

Summary
Emerging evidence suggests the tumor microbiome at gut-distal sites can modulate tumor immunity and response to cancer immunotherapy. However, detection of commensal bacteria at gut-distal tumor sites is challenging given their low abundance. Here, we present a culturomics approach to facilitate recovery of phylogenetically diverse live commensal bacteria within gut-distal melanoma tumors. We describe steps for media preparation, tissue isolation, tissue homogenization, and host cell lysis. We then detail broth expansion culture followed by agar culture and single-colony 16S rRNA sequencing.
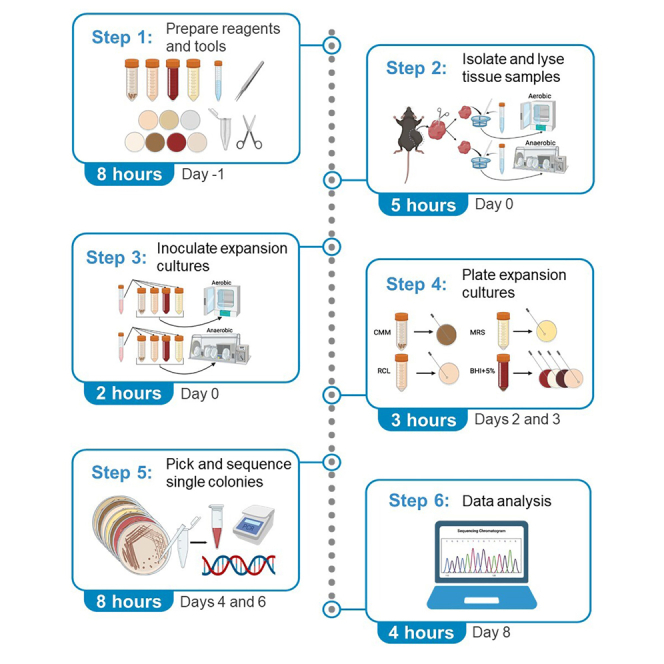
For complete details on the use and execution of this protocol, please refer to Bender and McPherson et al. (2023).1
Subject areas: Cancer, Sequencing, Microbiology, Molecular Biology
Graphical abstract
Highlights
-
•
Culturomics to detect translocated commensal bacteria in murine melanoma
-
•
Broth expansion of tumor homogenate facilitates detection of low abundance bacteria
-
•
Diverse media support growth of members of all major mammalian commensal phyla
-
•
Culturomics can be applied to other mouse and human tissues
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Emerging evidence suggests the tumor microbiome at gut-distal sites can modulate tumor immunity and response to cancer immunotherapy. However, detection of commensal bacteria at gut-distal tumor sites is challenging given their low abundance. Here, we present a culturomics approach to facilitate recovery of phylogenetically diverse live commensal bacteria within gut-distal melanoma tumors. We describe steps for media preparation, tissue isolation, tissue homogenization, and host cell lysis. We then detail broth expansion culture followed by agar culture and single-colony 16S rRNA sequencing.
Before you begin
This protocol describes specific steps for recovery of live commensal bacteria within murine melanoma tumors, as performed in our recent study.1 However, we have used this approach to profile bacterial populations in diverse samples including murine livers2 and human ocular corneal swabs (data not shown), highlighting the versatility of this protocol. This protocol includes directions for key steps including sterile tumor resection, tumor tissue homogenization, broth expansion culture, agar culture plating, and identification of recovered colonies by single-colony sequencing.
This protocol draws inspiration from cutting-edge culture approaches for recovery of commensal bacteria from gut-distal tissues utilized by other research teams, including a broth-expansion culture to grow tissue-resident bacteria to detectable concentrations, aerobic and anaerobic culture conditions, and agar plating on a nutritionally diverse set of bacterial media.3,4
We have selected the indicated bacterial broths and agars based on their ability to support growth of the four dominant bacterial phyla of the mammalian microbiome5: the highly abundant Bacillota and Bacteroidota and the less abundant Pseudomonadota and Actinomycetota. Our protocol utilizes a precise selection of bacterial media to recover and culture these and other bacterial taxa (Figure 1) without excessive redundancy, helping to make this approach scalable to preclinical laboratory experiments. Given findings by us1 and others4,6 that suggest the melanoma tumor microbiome in mice and humans consists of a heterogenous pool of bacteria including obligate anaerobic, facultative anaerobic, and aerobic bacteria; we culture melanoma tumor homogenate under both aerobic and anaerobic conditions.
