

Abstract
Normal tissues are essential for studying disease-specific differential gene expression. However, healthy human controls are typically available only in postmortal/autopsy settings. In cancer research, fragments of pathologically normal tissue adjacent to tumor site are frequently used as the controls. However, it is largely underexplored how cancers can systematically influence gene expression of the neighboring tissues. Here we performed a comprehensive pan-cancer comparison of molecular profiles of solid tumor-adjacent and autopsy-derived “healthy” normal tissues. We found a number of systemic molecular differences related to activation of the immune cells, intracellular transport and autophagy, cellular respiration, telomerase activation, p38 signaling, cytoskeleton remodeling, and reorganization of the extracellular matrix. The tumor-adjacent tissues were deficient in apoptotic signaling and negative regulation of cell growth including G2/M cell cycle transition checkpoint. We also detected an extensive rearrangement of the chemical perception network. Molecular targets of 32 and 37 cancer drugs were over- or underexpressed, respectively, in the tumor-adjacent norms. These processes may be driven by molecular events that are correlated between the paired cancer and adjacent normal tissues, that mostly relate to inflammation and regulation of intracellular molecular pathways such as the p38, MAPK, Notch, and IGF1 signaling. However, using a model of macaque postmortal tissues we showed that for the 30 min – 24-hour time frame at 4ºC, an RNA degradation pattern in lung biosamples resulted in an artifact “differential” expression profile for 1140 genes, although no differences could be detected in liver. Thus, such concerns should be addressed in practice.
Keywords: Cancer research, Molecular pathology, Autopsy, Tumor matched pathologically normal tissues, Healthy tissue controls, Differential gene expression analysis, RNA sequencing, Molecular pathways
Graphical Abstract
1. Introduction
Normal tissue controls are crucial for examining differential gene expression profiles associated with human pathology. However, obtaining healthy human norms is problematic for most of the tissues and available only for postmortal autopsy biosamples. Several projects were initiated to create reference banks of RNA sequencing (RNAseq), expression microarray, and proteomic profiles of healthy human tissues such as GTEX [1], ANTE [2], and CPTAC [3]. In GTEX project database, totally 979 paired RNAseq and microarray profiles for 54 human tissue types are available that correspond to multiple autopsy materials from donors who died from different reasons including diseases. Tissues were analyzed both by sequencing and by gene expression microarrays to enable a technology comparison [1]. Some GTEX RNAseq profiles were shown to contain signs of minor batch-specific cross-tissue contamination [4]. In turn, we created ANTE collection including only 196 RNAseq profiles for 20 tissue types, but they were obtained for the donors killed in road accidents that could be regarded relatively free from severe chronic diseases and, therefore, more likely represent true “healthy” tissue controls [2]. From CPTAC project repository, 907 Orbitrap proteomic profiles of 10 normal human tissues can be explored, but for smaller number of genes compared to the above transcriptomic databases: for 6755 proteins compared to ∼60 600 transcripts, respectively [1].
Alternatively, in cancer research, so-called tumor matched norms (fragments of pathologically normal tissue located adjacent to tumor site and removed during surgery/biopsy) are frequently used as the controls [5], [6]. Perhaps the most complete such data repository was provided by The Cancer Genome Atlas (TCGA) database which includes roughly 2900 RNAseq profiles for tumor matched pathologically normal samples of 33 tissue types [7].
However, cancer cells can influence neighboring tissues in many ways including by causing inflammation and by producing growth factors [8]. This can result in coordinated expression patterns between the cancer and neighboring “normal” tissue profiles. The latter was recently demonstrated for an impressive DNA repair pathway activation congruence between the cancerous and matched normal thyroid tissues [9]. Tumor cells can significantly influence adjacent normal tissues by altering their functions and forcing them to acquire new phenotypes and/or to produce molecular factors necessary for tumor growth and spread [10], [11] To do this, cancer cells can affect both the surrounding non-malignant cells and the extracellular matrix [12], [13]. Tumors can suppress immune editing or cause a pro-inflammatory condition due to the use of cancer associated fibroblasts (CAFs) and tumor associated macrophages (TAMs). They can also stimulate pericytes and endothelial cells to promote angiogenesis [14]. They can influence activities of intrinsic signaling pathways of the neighboring cells to better detach and invade in the form of circulating tumor cells [15]. The specific DNA repair pathway activation profiles there were strongly connected between the tumor and the corresponding matched normal tissues, which directly indicates their molecular interplay [9], [16]. Thus, tumor matched “normal” tissues can be pathologically biased. However, to the date this possibility remained largely underexplored at the transcriptome-wide level.
Here we investigated correlations of gene expression and molecular pathway activation patterns between the cancer and matched paired normal tissues in TCGA profiles for 22 cancer types, and experimentally validated the correlations found. Activities of few thousands of human molecular pathways can be algorithmically deduced using transcriptomic profiles of individual cancer and normal biosamples [17]. Pathway activation assay is the next-level way of gene expression data analysis, where positive/negative pathway activation levels (PALs) mean pathway activation/inhibition in a biosample compared to the control group, whereas zero PAL indicates no difference in the pathway activation [18], [19]. In addition, the extent of PAL quantitatively reflects up/downregulation of a pathway [20].
The first pan-cancer high-throughput comparison of molecular profiles of the tumor-adjacent pathologically normal tissues with the autopsy-derived “healthy” normal tissues was performed by D. Aran et al. The study revealed pro-inflammatory molecular peculiarities in tumor-adjacent normal tissues [8]. Here we analyzed experimental and literature datasets and demonstrate that compared to the autopsy-derived tissues, the tumor-adjacent norms have a number of systemic molecular differences including alterations of cancer drug targets.
These differences may be driven by molecular events that are correlated between the paired cancers and the adjacent non-cancer tissues. As a result, molecular targets of 32 and 37 cancer drugs appeared to be over- or underexpressed, respectively, in the tumor-adjacent compared to the “healthy” norms. Thus, the tumor-adjacent pathologically normal tissues cannot be considered as the fully adequate norms for the analysis of tumor molecular profiles, and autopsy tissue biosamples taken from the healthy donors can be a plausible alternative. However, we show that for the latter possibility an RNA degradation-induced bias in gene expression profiles has to be carefully taken into account.
2. Materials and methods
2.1. Patients and samples
All patients whose biosamples were included in this study have previously signed written informed consents to participate in the observational clinical investigation, and profiling of their biosamples by RNA sequencing using Illumina HiSeq3000 or Illumina NextSeq550 next generation sequencing platform. The patients signed agreement for publication of depersonalized RNAseq profiles of pairs of cancer and matched pathologically normal tissues for their biosamples, and for publication of study results in the form of gene activity profiles associated with age, sex, and diagnosis.
The study was planned and performed in accordance with the Declaration of Helsinki ethical principles. Local ethical committee at I.M. Sechenov First Moscow State Medical University and Vitamed clinic approved design of this study and its public presentation as the research paper. Pairs of biosamples where tumor matched pathologically normal tissue specimens were available were collected prospectively during the clinical trial Oncobox (NCT03724097) from February 2019 till December 2020. All biosamples were FFPE solid tumor blocks obtained from primary tumor sites and evaluated by pathologist, with no less than 60% of cancer cells, or matched pathologically normal tissue blocks with no detectable cancer cells (Table S1).
2.2. Animal biosamples
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the local Institutional Ethics Committee (protocol № 81/1, January 26, 2022) in the Research Institute of Medical Primatology, Sochi, Russia. Three adult Macaca Fascicularis monkeys were involved in the study. The animals were sacrificed by intravenous injection of 5.0 ml of 5% Anestofol (Interfarm LLC, Russia) with preliminary general anesthesia by intravenous injection of 0.10 ml/kg of 2% Xylazine (Interchemie Werken "de Adelaar" BV, Netherlands) and 0.05 ml/kg Zoletil (Virbac Sante Animale, France). After autopsy of lung and liver tissues, the aliquots of each sample were stored at 4ºC for 30 min, 3 h, 6 h, and 24 h, respectively, and then immediately frozen in liquid nitrogen prior to laboratory testing. The animals were: Animal 1 – 48 months-old male, body mass 2.96 kg; Animal 2 – 40 months-old male, body mass 2.58 kg; Animal 3 – 41 months-old male, body mass 3.4 kg.
2.3. RNA sequencing
RNA sequencing was performed at Department of Pathology and Laboratory Medicine, University of California Los Angeles, and at Laboratory of Clinical Genomic Bioinformatics, Sechenov First Moscow Medical University, according to [2], [21]. Library construction and depletion of ribosomal RNA were done using KAPA RNA Hyper with rRNA erase (HMR only) kit. For multiplexing of samples in one sequencing run different adaptors were used. Library concentrations were measured using Qubit ds DNA HS Assay kit (Life Technologies) and quality was assessed using Agilent Tapestation (Agilent). RNA sequencing was done using Illumina HiSeq 3000 engine for single-end sequencing, 50 bp read length, for at least 30 million (mln) raw reads per sample. Data quality check was done with Illumina SAV. De-multiplexing was performed using Illumina Bcl2fastq2 v 2.17 software.
2.4. Autopsy and tumor-adjacent normal tissue gene expression datasets
For comparison of tumor adjacent normal tissues vs normal tissues derived from healthy subjects we extracted 2911, 979 and 196 gene expression profiles from TCGA [7], GTEx [1] and experimental ANTE [2] databases, respectively (Table S2).
For studying correlation between tumors and adjacent normal tissues we extracted 715 gene expression profiles from TCGA [7] (Table S3).
For studying correlation between tumors and adjacent normal tissues at the proteomic level we extracted 306 protein expression profiles from the CPTAC database [22] (Table S4).
2.5. Processing of gene expression data
RNAseq FASTQ files were processed by STAR aligner (Dobin et al., 2013) using “GeneCounts” mode, Ensembl human transcriptome annotation (Build version GRCh38, transcript annotation GRCh38.89) or Ensembl Macaca fascicularis transcriptome annotation (Build version 6.0, transcript annotation 6.0.106). Ensembl gene IDs were converted to HGNC gene symbols using Complete HGNC dataset (https://www.genenames.org/, database version from 2017 July 13). Macaca fascicularis Ensembl gene symbols were converted to human Ensembl gene symbols using R biomaRt package. Overall, expression levels were determined for 36,596 genes with HGNC identifiers in case of human biosamples and 15,248 genes in case of Macaca fascicularis biosamples.
Differential gene expression analysis was performed using DESeq2. FDR-Adjusted P-value cut-off was set to 0.1, |Log2 fold change| cut-off was set to > 1.
2.6. Molecular pathway analysis
In this study we used a publicly available collection of molecular pathways extracted from Reactome [23], NCI [24], KEGG [25] and Qiagen (https://www.qiagen.com) databases, and algorithmically annotated for molecular functions of pathway components and nodes[17]. Using the Oncobox bioinformatics platform [19] we calculated pathway activation levels (PALs) for totally 2934 molecular pathways. For PAL calculations, each individual expression profile was normalized on mean geometrical levels of RNA expression for all samples in the dataset under analysis.
PAL approach considers the impact of each gene product on overall molecular pathway activation [18], PAL value for pathway in a given sample is calculated as follows:
where (case-to-normal ratio) is the ratio of gene expression level in the sample under investigation to the mean geometrical gene expression level in the group of control samples. The Boolean flag (beyond tolerance interval flag) is zero when the value has not passed the significance criterion: when the difference with the control group of samples is not significant, where . (activator/repressor role of gene n in pathway p) is the discrete value that equals to when gene product is a repressor of pathway p; , when gene product is an activator of pathway p; , when gene product has both activities of an activator and of a repressor of pathway p; and , respectively, when gene product is rather an activator or repressor of pathway .
2.7. Visualization
Pathway activation graphs were visualized using open.oncobox.com web-service [26]. Ggplot2 and pheatmap R packages were used for other plots. PCA was done for log transformed counts of all genes using prcomp R functions.
2.8. Permutation test
Statistical significance of the intersections shown on the Venn diagrams were investigated by randomly permutating gene/pathway names and by intersecting random gene groups (n = 10,000) of the same sizes as above for the actual data. Actual intersection was considered significant if higher number of genes/pathways was observed in less than 5% of random groups.
2.9. Experimental pathway and gene expression data availability
Experimental RNAseq data are available at NCBI Sequencing Read Archive under accession ID PRJNA905832. Pathway activation data calculated for experimental, TCGA, GTEX, and ANTE collections are given in Table S5.
3. Results and discussion
3.1. Design of the study
In this study we aimed to investigate major differences between human tumor-adjacent and healthy tissue normal biosamples at the transcriptomic level of gene expression and of molecular pathway activation at the pan-cancer level. Specific tasks were to explore (i) whether there are genes and molecular pathways that are regulated differently between the adjacent (tumor-matched) and healthy (autopsy-derived) norms, and (ii) whether there is a correlation between the cancers and their paired adjacent norms. Another task (iii) was to identify which of the correlated genes/pathways are connected with the patient identity or physiological conditions like infection, and which are due to the tumor influence on the neighboring tissues. To conclude (iv), we attempted to build a model of tumor transformation of the neighboring tissues at the level of major molecular mechanisms. In addition (v), we explored whether different times of postmortal biopsies can cause a bias in the gene expression/molecular pathway activation pattern on the primate model of crab-eating macaques.
3.2. Comparison of tumor-adjacent and healthy normal tissues
We explored differences between human healthy/autopsy and tumor-matched normal tissues at the levels of gene expression, molecular pathway activation, and cell type enrichment scores. Data sourсe of tumor-adjacent normal tissue was TCGA database, and healthy normal samples were extracted from GTEx and ANTE databases, respectively. We compared RNAseq profiles of 4086 samples: 2911 TCGA tumor-adjacent norms for colon (n = 723), kidney (n = 1032), and lung (n = 1156) with the autopsy-derived healthy tissue profiles from ANTE and GTEX databases (Table S2). The datasets were harmonized using two alternative approaches: Shambhala method [27], [28], or quantile normalization [29].
3.2.1. Gene level of data analysis
For every individual sample (tumor-adjacent normal or healthy normal) we assessed CNRs (Case-to-Normal-Ratios) as the major gene expression metric, i.e. fold-change of expression level in the individual sample relative to averaged expression in the healthy normal tissue (control group). Tumor-adjacent normal samples were extracted from TCGA database and healthy normal samples were extracted from GTEx or ANTE databases. A comparison was done for each of the three tissue types under investigation (colon, kidney, lung) separately. Two types of CNRs were calculated. First, a geometrical mean of GTEX samples was used as the control; second, a geometrical mean of ANTE group of samples was used as the control.
For statistical estimates of differential gene expression between TCGA and ANTE or GTEX samples, we used Student’s T-test between log2CNR of TCGA samples and of ANTE or GTEX samples, with the subsequent Benjamini-Hochberg FDR correction, significance threshold q < 0.05). We considered genes with mean log2CNR > 0, q < 0.05 as upregulated, and genes with log2CNR < 0, q < 0.05 as downregulated. The comparisons were done separately for the TCGA/GTEX, and TCGA/ANTE datasets. In order to exclude batch effect, comparisons were done separately for quantile normalized and Shambhala-harmonized expression profiles. Normalization methods can introduce bias, and we attempted to avoid bias specific to one of the normalization methods used. Thus, we intersected results of the two popular normalization methods which hopefully made our results free from distortion by one of the normalization algorithms. However, this approach has a limitation that some of true differential genes could be lost due to possible inconsistence between the two normalization methods used.
For the quantile normalized TCGA/ANTE comparison we totally found 3088/2990 differential up/downregulated genes for colon, 3070/3008 for lung, and 3088/2990 for kidney (Fig. S1). For the quantile normalized TCGA/GTEX comparison, there were 3631/3565 differential up/downregulated genes for colon, 3940/3256 for lung, and 3950/3246 for kidney (Fig. S1). For each of the three above human tissue types, we then intersected TCGA/ANTE and TCGA/GTEX differential gene expression profiles. In total, we found 2462/2347 common up/downregulated genes for colon, 2283/1936 genes for lung, and 1993/1663 genes for kidney. In every case, the intersections were non-random, as evidenced by the permutation test p < 0.0001, Fig. S1. This means that the experimentally observed intersection exceeded any of the 10,000 randomly generated intersections of the initial gene sets of the same size [30].
Similarly, for the intersection of the Shambhala normalized TCGA/ANTE and TCGA/GTEX comparisons (Fig. S1) we found 2761/2255 common up/downregulated genes for colon, 2316/1691 for lung, and 2103/1691 for kidney. In every case, the intersections were non-random with the permutation test p < 0.0001.
Finally, to obtain an overall differential profile of healthy vs tumor-adjacent tissues, we then intersected all the above differential profiles for the three tissue types under analysis, all datasets, and two expression normalization methods. In a total intersection, we detected 1056/818 common statistically significantly up/downregulated genes for the TCGA vs ANTE and GTEX datasets (Table S6). This overall intersection was non-random (permutation test p < 0.0001, Fig. 2A,B).
Fig. 2.

