

SUMMARY
Pancreatic ductal adenocarcinoma (PDA) orchestrates a suppressive tumor microenvironment that fosters immunotherapy resistance. Tumor-associated macrophages (TAMs) are the principal immune cell infiltrating PDA and are heterogeneous. Here, by employing macrophage fate-mapping approaches and single-cell RNA sequencing, we show that monocytes give rise to most macrophage subsets in PDA. Tumor-specific CD4, but not CD8, T cells promote monocyte differentiation into MHCIIhi anti-tumor macrophages. By conditional major histocompatibility complex (MHC) class II deletion on monocyte-derived macrophages, we show that tumor antigen presentation is required for instructing monocyte differentiation into anti-tumor macrophages, promoting Th1 cells, abrogating Treg cells, and mitigating CD8 T cell exhaustion. Non-redundant IFNγ and CD40 promote MHCIIhi anti-tumor macrophages. Intratumoral monocytes adopt a pro-tumor fate indistinguishable from that of tissue-resident macrophages following loss of macrophage MHC class II or tumor-specific CD4 T cells. Thus, tumor antigen presentation by macrophages to CD4 T cells dictates TAM fate and is a major determinant of macrophage heterogeneity in cancer.
In brief
Patterson et al. demonstrate monocyte differentiation into heterogeneous macrophage populations in pancreatic cancer. They identify that tumor-specific CD4 T cells instruct MHCIIhi anti-tumor macrophage differentiation through CD40 and IFNγ. Loss of CD4 T cells or MHC class II on monocyte-derived macrophages drives monocytes toward alternatively activated states and promotes tumor growth.
Graphical Abstract
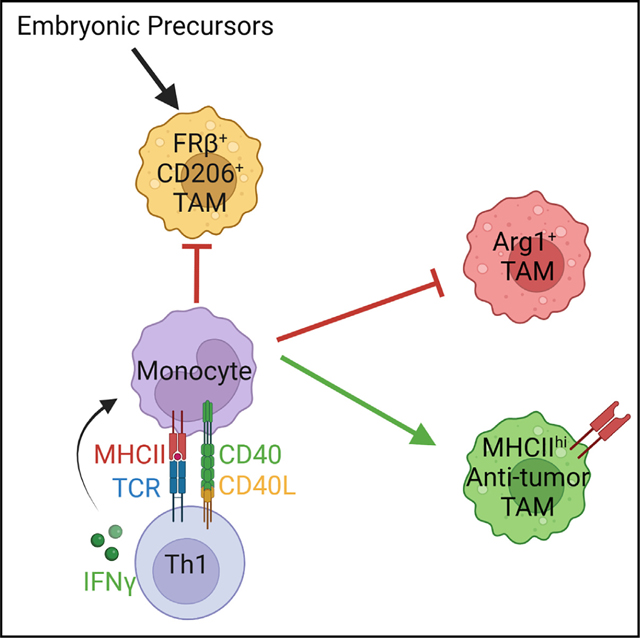
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is a lethal malignancy that resists current treatments.1 PDA orchestrates a suppressive and fibroinflammatory tumor microenvironment (TME) comprising numerous immune cells.2 The pancreatic TME interferes with the efficacy of chemotherapy3 and immunotherapy.2 However, stromal composition, including particular subsets of T cells and macrophages, correlates with disease-free and overall survival,4–7 supporting the idea that some aspects of the stromal response may be beneficial.
Pancreatic tumor cells produce numerous pro-myeloid factors, thereby promoting myeloid cell expansion and recruitment into the TME.2,8–12 Tumor-associated macrophages (TAMs) are abundant cells in PDA and often outnumber tumor cells.13 While TAM crosstalk with tumor cells and fibroblasts has a pivotal role in promoting carcinogenesis,14,15 the bidirectional interactions between TAMs and T cells that affect tumor growth are just beginning to be understood.16,17 In human PDA, TAMs colocalize with T cells, and tumors with an abundant T cell infiltrate also contain numerous TAMs.13 These data are consistent with an immunogenic subtype that constitutes a large fraction of the patient population.18
TAMs are diverse and participate in complex processes, including angiogenesis, metastasis, and inflammation.19,20 Circulating monocytes and embryonically seeded tissue-resident macrophages comprise the TAM pool.21–23 Macrophages derived from monocytes display both phenotypic and functional differences compared with tissue-resident TAMs, suggesting that ontogeny may partially account for heterogeneity.22,24 Tissue-resident TAMs promote extracellular matrix (ECM) deposition, whereas monocyte-derived macrophages act by shaping immunity.24 In the absence of a defined tumor-specific antigen, TAMs potently suppress T cell activation,8 and global TAM depletion using Csf1r blockade can be beneficial.25,26 In contrast, TAM depletion is not advantageous when a tumor-specific T cell response is engaged.26 Global TAM depletion also fails to promote engineered T cell functionality in autochthonous PDA.27 While clinical efforts to ablate TAMs have not shown benefit in PDA patients,28 promoting anti-tumor macrophages using CD40 agonist can exert transient anti-tumor effects.29 Together, TAMs may aid in antigen-specific T cell-mediated destruction of tumors if programmed appropriately. Thus, understanding events that program TAMs toward anti-tumor states could reveal novel therapeutic opportunities.
Here, using an in vivo monocyte fate-mapping approach, we temporally track monocyte differentiation in an orthotopic PDA mouse model that expresses a defined model neoantigen.30 Our study uncovers a critical role for monocyte-derived macrophage cognate tumor antigen presentation to CD4 T cells that dictates macrophage fate and tumor control. Deletion of major histocompatibility complex (MHC) class II in specifically monocyte-derived macrophages or loss of tumor-specific CD4 T cells drives monocytes to adopt a phenotypic and transcriptional state mirroring immunosuppressive pancreas tissue-resident macrophages. We posit that therapeutic resistance by poorly immunogenic tumors may derive from a de facto monocyte differentiation trajectory toward a pro-tumor state due to a failure to encounter tumor-specific CD4 T cells.
RESULTS
Fate mapping of monocyte-derived or tissue-resident macrophages in PDA
Given that both monocyte-derived and embryonically derived macrophages contribute to the TAM pool, distinguishing monocyte-derived macrophages in vivo has been challenging. Thus, we utilized the CCR2CreER R26Tdtomato mouse, which allows for specific labeling of individual waves of blood monocytes following tamoxifen treatment, enabling tracking of their differentiation upon entry into tissue.31–33 Tamoxifen treatment of CCR2CreER R26Tdtomato mice revealed robust classical monocyte labeling in the blood, but no labeling of pancreatic tissue-resident macrophages (Figures S1A–S1D), consistent with a reported lack of Ccr2.34 To track monocyte differentiation in PDA in the presence of tumor-specific T cells, we orthotopically implanted KPC2a tumor cells that express a model neoantigen click beetle red luciferase (CB)30 into the pancreas of CCR2CreER R26Tdtomato mice following a single dose of tamoxifen (Figure 1A). We analyzed mice at days 3, 7, and 14 after implantation and identified two distinct Tomato+ monocyte-derived macrophage subsets based on MHCIIhi and MHCIIlo expression (Figure 1B). MHCIIhi macrophages expressed higher CD86, consistent with an immunostimulatory phenotype, whereas MHCIIlo macrophages expressed alternative activation markers, including Arg-1 (Figure 1C). Intratumoral Tomato+ macrophages increased between days 3 and 7 and then decreased by day 14 (Figure 1D), consistent with replacement by subsequent monocyte waves. On day 3, most Tomato+ cells resembled undifferentiated monocytes (CD11b+Ly6C+F4/80−). However, by days 7 and 14, most Tomato+ cells expressed markers of differentiated macrophages (Figure 1E), indicating that recruited monocytes differentiate into macrophages as early as day 7 and are maintained for at least 1 week. At day 3, Tomato+ macrophages (CD64+F480+Ly6C−) were MHCIIlo, whereas by days 7 and 14, most Tomato+ macrophages were MHCIIhi (Figure 1F) Thus, monocytes undergo an initial differentiation into a transitory MHCIIlo state and progressively upregulate MHCII. Alternatively, MHCIIhi monocyte-derived macrophages may preferentially expand during tumor progression.
Figure 1. Fate mapping of monocyte-derived or tissue-resident macrophages in PDA.

(A) In vivo monocyte tracking approach in CCR2CreER R26TdTomato mice orthotopically implanted with KPC2a tumor cells and treated with tamoxifen. See also Figure S1.
(B) Gating strategy for Tomato+ MHCIIhi and MHCIIlo macrophages.
(C) Representative CD86 and Arg1 histograms gated on Tomato+ MHCIIhi or MHCIIlo TAMs.
(D) Frequency and number of Tomato+ TAMs. Each dot is an independent mouse. Data are mean ± SEM; n = 3–4 mice per group; **p < 0.005; one-way ANOVA with Tukey’s posttest.
(E) Proportion of intratumoral Tomato+ cells that are macrophages (CD64+F4/80+) or monocytes (F4/80−Ly6C+). Each dot is an independent mouse. Data are mean ± SEM; n = 3–4 mice per group; ****p < 0.0001; one-way ANOVA with Tukey’s posttest.
(F) Proportion of intratumoral Tomato+ MHCIIhi or MHCIIlo macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 3–4 mice per group; **p < 0.005; one-way ANOVA with Tukey’s posttest.
(G) In vivo monocyte tracking approach in CCR2CreER R26TdTomato mice implanted with KPC2a tumor cells and treated with tamoxifen twice.
(H) Representative plot gated on intratumoral F4/80+CD64+ macrophages on day 7. Proportion of total Tomato+ macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 4 mice per group; ****p < 0.0001; Student’s t test.
(I) Representative plots gated on day 7 and proportion of Tomato+ or Tomato− intratumoral MHCIIhi or MHCIIlo macrophages on day 7. Each dot is an independent mouse. Data are mean ± SEM; n = 4 mice per group; ***p < 0.001; Student’s t test.
(J) Representative plots gated on day 7 and proportion of Tomato+ and Tomato− MHCIIloCD206+FRβ+ macrophages on day 7. Each dot is an independent mouse. Data are mean ± SEM; n = 4 mice per group; ***p < 0.001; Student’s t test.
(K) PD-L1 staining and mean fluorescence intensity (MFI) of Tomato+ and Tomato− macrophages at day 7. Each dot is an independent mouse. Data are mean ±SEM; n = 4 mice per group; *p < 0.05; Student’s t test.
(L) Representative histograms and frequency of Ki67+ macrophages on day 7. Each dot is an independent mouse. Data are mean ± SEM; n = 4 mice per group; **p < 0.005; Student’s t test.
(M) Embryonic fate-mapping approach in CX3CR1CreER R26TdTomato. Briefly, pregnant mothers were gavaged with tamoxifen on embryonic day 14.5 and then progeny were implanted with tumors. See also Figure S2.