Figure 1.

Expansion broths and corresponding agars
(A) Expansion broth cultures will be plated onto corresponding agar plates as follows: CMM broth plated on CMM agar; RCL broth plated on RCL agar; MRS broth plated on MRS agar; BHI+5% broth plated on BHI+5% agar, CHA agar, TSB agar, and BBE agar. The bacterial phyla supported by each broth and agar combination and the unique properties of each agar are indicated.
Institutional permissions
All studies involving mice were approved by The University of Pittsburgh Animal Research Protection Office. Those utilizing this protocol must ensure approval by their corresponding institutional office.
Prepare bacterial broth media
Timing: 4–5 h [At least 1 day prior to culture]
-
1.Prepare 15 mL of each broth media type for each sample, plus 30 mL of each media type for contamination controls.
-
a.E.g., for 10 samples, prepare at least 180 mL of each broth media.
-
a.
-
2.Add an appropriate amount of powdered media to deionized water according to the manufacturer’s instructions (see Table 1 for manufacturer protocols).
-
a.For CMM, weigh out pellets into a sterile glass beaker with a capacity that is approximately 4× the volume of media being made. Submerge the pellets in deionized water per manufacturer instructions for at least 30 min with an aluminum foil cover.
-
a.
Note: CMM may boil vigorously in the autoclave and must be stored in a container of sufficient volume during the autoclave process to prevent leakage.
-
3.
Sterilize by autoclaving at 121°C at 15 pounds per square inch (psi) for 20 min.
-
4.Allow media to cool to 20°C–22°C prior to use.
-
a.While cooling, allow sheep’s blood to reach 20°C–22°C prior to adding to BHI media.
-
a.
CRITICAL: To prevent hemolysis, always allow autoclaved media to cool to <45°C and blood to reach 20°C–22°C before combining.
-
5.Aliquot 5 mL of media into 50 mL (aerobic) and 5 mL of media into 15 mL (anaerobic) conical tubes at least 1 day prior to culture.
-
a.1 tube for each sample in each broth type in each condition (aerobic and anaerobic).
-
b.1 tube for each broth type in each condition for a mock-sample control.
-
c.1 tube for each broth type in each condition for a media control.
-
d.For CMM, aliquot media using a serological pipette and an autoclaved spoon or weighing tool to ensure equal pellet distribution.
-
a.
Note: Media must be aliquoted at least 1 day prior to allow anaerobic media to deoxidize.
-
6.Label all culture tubes with the following information:
-
a.Broth Type.
-
b.Sample Number or Type of Contamination Control (Broth or Mock-Sample).
-
c.Culture Condition (Aerobic or Anaerobic).
-
a.
-
7.
One day prior to culture, transfer anaerobic broths into an anaerobic chamber and loosen the caps to de-oxygenate at 20°C–22°C for at least 12 h.
-
8.
Store aerobic broth aliquots at 20°C–22°C for at least 12 h.
Note: Bacterial broth media can be stored at 4°C for 1–2 months.
Table 1.
Bacterial media instructions
| Broth / Agar | Manufacturer and Web Link | Manufacturer instructions |
|---|---|---|
| Lactobacilli MRS Broth (MRS) | BD Biosciences | Suspend 55 g of the powder in 1 L of purified water. Mix thoroughly. Heat with frequent agitation and boil for 1 min to completely dissolve the powder. Autoclave at 121°C for 15 min. Test samples of the finished product for performance using stable, typical control cultures. |
| Reinforced Clostridial Medium (RCL) | Fisher Scientific | Suspend 38 g in 1 L of distilled water. Bring to the boil to dissolve completely. Sterilize by autoclaving at 121°C for 15 min. |
| Tryptic Soy Broth (TSB) | BD Bacto | Suspend 30.0 g of the powder in 1 L of purified water. Mix thoroughly. Warm slightly to completely dissolve the powder. Autoclave at 121°C for 15 min. Test samples of the finished product for performance using stable, typical control cultures. |
| Cooked Meat Media (CMM) | Fisher BD | Suspend 12.5 g of the powder in 100 mL of purified water (1.25 g/10 mL). Let stand until all particles are thoroughly wetted and form an even suspension. Autoclave at 121°C for 15 min. Reduce pressure slowly. Cool without agitation. If not used within 24 h, reheat (100°C) prior to use to drive off absorbed oxygen. Test samples of the finished product for performance using stable, typical control cultures. Final pH 7.2 + 0.2 |
| Chocolate agar (CHA) | Fisher Scientific | Comes premade. |
| Bacteroides Bile Esculin Agar (BBE) | Fisher Scientific | Comes premade. |
| Brain Heart Infusion (BHI) | Fisher Scientific | Add 37 g to 1 L of distilled water. Mix well and distribute into final containers. Sterilize by autoclaving at 121°C for 15 min. |
Prepare bacterial agar media
-
9.