Results of the permutation test for total intersection between colon, kidney and lung tissue types, two harmonization methods (quantile normalization and Shambhala), and all datasets under analysis (TCGA vs GTEX, and TCGA vs ANTE). Observed (red) and random model-expected (black) number of intersections is shown for TCGA-upregulated (A) and downregulated (B) genes; and for TCGA-upregulated (C) and downregulated (D) molecular pathways.
We then performed Gene Ontology (GO) terms enrichment analysis for the genes which were up/downregulated after such an overall intersection. We found that for the upregulated genes the major enriched processes deal with the neutrophil degranulation and activation, antigen processing and presentation; protein folding and targeting; endoplasmic reticulum to Golgi vesicle transport and vesicle organization; macroautophagy; mitochondrial movement and gene expression, cellular respiration and catabolism of organic molecules; Krebs cycle and secondary alcohol synthetic processes; telomerase localization to Cajal bodies (Fig. 1A).
Fig. 1.

Gene Ontology terms enriched among the list of upregulated (A) and downregulated (B) genes found in an overall intersection for TCGA vs GTEX and ANTE comparisons.
Here, activated antigen processing and presentation processes may point on enhanced presentation of tumor-specific antigens in the tumor-adjacent tissues. In turn, the neutrophil degranulation occurs after activation of pathogen recognition receptors such as TLRs, or as the response on proinflammatory cytokines such as IL-8, TNF, and N-formyl-methionyl-leucyl-phenylalanine (fMLP) [31]. This may suggest increased inflammatory status of the tumor-matched norms compared to the autopsy-derived tissues, as previously observed by Aran and coauthors [8]. Further focused studies are needed to analyze these mechanisms in-depth. Other implicated processes evidence the influence of tumors on the metabolism and cell physiology of the neighboring tissues. Furthermore, the process of telomerase localization to Cajal bodies is not normally observed in the healthy solid tissues but instead frequently occurs in tumors where it reflects production of an active telomerase including both RNA and protein components in cancer cells. Thus, this process activation in “normal” adjacent tissues may indicate physical presence there of proliferation-competent cancer cells [32].
In contrast, the downregulated genes resulted in another set of enriched processes (Fig. 1B). First of all, these indicated altered degradation of extracellular matrix and cell-substrate adhesion patterns in the tumor-adjacent tissues. Other processes were connected with the regulation of signal transduction via small GTPases (including Ras), and with the regulation of G2/M cell cycle transition. The latter two processes are strongly interconnected as the activated Ras is the major positive regulator of passing through the G2/M checkpoint in cell cycle progression [33]. Of note, among the downregulated genes there were 24 members of the “Ras protein signal transduction” GO term, but no Ras gene family members themselves. KRAS gene is mutated in a significant proportion of colorectal cancer patients. Thus, it may be of interest to perform a separate series of analyses focused on the comparison of tumor-adjacent tissues in KRAS mutated vs wild type patients in the future, as well as for the other hot spot mutations as well, such as BRAF in melanoma and thyroid cancer, EGFR in lung cancer, BRCA1–2 in breast cancer, et cetera.
3.2.2. Pathway level of data analysis
We then performed similar analysis at the level of molecular pathway activation assessment in order to complement GO analysis and obtain additional insights on difference between tumor adjacent and healthy normal tissues. To this end, based on the CNR values for each gene, we calculated pathway activation levels (PALs) for 2934 molecular pathways using OncoboxPD online pathway analysis tool [26]. We then intersected quantile normalized TCGA/ANTE and TCGA/GTEX differential pathway profiles separately for the three tissue types under analysis (Fig. S2). In total, we found 927/830 common up/downregulated pathways for colon, 782/802 for lung, and 735/620 for kidney.
In the same way we found for the intersected Shambhala normalized TCGA/ANTE and TCGA/GTEX differential pathway profiles (Fig. S2) 1181/595 common up/downregulated pathways for colon, 913/536 for lung, and 868/428 for kidney (Fig. S2). In every case, the intersections were non-random with the permutation test p < 0.0001 (Fig. S2).
Further overall intersection between all three tissue types and both normalization methods gave a non-random (p < 0.0001) set of differentially activated pathways, where 384 were up-, and 178 were downregulated in the tumor-adjacent tissues (TCGA) compared to the healthy norms (GTEX and ANTE) (Table S5; Fig. 2C,D). The resulting top-10 up- and downregulated pathways are shown on Fig. 3.
Fig. 3.

Pathway activation chart for the top differentially activated pathways in tumor adjacent (TCGA) vs healthy (GTEX and ANTE) normal tissues. Mean pathway activation level (PAL) is shown for each pathway. Upregulated pathways are shown in green, and downregulated - in red color.
Specifically, the set of the most strongly upregulated pathways indicated altered cell-cell and cell-intracellular matrix interactions via E cadherin-derived adherens junctions, beta 7 integrin cell surface interactions, EGF pathway regulation of cytoskeleton organization, and Arf6-mediated endocytosis processes including recycling of receptor molecules located on cell surface. Furthermore, upregulated CDC42-linked pathways are also in line with the enhanced cell migration, endocytosis and cell cycle progression processes [34].
In contrast, the top downregulated pathways indicated suppressed apoptotic signaling and inhibited negative regulation of cell growth; decreased formation of neuronal synapses; decreased C20 prostanoid biosynthesis; decreased regulation of cell migration by VEGFR3; inhibited branches of NOTCH and Hedgehog pathways. Similar to our previous findings of degraded extracellular matrix organization at the level of GO analysis for individual genes, we also detected here strongly downregulated pathways of syndecan 1-mediated cell-matrix interaction, beta 1 integrin cell surface interactions, collagen biosynthesis, and chondroitin and dermatan biosynthesis (data not shown).
3.2.3. Technical validation: comparison of tumor adjacent and healthy normal tissues profiled by RNA sequencing using the same reagents and equipment
We then investigated whether specific trends identified in large-scale comparisons of TCGA tumor-adjacent norms with ANTE or GTEX autopsy-derived healthy tissues represented true functional differences or were related to cross-platform normalization bias. We performed an additional comparison of a small experimental group of tumor-adjacent norms with autopsy-derived healthy tissues (presented early as the ANTE database) profiled using the same equipment, reagents, research team and protocols according to [2]. To this end, six experimental tumor-adjacent and nine healthy liver samples were analyzed. In this case, no cross-platform normalization was needed because of uniform technical nature of RNA sequencing profiles.
3.2.4. Gene level technical validation
All samples were normalized on geometrical mean of nine autopsy-derived healthy tissue profiles. We calculated CNRs in the tumor-adjacent norms relatively to averaged expression in the autopsy-derived healthy tissues. Statistical estimates were performed in the same way as for the above TCGA vs ANTE/GTEX analysis. Thus, we totally identified 1393/1019 differential up/downregulated genes (Table S7).
We then intersected the above differential genes with the total gene intersection set obtained for the TCGA vs ANTE/GTEX comparison and detected 121/58 common statistically significantly up/downregulated genes (Table S7). This overall intersection was non-random (permutation test p < 0.0001, Fig. 5A,B).
Fig. 5.

Results of the permutation test for total intersection between colon, kidney and lung tissue types, two harmonization methods (quantile normalization and Shambhala), and all datasets under analysis (TCGA vs GTEX, and TCGA vs ANTE), and new analysis (experimental tumor-adjacent vs healthy norms). Observed (red) and random model-expected (black) number of intersections is shown separately for upregulated (A) and downregulated (B) genes; and for upregulated (C) and downregulated (D) molecular pathways.
We then performed GO enrichment analysis for the common upregulated genes. In agreement with the previous results, we found a very similar set of the enriched terms: neutrophil degranulation and activation; antigen processing and presentation; endoplasmic reticulum to Golgi vesicle transport and vesicle organization; macroautophagy; telomerase localization to Cajal bodies (Fig. 4).
Fig. 4.

