight months, crawling by one-year, independent standing by one year six months, walking with support by two years and independent walking by two years six months. He was able to speak monosyllables by two years of age. Currently, at five years of age, he runs and climbs stairs. He is not yet toilet trained. He also has poor eye contact and no peer play.
At five years of age, his height was 108 cm (−0.5 SD), head circumference was 47 cm (−2.3 SD), and weight was 14.5 kg (−2 SD). He had scaphocephaly, tall forehead, low set ears with small ear lobes, high bridge of nose with overhanging columella, long philtrum, thin upper and lower vermillion borders and downturned corners of mouth (Figure 1C.i).
On central nervous system examination, his tone and reflexes were normal. On hematologic workup, his haemoglobin levels are 9 g/dL (Reference range: 11–13) with normochromic normocytic anaemia. His total white blood cell count was 4 × 103/μL (Reference range: 5–15). On immunologic work up, his serum immunoglobulin levels (IgG, IgA, IgM) were in the normal range except immunoglobulin E which was elevated [IgE > 2500 IU (Reference range: < 60 IU/mL)]. On magnetic resonance imaging of brain, non-specific white matter hyperintensities in bilateral frontoparietal area were noted on T2/FLAIR sequence. A Vineland Social Maturity Scale showed a social quotient of 87. On X-ray skull, antero-posterior and lateral view showed premature fusion of sagittal suture suggestive of sagittal suture craniosynostosis in him (Figure 1C. ii).
Chromosomal microarray did not reveal any clinically significant copy number variants. On singleton exome sequencing, a heterozygous frameshift deletion, c.2443del p.(Arg815GlyfsTer29) in exon 4 of BCL11B (NM_138576.4) was identified (ClinVar ID: VCV001804880.1). Sanger sequencing confirmed the de novo status of the variant in the family (Supplementary figure 3).
Discussion
BAF chromatin remodelling complex is composed of multi-component entities that govern nuclear architecture by altering nucleosome positioning and functioning (Mashtalir et al., 2020). BAF complex is composed of several proteins including BCL11A, BCL11B, SMARC1, BRD9, AIRD2. These subunits contain DNA and histone binding domains and thereby have a role in specific transcription factor recruitment, genome targeting, protein-protein and DNA-protein interactions, and thus an ability to modulate gene expression in a cell lineage restricted manner (Sokpor, Xie, Rosenbusch, & Tuoc, 2017). The disorders associated with genomic alterations in these genes include variable presentation of syndromic and/or non-syndromic intellectual disability (ID), growth delay, autism, ectodermal defects, and skeletal anomalies (Machol et al., 2019). BCL11B, an important component of BAF complex, is highly conserved and encodes for a Cys2His2 zinc finger transcription factor. (Lessel et al., 2018; Punwani et al., 2016) It is located at 14q32.3 and has four exons. BCL11B is composed of 894 amino acids with all the six zinc finger domains encompassing the exon 4 (Figure 1D). Of these, the second and the third zinc finger domains are known to mediate DNA binding. BCL11B has a role in both transcriptional activation and repression. BCL11B regulates expression of several downstream genes and thereby plays an essential role in physiological developmental processes (Kominami, 2012). It is also known to be involved in the proliferation, migration, and differentiation of neural stem cells, neurons, and granule cells (Lessel et al., 2018; Yang et al., 2020). The role of BCL11B in regulating early thymocyte development is also well studied (Avram & Califano, 2014).