Prepare 2 agar plates of each agar type for each sample, plus 4 agar plates of each agar type for contamination controls.
-
10.Add an appropriate amount of powdered media to deionized water according to the manufacturer’s instructions (see Table 1).
-
a.For CMM, weigh out pellets into a sterile glass beaker and let them sit in only enough deionized water to be submerged. Record the volume of water used to hydrate the pellets. Cover with aluminum foil and incubate at 20°C–22°C for at least 30 min.
-
b.Add the soaked pellets and the deionized water to a food processor and blend until uniform.
-
c.Transfer the blended pellets back to a glass beaker with a capacity that is 4 times the volume of media that is being produced (e.g., use a 2 L beaker to make 500 mL of CMM).
-
d.Subtract the volume of deionized water used to hydrate the pellets from the volume of deionized water required per the manufacturer’s instructions.
-
a.
Note: CMM may boil vigorously in the autoclave and must be stored in a container of sufficient volume during the autoclave process to prevent leakage.
-
11.
Wash the food processor with the volume of deionized water calculated in step 10d and transfer it to the original glass beaker. Add 1.5% agar to the media (15 g per L of media).
-
12.
Mix and heat media according to the manufacturer’s instructions (see Table 1).
-
13.Sterilize by autoclaving at 121°C at 15 psi for 20 min.
-
a.Cool BHI agar to 45°C–50°C and warm sheep’s blood to 20°C–22°C prior to combining.
-
a.
-
14.Pour 15–20 mL agar plates sterilely within a biosafety cabinet.
-
a.Allow plates to dry for 20–30 min or until there is minimal surface moisture remaining.
-
a.
-
15.
Place plates agar side up in plastic sleeves sealed with tape.
Note: Plates can be sealed and then stored at 4°C for 1–2 months.
-
16.
One day prior to plating, transfer sleeves of anaerobic plates into the anaerobic chamber and acclimate at 20°C–22°C for at least 12 h to de-oxidize.
-
17.
Allow aerobic plates to acclimate at 20°C–22°C for at least 12 h.
Prepare other reagents and tools
-
18.Dilute stock NP-40 with autoclaved deionized water to make 0.05% NP-40.
-
a.After diluting, filter the solution through a 0.22 μm sterile filter.
-
a.
Note: We use 0.05% NP-40 stored at 20°C–22°C for up to 4 weeks
-
19.One day prior to culture, prepare aliquots of reagents to acclimate in the anaerobic chamber. You will need one aliquot of 0.05% NP-40 (3 mL/sample) and one aliquot of phosphate-buffered saline (PBS) (6 mL/sample). Loosely cap these aliquots and transfer them into the anaerobic chamber to de-oxidize.
-
a.Store at 20°C–22°C for at least 12 h.
-
a.
-
20.Autoclave 1 large dissection scissor, 2 medium dissection scissors, and 2 forceps per mouse being used in the experiment at 121°C at 15 psi for 20 min.
-
a.Store tools in sterile wrapping prior to use.
-
a.
Optional: Autoclave 1 spoon per mouse to help collect large non-solid tumors as well as extra tools in case of accidental contamination.
-
21.Prepare colony selection tubes (Figure 2).
-
a.Gather a 250 μL pipette tip.
-
b.Cut the wide end of the pipette tip so that the tip fits within a closed 1.5 mL flip-cap tube.
