

Abstract
The transmembrane α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) receptor regulatory protein γ−8 (TARP γ−8) constitutes an auxiliary subunit of AMPA receptors, which mediates various brain functions including learning and memory. TARP γ−8 has emerged as a promising therapeutic target for central nervous system disorders. Despite considerable efforts, previously reported TARP γ−8 PET radioligands, such as [11C]TARP-1903 or the [11C]TARP-1811 series, were plagued by limited brain uptake and/or high nonspecific binding in vivo. Herein, we developed two novel 11C-labeled probes, [11C]8 and [11C]15 (also named as [11C]TARP-2105), of which the latter exhibited reasonable brain uptake as well as specific binding towards TARP γ−8 both in vitro and in vivo, as confirmed for the TARP γ−8-rich hippocampus by blocking experiments with the commercially available TARP γ−8 inhibitor, JNJ-55511118. Overall, [11C]15 exhibited promising tracer characteristics and proved to be a lead positron-emission tomography (PET) ligand for non-invasive quantification of TARP γ−8 in the mammalian brain.
Keywords: α-Amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, transmembrane AMPA receptor regulatory proteins (TARPs), γ−8 dependent TARPs, positron emission tomography (PET), carbon-11
Graphical Abstract

INTRODUCTION
α-Amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) constitute glutamate-gated ion channels that mediate fast excitatory synaptic transmission throughout the mammalian central nervous system (CNS).1 Notably, the regulation of AMPARs is critical for synaptic plasticity and as such, physiological AMPARs orchestrate cognitive functions such as learning and memory.2 Conversely, malfunction of these receptors has been implicated in a number of neurological and psychiatric disorders, such as schizophrenia and epilepsy.3,4 The broad-spectrum AMPAR-targeted antagonist, perampanel, has recently been approved by the FDA for the treatment of epilepsy patients.5–7 However, this compound has been associated with unwanted side effects, such as dizziness, sedation and ataxia, potentially owing to the suppression of AMPAR activity that would be required for physiological brain function.8 Accordingly, more recent AMPAR-targeted drug discovery efforts have shifted towards the discovery of subtype-selective modulators and prompted the identification of a number of potent inhibitors that are selective for the transmembrane AMPAR regulatory protein γ−8 (TARP γ−8).9–12
AMPARs form a complex with their auxiliary subunits,13–15 whereas these auxiliary subunits are involved in receptor trafficking, gating kinetics, and pharmacology.16–18 Transmembrane AMPAR regulatory proteins (TARPs), the first identified receptor auxiliary subunits, are categorized into three subgroups based on the sequence homology and modulatory action: type 1a (γ−2 and γ−3), type 1b (γ−4 and γ−8), and type 2 (γ−5 and γ−7).15, 19, 20 Generally speaking, TARP isoforms are expressed in distinct brain regions and promote slow gating kinetics, thereby prolonging receptor activation. For instance, TARP γ−8 is enriched in the hippocampus of murine brain, which offers an opportunity to selectively regulate AMPAR activity in specific brain circuits without eliciting the aforementioned side effects of perampanel.21 Several promising TARP γ−8-dependent antagonists have recently been discovered, including JNJ-55511118,8 LY-3130481,10, 22, 23 and JNJ-61432059 (Figure 1A).11, 24
Figure 1.

Representative TARP γ−8-selective AMPAR antagonists and PET tracers. (A) TARP γ8-dependent AMPAR antagonists; (B) TARP γ8-selective AMPAR tracers; (C) this work.
Non-invasive molecular imaging by positron emission tomography (PET) constitutes an ideal tool to study TARP γ−8 in vivo, and can be harnessed to quantitatively assess TARP γ−8 abundancy. Accordingly, the availability of a clinically validated TARP γ−8 PET radioligand would allow scientists to obtain in-depth knowledge of TARP γ−8 related pathological changes between normal and disease states. Further, PET imaging holds promise to facilitate the development of novel TARP γ−8 inhibitors by means of target engagement studies with promising clinical drug candidates. Considerable efforts have been devoted to the development of a suitable TARP γ−8 PET ligand in the past few years. We have recently reported on an initial attempt of TARP γ−8-targered PET imaging with several 11C-labeled TARP γ−8 inhibitors including [11C]TARP-1811 series (Figure 1B).25 Nonetheless, all these probes had suffered from limited brain uptake and/or high nonspecific binding in vivo, impeding their further evaluation in higher species. In a second attempt, a structurally distinct TARP γ−8-targeted PET ligand that was derived from the TARP γ−8 antagonist, LY3130481/CERC-611, has recently been disclosed ([11C]TARP-1903, Figure 1B).10,26 Unfortunately, the development of [11C]TARP-1903 was discontinued due to poor in vivo specificity in the TARP γ−8-rich hippocampus. To date, there is an unmet need for the development of TARP γ−8-specific and -selective AMPAR PET radioligands with favorable stability attributes and brain uptake kinetics.
As part of our TARP γ−8 medicinal chemistry program,25–28 we designed and synthesized a new generation of TARP γ−8 inhibitors with high potency and amenability for radiolabeling. Our previous scaffolds including phenotypes in [11C]TARP-1811 and [11C]TARP-1903 series showed good to excellent potency but suffered from low brain uptake. We postulate that the poor CNS permeability could be attributed to urea or thiazolone moiety in the early generation TARP γ−8 antagonists, in which the carbonyl group might cause extensive interaction with efflux transporters at the blood-brain barrier. We therefore design a structural hopping strategy and comparison approach from benzothiazoline to indazole by removing the carbonyl group and test our hypothesis in two series of novel TARP γ−8 scaffolds amenable for 11C-labeling. In this study, we describe the chemical synthesis and pharmacological evaluation of these novel classes of compounds that are based either on a benzothiazolone or indazole scaffold, respectively (Scheme 1). Pharmacological studies and physicochemical evaluations led to the identification of compounds 8 and 15 as the most promising TARP γ−8 inhibitors, which were selected for radiolabeling, yielding [11C]8 (also named as [11C]TARP-2103) and [11C]15 (also named as [11C]TARP-2105; Figure 1C). To assess the suitability of [11C]8 and [11C]15 as PET ligands, we conducted in vitro autoradiography, ex vivo metabolite analysis and whole-body biodistribution as well PET imaging and dosimetry experiments. [11C]15 exhibited moderate brain uptake with high-level specific binding to TARP γ−8 in the brain and was served as a promising lead to develop suitable PET ligands for TARP γ−8-targered PET imaging.
Scheme 1.

Synthesis of two series of TARP γ−8 inhibitors: (A) benzothiazolone scaffold 5–8 and (B) indazole scaffold 13–15. Reagents and Conditions: (i) 10% Pd/C, H2, EtOH, rt, 16 h; (ii) 2-chloro-6-methyl-3-nitropyridine, NEt3, 1,4-dioxane, 100 °C, 16 h, 53% in two steps; (iii) 10% Pd/C, H2, EtOH, rt, 16 h, 94%; (iv) carbaldehydes, Cu(OAc)2, HOAc, rt, 16 h, 30–59%; (v) 10% Pd/C, H2, EtOH, rt, 16 h; (vi) 2-chloro-6-methyl-3-nitropyridine, NEt3, 1,4-dioxane, 100 °C, 16 h, 81% in two steps; (vii) 10% Pd/C, H2, EtOH, rt, 16 h, 91%; (viii) carboxyl acids, HATU, DIEA, DMF, rt; (ix) HOAc, 80 °C; (x) K2CO3, CH3OH, rt, 1 h, 39–48% in three steps. HATU = 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate, DIEA = N,N-diisopropylethylamine, DMF = N,N-dimethylformamide.
RESULTS AND DISCUSSION
Chemistry and Pharmacology.
Based on recent discovery of clinical-stage-ready TARP γ−8 targeted therapeutic agents from Eli Lilly/CREC and Janssen Pharmaceuticals,11, 29–32 as a continuation of our efforts towards PET ligand development, two focused libraries of 6-(6-methyl-1H-benzo[d]imidazol-1-yl)benzo[d]thiazol-2(3H)-one 5-8 and 5-(6-methyl-1H-benzo[d]imidazol-1-yl)-1H-indazole 13-15 as TARP γ−8 inhibitor candidates with amenability for 11C-labeling were designed and synthesized. We anticipated these second-generation radiolabeled TARP γ−8 antagonists with high potency and optimal lipophilicity would overcome the challenges associated with low brain uptake and/or high non-specific binding in vivo. As summarized in Scheme 1A, 6-aminobenzo[d]thiazol-2(3H)-one 2 was obtained in 93% yield by reducing commercially available nitro compound 1 in the presence of 10% Pd/C and H2.The synthesis of key intermediate 3 was achieved in 76% through aromatic nucleophilic substitution reactions between 2-chloro-6-methyl-3-nitropyridine and aminobenzothiazolone 2. After second Pd-catalyzed hydrogenation, the ensuing compound 4 was reacted with different aldehydes under Cu(OAc)2/HOAc mediated condensation and oxidation reactions to afford a series of TARP γ−8 inhibitor candidates 5-8 in 30% to 59% yields over 2 steps. For the preparation of TARP γ−8 inhibitor candidates 13–15, by replacing 6-aminobenzo[d]thiazol-2(3H)-one 2 to 1H-indazol-5-amine 7, we applied the similar experimental procedures as shown in Scheme 1B. Owing to the different electronic properties of 6-methyl-N2-(7-H/methyl-1H-indazol-5-yl)pyridine-2,3-diamine 12 from 6-((3-amino-6-methylpyridin-2-yl)amino)benzo[d]thiazol-2(3H)-one 4, a cascade 3-step-synthesis strategy was employed to provide compounds 13–15 in a range of yields from 39% to 45% by reacting key intermediate 12 with the corresponding carboxylic acids.