Gene Ontology terms significantly enriched among the list of upregulated genes found in an overall intersection of TCGA vs GTEx/ANTE and experimental comparison differential gene lists.
Overall, this indicates congruent trends with the results obtained in the large-scale TCGA vs ANTE/GTEx comparison.
3.2.5. Pathway level technical validation
On the level of molecular pathway activation, we found 313/48 up/downregulated pathways (Table S7). Further intersection of these differential pathways with the total pathway intersection set obtained for the TCGA vs ANTE/GTEX comparison returned a list of non-random (p = 0.016) common 52 upregulated pathways and borderline significance (p = 0.055) list of six downregulated pathways (Table S7; Fig. 5C,D).
The top-10 upregulated pathways are shown in Table 1. Specifically, this set suggests activated DNA repair and related G2M checkpoint mechanism; CD28-mediated Vav1 activation which in turn activates the mitogen-activated protein kinases JNK and p38 [35]; processing of endosomal Toll-like receptors; T-cellular receptor signal transduction; activation of the BCR signaling promoting survival [36]; increased PD-1 signaling responsible for immune checkpoint inhibition, related with higher response rate and more prolonged progression-free survival [37]; and plasmalogen biosynthesis pathway strongly connected with inflammation [38].
Table 1.
Common differential pathways between overall intersection for TCGA vs GTEx/ANTE and within ANTE comparisons. Top-10 upregulated pathways are shown.
| Pathway ID | PAL |
|---|---|
| NCI Fanconi anemia Pathway (DNA repair) | 66.4 |
| reactome Chk1 Chk2 Cds1 mediated inactivation of Cyclin B Cdk1 complex Main Pathway | 61.1 |
| reactome G2 M DNA replication checkpoint Main Pathway | 59.0 |
| reactome CD28 dependent Vav1 Main Pathway | 52.3 |
| reactome Trafficking and processing of endosomal TLR Main Pathway | 48.7 |
| reactome Phosphorylation of CD3 and TCR zeta chains Main Pathway | 47.0 |
| reactome Regulation of the Fanconi anemia Main Pathway | 43.1 |
| NCI BCR signaling Pathway (cell survival) | 42.9 |
| reactome PD 1 signaling Main Pathway | 42.4 |
| reactome Plasmalogen biosynthesis Main Pathway | 42.4 |
Major pathways related to cell-cell and cell-intracellular matrix interactions revealed in the previous large-scale cross-platform analysis were also detected here but were not among the top-10 processes identified (Table S7).
Thus, at the pathway level we obtained congruent activation patterns with the cross-platform comparison. Taken together, this confirms the results obtained in the previous large-scale TCGA vs ANTE/GTEX comparison on both gene and pathway levels.
3.2.6. Cell type enrichment assay
We then used gene expression data to assess the cell type content of the tumor-adjacent and healthy norms. We performed bioinformatic cell type deconvolution using xCell method [39]. An overall intersection for all comparisons (TCGA/ANTE and TCGA/GTEX), tissue types (lung, kidney, and colon), and normalization methods (quantile or Shambhala) was non-random (permutation test p < 0.0001), and gave a figure of two cell types overrepresented in the tumor adjacent tissues compared to the healthy norms: (i) megakaryocyte erythroid progenitor (MEP) cells and (ii) natural killer T (NKT) cells (Fig. S3).
Interestingly, megakaryocyte erythroid progenitor cells were reported to be associated with tumor microenvironment and their content correlated with worse outcome in pancreatic cancer [40]. In turn, altered content of NKT cells in tumor microenvironment was also frequently mentioned in the literature (e.g. [41]).
Thus, when comparing the tumor-adjacent and healthy normal tissues for kidney, colon, and lung, we identified a number of statistically significantly differential gene expression patterns, molecular pathways and Gene Ontology terms, and also identified two associated cell types.
Such differences can be explained by the following three major reasons. First, alterations directly influenced by the tumors. Second, influence caused by the complex organismic reaction on tumors. Third, a possibility of an artifact component linked with the specific features of postmortal biosampling in “healthy” controls theoretically cannot be excluded.
We then attempted to further explore possible impacts of those factors in more detail. For the first two components (reciprocal influence of tumors and the adjacent tissues) we assessed genes and molecular pathways which expression/activation levels statistically significantly correlate between the tumors and the patient-matched adjacent norms (Fig. 6).
Fig. 6.

Correlation of gene expression and pathway activation level among TCGA-male, TCGA-female, and experimental group of paired tumor/normal samples. X-axis, Spearman correlation coefficient; Y-axis, negative logarithm of Benjamini-Hochberg FDR-corrected q-value. Color scale reflects the extent of the Spearman correlation coefficient value.
For the possible artifact component, we explored gene expression and pathway activation features of the experimental postmortal samples of crab-eating macaque tissues taken 30 min - 24 h after the animal death. We hope that the comparable size and the primate origin of these organisms makes them an adequate model of time-dependent RNA degradation in human tissues under analysis.
3.3. Genes and molecular pathways with coordinated activities between tumors and adjacent tissues
In order to quantitatively characterize associations between the paired tumor and matched/adjacent pathologically normal tissues we took 715 available pairs of matched cancer-normal RNA sequencing profiles from the TCGA database. The samples represented 23 cancer types and the corresponding normal tissues (Table S4). For all tests, we considered male (n = 349) and female (n = 366) biosamples separately and then merged the results to exclude sex-specific bias from the final results of gene/pathway activation analysis.
To explore which genes and pathways were significantly correlated between the paired tumor/normal biosamples, we calculated correlations for CNRs of separately taken genes between all cancer and all normal samples among the matched pairs under study. CNR for the tumor sample was calculated as a gene expression level in the tumor divided by geometric mean expression in all normal samples, except this patient. CNR for the normal sample was calculated as a gene expression level in the normal sample divided by geometric mean expression in all normal samples, except this patient.
On the correlation plots, dots were CNRs for an individual gene measured for paired cancer (X-axis) and normal (Y-axis) samples. The same analysis was done also for the pathway activation levels (PALs) of individual molecular pathways. We considered significant Spearman correlations that would be both (i) statistically significant after Benjamini-Hochberg FDR correction (q < 0.05), and (ii) either exceeding 0.2 threshold for positive correlations, or less than − 0.2 for negative correlations (Fig. 6).
In such a way we tested all available paired TCGA samples, and also an experimental cohort of 28 paired tumor/normal samples to validate the results (Fig. 5). Twenty-eight experimental pairs of tumor and adjacent pathologically normal samples were compared here with the control group of experimental healthy postmortal tissues that were obtained by us and published separately [42]. The current normal tissue adjacent to the tumor (NAT) and ANTE expression profiles were obtained by the same team, and sequenced using the same protocol, reagents and equipment. To our knowledge, ANTE database is unparalleled because for this collection only the tissues taken from healthy donors killed in road accidents were included. It contrasts with the samples deposited in GTEx database, where the patients normally died in the hospitals after disease [43].
Interestingly, we identified only positive, but no negative statistically significant correlations at the levels of both individual genes and molecular pathways, in both male and female paired tumor/normal samplings, and also in the experimental group (Table S8, Fig. 3). After triple intersection of all three groups of paired samples (male, female, and experimental), we found 1620 common genes and 12 common pathways (Table S9), all positively correlated, permutation test p < 0.0001 for genes and 0.0004 for pathways (Fig. 7).
Fig. 7.

Venn diagrams and permutation tests for triple intersection (TCGA-male vs TCGA-female vs experimental). A, Venn diagram for triple intersection at the gene level. B, Venn diagram for triple intersection at the pathway level. Results obtained with random permutations for genes (C) and pathways (D) are shown in black, red line corresponds to the experimentally observed numbers of intersected items.
Thus, we did a pan-cancer screen for the congruently regulated genes and pathways between tumors and paired norms, which are common in both male and female patients. We then investigated these common genes and pathways in more detail.
3.3.1. Coordinated gene expression patterns
The GO terms enrichment analysis showed that the triple-intersected gene set was most strongly enriched by the terms dealing with the chemosensory perception and olfaction, epithelial development, keratinocyte differentiation, regulation of JAK-STAT signaling and, specifically, phosphorylation of STAT, defense response to bacteria, and response to exogenous double-stranded RNA (dsRNA) (Fig. 8).
Fig. 8.

Gene Ontology terms enrichment analysis for triple-intersected set of the correlated paired tumor/normal genes.
We hypothesize that the correlation of processes dealing with defense against infectious agents like response to foreign dsRNA and response to bacteria can be explained by the common protective reaction of both cancer and adjacent non-cancer tissues on pathogen invasion.
The epithelial development and differentiation of keratinocytes can be related to many common processes in cancer and adjacent tissues such as the tumor encapsulation and the transforming influence of cancer cells e.g. through the production of growth factors [44], [45]. The correlated genes related to keratinization are listed in Table S10, and top-30 correlated keratinization genes are shown on Fig. 9A.
Fig. 9.

Top-30 genes with highest mean correlation among pairs of tumors and matched norms in TCGA male, TCGA female, and experimental samples. A, genes involved in epithelial/keratinocyte development Gene Ontology (GO) terms. B, genes involved in chemosensory perception GO terms. Color scale reflects correlation coefficients of each gene product between tumor and adjacent normal tissue samples.
For the chemosensory perception and olfaction GO terms, the significantly correlated gene products identified are shown in Table S11, top-30 genes shown on Fig. 9B. The link between aberrant expression of olfactory receptors and cancer development, progression, and metastasis was previously established for many cancer types [46], [47], [48]. However, to our knowledge there were no previous reports on the association of their profiles in tumors with the adjacent pathologically normal tissues. Since chemosensory perception and olfaction represented six most robust clusters of the correlated genes, this phenomenon may have a considerable yet poorly investigated significance in tumor biology.
In turn, the JAK-STAT signaling pathway activity and STAT phosphorylation are directly linked with the immunity and tumorigenesis [49]. This pathway primarily regulates events following cytokine binding to the immune cells. Specifically, the binding of interferons and interleukins to cell-surface receptors on the immune cells results in dimerization of the receptors, which are complexed with JAK proteins [50]. This brings JAKs from the two receptor molecules into close proximity, they are then reciprocally phosphorylated by each other at tyrosine residues, which additionally activates their kinase domains [51]. Activated JAKs then phosphorylate tyrosine residues of the receptor molecule, which creates binding platform for STAT proteins which are, in turn, phosphorylated by JAKs. This causes dissociation of STATs from the receptor complex, and their activation as the nuclear transcriptional regulators [52]. Thus, these signaling events can be directly implicated in the inflammation and tumor niche formation processes, which are all connected with the infiltration of tumor and neighboring tissues by the immune cells [53], [54]. The most strongly impactful nodes of the JAK-STAT pathway are shown on Fig. 10.
Fig. 10.

Pathway activation graph for JAK-STAT signaling pathway. Color depth of the pathway nodes is proportionate to averaged correlation of the respective gene products between tumors and matched norms in paired samples, for (A) TCGA male, (B) TCGA female, and (C) experimental samplings. Color scale denotes correlation of the respective node-forming gene product(s). Green arrows indicate activating interactions.
We then tried to perform such an assay at the proteomic level. To this end we took five publicly available datasets of paired tumor-normal proteomic profiles (totally 309 pairs of samples) from the CPTAC database. We collected profiles for totally 139 female and 170 male patients with clear cell renal cell carcinoma, colon cancer, lung adenocarcinoma, breast and ovarian cancers (Table S12). At the level of gene expression, we found 76 common correlated genes with the same thresholds as before for the transcriptomic data analysis (non-random overlap, p < 0.0001, Fig. 12A). This figure is less than what was observed before for the RNA sequencing data, which can be explained in part by lower number of genes included in the proteome profiling for all the datasets under analysis (6755 genes for proteomic and 20,501 genes for transcriptomic correlation assay). Four correlated genes (FKBP5, GSTM1, MRI1, MX1) were also identified before by transcriptomic data.
Fig. 12.

Intersection of paired tumor-normal correlated genes (A) and pathways (B) identified in CPTAC proteomic datasets (left) and representation of the results of random permutation test (right). Results obtained with random permutations are shown in black, red line corresponds to the really observed number of intersected items.
Interestingly, the most strongly correlated gene in this analysis was GSTM1 (Table 2), which was also the most strongly correlated gene in the RNA sequencing data. Of note, GSTM1 function deals with the detoxification of electrophilic compounds, including carcinogens, therapeutic drugs, environmental toxins, and products of oxidative stress, by conjugation with glutathione [55].
Table 2.
Top-10 intersected genes with correlated activation levels in paired tumor-normal samples in transcriptomic and proteomic samplings (TCGA-male, TCGA-female, CPTAC-male, CPTAC-female, and experimental) sorted by mean correlation.
| Transcriptomic data | ||||
|---|---|---|---|---|
| Gene ID | Correlation, TCGA-Male | Correlation, TCGA-Female | Correlation, Experimental | Relation to patient identity |
| GSTM1 | 0.91 | 0.87 | 0.90 | yes |
| RPS28 | 0.86 | 0.84 | 0.98 | no |
| RPL9 | 0.84 | 0.81 | 0.98 | yes |
| ERAP2 | 0.79 | 0.78 | 0.81 | yes |
| FKBP1AP1 | 0.70 | 0.65 | 1.00 | no |
| GSTT2 | 0.71 | 0.66 | 0.90 | yes |
| POMZP3 | 0.70 | 0.73 | 0.79 | yes |
| XRRA1 | 0.72 | 0.59 | 0.90 | yes |
| POM121L10P | 0.60 | 0.58 | 0.93 | no |
| CTAG2 | 0.47 | 0.71 | 0.95 | no |
| Proteomic data | ||||
| Gene ID | Correlation, CPTAC-Male | Correlation, CPTAC-Female | Relation to patient identity | |
| GSTM1 | 0.76 | 0.74 | yes | |
| GSTT1 | 0.74 | 0.69 | no | |
| GSTM4 | 0.7 | 0.62 | no | |
| OAS1 | 0.63 | 0.66 | no | |
| NUDT2 | 0.54 | 0.59 | no | |
| LBP | 0.61 | 0.52 | no | |
| CPS1 | 0.48 | 0.57 | no | |
| SAA2-SAA4 | 0.5 | 0.52 | no | |
| SQSTM1 | 0.71 | 0.26 | no | |
| NTPCR | 0.45 | 0.5 | no | |
GO enrichment analysis of the 76 common correlated genes revealed the following functional groups: organic acid biosynthesis, inflammatory response, neutrophil activation and degranulation, regulation of complement activation, response to toxic substances, lipoprotein regulation, glutathione biosynthetic processes, and zymogen activation (Fig. 11). Overall, about 37% of all top GO terms identified deal with the immunity and inflammatory reactions.
Fig. 11.

