

Abstract
KRAS mutations are one of the most common oncogenic driver mutations in human cancers, including non-small cell lung cancer (NSCLC), and have established roles in cancer pathogenesis and therapeutic resistance. The development of effective inhibitors of mutant KRAS represents a significant challenge. Three-way junction (3WJ)-based multi-functional RNA nanoparticles have the potential to serve as an effective in vivo siRNA delivery platform with the ability to enhance tumor targeting specificity and visualize biodistribution through an imaging moiety. Herein, we assembled novel EGFRapt-3WJ-siKRASG12C mutation targeted nanoparticles to target EGFR-expressing human NSCLC harboring a KRASG12C mutation to silence KRASG12C expression in a tumor cell-specific fashion. We found that EGFRapt-3WJ-siKRASG12C nanoparticles potently depleted cellular KRASG12C expression, resulting in attenuation of downstream MAPK pathway signaling, cell proliferation, migration/invasion ability, and sensitized NSCLC cells to chemoradiotherapy. In vivo, these nanoparticles induced tumor growth inhibition in KRASG12C NSCLC tumor xenografts. Together, this study suggests that the 3WJ pRNA-based platform has the potential to suppress mutant KRAS activity for the treatment of KRAS-driven human cancers, and warrants further development for clinical translation.
Keywords: MT: Delivery Strategies, RNA nanotechnology, pRNA, three-way junction, 3WJ, EGFR RNA aptamer, siRNA, KRASG12C, non-small cell lung cancer, NSCLC
Graphical abstract
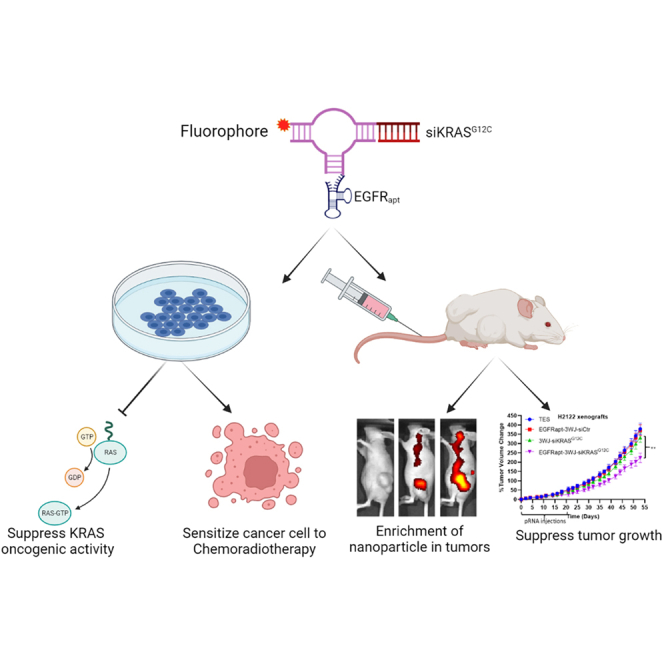
Williams and colleagues assembled EGFRapt-3WJ-siKRASG12C RNA nanoparticles, which efficiently suppressed KRAS expression in NSCLC, leading to suppression of MAPK pathway and tumor growth both in vitro and in vivo. The data support that 3WJ RNA nanoparticle is a potentially effective siRNA delivery platform for oncogene-driven human cancers treatment.
Introduction
Kirsten rat sarcoma viral oncogene homolog (KRAS) is a member of the human Ras gene family, encoding a small GTPase membrane-bound protein. KRAS functions as a binary molecular switch, cycling between a GDP-bound inactive and GTP-bound active state. KRAS mutations are widespread in human cancers, especially in three of the most lethal cancers: lung cancer, colorectal cancer, and pancreatic cancer.1 Missense mutations of KRAS, most commonly at codons 12 and 13, aberrantly attenuate GTPase activity, resulting in accumulation of GTP-bound activated KRAS. The constitutively activated KRAS oncoproteins initiate downstream cellular signal transduction cascades irrespective of input from extracellular signals including mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase pathways, leading to uncontrolled cell proliferation and abnormal cell survival, which are hallmarks of cancer.2,3,4,5 Lung cancer is the leading cause of cancer-related deaths worldwide, with non-small cell lung cancer (NSCLC) accounting for 84% of all lung cancer diagnoses.6 KRAS mutations occur in 20%–25% of NSCLC (most commonly in adenocarcinoma subtype), with KRASG12C mutation being the most frequent mutation variant, accounting for approximately 39% of all KRAS mutations in NSCLC.7,8,9,10
A large body of research has reported that the presence of KRAS mutations promotes tumor cell-autonomous-mediated resistance to cancer therapeutics. For instance, cetuximab and panitumumab (monoclonal antibodies to EGFR) and EGFR tyrosine kinase inhibitors, such as erlotinib and gefitinib, are ineffective in KRAS mutant colorectal cancer and NSCLC, respectively, restricting their use to KRAS wild-type colorectal cancer and EGFR-mutant NSCLC.11,12,13,14,15,16,17 While erlotinib is approved in combination with gemcitabine for pancreatic adenocarcinoma treatment, the combination produces minimal benefit in outcomes (only 2 weeks improved survival), likely due to the presence of KRAS mutations in 90%–95% of tumors leading to constitutive signaling downstream of the point of inhibition at EGFR.18 In radiotherapy, a preclinical study in 1988 first documented that hyperactivation of Ras isoforms (including KRAS) leads to tumor-intrinsic radioresistance.19 Emerging clinical evidence supports that KRAS mutations are associated with radiation resistance in colorectal and lung cancer.20,21,22 Our recent studies and those of others have detailed some of the molecular mechanisms involved in KRAS activation promoting radiation resistance in colorectal and lung cancer.22,23
Due to the high prevalence and importance of KRAS mutations in human cancer, pharmaceutical companies and academic laboratories have tried for decades to identify small-molecule inhibitors of KRAS mutation, but little progress had been achieved and many had labeled KRAS as “undruggable.” However, beginning in 2013, Shokat and co-workers reported the identification of covalent small-molecule compounds to target the reactive cysteine-12 of KRASG12C.24 Based on this finding, Janes et al. developed a covalent KRASG12C inhibitor ARS-1620, which could induce NSCLC tumor regression in in vivo models.25 Furthermore, Canon et al. expanded on the success of ARS-1620 to develop the compound named AMG 510,26 which is the first molecule to enter the clinic (NCT03600883) and ultimately receive FDA approval for the treatment of KRASG12C tumors. Recently, several other KRASG12C covalent inhibitors are also in phase I/II clinical trials,27 such as MRTX849, ARS-3248, and LY3499446 (NCT03785249, NCT04006301, and NCT04165031). As these covalent inhibitors require KRASG12C to be in the GDP bound state, drug resistance could be induced by disabling the GTPase activity or promoting the guanine exchange of GDP for GTP.27 Currently, small-molecular inhibitors that can target the plethora of other KRAS mutations (e.g., G12D, G12V, G12R, G13D, etc.) are either under-studied or in preclinical development.