-
c.Place the shortened pipette tip into a previously autoclaved 1.5 mL tube.Note: Use previously autoclaved flip-cap tubes when making colony selection tubes to reduce moisture buildup and allow for better bacterial colony selection and storage.
-
d.Close the tube and sterilize it by autoclaving it at 121°C at 15 psi for 20 min.
-
e.Prepare at least 10–15 colony selection tubes per agar plate used in the experiment.
-
a.
-
22.
Prepare ATE buffer (200 μL/sample) by combining the following reagents in sterile nuclease-free water at the indicated final concentration. Pass solution through a 0.22 μm filter to achieve sterility.
ATE Buffer
| Reagent | Final concentration |
|---|---|
| Tris-Cl pH 8.3 | 10 mM |
| EDTA pH 8.0 | 0.1 mM |
| Sodium Azide | 0.04% |
Note: ATE buffer can be stored at 20°C–22°C for up to 3 months.
Figure 2.

Preparation of colony selection tubes
(A) Gather a 250 μL pipette tip.
(B) Cut the wide end of a pipette tip so the tip fits within a closed 1.5 mL flip-cap tube.
(C) Place the shortened pipette tip into a previously autoclaved 1.5 mL tube.
(D) Close the tube and sterilize it by autoclaving at 121°C at 15 Psi for 20 min.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Lactobacilli MRS broth (De Mann Rosa Sharpe) | BD Biosciences | Cat# DF0881-17-5 |
| Tryptic soy broth | BD Bacto | Cat# DF0370-17-3 |
| Cooked meat broth | Fisher BD | Cat# 226730 |
| Reinforced clostridial broth | Fisher Scientific | Cat# OXCM0149B |
| Brain heart infusion broth | Fisher Scientific | Cat# CM1135B |
| Chocolate agar | Fisher Scientific | Cat# R01300 |
| Bacteroides Bile Esculin agar | Fisher Scientific | Cat# NC0998155 |
| Defibrinated Sheep Blood | Fisher Scientific | Cat# 50-863-753 |
| Agar | Fisher BD | Cat# DF0140-01-0 |
| ExoSAP-IT™ PCR Product Cleanup Reagent | Applied Biosystems | Cat# 78201.1.ML |
| NP-40 (IGEPAL® CA-630) | Sigma-Aldrich | Cat# 18896 |
| Bullseye Red TAQ DNA Polymerase Mastermix∗ | MIDSCI | Cat# BE180303 |
| Software and algorithms | ||
| BioRender | BioRender | N/A |
| Other | ||
| 16s primer: 27F, 5′-AGAGTTTGATCMTGGCTCAG-3′ | N/A | N/A |
| 16s primer: 1525R, 5′-AAGGAGGTGATCCAGCC-3′ | N/A | N/A |
∗Cannot be substituted. Can be purchased at: https://midsci.com/item/ASPCRREAG3/PREMIUM-Bullseye-Taq-DNA-Polymerase-Master-Mix/.
Step-by-step method details
Sterile tumor tissue isolation
This step will describe our method for the sterile resection of a subcutaneously engrafted murine melanoma tumor.
-
1.
Euthanize mouse and generously spray the mouse with 70% ethanol (Figure 3A).
-
2.
Place the mouse in the prone position and pin it down at the shoulders (axillary) on an autoclaved paper towel on a dissection board.
-
3.
Make an initial incision through the skin approximately 1–2 cm above the hind-flank tumor using the big scissors (Figure 3B).
Note: These big scissors can be used for multiple mice.
-
4.Use the non-dominant hand to hold the mouse firmly against the dissection board. Use the dominant hand to grab the bottom edge of the skin at the incision point and pull continuously until the skin is peeled back past the tumor (Figure 3C).
-
a.Pin the pulled-back skin-flap and legs down to the board.
-
b.Ensure that the peritoneal cavity does not open and contaminate the tumor.
-
a.
-
5.
With a new pair of sterile medium scissors and forceps, carefully remove the tumor, ensuring no contact with mouse fur, and transfer the tumor into a sterile 40 μm filter in a sterile 6-well plate (Figure 3D).
-
6.
Sterilely divide the sample in half using sterile medium scissors and forceps and transfer it to another sterile filter inside a 6-well plate.