Gene Ontology terms enrichment analysis for double-intersected set of the correlated paired tumor/normal genes (proteomic profiles).
3.3.2. Coordinated pathway activation patterns
We then performed paired tumor-norm correlation assay at the level of molecular pathway activation. At the transcriptomic level, we totally found twelve pathways with strongly correlated PAL values (Table 3).
Table 3.
Molecular pathways with correlated activation levels in paired tumor-normal samples in transcriptomic and proteomic samplings (TCGA-male, TCGA-female, CPTAC-male, CPTAC-female, and experimental) sorted by mean correlation.
| RNA sequencing data | ||||
|---|---|---|---|---|
| Pathway ID | Correlation, TCGA-Male | Correlation, TCGA-Female | Correlation, Experimental | Relation to patient identity |
| KEGG_Circadian_rhythm_Main_Pathway | 0.31 | 0.25 | 0.74 | yes |
| KEGG_Glutathione_metabolism_Main_Pathway | 0.26 | 0.25 | 0.9 | no |
| Reactome_Synthesis_of_PIPs_at_the_early_endosome_membrane_Main_Pathway | 0.24 | 0.26 | 0.83 | no |
| Reactome_p38MAPK_events_Main_Pathway | 0.23 | 0.26 | 0.74 | no |
| Guanosine_nucleotides_de_novoi_biosynthesis | 0.2 | 0.23 | 0.95 | no |
| Reactome_Synthesis_of_Leukotrienes_and_Eoxins_Main_Pathway | 0.24 | 0.23 | 0.71 | no |
| KEGG_RNA_polymerase_Main_Pathway | 0.22 | 0.23 | 0.71 | no |
| Circadian_Pathway | 0.22 | 0.22 | 0.76 | yes |
| Guanosine_deoxyribonucleotides_de_novoi_biosynthesis | 0.21 | 0.2 | 0.86 | no |
| Pyrimidine_deoxyribonucleotides_de_novoi_biosynthesis | 0.23 | 0.2 | 0.74 | no |
| NCI_Notch_mediated_HES_HEY_network_Main_Pathway | 0.21 | 0.21 | 0.76 | no |
| NCI_IGF1_Main_Pathway | 0.21 | 0.2 | 0.74 | no |
| Proteomic data | ||||
| Pathway ID | Correlation, CPTAC-Male | Correlation, CPTAC-Female | Relation to patient identity | |
| NCI Endogenous TLR signaling Pathway (regulation of granulocyte colony stimulating factor production) | 0.72 | 0.83 | no | |
| NCI Endogenous TLR signaling Pathway (regulation of interleukin 10 production) | 0.72 | 0.83 | no | |
| Reactome Formyl peptide receptors bind formyl peptides and other ligands Main Pathway | 0.46 | 0.55 | no | |
| Thyroid hormone metabolism II via conjugation and/or degradation | 0.33 | 0.41 | no | |
| Reactome Acyl chain remodeling of PI Main Pathway | 0.34 | 0.39 | no | |
| Reactome STING mediated induction of host immune responses Main Pathway | 0.36 | 0.33 | no | |
| Akt Signaling Pathway Proto-Oncogenic and RTK-signaling | 0.38 | 0.31 | no | |
| Dermatan sulfate biosynthesis | 0.36 | 0.3 | no | |
| Reactome LDL endocytosis Main Pathway | 0.28 | 0.39 | no | |
| Akt Signaling Pathway Enhancement of Breast Epithelial | 0.35 | 0.31 | no | |
Among these correlated pathways, presence of two versions of circadian clock pathway (Table 3) may be considered as the internal positive control of our analytic approach of finding commonly regulated processes in the paired samples. Indeed, this pathway controls intracellular molecular clock that maintains daily rhythms and thereby regulates cellular physiology [56], [57]. Circadian rhythms are common to all cells of the body and the circadian clock works concordantly for different tissues of the same individual [58]. Thus, all cells of the body are thought to be synchronized through circadian clock pathway activation [59]. In turn, circadian clock pathway activation patterns are expected to be similar in all tissues taken postmortally from the same donor. High correlation among the paired tissue samples for this pathway can be explained by the same time of obtaining biosamples both for tumors and for their adjacent norms. In turn, the correlated activation of KEGG RNA polymerase main pathway may be a direct consequence of the circadian clock-associated transcriptional regulation [60], [61]. The genomic targets of circadian clocks are numerous and are intimately linked to the regulation of cell growth and metabolism [60]. Among the tumor-norm correlated metabolic pathways there were three responsible for guanosine and pyrimidine nucleotide biosynthesis, and one – for glutathione metabolism. Congruent activation patterns of these pathways can be also explained in part as the consequence of common circadian rhythm regulation [56], [62], [63]. Alternatively, their activation can be controlled by the proliferative status of tumor and adjacent cells, which is clearly to certain extent regulated by the cancer itself [64].
Furthermore, another metabolic pathway of leukotrienes and eoxins biosynthesis is strongly connected with the inflammation [65]. Again, this indicates congruent inflammatory status for the cancer and adjacent “normal” tissue. In turn, common activation of the p38 MAPK pathway in cancers and adjacent tissues can be a direct consequence of building cancer niche [66]. The remaining three correlated pathways dealing with the phosphatidylinositol phosphate synthesis, Notch and IGF1 signaling can be each the consequence of any of the above tumor niche or circadian clock interplay or may represent a specific phenomenon.
At the proteomic level of pathway analysis, we found 40 non-randomly intersected correlated molecular pathways (p < 0.0001, Fig. 12B). The top-10 correlated pathways (Table 3) did not overlap with the transcriptomic findings possibly due to technical reason of considerably lower number of genes interrogated in the proteomic assay. For example, proteomic datasets contained data for only 5 genes of the Circadian clock pathway, compared to 18 in the transcriptomic datasets. However, none out of top-10 “proteomic” pathways could be identified as the significantly correlated in our transcriptomic assay.
Functionally, the top correlated pathways by proteomic data (Table 3) deal with the inflammatory response (TLR signaling pathways, formyl peptide receptor pathway, STING-mediated induction of host immune response), proliferative signaling (remodeling of acyl residues in phosphatidylinositol, AKT signaling), remodeling of extracellular matrix (dermatan sulfate biosynthesis), cellular import of fatty acids from blood flow (LDL endocytosis pathway), and with thyroid hormone metabolism (Table 3).
Thus, in this pan-cancer study we identified a fraction of genes and molecular pathways which activation is coordinated between the tumors and the non-tumor tissues from the same site. A fraction of these genes and pathways can be explained by the common circadian clock regulation, and by the overlapping pathogen response pattern in tissues taken from the same individual and located nearby. However, the others clearly represent signatures of proliferative signaling, and of inflammation and building cancer niche. This trend was congruent for both transcriptomic and proteomic data and can explain at least some of molecular differences observed between the tumor-matched and “healthy” norms.
3.3.3. Molecular processes linked with the patient identity
We then investigated the finding that the molecular features of an adjacent normal tissue may correlate with those of a paired tumor not due to tumor impact, but because of individual peculiarities of a patient. To discriminate such molecular features from those caused by the tumor, we correlated gene expression profiles of different postmortal healthy tissues collected from the same individual donor. For this analysis we selected seven tissue types that allowed at least 70 samples in each pairwise correlation test (Fig. S4): esophagus, pancreas, stomach, breast, thyroid, colon, and lung.
We then did pairwise correlation tests for all the samples from the same donor available for the above seven healthy tissue types. Using the same criteria as before for the correlated items, we considered that the feature was related to patient identity when correlation coefficients in pairwise comparisons in all groups exceeded 0.2 and were statistically significant (q-value < 0.05). In order to exclude the influence of gender, the comparison was made separately for male and female donors, and only the features that coincided in both sets were selected for further analysis.
Among the previously identified paired tumor-norm correlated hits, we found 52 genes (Fig. 13A) and one molecular pathway (Fig. 13B) which expression/activation level were found to be associated with the individual patient/donor identity, but most likely not with the tumor impact (Table S13). Triple intersections were non-random both in the gene level and pathway level, as evidenced by the permutation test p < 0.0001 and p = 0.0035 (Fig. 13C, D).
Fig. 13.

Venn diagrams (A, B) and permutation tests (C, D) for individual genes (A, C) and molecular pathways (B, D) correlated with the subject identity (shown separately for male and female donors), and between the paired tumor and normal tissues.
Interestingly, 6/10 top correlated transcripts and 1/10 top correlated proteins that were obtained from RNA sequencing and proteomic data, respectively, appeared to be related to patient identity (Table 3). Thus, these entries most likely correlated with the paired tumors because of intrinsic physiological patterns. This finding also showed adequacy of our analytic approach for refinement of the lists of tumor-linked molecular factors.
In contrast, the other top correlated genes involved in epithelial/keratinocyte development, chemosensory perception and JAK-STAT pathway were not connected with patient identity and, therefore, should be considered as those influenced by the tumorigenesis.
The only subject identity-linked molecular pathway identified here was the Circadian rhythm pathway which was also among the list of paired tumor-normal correlated items and can be explained by the coordinated work of molecular clock in all tissues of the same individual.
3.3.4. Time-dependent alterations in healthy tissues after sample collection during autopsy
We then experimentally tested the hypothesis that gene expression profiles in healthy tissue autopsies contain an artifact component linked with the time of obtaining biosample after death which could be due to transcription in postmortal tissues or RNA degradation. To this end, we used the model of crab-eating macaque (Macaca fascicularis) as the mammalian species of a common primate ancestry and of a comparable size with human.
Three male Macaca fascicularis animals (age 3, 3, and 4 years) were sacrificed for the experiments unrelated to this study. We then immediately isolated lung and liver tissue samples and stored them at 4ºC for 30 min, 3 h, 6 h, and 24 h. The 4ºC regimen roughly corresponds to the average temperature conditions in the Moscow region from November till March and storage conditions in morgue. These conditions were previously applied for obtaining our experimental collection ANTE of healthy tissue RNA sequencing profiles obtained from biosamples of donors killed in road accidents [2].
We profiled gene expression in macaque tissues by RNA sequencing and compared the molecular profiles corresponding to the different sample storage timepoints. On the principal component analysis (PCA) plot of the biosamples obtained (Fig. 14A) the profiles most strongly clustered according to the tissue type (lung or liver; explains ∼74% of standard deviation) and then by the animal identity (explains ∼4% of standard deviation).
Fig. 14.

Comparison of gene expression profiles in crab-eating macaque (Macaca fascicularis) tissues at the different sample storage timepoints. (A) Principal component analysis of gene expression profiles for postmortal liver and lung specimens stored at 4ºC for 30 min, 3 h, 6 h, and 24 h. Percent of standard deviation explained by the principal components is shown in brackets. Color indicates tissue type, shape – sample storage time. Each dot is marked with the animal identifier; (B) RIN (top) and DV200 (bottom) values for RNA samples obtained at different time points for lung and liver tissues.
We then investigated whether postmortal transcription or RNA degradation may alter the results of further differential gene expression analysis. First, we compared RIN and DV200 values in all samples (Fig. 14B). RNA extracted from the liver tissue showed higher RIN and DV200 levels compared to the lung tissue. RIN, but not DV200 values decreased over the storage time for all three liver samples. DV200 values for the lungs decreased over time for all samples, while RIN changes showed no clear trend. We then performed differential expression analysis using DESeq2 software with a group of 30-minute samples as the reference, Table S14. Virtually no differential expression was detected in liver samples for all types of comparison (30 min vs 3 h; vs 6 h; vs 24 h), Fig. 15.
Fig. 15.