The field of RNA nanotechnology has advanced rapidly during recent decades. It was first introduced in 1998, and the field encompasses the design, fabrication, and application of nanometer-scale RNA architectures.28,29 RNA nanoparticles have the simplistic characteristic of DNA canonical base pairing, while containing the structural flexibility and functional diversity characteristics of proteins. Noncanonical base pairing, base stacking, and elaborate networks of tertiary contacts further expand RNA structure versatility while also increasing thermodynamic stability.29 Yet the dynamic nature of RNA allows RNA nanoparticles to remain deformative and motile to lead to high tumor accumulation and rapid renal clearance for maximized therapeutic dose delivery and reduced toxicities.30,31 Three-way junction (3WJ) packaging RNA (pRNA) is a novel type of RNA nanotechnology derived from the pRNA of the bacteriophage phi29 DNA packaging motor. 3WJ pRNA has been extensively studied to fabricate various RNA nanoparticles with precise control of shape, size, and stoichiometry. The extending arms of 3WJ pRNA structures could be intelligently replaced with small interfering RNAs (siRNAs), miRNAs, riboswitches, and RNA aptamers, and conjugated with fluorescent probes or other moieties to construct multi-functional pRNA nanoparticles.32 Due to their great plasticity and stability, 3WJ pRNA nanoparticles are emerging as a highly desirable in vivo delivery system for targeted gene therapy in human cancers.28,29,32
In this study, we report the development of a multifunctional RNA nanoparticle, EGFRapt-3WJ-Alexa647-siKRASG12C, to explore the potential of 3WJ pRNA nanoparticles to deliver siRNA targeting KRASG12C into NSCLC cells. Overexpression of EGFR has been reported in many human cancers, including NSCLC.33 Thus, we have engineered EGFR targeting RNA aptamers (EGFRapt) to further enhance the tumor specificity of the technology. We find that our EGFRapt-3WJ-siKRASG12C nanoparticles exhibit high thermostability and demonstrate high efficiency to specifically silence KRASG12C and suppress downstream signaling pathways. Importantly, these nanoparticles sensitized NSCLC cells to chemotherapy and radiation therapy, inhibited proliferation, migration, invasion, and suppressed tumor growth by systemic delivery in a heterotopic mouse model. This platform technology has the potential to be adapted to other RAS mutations, enabling high tumor specificity.
Results
Construction and characterization of EGFRapt-3WJ-siKRASG12C nanoparticles
Utilizing the 3WJ of phi29 pRNA (3WJ pRNA) as the core scaffold, multifunctional EGFRapt-3WJ-siKRASG12C pRNA nanoparticles were constructed, harboring EGFR-targeting RNA aptamer, therapeutic KRASG12C siRNA, and Alexa 647 as the imaging module (Figure 1A). When the four nanoparticle component strands were mixed in equal molar ratio in TMS buffer, the pRNA nanoparticles assembled with very high efficiency, as indicated by agarose gel shift assays, showing stepwise assembly of the pRNA nanoparticle (Figure 1B). Dynamic light scattering (DLS) analysis determined the diameter of the EGFRapt-3WJ-siKRASG12C RNA nanoparticles (RNPs) as 7.2 ± 0.5 nm (Figure 1C) compared with 4.2 ± 1.1 nm for the 3WJ pRNA core scaffold.34 To assess thermodynamic parameters, the Alexa 647-labeled EGFRapt-3WJ-siKRASG12C RNPs were subjected to temperature gradient gel electrophoresis (TGGE). Alexa 647-labeled strands were used to determine the percentage of remaining intact nanoparticles as temperature gradient increased from 25°C to 80°C. We determined the melting temperature (Tm) of EGFRapt-3WJ-siKRASG12C nanoparticles as 51.77°C (Figure 1D), indicating that the constructed RNPs with all functional modules are thermostable.
Figure 1.

Construction and characterization of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles
(A) Schematic of Alexa 647-labeled EGFRapt-3WJ-siKRASG12C pRNA structure. (B) Native PAGE showing stepwise assembly of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles. (C) DLS assay to assess hydrodynamic size of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles. (D) TGGE assay determined Tm value of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles.
EGFRapt-3WJ-siKRASG12C nanoparticles efficiently bind and deliver KRASG12C siRNA to NSCLC cells
We next examined the EGFR-targeting capabilities of Alexa 647-conjugated EGFRapt-3WJ-siKRASG12C nanoparticles in KRASG12C NSCLC cell lines H2030 and H2122. Flow cytometric analysis illustrated a significantly higher level of target cell association using the EGFRapt-3WJ-siKRASG12C nanoparticles compared with 3WJ-siKRASG12C nanoparticles and scrambled aptamer-conjugated pRNA nanoparticles (Figures 2A and S1A). To further confirm whether the observed improvement in pRNA nanoparticle uptake is dependent on EGFR aptamer, we evaluated the binding ability of pRNA nanoparticles to NSCLC after silencing EGFR expression by siRNA. We found that, compared with the non-targeting silencing control, the EGFR silencing induced by siRNA (confirmed in Figure S1B) reduced the binding ability of EGFRapt-3WJ-siKRASG12C nanoparticles by 30% and 38% in H2030 and H2122 cell lines, respectively (Figures 2B and S1C). Interestingly, EGFR depletion also slightly reduced binding of SCRapt-3WJ-siKRASG12C nanoparticles by 20% and 15% in H2030 and H2122 cell lines, respectively, perhaps indicating that there are other non-EGFR-dependent properties of the aptamers that confer increased binding to these NSCLC cell lines.
Figure 2.