Heterozygous variants in BCL11B are associated with a diverse phenotypic spectrum of neurodevelopmental defects, craniofacial defects and immunodeficiency (Table 1, Supplementary information). The first family with a germline variant in BCL11B was reported by Punwani et al., in a male infant with severe developmental delay, absence of corpus callosum, facial dysmorphism and milder form of severe combined immunodeficiency (SCID). Exome sequencing revealed a de novo missense variant, c.1323T>G: p.(Asn441Lys) in exon 4 of BCL11B. This variant is located in the second zinc finger DNA binding domain. Another report by Lessel et al., in 2018, described 12 individuals with genomic alterations in BCL11B. Of these, one was a missense variant, nine were truncating variants and two de novo balanced chromosomal translocation. All reported individuals had intellectual disability, developmental delay and impairment of T‐cell development. The immune function was evaluated by immune compartment analysis in eight of 12 individuals due to known role of BCL11B in immune function and was found to be impaired. However, none of the affected individuals exhibited clinical signs of immune deficiency, craniosynostosis or any craniofacial deformities (Lessel et al., 2018). Here, the authors proposed haploinsufficiency as the disease mechanism underlying the truncating variants. Notably, the phenotype of the individual with the missense variant in this cohort was reported to be more severe than other heterozygous loss-of-function variants. In a recent report by Qiao et al., the proband presented with neurodevelopmental defects and abnormal white matter changes on magnetic resonance imaging of the brain. On exome sequencing, a de novo frameshift variant, c.2190_2200del p.(Thr730Thrfs*151) in exon 4 of BCL11B was identified (Qiao et al., 2019). In a case series on autoimmune diseases by Baxter et al., two individuals with missense variants in BCL11B were reported. Affected individual, S025 presented with failure to thrive, nutritional deficiencies, chronic emesis, pyloric stenosis and focal villous blunting of duodenum. On exome sequencing analysis, a missense variant, c.779C>T p.(Thr260Met) in exon 4 of BCL11B is identified. The second affected individual (S029) from this cohort had severe dermatitis and harboured a heterozygous missense variant, c.2421C>G p.(Asn807Lys) in exon 4 of BCL11B was identified in de novo state (Baxter et al., 2022).
In contrast to the multisystem abnormalities reported in most individuals with this disorder, Goos et al. reported a male proband with isolated coronal suture craniosynostosis. On genome sequencing, a de novo heterozygous variant, c.7C>A; pArg3Ser in exon 1 of BCL11B was identified. There was no history of immunodeficiency, recurrent infections or neurodevelopmental defects. The authors demonstrated isolated craniosynostosis as one of the clinical presentations of BCL11B associated disorder with haploinsufficiency as the mechanism of the disease (Goos et al., 2019).
Interestingly, dominant negative, gain of function and haploinsufficiency have been proposed as disease mechanisms for BCL11B related disorder. We queried all the disease-causing variants reported in the literature and the pathogenic/likely pathogenic variants from ClinVar to review the current understanding of disease mechanisms and also attempt genotype-phenotype correlation. Currently, there are 53 disease-causing variants including single nucleotide variants, indels and two balanced translocations for BCL11B-related BAFopathy (Figure 1D). The detailed phenotype of individuals is available only for published reports. The phenotypic entries associated with variants in BCL11B in ClinVar are variable and includes immunodeficiency 49 (OMIM# 617237), intellectual developmental disorder with dysmorphic facies, speech delay, and T-cell abnormalities (OMIM# 618092), inborn genetic diseases and BCL11B-related BAFopathy.
Haploinsufficiency has been proposed as the disease mechanism for truncating variants in BCL11B. Overall, 43/53 variants in BCL11B are truncating (81%). Of note, 38/43 (88%) of these variants are located in the last exon i.e exon 4 and hence predicted to escape nonsense mediated decay and result in a truncated protein with variable number of intact DNA binding domains. However, no clear genotype-phenotype correlation is seen within individuals with truncating variants at different locations in the transcript. Two individuals who harboured balanced translocations and with phenotypes comparable to the truncating variants were shown to have decreased levels of BCL11B mRNA which also favours haploinsufficiency as the disease mechanism (Lessel et al., 2018).
A total of eight missense variants in nine affected individuals are reported as disease causing in literature and/or ClinVar. Punwani et al. demonstrated that the p.(Asn441Lys) mutation results in impaired BCL11B binding to known target DNA sites and in addition also promotes binding to novel DNA binding sites with a dominant negative mechanism of disease (Punwani et al., 2016). However, haploinsufficiency has been proposed as the disease mechanism for the missense variant, c.7C>A in exon 1, which is reported in the individual with isolated craniosynostosis (Goos et al., 2019). The missense variant, p.(Asn807Lys) reported by Lessel et al. is predicted to have a possible gain of function mechanism (Lessel et al., 2018). The p.Thr260Met, p.Arg262Gln, p.Ser836Asn, p.Ser838Asn, p.His846Asp substitutions in BCL11B have not yet been evaluated for the underlying disease mechanism.