Differential gene expression analysis of liver (A) and lung (B) Macaca fascicularis tissues. Group of 30-minute storage timepoint samples was used as the reference, and compared with samples stored for 3 h (top), 6 h (middle) and 24 h (bottom). Adjusted p-value cut-off was set 0.05, |Log2fold change| cut-off was set higher than 1. Statistics of significantly differentially regulated genes is shown at the bottom of each volcano plot. Differentially up-/downregulated molecular drug targets are shown on the left/right side of each volcano plot.
However, for the lung samples we observed dramatically different results; 31, 6, and 300 genes were significantly upregulated, and 97, 10, and 696 genes were downregulated in samples stored for 3, 6, and 24 h, respectively (Fig. 15). For 3 h timepoint we observed even bigger number of differential genes than for the 6 h point which can be related to sampling bias or relatively soft statistical criteria used. Among them 0, 1, and 1 drug target genes were upregulated, while 3, 0, and 7 were downregulated at 3, 6, and 24-hour timepoints, respectively. This indicates that in lungs the expression profiles were altered with storage time for a significant fraction of genes (up to 1140 genes as for 24 h at 4ºC).
Since there were no such common artifact differential genes for the macaque lung and liver tissues at any time point, these results evidence in favor of the adequacy of the analytic approach used here for the assessment of human tissues, where we considered common differential genes for all tissue types under analysis. Extrapolation of the above test shows that this should have eliminated all possible RNA degradation-introduced artifacts.
We also calculated pathway activation level values to identify putative differential molecular pathways. No differential pathways were found for all comparisons with liver samples, and for the lung samples stored for 3 h. For 6-hour and 24-hour lung samples, the differential pathways were identified: 2/1 and 2/2 up/downregulated pathways, respectively, although there were no common differential pathways for the different timepoints.
Furthermore, although there was a number of common differential genes for the different timepoints in lung tissues, the intersection with the liver tissue patterns could give no common differential genes. This gives hope that our analysis of common pan-cancer trends was not significantly impacted by the times of exposure for the postmortal biosamples used.
However, our results evidence that differential expression analysis in some tissue types may be relatively strongly affected by the times of storage of biomaterials (e.g. ∼1000 affected genes in lungs for 24 h of exposure at 4ºC, Fig. 15). Moreover, we identified 9 cancer drug target genes among such artifact differential genes, and highlighted drugs approved for lung cancer treatment (Table S15). Although most of these targets are not analyzed at RNA level as routine predictive biomarkers, evidence is provided that their transcriptomic profiling is useful for clinical settings. For instance, transcriptomic profiling, compared to DNA mutation-based approach, was reported to substantially extend the cohort of patients who could receive benefit from personalized molecular diagnostics, and prescription of the corresponding targeted therapies. This could increase median overall survival, as well as the speed and efficiency of clinical trials [67], [68], [69].
Overall, these results are in line with the previous reports showing that different storage/time conditions of the autopsy or operational or animal tissue materials can lead to artifact bias of the gene expression profiles measured by RNA sequencing [70], [71], [72], [73].
4. Discussion
Implications in cell and tissue physiology.
Based on the results obtained, we tried to build a simplified pan-cancer model of solid tumor influence on gene expression in adjacent tissues. It has a limitation of being derived from the molecular profiles of normal tissues for 22 cancer types and, therefore, represents an overall averaging, which does not take into consideration all possible tissue-specific patterns.
4.1. Differentially activated processes between a tumor-adjacent and healthy normal tissues
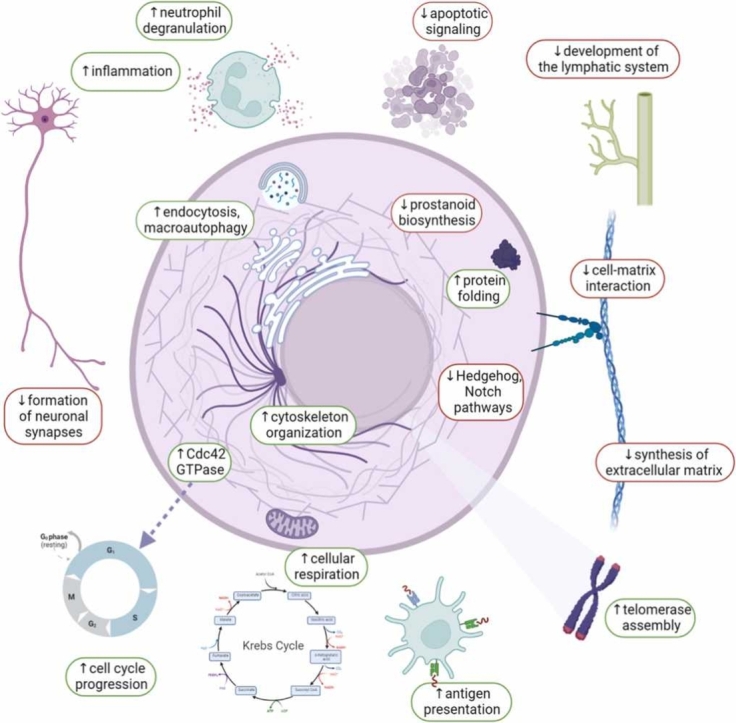
We found the following differentially activated processes in tumor-adjacent normal tissues when compared to postmortal pathologically healthy tissues: enhanced neutrophil degranulation and activation; antigen processing and presentation; protein folding and targeting; endocytosis and recycling of receptor molecules from cell surface; vesicle organization and transfer from endoplasmic reticulum to Golgi; macroautophagy; mitochondrial movement and gene expression; cellular respiration, catabolism of organic molecules, Krebs cycle and secondary alcohol synthetic processes; telomerase localization to Cajal bodies; CDC42 and EGF pathway regulation of cytoskeleton organization; degradation of extracellular matrix and cell-substrate adhesion patterns through the decrease of syndecan 1-mediated cell-matrix interaction, beta 1 integrin cell surface interactions, collagen biosynthesis, and chondroitin and dermatan biosynthesis (Fig. 16A).
Fig. 16.