Utilizing fluorescence imaging plate reader (FLIPR) assay of heterologous cells, two classes of compounds 5–8 and 13–15 were subsequently tested for their potency and selectivity toward TARP γ−8 as shown in Table 1. GluA1 subunit, one of the major AMPARs in the hippocampus, was utilized here for in vitro pharmacology assay studies. Specifically, the GluA1 flop splicing isoform (GluA1o) was utilized in our assay, due to its faster desensitization compared to other splice variant flip isoforms as well as the increased modulation by TARP. Briefly, GluA1o and TARP γ−8 were co-expressed in human embryonic kidney 293 (HEK-293) cells, followed by assessment of the ability of candidate compounds to block 100 μM glutamate-induced calcium influx via FLIPR assays. As a result, compound 8 and compound 15, containing the same cyclobutyl group, demonstrated the most promising potency among their own series. The inhibition profiles for compound 8 and compound 15 toward TARP γ−8 activity by IC50 values were 0.02 nM and 1.26 nM, respectively, while all the other candidates 5–7 and 13–14 indicated inferior potency. Both, compound 8 and 15, fall into the optimal range of preferred parameters for CNS ligands,33–35 i.e., molecular weight (≤350); lipophilicity (log P 2–3.5) and reasonable tPSA values (Table 1). Measured by the ‘shake-flask’ method, we determined the logD7.4 value of compounds 8 and 15 to be 2.3±0.06 and 2.5±0.01, respectively. In addition, compound 15 showed a higher probability of brain penetration (logBBcompound 15 = 0.12) compared to that of compound 8 (logBBcompound 8 = −0.30) based on ACD Percepta® prediction. To assess compound selectivity, an off-target counter screen of compound 15 was carried out at a testing concentration of 10 μM towards ionotropic glutamate receptors (iGluR) and other major CNS targets, including common GPCRs, ion channels, enzymes and transporters (Figure S1). While the results exhibited that compound 15 has no substantial interaction with related iGlu receptor family, there were three targets, namely 5-HT1A, Alpha2C and DOR (delta opioid receptor), identified with greater than 50% target inhibition in the comprehensive CNS screening. Follow-up studies for 5-HT1A, Alpha2C and DOR binding assays indicated greater than 750-fold selectivity towards TARP γ−8 among any other tested CNS targets (Figure S1).
Table 1.
Potency and physicochemical properties of two classes of compounds 5–8, 13–15, TARP-1811 and TARP-1903
| Compound | TARP γ−8 IC50 (nM)a | MWb | tPSAb | clogPc | Compound | TARP γ−8 IC50 (nM)a | MWb | tPSAb | clogPc |
|---|---|---|---|---|---|---|---|---|---|
| 5 | 4.50 | 324.4 | 57.06 | 3.28 | 13 | 11.3 | 291.4 | 52.35 | 3.72 |
| 6 | 1.50 | 324.4 | 57.06 | 3.35 | 14 | 3.40 | 289.3 | 52.35 | 2.92 |
| 7 | 1.41 | 322.4 | 57.06 | 2.71 | 15 | 1.26 | 303.4 | 52.35 | 3.36 |
| 8 | 0.02 | 336.4 | 57.06 | 3.49 | TARP-1811 | 19.5 | 283.7 | 64.92 | 3.48 |
| TARP-1903 | 16.0 | 340.4 | 57.06 | 2.88 |
Notes:
determined by FLIPR assays in human TARP γ−8 expressing HEK cell lines;
calculated by ChemBioDraw Ultra 14.0;
calculated by ACD/Labs Percepta 2021.
Radiochemistry.
Among two series of TARP γ−8 inhibitor candidates, benzothiazolone 8 and indazole 15 were selected for radiochemistry development attributed to their high potency, optimal physiochemical property, and predicted brain permeability in each category, respectively. Interestingly, the most commonly-used 11C-methylation method by SN2 displacement35 was not suitable for producing radioligands [11C]8 and [11C]15. Therefore, inspired by previously reported methodology,36, 37 we developed a Stille coupling-based labeling methodology from the corresponding stannyl precursors to achieve 11C-methylation on the common chemotype of 3-substituted 5-methyl-3H-imidazo[4,5-b]pyridine. In particular, the preparation of the corresponding stannyl precursors 22 and 27 were carried out as demonstrated in Scheme 2. In comparison to the synthesis of 4, Fe/NH4Cl instead of Pd/C-H2 system offered mild conditions to afford chemoselectivity (nitro reduction to amine) in the presence of bromo-substitution. 6-((3-Amino-6-bromopyridin-2-yl)amino)benzo[d]thiazol-2(3H)-one 20 was thus obtained in 81% yield from bromide 19, which underwent condensation with cyclobutanecarbaldehyde, followed by oxidation, to afford indazole 21. The subsequent Pd catalyzed Stannylation generated 22 as a precursor in the yield of 36% (Scheme 2A). Similarly, the other precursor 27 was obtained from commercially available starting material of 1H-indazol-5-amine 10 in 6 steps (Scheme 2B).
Scheme 2.

Synthetic chemistry and radiolabeling. (A) Synthesis of radiolabeling precursor 22 and radiosynthesis of [11C]8. Reagents and conditions: (i) 2,6-dibromo-3-nitropyridine, Et3N, 1,4-dioxane, 100 °C, 16 h, 81%; (ii) Fe, sat. NH4Cl, EtOH, reflux, 4 h, 76%; (iii) cyclobutanecarbaldehyde, Cu(OAc)2, HOAc, rt, 16 h, 82%; (iv) (nBu3Sn)2, Pd(PPh3)4, toluene, reflux, 16 h, 36%; (v) [11C]CH3I, Pd2(dba)3, P(o-tol)3, CuBr, K2CO3, DMF, 130 °C, 5 min, 9% RCY (decay-corrected). (B) Synthesis of radiolabeling precursor 27 and radiosynthesis of [11C]15. Reagents and conditions: (vi) 2,6-dibromo-3-nitropyridine, NEt3, 1,4-dioxane, 100 °C, 16 h, 81%; (vii) Fe, sat. NH4Cl, EtOH, reflux, 4 h, 86%; (viii) cyclobutanecarboxylic acid, HATU, DIEA, DMF, rt; (ix) HOAc, 80 °C; (x) K2CO3, CH3OH, rt, 1 h, 54% over three steps; (xi) (nBu3Sn)2, Pd(PPh3)4, toluene, reflux, 16 h, 15%; (xii) [11C]CH3I, Pd2(dba)3, P(o-tol)3, CuBr, K2CO3, DMF, 130 °C, 5 min, 22% RCY (decay-corrected); dba = dibenzylideneacetone, HATU = 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-Oxide Hexafluorophosphate, DIEA = N,N-Diisopropylethylamine, DMF = N,N-Dimethylformamide, dba = dibenzylideneacetone.
Subsequently, the radiosynthesis of [11C]8 and [11C]15 from the corresponding precursors were carried out. The reactions between [11C]CH3I and precursor 22 or 27 were carried out by heating a mixture of the precursor (0.8 mg), [11C]CH3I, Pd2(dba)3 (0.2 mg), P(o-tol)3 (6.0 mg), CuBr (1.0 mg) and K2CO3 (1.0 mg) in DMF (0.45 mL) at 130 °C for 5 min. As a result, [11C]8 and [11C]15 were obtained in an average radiochemical yield (RCY) of 9% and 22%, respectively (relative to [11C]CO2, decay corrected) with both high radiochemical purity (>99%) and excellent molar activity (>150 GBq/μmol at the end of synthesis (EOS)) with total synthesis time 27 minutes. Both radioligands [11C]8 and [11C]15 exhibited excellent in vitro stability (>95%) up to 90 min after formulation. The novel radiosynthesis, combined with high radiochemical purity and molar activities of [11C]8 and [11C]15 allowed us to perform the subsequent in vitro and in vivo evaluation studies.
Autoradiography study of TARP γ−8 tracer.
The autoradiography (ARG) studies for [11C]8 and [11C]15 were conducted to evaluate target binding specificity and selectivity. As shown in Figure 2A and 2B, autoradiograms on sagittal sections of rat brains (baseline) indicated heterogeneous regional distribution pattern from high to low in the order of hippocampus, frontal cortex, striatum, thalamus and cerebellum in [11C]8 and [11C]15 ARG studies. The distribution profile was consistent with the mRNA expression pattern of TARP γ−8 with the highest signal level in the hippocampus and the lowest level in the cerebellum.38, 39 As for [11C]8, blocking studies by non-radioactive compound 8 (1 μM) led to a markedly decreased bound radioactivity (83% reduction) in the TARP γ−8 rich hippocampus (Figure 2C). Concurrently, no notable change was observed for the reference region, i.e. the cerebellum. Similarly, the autoradiography studies for [11C]15 revealed baseline autoradiograms with heterogeneous distributions on sagittal sections of rat brain. For ligand [11C]15, blocking studies with 15 (1 μM) were carried out, yielding a specificity value of 86% in the hippocampus (Figure 2D). Furthermore, a validated TARP γ−8 inhibitor JNJ-55511118 (1 μM)30 was employed as structurally distinct blocking reagent and resulted in 77% reduction of brain uptake in the hippocampus (Figure 2E). Both blocking studies of [11C]15 successfully abolished heterogeneity of tracer binding in the different brain regions. These results indicated that [11C]8 and [11C]15 exhibited high in vitro specificity towards TARP γ−8, which encouraged us to advance these ligands to PET studies.
Figure 2.

In vitro autoradiography of [11C]8 and [11C]15 in rat brain sections and quantification. (A) Representative image for baseline studies of [11C]8; (B) Representative image for baseline studies of [11C]15; (C) Representative image for blocking studies (8, 1 μM) with [11C]8; (D) Representative image for blocking studies (15, 1 μM) with [11C]15; (E) Representative image for blocking studies (JNJ-55511118, 1 μM) with [11C]15; (F) Quantification of autoradiography studies for baseline and blocking (8) with [11C]8; (G) Quantification of autoradiography studies for baseline and blocking (8 or JNJ-55511118) with [11C]8. All data were referred as mean ± SD, n ≥ 3. Statistical analysis was achieved by use of a student’s t-test with asterisks indication the statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
PET Imaging Study of TARP γ−8 Tracers.