The truncating variants have largely been proposed to result in impairment of interaction with known target binding sites of BCL11B while the severe phenotype due to missense variants has been attributed to disruption of known binding sites as well as creation of novel DNA binding sites for BCL11B. It appears that the phenotypic variability in BCL11B related disorder may arise due to a combination of disease mechanisms ranging from haploinsufficiency as seen in individuals with reduced expression to those with dominant negative impact where a truncated protein might disrupt dimerization with normal protein or a missense change conferring novel DNA binding sites to the altered protein, all resulting in differential perturbation of the functioning and binding of the downstream genes.
Among the three affected individuals from the present study, coronal suture craniosynostosis was assessed clinically in proband 1 while in proband 2 and proband 3, sagittal suture craniosynostosis was noted on computed tomography of head and skull radiographs respectively besides the characteristic findings of neurodevelopmental delay, anaemia and recurrent infections. Also, all three of them carried de novo heterozygous truncating variants in exon 4 of BCL11B. All the three variants are rare and not reported in heterozygous state in population database, gnomAD and our in-house data of 2579 individuals. With this, all the variants are classified as pathogenic according to the American College of Medical Genetics and Genomics guidelines.
Proband 1 in the current study also harboured de novo heterozygous variants in MORC2 and KRIT1. Monoallelic variants in MORC2 are associated with two overlapping phenotypes of Charcot-Marie-Tooth disease, axonal, type 2Z, (OMIM# 616688) and developmental delay, impaired growth, dysmorphic facies, and axonal neuropathy (OMIM# 619090). Proband 1 had developmental delay which overlaps with the phenotypes associated with both BCL11B as well as MORC2-related disorder. Significant growth failure on the other hand can be possibly attributed to variants in MORC2. However, no overt signs of neuropathy were observed in her. Monoallelic variants in KRIT1 are associated with cavernous malformations of central nervous system and retina, (OMIM# 116860), cerebral cavernous malformations-1, (OMIM# 116860), hyperkeratotic cutaneous capillary-venous malformations associated with cerebral capillary malformations, (OMIM# 116860). The age of onset of KRIT1 associated disorders is around second to third decade of life. At the age of eleven months of age, there were no signs of any vascular malformation on clinical evaluation or in the magnetic resonance imaging of brain.
To conclude, this report presents three additional cases of BCL11B-related BAFopathy manifesting a combination of clinical findings not reported earlier. This disorder should be considered as a clinical possibility in individuals presenting with findings of anaemia, recurrent infections/immunodeficiency, neurodevelopmental delay and craniosynostosis. Further reports of affected individuals with pathogenic variants in BCL11B will help to establish the phenotypic spectrum and also provide deeper insight into the underlying patho-mechanisms.
Supplementary Material
Supplementary figure 3. Pedigree of family 3. Sanger chromatograms depicting a de novo heterozygous variant, c.2443del p.(Arg815GlyfsTer29) in exon 4 of BCL11B (NM_138576.4) in proband, III.1 with a wild type phenotype in his parents, II.2 and II.1.
Supplementary figure 1. A. Pedigree of family 1. B. Sanger chromatograms depicting the de novo heterozygous variant, c.1662_1668 in exon 4 of BCL11B (NM_138576.3) in the proband (IV.1) and wild type genotypes in parents (II.3 and III.1) C. Sanger chromatograms depicting the de novo heterozygous variant, c.328C>T in exon 6 of MORC2 (NM_001303256.3) in the proband 1 (IV.1) and wild type genotypes in parents (II.3 and III.1).
Supplementary figure 2. Pedigree of family 2. Sanger chromatograms depicting a de novo heterozygous variant, c.2605del p.(Leu869TrpfsTer5) in exon 4 of BCL11B (NM_138576.4) in proband, II.2 with wild type genotype in her parents, I.1 and I.2 and in elder sibling, II.1.
Acknowledgement
We would like to acknowledge the families for their consent and participation in the study. We would also like to acknowledge and thank the National Institutes of Health, United States for funding the study, “Genetic Diagnosis of Neurodevelopmental Disorders in India” (1R01HD093570-01A1). This grant is awarded to AS, KMG and SB. We also thank the Indian council of Medical Research for awarding the Nurturing Clinical Scientist Fellowship (HRD/NCS-2019-03) to SP.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Data availability statement
The data providing the evidence of the study is available from the corresponding author upon reasonable request.