(N) Representative plot gated on intratumoral F4/80+CD64+ macrophages on day 7 from (M). Proportion of each cell subtype that is Tomato+ is shown. Each dot is an independent mouse. Data are mean ± SEM; n = 3 mice per group; **p < 0.005, ****p < 0.0001; one-way ANOVA with Tukey’s post test.
(O) Representative plots gated on day 7 and proportion of Tomato+ and Tomato− MHCIIloCD206+FRβ+ tumor macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 3 mice per group; **p < 0.005; Student’s t test.
(P) Experimental schematic to label the initial wave of recruited monocytes (purple) or subsequent waves of recruited monocytes (green).
(Q) Proportion of each monocyte wave that gives rise to MHCIIhi or MHCIIlo macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 3–4 mice per group; **p < 0.005; Student’s t test.
(R) Proportion of each monocyte wave that gives rise to CD206+ macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 3–4 mice per group;****p < 0.0001; Student’s t test.
(S) Proportion of each monocyte wave that gives rise to FRβ+ macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 3–4 mice per group.
We next compared the phenotype of Tomato+ monocyte-derived macrophages with that of Tomato− tissue-resident macrophages. We administered tamoxifen at both prior to and following tumor implantation (Figure 1G), which resulted in complete monocyte labeling for 7 days (Figure S1E) and resulting in 80% of TAMs that were Tomato+ (Figure 1H). Consistent with a prior report,24 non-monocyte-derived TAMs primarily adopted an MHCIIlo phenotype (Figure 1I). MHCIIlo Tomato− macrophages also expressed markers of alternative activation including CD206 and FRβ. In contrast, Tomato+ macrophages rarely adopted a MHCIIloCD206+FRβ+ phenotype (Figure 1J) and instead expressed high PD-L1 (Figure 1K) and were proliferative (Figure 1L). Macrophages in healthy pancreas were also MHCIIloCD206+FRβ+ (Figures S2A and S2B). These data highlight immune modulatory and proliferation differences based on macrophage ontogeny. To investigate the developmental origin of pancreatic tissue-resident macrophages in PDA, we performed embryonic pulse-chase experiments in tumor-bearing CX3CR1CreER R26TdTomato mice. Given that CX3CR1 is turned on in embryonic-derived macrophages during development,35 in utero treatment of tamoxifen allows for specific labeling of embryonic macrophages.32 Pregnant CX3CR1CreER R26TdTomato mice were administered tamoxifen on embryonic day 14.5 and then their progeny were implanted with tumors and assessed 7 days after (Figure 1M). Nearly 100% of microglia were Tomato+, while labeling in the blood was negligible (Figure 1N), validating the specificity of this approach. Approximately 10% of TAMs were Tomato+ (Figure 1N), confirming that most TAMs are of monocytic origin. Tomato+ TAMs primarily adopted an MHCIIloCD206+FRβ+ phenotype, while a much lower frequency of Tomato− TAMsadopted this phenotype (Figure 1O).
We next compared monocyte-derived macrophage fate during early invasive tumor growth with monocytes recruited at a later time point (Figure 1P). Monocytes recruited later during tumor growth preferentially differentiated into MHCIIhi macrophages compared with initially recruited monocytes (Figure 1Q). For monocytes in larger tumors, a subset upregulated CD206 (Figure 1R), suggesting acquisition of an alternatively activated phenotype. At both time points, Tomato+ macrophages failed to upregulate FRβ (Figure 1S). Together, the data support that monocytes comprise most TAMs in neoantigen+ PDA and that the MHCIIloCD206+FRβ+ phenotype may identify pancreas-resident macrophages.
CD4 T cells govern fate decisions in tumor-infiltrating monocytes
T cells colocalize with macrophages in resected human PDA,13 and engineered T cell therapy recruits monocytes into KPC PDA.27 Given these observations, we hypothesized that T cells may instruct monocyte differentiation. Therefore, we depleted CD4 or CD8 T cells in CCR2creER R26tdTomato mice bearing orthotopic tumors and assessed monocyte differentiation at day 7 (Figure 2A). CD8 T cell depletion led to a modest but significant increase in number of Tomato+ macrophages (Figure 2B), suggesting regulation of monocyte recruitment and/or survival. While CD8 T cell depletion had minimal effect on the Tomato+ macrophage phenotype, CD4 T cell depletion led to a dramatic reduction in MHCIIhi macrophages and a corresponding increase in MHCIIlo macrophages (Figure 2C). In contrast, CD8 T cell depletion did not have an impact on monocyte phenotype at day 7 (Figure 2C) or following extended depletion at day 14 (Figures S3A–S3C). To test if CD4 T cells were merely modulating MHC class II or instead shifting macrophage subsets, we developed an expanded antibody panel to delineate macrophage subpopulations (Figure 2D). At day 14, tumor weights were significantly increased in CD4 T cell-depleted mice compared with control mice, indicating that CD4 T cells control tumor growth (Figure 2E). Moreover, CD4 T cell depletion increased Tomato+ MHCIIlo, Arg1+, CD206+, and/or FRβ+ macrophages (Figures 2F–2H) while decreasing PD-L1 (Figure 2I). Unexpectedly, CD4 T cell depletion biased monocyte-derived macrophages toward an MHCIIloCD206+FRβ+ tissue-resident phenotype (Figure 2J). Finally, CD4 T cell depletion increased Tomato− tissue-resident macrophage proliferation (Figure 2K). Thus, CD4 T cells instruct monocytes toward an MHCIIhi state and limit differentiation into Arg1+ or CD206+FRβ+ TAMs, the latter indistinguishable from tissue-resident TAMs.
Figure 2. CD4 T cells govern fate decisions of tumor-infiltrating monocytes.

(A) Experimental approach to test the role of T cell subsets on monocyte fate.
(B) Proportion (left) and number (right) of Tomato+ intratumoral macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 3–5 mice per group; *p < 0.05; one-way ANOVA with Tukey’s posttest.
(C) Proportion of MHCIIhi or MHCIIlo Tomato+ macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 3–5 mice per group; ****p < 0.0001; one-way ANOVA with Tukey’s posttest.
(D) Experimental approach to test the impact of prolonged CD4 T cell depletion in tumor-bearing CCR2CreER R26TdTomato mice. See also Figure S3.
(E) Tumor weight from mice in (D); n = 3–6 mice per group. Data are mean ± SEM; **p < 0.005; Student’s t test for each time point.
(F) Proportion of Tomato+ macrophages from (D) that are MHCIIhi or MHCIIlo. Data are mean ± SEM; n = 3–6 mice per group; ***p < 0.001, ****p < 0.0001; Student’s t test for each time point.
(G) Representative histograms gated on Tomato+ macrophages isolated from PBS- or αCD4-treated mice.
(H) Proportion of Tomato+ macrophages expressing Arg1, CD206, and FRβ from PBS- or αCD4-treated mice. Data are mean ± SEM; n = 3–6 mice per group;*p < 0.05, **p < 0.005, ****p < 0.0001; Student’s t test for each time point.
(I) Representative PD-L1 staining (top) and MFI of Tomato+ macrophages from PBS- or αCD4-treated mice (bottom); n = 3–6 mice per group; *p < 0.05; Student’s t test.
(J) Representative plots and proportion of Tomato+ MHCIIlo macrophages that coexpress CD206 and FRβ from mice in (D). Data are mean ± SEM; n = 3–6 mice per group; ****p < 0.0001; Student’s t test for each time point.
(K) Representative histograms and proportion of Tomato− macrophages that are Ki67+. Data are mean ± SEM; n = 3–4 mice per group; **p < 0.005; Student’s t test.
(L) Experimental approach to test the impact of T cell antigen specificity on monocyte differentiation. See also Figures S4A–S4F.
(M) Tumor weight from (L). Data are mean ± SEM; n = 4 mice per group; **p < 0.005; Student’s t test.
(N) Proportion of MHCIIhi or MHCIIlo macrophages from (L). Data are mean ± SEM; n = 4 mice per group; ****p < 0.0001; Student’s t test.
(O) Proportion of Arg1+ macrophages from (L). Data are mean ± SEM; n = 4 mice per group; ***p < 0.001; Student’s t test.
(P) Proportion of MHCIIloCD206+FRβ+ macrophages from (L). Data are mean ± SEM; n = 4 mice per group; ****p < 0.0001; Student’s t test.
(Q) Experimental approach to test if transferred tumor-specific (OTII) or non-specific (SM1) CD4 T cells modulate monocyte differentiation in KPC-OVA-bearing Rag−/− mice. See also Figures S4G–S4I.
(R) Proportion of MHCIIhi or MHCIIlo macrophages from (Q). Data are mean ± SEM; n = 4 mice per group; ***p < 0.001; Student’s t test.
(S) Proportion of Arg1+ macrophages from (Q). Data are mean ± SEM; n = 4 mice per group; **p < 0.005; Student’s t test.
(T) Proportion of MHCIIloCD206+FRβ+ macrophages from (Q). Data are mean ± SEM; n = 4 mice per group; **p < 0.005; Student’s t test. See also Figure S5.
Tumor-specific CD4 T cells drive MHCIIhi macrophage differentiation
To test if T cell antigen specificity affects macrophage differentiation, KPC2a cells were implanted into wild-type (WT) or SM1 Rag1−/− mice, in which CD4 T cells express a monoclonal T cell receptor (TCR) specific to a Salmonella antigen36 (Figure 2L), thereby eliminating endogenous tumor-reactive T cells. Tumors were larger in SM1 compared with WT mice (Figure 2M). Frequency of intratumoral MHCIIlo (Figure 2N), Arg1+ (Figure 2O), and MHCloCD206+FRβ+ (Figure 2P) TAMs was also increased in SM1 mice. Intratumoral CD4 T cell number and percentage were not significantly different between WT and SM1 mice (Figures S4A–S4C). However, the frequency of CD4 T cells that expressed T-bet, CD44, or Klrg1 was reduced in SM1 mice (Figures S4D–S4F). To determine if antigen-specific CD4 T cells were sufficient to promote MHCIIhi TAMs, tumor-specific OTII or non-specific SM1 CD4 T cells were infused into Rag1−/− mice bearing orthotopic KPC-OVA tumors (Figure 2Q). OTII cells tended to reduce tumor mass (Figure S4G) and significantly increased MHCIIhi TAMs while decreasing Arg1+ and MHCloCD206+FRβ+ TAMs (Figures 2R–2T). Intratumoral OTII cells also trended to be increased compared with SM1 T cells (Figures S4H and S4I). CD4 T cell depletion failed to alter the bias toward suppressive TAMs in the neoantigen-negative (CB−) parental KPC2 tumors in CCR2CreER R26Tdtomato mice (Figure S5). Thus, tumor-specific CD4 T cells promote monocyte differentiation into MHCIIhi TAMs, and in their absence, monocytes default to suppressive states.