EGFRapt-3WJ-siKRASG12C pRNA nanoparticles target EGFR-expressing cells and suppress KRAS expression in human lung cancer
(A and B) Cellular uptake of AF647-labeled pRNA nanoparticles after 1 h incubation was determined by flow cytometry assay. (C) KRAS mRNA expression was quantified by qRT-PCR assay 48 h post-treatment with pRNA nanoparticles. (D) KRAS protein expression and activation status of KRAS downstream pathway intermediates was assessed by immunoblotting assay 48 h post-treatment with pRNA nanoparticles. Error bars represent SD. Significance was calculated using Student’s t test: ∗∗p < 0.001.
We next examined the ability of EGFRapt-3WJ-siKRASG12C nanoparticles to silence the expression of KRASG12C in H2030 and H2122 cells, and used KRAS wild-type H1299 NSCLC cells as a negative control. To optimize the dose of pRNA for in vitro treatment, we treated H2122 and H2030 cells with increasing doses of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles for 48 h. We found that KRAS expression was suppressed in a dose-dependent manner, with 50 nM of pRNA nanoparticle serving as the lowest, most effective dose (Figure S2). To further determine the optimal pRNA treatment time, we treated H2122 and H2030 cells with 50 nM of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles and collected cells at various time points after treatment. We found that KRAS mRNA knockdown efficacy is approximately equivalent between 48 and 72 h after treatment with 50 nM of pRNAs, with slightly improved efficacy over 24 h (Figure S2). Thus, in this study, we chose 50 nM of pRNAs for 48 h as the optimal treatment conditions for functional assays. Sanger sequencing of KRAS exon 2 was performed to confirm KRAS mutation status in all of these cell lines, with H2122 and H2030 harboring homozygous KRASG12C mutations as expected (Figure S3). Then, we treated H2122 and H2030 cells with 50 nM of EGFRapt-3WJ-siKRASG12C nanoparticles along with control RNPs (PBS, EGFRapt-3WJ-siScramble, and 3WJ-siKRASG12C) for 48 h. We found that 3WJ-siKRASG12C nanoparticles significantly suppressed KRAS mRNA expression in KRAS mutant cells, and that EGFR aptamer-conjugated EGFRapt-3WJ-siKRASG12C nanoparticles significantly further suppressed KRAS expression in KRASG12C NSCLCL cell lines (Figure 2C). However, neither 3WJ-siKRASG12C or EGFRapt-3WJ-siKRASG12C nanoparticles were able to significantly decrease KRAS expression in KRAS wild-type H1299 cells (Figure S4, top panel). Furthermore, by immunoblotting we confirmed that EGFRapt-3WJ-siKRASG12C nanoparticles suppressed protein expression of KRAS and activation of its downstream signaling components p-MEK-1/2, p-ERK-1/2 in KRASG12C NSCLC cells (Figure 2D), but not KRAS wild-type NSCLC cells (Figure S4, bottom panel). These data demonstrated that EGFRapt-3WJ-siKRASG12C nanoparticles efficiently silence KRASG12C expression only in KRASG12C mutant NSCLC cells.
EGFRapt-3WJ-siKRASG12C nanoparticles suppress KRAS activity, cell growth, migration, and invasion capabilities of NSCLC cells
To investigate whether KRASG12C-specific knockdown might result in reduced KRAS activity, we treated H2122 and H2030 cells with 50 nM of EGFRapt-3WJ-siKRASG12C nanoparticles along with control RNPs (PBS, EGFRapt-3WJ-siScramble, and 3WJ-siKRASG12C) for 24 h followed by assessment of KRAS functional activation. In both cell lines, we confirmed that 3WJ-siKRASG12C can suppress KRAS activity, which was maximal when cells were treated with EGFRapt-3WJ-siKRASG12C nanoparticles (Figure 3A), which is consistent with immunoblotting data showing that EGFRapt-3WJ-siKRASG12C nanoparticles suppressed activation of MEK-1/2 and ERK-1.2 (Figure 2D).
Figure 3.

Silencing of KRAS by EGFRapt-3WJ-siKRASG12C pRNA nanoparticles suppressed KRAS activity, cell growth, and metastatic capabilities of NSCLC cells
(A) KRAS activity was determined by KRAS activation ELISA assay on H2030 and H2122 cells. (B) Effects of KRAS silencing by pRNA nanoparticles on cell growth was determined by IncuCyte cell proliferation assay (cell number reflects percent confluency). (C) Quantification of transwell migration and invasion assays (top panels) and calculated invasion index (bottom panels). Error bars represent SD. Significance (A and C) was calculated using Student’s t test; significance (B) was calculated using one-way ANOVA analysis followed by Tukey’s post-hoc test: ∗p < 0.05, ∗∗p < 0.001.
Oncogenic KRAS results in persistent stimulation of its downstream signaling intermediates, which results in many of the phenotypic hallmarks of cancer including increased proliferation and metastasis. To assess the effects of depletion of oncogenic KRASG12C by siRNA delivered our RNPs on cell growth, IncuCyte cell proliferation assays were carried out. As shown in Figure 3B, 3WJ-siKRASG12C nanoparticles suppressed NSCLC cell growth, and the presence of EGFR aptamer maximally suppressed cell growth, while the control (scrambled) siRNA RNPs had no effect on cell proliferation. To investigate the influence of KRASG12C knockdown on NSCLC cell metastasis, migration ability, and invasion ability were evaluated by transwell assay and Matrigel assay, respectively. As shown in Figures 3C and S5 (top panels), the number of cells that migrated or invaded into the lower chamber decreased significantly in cells treated with EGFRapt-3WJ-siKRASG12C nanoparticles compared with the control siRNA group. The calculated invasion index also confirmed that KRAS knockdown decreased cell invasion ability independent of differences in migration (Figure 3C, bottom panels).