PET imaging studies for in vivo brain kinetics and binding specificity of radioligands [11C]8 and [11C]15 were performed in Sprague-Dawley rats for 60 min. Representative PET images of [11C]8 and [11C]15 in the brain (coronal, summed images 0–60 min) and time-activity curves (TACs) are shown in Figure S2 and Figure 3, respectively. Preliminary results suggested that compound [11C]8 failed to penetrate the blood brain barrier (BBB) with a maximum standardized uptake value (SUV) of 0.4 at 2.5 min in the brain. In addition, no significant heterogeneous distribution in the hippocampus, frontal cortex, and cerebellum was observed in the baseline study of [11C]8. Conversely, and as shown in Figure 4A, the TACs of compound [11C]15 exhibited improved BBB penetration with a maximum SUV of ca. 0.7 at 2.5 min in the brain hippocampus (Figure 3D). The brain uptake was low to moderate, inconsistent with our predicted brain permeability. We suspected that this discrepancy could be attributed to the ligand’s in vivo interaction with multiple efflux transporters such as Pg-P and BCPR in the murine brain. Further efflux mechanistic investigation will be needed using efflux transporter knock mice. The radioactivity uptake showed heterogeneous distribution in the brain, including hippocampus, frontal cortex, striatum, and cerebellum. Particularly, the regional brain uptake of [11C]15 was substantially higher in the hippocampus (TARP γ−8 rich region) than in the cerebellum (TARP γ−8 deficient area), which was in agreement with the known TARP γ−8 expression patterns in the rodent brain.38, 39 Pretreatment with cold compound 15 (1 mg/kg; Figure 3B and 3E) and a validated TARP γ−8 antagonist, JNJ-55511118 (1 mg/kg; Figure 3C and 3F), demonstrated decreased uptake of radioactivity in the hippocampus (43–53% and 35–47% reduction based on area under curve (AUC), respectively; Figure S3). In addition, the TACs of [11C]15 in the hippocampus were compared under baseline and blocking conditions in Figure 4G, and substantial reduction of [11C]15 uptake in blocking studies were observed. These collective results indicated excellent in vivo binding specificity of [11C]15 toward TARP γ−8.
Figure 3.

Representative PET/MRI fused coronal images (summed at 0–10 min, 15–30 min and 30–60 min) and time-activity curves (TACs) of [11C]15 under baseline and blocking conditions in SD rat brain. (A) Representative summed PET images of [11C]15 in various brain regions of interest under baseline condition; (B) Representative summed PET images of [11C]15 under blocking condition with 15; (C) Representative summed PET images of [11C]15 under blocking condition with JNJ-55511118; (D) TACs of [11C]15 in various brain regions of interest under baseline condition; (E) TACs of [11C]15 under blocking condition with 15; (F) TACs of [11C]15 under blocking condition with JNJ-55511118; (G) TACs of [11C]15 in hippocampus region under baseline and blocking conditions. Blocking conditions: 15 (1 mg/kg) or JNJ-55511118 (1 mg/kg, iv). Data are presented as mean ± SD (n = 3).
Figure 4.

Parametric mapping and binding potentials of [11C]15 in rat brains. (A) Baseline studies of [11C]15; (B) Blocking conditions: 15 (1 mg/kg, iv); (C) Blocking conditions: JNJ-55511118 (1 mg/kg, iv); (D) Quantification of BPND for baseline and blocking conditions. Data are presented as mean ± SD (n = 3) and analyzed by use of a student’s t-test with asterisks indication the statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To further quantify the specific binding of [11C]15, we also analyzed the non-displaceable binding potential (BPND) values by the simplified reference tissue model (SRTM).40, 41 The cerebellum was used as the pseudo reference region because of its lowest and consistent brain uptake in TACs profile between baseline and blocking conditions. Figure 4 demonstrated that the BPND map and bound signal of [11C]15 in rat brain in a decreasing order in the hippocampus (1.22), frontal cortex (0.94), and striatum (0.65) under baseline conditions. The distribution pattern was consistent with the expression of TARP γ−8 in murine brain22, 38, 39 as well as in vitro autoradiography results in Figure 2. It was observed that the BPND values were decreased substantially under blocking conditions by the pretreatment with 15 (93–99% reduction, 1 mg/kg) and JNJ-55511118 (66–81% reduction, 1 mg/kg). The discrepancy of blocking effects using two different TARP γ−8 inhibitors could be explained, at least in part, by the poor solubility of JNJ-55511118, which limits its blocking dose higher than 1 mg/kg and poor brain permeability due to urea structure with two hydrogen-bonding interactions. Although ligand 15 demonstrated high specific binding both in vitro and in vivo, further studies using TARP γ−8 knockout mice will be needed to exclude off-target binding. In addition, the parametric brain mapping demonstrated that the signal heterogeneity was abolished completely under these blocking conditions. Collectively, the uptake differences of radioactivity in the detailed brain regions were visualized clearly by parametric BPND images using the SRTM method, and the high specific binding of [11C]15 in vivo between baseline and blocking conditions was also confirmed.
Whole-body biodistribution study and estimated human dosimetry.
Whole-body ex vivo biodistribution of [11C]15 was investigated in ddY mice at five time points (1, 5, 15, 30, and 60 min) after radioligand injection. As shown in Figure 5, initial high uptake of radioactivity (≥5 %ID/g at 1 min) was observed in the lungs and heart, followed by fast clearance (ratio of %ID/g1min/60min: 10.5 in lungs, 3.6 in heart). Rapid washout of [11C]15 from blood (%ID/g1min/60min ratio of 2.9) was observed. High radioactivity was accumulated in the liver, kidneys, and small intestine at later time point, which indicated possible hepatobiliary and urinary elimination of [11C]15. The whole-body biodistribution of [11C]15 in mice were then translated into radiation dose estimates for human subjects by calculation (male and female models), shown in Table 2. The effective dose of [11C]15 was 0.0044 mSv/MBq for a standard human (Male = 0.0040 mSv/MBq, Female = 0.0049 mSv/MBq), which is similar to that (0.005 mSv/MBq) of the widely used [11C]methionine.42 These results indicate that clinical PET with [11C]15 would be safe with respect to radiation exposure.
Figure 5.
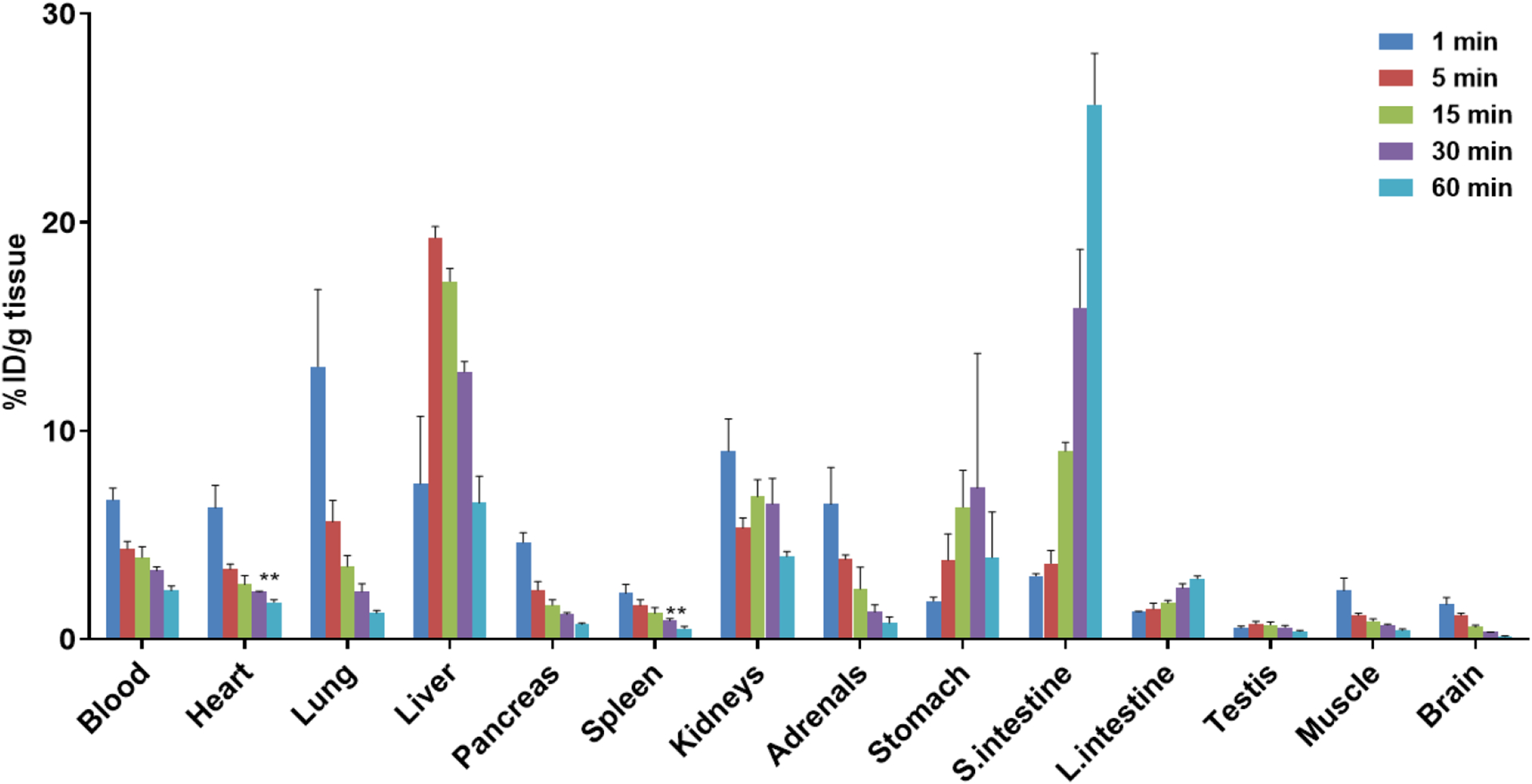
Whole-body ex vivo biodistribution studies in mice at five different time points (1, 5, 15, 30 and 60 min) post injection of [11C]15. Data are expressed as %ID/g (mean ± SD, n = 3). %ID/g = injected dose per gram of wet tissue.