MHC class II on monocyte-derived TAMs promotes Th1 CD4 T cells and anti-tumor immunity
To investigate the role of monocyte-derived macrophage antigen presentation in tumors, we generated CCR2CreER MHCIIfl/fl mice to conditionally delete MHC class II on monocyte-derived macrophages (Figures 3A and 3B). Loss of MHC class II on monocyte-derived TAMs increased tumor weight and Arg1+ and MHCloCD206+FRβ+ TAMs (Figures 3C–3E). Remaining MHCII+ TAMs in tamoxifen-treated CCR2CreER MHCIIfl/fl mice were MHCIIloCD206+FRβ+ tissue-resident phenotype, confirming the targeting approach specificity (Figure S6A). Furthermore, dendritic cell (DC) MHC class II was maintained (Figures S6B and S6C), supporting the specificity of this approach. Loss of MHC class II on monocyte-derived TAMs reduced intratumoral CD4 T cell frequency and number (Figures 3F and 3G) and intratumoral CD8 T cell number (Figure 3H). Strikingly, MHC class II loss on monocyte-derived TAMs also increased T regulatory cell (Treg) frequency (Figure 3I) and decreased T-bet+ and IFNγ-producing CD4 T cells (Figures 3J and 3K). Finally, MHC class II loss increased the frequency of CD8 T cells coexpressing PD1 and Lag3 (Figure 3L), markers that identify exhausted T cells30 and may be a result of increased Treg and/or suppressive TAMs in this context. Thus, MHC class II on monocyte-derived TAMs is critical for promoting anti-tumor Th1 cells and antitumor immunity.
Figure 3. MHC class II on monocyte-derived TAMs promotes Th1 CD4 T cells and anti-tumor immunity.

(A) Experimental approach to test the impact of MHC class II deletion in monocyte-derived macrophages. See also Figure S6.
(B) MHC class II staining gated on TAMs at day 12. Data are mean ± SEM; n = 6 mice per group; ****p < 0.0001; Student’s t test.
(C) Tumor weights. Data are mean ± SEM; n = 6 mice per group; *p < 0.05; Student’s t test.
(D) Proportion of Arg1+ macrophages. Data are mean ± SEM; n = 6 mice per group; ****p < 0.0001; Student’s t test.
(E) Proportion of MHCIIloCD206+FRβ+ macrophages. Data are mean ± SEM; n = 6 mice per group; ****p < 0.0001; Student’s t test.
(F) Frequency and number of T cells. Data are mean ± SEM; n = 6 mice per group; *p < 0.05, **p < 0.005; Student’s t test for each tissue.
(G) Frequency and number of CD4 T cells. Data are mean ± SEM; n = 6 mice per group; *p < 0.05, **p < 0.005; Student’s t test for each tissue.
(H) Frequency and number of CD8 T cells. Data are mean ± SEM; n = 6 mice per group; **p < 0.005; Student’s t test for each tissue.
(I) Representative Foxp3 staining gated on CD4 T cells and frequency. Data are mean ± SEM; n = 6 mice per group; ***p < 0.001; Student’s t test for each tissue.
(J) Frequency of CD4 T cells that are T-bet+. Data are mean ± SEM; n = 6 mice per group; **p < 0.005; Student’s t test for each tissue.
(K) Frequency of CD4 T cells producing IFNγ after ex vivo restimulation. Data are mean ± SEM; n = 6 mice per group; **p < 0.005, Student’s t test for each tissue.
(L) Proportion of CD8 T cells coexpressing Lag3 and PD1. Data are mean ± SEM; n = 6 mice per group; ****p < 0.0001; Student’s t test for each tissue.
Single-cell fate mapping of monocyte differentiation in PDA
We next sought to interrogate how CD4 T cells affect monocyte differentiation trajectories. We performed single-cell RNA sequencing (scRNA-seq) combined with cellular indexing of transcriptomes and epitopes sequencing (CITE-seq) of sorted CD45+Tomato+ and CD45+Tomato− immune cells from orthotopic tumors from control or CD4-depleted CCR2CreER R26Td tomato mice (Figure S7A). Given the overlap in phenotype of tumor-infiltrating TAMs following CD4 T cell depletion at days 7 and 14, we selected the day 7 time point because tumor weight was similar (Figure 2E). Control tumors were harvested at days 3 and 7, and CD4 T cell-depleted tumors were harvested at day 7 post monocyte labeling and tumor implantation (Figure S7A). Clustering of integrated data at the two time points revealed distinct immune populations (Figures 4A and S7B). Subclustering of monocyte/macrophage populations revealed five populations (Figure 4B). Cluster 1 was enriched for monocyte-specific genes, including Ly6c2, Ms4a4c, and Plac8 (Figure 4C). Clusters 2, 3, and 4 expressed macrophage-specific genes such as Adgre1, Cd68, and Fcgr1, suggesting three distinct macrophage populations (Figure S8A). Cluster 2 was enriched for Folr2, Lyve1, and Cd206 (Figures 4C and S8A), which are elevated in tissue-resident macrophages.37,38 Cluster 3 was enriched for MHC class II antigen presentation (H2-Ab1, H2-Aa, Cd74), and cluster 4 expressed immunosuppressive genes like Arg1 and Spp1 (Figure 4C), mirroring the MHCIIhi or MHCIIlo Arg-1+ populations (Figure 1B). Both Arg139 and Spp140 have immunosuppressive properties, suggesting pro-tumorigenic functions. Cluster 5 consisted of proliferating cells, as shown by abundant Mki67 and Top2a (Figure 4C). Based on CITE-seq analysis, MHC class II protein was almost exclusively restricted to cluster 3, while FRβ was primarily unique to cluster 2 (Figure 4D). PD-L1 was expressed by both clusters 3 and 4, yet was absent from cluster 2. We next compared gene expression in macrophage clusters with canonical pro-inflammatory or anti-inflammatory macrophage gene sets. Cluster 3 (MHCIIhi) expressed more genes associated with a pro-inflammatory state, including IFNγ-inducible chemokine ligands Cxcl9 and Cxcl10. In contrast, cluster 2 (Folr2+) and cluster 4 (MHCIIlo Arg1+) were enriched for anti-inflammatory genes such as Il10 (Figure 4E).
Figure 4. Single-cell fate mapping of monocyte differentiation in PDA.

(A) t-distributed stochastic neighbor embedding (t-SNE) plots of intratumoral immune cells isolated on days 3 and 7 ± αCD4 post tumor (n = 4 orthotopic KPC2a tumors per group). See also Figure S7.
(B) t-SNE plots of only monocytes and macrophage clusters.
(C) Heatmap showing normalized cluster-specific gene expression. See also Figure S8A.
(D) Normalized CITE-seq protein in t-SNE plot.
(E) Gene set enrichment analysis of canonical pro-inflammatory and anti-inflammatory genes split by monocyte/macrophage cluster defined in (B).
(F) t-SNE plot showing Tomato+ and Tomato− cells from days 3 and 7 untreated, and proportion of each monocyte/macrophage cluster among Tomato+ and Tomato− myeloid cells from clusters defined in (B). See also Figure S8B.
(G) t-SNE plot and proportion of each monocyte/macrophage cluster at days 3 and 7. See also Figure S8C.
We next assessed the relative contribution of monocytes to the TAM pool (Figure S8B). Most Tomato− macrophages clustered within the Folr2+ cluster, whereas Tomato+ macrophages clustered within the monocyte, MHCIIhi, and Arg1+ clusters (Figure 4F). Together, these data support that, at steady state, MHCIIhi and Arg1+ macrophages are derived from monocytes, and FRβ+ (Folr2+) macrophages are derived from tissue-resident macrophages.
To examine temporal changes, we compared the proportions of each cluster between days 3 and 7. At day 3, most macrophages clustered within the Folr2+ or Arg1+ population. However, by day 7, macrophages adopting the MHCIIhi phenotype increased (Figures 4G and S8C), suggestive of monocyte skewing toward MHCIIhi over time (Figure 1F) and corresponding to timing of developing an antigen-specific CD4 T cell response.
CD4 T cell depletion rewires monocyte differentiation toward a pro-tumor state
To further assess the role of CD4 T cells in monocyte fate, we clustered intratumoral Tomato+ cells from control or CD4 T cell-depleted mice. Most Tomato+ cells clustered in the MHCIIhi population in tumors from control mice (Figures 5A and 5B). In contrast, CD4 T cell depletion increased Tomato+ cells clustering with the Arg1+ or Folr2+ population, suggesting acquisition of a pro-tumor macrophage transcriptional profile, while impairing acquisition of an MHCIIhi state (Figures 5A and 5B). Furthermore, Tomato+ cells upregulated genes associated with tissue-resident macrophages, including Lyve1,38 in CD4 T cell-depleted tumors (Figure S8D). Using pseudotime,41 Folr2+ macrophages were predicted to be the most terminally differentiated population (Figures 5C and 5D). In the presence of CD4 T cells, monocytes gave rise to MHCIIhi or Arg1+ macrophages. In contrast, CD4 T cell depletion resulted in MHCIIhi and Arg1+ macrophages adopting a Folr2+ macrophage fate (Figure 5D). Kinetic analysis of cluster-defining genes showed that the upregulation of Arg1 and Folr2 was accelerated and H2-Aa was blunted in macrophages from CD4 T cell-depleted tumors (Figure S9). Together, the data support that tumor-specific CD4 T cells may halt MHCIIhi macrophage transition to a suppressive state.
Figure 5. CD4 T cell depletion rewires monocyte differentiation toward a pro-tumor state.

(A) t-SNE plot of intratumoral Tomato+ monocytes/macrophages from PBS- or αCD4-treated mice. See also Figure S8D.
(B) Proportion of each cluster among intratumoral Tomato+ monocyte/macrophages from PBS- or αCD4-treated mice.
(C) Pseudotime analysis of Tomato+ monocyte/macrophages split by pseudotime. See also Figure S9.
(D) Pseudotime analysis of Tomato+ monocyte/macrophages split by cluster.