EGFRapt-3WJ-siKRASG12C nanoparticles sensitize NSCLC cells to chemotherapy and radiation therapy in vitro
It has been well documented that hyperactivation of KRAS can lead to development of intrinsic chemotherapy and radiation therapy resistance in tumor cells.19,20,23 To test whether KRAS suppression by EGFRapt-3WJ-siKRASG12C nanoparticles sensitizes NSCLC to radiation therapy, we performed radiation clonogenic assays using 2 Gy ionizing radiation in H2122 and H2030 cell lines at 24 h after pRNA nanoparticle treatment. As shown in Figure 4A, 3WJ-siKRASG12C nanoparticles significantly sensitized cells to radiation treatment, as observed by lower cell surviving fraction after 2 Gy compared with treatment with pRNA nanoparticles containing control siRNA. EGFRapt-3WJ-siKRASG12C nanoparticles further sensitized NSCLC cells to radiation therapy. We further investigated whether EGFRapt-3WJ-siKRASG12C nanoparticles could sensitize cells to cisplatin, a common first-line chemotherapy drug for NSCLC. Cells were pretreated with EGFRapt-3WJ-siKRASG12C nanoparticles for 24 h, followed by 72 h cisplatin treatment. alamarBlue cytotoxicity assays were used to determine whether the RNPs could alter the half-maximal inhibitory concentration (IC50) values to cisplatin. As shown in Figure 4B, EGFRapt-3WJ-siKRASG12C nanoparticles significantly lowered the IC50 values to cisplatin compared with control siRNA RNPs, decreasing from 7.6 to 3.8 μM and 6.6 to 1.3 μM in H2030 and H2122 cell lines, respectively. This finding was corroborated by clonogenic assays (Figure 4C), revealing reduced cell surviving fraction in both cell lines during treatment with increasing doses of cisplatin and EGFRapt-3WJ-siKRASG12C nanoparticles. Together, these data support that KRAS mutation-specific silencing by EGFRapt-3WJ-siKRASG12C nanoparticles sensitizes NSCLC cells to chemoradiation therapy in vitro.
Figure 4.

EGFRapt-3WJ-siKRASG12C pRNA nanoparticles sensitized NSCLC cells to radiotherapy and chemotherapy
(A) Radiation clonogenic assay was performed and normalized surviving fraction was calculated to evaluate sensitivity of NSCLC cells to radiation treatment after 48 h pRNA treatment. (B and C) Effects of 48 h treatment of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles on cisplatin cytotoxicity on NSCLC cells assessed by both alamarBlue (B) and colony formation assays (C). Error bars represent SD. Significance was calculated using Student’s t test: ∗p < 0.05, ∗∗p < 0.001.
EGFRapt-3WJ-siKRASG12C nanoparticles are preferentially taken up by tumors and significantly attenuate tumor growth in vivo
We further investigated the potential therapeutic effects of EGFRapt-3WJ-siKRASG12C nanoparticles in vivo. First, to determine the in vivo siRNA delivery effects of EGFRapt-3WJ-siKRASG12C nanoparticles, we established heterotopic tumor xenografts with H2122 cells in nude mice. To decrease photo-toxicity, we conjugated pRNA nanoparticles with Alexa 750, and systemically delivered Alexa 750-conjugated pRNA nanoparticles in TES via tail vein injection. At 24 h post-injection, an IVIS lumina imaging system was utilized to assess pRNA biodistribution in live animals and in ex vivo organs. Live imaging demonstrated that Alexa 750-conjugated 3WJ-siKRASG12C nanoparticles exhibited intense fluorescence in the areas of the tumors, and the presence of EGFR aptamer further increased the accumulation of RNPs in the tumor (Figure 5A, left top). Furthermore, ex vivo organ biodistribution analysis confirmed that EGFR aptamer facilitated pRNA nanoparticle accumulation in the tumors, with minimal uptake in some normal organs including lung, spleen, and heart (Figure 5A, left bottom). We also compared the in vivo biodistribution of EGFRapt-3WJ-siKRASG12C and SCRapt-3WJ-siKRASG12C pRNA nanoparticles. As shown in Figure S6, EGFRapt-pRNA can more efficiently accumulate in tumor xenografts compared with SCRapt-pRNA. To identify the optimal dose of RNPs for in vivo treatment, serial doses of EGFRapt-3WJ-siKRASG12C nanoparticles were intravenously administered to mice bearing H2122 xenografts every 2 days, four times over 7 days, and tumors were isolated for extraction of total RNA and protein (Figure 5B). We found that KRAS RNA (Figure 5C) and ERK activation (Figure 5D) were suppressed in a dose-dependent manner. One micromole per kilogram dose (1 μmol/kg) of pRNA nanoparticle was the most effective dose for suppression of KRAS in vivo. To assess whether systemically delivered EGFRapt-3WJ-siKRASG12C nanoparticles could suppress tumor growth, mice bearing H2122 or H2030 tumors were injected with 1 μmol/kg of the indicated pRNA nanoparticles twice a week for 3 weeks. We noted suppression of tumor growth with EGFRapt-3WJ-siKRASG12C, but not 3WJ-siKRASG12C nanoparticles (Figures 5E and 5F). These data confirm that EGFRapt-3WJ-siKRASG12C nanoparticles are more effective RNPs for targeting KRASG12C NSCLC cells in vivo relative to 3WJ-siKRASG12C RNPs, in alignment with the results obtained from our in vitro cell-based experiments.
Figure 5.

In vivo evaluation of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles on NSCLC tumor xenograft models
(A) IVIS lumina imaging system was used to study the biodistribution of Alexa 750-labeled EGFRapt-3WJ-siKRASG12C pRNA nanoparticles in live mice (top panels) and dissected organs ex vivo (bottom panels). (B) Dosing schematic of pRNA nanoparticle injection and tumor isolation for dose optimization study. (C and D) After four intravenous administrations of increasing doses of EGFRapt-3WJ-siKRASG12C pRNA nanoparticles, KRAS mRNA expression (C) in tumor xenografts was quantified by qRT-PCR, and activation status of KRAS downstream MAPK pathway signaling was evaluated by p-ERK immunoblotting (D). (E and F) Tumor growth curve of H2122 and H2030 xenograft mouse models, which were treated with pRNA nanoparticles two times a week for 3 weeks (n = 10 mice per group). Error bars represent SEM. Significance was calculated using one-way ANOVA analysis followed by Tukey’s post-hoc test: ∗∗p < 0.001.
Discussion
For more than 30 years, development of effective therapeutics targeting oncogenic RAS has eluded the field and RAS was thought to be “undruggable.”27 Although breakthroughs have been made recently with the development of covalent inhibitors targeting KRASG12C, such as AMG510 (sotorasib), there are still challenges and questions regarding the development and prevention of drug resistance, and the ability to successfully target other KRAS mutations.27 Other strategies targeting KRAS have been attempted. For example, systemic delivery of KRAS siRNA in nanoparticles suppressed pancreatic, lung, and colorectal cancer xenografts in various mouse models.35,36,37,38 SiG12D-LODER, is a specific siRNA targeting KRASG12D, and is in a phase II trial to evaluate the clinical efficacy of siG12D LODER in combination with gemcitabine and nab-paclitaxel in pancreatic cancer patients with KRASG12D mutation (NCT01676259).27,39 AZD4785, a chemically modified antisense oligonucleotide, significantly depleted cellular KRAS mRNA and protein in a preclinical study,40 but failed to suppress KRAS expression in patients in a clinical trial (NCT03101839). Further studies are ongoing to improve delivery efficiency, uptake, and internalization to make this approach more effective.