Table 2.
Radiation dosimetry estimates for human
| Target Organ | Radiation dose(μSv/MBq) | ||
|---|---|---|---|
| Male | Female | Standard human | |
| Adrenals | 3.4 | 4.1 | 3.8 |
| Drain | 1.0 | 1.2 | 1.1 |
| Breasts | 2.0 | 2.5 | 2.3 |
| Gallbladder wall | 4.0 | 4.8 | 4.4 |
| Lower large intestine wall | 5.0 | 5.9 | 5.5 |
| Small Intestine | 13.7 | 16.0 | 14.9 |
| Stomach wall | 5.3 | 6.4 | 5.8 |
| Upper large intestine wall | 5.2 | 6.3 | 5.8 |
| Heart wall | 4.4 | 5.4 | 4.9 |
| Kidneys | 5.6 | 6.3 | 5.9 |
| Liver | 11.1 | 14.7 | 12.9 |
| Lungs | 3.6 | 4.6 | 4.1 |
| Muscle | 1.8 | 2.4 | 2.1 |
| Ovaries | 3.4 | 4.2 | 3.8 |
| Pancreas | 3.1 | 3.7 | 3.4 |
| Red marrow | 2.3 | 2.7 | 2.5 |
| Osteogenic cells | 2.8 | 3.6 | 3.2 |
| Skin | 1.8 | 2.2 | 2.0 |
| Spleen | 2.1 | 2.6 | 2.4 |
| Testes/Uterus | 0.8 | 4.0 | 2.4 |
| Thymus | 2.3 | 2.9 | 2.6 |
| Thyroid | 2.1 | 2.5 | 2.3 |
| Urinary bladder wall | 2.6 | 2.8 | 2.7 |
| Total Body | 2.9 | 3.6 | 3.2 |
| Effective dose | 4.0 | 4.9 | 4.4 |
Radiometabolite analysis.
To investigate the in vivo stability of [11C]15, the radiometabolic analysis at 10 and 30 min post injection of the radiotracer in rat brain and plasma homogenate was performed (Figure 6). The fractions of the remaining parent [11C]15 were determined by radio-HPLC. The values at 10 min in brain and plasma were 98% and 55% respectively, and at 30 min post injection, the parent compound remained 92% and 26% in the brain and plasma, respectively, which suggested reasonable metabolic stability in vivo.
Figure 6.

Radiometabolite analysis of [11C]15 in rats (n=2)
CONCLUSION
Substantial progress has been made in delineating the mechanisms by which TARP γ−8 is involved in neurophysiology and there is an accumulating body of evidence supporting its implication in various neurological disorders. Notwithstanding the stannous efforts among several PET ligand discovery programs, a clinically validated TARP γ−8 PET radioligand is currently lacking. In the present work, we report on the first TARP γ−8-targeted PET probe, [11C]15 (also named as [11C]TARP-2105), with promising tracer characteristics in vitro as well as in vivo. Starting from a focused medicinal chemistry library comprising newly designed and synthesized benzothiazolones and indazoles, target compounds 8 and 15 were selected as best in class candidates for radiolabeling and biological evaluation. Their carbon-11 labeled analogues, [11C]8 and [11C]15, both exhibited high TARP γ−8 selectivity by in vitro autoradiography on rodent brain sections. While [11C]8 did not reach sufficient brain exposure, [11C]15 exhibited moderate to good brain uptake and a slow washout from the rodent brain while showing an effective radiation dose that was within the range of routinely used clinical PET ligands, as determined by ex vivo biodistribution and dosimetry experiments. Further kinetic modeling in nonhuman primates, efflux mechanism study in Pgp/BCRP knockout mice and/or binding specificity in TARP γ−8 knockout mice are necessary steps to support future clinical translation. We envision that the availability of a clinically validated probe would enable non-invasive mapping of TARP γ−8 in patients and healthy subjects, thereby shedding light on its role in the various pathologies and allowing personalized treatment regimens for every individual patient.
Experimental Section
General information
The experimental procedures were conducted as previous reports with minor modifications.40, 41, 43, 44 All chemicals were ordered from commercial suppliers and used in the synthesis without further purification. The NMR spectra were collected on a 300 MHz Bruker and/or 500 MHz Varian spectrometers at room temperature, and 1H NMR spectra and 13C NMR spectra were obtained at 300MHz/75MHz and 500MHz/126MHz, respectively. 1H NMR chemical shifts were determined relative to internal (CH3)4Si (TMS) at δ 0.00 ppm. 13C NMR chemical shifts were determined relative to the signal of the solvent: CDCl3 δ 77.16 ppm, CD3OD δ 49.00 ppm, or DMSO-d6 δ 39.52 ppm. Chemical shifts (δ) were reported in ppm and coupling constants are reported in Hz. The multiplicities are abbreviated as follows: s (singlet), d (doublet), t (triplet), m (multiplet), br (broad signal), dd (doublet of doublets), and so forth. The purities of key compounds are > 95%, which were determined by reverse-phase HPLC (Agilent 1200). Mass spectra was obtained on Agilent 6430 Triple Quad LC/MS mass spectrometer with ESI as the ionization approach. High-resolution mass data were recorded on a high resolution mass spectrometer in the ESI mode. All animal studies were approved by the institutional animal care and use committee (IACUC) and institutional biosafety committee (IBC) at the Massachusetts General Hospital. DdY mice (female, 22−24 g, 7–9 weeks), SD rats (male, 210−230 g, 7–9 weeks) were fed ad libitum with food and water under 12 h light/12 h dark cycle.
Synthesis of TARP ɣ−8 inhibitors 5–8
6-((6-Methyl-3-nitropyridin-2-yl)amino)benzo[d]thiazol-2(3H)-one (3).
Step 1: 6-Nitrobenzo[d]thiazol-2(3H)-one (1, 0.30 g, 1.5 mmol) was subjected to the mixture of 10% Pd/C (0.15 g) in ethanol (15 mL) and stirred at room temperature under H2 atomsphere for 16 hours. The mixture was filtered through Celite and concentrated in vacuum for the next step directly. Step 2: To the crude 6-aminobenzo[d]thiazol-2(3H)-one (2, 1.5 mmol) in 1,4-dioxane (15 mL) was added 2-chloro-6-methyl-3-nitropyridine (0.26 g, 1.5 mmol) and Et3N (0.62 mL, 4.5 mmol). Then the mixture was heated at 100 °C for 16 hours. The reaction was monitored by TLC. After the reaction completed, the solvent was removed under reduced pressure and the residue purified by silica gel column to give 3 (240.1 mg, 53% in two steps) as a red solid. 1H NMR (300 MHz, DMSO-d6) δ 11.81 (brs, 1H), 10.01 (s, 1H), 8.40 (d, J = 8.5 Hz, 1H), 7.92 (d, J = 2.1 Hz, 1H), 7.49 (dd, J = 8.6, 2.2 Hz, 1H), 7.09 (d, J = 8.6 Hz, 1H), 6.81 (d, J = 8.5 Hz, 1H), 2.40 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 170.4, 166.1, 149.5, 136.1, 133.9, 133.4, 126.7, 123.8, 122.3, 117.6, 114.7, 111.8, 25.1. Melting point = 223–226 °C. m/z calcd for C13H10N4O3S: 302.0; found 303.1 (M + H+).
6-((3-Amino-6-methylpyridin-2-yl)amino)benzo[d]thiazol-2(3H)-one (4).
Compound 3 (0.36 g, 1.2 mmol) was subjected to the mixture of 10% Pd/C (0.12 g) in ethanol (12 mL) and stirred at room temperature under H2 atomsphere for 16 hours. The mixture was filtered through Celite and concentrated in vacuum to yield 4 in 94% as a yellow solid without further purification. 1H NMR (300 MHz, DMSO-d6) δ 8.03 (d, J = 2.1 Hz, 1H), 7.73 (s, 1H), 7.45 (dd, J = 8.7, 2.2 Hz, 1H), 6.99 (d, J = 8.6 Hz, 1H), 6.79 (d, J = 7.6 Hz, 1H), 6.43 (d, J = 7.6 Hz, 1H), 4.82 (s, 2H), 2.22 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 170.1, 143.6, 142.5, 138.5, 129.8, 129.2, 123.7, 121.2, 117.6, 114.5, 111.95, 111.8, 23.6. Melting point = 121–123 °C. m/z calcd for C13H12N4OS: 272.1; found 273.2 (M + H+).
6-(5-Methyl-2-propyl-3H-imidazo[4,5-b]pyridin-3-yl)benzo[d]thiazol-2(3H)-one (5).
The mixture of 4 (82.2 mg, 0.3 mmol), n-butyraldehyde (28.8 mg, 0.4 mmol) and Cu(OAc)2 (163.1 mg, 0.9 mmol) in HOAc (30 mL) was stirred at room temperature for 16 h under N2. After completion of the reaction, the solvent was removed under reduced pressure and the residue was purified by flash column chromatography on silica gel to give 5 (40.8 mg, 42%) as an off-white solid. 1H NMR (300 MHz, CD3OD) δ 7.91 (d, J = 7.8 Hz, 1H), 7.64 (s, 1H), 7.35 (s, 2H), 7.21 (d, J = 8.0 Hz, 1H), 2.78 (t, J = 7.4 Hz, 2H), 2.53 (s, 3H), 1.79–1.72 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CD3OD) δ 171.7, 156.5, 153.4, 148.3, 137.2, 131.8, 129.0, 126.4, 126.3, 125.3, 122.4, 118.7, 111.9, 29.3, 22.3, 20.3, 12.6. Melting point = 243–244 °C. MS (ESI, m/z): 325.2 (M + H+). HRMS (ESI): exact mass calcd for C17H17N4OS (M + H+): 325.1123, found: 325.1122.
6-(2-Isopropyl-5-methyl-3H-imidazo[4,5-b]pyridin-3-yl)benzo[d]thiazol-2(3H)-one (6).