Ifngr1 and CD40 signaling non-redundantly drive anti-tumor TAM differentiation
We utilized NicheNet42 to predict downstream ligand gene interactions driving anti-tumor TAM fate. By setting CD4 T cells as the donor cluster and MHCIIhi macrophages as the acceptor cluster, several predicted ligand-gene interactions, including Ifng, Tnf, and Cd40l, that may drive MHCIIhi macrophage differentiation were identified (Figure 6A). Both IFNγ and CD40L promote anti-tumor immunity43 and may be more highly expressed by CD4 T cells rather than CD8 T cells, in KPC2a tumors (Figure S10). MHCIIhi macrophages exhibited increased Stat1 compared with MHCIIlo macrophages (Figure 6B), consistent with enhanced downstream IFNγR signaling.44 Ifngr1−/− mice had significantly larger tumors at day 14 (Figure 6C) that correlated with expanded MHCIIlo and decreased MHCIIhi TAMs (Figure 6D). As predicted from NicheNet, tumors from Ifngr1−/− mice displayed an expansion of Arg1+ TAMs (Figure 6E) and MHCIIloCD206+FRβ+ TAMs compared with tumors from WT mice (Figure 6F), suggesting that IFNγR signaling mediates TAM fate decision toward an anti-tumor state. Contradictory to other models,45,46 PD-L1 was unchanged at day 7 and only slightly reduced at day 14 in Ifngr1−/− mice (Figure 6G), indicating IFNγR-independent mechanisms for driving PD-L1.
Figure 6. Ifngr1 and CD40 signaling non-redundantly drive anti-tumor TAM fate.

(A) NicheNet analysis from scRNA-seq data from Figure 4 using CD4 T cells as donor cells and MHCIIhi macrophages as acceptor cells. See also Figure S10.
(B) Stat1 staining and MFI in MHCIIhi and MHCIIlo intratumoral macrophages at day 7. Data are mean ± SEM; n = 4 mice per group; ****p < 0.0001; Student’s t test.
(C) Tumor weights from WT or Ifngr1−/− mice; n = 4–8 mice per group; ****p < 0.0001; Student’s t test for each time point.
(D) Proportion of MHCIIhi or MHCIIlo macrophages from WT or Ifngr1−/− mice. Data are mean ± SEM; n = 4–8 mice per group; ****p < 0.0001; Student’s t test for each time point.
(E) Proportion of Arg1+ macrophages on day 7. Data are mean ± SEM; ****p < 0.0001; Student’s t test.
(F) Proportion of MHCIIloCD206+FRβ+ macrophages from WT or Ifngr1−/− mice. Data are mean ± SEM; n = 4–8 mice per group; ****p < 0.0001; Student’s t test for each time point.
(G) PD-L1 MFI gated on total macrophages from WT or Ifngr1−/− mice. Data are mean ± SEM; **p < 0.005; Student’s t test for each time point.
(H) Normalized expression of selected genes from bulk RNA sequencing from sorted live MHCIIhi or MHCIIlo tumor macrophages from WT and Ifngr1−/− mice at day 14. See also Figure S11.
(I) Gene set enrichment analysis (GSEA) of the top 300 differentially expressed genes from sorted cells in (H).
(J) Orthotopic KPC2a tumor weights at day 7 from WT control mice, WT mice treated with CD40L blockade (αCD40L), or Cd40−/− mice. *p < 0.05; one way ANOVA with Tukey’s post test. See also Figure S12.
(K) Proportion of MHCIIhi or MHCIIlo macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 4–5 mice per group; ***p < 0.001; one-way ANOVA with Tukey’s posttest.
(L) Proportion of Arg1+ macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 4–5 mice per group; ***p < 0.001; one-way ANOVA with Tukey’s posttest.
(M) Proportion of MHCIIloCD206+FRβ+ macrophages. Each dot is an independent mouse. Data are mean ± SEM; n = 4–5 mice per group; ***p < 0.001; one-way ANOVA with Tukey’s posttest. See also Figure S13.
To elucidate transcriptomic changes driven by IFNγR signaling on TAMs, we performed bulk RNA sequencing on sorted MHCIIhi and MHCIIlo tumor macrophages from WT or Ifngr1−/− mice (Figure S11). Differentially expressed gene (DEG) analysis showed a downregulation in IFNγ-stimulated and pro-inflammatory genes in both MHCIIhi and MHCIIlo populations from Ifngr1−/− macrophages, with a compensatory increase in anti-inflammatory genes (Figure 6H). Moreover, IL1b, Pdgfa, and Fgf1, which promote tumor growth,47–49 were increased in Ifngr1−/− macrophages. The MHCIIhi population downregulated cell-cycle genes in the absence of IFNγR signaling (Figure 6H), suggesting that, in contrast to a well-established anti-proliferative role, IFNγ can promote MHCIIhi macrophage proliferation. Furthermore, there was a decrease in glycolytic genes in Ifngr1−/− macrophages, particularly in the MHCIIhi population, suggesting that IFNγ promotes MHCIIhi macrophage glycolysis. Pathway analysis indicated upregulation of cholesterol metabolism in Ifngr1−/− MHCIIhi macrophages, consistent with metabolic reprogramming (Figure 6I). Pathway analysis of the MHCIIlo subsets revealed an enrichment of processes involved in proliferation in Ifngr1−/− macrophages (Figure 6I). Overall, the data suggest that IFNγR signaling is a key regulator of monocyte fate specification and has differential effects on macrophage proliferation and metabolism in a subset-specific manner.
Signaling downstream of CD40 was enriched in fate-mapped, monocyte-derived MHCIIhi macrophages (Figure 6A). To determine if endogenous CD40/CD40L between CD4 T cells and macrophages promotes anti-tumor TAMs, we blocked this pathway using either anti-CD40L or Cd40−/− mice. Genetic loss of CD40 or CD40L blockade caused an increase in tumor weight (Figure 6J). Abrogating CD40/CD40L resulted in TAM populations mirroring those of CD4 T cell-depleted animals, including a decrease in MHCIIhi macrophages, an increase in MHCIIlo macrophages (Figure 6K), and an expansion of both Arg1+ (Figure 6L) and MHCIIloCD206+FRβ+ (Figure 6M) macrophages. Such changes were not due to a decrease in CD4 T cell infiltration, activation, or IFNγ production (Figure S12).
Given the proposed role of TNF-α in driving inflammatory macrophages in other models50 and NicheNet (Figure 6A), we tested if TNF receptor 1 signaling affected macrophage fate by implanting KPC2a tumors into Tnfr1−/− mice. Although we observed a minor trend for enrichment of MHCIIhi macrophages, this was not significant, and the proportion and number of TAM subpopulations were largely similar at day 7 (Figure S13). Together, IFNγ and CD40 rather than Tnfr1 promote MHCIIhi anti-tumor TAMs.
Human PDA TAM profiling
To compare our findings with human PDA, we performed a deep characterization of myeloid cells from publicly available human PDA scRNA-seq data.51 After data integration of human tumors, 14 distinct cell populations were identified (Figures 7A and S14A). We noted abundant populations of both CD4 and CD8 T cells in human PDA, like in KPC2a tumors (Figure 4A). Clustering of myeloid cells generated seven unique clusters (Figure 7B). Cluster 4 expressed genes associated with undifferentiated monocytes (PLAC8, CD115) and lacked granulocytic genes (ELANE, CXCR2) (Figure S14B), while clusters 0, 1, 2, 3, 5, and 7 expressed genes of differentiated myeloid cells (CD68, CSFR1) (Figures 7C and S14B). Cluster 0 expressed genes similar to those of the murine Arg1+ population (SPP1, FN1) (Figure 7C), but lacked ARG1 expression. Cluster 3 expressed genes associated with antigen presentation (HLA-DQB2), paralleling the murine MHCIIhi macrophages. Cluster 3 also expressed CD1C and CD1D (Figure 7C), consistent with a phenotypical relationship to monocyte-derived DCs (MoDCs), which have been well defined in humans.52 Notably, cluster 3 was also enriched for CD14 and CSF1R, which are well-established markers on cells of monocyte lineage52 (Figure S14B). Cluster 1 was enriched for FOLR2 (Figure 7C), resembling tissue-resident macrophages in the KPC2a tumors and in human breast cancer.37 Both clusters 2 and 6 were enriched for TREM2 (Figure 7D), which may mark monocyte-derived TAMs.37 Cluster 2 expressed higher transcript levels of complement-associated genes like C1QB, while cluster 6 expressed higher levels of GPNMB (Figures 7C and 7D), supporting heterogeneity among TREM2+ TAMs. Next, we performed pseudotime analysis to map monocyte differentiation trajectories. Using monocytes as the origin, this model predicted two endpoint trajectories, either to FOLR2+ and TREM2+ TAMs or to MHCIIhi MoDCs (Figure 7E). Given that our mouse data suggested that CD4 T cells instruct monocyte differentiation toward a more immunostimulatory phenotype, we next predicted potential interactions between CD4 T cells and MoDCs using NicheNet. This algorithm predicted that TNF and CD40/CD40L signaling was enriched between these two populations (Figure 7F).
Figure 7. Profiling of TAMs from human PDA.

(A) Uniform manifold approximation and projection (UMAP) of scRNA-seq data from six merged human tumors from Elyada et al.51 See also Figure S14.
(B) UMAP reclustering of myeloid clusters in (A).
(C) Heatmap of normalized gene expression of the top differentially expressed genes.
(D) Violin plots of selected genes.
(E) Pseudotime of myeloid clusters using Monocle 3 split by pseudotime and by cluster.
(F) NicheNet analysis using CD4 T cells as the donor cluster and MoDCs as the acceptor cluster.
(G) Gating scheme and frequency of myeloid subpopulations from resected human PDA (n = 4) and normal adjacent (n = 3) tissue. Gated on live, CD15− cells. Data are mean ± SEM; *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001; one-way ANOVA with Tukey’s posttest.
(H) Representative HLA-DR staining and HLA-DR MFI from CD11bhiFRβ− and CD11bloFRβ+ macrophages from human PDA and normal adjacent tissue. Data are mean ± SEM. Each dot is an independent sample.
(I) Representative PD-L1 staining and PD-L1 MFI of each population isolated from human PDA.
(J) Correlation of CD4 gene signatures with macrophage subpopulation gene signatures in human PDA. TCGA datasets were determined using GEPIA2. Gene signatures were derived from the top 5 DEGs from (B).
To examine the relative contributions of TAM subsets, we performed flow cytometry on samples from resected PDA and normal adjacent human pancreas. We identified two major macrophage populations in normal adjacent tissue and tumor: CD11bhiFRβ− or CD11bloFRβ+ macrophages (Figure 7G). Given that embryonically derived macrophages express lower levels of CD11b,53 CD11bloFRβ+ may be tissue-resident TAMs. Consistent with this, most macrophages in normal adjacent tissue adopted a CD11bloFRβ+ phenotype; however, within the tumor, more macrophages were CD11bhiFRβ− (Figure 7G), suggesting most of the TAM pool in human PDA is derived from monocytes. Unlike in our mouse model, FRβ+ TAMs expressed similar amounts of HLA-DR compared with FRβ− cells (Figure 7H), consistent with FRβ-expressing TAMs in human breast cancer.37 However, like the KPC2a orthotopic PDA model, FRβ− macrophages tended to express higher PD-L1 (Figure 7I), suggesting monocyte-derived CD11bhiFRβ− macrophages may regulate immunity. Finally, to determine if CD4 T cells influence immunostimulatory monocyte differentiation in human PDA, we performed correlation analysis using transcriptomic data of human PDA in GEPIA. Gene signatures for CD4 T cells were derived from our integrated dataset and compared with gene signatures from each macrophage population. CD4 T cell gene signature showed the strongest correlation with MoDCs, while immunosuppressive TREM2+ TAMs showed a much weaker correlation (Figure 7J). Thus, while differences in other factors may confound this analysis, the data suggest a link between activated CD4 T cells and MoDC differentiation over immunosuppressive TAM formation.