In this study, we took a mutation-specific gene-silencing approach by constructing novel multifunctional EGFRapt-3WJ-siKRASG12C nanoparticles and explored the potential of these RNPs carrying KRASG12C siRNA to target KRAS in NSCLC cells in a mutation-specific fashion. Furthermore, we added EGFRapt to the RNP to enhance tumor cell targeting. Our results demonstrate that EGFRapt-3WJ-siKRASG12C nanoparticles were successfully delivered to (and enriched in) NSCLC cells both in vitro and in vivo. These RNA nanoparticles effectively silenced KRAS expression only in KRASG12C mutant cells, attenuated activation of the MAPK signaling pathway, suppressed cancer cell migration and invasion properties, enhanced chemotherapy and radiotherapy sensitivity, and showed some tumor growth inhibitor properties in tumor xenograft mouse models.
Currently, a variety of nanocarriers are being investigated for siRNA delivery, such as polymers, dendrimers, liposomes, exosomes, and RNA nanoparticles.28,32,41,42,43 Among them, 3WJ pRNA nanoparticles are showing great potential as an efficient siRNA delivery system, harnessing beneficial characteristics, which include a relatively uniform nanoscale size, precise stoichiometry, ultrastability, and a high degree of biocompatibility compared with other therapeutics (e.g., liposomal nanoparticles). In addition, the branches/arms of the 3WJ motif could be easily modified with different subunits, such as multiple sequence-independent siRNAs, different RNA aptamers to improve specificity, or fluorescent dyes, without disrupting the stability and conformation.29 This technology enables multiple functional units for targeting, therapy, and tracking, which can all be combined into one nanoparticle. In our study, EGFRapt-3WJ-siKRASG12C nanoparticles are ultracompact with 7.2 + 0.5 nm diameter, and thermostable with a Tm of 51.77°C. Alexfluo647 was conjugated the RNP to form Alexa 647-EGFRapt-3WJ-siKRASG12C nanoparticles for in vitro cellular binding and in vivo biodistribution analysis. Alternatively, multiple units of the same function, such as different siRNAs targeting the same gene or different genes, can be combined on the same 3WJ pRNA nanoparticle for enhanced therapeutic effects. For example, as resistance to AMG510 has been shown to be associated with intratumoral heterogeneity and evolution of new non-G12C mutations,44 one could envision putting on multiple mutation-specific siRNAs targeting KRAS G12C, G12D, G12R, G12V, etc., to suppress development of resistance to these RNPs. Conversely, we could fashion these EGFRapt-3WJ-siKRASG12C nanoparticles to be more efficient at suppressing KRASG12C activity by adding another KRASG12C siRNA on another branch of 3WJ pRNA to increase dose of gene silencing.
Overexpression of EGFR has been reported in many human malignancies and is detected in up to 85% in NSCLC patients.33,45 EGFR monoclonal antibodies including cetuximab and necitumumab have been used to treat NSCLC patients combined with cytotoxic chemotherapeutics.46 Recently, RNA aptamers are emerging as promising targeting moieties, analogous to monoclonal antibodies. EGFR aptamers are selected through SELEX methods (systematic evolution of ligands by exponential enrichment), and have been utilized to decorate nanocarriers for more targeted delivery.34,43 In our study, the addition of EGFRapt to 3WJ-siKRASG12C showed obvious enhancement on cellular uptake and gene knockdown in both in vitro and in vivo experiments. Although EGFR depletion by EGFR siRNA did not completely abolish EGFRapt pRNA binding (Figure 2B), this observation might be due to insufficient EGFR knockdown or, more likely, that EGFRapt may nonspecifically bind to the membrane or other membrane receptors/components. This represents an opportunity to further optimize EGFRapt binding specificity in the future through re-engineering of EGFRapt or utilizing other targeting moieties.
Upregulation of KRAS-mediated signaling pathways is one of the mechanisms of chemotherapy resistance. For example, KRAS mutations have been shown to activate the anti-oxidant NRF2 pathway in NSCLC, thereby decreasing cisplatin-induced reactive oxygen species within the tumor cells, ultimately leading to cisplatin resistance.47 Studies have also shown that KRAS mutations render patients resistant to gefitinib in NSCLC and cetuximab resistance in colorectal cancer.48,49 In addition, KRAS mutations are also associated with radiation resistance in various cancers, including NSCLC and colorectal cancer.22,23 Herein, we document that EGFRapt-3WJ-siKRASG12C nanoparticles can sensitize NSCLC cells to radiation therapy or the first-line chemotherapy agent cisplatin (Figure 4). The results show the potential of 3WJ RNPs for allowing reduction in the doses of chemotherapy or radiotherapy leading to reduced normal tissue toxicity or, conversely, allowing enhancement of therapeutic efficacy if doses are kept the same.
In conclusion, we have demonstrated that EGFRapt-3WJ-siKRASG12C RNPs efficiently suppressed KRAS mRNA expression, resulting in suppression of downstream effector pathways in KRASG12C NSCLC cells, leading to suppression of tumor cell proliferation, migration/invasion ability, and sensitized NSCLC cells to chemoradiotherapy. Furthermore, our animal modeling showed excellent biodistribution of the RNPs to the tumor, effective KRAS silencing, and subsequent tumor growth inhibition. In addition, the 3WJ pRNA motif is universal and can be easily applied to target other KRAS mutant variants. Overall, these data support that 3WJ pRNA is an attractive RNA nanotechnology platform to deliver KRAS mutation-specific siRNA for the treatment of KRAS-driven human cancers that could lead to a widened therapeutic index by being a more tumor-selective therapy.