Compound 6 was prepared in a similar manner to that described for 5, except that isobutyraldehyde was used. The product 6 was obtained in 31% yield as an off-white solid. 1H NMR (300 MHz, CDCl3) δ 11.66 (s, 1H), 8.03 (d, J = 6.0 Hz, 1H), 7.42 (s, 1H), 7.16 (d, J = 7.2 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 3.08–3.03 (m, 1H), 2.66 (s, 3H), 1.35 (d, J = 5.5 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 171.2, 160.9, 153.1, 148.8, 136.7, 132.6, 129.2, 127.9, 126.0, 125.9, 122.2, 119.1, 112.7, 26.9, 23.6, 21.4. Melting point > 300 °C. MS (ESI, m/z): 325.2 (M + H+). HRMS (ESI): exact mass calcd for C17H17N4OS (M + H+): 325.1123, found: 325.1122.
6-(2-Cyclopropyl-5-methyl-3H-imidazo[4,5-b]pyridin-3-yl)benzo[d]thiazol-2(3H)-one (7).
Compound 7 was prepared in a similar manner to that described for 5, except that cyclopropanecarbaldehyde was used. The product 7 was obtained in 59% yield as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.17 (s, 1H), 7.83–7.80 (m, 2H), 7.42 (dd, J = 8.4, 2.0 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 7.09 (d, J = 8.1 Hz, 1H), 2.43 (s, 3H), 1.87–1.76 (m, 1H), 1.13–1.06 (m, 2H), 0.99 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 170.5, 157.6, 151.9, 149.2, 137.1, 132.5, 129.4, 127.0, 126.4, 124.8, 123.1, 118.5, 112.5, 24.2, 9.7, 8.6. Melting point = 229–231 °C. MS (ESI, m/z): 323.2 (M + H+). HRMS (ESI): exact mass calcd for C17H15N4OS (M + H+): 323.0967, found: 323.0967.
6-(2-Cyclobutyl-5-methyl-3H-imidazo[4,5-b]pyridin-3-yl)benzo[d]thiazol-2(3H)-one (8).
Compound 8 was prepared in a similar manner to that described for 5, except that cyclobutanecarbaldehyde was used. The product 8 was obtained in 30% yield as an off-white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.16 (s, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.71 (s, 1H), 7.29–1.27 (m, 2H), 7.13 (d, J = 8.1 Hz, 1H), 3.68 – 3.46 (m, 1H), 2.44 (s, 3H), 2.43–2.38 (m, 2H), 2.10–2.00 (m, 2H), 1.95 – 1.79 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 170.5, 158.6, 152.5, 149.4, 137.1, 132.5, 129.5, 127.0, 126.9, 124.6, 122.9, 118.5, 112.4, 32.7, 27.3, 24.3, 18.4. Melting point = 226–228 °C. MS (ESI, m/z): 337.2 (M + H+). HRMS (ESI): exact mass calcd for C18H17N4OS (M + H+): 337.1123, found: 337.1124.
Synthesis of TARP ɣ−8 inhibitors 13–15
N2-(1H-Indazol-5-yl)-6-methylpyridine-2,3-diamine (12).
Step 1: 6-Nitrobenzo[d]thiazol-2(3H)-one (9, 0.25 g, 1.5 mmol) was subjected to the mixture of 10% Pd/C (0.15 g) in ethanol (15 mL) and stirred at room temperature under H2 atomsphere for 16 hours. The mixture was filtered through Celite and concentrated in vacuum for the next step directly. Step 2: To the crude 1H-indazol-5-amine (10, 1.5 mmol) in 1,4-dioxane (15 mL) was added 2-chloro-6-methyl-3-nitropyridine (0.26 g, 1.5 mmol) and Et3N (0.62 mL, 4.5 mmol). Then the mixture was heated at 100 °C for 16 hours. The reaction was monitored by TLC. After the reaction completed, the solvent was removed under reduced pressure and the residue purified by silica gel column to give 11 (336 mg, 83% in two steps) as a red solid. Step 3: Compound 11 (0.32 g, 1.2 mmol) was subjected to the mixture of 10% Pd/C (0.12 g) in ethanol (12 mL) and stirred at room temperature under H2 atomsphere for 16 hours. The mixture was filtered through Celite and concentrated in vacuum to give 12 (181 mg, 91%) as a yellow solid. Melting point = 120–123 °C. m/z calcd for C13H13N5: 239.1; found 240.2 (M + H+).
3-(1H-Indazol-5-yl)-2-isopropyl-5-methyl-3H-imidazo[4,5-b]pyridine (13).
Step 1: The mixture of 12 (71.7 mg, 0.3 mmol), isobutyric acid (79.2 mg, 0.9 mmol), HATU (114.1 mg, 0.3 mmol) and DIEA (150.4 μL, 0.9 mmol) in DMF (30 mL) was stirred at room temperature for 16 h under N2. Step 2: After completion of the reaction, the solvent was removed under reduced pressure and the residue was resolved in HOAc (30 mL) and stirred at 80 °C for 16 hours. Then, the solvent was was removed under reduced pressure. Step 3: After completion of the reaction, and the residue was resolved in CH3OH (30 mL) and K2CO3 (207.3 mg, 1.5 mmol) was added. The mixture was stirred at room temperature for 16 hours. Then, the solid was filtered and washed with CH3OH/CH2Cl2 (v/v = 1/1). The filtrate was concentrated under reduced pressure, and the residue was purified by flash column chromatography on silica gel to give 13 (34.1 mg, 39%) as an off-white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.97 – 7.91 (m, 2H), 7.72 (d, J = 8.7 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 7.9 Hz, 1H), 3.08 – 2.98 (m, 1H), 2.41 (s, 3H), 1.20 (d, J = 6.8 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 160.83, 153.20, 149.05, 140.08, 134.72, 127.23, 126.90, 126.59, 123.49, 121.21, 121.08, 118.96, 111.82, 26.91, 24.35, 21.65, 21.44. Melting point = 284–285 °C. MS (ESI, m/z): 292.3 (M + H+). HRMS (ESI): exact mass calcd for C17H18N5 (M + H+): 292.1562, found: 292.1562.
2-Cyclopropyl-3-(1H-indazol-5-yl)-5-methyl-3H-imidazo[4,5-b]pyridine (14).
Compound 14 was prepared in a similar manner to that described for 13, except that cyclopropanecarboxylic acid was used. The product 14 was obtained in 45% yield as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.38 (s, 1H), 8.21 (s, 1H), 7.97–7.94 (m, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.75 (d, J = 8.7 Hz, 1H), 7.45 (dd, J = 8.7, 1.9 Hz, 1H), 7.09 (d, J = 8.1 Hz, 1H), 2.42 (s, 3H), 1.86–1.78 (m, 1H), 1.13–1.08 (m, 2H), 0.99–0.95 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 157.8, 151.8, 149.5, 139.8, 134.6, 132.5, 127.5, 126.6, 126.3, 123.4, 120.7, 118.4, 111.5, 24.2, 9.6, 8.7. Melting point > 300 °C. MS (ESI, m/z): 290.2 (M + H+). HRMS (ESI): exact mass calcd for C17H16N5 (M + H+): 290.1406, found: 290.1407.
2-Cyclobutyl-3-(1H-indazol-5-yl)-5-methyl-3H-imidazo[4,5-b]pyridine (15).
Compound 15 was prepared in a similar manner to that described for 13, except that cyclobutanecarboxylic acid was used. The product 15 was obtained in 48% yield as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.04 (d, J = 8.1 Hz, 1H), 7.91 (s, 1H), 7.55 (d, J = 1.2 Hz, 1H), 7.21–7.16 (m, 1H), 7.06 (dd, J = 8.7, 1.8 Hz, 1H), 3.55–3.50 (m, 1H), 2.69 (s, 3H), 2.65–2.49 (m, 2H), 2.21–2.04 (m, 2H), 2.15–1.96 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 159.1, 152.9, 149.6, 139.5, 134.7, 133.0, 127.5, 127.1, 125.4, 123.2, 120.3, 118.6, 111.3, 32.8, 27.6, 23.9, 18.5. Melting point > 300 °C. MS (ESI, m/z): 304.3 (M + H+). HRMS (ESI): exact mass calcd for C18H18N5 (M + H+): 304.1562, found: 304.1563.
Synthesis of labeling precursor 22
6-((6-Bromo-3-nitropyridin-2-yl)amino)benzo[d]thiazol-2(3H)-one (19).
To 2 (249.1 mg, 1.5 mmol) in 1,4-dioxane (15 mL) was added 2,6-dibromo-3-nitropyridine (423.7 mg, 1.5 mmol) and Et3N (0.62 mL, 4.5 mmol). Then the mixture was heated at 100 °C for 16 hours. The reaction was monitored by TLC. After the reaction completed, the solvent was removed under reduced pressure and the residue purified by silica gel column to give 19 (445.9 mg, 81%) as a red solid. 1H NMR (300 MHz, DMSO-d6) δ 11.92 (s, 1H), 10.08 (s, 1H), 8.37 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 1.7 Hz, 1H), 7.41 (dd, J = 8.5, 1.9 Hz, 1H), 7.13–7.07 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 170.3, 149.8, 146.5, 138.5, 134.1, 133.0, 128.5, 123.8, 123.3, 118.7, 118.1, 111.8. Melting point = 200–203 °C. m/z calcd for C12H779BrN4O3S: 365.9; found 367.0 (M + H+).
6-((3-Amino-6-bromopyridin-2-yl)amino)benzo[d]thiazol-2(3H)-one (20).
Compound 19 (0.37 g, 1.0 mmol) was subjected to the mixture of Fe powder (0.56 g, 10.0 mmol) in satuarated NH4Cl (5 mL) and ethanol (5 mL) and stirred in refluxed under N2 for 4 hours. The mixture was filtered through Celite and concentrated in vacuum to yield 20 as a grey solid (256.1 mg, 76%) without further purification. 1H NMR (300 MHz, DMSO-d6) δ 11.71 (s, 1H), 8.03 (s, 1H), 7.85 (s, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 7.6 Hz, 1H), 6.74 (d, J = 7.6 Hz, 1H), 4.63 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 170.1, 143.9, 137.1, 131.2, 130.8, 123.8, 123.4, 122.8, 118.4, 118.2, 112.9, 112.0. Melting point = 171–173 °C. m/z calcd for C12H979BrN4OS: 336.0; found 337.1(M + H+).