DISCUSSION
Here, we identify bidirectional crosstalk between macrophages and antigen-specific CD4 T cells that orchestrate anti-tumor immunity in PDA. Our data uncover that tumor-specific CD4 T cells instruct monocyte differentiation into anti-tumor macrophages through cognate antigen recognition and downstream CD40:CD40L and IFNγ pathways. Critically, in the absence of tumor-specific CD4 T cells or MHC class II on monocyte-derived macrophages, monocytes adopt a phenotypic and transcriptomic profile indistinguishable from that of tissue-resident macrophages. Our study uncovers that monocytes can masquerade as tissue-resident TAMs in tumors that lack an MHC class II-restricted tumor antigen, and CD4 T cell recognition of tumor antigen presented by monocyte-derived macrophages may contribute to macrophage heterogeneity in cancer.
Using macrophage fate-mapping approaches in a setting in which tumor-specific T cells are engaged, we show that monocyte and embryonically derived TAMs have distinct phenotypes. We find that most TAMs are derived from monocytes and confirm that embryonically derived TAMs are phenotypically distinct from monocyte-derived TAMs, consistent with a prior study.24 As tumor-reactive T cells can accelerate monocyte recruitment,17,27 the contribution of monocytes to the overall TAM pool may be dependent on tumor antigenicity and vary among cancers.
Through direct recognition of tumor cells that express MHC class II, or by effector cytokine production, CD4 T cells can promote tumor rejection in mouse models.54,55 The adoptive transfer of neoantigen-specific CD4 T cells can lead to solid tumor regressions in advanced cancer patients.56–58 Here we show that CD4 T cell recognition of tumor antigens presented by monocyte-derived macrophages initiates a cascade of events in the TME that likely work in concert to impede tumor cell growth. Not only did monocytes differentiate into anti-tumor TAMs when they expressed MHC class II, but also overt changes were detected in the T cell compartments, including increasing intratumoral accumulation of Th1 T cells and decreasing Treg and exhausted CD8 T cells. These data highlight that strategies to amplify CD4 T cells that express tumor-specific MHC class II-restricted TCRs are a potent approach for modulating the TME for controlling pancreatic tumor growth.
In other cancer types, CD8 T cell engagement with TAMs has been suggested to promote CD8 T cell exhaustion.17 In clear cell renal carcinoma, exhausted CD8 T cells and anti-inflammatory macrophages express ligands and receptors that support T cell dysfunction.16 Here, we show that antigen-specific CD4 T cells promote pro-inflammatory cues within the TME leading to monocyte differentiation into anti-tumor macrophages. CD8 T cells are dispensable for monocyte fate, consistent with the fact that macrophages are not efficient at cross presentation, which is mainly carried out by specific conventional DC subsets in vivo.59 Type I DCs, but not macrophages, are critical for priming and maintaining tumor-specific CD8 T cells in the KPC2a model.26 Type 2 DCs prime naive CD4 T cells in other contexts.60,61 Thus, monocyte-derived macrophage presentation of tumor antigen locally likely promotes recruitment, proliferation, and/or survival of recently primed CD4 T cells. Reactivation of CD4 T cells by monocyte-derived macrophages may also imprint Th1 lineage commitment.
Our data support that tumor-specific CD4 T cell instruction of monocyte fate rather than stochastic cell-intrinsic fate choices underlies macrophage heterogeneity in cancer. Monocytes can differentiate into immunostimulatory monocyte-derived DCs or immunosuppressive TAMs in human PDA,62 supporting our findings here. CD4-TAM interplay occurs in other models where infusion of tumor-reactive T cells leads to upregulation of inflammatory markers on TAMs63; however, it was unclear if this was a repolarization event or changes in tumor-infiltrating monocyte fate decisions. Th2 CD4 T cells can skew macrophages toward an anti-inflammatory state within tumors,64 highlighting the linkage between macrophages and CD4 T cells. Since macrophages acquire tumor debris within the TME,26 and fate is governed by antigen-specific CD4 T cells, it is likely that T cell-mediated instruction of macrophage fate is occurring intratumorally.
Resident macrophages appear predisposed to adopt a protumor state and contribute to tumor growth.24,65–67 Our observations suggest that tissue-resident macrophages are resistant toward rewiring to an immunostimulatory state and that, in the absence of tumor-specific CD4 T cells, monocytes adopt tissue-resident features. Thus, a fate-mapping approach is likely critical for distinguishing monocyte-derived from embryonic-derived TAMs. As embryonically derived macrophages become highly proliferative following CD4 T cell depletion, they may produce factors that direct infiltrating monocyte fate toward a similar tissue-resident-like state.
Human PDA has been stratified into squamous, pancreatic progenitor, immunogenic, or aberrantly differentiated endocrine exocrine genomic subtypes.18 The immunogenic subset contains genes enriched for T cells and myeloid cells,18 consistent with our cellular analysis of tumor-infiltrating immune cells in resected human PDA13 and the KPC2a model used herein.30 While the nature of the neoepitopes in human PDA is unknown, neoantigen quality correlates with long-term survival in PDA,7,68 human PDA can harbor neoantigen-reactive CD4 T cells,69 and a personalized neoantigen vaccine containing both MHC class I and MHC class II epitopes can stimulate T cells in human PDA.70 Together, our findings supporta pivotal role for tumor-specific CD4 T cells in governing anti-tumor macrophage fate and inform immunotherapy design by directing efforts to engage anti-tumor CD4 T cells.
Limitations of the study
While Arg1+ 39,71 and Lyve1+ 67 macrophages are immunosuppressive and promote tumor progression in other cancer models, we did not directly test if deletion of these subsets led to improved outcomes in PDA. While CD40L is reported to be mostly restricted to CD4 T cells72,73 and CD4 T cells are potent producers of IFNγ,74,75 we did not test the CD4 T cell intrinsic role of these molecules on macrophage phenotype. Our study is focused on invasive rather than preinvasive disease, and thus our claims apply to understanding advanced disease.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information should be directed to and will be fulfilled by the lead contact, Ingunn M. Stromnes (ingunn@ umn.edu ).
Materials availability
Further information and requests for resources and reagents generated in this report should be directed to and will be fulfilled by the lead contact, Ingunn M. Stromnes (ingunn@umn.edu).
Data and code availability
Single-cell RNA sequencing data and BulkRNA sequencing data of tumor associated macrophages have been deposited on the Gene Expression Omnibus (GEO) under the accession number GSE233068 and are publicly available.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
Human resected tumor and normal adjacent tissues were obtained from BioNET, a University of Minnesota IRB-approved protocol and tissue bank for investigators. All patients gave informed consent. Tumor and normal adjacent sample 1 were from a 74 year old white male. Tumor and normal adjacent sample 2 were from a 67 year old white female. Tumor and normal adjacent sample 3 were from a 63 year old white female. Tumor sample 4 was from a 61 year old white male.
Animals
University of Minnesota Institutional Animal Care and Use Committee (IACUC) approved all animal studies. 6- to 12-wk-old female and male C57BL/6J (000664), Ifngr1−/− (003288), Tnfrsf1a−/− (003242), Cd40−/− (002928), and CX3CR1CreER (020940) mice were purchased from The Jackson Laboratory and on a C57Bl/6 background. Sm1xRag1−/− mice77, CCR2-CreER-GFP (C57BL/6-Ccr2em1(icre/ERT2)Peng/J) x I-AB-flox (B6.129X1-H2-Ab1tm1Koni/J), OT-II (B6.Cg-Tg(TcraTcrb)425Cbn/J) and Rag−/− (B6.Cg-Rag2tm1.1Cgn/J) were kindly provided by Dr. Marc Jenkins (University of Minnesota). CCR2creERT2 [C57BL/6NTac-Ccr2tm2982(T2A-Cre7ESR1-T2A-mKate2)] reporter mice76 were kindly provided by Burkhart Becker (University of Zurich) and were crossed to R26-tdTomato reporter mice (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J). Animals were maintained in SPF conditions at the University of Minnesota Research Animals Resources facility with free access to food and water and kept on a 12-hour light-dark cycle.
Tumor cell lines
The KPC2a cell line was transduced to express click beetle red luciferase linked to eGFP (CB-eGFP)30. Tumor cells were cultured in Basic media: DMEM (Life Technologies) + 10% FBS (Life Technologies) + 2.5 mg/ml amphotericin B (Life Technologies) + 100 mg/ml penicillin/streptomycin (Life Technologies) + 2.5 g dextrose (Fisher Chemical) at 37˚C + 5% CO2. Medium was sterile filtered and stored in the dark at 4˚C. Cell lines used for experiments were maintained below passage 15 and 0.25% trypsin-EDTA (Thermo Fisher) was used for cell passage.
METHOD DETAILS
Orthotopic tumor cell implantation
For orthotopic tumor implantation, mice received 1 mg/kg slow-release buprenorphine injected subcutaneously prior to surgery for analgesia. Mice were anesthetized using continuous flow of 2–5% isoflurane. Hair was removed using clippers and Nair (Church & Dwight Co., Inc.) and the abdomen was sterilized using a series of 100% EtOH and Betadine washes. Once mice reached surgical plane anesthesia, a small incision was made in the abdomen followed by a small incision in the peritoneum to access the pancreas. 1×105 KPC2a cells in 20 μl of 60% Matrigel (Discovery Labware) were injected into the pancreas using an insulin syringe (Covidien)30. Sutures were used to close the peritoneum (Ethicon) and skin was closed using wound clips (CellPoint Scientific). Mice were monitored daily for 5 days to ensure healing of outer skin.
Tamoxifen administration
For fate mapping studies, Ccr2 reporter mice were gavaged orally with 250 μl of Tamoxifen (Sigma Aldrich Cat: T5648) at 20 mg ml−1 in corn oil on the day of tumor implantation or 1 day prior to tumor implantation or as indicated.