Materials and methods
Construction of EGFRapt-3WJ-siKRASG12C RNA nanoparticles
Multifunctional 3WJ pRNA nanoparticles (RNPs) were prepared using a bottom-up self-assembly approach as described previously.50,51,52 The EGFRapt-3WJ-Alexa647-siKRASG12C consisted of four components attached to the 3WJ core motif (Figure 1A), harboring EGFR-targeting RNA aptamer (EGFRapt) as a targeting ligand; Alexa Fluor 647 or 750 (Thermo Fisher Scientific, Waltham, MA) as a fluorescent imaging module; a KRASG12C sense strand, and a KRASG12C anti-sense strand, as the therapeutic module. The controls include RNPs without targeting EGFRapt ligand (denoted as 3WJ-siKRASG12C), with scramble aptamer ligand (denoted as SCRapt-3WJ-siKRASG12C), without therapeutic module (denoted as EGFRapt-3WJ-siScramble), or without therapeutic and targeting modules (denoted as 3WJ). The sequences of the four strands of EGFRapt-3WJ-siKRASG12C RNPs are as below: strand “a” (5′-UUG CCA UGU GUA UGU GGG AGU UGG AGC UUG UGG CGU AGU U-3′), strand “b” (5′-CCC ACA UAC UUU GUU GAU CC-EGFRapt-3′), strand “c” (5′-GGA UCA AUC AUG GCA A-3′), strand “d” (5′-CUA CGC CAC AAG CUC CAA C-3′). The complete pRNA sequences are provided in Table S1. The KRASG12C siRNA sequence is derived from a previous publication that demonstrated that this sequence could knockdown oncogenic KRAS but not wild-type KRAS.53 The RNA fragments were either synthesized via standard phosphoramidite chemistry by ourselves, or purchased from Trilink (San Diego, CA), and strands a, b, and c are 2′-F modified at cytosine (C) and uracil (U) nucleotides to make the RNPs resistant to RNase degradation. For the experiments involving the detection of RNPs (binding assay), strand c was conjugated to fluorophore Alexa 647 at the 3′ end. The RNP was formed through one-step self-assembly by mixing the four RNA module strands at equal molar ratios in annealing buffer (10 mM Tris [pH 7.5–8.0], 50 mM NaCl, 1 mM EDTA) and heated to 95°C for 5 min and slowly cooled to 4°C over 45 min. Stepwise assembly of RNPs was verified on a native 10% PAGE running in 1× TBE buffer (89 mM Tris-borate, 2 mM EDTA), and imaged by Typhoon FLA7000 (GE Healthcare) under the ethidium bromide channel. The self-assembled EGFRapt-3WJ-Alexa647-siKRASG12C RNPs were purified from 8 M urea-containing PAGE and stored at −80°C until use. The RNPs were freshly reconstituted in PBS before each use.
Characterization of the assembled pRNA-3WJ nanoparticle
The molecular size of assembled 3WJ pRNA nanoparticles was confirmed by native 10% PAGE gel electrophoresis. The hydrodynamic diameter of pRNA nanoparticles was assessed by DLS using a Zetasizer nano ZS (Malvern Instruments) at 25°C via a laser wavelength at 633 nm. The thermodynamic stability of the pRNA nanoparticles was studied using the TGGE system (Biometra, Germany), as described previously. The apparent Tm of the pRNA nanoparticle was determined as the temperature at which 50% of the pRNA nanoparticle remained assembled. Data shown are representative of three independent measurements.
Cell culture, chemicals, and antibodies
Human NSCLC cell lines H2122, H2030, and H1299 were obtained from the American Type Culture Collection (ATCC) and maintained in RPMI 1640 medium (Thermo Fisher Scientific), with 10% FBS (GE Healthcare, Chicago, IL) and 1% penicillin/streptomycin (Life Technologies, Carlsbad, CA). Cells were cultured in 37°C with 5% CO2. Typically, cells were kept in culture for a minimum of two passages prior to and a maximum of 20 passages during experiments. The identity of all cell lines was confirmed by STR genotyping (Identifier Kit, Applied Biosystems, Carlsbad, CA). For the detection of mycoplasma in cell culture, the Universal Mycoplasma Detection Kit (ATCC) was used. Cisplatin (Sigma, St. Louis, MO) was dissolved in dimethyl formamide (Sigma). Anti-KRAS antibody was purchased from Santa Cruz Biotechnology (Dallas, TX); anti-p-MEK-1/2, p-ERK-1/2, EGFR, and GAPDH primary antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-rabbit and anti-mouse secondary antibodies were purchased from Li-Cor Bioscience (Lincoln, NE).
DNA extraction and KRAS mutation analysis
Genomic DNA was isolated by using a QIAamp DNA mini kit (QIAGEN) according to the manufacturer’s instructions. Exon 2 of KRAS gene was amplified by polymerase chain reaction (PCR) using the following primers: forward 5-TGA CAT GTT CTA ATA TAG TCA G-3 and reverse: 5-ACA AGA TTT ACC TCT ATT GTT G-3. PCR was performed as described previously.23 PCR products were purified using a DNA purification kit (Zymo Research, Irvine, CA) and direct Sanger sequencing was performed on capillary electrophoresis using an Applied Biosystems 3730 DNA Analyzer (Thermo Fisher Scientific).
In vitro pRNA nanoparticle cellular binding assay
Specific binding of pRNA nanoparticles to NSCLC cells was assessed in vitro by flow cytometry. In brief, 1 × 105 H2122 and H2030 cells were resuspended in 100 μL PBS, and incubated with 50 nM of Alexa 647-labeled EGFRapt-3WJ-siKRASG12C and 3WJ-siKRASG12C pRNA nanoparticles at 37°C for 1 h. After washing three times with PBS, the cells were subjected to flow cytometry analysis using the BD LSR Fortessa flow cytometer (Becton Dickinson). Data were analyzed using the FlowJo 7.6.1 software (Tree Star). For EGFR-dependent selectivity assessment, cells were treated with EGFR-siRNA to knock down EGFR expression, followed 24 h later by pRNA nanoparticle cellular binding assay. Data shown are representative of three independent experiment.
Real-time qPCR analysis
KRAS gene silencing was detected by real-time quantitative PCR assay. Total cellular RNA was isolated using TRIzol (Thermo Fisher Scientific), and 1 μg of total RNA was reverse-transcribed using SuperScript reverse transcriptase (Bio-Rad). PCR was performed on iCYCLER real-time PCR machine (Bio-Rad) using SYBR Green chemistry (Bio-Rad). The genes expression levels were normalized to housekeeping gene GAPDH. The primer sequences are as follows: KRAS forward: 5′-GAC TCT GAA GAT GTA CCT ATG GTC CTA-3′ and reverse: 5′-CAT CAT CAA CAC CCT GTC TTG TC-3′; GAPDH forward: 5′-AAC GGG AAG CTT GTC ATC AAT GGA AA-3′ and reverse: 5′-GCA TCA GCA GAG GGG GCA GAG-3′. Experiments were performed three times.