Schematic representation of the processes differentially activated in tumor-adjacent norms compared to the postmortal pathologically healthy human tissues (A) and major functional categories of molecular processes which activities correlate among the paired tumor and adjacent normal tissues (B). Visualized with BioRender.com.
At the same time, the following molecular processes were differentially downregulated: apoptotic signaling and negative regulation of cell growth, formation of neuronal synapses, C20 prostanoid biosynthesis, regulation of cell migration by VEGFR3, and specific branches of Notch and Hedgehog pathways, signaling by small GTPases including Ras, and regulation of G2/M cell cycle transition (Fig. 16A). The latter two processes are strongly interconnected as the activated Ras is known as the major positive regulator of passing through the G2/M checkpoint [33].
4.2. Coordinated processes between paired tumor and tumor-adjacent normal tissues
We then focused on the processes which are statistically significantly correlated between the paired tumor and normal tissues. We found that adjacent normal tissues share with tumors the common patterns of defense response to bacteria and viruses, regulation of JAK-STAT signaling, chemosensory perception and olfaction, activation of RNA polymerase, epithelial development and keratinocyte differentiation, regulation of the p38 MAPK, Notch and IGF1 pathways, activities of metabolic pathways for nucleotide biosynthesis, for glutathione metabolism, for leukotrienes and eoxins biosynthesis. In this analysis we have filtered out the processes and genes related to the patient identity, e.g. regulation of circadian clock. Interestingly, the chemosensory perception-related processes formed the biggest clusters during the Gene Ontology analysis of the correlated genes.
Overall, most of the processes identified can be attributed to the following three main categories (Fig. 16B): (i) chemosensory perception; (ii) antiviral and bacterial defense mechanisms; (iii) inflammation; (iv) regulation of intracellular molecular pathways;.
Thus, we suggest that the differences seen between the tumor-adjacent and “healthy” norms may be due to the above molecular processes correlated among the cancer and paired normal tissues, which can act as the drivers of the apparent phenotypic remodeling (Fig. 16).
4.3. Possible clinical significance
In this study we confirmed that human cancers exhibit strong transforming activities on gene expression of the adjacent pathologically normal tissues. Compared to the “healthy” norms obtained from the autopsies, we detected in tumor-matched norms statistically significant upregulation of target genes for 33 clinically approved cancer drugs, and downregulation of target genes for 37 drugs (Table 4). This indicates that the differential analysis of gene expression, which can guide targeted therapy prescription (e.g., exemplified in [67], [74], [75], will be significantly biased for at least 52 cancer drugs, if the tumor-adjacent tissues are used as the controls (Table 4).
Table 4.
Cancer drug target genes which are differentially regulated between tumor-matched norms and autopsy-derived “healthy” tissue norms at the level of pan-cancer comparison.
| Drug target gene ID | Up/down-regulated in tumor adjacent norms | Cancer drug generic names | Approved cancer types |
|---|---|---|---|
| TUBA1C | Up | Ado-trastuzumab emtansine, enfortumab vedotin, vinblastine, vincristine, vindesine, vinorelbine | Breast cancer, non-small cell lung cancer (NSCLC), bladder cancer, ovarian cancer |
| IDH1 | Up | Ivosidenib | Acute myeloid leukemia |
| NFKB1 | Up | Thalidomide | Multiple myeloma |
| TUBB4B | Up | Cabazitaxel, docetaxel, eribulin, ixabepilone, paclitaxel | Prostate cancer, bladder cancer, breast cancer, NSCLC, ovarian cancer, stomach cancer, endometrial cancer, cervical cancer, kidney cancer |
| CSF1R | Up | Dovitinib, sunitinib | Kidney cancer, thyroid cancer |
| PSMB5 | Up | Bortezomib, carfilzomib, ixazomib (MLN9708) | Multiple myeloma |
| HDAC2 | Up | Belinostat, panobinostat, romidepsin, vorinostat | T-cell lymphoma, multiple myeloma |
| TUBB | Up | Ado-trastuzumab emtansine, brentuximab vedotin, cabazitaxel, docetaxel, enfortumab vedotin, eribulin, ixabepilone, paclitaxel, Vinblastine, Vincristine, Vindesine, Vinorelbine | Breast cancer, NSCLC, prostate cancer, bladder cancer, ovarian cancer, stomach cancer, endometrial cancer, cervical cancer, kidney cancer |
| IDH2 | Up | enasidenib | Acute Myeloid Leukemia |
| TUBA1B, TUBA4A | Up | Ado-trastuzumab emtansine, brentuximab vedotin, enfortumab vedotin, vinblastine, vincristine, vindesine, vinorelbine | Breast cancer, NSCLC, bladder cancer, ovarian cancer |
| CDK4 | Up | Abemaciclib (LY2835219), flavopiridol (alvocidib), palbociclib, ribociclib | Breast cancer |
| LYN | Up | Masitinib | Pancreatic cancer, melanoma, multiple myeloma, peripheral T-cell lymphoma, gastrointestinal stromal tumor, gastric cancer, colorectal cancer, esophageal cancer |
| ERBB2 | Up | Ado-trastuzumab emtansine, afatinib, lapatinib, pertuzumab, trastuzumab | Breast cancer, NSCLC, stomach cancer, endometrial cancer |
| FGFR1 | Down | Dovitinib, erdafitinib, lenvatinib, nintedanib (BIBF 1120), regorafenib, sorafenib | Bladder cancer, kidney cancer, hepatocellular carcinoma, thyroid cancer, colorectal cancer, ovarian cancer |
| MAPK11 | Down | Regorafenib | Colorectal cancer, hepatocellular carcinoma |
| PGF | Down | Aflibercept | Colorectal cancer |
| ABL1 | Down | Bosutinib, dasatinib, imatinib, nilotinib, ponatinib, regorafenib | Colorectal cancer, hepatocellular carcinoma |
| FLT4 | Down | Axitinib, dovitinib, foretinib, lenvatinib, nintedanib (BIBF 1120), pazopanib, regorafenib, sorafenib, sunitinib, tivozanib, vandetanib | Kidney cancer, thyroid cancer, hepatocellular carcinoma, colorectal cancer, ovarian cancer, NSCLC |
| PDGFRA | Down | Imatinib, lenvatinib, masitinib, midostaurin, nintedanib (BIBF 1120), olaratumab, pazopanib, regorafenib, sunitinib, tivozanib | Kidney cancer, hepatocellular carcinoma, thyroid cancer, colorectal cancer |
| TUBB3 | Down | Ado-Trastuzumab Emtansine, Brentuximab vedotin, Cabazitaxel, Enfortumab vedotin, Eribulin, Ixabepilone, Vinblastine, Vincristine, Vindesine | Breast cancer, NSCLC, prostate cancer, bladder cancer |
| HDAC4 | Down | Belinostat, Vorinostat | T-cell lymphoma |
| NTRK3 | Down | Entrectinib, Larotrectinib | Pan-cancer |
| RARG | Down | Acitretin, Alitretinoin, Tretinoin | Nonmelanoma skin cancers (chemoprevention), Kaposi’s sarcoma |
| PDGFRB | Down | Dovitinib, Imatinib, Lenvatinib, Masitinib, Midostaurin, Nintedanib (BIBF 1120), Pazopanib, Regorafenib, Sorafenib, Sunitinib, Tivozanib | Kidney cancer, Hepatocellular carcinoma, thyroid cancer, colorectal cancer, ovarian cancer |
| HDAC7 | Down | Belinostat | T-cell lymphoma |
In particular, this list includes tyrosine kinase inhibitors axitinib, imatinib, dovitinib, imatinib, lenvatinib, masitinib, nintedanib, pazopanib, regorafenib, ponatinib, sorafenib, sunitinib, tivozanib, entrectinib, larotrectinib, afatinib, lapatinib, and ivosidenib; monoclonal therapeutic antibodies ado-trastuzumab, brentuximab, olaratumab, pertuzumab, and trastuzumab. Thus, using tumor-matched normal tissues as the controls can result in significantly biased predicted personalized drug activity profiles, constructed based on RNA expression data [67], [76].
Furthermore, we also found statistically significant correlation between tumors and adjacent normal tissues for the expression of genes TNF, MAP2K2, and PIK3CA which serve as the targets for 6 cancer drugs (Table 5). This association needs to be further investigated but theoretically can be another factor that can bias results of the differential expression assay with such normalization.
Table 5.
Cancer drug target genes which are correlated among the paired tumor and adjacent normal tissues.
| Drug target gene ID | Mean correlation coefficient | Mean q-value | Cancer drug generic names | Approved cancer type |
|---|---|---|---|---|
| TNF | 0.412 | 0.009 | Pomalidomide, thalidomide | Multiple myeloma |
| MAP2K2 | 0.464 | 0.002 | Binimetinib, selumetinib, trametinib | Мelanoma |
| PIK3CA | 0.405 | 0.012 | Alpelisib | Breast cancer |
4.4. Technical validation of data consistency
Experimental tumor samples, tumor-adjacent normal samples, and healthy normal samples investigated in this study were manipulated by the same group of researchers using uniform standard protocol and reagent settings. We investigated consistency of the method used here for obtaining experimental RNA sequencing profiles in the following assays. First, we did technical replicates and assessed correlation coefficients between gene expression profiles among the replicates. Second, we analyzed congruence of the RNA expression and immunihistochemical (IHC) profiles in the same biosamples for selected biomarker genes.
In our experiments, 0.95 Spearman correlation coefficient was obtained for the comparison of tumor tissue replicates [77], and the replicates demonstrated tight clustering on the dendrograms [77]. The same correlation coefficient of 0.95 was also demonstrated in our experiments for the quadruplicates of postmortal normal human tissues [42].
The correlation between IHC and RNA sequencing profiles was assessed for the HER2/ERBB2, ESR1 and PGR biomarkers in breast cancer, and for the PD-L1 biomarker in lung cancer specimens. For HER2, ESR1, and PGR correlations (Spearman’s rho) were 0.798 (p = 6.9 × 10−10); 0.777 (p = 3.8 × 10−9), and 0.653 (p = 4.9 ×10−6), accordingly. For PD-L1, correlation was 0.797 (p = 4.4 × 10−5) [78].
4.5. Functional assessment of the results
In this study we found statistically significant correlation of the expression of MAPK1 gene for ERK2 regulatory kinase in tumors and tumor-adjacent norms. In addition, MAPK1 was found upregulated in tumor-matched compared with healthy normal tissues. In our separately published experiments, we added ERK2 inhibitors (SCH772984, ravoxertinib, LY3214996, ulixertinib, and VX-11e) in combination with targeted tyrosine kinase inhibitors, which had a synergistic effect on inhibiting cancer cell survival and proliferation [79], [80], [81].
4.6. Future directions
In this study, we attempted to identify general characteristics of tumor-adjacent pathologically normal tissues. Here, all the patients investigated were adults, of similar age in both experimental (mean 59.7, sd 14.8 y.o.) and in the TCGA (mean 59.1, sd 14.4 y.o.) [82] cohorts. We sought to exclude the influence of gender on the results by intersecting subsets of differential genes obtained separately for the groups of male and female patients. In addition, our study most likely reflects features specific for the later cancer stages, as those were overrepresented in the experimental group. In the future, it will be important to investigate in-depth also specific relevant factors connected with tumor type, stage, gender, age, separately for the adult and pediatric cancers. This will be a matter of our further studies.
4.7. Potential limitations
In total, we analyzed 252 experimental samples (196 postmortal healthy tissues, 28 pairs of tumor and normal NAT samples). The postmortal samples were published by us earlier in the form of ANTE database. The sampling size of NATs is limited yet we believe it is sufficient for the statistical methods used here, as can be exemplified by the following published papers with smaller sampling assessed with the same statistical criteria: [83], [84], [85], [86], [87], [88].
Another point that has to be discussed here is the potential presence of cancer cells in the tumor-adjacent normal tissues. We presume that part of the differential genes found between the tumor-adjacent and healthy normal tissues can be associated with the residual tumor cells that could not be identified by a pathologist. This leads to certain limitations of the current study and suggests innovative technological approaches such as utilizing single-cell sequencing, to gain deeper insights into the impact of cancer cells on surrounding normal tissues in the future.
5. Conclusion
Here we demonstrate that compared to the autopsy-derived pathologically healthy tissues, the tumor-adjacent tissues have a number of systemic molecular differences. These differences relate to activation of the immune cells, intracellular vesicle transport and autophagy, cellular respiration, activation of telomerase, activation of p38 signaling, cytoskeleton remodeling, and reorganization of the extracellular matrix. The tumor-adjacent tissues are deficient in apoptotic signaling and negative regulation of cell growth including G2/M cell cycle transition checkpoint. In addition, there is an extensive rearrangement of the chemical perception network that is strongly connected with the neighboring tumor development.
We showed that these processes may be driven by another group of molecular events that are correlated between the paired cancers and the adjacent non-cancer tissues, that mostly relate to inflammation and regulation of intracellular molecular pathways such as the p38, MAPK, Notch and IGF1 signaling.