References
- Avram D, & Califano D (2014). The multifaceted roles of Bcl11b in thymic and peripheral T cells: impact on immune diseases. J Immunol, 193(5), 2059–2065. doi: 10.4049/jimmunol.1400930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter SK, Walsh T, Casadei S, Eckert MM, Allenspach EJ, Hagin D, . . . King MC. (2022). Molecular diagnosis of childhood immune dysregulation, polyendocrinopathy, and enteropathy, and implications for clinical management. J Allergy Clin Immunol, 149(1), 327–339. doi: 10.1016/j.jaci.2021.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goos JAC, Vogel WK, Mlcochova H, Millard CJ, Esfandiari E, Selman WH, . . . Twigg SRF. (2019). A de novo substitution in BCL11B leads to loss of interaction with transcriptional complexes and craniosynostosis. Hum Mol Genet, 28(15), 2501–2513. doi: 10.1093/hmg/ddz072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominami R (2012). Role of the transcription factor Bcl11b in development and lymphomagenesis. Proc Jpn Acad Ser B Phys Biol Sci, 88(3), 72–87. doi: 10.2183/pjab.88.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Gehbauer C, Bramswig NC, Schluth-Bolard C, Venkataramanappa S, van Gassen KLI, . . . Kubisch C. (2018). BCL11B mutations in patients affected by a neurodevelopmental disorder with reduced type 2 innate lymphoid cells. Brain, 141(8), 2299–2311. doi: 10.1093/brain/awy173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machol K, Rousseau J, Ehresmann S, Garcia T, Nguyen TTM, Spillmann RC, . . . Campeau PM. (2019). Expanding the Spectrum of BAF-Related Disorders: De Novo Variants in SMARCC2 Cause a Syndrome with Intellectual Disability and Developmental Delay. Am J Hum Genet, 104(1), 164–178. doi: 10.1016/j.ajhg.2018.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashtalir N, Suzuki H, Farrell DP, Sankar A, Luo J, Filipovski M, . . . Kadoch C. (2020). A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell, 183(3), 802–817.e824. doi: 10.1016/j.cell.2020.09.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A, . . . Puck JM. (2016). Multisystem Anomalies in Severe Combined Immunodeficiency with Mutant BCL11B. N Engl J Med, 375(22), 2165–2176. doi: 10.1056/NEJMoa1509164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao F, Wang C, Luo C, Wang Y, Shao B, Tan J, . . . Xu Z. (2019). A De Novo heterozygous frameshift mutation identified in BCL11B causes neurodevelopmental disorder by whole exome sequencing. Mol Genet Genomic Med, 7(9), e897. doi: 10.1002/mgg3.897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokpor G, Xie Y, Rosenbusch J, & Tuoc T (2017). Chromatin Remodeling BAF (SWI/SNF) Complexes in Neural Development and Disorders. Front Mol Neurosci, 10, 243. doi: 10.3389/fnmol.2017.00243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Kang Q, Hou Y, Wang L, Li L, Liu S, . . . Xiao Z. (2020). Mutant BCL11B in a Patient With a Neurodevelopmental Disorder and T-Cell Abnormalities. Front Pediatr, 8, 544894. doi: 10.3389/fped.2020.544894 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 3. Pedigree of family 3. Sanger chromatograms depicting a de novo heterozygous variant, c.2443del p.(Arg815GlyfsTer29) in exon 4 of BCL11B (NM_138576.4) in proband, III.1 with a wild type phenotype in his parents, II.2 and II.1.
Supplementary figure 1. A. Pedigree of family 1. B. Sanger chromatograms depicting the de novo heterozygous variant, c.1662_1668 in exon 4 of BCL11B (NM_138576.3) in the proband (IV.1) and wild type genotypes in parents (II.3 and III.1) C. Sanger chromatograms depicting the de novo heterozygous variant, c.328C>T in exon 6 of MORC2 (NM_001303256.3) in the proband 1 (IV.1) and wild type genotypes in parents (II.3 and III.1).
Supplementary figure 2. Pedigree of family 2. Sanger chromatograms depicting a de novo heterozygous variant, c.2605del p.(Leu869TrpfsTer5) in exon 4 of BCL11B (NM_138576.4) in proband, II.2 with wild type genotype in her parents, I.1 and I.2 and in elder sibling, II.1.
Data Availability Statement
The data providing the evidence of the study is available from the corresponding author upon reasonable request.