Immunoblotting
Immunoblotting was performed as described previously.54 In brief, cell lysates were prepared using RIPA buffer (Thermo Fisher Scientific) supplemented with 1× protease inhibitors (Complete, Roche, Indianapolis, IN) and phosphatase inhibitors (PhosSTOP, Roche) followed by protein quantification with the DC protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein were loaded and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Membranes were incubated in 5% bovine serum albumin in Tris-buffered saline with 0.1% Tween 20 (TBST) blocking buffer for 1 h at room temperature. Primary antibodies with dilution of 1:200–1,000 were allowed to bind overnight at 4°C, or for 2 h at room temperature. After washing in TBST, the membranes were incubated with immunofluorescent secondary antibodies at a 1:5,000 dilution for 1 h at room temperature. Membranes were washed with TBST and allowed to air dry prior to imaging via Li-Cor Odyssey CLx Imaging System (Thermo Fisher Scientific). Immunoblotting data represent three independent experiments.
KRAS activity assay
As previous reported,23 KRAS activity was detected using a Ras Activation ELISA assay kit (Millipore) according to the manufacturer’s instructions. GST-Raf-RBD was used to pull down RAS-GTP from 50 μg of cell lysate prepared the same way as for immunoblotting, and then a primary antibody for KRAS was added, followed by incubation with an HRP-conjugated secondary antibody. After addition of developing reagent, chemiluminescent reaction was determined with a Fluoroskan Ascent FL luminometer. Experiments were repeated three times.
IncuCyte cell proliferation assay
Cells were treated with various pRNA nanoparticles accordingly for 48 h, and then seeded at 1,000–2,000 cells per well in 96-well plates. Cell confluence as a measure of cell growth over time was monitored every 4 h for up to 6 days using the IncuCyte ZOOM Live-Cell Imaging System (Essen Biosciences) until cells reached about 80% confluence. Cell proliferation curves were plotted using GraphPad Prism v.9.0 for Windows (La Jolla, CA). Experiments were repeated three times.
Cell migration and invasion assays
In the migration assay, cells were treated for 48 h with nanoparticles, then 2 × 104 cells were resuspended in 300 μL cell culture medium with 0.5% FBS and placed in the upper transwell chamber (8 μm pore size, BD Biosciences). The upper chamber was placed in a 24-well culture dish containing 1 mL of complete cell culture medium (with 10% FBS). After 48 h incubation, non-migrated cells on the upper membrane were removed with a cotton swab. Migrated cells on the bottom surface were fixed with methanol (−20°C) and stained with 0.5% crystal violet. Four fields of each well were photographed, and the cells were counted. In the invasion assay, Matrigel-coated transwell chambers (BD Biosciences) were used. Percentage invasion was calculated as the number of invaded cells in comparison with the number of migrated cells. All experiments were repeated three times.
Chemotherapy and radiation sensitivity assays
To study the effects of pRNA nanoparticles on chemoradiation sensitivity in NSCLC, a clonogenic assay was performed as reported previously.23 In brief, cells were treated with the indicated pRNA nanoparticles for 24 h, and then harvested to generate a single-cell suspension and seeded onto 60-mm tissue culture plates in triplicate. After 24 h, for the radiation sensitivity assay, cells were irradiated with 0 or 2 Gy X-rays using a Radsource RS2000 biological irradiator (RadSource, GA). For the chemotherapy sensitivity assay, cells were treated with increasing doses of cisplatin (0, 0.5, and 1 μM). Twenty-four hours later, the medium was changed to remove cisplatin. Ten to 14 days after irradiation or cisplatin treatment, colonies were fixed with methanol/acetic acid, stained with 0.5% crystal violet, and the numbers of colony-forming units containing at least 50 cells were counted using a dissecting microscope (Leica Microsystems, Buffalo Grove, IL) and surviving fractions were calculated. Experiments were performed three independent times.
Cisplatin cytotoxicity was also assessed by alamarBlue assay (Bio-Rad). In brief, cells were treated with pRNA nanoparticles as indicated for 24 h, and then collected and seeded in 96-well plates in 4 replicates at a density of 2,000 cells per well in 100 μL medium. The next day (∼48 h after adding pRNA), cells were treated with cisplatin at various concentrations. After 72 h, alamarBlue reagent was added to cells at 37°C for 4 h, and absorbance was measured at 490 nm. IC50 was determined using the nonlinear four-parameter regression function in GraphPad Prism. Experiments were performed three independent times.
Biodistribution and antitumor activity of pRNA nanoparticles in vivo
Animal studies were conducted in accordance with an approved protocol adhering to the IACUC policies and procedures at The Ohio State University. Six- to 8-week-old male athymic nude mice (Taconic Farms, NY) were caged in groups of five or less and fed with a diet of animal chow and water ad libitum. H2122 and H2030 cells were injected subcutaneously into the flanks of each mouse at 5 × 106 cells per injection. When the tumor size reached 150–200 mm3, the mice were randomly divided into 4 groups with 10 mice per group and injected with pRNA nanoparticles via tail vein twice a week for 3 weeks. The tumor volume was monitored 3 times a week, and tumor size was calculated using the formula: V = , where V (volume) is determined by length (L) and width (W). For in vivo pRNA nanoparticle targeting and tumor imaging, mice were tail vein injected with 100 L of 20 M of RNA Alexa 750-labeled pRNA nanoparticles, and animal or tissue were imaged using the IVIS lumina imaging system with Living Images 3.0 software (Caliper Life Sciences).
Data analysis
Data are presented as the mean ± standard deviation (SD) or standard error of the mean (SEM), with a representative experiment from at least three independent experiments shown. The difference among groups was calculated using Student’s t test or one-way ANOVA analysis followed by Tukey’s post-hoc test (GraphPad Prism).
Data and code availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information. Additional data related to this paper may be requested from the authors.
Acknowledgments
This work was supported by the following grants: The Ohio State University Comprehensive Cancer Center (OSU-CCC) National Institutes of Health (P30 CA016058), and American Cancer Society RSG-17-221-01-TBG (to T.M.W.).