As the consequence, molecular targets of 32 and 37 cancer drugs appear to be over- or underexpressed, respectively, in the tumor-adjacent compared to the “healthy” norms. However, using a model of crab-eating macaque postmortal tissues (liver and lung) we showed that for the 30 min – 24 h time frame of storage at 4ºC, there was a significant RNA degradation pattern in lung biosamples that resulted in an artifact “differential” expression profile for 1140 genes, including molecular targets of 9 cancer drugs. This effect was not seen for the liver samples.
Taken together, our results evidence that tumor-adjacent pathologically normal tissues cannot be considered as the fully adequate norms for the analysis of cancer molecular profiles. The alternative solution could be the autopsy tissue biosamples taken from the healthy donors, yet for the latter possibility an RNA degradation-induced bias in gene expression profiles has to be carefully considered. In addition, normal adjacent tumor tissue is not guaranteed for being free from cancer cells. Thus, comparing tumor adjacent tissue with healthy controls may uncover differential expression of residual cancer cells. In the future, single-cell sequencing approach may provide deeper insights on the impact of cancer cells on surrounding normal tissues. However, at present several technical aspects of this technology seem to be underdeveloped for such a large-scale analysis including poor compatibility of the results/strong batch effect and low RNA sequencing reads coverage per cell [89].
CRediT authorship contribution statement
Maksim Sorokin: Writing − original draft, Visualization, Data curation. Anton Buzdin: Writing − original draft, Conceptualization, Methodology, Supervision. Anastasia Guryanova: Writing − original draft, Formal analysis, Visualization. Victor Efimov: Investigation, Visualization. Maria Suntsova: Investigation. Marianna Zolotovskaia: Investigation. Elena Koroleva: Investigation. Marina. Sekacheva: Resources. Victor Tkachev: Formal analysis. Andrew Garazha: Project administration, Investigation. Kristina Kremenchutckaya: Formal analysis. Aleksey Drobyshev: Investigation. Aleksander Seryakov: Resources. Alexander Gudkov: Investigation. Irina Alekseenko: Investigation. Olga Rakitina: Investigation. Maria Kostina: Investigation. Uliana Vladimirova: Investigation. Aleksey Moisseev: Resources. Dmitry Bulgin: Investigation. Elena Radomskaya: Investigation. Viktor Shestakov: Investigation. Vladimir P. Baklaushev: Investigation. Vladimir Prassolov: Investigation. Petr Shegay: Resources. Xinmin Li: Investigation. Elena Poddubskaya: Conceptualization, Writing − original draft. Nurshat Gaifullin: Conceptualization, Writing − original draft, Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research was funded by the Russian Science Foundation, grant number 22-14-00074. The contribution of Mikhail Raevskiy, Marina Sekacheva, and Anton Buzdin was financed by the Ministry of Science and Higher Education of the Russian Federation within the framework of state support for the creation and development of World-Class Research Centers “Digital biodesign and personalized healthcare” No 075-15-2022-304. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Data deposition and access
RNA sequencing data were deposited in NCBI Sequencing Read Archive (SRA) under accession ID PRJNA905832.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2023.07.040.
Contributor Information
Maksim Sorokin, Email: sorokin@oncobox.com.
Elena V. Poddubskaya, Email: poddubskaya_e_v@staff.sechenov.ru.
Nurshat Gaifullin, Email: gaifulin@rambler.ru.
Appendix A. Supplementary material
Supplementary material
.
Supplementary material
.
Supplementary material
{kind=link}
.
Supplementary material
{kind=link}
.
Supplementary material
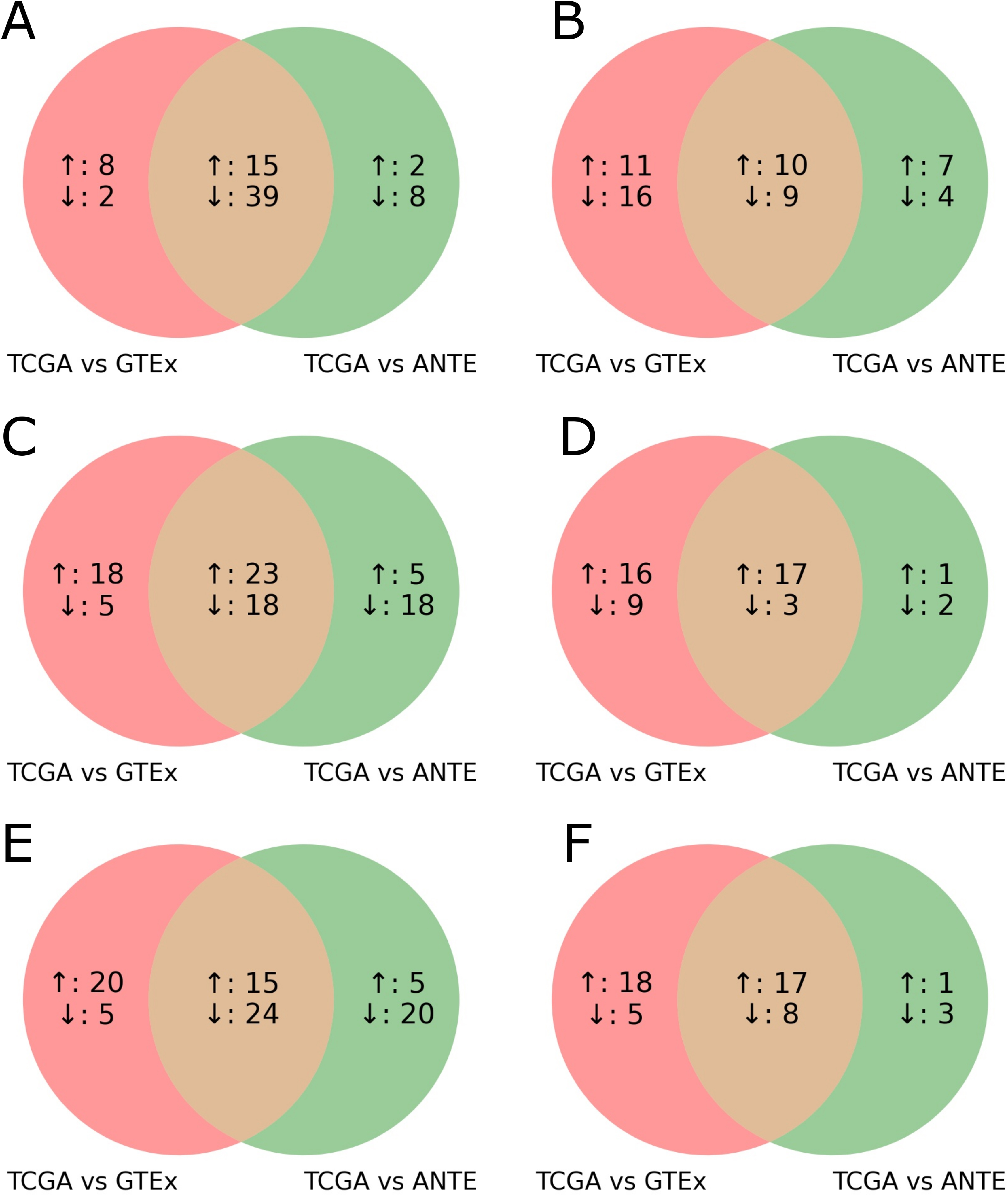{kind=link}
.
Supplementary material
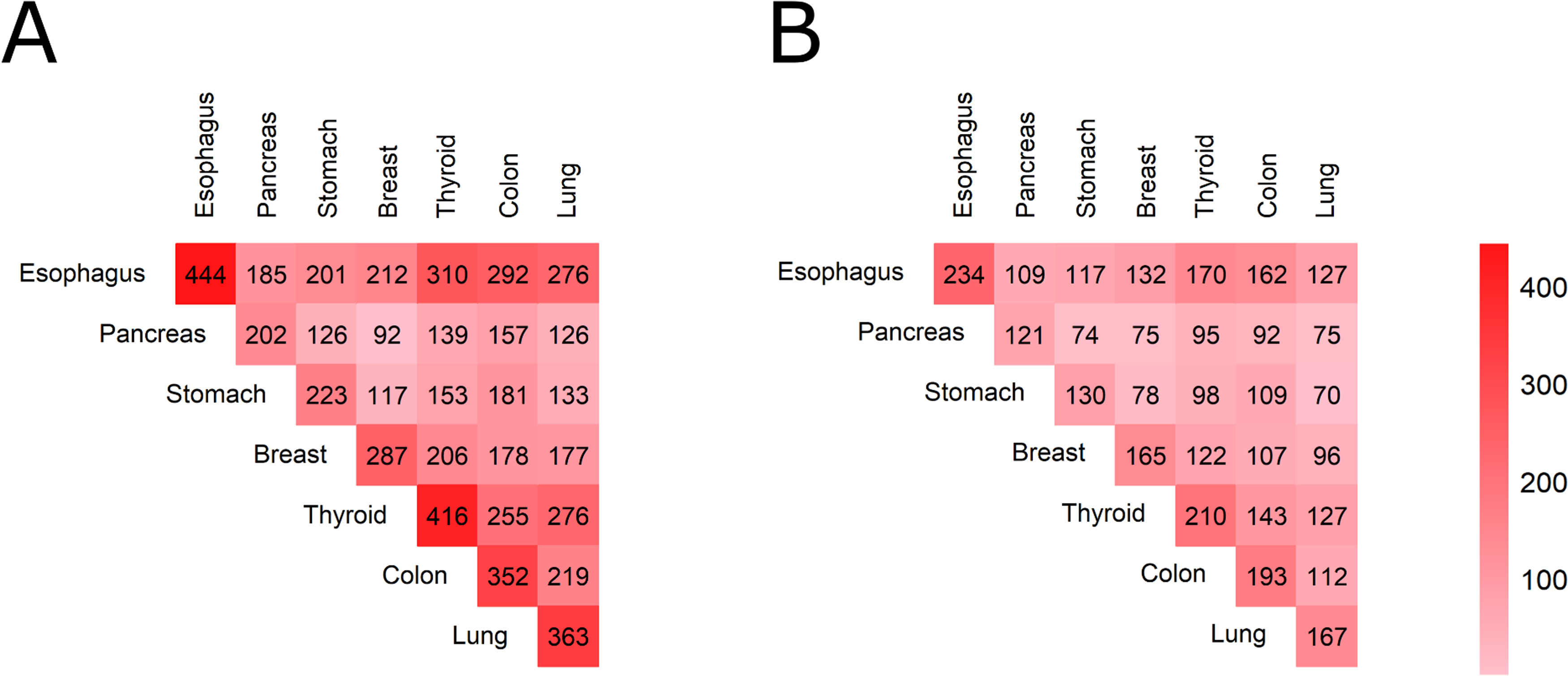{kind=link}
.
References
- 1.Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/NG.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suntsova M., Gaifullin N., Allina D., Reshetun A., Li X., Mendeleeva L., et al. Atlas of RNA sequencing profiles for normal human tissues. Sci Data. 2019;6 doi: 10.1038/S41597-019-0043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis M.J., Gillette M., Carr S.A., Paulovich A.G., Smith R.D., Rodland K.K., et al. Connecting genomic alterations to cancer biology with proteomics: the NCI clinical proteomic tumor analysis consortium. Cancer Discov. 2013;3:1108–1112. doi: 10.1158/2159-8290.CD-13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nieuwenhuis T.O., Yang S.Y., Verma R.X., Pillalamarri V., Arking D.E., Rosenberg A.Z., et al. Consistent RNA sequencing contamination in GTEx and other data sets. Nat Commun. 2020;11 doi: 10.1038/S41467-020-15821-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gross A.M., Kreisberg J.F., Ideker T. Analysis of matched tumor and normal profiles reveals common transcriptional and epigenetic signals shared across cancer types. PLoS One. 2015;10 doi: 10.1371/JOURNAL.PONE.0142618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang X., Stern D.F., Zhao H. Transcriptional profiles from paired normal samples offer complementary information on cancer patient survival – evidence from TCGA pan-cancer data. Sci Rep. 2016;6 doi: 10.1038/SREP20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomczak K., Czerwińska P., Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Conte Oncol (Pozn, Pol) 2015;19:A68–A77. doi: 10.5114/WO.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aran D., Camarda R., Odegaard J., Paik H., Oskotsky B., Krings G., et al. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat Commun. 2017;81:2017. doi: 10.1038/s41467-017-01027-z. 8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vladimirova U., Rumiantsev P., Zolotovskaia M., Albert E., Abrosimov A., Slashchuk K., et al. DNA repair pathway activation features in follicular and papillary thyroid tumors, interrogated using 95 experimental RNA sequencing profiles. Heliyon. 2021;7 doi: 10.1016/J.HELIYON.2021.E06408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominiak A., Chełstowska B., Olejarz W., Nowicka G. Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers (Basel) 2020;12 doi: 10.3390/CANCERS12051232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jahanban-Esfahlan R., Seidi K., Zarghami N. Tumor vascular infarction: prospects and challenges. Int J Hematol. 2017;105:244–256. doi: 10.1007/S12185-016-2171-3. [DOI] [PubMed] [Google Scholar]
- 12.Jahanban-Esfahlan R., de la Guardia M., Ahmadi D., Yousefi B. Modulating tumor hypoxia by nanomedicine for effective cancer therapy. J Cell Physiol. 2018;233:2019–2031. doi: 10.1002/JCP.25859. [DOI] [PubMed] [Google Scholar]
- 13.Seidi K., Neubauer H.A., Moriggl R., Jahanban-Esfahlan R., Javaheri T. Tumor target amplification: Implications for nano drug delivery systems. J Control Release. 2018;275:142–161. doi: 10.1016/J.JCONREL.2018.02.020. [DOI] [PubMed] [Google Scholar]
- 14.Rozenberg J.M., Buzdin A.A., Mohammad T., Rakitina O.A., Didych D.A., Pleshkan V.V., et al. Molecules promoting circulating clusters of cancer cells suggest novel therapeutic targets for treatment of metastatic cancers. Front Immunol. 2023;14:1099921. doi: 10.3389/FIMMU.2023.1099921/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baghban R., Roshangar L., Jahanban-Esfahlan R., Seidi K., Ebrahimi-Kalan A., Jaymand M., et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020;18 doi: 10.1186/S12964-020-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zolotovskaia M.A., Modestov A.A., Suntsova M.V., Rachkova A.A., Koroleva E.V., Poddubskaya E.V., et al. Pan-cancer antagonistic inhibition pattern of ATM-driven G2/M checkpoint pathway vs other DNA repair pathways. DNA Repair (Amst) 2023;123 doi: 10.1016/J.DNAREP.2023.103448. [DOI] [PubMed] [Google Scholar]
- 17.Sorokin M., Borisov N., Kuzmin D., Gudkov A., Zolotovskaia M., Garazha A., et al. Algorithmic Annotation of Functional Roles for Components of 3,044 Human Molecular Pathways. Front Genet. 2021;12 doi: 10.3389/FGENE.2021.617059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borisov N., Sorokin M., Garazha A., Buzdin A. Quantitation of molecular pathway activation using RNA sequencing data. Methods Mol Biol. 2020;2063:189–206. doi: 10.1007/978-1-0716-0138-9_15. [DOI] [PubMed] [Google Scholar]
- 19.Buzdin A., Tkachev V., Zolotovskaia M., Garazha A., Moshkovskii S., Borisov N., et al. Using proteomic and transcriptomic data to assess activation of intracellular molecular pathways. Adv Protein Chem Struct Biol. 2021;127:1–53. doi: 10.1016/BS.APCSB.2021.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Buzdin A., Sorokin M., Garazha A., Sekacheva M., Kim E., Zhukov N., et al. Molecular pathway activation - New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin Cancer Biol. 2018;53:110–124. doi: 10.1016/J.SEMCANCER.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Sorokin M., Ignatev K., Poddubskaya E., Vladimirova U., Gaifullin N., Lantsov D., et al. RNA sequencing in comparison to immunohistochemistry for measuring cancer biomarkers in breast cancer and lung cancer specimens. Biomedicines. 2020;8 doi: 10.3390/BIOMEDICINES8050114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards N.J., Oberti M., Thangudu R.R., Cai S., McGarvey P.B., Jacob S., et al. The CPTAC data portal: a resource for cancer proteomics research. J Proteome Res. 2015;14:2707–2713. doi: 10.1021/PR501254J. [DOI] [PubMed] [Google Scholar]
- 23.Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2016;44:D481–D487. doi: 10.1093/NAR/GKV1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaefer C.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay T., et al. PID: the pathway interaction database. Nucleic Acids Res. 2009;37 doi: 10.1093/NAR/GKN653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/NAR/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zolotovskaia M.A., Tkachev V.S., Guryanova A.A., Simonov A.M., Raevskiy M.M., Efimov V.V., et al. OncoboxPD: human 51 672 molecular pathways database with tools for activity calculating and visualization. Comput Struct Biotechnol J. 2022;20:2280–2291. doi: 10.1016/J.CSBJ.2022.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borisov N., Shabalina I., Tkachev V., Sorokin M., Garazha A., Pulin A., et al. Shambhala: a platform-agnostic data harmonizer for gene expression data. BMC Bioinforma. 2019;20 doi: 10.1186/S12859-019-2641-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borisov N., Buzdin A. Transcriptomic harmonization as the way for suppressing cross-platform bias and batch effect. Biomedicines. 2022;10 doi: 10.3390/BIOMEDICINES10092318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolstad B.M., Irizarry R.A., Åstrand M., Speed T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/BIOINFORMATICS/19.2.185. [DOI] [PubMed] [Google Scholar]
- 30.Sorokin M., Ignatev K., Barbara V., Vladimirova U., Muraveva A., Suntsova M., et al. Molecular Pathway Activation Markers Are Associated with Efficacy of Trastuzumab Therapy in Metastatic HER2-Positive Breast Cancer Better than Individual Gene Expression Levels. Biochem (Mosc) 2020;85:758–772. doi: 10.1134/S0006297920070044. [DOI] [PubMed] [Google Scholar]
- 31.Mollinedo F. Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol. 2019;40:228–242. doi: 10.1016/J.IT.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y., Tomlinson R.L., Lukowiak A.A., Terns R.M., Terns M.P. Telomerase RNA accumulates in Cajal bodies in human cancer cells. Mol Biol Cell. 2004;15:81–90. doi: 10.1091/MBC.E03-07-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knauf J.A., Ouyang B., Knudsen E.S., Fukasawa K., Babcock G., Fagin J.A. Oncogenic RAS induces accelerated transition through G2/M and promotes defects in the G2 DNA damage and mitotic spindle checkpoints. J Biol Chem. 2006;281:3800–3809. doi: 10.1074/JBC.M511690200. [DOI] [PubMed] [Google Scholar]
- 34.Qadir M.I., Parveen A., Ali M. Cdc42: role in cancer management. Chem Biol Drug Des. 2015;86:432–439. doi: 10.1111/CBDD.12556. [DOI] [PubMed] [Google Scholar]
- 35.Hehner S.P., Hofmann T.G., Dienz O., Dröge W., Schmitz M.L. Tyrosine-phosphorylated Vav1 as a point of integration for T-cell receptor- and CD28-mediated activation of JNK, p38, and interleukin-2 transcription. J Biol Chem. 2000;275:18160–18171. doi: 10.1074/JBC.275.24.18160. [DOI] [PubMed] [Google Scholar]
- 36.Bagnara D., Mazzarello A.N., Ghiotto F., Colombo M., Cutrona G., Fais F., et al. Old and new facts and speculations on the role of the B cell receptor in the origin of chronic lymphocytic leukemia. Int J Mol Sci. 2022;23 doi: 10.3390/IJMS232214249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ott P.A., Bang Y.J., Piha-Paul S.A., Abdul Razak A.R., Bennouna J., Soria J.C., et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J Clin Oncol. 2019;37:318–327. doi: 10.1200/JCO.2018.78.2276. [DOI] [PubMed] [Google Scholar]