Figure 3.

Sterile resection of subcutaneously engrafted murine melanoma tumor
(A) Thoroughly wet euthanized mouse with 70% ethanol.
(B) Make a single incision through the skin 1–2 cm above the tumor.
(C) Pull skin back far enough to reveal tumor. Pin down skin and back legs.
(D) Remove all tumors sterilely.
Sterile tumor lysis
This section describes our process for tumor tissue homogenization and lysis to extract live bacteria from the tumor.
Optional: Weigh individual tissue samples by placing the filter on a scale covered with a sterile paper towel. Return filters to the 6-well plate.
-
7.Set up contamination controls for both aerobic and anaerobic culture plates.
-
a.Set aside a single well in two of the 6-well plates for a mock-sample control (one should be placed in an aerobic plate, and one in an anaerobic plate).
-
b.Add a sterile 40 μm filter to each well.
-
c.Wave a set of sterile tools in the air and rub them on the filter to account for potential contamination during tissue isolation.
-
d.Perform all steps indicated in the remainder of the protocol upon these mock-sample control wells in addition to your sample wells.
-
a.
Note: Without the sample control, one cannot confidently affirm that the bacterial growth was not due to contamination encountered during tissue processing.
-
8.
Transfer anaerobic samples to the anaerobic chamber. Transfer aerobic samples to the biosafety cabinet.
-
9.Add an appropriate volume of 0.05% NP-40 lysis solution (see Table 2) and record the volume of 0.05% NP-40 used for each tumor.
-
a.For control wells, add 2 mL 0.05% NP-40.
-
a.
-
10.Mash tissue through the filter using a sterile 1 mL syringe plunger.
-
a.For control wells, rub a sterile syringe plunger in the filter with a mock mashing motion.
-
b.For large solid tumors (>1000 mg), use a set of sterile dissection scissors to mince tumor tissue into pieces ∼ 5 × 5 mm in size prior to mashing it through the filter.
-
a.
-
11.
Incubate at 37°C for 3 h in either aerobic or anaerobic condition.
-
12.
Dilute 0.05% NP-40 lysis-tissue homogenate with double the volume of sterile PBS that is at 20°C–22°C (e.g., for a tumor <500 mg and 2 mL 0.05% NP40, add 4 mL of PBS).
-
13.
Pipette homogenate up and down with a serological pipette around 10 times to further homogenize tissue lysate and collect sample that is out of solution.
-
14.
Using sterile tweezers, carefully remove the filter from each well.
-
15.
Transfer tumor homogenate to a 15 mL conical tube.
-
16.Centrifuge 4700 × g for 10 min to pellet all bacteria within the solution.
-
a.Ensure all tube lids are tight to maintain their aerobic or anaerobic atmosphere.
-
a.
-
17.
Transfer aerobic tubes back to the biosafety cabinet. Transfer anaerobic tubes back to the anaerobic chamber.
-
18.
Sterilely aspirate aerobic supernatant. Gently pour anaerobic supernatant into a waste cup or use a serological pipette to gently remove supernatant if the pelleted tumor homogenate is unstable.
-
19.Resuspend samples in 1 mL sterile PBS.
-
a.For large pellets, if 1 mL is not enough volume to completely resuspend sample, add additional PBS in 500 μL increments and record the final volume.
-
a.
Table 2.
NP 40 volume per Tumor Weight
| Tumor weight: | 0.05% NP-40 volume: |
|---|---|
| <500 mg | 2 mL |
| 500–1500 mg | 2.5 mL |
| >1500 mg | 3 mL |
Culture procedure
This section describes our process for the live culture of tumor homogenate to facilitate the growth of bacterial populations.
-
20.Seed expansion cultures by adding 200 μL of tumor homogenate suspended in PBS to all 5 mL broth aliquots.
-
a.If more than 1 mL was used for resuspension, add 1/5 of the total volume to each of the broth aliquots (see Table 3).
-
b.Vortex at medium speed for 10 s to mix thoroughly.
-
c.Contamination control: Designate one aliquot of expansion broth of each broth type under aerobic and anaerobic conditions as a broth control to identify broth media contamination. This aliquot should be loosely capped and incubated alongside samples and the sample control.
-
a.