6-(5-Bromo-2-cyclobutyl-3H-imidazo[4,5-b]pyridin-3-yl)benzo[d]thiazol-2(3H)-one (21).
The mixture of 20 (101.2 mg, 0.3 mmol), n-butyraldehyde (28.8 mg, 0.4 mmol) and Cu(OAc)2 (163.1 mg, 0.9 mmol) in HOAc (30 mL) was stirred at room temperature for 16 h under N2. After completion of the reaction, the solvent was removed under reduced pressure and the residue was purified by flash column chromatography on silica gel to give 21 (98.6 mg, 82%) as an off-white solid. 1H NMR (300 MHz, CDCl3) δ 10.27 (s, 1H), 7.98 (d, J = 8.3 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.38 (s, 1H), 7.18–7.07 (m, 2H), 3.63–3.52 (m, 1H), 2.68–2.49 (m, 2H), 2.28–2.15 (m, 2H), 2.04–1.96 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 171.1, 159.9, 1491, 136.2, 135.1, 134.0, 129.5, 128.8, 125.8, 125.6, 122.7, 121.8, 112.3, 32.7, 27.6, 18.5. Melting point = 186–189 °C. MS (ESI, m/z): 401.1 (M + H+). HRMS (ESI): exact mass calcd for C17H1479BrN4OS (M + H+): 401.0072, found: 401.0071.
6-(2-Cyclobutyl-5-(tributylstannyl)-3H-imidazo[4,5-b]pyridin-3-yl)benzo[d]thiazol-2(3H)-one (22).
The mixture of 21 (40.1 mg, 0.1 mmol), Pd(PPh3)4 (11.6 mg, 0.01 mmol) and 1,1,1,2,2,2-hexabutyl-distannane (153.2 μL, 0.3 mmol) in toluene (3.0 mL) was stirred in relux for 16 h under N2. After completion of the reaction, the solvent was removed under reduced pressure and the residue was purified by flash column chromatography on silica gel to give 22 (22.0 mg, 36%) as slurry. 1H NMR (300 MHz, CDCl3) δ 10.80 (s, 1H), 7.98 (d, J = 7.5 Hz, 1H), 7.42–7.39 (m, 2H), 7.22–7.15 (m, 1H), 7.11 (d, J = 8.4 Hz, 1H), 3.64–3.58 (m, 1H), 2.72–2.52 (m, 2H), 2.23–2.18 (m, 2H), 2.3–1.96 (m, 2H), 1.67–1.43 (m, 6H), 1.36–1.20 (m, 6H), 1.10–1.05 (m, 6H), 0.82 (t, J = 7.2 Hz, 9H). 13C NMR (75 MHz, CDCl3) δ 171.3, 166.8, 158.3, 150.4, 135.7, 133.8, 129.9, 127.5, 125.6, 125.3, 124.6, 121.8, 112.1, 32.8, 29.0, 27.7, 27.3, 18.5, 13.6, 10.2. m/z calcd for C29H40N4OS120Sn: 612.2; found 613.3 (M + H+).
Synthesis of labeling precursor 27
N-(6-Bromo-3-nitropyridin-2-yl)-1H-indazol-5-amine (24).
To 10 (199.5 mg, 1.5 mmol) in 1,4-dioxane (15 mL) was added 2,6-dibromo-3-nitropyridine (423.7 mg, 1.5 mmol) and Et3N (0.62 mL, 4.5 mmol). Then the mixture was heated at 100 °C for 16 hours. The reaction was monitored by TLC. After the reaction completed, the solvent was removed under reduced pressure and the residue purified by silica gel column to give 24 (405.8 mg, 81%) as a red solid. 1H NMR (300 MHz, DMSO-d6) δ 13.11 (s, 1H), 10.17 (s, 1H), 8.39 (d, J = 8.6 Hz, 1H), 8.08 (s, 1H), 7.91 (s, 1H), 7.55 (d, J = 8.8 Hz, 1H), 7.44 (dd, J = 8.9, 1.6 Hz, 1H), 7.08 (d, J = 8.5 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 150.3, 146.6, 138.5, 138.3, 134.0, 130.8, 128.3, 124.7, 123.2, 117.7, 115.4, 110.5. Melting point = 227–229 °C. m/z calcd for C12H879BrN5O2: 333.0; found 334.1 (M + H+).
5-Bromo-2-cyclobutyl-3-(1H-indazol-5-yl)-3H-imidazo[4,5-b]pyridine (26).
Step 1: Compound 24 (0.33 g, 1.0 mmol) was subjected to the mixture of Fe powder (0.56 g, 10.0 mmol) in saturated NH4Cl (5 mL) and ethanol (5 mL) and stirred at refluxed under N2 for 4 hours. The mixture was filtered through Celite and concentrated in vacuum to yield 6-bromo-N2-(1H-indazol-5-yl)pyridine-2,3-diamine (25) as a grey solid (261.4 mg, 86%) without further purification. Step2: The mixture of 25 (91.2 mg, 0.3 mmol), cyclobutanecarboxylic acid (100.1 mg, 0.9 mmol), HATU (114.1 mg, 0.3 mmol) and DIEA (150.4 μL, 0.9 mmol) in DMF (30 mL) was stirred at room temperature for 16 h under N2. Step 3: After completion of the reaction, the solvent was removed under reduced pressure and the residue was resolved in HOAc (30 mL) and stirred at 80 °C for 16 hours. Then, the solvent was was removed under reduced pressure. Step 4: After completion of the reaction, and the residue was resolved in CH3OH (30 mL) and K2CO3 (207.3 mg, 1.5 mmol) was added. The mixture was stirred at room temperature for 16 hours. Then, the solid was filtered and washed with CH3OH/CH2Cl2 (v/v = 1/1). The filtrate was concentrated under reduced pressure, and the residue was purified by flash column chromatography on silica gel to give 26 (59.6 mg, 54% in 3 steps) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.41 (s, 1H), 8.21 (s, 1H), 8.06 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 1.4 Hz, 1H), 7.73 (d, J = 8.7 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.36 (dd, J = 8.7, 1.8 Hz, 1H), 3.64–3.53 (m, 1H), 2.44–2.38 (m, 2H), 2.13–2.01 (m, 2H), 1.93–1.78 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 160.4, 149.7, 140.0, 134.6, 134.3, 133.9, 129.7, 126.7, 126.2, 123.3, 122.2, 120.7, 111.6, 32.7, 27.2, 18.3. Melting point = 252–253 °C. MS (ESI, m/z): 368.2 (M + H+). HRMS (ESI): exact mass calcd for C17H1579BrN5 (M + H+): 368.0511, found: 368.0512.
2-Cyclobutyl-3-(1H-indazol-5-yl)-5-(tributylstannyl)-3H-imidazo[4,5-b]pyridine (27).
The mixture of 26 (36.7 mg, 0.1 mmol), Pd(PPh3)4 (11.6 mg, 0.01 mmol) and 1,1,1,2,2,2-hexabutyl-distannane (153.2 μL, 0.3 mmol) in toluene (3.0 mL) was stirred at relux for 16 h under N2. After completion of the reaction, the solvent was removed under reduced pressure and the residue was purified by flash column chromatography on silica gel to give 27 (8.7 mg, 15%) as slurry. 1H NMR (300 MHz, CDCl3) δ 12.38 (s, 1H), 8.03 (d, J = 7.7 Hz, 1H), 7.93 (s, 1H), 7.58 (s, 1H), 7.44 (d, J = 7.9 Hz, 1H), 7.14 (d, J = 8.7 Hz, 1H), 7.05 (d, J = 8.6 Hz, 1H), 3.62–3.50 (m, 1H), 2.70–2.50 (m, 2H), 2.18–2.12 (m, 2H), 1.99–1.93 (m, 2H), 1.55–1.41 (m, 6H), 1.30–1.18 (m, 6H), 1.11–1.01 (m, 6H), 0.79 (t, J = 7.2 Hz, 9H). 13C NMR (75 MHz, CDCl3) δ 166.5, 158.9, 151.1, 139.4, 134.4, 134.1, 127.4, 127.3, 125.3, 124.7, 123.2, 119.9, 110.8, 32.8, 29.0, 27.7, 27.2, 18.5, 13.6, 10.2. m/z calcd for C29H41N5120Sn: 579.2; found 580.3 (M + H+).
Radiosynthesis of TARP ɣ−8 ligand [11C]8 and [11C]15
The procedure for [11C]CH3I formation was modified in this work according to the previous reports.35–37 In brief, [11C]CH3I was yielded from cyclotron-produced [11C]CO2 by 14N(p, α)11C nuclear reaction. In this study, [11C]CO2 from cyclotron was bubbled into the 0.4 M solution of LiAlH4 in THF (300 μL). After evaporation, 57% of hydroiodic acid aqueous solution (300 μL) was added into the remaining reaction mixture. With heating, the resulting [11C]CH3I was then transferred under helium gas into a precooled (−15 to −20 °C) reaction vessel (Wheaton V-Vial, 1 mL; sealed with PTFE septum) containing the precursor 22 (0.8 mg), Pd2(dba)3 (0.2 mg), P(o-tol)3 (6.0 mg), CuBr (1.0 mg) and K2CO3 (1.0 mg) and anhydrous DMF (0.45 mL). When the radioactivity of this mixture reached a plateau, this reaction mixture was heated to 130 °C and maintained for 5 min. After the reaction completed, the mixture was neutralized by a buffer solution and this mixture was loaded into a preparative HPLC column. HPLC purification was completed on a CAPCELL PAK UG80 column (10 mm ID × 250 mm) using the mobile phase of CH3CN/50 mM NH4OAc (50/50, v/v) at a flow rate of 5 mL/min. The fraction corresponding to [11C]8 (retention time = 7 min; See supporting information Fig. S5) was collected into a flask containing Tween 80 (75 μL) and ethanol (150 μL), evaporated to dryness in vacuo, re-dissolved in 5 mL of sterile normal saline and passed through a 0.22 μm Millipore filter for analysis and application experiments. The synthesis time was 30 min. The radiochemical yields (RCY, decay-corrected) was 9% for [11C]8 based on [11C]CO2 with > 99% radiochemical purity and >95% chemical purity (retention time = 6.4 min; See supporting information Fig. S6). The molar activity of [11C]8 was determined by comparison of the assayed radioactivity to the mass associated with the carrier UV peak at 254 nm and was greater than 150 GBq/μmol at the end of synthesis (EOS). [11C]15 (retention time = 8.2 min; See supporting information Fig. S7) was radiosynthesized from the precursor 27 via the same method as the radiosynthesis of [11C]8 with decay-corrected RCY (22%), high molar activity (>150 GBq/μmol), high radiochemical purity (>99%) and high chemical purity (>95%; See supporting information Fig. S8). Both [11C]8 and [11C]15 were stable in the formulated solution and no radiolysis was observed up to 90 min (See supporting information Fig. S9).