In vivo antibody treatments
For T cell depletion studies, mice were injected intraperitoneally (I.P.) with 200 μg of either anti-CD4 (BioXcell, Cat#-BE0003–1) or anti-CD8 (BioXcell, Cat#-BE0061) on days −1, +2 and +10 post tumor implantation. For CD40L blockade experiments, 500 μg of anti-CD40L (BioXcell, Cat#-BP0017–1) was injected I.P. on days −1 and +2 post tumor implantation.
Preparation of mononuclear cells from tissues
Spleens were mechanically dissociated to single cells followed by RBC lysis in 1 ml of Tris-ammonium chloride (ACK) lysis buffer (Life Technologies) for 2 min at room temperature (rt). RBC lysis was quenched by addition of 9 ml of T cell media. Splenocytes were centrifuged at 1400 rpm for 5 min, resuspended in T cell media (DMEM, 10% FBS, 2 μM L-glutamine, 100 U/ml penicillin/streptomycin, 25 μM 2-β-mercaptoethanol) and kept on ice until further analysis. Tumors were collagenase digested at 37°C for 15 minutes then mechanically digested to single cell suspensions and washed twice to remove cell debris and pancreatic enzymes.
Cell surface staining
Cells were stained in the presence of 1:500 Fc block (CD16/32, Tonbo) and antibodies diluted 1:200 in FACs buffer (PBS+2.5% FBS) for 45 minutes in the dark at 4°C. Ghost viability dye BV540 (Tonbo) was used to exclude dead cells at 1:500. Cells were fixed in 2% PFA or fixation buffer (Tonbo) for 10–15 at room temperature in the dark prior to data acquisition. Cells were acquired within 24 h using a Cytek Aurora.
Intracellular staining
The Foxp3 intracellular staining kit (Tonbo) was used for detecting intracellular transcription factors and proteins. Following cell surface staining, cells were washed 2X in FACs buffer, fixed for 30 min at 4°C, washed 2X in permeabilization buffer, stained with antibodies diluted 1:100 in permeabilization buffer for 1–2 hours in the dark at 4°C. Cells were washed 2X in permeabilization buffer, resuspended in FACs buffer and acquired within 24 h on a Cytek Aurora flow cytometer following addition of cell counting beads (Sigma).
PMA/Ionomycin restimulation
To determine ex vivo T cell functionality from tumor-bearing mice, single cell suspensions from spleen and tumor were obtained and activated in vitro30. Briefly, mononuclear cells were restimulated with 1X Cell Stimulation Cocktail (eBioscience) in the presence of Golgiplug and Golgistop (BD) according to manufacturer’s instructions in T cell media. 4–5 hours later, cells were stained with live/dead ghost dye at 1:500 (Tonbo) and the following antibodies at diluted in FACs buffer at 1:200 against CD45 (30F-11, BD), CD3 (17A2, Biolegend), CD4 (RM4.5, Tonbo), CD8 (53–6.7, Tonbo), Klrg1 (2F1, eBioscience), and CD44 (IM7, Tonbo) for 30 minutes at 4°C in the dark. Cells were washed 2X in FACs buffer, fixed/permeabilized using the BD cytofix/cytoperm kit (BD) and stained with anti-IFNγ (XMG1.2, Biolegend) diluted 1:100 in perm/wash buffer for 1 h at 4°C. Cells were washed 2X in perm/wash buffer, resuspended in FACs buffer and stored overnight at 4°C in the dark. Cells were acquired the following day on a Fortessa 1770 flow cytometer following the addition of counting beads (Sigma) and analyzed using FlowJo software (version 10).
Murine tumor scRNAseq sample acquisition and data analysis
For scRNAseq, 4 tumors from each group were harvested and processed to generate single cell suspensions. Live CD45+ Tomato+ cells and CD45+ Tomato-cells were FACS sorted using a BD FACSAria II. Each population was then stained with hashtag oligo antibodies (BD Biosciences; HTO#9 and HTO#10) and BD Bioscience CITEseq antibodies: CD274 (Cat:153604), IA-IE (Cat:107653), CD11b (Cat: 101265), Folate receptor beta (Cat: 153307) for 30 mins at 4°C (1:500) then recombined at a 1:1 mix. Sorted cells were resuspended in a final concentration of 100 cells per μl in 1X PBS containing 0.04% BSA for single cell capture of approximately 20,000 cells per group. Cells were submitted to University of Minnesota Genomics Core (UMGC) for single cell 10X Chromium 3’ GEX Capture and NovaSeq 2 ×150 S4 sequencing targeting ~50,000 reads per cell.
For preprocessing of the mouse scRNAseq data, we removed genes detected in less than 10 cells, potential empty cells with less than 200 feature counts, and apoptotic cells possessing more than 25% mitochondrial mRNA content. We then utilized DoubletFinder78 to perform a more elegant doublet removal independently for each sequence capture prior to data merging and integration. NormalizeData and ScaleData functions from Seurat (v4.0.1) were used for normalization and scaling. Variable features were extracted using FindVaribleFeatures function. For integration purpose, we chose Harmony package79. The first 20 principle components were used to generate uniform manifold approximation and projection (UMAP) and t-distributed stochastic neighbor embedding (tSNE). For gene set scores, Seurat AddModuleScore function was used, where pro-inflammatory (H2-Aa, Cxcl9, Il1b, Cxcl10, Tnf) and anti-inflammatory (Mrc1, Il10, Siglec1, FRβ, Arg1) gene sets were used. SingleR (v1.6.1)80 was used as an unbiased computational method for immune cell type annotation. Clusters generated using resolution 0.1 that were identified as SingleR monocyte and macrophage were extracted for further sub-clustering, pseudo-time trajectory and intercellular interaction analysis. Trajectory analysis were performed with support of Monocle3 (v1.0.0)41. In order to avoid batch effects across captures, we applied “harmony” as the base reduction method for all Monocle3 functions. Cells in the monocyte cluster were used as the root population to calculate pseudo-time inference of monocyte-macrophage differentiation. Pseudo-time parameter from ordered cells was extracted for visualization in “harmony” embeddings. For intercellular communication analysis, we utilized NicheNet (v1.0.0)42. All NicheNet models were first converted to mouse gene symbols using convert_human_to_mouse_symbols function.
Bulk RNA sequencing collection and analysis
Tumor single cell suspensions were isolated from 4 WT and 4 Ifngr1−/− mice and CD45+ CD11b+ F4/80+ MHCIIhi and MHCIIlo macrophages were FACS sorted into Trizol for RNA extraction. A minimum of 10,000 cells were sorted and submitted to UMGC for RNA isolation and sequencing using the Novaseq platform. Bulk RNAseq processing was performed using CHURP pipeline developed by the Minnesota Supercomputing Institute, which implemented and integrated Trimmomatic, HISAT2, SAMTools and featureCounts. Mus musculus GRCm38 (Ensembl release 102) was used as mouse reference genome. Differential expression analysis was adopted from DEseq2 (v.1.32.0). Pathway analyses were performed using fgsea function from the fgsea package (v.1.18.0).
Human PDA samples
De-identified and resected human tumor and normal adjacent tissues were obtained from BioNET, a University of Minnesota IRB-approved protocol and tissue bank for investigators. Tumors were from patients diagnosed with PDA. Mononuclear cells were stored at −80°C in Cryostor and thawed for staining and flow cytometric analysis like mouse samples.
Human PDA scRNAseq data analysis
Publicly available scRNAseq data from tumors from 6 PDA patients available from Elyada et al51 was downloaded after NIH approval at dbGaP (accession number phs001840.v1.p1). Filtered count matrices for 6 human tumor samples (SRR9274536, SRR9274537, SRR9274538, SRR9274539, SRR9274542, SRR9274544) were used as input data. Analytic tools used for human PDA scRNAseq data were identical to that of mouse scRNAseq described above, with the exception that SingleR model training was performed using HumanPrimaryCellAtlasData from Celldex.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad software (version 9.0). Mouse experiments include n=3–8 mice per group. Unpaired, two-tailed Student’s T test was used to compare two-group data. One-way ANOVA and Tukey posttest were used for comparing >2-group data. Data are presented as mean ± standard error of the mean (SEM), and p<0.05 was considered significant. *p<0.05, **p<0.005, ***p<0.0005, and ****p<0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| anti-mouse CD45 (clone 30-F11) | BD Bioscience | Cat#: 566168; RRID: AB_2739565 |
| anti-mouse Ly6C (clone HK1.4) | Biolegend | Cat#: 128031; AB_2562177 |
| anti-mouse CD11b (clone M1/70) | Biolegend | Cat#: 101237; RRID: AB_11126744 |
| anti-mouse F4/80 (clone BM8) | Biolegend | Cat#: 123149; RRID: AB_2564589 |
| anti-mouse Ly6G (clone 1A8) | Biolegend | Cat#: 127645; RRID: AB_2566317 |
| anti-mouse CD206 (clone C068C2) | Biolegend | Cat#: 141715; AB_2561991 |
| anti-mouse PD-L1 (clone 10F.9G2) | Biolegend | Cat#: 124313; RRID: AB_10639934 |
| anti-mouse FRβ (clone 10F.9G2) | Biolegend | Cat#: 153305; RRID: AB_2721312 |
| anti-mouse Arg1 (clone A1exF5) | ThermoFisher | Cat#: 25369782: RRID: AB_2734841 |
| anti-mouse I-A/I-E (clone M5/114.15.2) | Biolegend | Cat#: 107651; RRID: AB_2616728 |
| anti-mouse Ki67 (clone 16A6) | Biolegend | Cat#: 652420; RRID: AB_2564285 |
| anti-mouse CD3 (clone 17A2) | BD Bioscience | Cat#: 740530: RRID:AB_2740239 |
| anti-mouse CD8 (clone 53–6.7) | Biolegend | Cat#: 100759; RRID: AB_2563510 |
| anti-mouse CD44 (clone IM7) | Tonbo Bioscience | Cat#: 65-0041; RRID:AB_2621847 |
| anti-mouse CD4 (clone RM4-5) | ThermoFisher | Cat#: 56004282; RRID: AB_494000 |
| anti-mouse Klrg1 (clone 2F1) | eBioscience | Cat#: 25589382; RRID: AB_1518768 |
| anti-mouse Foxp3 (clone FJK-16s) | eBioscience | Cat#: 48577382; RRID: AB_1518812 |
| anti-mouse T-bet (clone 4B10) | Biolegend | Cat#: 644810; RRID: AB_2200542 |
| anti-mouse Stat1 (clone D1K9Y) | Cell Signaling | Cat#: 80916S |
| anti-mouse IFNγ (clone XMG1.2) | Invitrogen | Cat#: 24731141 |
| anti-mouse CD19 (clone 1D3) | Biolegend | Cat#: 152403; RRID: AB_2629812 |
| anti-mouse CD115 (clone AFS98) | Biolegend | Cat#:135525: RRID: AB_2566461 |
| anti-mouse Lyve1 (clone ALY7) | ThermoFisher | Cat#: 14-0443-82 |
| anti-human CD11b (clone ICRF44) | Biolegend | Cat#: 301310; RRID:AB_314162 |
| anti-human CD11c (clone 3.9) | eBioscience | Cat#: 25016642 |
| anti-human CD64 (clone 10.1) | Biolegend | Cat#: 305036; RRID: AB_2650834 |
| anti-human CD14 (clone 61D3) | ThermoFisher | Cat#: 11014942; RRID:AB_10597597 |
| anti-human CD15 (clone HI98) | BD Bioscience | Cat#: 564232; RRID: AB_2738686 |
| anti-human FRβ (clone 94b) | Biolegend | Cat#: 391703; RRID: AB_2721335 |
| anti-human HLA-DR (clone L243) | BD Bioscience | Cat#: 552764; RRID: AB_394453 |
| anti-human PD-L1 (clone 29E.2A3) | Biolegend | Cat#: 329722; RRID: AB_2565764 |
| anti-human CD163 (clone GHI/61) | Biolegend | Cat#: 333632; RRID: AB_2728288 |