-
21.Ensure broth aliquots in both conditions are loosely capped.
-
a.Aerobic expansion cultures should be incubated on a shaker incubator set to 150 rpm in 50 mL conical tubes, lids loosely affixed with a small piece of tape.
-
b.Anaerobic expansion cultures should be incubated stationary in 15 mL conical tubes, loosely capped.
-
a.
-
22.
Incubate at 37°C for either 2 days aerobically or 3 days anaerobically.
-
23.Plate the expansion cultures onto appropriate agar plates (Figure 1).
-
a.Tightly cap the expansion culture tube and vortex at medium speed for 10 s.
-
b.Dip a sterile inoculation loop into the expansion culture.
-
c.Streak across the corresponding agar plate(s) for single colony isolation.
-
i.Streak both the sample and broth controls across the corresponding agar plate(s) as well.
-
i.
-
a.
-
24.
Culture a bacterial strain of known identity on the appropriate agar media for use as a positive control during single colony 16S rRNA sequencing.
-
25.
Incubate at 37°C for either 2 days aerobically or 3 days anaerobically.
-
26.Pick colonies of at least 1 mm in size using the sterile pipette tip from the colony selection tube.
-
a.Pick 2–3 colonies per morphology per agar per sample.
-
b.If there was contamination on control plates, pick those colonies as well.
-
c.Ensure colony selection tubes are labeled with agar type, collection time, and condition type (aerobic or anaerobic).
-
d.Store tubes containing colonies at −80°C.
-
a.
Optional: If desired, wrap agar plates tightly with parafilm and store at 4°C agar-side up.
Table 3.
Tumor homogenate aliquot volumes
| Total resuspension volume | Volume to add per broth aliquot |
|---|---|
| 1 mL | 200 μL |
| 1.5 mL | 300 μL |
| 2.0 mL | 400 μL |
16S sequencing
This step will describe our method for PCR amplification of the 16S rRNA region of individual bacterial colonies. These PCR products will be used for Sanger sequencing to identify individual colonies.
-
27.Remove colony selection tubes from the −80°C freezer.
-
a.Include a positive control colony selection tube containing a bacterial colony of known identity and a negative control colony selection tube containing no bacterial colony.
-
b.Add 20 μL of sterile ATE buffer directly into the pipette tip that was used to pick the colony.
-
c.Let incubate at 20°C–22°C for 5 min.
-
d.Vortex for 5 s at medium speed.
-
e.Quick spin the tubes using a bench-top centrifuge.
-
f.Make a key identifying colony ID to sample #.
-
a.
-
28.Using the cut pipette tip that was used to pick the colony, pipette the ATE solution up and down to mix the sample and transfer 10 μL of the colony suspension into a sterile 8-strip PCR microtube.
-
a.Place the 8-strip PCR microtubes in a bench-top centrifuge and give a quick spin.
-
a.
-
29.Lyse bacteria by placing 8-strip PCR microtube in a thermocycler following the parameters below:
-
a.The remaining un-lysed sample can be placed back into −80°C freezer for storage and can be used to grow monoclonal stocks of the recovered bacterial strain if desired.
-
a.
PCR cycling conditions for lysis
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Lyse | 95°C | 10 min | 1 |
| Hold | 4°C | Infinite Hold | |
-
30.
During sample lysis, prepare a master mix for PCR amplification, as seen in Table 4.
-
31.
Transfer 2 μL of bacterial lysate to new sterile 8-strip PCR microtubes.
-
32.Add 8 μL of the master mix.
-
a.Quick spin to bring down contents of each tube.
-
a.
-
33.
Amplify 16S rRNA gene using the following parameters:
PCR cycling conditions for 16S rRNA Amplification
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 95°C | 5 min | 1 |
| Denaturation | 95°C | 30 s | 35 cycles |
| Annealing | 55°C | 30 s | |
| Extension | 72°C | 2 min | |
| Final extension | 72°C | 20 min | 1 |
| Hold | 4°C | Infinite Hold | |
Optional: Combine 2 μL of amplified PCR mix with 8 μL PCR grade water and run 1% agarose gel with 6 μL of ethidium bromide at 150 V for 20 min. If the reaction was successful, a band at 1500 base pairs should be seen.
-
34.