TARP γ−8 inhibitor screening assay
The procedure for pharmacological analysis of candidate compounds was slightly modified according to the previous reports.25 Human TARP γ−8 and GluA1o were co-transfected into HEK cells. Two days after transfection, cells were washed with HBSS and incubated with FLIPR calcium 5 dye (Molecular Device) for one hour. Cells were pre-incubated with compounds and stimulated with 100 μM glutamate with 20 μM cyclothiazide to slow desensitization, and changes in signals were measured by the FLIPR tetra system (Molecular Device).
In vitro autoradiography
The procedure for in vitro autoradiography was slightly modified according to the previous reports.37, 40, 41, 43, 44 Rat brain was cut into 20 μM sections and stored at −80°C until they were used for experiment. The rat brain sections were pre-incubated with Tris-HCl buffer (50 mM) for 20 min at ambient temperature, followed by incubation with [11C]8 or [11C]15. For blocking studies, 8 (1 μM), 15 (1 μM), or JNJ-55511118 (1 μM) was added to incubation solution in advance to determine the specificity of radioligand binding. Then brain sections were washed with ice-cold buffer for three times, dipped in cold distilled water for 10 seconds. The brain sections were dried on cold air, and then placed on imaging plates (BAS-MS2025, GE Healthcare, NJ, USA). Autoradiograms were obtained and ROIs were carefully drawn with the reference of naked-eye observation. Radioactivity was expressed as photostimulated luminescene values per unit area (PSL/mm2) and measured by a Bio-Imaging analyzer system (BAS5000, Fujifilm).
Small-animal PET imaging studies
The imaging studies were conducted according to the previous literature37, 40, 41, 43, 44 with minor modification in this work. Sprague-Dawley rats were kept under anesthesia with 1–2% (v/v) isoflurane during the scan. The radiotracer [11C]15 (38.5 ± 2.8 MBq, 0.3 ± 0.2 nmol) was injected via a preinstalled catheter into the tail vein. A dynamic scan in 3D mode was acquired for 60 min. For blocking studies, a solution of cold compound 15 (1 mg/kg) or JNJ-55511118 (1 mg/kg) in 300 μL saline containing 10% ethanol and 5% Tween® 80 was injected at 5 min via the pre-embedded tail vein catheter before PET tracer injection. An MR template of SD rats was applied for PET/MR merged images. The radioactivity was decay-corrected and expressed as the standardized uptake value (SUV). SUV = (radioactivity per mL tissue / injected radioactivity) × body weight.
Ex vivo whole body biodistribution of [11C]15 in mice
Biodistribution experiments were conducted according to previous literature.37, 40, 41, 43, 44 Briefly, [11C]15 (3.7 MBq/100 μL) was intravenously injected via the tail vein of ddY mice. At different time points (1, 5, 15, 30 and 60 min) after injecting [11C]15, the mice were sacrificed and organs of interest were collected and weighted and radioactivity in each organ was measured.
Radiometabolite analysis
The radiometabolite analysis was conducted according to the previous literature with minor modifications.37 Radiometabolite analysis in rats: Following the intravenous injection of [11C]15 (74 MBq, 0.5 nmol/head), Sprague-Dawley rats (n=2) were sacrificed at 10 and 30 min post injection. The rat brain and blood were quickly removed and homogenized. The homogenate from brain sample was centrifuged and the supernatant was analyzed by the radio-HPLC. The percentage of [11C]15 to total radioactivity (corrected for decay) on the HPLC charts was calculated as (peak area for [11C]15/total peak area) × 100%. The same procedure was used for metabolite analysis in the plasma at 10 and 30 min post-injection as well.
Supplementary Material
Acknowledgments
We thank the Division of Nuclear Medicine and Molecular Imaging, Radiology, MGH and Harvard Medical School, USA for general support. We also thank the National Institute of Mental Health’s Psychoactive Drug Screening Program (NIMH PDSP; directed by Bryan L. Roth at the University of North Carolina at Chapel Hill and Jamie Driscoll at NIMH, USA) for CNS off-target counter screening. We also gratefully acknowledge the financial support from the NIH grants (MH120197, AG073428, AG074218 to S.H.L. and MH078939 to S.T.), JSPS KAKENHI (Grant No 20H03635 to M-R. Z.), AMED Moonshot Research and Development Program (Grant No 21zf0127003h001 to M-R. Z.), and the Swiss National Science Foundation for postdoctoral fellowship to Dr. Achi Haider.
Abbreviations used
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic
- AMPARs
AMPA receptors
- ARG
autoradiography
- AUC
area under curve
- BPND
non-displaceable binding potential
- CNS
central nervous system
- dba
dibenzylideneacetone
- DIEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- DOR
delta opioid receptor
- EOS
end of synthesis
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- %ID/g
the percentage of injected dose per gram of wet tissue
- NMDA
N-methyl-D-aspartate
- PET
Positron-emission tomography
- RCY
radiochemical yield
- SRTM
simplified reference tissue model
- SUV
standardized uptake value
- TAC
time−activity curve
- TARP γ−8
transmembrane AMPA receptor regulatory protein γ−8
- TARPs
transmembrane AMPAR regulatory proteins
- tPSA
topological polar surface areas
Footnotes
Supporting figures; Synthetic routes; HPLC traces of key compounds and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors have no conflicts of interest to declare.
References
- 1.Compans B; Choquet D; Hosy E Review on the role of AMPA receptor nanoorganization and dynamic in the properties of synaptic transmission. Neurophotonics 2016, 3, 041811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kessels HW; Malinow R Synaptic AMPA receptor plasticity and behavior. Neuron 2009, 61, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calabresi P; Cupini LM; Centonze D; Pisani F; Bernardi G Antiepileptic drugs as a possible neuroprotective strategy in brain ischemia. Ann. Neurol 2003, 53, 693–702. [DOI] [PubMed] [Google Scholar]
- 4.Chang PK; Verbich D; McKinney RA AMPA receptors as drug targets in neurological disease--advantages, caveats, and future outlook. Eur. J. Neurosci 2012, 35, 1908–1916. [DOI] [PubMed] [Google Scholar]
- 5.French JA; Krauss GL; Steinhoff BJ; Squillacote D; Yang H; Kumar D; Laurenza A Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia 2013, 54, 117–125. [DOI] [PubMed] [Google Scholar]
- 6.Zwart R; Sher E; Ping X; Jin X; Sims JR Jr.; Chappell AS; Gleason SD; Hahn PJ; Gardinier K; Gernert DL; Hobbs J; Smith JL; Valli SN; Witkin JM Perampanel, an antagonist of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, for the treatment of epilepsy: studies in human epileptic brain and nonepileptic brain and in rodent models. J. Pharmacol. Exp. Ther 2014, 351, 124–133. [DOI] [PubMed] [Google Scholar]
- 7.Ko D; Yang H; Williams B; Xing D; Laurenza A Perampanel in the treatment of partial seizures: time to onset and duration of most common adverse events from pooled Phase III and extension studies. Epilepsy Behav. 2015, 48, 45–52. [DOI] [PubMed] [Google Scholar]
- 8.Maher MP; Wu N; Ravula S; Ameriks MK; Savall BM; Liu C; Lord B; Wyatt RM; Matta JA; Dugovic C; Yun S; Ver Donck L; Steckler T; Wickenden AD; Carruthers NI; Lovenberg TW Discovery and characterization of AMPA receptor modulators selective for TARP-gamma8. J. Pharmacol. Exp. Ther 2016, 357, 394–414. [DOI] [PubMed] [Google Scholar]
- 9.Kato AS; Burris KD; Gardinier KM; Gernert DL; Porter WJ; Reel J; Ding C; Tu Y; Schober DA; Lee MR; Heinz BA; Fitch TE; Gleason SD; Catlow JT; Yu H; Fitzjohn SM; Pasqui F; Wang H; Qian Y; Sher E; Zwart R; Wafford KA; Rasmussen K; Ornstein PL; Isaac JT; Nisenbaum ES; Bredt DS; Witkin JM Forebrain-selective AMPA-receptor antagonism guided by TARP gamma-8 as an antiepileptic mechanism. Nat. Med 2016, 22, 1496–1501. [DOI] [PubMed] [Google Scholar]
- 10.Gardinier KM; Gernert DL; Porter WJ; Reel JK; Ornstein PL; Spinazze P; Stevens FC; Hahn P; Hollinshead SP; Mayhugh D; Schkeryantz J; Khilevich A; De Frutos O; Gleason SD; Kato AS; Luffer-Atlas D; Desai PV; Swanson S; Burris KD; Ding C; Heinz BA; Need AB; Barth VN; Stephenson GA; Diseroad BA; Woods TA; Yu H; Bredt D; Witkin JM Discovery of the first alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist dependent upon transmembrane AMPA receptor regulatory protein (TARP) gamma-8. J. Med. Chem 2016, 59, 4753–4768. [DOI] [PubMed] [Google Scholar]