- 38.Bozelli J.C., Azher S., Epand R.M. Plasmalogens and chronic inflammatory diseases. Front Physiol. 2021;12 doi: 10.3389/FPHYS.2021.730829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aran D., Hu Z., Butte A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18 doi: 10.1186/S13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X., Li L., Yang Y., Fan L., Ma Y., Mao F. Reveal the heterogeneity in the tumor microenvironment of pancreatic cancer and analyze the differences in prognosis and immunotherapy responses of distinct immune subtypes. Front Oncol. 2022;12 doi: 10.3389/FONC.2022.832715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takahashi K., Kurashina K., Yamaguchi H., Kanamaru R., Ohzawa H., Miyato H., et al. Altered intraperitoneal immune microenvironment in patients with peritoneal metastases from gastric cancer. Front Immunol. 2022;13 doi: 10.3389/FIMMU.2022.969468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suntsova M., Gaifullin N., Allina D., Reshetun A., Li X., Mendeleeva L., et al. Atlas of RNA sequencing profiles for normal human tissues. Sci Data. 2019;6 doi: 10.1038/S41597-019-0043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. 456 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witsch E., Sela M., Yarden Y. Roles for growth factors in cancer progression. Physiol (Bethesda) 2010;25:85–101. doi: 10.1152/PHYSIOL.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X., Nie D., Chakrabarty S. Growth factors in tumor microenvironment. Front Biosci (Landmark Ed. 2010;15:151–165. doi: 10.2741/3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masjedi S., Zwiebel L.J., Giorgio T.D. Olfactory receptor gene abundance in invasive breast carcinoma. Sci Rep. 2019;9 doi: 10.1038/S41598-019-50085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shibel R., Sarfstein R., Nagaraj K., Lapkina-Gendler L., Laron Z., Dixit M., et al. The Olfactory Receptor Gene Product, OR5H2, Modulates Endometrial Cancer Cells Proliferation via Interaction with the IGF1 Signaling Pathway. Cells. 2021;10 doi: 10.3390/CELLS10061483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li M., Schweiger M.W., Ryan D.J., Nakano I., Carvalho L.A., Tannous B.A. Olfactory receptor 5B21 drives breast cancer metastasis. IScience. 2021;24 doi: 10.1016/J.ISCI.2021.103519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schindler C., Levy D.E., Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/JBC.R700016200. [DOI] [PubMed] [Google Scholar]
- 50.Haan C., Kreis S., Margue C., Behrmann I. Jaks and cytokine receptors--an intimate relationship. Biochem Pharm. 2006;72:1538–1546. doi: 10.1016/J.BCP.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 51.Feng J., Witthuhn B.A., Matsuda T., Kohlhuber F., Kerr I.M., Ihle J.N. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol. 1997;17:2497–2501. doi: 10.1128/MCB.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kisseleva T., Bhattacharya S., Braunstein J., Schindler C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/S0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- 53.Thomas S.J., Snowden J.A., Zeidler M.P., Danson S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 2015;113:365–371. doi: 10.1038/bjc.2015.233. 1133 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brooks A.J., Putoczki T. JAK-STAT signalling pathway in cancer. Cancers (Basel) 2020;12:1–3. doi: 10.3390/CANCERS12071971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pita-Oliveira M., Rodrigues-Soares F. Influence of GSTM1, GSTT1, and GSTP1 genetic polymorphisms on disorders in transplant patients: a systematic review. Drug Metab Pers Ther. 2021;37:123–131. doi: 10.1515/DMPT-2021-0165. [DOI] [PubMed] [Google Scholar]
- 56.Buhr E.D., Takahashi J.S. Molecular components of the Mammalian circadian clock. Handb Exp Pharm. 2013;217:3–27. doi: 10.1007/978-3-642-25950-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hogenesch J.B., Herzog E.D., Merrow M., Brunner M. Intracellular and intercellular processes determine robustness of the circadian clock. FEBS Lett. 2011;585:1427–1434. doi: 10.1016/J.FEBSLET.2011.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Husse J., Eichele G., Oster H. Synchronization of the mammalian circadian timing system: Light can control peripheral clocks independently of the SCN clock: Alternate routes of entrainment optimize the alignment of the body’s circadian clock network with external time. Bioessays. 2015;37:1119. doi: 10.1002/BIES.201500026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fagiani F., Di Marino D., Romagnoli A., Travelli C., Voltan D., Mannelli L.D.C., et al. Molecular regulations of circadian rhythm and implications for physiology and diseases. Signal Transduct Target Ther. 2022;7:1–20. doi: 10.1038/s41392-022-00899-y. 71 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takahashi J.S. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18:164–179. doi: 10.1038/NRG.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trott A.J., Menet J.S. Regulation of circadian clock transcriptional output by CLOCK:BMAL1. PLoS Genet. 2018;14 doi: 10.1371/JOURNAL.PGEN.1007156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinoshita C., Aoyama K., Nakaki T. Neuroprotection afforded by circadian regulation of intracellular glutathione levels: A key role for miRNAs. Free Radic Biol Med. 2018;119:17–33. doi: 10.1016/J.FREERADBIOMED.2017.11.023. [DOI] [PubMed] [Google Scholar]
- 63.Beaver L.M., Klichko V.I., Chow E.S., Kotwica-Rolinska J., Williamson M., Orr W.C., et al. Circadian regulation of glutathione levels and biosynthesis in Drosophila melanogaster. PLoS One. 2012;7 doi: 10.1371/JOURNAL.PONE.0050454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46. doi: 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 65.Claesson H.E. On the biosynthesis and biological role of eoxins and 15-lipoxygenase-1 in airway inflammation and Hodgkin lymphoma. Prostaglandins Other Lipid Mediat. 2009;89:120–125. doi: 10.1016/J.PROSTAGLANDINS.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Brichkina, Bertero A., Loh T., Nguyen NTM H.M., Emelyanov A., Rigade S., et al. P38MAPK builds a hyaluronan cancer niche to drive lung tumorigenesis. Genes Dev. 2016;30:2623–2636. doi: 10.1101/GAD.290346.116/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rodon J., Soria J.C., Berger R., Miller W.H., Rubin E., Kugel A., et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25:751–758. doi: 10.1038/S41591-019-0424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lazar V., Zhang B., Magidi S., Le Tourneau C., Raymond E., Ducreux M., et al. A transcriptomics approach to expand therapeutic options and optimize clinical trials in oncology. Ther Adv Med Oncol. 2023;15 doi: 10.1177/17588359231156382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.A B., M S., A G., A G., A A., E P., et al. RNA sequencing for research and diagnostics in clinical oncology. Semin Cancer Biol. 2020;60:311–323. doi: 10.1016/J.SEMCANCER.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 70.Zhu Y., Wang L., Yin Y., Yang E. Systematic analysis of gene expression patterns associated with postmortem interval in human tissues. Sci Rep. 2017;7:1–12. doi: 10.1038/s41598-017-05882-0. 71 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferreira P.G., Muñoz-Aguirre M., Reverter F., Sá Godinho C.P., Sousa A., Amadoz A., et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat Commun. 2018;9:1–15. doi: 10.1038/s41467-017-02772-x. 91 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carlström E.L., Niazi A., Etemadikhah M., Halvardson J., Enroth S., Stockmeier C.A., et al. Transcriptome Analysis of Post-Mortem Brain Tissue Reveals Up-Regulation of the Complement Cascade in a Subgroup of Schizophrenia Patients. Genes. 2021;Vol 12:1242. doi: 10.3390/GENES12081242. Page 1242 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lam S., Kommadath A., López-Campos Ó., Prieto N., Aalhus J., Juárez M., et al. Evaluation of RNA quality and functional transcriptome of beef longissimus thoracis over time post-mortem. PLoS One. 2021;16 doi: 10.1371/JOURNAL.PONE.0251868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gudkov A., Shirokorad V., Kashintsev K., Sokov D., Nikitin D., Anisenko A., et al. Gene Expression-Based Signature Can Predict Sorafenib Response in Kidney Cancer. Front Mol Biosci. 2022;9 doi: 10.3389/FMOLB.2022.753318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sorokin M., Poddubskaya E., Baranova M., Glusker A., Kogoniya L., Markarova E., et al. RNA sequencing profiles and diagnostic signatures linked with response to ramucirumab in gastric cancer. Cold Spring Harb Mol Case Stud. 2020;6 doi: 10.1101/MCS.A004945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Poddubskaya E., Sorokin M., Garazha A., Glusker A., Moisseev A., Sekacheva M., et al. Clinical use of RNA sequencing and oncobox analytics to predict personalized targeted therapeutic efficacy. Https://DoiOrg/101200/JCO20203815supplE13676 2020;38:e13676–e13676. https://doi.org/10.1200/JCO.2020.38.15SUPPL.E13676.
- 77.Vladimirova U., Rumiantsev P., Zolotovskaia M., Albert E., Abrosimov A., Slashchuk K., et al. DNA repair pathway activation features in follicular and papillary thyroid tumors, interrogated using 95 experimental RNA sequencing profiles. Heliyon. 2021;7 doi: 10.1016/J.HELIYON.2021.E06408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sorokin M., Ignatev K., Poddubskaya E., Vladimirova U., Gaifullin N., Lantsov D., et al. RNA Sequencing in Comparison to Immunohistochemistry for Measuring Cancer Biomarkers in Breast Cancer and Lung Cancer Specimens. Biomedicines. 2020;8 doi: 10.3390/BIOMEDICINES8050114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lebedev T., Vagapova E., Spirin P., Rubtsov P., Astashkova O., Mikheeva A., et al. Growth factor signaling predicts therapy resistance mechanisms and defines neuroblastoma subtypes. Oncogene. 2021;40:6258–6272. doi: 10.1038/S41388-021-02018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lebedev T.D., Khabusheva E.R., Mareeva S.R., Ivanenko K.A., Morozov A.V., Spirin P.V., et al. Identification of cell type-specific correlations between ERK activity and cell viability upon treatment with ERK1/2 inhibitors. J Biol Chem. 2022;298 doi: 10.1016/J.JBC.2022.102226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lebedev T., Buzdin A., Khabusheva E., Spirin P., Suntsova M., Sorokin M., et al. Subtype of Neuroblastoma Cells with High KIT Expression Are Dependent on KIT and Its Knockdown Induces Compensatory Activation of Pro-Survival Signaling. Int J Mol Sci. 2022;23 doi: 10.3390/IJMS23147724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang X., Steensma J.T., Bailey M.H., Feng Q., Padda H., Johnson K.J. Characteristics of The Cancer Genome Atlas cases relative toU.S. general population cancer cases. Br J Cancer. 2018;119:885. doi: 10.1038/S41416-018-0140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vollmers A.C., Covarrubias S., Kuang D., Shulkin A., Iwuagwu J., Katzman S., et al. A conserved long noncoding RNA, GAPLINC, modulates the immune response during endotoxic shock. Proc Natl Acad Sci USA. 2021;118 doi: 10.1073/PNAS.2016648118/SUPPL_FILE/PNAS.2016648118.SD01.XLSX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ibarra-Soria X., Nakahara T.S., Lilue J., Jiang Y., Trimmer C., Souza M.A.A., et al. Variation in olfactory neuron repertoires is genetically controlled and environmentally modulated. Elife. 2017;6 doi: 10.7554/ELIFE.21476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arbyn M., Weiderpass E., Bruni L., de Sanjosé S., Saraiya M., Ferlay J., et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Heal. 2020;8:e191–e203. doi: 10.1016/S2214-109X(19)30482-6/ATTACHMENT/FD42A35C-794D-4608-B131-ED8B7406F3C5/MMC1.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leung S.K., Jeffries A.R., Castanho I., Jordan B.T., Moore K., Davies J.P., et al. Full-length transcript sequencing of human and mouse cerebral cortex identifies widespread isoform diversity and alternative splicing. Cell Rep. 2021;37 doi: 10.1016/J.CELREP.2021.110022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Imada E.L., Strianese D., Edward D.P., alThaqib R., Price A., Arnold A., et al. RNA-sequencing highlights differential regulated pathways involved in cell cycle and inflammation in orbitofacial neurofibromas. Brain Pathol. 2022;32 doi: 10.1111/BPA.13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cnop M., Abdulkarim B., Bottu G., Cunha D.A., Igoillo-Esteve M., Masini M., et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes. 2014;63:1978–1993. doi: 10.2337/DB13-1383. [DOI] [PubMed] [Google Scholar]
- 89.Wang Y., Mashock M., Tong Z., Mu X., Chen H., Zhou X., et al. Changing technologies of RNA sequencing and their applications in clinical oncology. Front Oncol. 2020;10 doi: 10.3389/fonc.2020.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Data Availability Statement
RNA sequencing data were deposited in NCBI Sequencing Read Archive (SRA) under accession ID PRJNA905832.