After PCR amplification (2.5 h), use Exosap-IT to purify PCR products.
-
35.
Add 2 μL of Exosap-IT to a new set of sterile 8-strip PCR microtubes.
-
36.Add 5 μL of the amplified PCR mix to the PCR tubes containing Exosap-IT.
-
a.Quick spin to bring down contents.
-
a.
-
37.
Purify PCR products according to PCR cycling conditions for Exosap Purification using the following parameters:
PCR cycling conditions for Exosap Purification
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 37°C | 15 min | 1 |
| Denaturation | 80°C | 15 min | 1 |
| Hold | 4°C | Infinite Hold | |
-
38.After purification, prepare the sample according to desired sequencing company’s instructions (i.e., dilute to proper DNA concentration, transfer to a specific size tube, etc.).
-
a.There are several commercial options available for targeted sequencing, as well as core facilities at many universities that are also qualified to perform this sequencing step.
-
a.
Table 4.
PCR reaction master mix
| Reagent | Amount |
|---|---|
| Bullseye Red TAQ Polymerase Mastermix | 6 μL × # of samples |
| 27F Primer: 5′-AGAGTTTGATCMTGGCTCAG-3′ | 0.25 μL × # of samples |
| 1525R Primer: 5′-AAGGAGGTGATCCAGCC-3′ | 0.25 μL × # of samples |
| PCR grade water | 1.5 μL × # of samples |
Expected outcomes
We expect visible growth in bacterial broth and agar media inoculated with tissue samples and no detectable growth in bacterial broth and agar media inoculated with broth controls or mock-sample controls. Additionally, across the bacterial agars, multiple colony morphologies should be apparent. Once picked and sequenced, 16S sequences derived from these colonies should yield substantial alignment to reference genomes. To see results derived from this protocol, please refer to our recent study.1
Quantification and statistical analysis
After sequencing, use NCBI Nucleotide Basic Local Alignment Search Tool (BLAST) 16S ribosomal RNA sequences database to match FASTA sequences to existing 16S rRNA reference sequences (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Record 16S sequence matches at the species and/or genera level primarily based on the percent identity and query cover, which indicate the strength of the sequence alignment. We consider sequence alignments to be reliable if the percent identity is greater than 98% (species level identification) or is greater than 95% (genus level identification). We record significant sequence alignments at the species and genus level and save raw FASTA sequences for future reference.
To address potential contaminants, check control agar plates for bacterial growth. If there is growth from either the mock-sample control or broth control, sequence and identify those colonies as described above and remove the identified species from your experimental sample results. For example, if the MRS broth control in anaerobic conditions grew E. coli, then any E. coli growth in anaerobic MRS experimental samples must be excluded, as we assume this species was a contaminant present in all anaerobic MRS broth cultures. Species and genera identified on experimental sample agars that are NOT identified on control treatment agars are live bacteria present within the original tissue sample. These results can only be used to identify presence or absence of bacterial taxa and are not quantitative. Thus, visualization of results should be performed carefully to demonstrate the qualitative recovery of live bacterial species and not mislead readers to make quantitative conclusions.
Limitations
While this culturomics approach enables the detection of low-abundance bacterial populations and supports the recovery of many diverse bacterial taxa, many commensal bacteria cannot be cultured ex vivo.7 This protocol cannot be used if a tumor has ulcerated, as the tumor microenvironment is no longer protected from environmental contamination. This protocol only provides a qualitative assessment of bacteria present within tissue and cannot be used to quantify bacterial populations. Further, the identification of bacterial species through this approach is limited by which reference bacterial strain sequences are available through the NCBI nucleotide BLAST database. Lastly, this approach is labor intensive and will yield the best recovery when performed by a team of at least 2 researchers to ensure proper timing of all lysis and culture steps across both aerobic and anaerobic conditions.
Troubleshooting
Problem 1
Failure to recover isolated single colonies upon agar culture (Culture Procedure Step 23).
Potential solution
-
•
Dry agar plates in biosafety cabinet to remove excess moisture prior to overnight acclimation.
-
•
Increase agar content to 2%.
Problem 2
No bacterial growth or limited bacterial diversity upon agar culture (Culture Procedure Step 23).
Potential solution
-
•
Increase the quantity of individual samples.