- 11.Savall BM; Wu D; Swanson DM; Seierstad M; Wu N; Vives Martinez J; Garcia Olmos B; Lord B; Coe K; Koudriakova T; Lovenberg TW; Carruthers NI; Maher MP; Ameriks MK Discovery of Imidazo[1,2-a]pyrazines and pyrazolo[1,5-c]pyrimidines as TARP gamma-8 selective AMPAR negative modulators. ACS Med. Chem. Lett 2019, 10, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azumaya CM; Days EL; Vinson PN; Stauffer S; Sulikowski G; Weaver CD; Nakagawa T Screening for AMPA receptor auxiliary subunit specific modulators. PLoS One 2017, 12, e0174742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw AS; Filbert EL Scaffold proteins and immune-cell signalling. Nat. Rev. Immunol 2009, 9, 47–56. [DOI] [PubMed] [Google Scholar]
- 14.Yan D; Tomita S Defined criteria for auxiliary subunits of glutamate receptors. J. Physiol 2012, 590, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson AC; Nicoll RA The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron 2011, 70, 178–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hashimoto K; Fukaya M; Qiao X; Sakimura K; Watanabe M; Kano M Impairment of AMPA receptor function in cerebellar granule cells of ataxic mutant mouse stargazer. J. Neurosci 1999, 19, 6027–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L; Chetkovich DM; Petralia RS; Sweeney NT; Kawasaki Y; Wenthold RJ; Bredt DS; Nicoll RA Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000, 408, 936–943. [DOI] [PubMed] [Google Scholar]
- 18.Tomita S Regulation of ionotropic glutamate receptors by their auxiliary subunits. Physiology (Bethesda) 2010, 25, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Straub C; Tomita S The regulation of glutamate receptor trafficking and function by TARPs and other transmembrane auxiliary subunits. Curr. Opin. Neurobiol 2012, 22, 488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato AS; Gill MB; Yu H; Nisenbaum ES; Bredt DS TARPs differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci. 2010, 33, 241–248. [DOI] [PubMed] [Google Scholar]
- 21.Gill MB; Bredt DS An emerging role for TARPs in neuropsychiatric disorders. Neuropsychopharmacology 2011, 36, 362–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee MR; Gardinier KM; Gernert DL; Schober DA; Wright RA; Wang H; Qian Y; Witkin JM; Nisenbaum ES; Kato AS Structural Determinants of the gamma-8 TARP Dependent AMPA Receptor Antagonist. ACS Chem. Neurosci 2017, 8, 2631–2647. [DOI] [PubMed] [Google Scholar]
- 23.Witkin JM; Li J; Gilmour G; Mitchell SN; Carter G; Gleason SD; Seidel WF; Eastwood BJ; McCarthy A; Porter WJ; Reel J; Gardinier KM; Kato AS; Wafford KA Electroencephalographic, cognitive, and neurochemical effects of LY3130481 (CERC-611), a selective antagonist of TARP-gamma8-associated AMPA receptors. Neuropharmacology 2017, 126, 257–270. [DOI] [PubMed] [Google Scholar]
- 24.Dohrke JN; Watson JF; Birchall K; Greger IH Characterizing the binding and function of TARP gamma8-selective AMPA receptor modulators. J. Biol. Chem 2020, 295, 14565–14577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z; Mori W; Zhang X; Yamasaki T; Dunn PJ; Zhang G; Fu H; Shao T; Zhang Y; Hatori A; Ma L; Fujinaga M; Xie L; Deng X; Li H; Yu Q; Rong J; Josephson L; Ma JA; Shao Y; Tomita S; Zhang MR; Liang SH Synthesis, pharmacology and preclinical evaluation of 11C-labeled 1,3-dihydro-2H-benzo[d]imidazole-2-ones for imaging gamma8-dependent transmembrane AMPA receptor regulatory protein. Eur. J. Med. Chem 2018, 157, 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Q; Kumata K; Li H; Zhang Y; Chen Z; Zhang X; Shao T; Hatori A; Yamasaki T; Xie L; Hu K; Wang G; Josephson L; Sun S; Zhang MR; Liang SH Synthesis and evaluation of 6-(11C-methyl(4-(pyridin-2-yl)thiazol-2-yl)amino)benzo[d]thiazol-2(3H)-one for imaging gamma-8 dependent transmembrane AMPA receptor regulatory protein by PET. Bioorg. Med. Chem. Lett 2020, 30, 126879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Q; Chen Z; Kumata K; Shao T; Wey HY; Shao Y; Josephson L; Zhang MR; Liang S Radiosynthesis of 6-(2-cyclobutyl-5-[11C]methyl-3H-imidazo[4,5-b]pyridin-3-yl)benzo[d]thiazol-2(3H)-one as a novel PET tracer for imaging of γ-o dependent transmembrane AMPA receptor regulatory protein. J. Nucl. Med 2019, 60, 1604. [Google Scholar]
- 28.Yu Q; Chen Z; Shao T; Shao Y; Zhang MR; Liang S Radiosynthesis of 6-(11C-methyl(4-(pyridin-2-yl)thiazol-2-yl)amino)benzo[d]thiazol-2(3H)-one as a novel PET tracer towards imaging of γ−8 dependent transmembrane AMPA receptor regulatory protein. J. Nucl. Med 2020, 61, 1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savall BM; Swanson DM; Ameriks MK Imidazopyrazines and pyrazolopyrimidines and their use as ampa receptor modulators. WO2016176457 2016.
- 30.Ravula S; Swanson DM; Savall BM; Ameriks MK Benzimdazoleone and and benzothiazolone compounds and their use as AMPA receptor mudulators. WO2016176449 2016.
- 31.Berry CGB; Chen G; Jourdan FL; Lebold TP; Pena PM; Savall BM; Swanson DM; Wu D; Zhang W; Ameriks MK Azabenzimidazoles and their use as AMPA receptor modulators. WO2016176460 2016.
- 32.Ameriks MK; Ravula S; M. SB; Swanson DM; Ziff JM; T. SB Indolone compounds and their use as AMPA receptor mudulators. WO2016176463 2016.
- 33.Pike VW Considerations in the development of reversibly binding PET radioligands for brain imaging. Curr. Med. Chem 2016, 23, 1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waterhouse RN Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol 2003, 5, 376–389. [DOI] [PubMed] [Google Scholar]
- 35.Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH Chemistry for positron emission tomography: recent advances in 11C-, 18F-, 13N-, and 15O-labeling reactions. Angew. Chem. Int. Ed 2019, 58, 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y; Narendran R; Bischoff F; Guo N; Zhu Z; Bae SA; Lesage AS; Laruelle M A positron emission tomography radioligand for the in vivo labeling of metabotropic glutamate 1 receptor: (3-ethyl-2-[11C]methyl-6-quinolinyl)(cis- 4-methoxycyclohexyl)methanone. J. Med. Chem 2005, 48, 5096–5099. [DOI] [PubMed] [Google Scholar]
- 37.Shimoda Y; Yamasaki T; Fujinaga M; Ogawa M; Kurihara Y; Nengaki N; Kumata K; Yui J; Hatori A; Xie L; Zhang Y; Kawamura K; Zhang MR Synthesis and evaluation of novel radioligands based on 3-[5-(pyridin-2-yl)-2H-tetrazol-2-yl]benzonitrile for positron emission tomography imaging of metabotropic glutamate receptor subtype 5. J. Med. Chem 2016, 59, 3980–90. [DOI] [PubMed] [Google Scholar]
- 38.Tomita S; Chen L; Kawasaki Y; Petralia RS; Wenthold RJ; Nicoll RA; Bredt DS Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol 2003, 161, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukaya M; Yamazaki M; Sakimura K; Watanabe M Spatial diversity in gene expression for VDCCgamma subunit family in developing and adult mouse brains. Neurosci Res 2005, 53, 376–383. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X; Zhang Y; Chen Z; Shao T; Van R; Kumata K; Deng X; Fu H; Yamasaki T; Rong J; Hu K; Hatori A; Xie L; Yu Q; Ye W; Xu H; Sheffler DJ; Cosford NDP; Shao Y; Tang P; Wang L; Zhang MR; Liang SH Synthesis and preliminary studies of 11C-labeled tetrahydro-1,7-naphthyridine-2-carboxamides for PET imaging of metabotropic glutamate receptor 2. Theranostics 2020, 10, 11178–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L; Cheng R; Fujinaga M; Yang J; Zhang Y; Hatori A; Kumata K; Yang J; Vasdev N; Du Y; Ran C; Zhang MR; Liang SH A facile radiolabeling of [18F]FDPA via spirocyclic Iodonium ylides: preliminary PET imaging studies in preclinical models of neuroinflammation. J. Med. Chem 2017, 60, 5222–5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deloar HM; Fujiwara T; Nakamura T; Itoh M; Imai D; Miyake M; Watanuki S Estimation of internal absorbed dose of L-[methyl-11C]methionine using whole-body positron emission tomography. Eur. J. Nucl. Med 1998, 25, 629–633. [DOI] [PubMed] [Google Scholar]
- 43.Cheng R; Mori W; Ma L; Alhouayek M; Hatori A; Zhang Y; Ogasawara D; Yuan G; Chen Z; Zhang X; Shi H; Yamasaki T; Xie L; Kumata K; Fujinaga M; Nagai Y; Minamimoto T; Svensson M; Wang L; Du Y; Ondrechen MJ; Vasdev N; Cravatt BF; Fowler C; Zhang MR; Liang SH In vitro and in vivo evaluation of 11C-labeled azetidinecarboxylates for imaging monoacylglycerol lipase by PET imaging studies. J. Med. Chem 2018, 61, 2278–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rong J; Mori W; Xia X; Schafroth MA; Zhao C; Van RS; Yamasaki T; Chen J; Xiao Z; Haider A; Ogasawara D; Hiraishi A; Shao T; Zhang Y; Chen Z; Pang F; Hu K; Xie L; Fujinaga M; Kumata K; Gou Y; Fang Y; Gu S; Wei H; Bao L; Xu H; Collier TL; Shao Y; Carson RE; Cravatt BF; Wang L; Zhang MR; Liang SH Novel reversible-binding PET ligands for imaging monoacylglycerol lipase based on the piperazinyl azetidine scaffold. J. Med. Chem 2021, 64, 14283–14298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.