| InVivoMAb anti-mouse CD4 (clone GK1.5) | BioXcell | Cat#:BE0003-1; RRID: AB_1107636 |
| InVivoMAb anti-mouse CD8 (clone 2.43) | BioXcell | Cat#:BE0061; RRID: AB_1125541 |
| InVivoMAb anti-mouse CD40L (clone MR1) | BioXcell | Cat#:BE0017-1; RRID: AB_1107601 |
| TotalSeq™-A0190 anti-mouse CD274 | Biolegend | Cat#:153604; RRID: AB_2783125 |
| TotalSeq™-A0564 anti-mouse FRβ | Biolegend | Cat#:153307; RRID: AB_2800690 |
| TotalSeq™-A0117 anti-mouse I-A/I-E | Biolegend | Cat#: 107653; RRID: AB_2750505 |
|
| ||
| Biological samples | ||
|
| ||
| Patient PDA Samples | University of Minnesota BioNet | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| PFA 4% | VWR International | Cat#: 9713.1000 |
| Tamoxifen | Sigma | Cat#: T5648 |
| DMEM medium | Life Technologies | Cat#: 21875091 |
| Red blood cell lysis buffer | BD Bioscience | Cat#: 555899 |
| Fetal bovine serum | ThermoFisher | Cat#: 12350273 |
| Amphotericin B | GIBCO | Cat#: 15290018 |
| Penicillin/streptomycin | GIBCO | Cat#: 10378016 |
| Dextrose | ThermoFisher | Cat#: D15-500 |
| EDTA | ThermoFisher | Cat#: 15575020 |
| L-Glutamate | ThermoFisher | Cat#: 25030081 |
| β-mercaptoethanol | GIBCO | Cat#: M6250 |
| Ghost viability dye BV540 | Tonbo | Cat#: 13-0879-T100 |
| Golgiplug | BD Bioscience | Cat#: 555029 |
| Collagenase IV | Sigma Aldrich | Cat#: V900893 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Foxp3 fixation and permeabilization kit | Tonbo | Cat#: TNB-0607-KIT |
| CD4+ T Cell Isolation Kit, mouse | Miltenyi | Cat# 130-092-916 |
| Cell Stimulation Cocktail (500X) | eBioscience | Cat#: 00-4970-03 |
|
| ||
| Deposited data | ||
|
| ||
| Mouse scRNAseq | GEO | GSE233068 |
| Mouse Bulkseq | GEO | GSE233068 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| KPC2a cells | Burrack et. al.30 | N/A |
| KPC OVA cells | Burrack et. al.30 | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | Jackson Labs | 000664 |
| Ifngr1−/− | Jackson Labs | 003288 |
| Tnfrsf1a−/− | Jackson Labs | 003242 |
| Cd40−/− | Jackson Labs | 002928 |
| CX3CR1CreER (B6.129P2(C)-Cx3cr1tm2.1 (cre/ERT2)Jung/J) | Jackson Labs | 020940 |
| CCR2creERT2 [C57BL/6NTac-Ccr2tm2982 (T2A-Cre7ESR1-T2A-mKate2)] | Croxford et. al.76 | N/A |
| R26-tdTomato (B6.Cg-Gt(ROSA)26Sortm9 (CAG-tdTomato)Hze/J). | Jackson Labs | 007909 |
| CCR2-CreER-GFP (C57BL/6-Ccr2em1 (icre/ERT2)Peng/J) x I-AB-flox (B6.129X1-H2-Ab1tm1Koni/J) | This report | N/A |
| Sm1xRag1−/− | Srinivasan et. al.36 | N/A |
| Rag1−/− (B6.Cg-Rag2tm1.1Cgn/J) | Jackson Labs | 008309 |
| OTII (B6.Cg-Tg(TcraTcrb)425Cbn/J) | Jackson Labs | 004194 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo version 10 | FlowJo | N/A |
| FACS Diva | BD | N/A |
| GraphPad | Prism | N/A |
|
| ||
| Other | ||
|
| ||
| Matrigel | Discovery Labware | 08-774-552 |
| 4.0 Sutures | Ethicon | 1611G |
| 5.0 Sutures | Ethicon | 8580H |
Highlights.
Monocytes are progenitors to most heterogeneous macrophage subsets in PDA
Tumor-specific CD4 T cells drive anti-tumor MHCIIhi macrophages
Loss of MHC class II on monocyte-derived macrophages promotes tumor progression
IFNγ and CD40 are non-redundant drivers of MHCIIhi antitumor macrophages
ACKNOWLEDGMENTS
We acknowledge the University of Minnesota Flow Cytometry Core and Research Animal Resource facility. M.T.P. is supported by an AHA predoctoral fellowship (903380). Z.C.S. is supported by a NIH T32 AG029796 followed by NIH F31CA275289. M.M.F. was supported by the Ministry of Science and Higher Education of the Russian Federation (agreement 075-15-2022-301). K.Z. was supported by the Priority 2030 Federal Academic Leadership Program. J.W.W. is supported by NIH R01AI165553 and AHA CDA 855022. I.M.S. is supported by NIH R01CA249393, R01CA255039, and P01CA254849; Department of Defense PA200286; a PanCAN-AACR Catalyst Award (19-35-STRO); and an American Cancer Society Institutional Research Grant (124166-IRG-58-001-55-IRG65).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112732.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Siegel RL, Miller KD, Fuchs HE, and Jemal A.(2022). Cancer statistics, 2022. Ca - Cancer J. Clin 72, 7–33. 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Schmiechen ZC, and Stromnes IM (2020). Mechanisms governing immunotherapy resistance in pancreatic ductal adenocarcinoma. Front. Immunol 11, 613815. 10.3389/fimmu.2020.613815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, and Tuveson DA (2012). The pancreas cancer microenvironment. Clin. Cancer Res 18, 4266–4276. 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiryu S, Ito Z, Suka M, Bito T, Kan S, Uchiyama K, Saruta M, Hata T, Takano Y, Fujioka S, et al. (2021). Prognostic value of immune factors in the tumor microenvironment of patients with pancreatic ductal adenocarcinoma. BMC Cancer 21, 1197. 10.1186/s12885-021-08911-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, and Hiraoka N.(2013). Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 108, 914–923. 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahajan UM, Langhoff E, Goni E, Costello E, Greenhalf W, Halloran C, Ormanns S, Kruger S, Boeck S, Ribback S, et al. (2018). Immune cell and stromal signature associated with progression-free survival of patients with resected pancreatic ductal adenocarcinoma. Gastroenterology 155, 1625–1639.e2. 10.1053/j.gastro.2018.08.009. [DOI] [PubMed] [Google Scholar]
- 7.Balachandran VP, quksza M, quksza M, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. (2017). Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516. 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, and Hingorani SR (2014). Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 63, 1769–1781. 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stromnes IM, DelGiorno KE, Greenberg PD, and Hingorani SR (2014). Stromal reengineering to treat pancreas cancer. Carcinogenesis 35, 1451–1460. 10.1093/carcin/bgu115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, and Vonderheide RH (2012). Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835. 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, and Bar-Sagi D.(2012). Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847. 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, and Vonderheide RH (2007). Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 67, 9518–9527. 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 13.Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, and Hingorani SR (2017). T-Cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol. Res 5, 978–991. 10.1158/2326-6066.CIR-16-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee H-J, Thurston G, Zhang Y, Lazarus J, Sajjakulnukit P, et al. (2019). Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metabol. 29, 1390–1399.e6. 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. (2013). Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73, 1128–1141. 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braun DA, Street K, Burke KP, Cookmeyer DL, Denize T, Pedersen CB, Gohil SH, Schindler N, Pomerance L, Hirsch L, et al. (2021). Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 39, 632–648.e8. 10.1016/j.ccell.2021.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kersten K, Hu KH, Combes AJ, Samad B, Harwin T, Ray A, Rao AA, Cai E, Marchuk K, Artichoker J, et al. (2022). Spatiotemporal co-dependency between macrophages and exhausted CD8+ T cells in cancer. Cancer Cell 40, 624–638.e9. 10.1016/j.ccell.2022.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJC, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 19.Ma R-Y, Black A, and Qian B-Z (2022). Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 43, 546–563. 10.1016/j.it.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Chijimatsu R, Kobayashi S, Takeda Y, Kitakaze M, Tatekawa S, Arao Y, Nakayama M, Tachibana N, Saito T, Ennishi D, et al. (2022). Establishment of a reference single-cell RNA sequencing dataset for human pancreatic adenocarcinoma. iScience 25, 104659. 10.1016/j.isci.2022.104659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christofides A, Strauss L, Yeo A, Cao C, Charest A, and Boussiotis VA (2022). The complex role of tumor-infiltrating macrophages. Nat. Immunol 23, 1148–1156. 10.1038/s41590-022-01267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, Brown M, Chang C, Troncoso L, Chen ST, et al. (2021). Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 595, 578–584. 10.1038/s41586-021-03651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franklin RA, and Li MO (2016). Ontogeny of tumor-associated macrophages and its implication in cancer regulation. Trends Cancer 2, 20–34. 10.1016/j.trecan.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et al. (2017). Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338.e6. 10.1016/j.immuni.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, and DeNardo DG (2014). CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069. 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burrack AL, Schmiechen ZC, Patterson MT, Miller EA, Spartz EJ, Rollins MR, Raynor JF, Mitchell JS, Kaisho T, Fife BT, and Stromnes IM (2022). Distinct myeloid antigen-presenting cells dictate differential fates of tumor-specific CD8+ T cells in pancreatic cancer. JCI Insight 7, e151593. 10.1172/jci.insight.151593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stromnes IM, Burrack AL, Hulbert A, Bonson P, Black C, Brockenbrough JS, Raynor JF, Spartz EJ, Pierce RH, Greenberg PD, and Hingorani SR (2019). Differential effects of depleting versus programming tumor-associated macrophages on engineered T cells in pancreatic ductal adenocarcinoma. Cancer Immunol. Res 7, 977–989. 10.1158/2326-6066.CIR-18-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho WJ, and Jaffee EM (2021). Macrophage-targeting by CSF1/1R blockade in pancreatic cancers. Cancer Res. 81, 6071–6073. 10.1158/0008-5472.CAN-21-3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Byrne KT, and Vonderheide RH (2016). CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. 15, 2719–2732. 10.1016/j.celrep.2016.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burrack AL, Spartz EJ, Raynor JF, Wang I, Olson M, and Stromnes IM (2019). Combination PD-1 and PD-L1 blockade promotes durable neoantigen-specific T cell-mediated immunity in pancreatic ductal adenocarcinoma. Cell Rep. 28, 2140–2155.e6. 10.1016/j.celrep.2019.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Y, Schrank PR, and Williams JW (2022). Macrophage fate mapping. Curr. Protoc 2, e456. 10.1002/cpz1.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolfi B, Gallerand A, Firulyova MM, Xu Y, Merlin J, Dumont A, Castiglione A, Vaillant N, Quemener S, Gerke H, et al. (2022). Unravelling the sex-specific diversity and functions of adrenal gland macrophages. Cell Rep. 39, 110949. 10.1016/j.celrep.2022.110949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallerand A, Stunault MI, Merlin J, Luehmann HP, Sultan DH, Firulyova MM, Magnone V, Khedher N, Jalil A, Dolfi B, et al. (2021). Brown adipose tissue monocytes support tissue expansion. Nat. Commun 12, 5255. 10.1038/s41467-021-25616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, Murphy KM, Yokoyama WM, Randolph GJ, and Unanue ER (2015). The pancreas anatomy conditions the origin and properties of resident macrophages. J. Exp. Med 212, 1497–1512. 10.1084/jem.20150496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srinivasan A, Foley J, Ravindran R, and McSorley SJ (2004). Low-dose Salmonella infection evades activation of flagellin-specific CD4 T cells. J. Immunol 173, 4091–4099. 10.4049/jimmunol.173.6.4091. [DOI] [PubMed] [Google Scholar]