-
•
Increase the duration of the expansion culture to 5 days.
-
•
Ensure media and agar are functional by culturing known bacterial strains.
Problem 3
Bacterial growth on control agar plates (Sterile tumor tissue isolation & Sterile tumor lysis steps).
Potential solution
-
•
Increase vigilance with sterility. Use proper aseptic technique.
-
•
Culture all reagents used individually to identify contaminated reagents.
Problem 4
Failure to amplify 16S rRNA region by PCR (16s sequencing steps).
Potential solution
-
•
Consider extracting DNA from individual colonies (e.g., with a commercially available kit).
-
•
Try using a different Taq polymerase for PCR amplification of the 16S region.
-
•
Ensure enough bacteria are removed from agar plates when using colony selection tubes. Ideally, the bacterial colony will be visible on the pipette tip used for colony selection.
-
•
Perform 16s rRNA amplification on known bacteria to check for problems with PCR reagents.
Problem 5
Limited alignment of bacterial 16S sequences of picked bacterial colonies with reference genomes (Quantification and Statistical Analysis).
Potential solution
-
•
Perform full-length 16S rRNA sequencing to increase sequence coverage when aligning with reference genomes.
-
•
Consider using a kit for DNA purification to enhance the purity of the final PCR products.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marlies Meisel (marlies@pitt.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by an Investigator Start-up Fund, Department of Immunology, University of Pittsburgh School of Medicine to M.M; a Hillman Developmental Pilot Award (NIH/NCI P30 CA047904) to M.M.; a NIH/NIDDK R01 DK130897, a NIH/NCI R21 CA259636, a Melanoma Research Alliance award https://doi.org/10.48050/pc.gr.143738 (820677), and NIH U24 EY035102-01 to M.M.; T32 CA082084 to C.M.P.; and a University of Pittsburgh Microbiology and Immunology Diversity Scholar fellowship, School of Medicine Summer Undergraduate Research Program fellowship, and Frederick Honors College Brackenridge Fellowship to J.H.S.
Figure 1 and components of the graphical abstract were created using Bio-Render (https://www.biorender.com/).
Author contributions
All authors designed and analyzed experiments. C.M.P and J.H.S. performed most experiments. C.R.L. performed experiments to optimize single colony sequencing. C.M.P., J.H.S., and M.M. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate any unique datasets or code.
References
- 1.Bender M.J., McPherson A.C., Phelps C.M., Pandey S.P., Laughlin C.R., Shapira J.H., Medina Sanchez L., Rana M., Richie T.G., Mims T.S., et al. Dietary tryptophan metabolite released by intratumoral Lactobacillus reuteri facilitates immune checkpoint inhibitor treatment. Cell. 2023;186:1846–1862.e26. doi: 10.1016/j.cell.2023.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pandey S.P., Bender M.J., McPherson A.C., Phelps C.M., Sanchez L.M., Rana M., Hedden L., Sangani K.A., Chen L., Shapira J.H., et al. Tet2 deficiency drives liver microbiome dysbiosis triggering Tc1 cell autoimmune hepatitis. Cell Host Microbe. 2022;30:1003–1019.e10. doi: 10.1016/j.chom.2022.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manfredo Vieira S., Hiltensperger M., Kumar V., Zegarra-Ruiz D., Dehner C., Khan N., Costa F.R.C., Tiniakou E., Greiling T., Ruff W., et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359:1156–1161. doi: 10.1126/science.aar7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nejman D., Livyatan I., Fuks G., Gavert N., Zwang Y., Geller L.T., Rotter-Maskowitz A., Weiser R., Mallel G., Gigi E., et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020;368:973–980. doi: 10.1126/science.aay9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckburg P.B., Bik E.M., Bernstein C.N., Purdom E., Dethlefsen L., Sargent M., Gill S.R., Nelson K.E., Relman D.A. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poore G.D., Kopylova E., Zhu Q., Carpenter C., Fraraccio S., Wandro S., Kosciolek T., Janssen S., Metcalf J., Song S.J., et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579:567–574. doi: 10.1038/s41586-020-2095-1. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Stewart E.J. Growing unculturable bacteria. J. Bacteriol. 2012;194:4151–4160. doi: 10.1128/JB.00345-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.