- 37.Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Nú ñ ez NG, Tosello Boari J, Richer W, Menger L, Denizeau J, et al. (2022). Tissue-resident FOLR2+ macrophages associate with CD8+ T cell infiltration in human breast cancer. Cell 185, 1189–1207.e25. 10.1016/j.cell.2022.02.021. [DOI] [PubMed] [Google Scholar]
- 38.Dawson CA, Pal B, Vaillant F, Gandolfo LC, Liu Z, Bleriot C, Ginhoux F, Smyth GK, Lindeman GJ, Mueller SN, et al. (2020). Tissue-resident ductal macrophages survey the mammary epithelium and facilitate tissue remodelling. Nat. Cell Biol 22, 546–558. 10.1038/s41556-020-0505-0. [DOI] [PubMed] [Google Scholar]
- 39.Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, and Weissleder R.(2018). Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 8, 5842–5854. 10.7150/thno.26888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P, et al. (2021). A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e23. 10.1016/j.cell.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 41.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Browaeys R, Saelens W, and Saeys Y.(2020). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162. 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 43.Burrack AL, Rollins MR, Spartz EJ, Mesojednik TD, Schmiechen ZC, Raynor JF, Wang IX, Kedl RM, and Stromnes IM (2021). CD40 agonist overcomes T cell exhaustion induced by chronic myeloid cell IL-27 production in a pancreatic cancer preclinical model. J. Immunol 206, 1372–1384. 10.4049/jimmunol.2000765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlin AF, Vizcarra EA, Branche E, Viramontes KM, Suarez-Amaran L, Ley K, Heinz S, Benner C, Shresta S, and Glass CK (2018). Deconvolution of pro- and antiviral genomic responses in Zika virus-infected and bystander macrophages. Proc. Natl. Acad. Sci. USA 115, E9172–E9181. 10.1073/pnas.1807690115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mimura K, Teh JL, Okayama H, Shiraishi K, Kua L-F, Koh V, Smoot DT, Ashktorab H, Oike T, Suzuki Y, et al. (2018). PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 109, 43–53. 10.1111/cas.13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201. 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Das S, Shapiro B, Vucic EA, Vogt S, and Bar-Sagi D.(2020). Tumor cell-derived IL1β promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 80, 1088–1101. 10.1158/0008-5472.CAN-19-2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahraei M, Roy LD, Curry JM, Teresa TL, Nath S, Besmer D, Kidiyoor A, Dalia R, Gendler SJ, and Mukherjee P.(2012). MUC1 regulates PDGFA expression during pancreatic cancer progression. Oncogene 31, 4935–4945. 10.1038/onc.2011.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhattacharyya S, Oon C, Kothari A, Horton W, Link J, Sears RC, and Sherman MH (2020). Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J. Exp. Med 217, e20191805. 10.1084/jem.20191805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mysore V, Tahir S, Furuhashi K, Arora J, Rosetti F, Cullere X, Yazbeck P, Sekulic M, Lemieux ME, Raychaudhuri S, et al. (2022). Monocytes transition to macrophages within the inflamed vasculature via monocyte CCR2 and endothelial TNFR2. J. Exp. Med 219, e20210562. 10.1084/jem.20210562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123. 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heger L, Hofer TP, Bigley V, de Vries IJM, Dalod M, Dudziak D, and Ziegler-Heitbrock L.(2020). Subsets of CD1c+ DCs: dendritic cell versus monocyte lineage. Front. Immunol 11, 559166. 10.3389/fimmu.2020.559166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SEW, Pollard JW, et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 54.Poncette L, Bluhm J, and Blankenstein T.(2022). The role of CD4 T cells in rejection of solid tumors. Curr. Opin. Immunol 74, 18–24. 10.1016/j.coi.2021.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, Meng W, Lichti CF, Esaulova E, Vomund AN, et al. (2019). MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574, 696–701. 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645. 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, Behjati S, Velds A, Hilkmann H, Atmioui DE, et al. (2015). High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med 21, 81–85. 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- 58.Veatch JR, Lee SM, Fitzgibbon M, Chow I-T, Jesernig B, Schmitt T, Kong YY, Kargl J, Houghton AM, Thompson JA, et al. (2018). Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J. Clin. Invest 128, 1563–1568. 10.1172/JCI98689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joffre OP, Segura E, Savina A, and Amigorena S.(2012). Cross-presentation by dendritic cells. Nat. Rev. Immunol 12, 557–569. 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 60.Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, et al. (2019). Unleashing type-2 dendritic cells to drive protective antitumor CD4+ T cell immunity. Cell 177, 556–571.e16. 10.1016/j.cell.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee H-W, Park CG, et al. (2007). Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111. 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez E, Boelaars K, Brown K, Eveline Li RJ, Kruijssen L, Bruijns SCM, van Ee T, Schetters STT, Crommentuijn MHW, van der Horst JC, et al. (2021). Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat. Commun 12, 1270. 10.1038/s41467-021-21550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perez-Diez A, Liu X, and Matzinger P.(2022). Neoantigen presentation and IFNγ signaling on the same tumor-associated macrophage are necessary for CD4 T cell-mediated antitumor activity in mice. Cancer Res. Commun 2, 316–329. 10.1158/2767-9764.crc-22-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, and Coussens LM (2009). CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102. 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Loyher P-L, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, Torstensson S, Bercovici N, Baudesson de Chanville C, Combadière B, et al. (2018). Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med 215, 2536–2553. 10.1084/jem.20180534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Etzerodt A, Moulin M, Doktor TK, Delfini M, Mossadegh-Keller N, Bajenoff M, Sieweke MH, Moestrup SK, Auphan-Anezin N, and Lawrence T.(2020). Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med 217, e20191869. 10.1084/jem.20191869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baer JM, Zuo C, Kang L-I, Alarcon de la Lastra A, Borcherding NC, Knolhoff BL, Bogner SJ, Zhu Y, Lewis MA, Zhang N, et al. (2022). Pancreas resident macrophage-induced fibrosis has divergent roles in pancreas inflammatory injury and PDAC. Preprint at bioRxiv. 10.1101/2022.02.09.479745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Łuksza M, Sethna ZM, Rojas LA, Lihm J, Bravi B, Elhanati Y, Soares K, Amisaki M, Dobrin A, Hoyos D, et al. (2022). Neoantigen quality predicts immunoediting in survivors of pancreatic cancer. Nature 606, 389–395. 10.1038/s41586-022-04735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meng Q, Valentini D, Rao M, Moro CF, Paraschoudi G, Jäger E, Dodoo E, Rangelova E, del Chiaro M, and Maeurer M.(2019). Neoepitope targets of tumour-infiltrating lymphocytes from patients with pancreatic cancer. Br. J. Cancer 120, 97–108. 10.1038/s41416-018-0262-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, Lihm J, Ceglia N, Guasp P, Chu A, et al. (2023). Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 618, 144–150. 10.1038/s41586-023-06063-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fu Y, Pajulas A, Wang J, Zhou B, Cannon A, Cheung CCL, Zhang J, Zhou H, Fisher AJ, Omstead DT, et al. (2022). Mouse pulmonary interstitial macrophages mediate the pro-tumorigenic effects of IL-9. Nat. Commun 13, 3811. 10.1038/s41467-022-31596-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grewal IS, Xu J, and Flavell RA (1995). Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature 378, 617–620. 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- 73.Elgueta R, Benson MJ, De Vries VC, Wasiuk A, Guo Y, and Noelle RJ (2009). Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev 229, 152–172. 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ngai P, McCormick S, Small C, Zhang X, Zganiacz A, Aoki N, and Xing Z.(2007). Gamma interferon responses of CD4 and CD8 T-cell subsets are quantitatively different and independent of each other during pulmonary Mycobacterium bovis BCG infection. Infect. Immun 75, 2244–2252. 10.1128/IAI.00024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Green AM, DiFazio R, and Flynn JL (2013). IFN-G from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J. Immunol 190, 270–277. 10.4049/jimmunol.1200061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, and Becher B.(2015). The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 43, 502–514. 10.1016/j.immuni.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 77.Reiley WW, Shafiani S, Wittmer ST, Tucker-Heard G, Moon JJ, Jenkins MK, Urdahl KB, Winslow GM, and Woodland DL (2010). Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 107, 19408–19413. 10.1073/pnas.1006298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e4. 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, and Raychaudhuri S.(2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172. 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA sequencing data and BulkRNA sequencing data of tumor associated macrophages have been deposited on the Gene Expression Omnibus (GEO) under the accession number GSE233068 and are publicly available.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact on request.