Author contributions
L.Y., Z.L., P.G., and T.M.W. designed the study, participated in the supervision, and coordination of the study. L.Y., Z.L., and T.M.W. conceived and designed the experiments. L.Y. and Z.L. performed most of the experiments. All authors analyzed the data, contributed to the writing, review, and revision of the manuscript, and read and approved the final manuscript.
Declaration of interests
P.G. is the consultant of Oxford Nanopore Technologies; the cofounder of Shenzhen P&Z Bio-medical Co. Ltd., as well as the cofounder of ExonanoRNA, LLC, and its subsidiary Weina Biomedical (Guangdong), Ltd.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.07.027.
Supplemental information
References
- 1.Bos J.L. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Karnoub A.E., Weinberg R.A. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lito P., Solomon M., Li L.S., Hansen R., Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351:604–608. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patricelli M.P., Janes M.R., Li L.S., Hansen R., Peters U., Kessler L.V., Chen Y., Kucharski J.M., Feng J., Ely T., et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 6.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 7.Clinical Lung Cancer Genome Project CLCGP. Network Genomic Medicine NGM A genomics-based classification of human lung tumors. Sci. Transl. Med. 2013;5:209ra153. doi: 10.1126/scitranslmed.3006802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dogan S., Shen R., Ang D.C., Johnson M.L., D'Angelo S.P., Paik P.K., Brzostowski E.B., Riely G.J., Kris M.G., Zakowski M.F., Ladanyi M. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012;18:6169–6177. doi: 10.1158/1078-0432.CCR-11-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Osta B., Behera M., Kim S., Berry L.D., Sica G., Pillai R.N., Owonikoko T.K., Kris M.G., Johnson B.E., Kwiatkowski D.J., et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019;14:876–889. doi: 10.1016/j.jtho.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheffler M., Ihle M.A., Hein R., Merkelbach-Bruse S., Scheel A.H., Siemanowski J., Brägelmann J., Kron A., Abedpour N., Ueckeroth F., et al. K-ras Mutation Subtypes in NSCLC and Associated Co-occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. 2019;14:606–616. doi: 10.1016/j.jtho.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 11.Linardou H., Dahabreh I.J., Kanaloupiti D., Siannis F., Bafaloukos D., Kosmidis P., Papadimitriou C.A., Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 12.Mao C., Qiu L.X., Liao R.Y., Du F.B., Ding H., Yang W.C., Li J., Chen Q. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer. 2010;69:272–278. doi: 10.1016/j.lungcan.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi S., Boggon T.J., Dayaram T., Jänne P.A., Kocher O., Meyerson M., Johnson B.E., Eck M.J., Tenen D.G., Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 14.Pao W., Miller V., Zakowski M., Doherty J., Politi K., Sarkaria I., Singh B., Heelan R., Rusch V., Fulton L., et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobs S.A., Lee J.J., George T.J., Wade J.L., 3rd, Stella P.J., Wang D., Sama A.R., Piette F., Pogue-Geile K.L., Kim R.S., et al. Neratinib-Plus-Cetuximab in Quadruple-WT (KRAS, NRAS, BRAF, PIK3CA) Metastatic Colorectal Cancer Resistant to Cetuximab or Panitumumab: NSABP FC-7, A Phase Ib Study. Clin. Cancer Res. 2021;27:1612–1622. doi: 10.1158/1078-0432.CCR-20-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lièvre A., Bachet J.B., Le Corre D., Boige V., Landi B., Emile J.F., Côté J.F., Tomasic G., Penna C., Ducreux M., et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 17.Peeters M., Douillard J.Y., Van Cutsem E., Siena S., Zhang K., Williams R., Wiezorek J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin. Oncol. 2013;31:759–765. doi: 10.1200/JCO.2012.45.1492. [DOI] [PubMed] [Google Scholar]
- 18.Moore M.J., Goldstein D., Hamm J., Figer A., Hecht J.R., Gallinger S., Au H.J., Murawa P., Walde D., Wolff R.A., et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 19.Sklar M.D. The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science. 1988;239:645–647. doi: 10.1126/science.3277276. [DOI] [PubMed] [Google Scholar]
- 20.Duldulao M.P., Lee W., Nelson R.A., Li W., Chen Z., Kim J., Garcia-Aguilar J. Mutations in specific codons of the KRAS oncogene are associated with variable resistance to neoadjuvant chemoradiation therapy in patients with rectal adenocarcinoma. Ann. Surg Oncol. 2013;20:2166–2171. doi: 10.1245/s10434-013-2910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mak R.H., Hermann G., Lewis J.H., Aerts H.J.W.L., Baldini E.H., Chen A.B., Colson Y.L., Hacker F.H., Kozono D., Wee J.O., et al. Outcomes by tumor histology and KRAS mutation status after lung stereotactic body radiation therapy for early-stage non-small-cell lung cancer. Clin. Lung Cancer. 2015;16:24–32. doi: 10.1016/j.cllc.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang M., Han J., Marcar L., Black J., Liu Q., Li X., Nagulapalli K., Sequist L.V., Mak R.H., Benes C.H., et al. Radiation Resistance in KRAS-Mutated Lung Cancer Is Enabled by Stem-like Properties Mediated by an Osteopontin-EGFR Pathway. Cancer Res. 2017;77:2018–2028. doi: 10.1158/0008-5472.CAN-16-0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L., Shen C., Estrada-Bernal A., Robb R., Chatterjee M., Sebastian N., Webb A., Mo X., Chen W., Krishnan S., Williams T.M. Oncogenic KRAS drives radioresistance through upregulation of NRF2-53BP1-mediated non-homologous end-joining repair. Nucleic Acids Res. 2021;49:11067–11082. doi: 10.1093/nar/gkab871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostrem J.M., Peters U., Sos M.L., Wells J.A., Shokat K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janes M.R., Zhang J., Li L.S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S.J., Darjania L., et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172:578–589.e17. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C.G., Koppada N., et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 27.Moore A.R., Rosenberg S.C., McCormick F., Malek S. RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 2020;19:533–552. doi: 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo P. The emerging field of RNA nanotechnology. Nat. Nanotechnol. 2010;5:833–842. doi: 10.1038/nnano.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jasinski D., Haque F., Binzel D.W., Guo P. Advancement of the Emerging Field of RNA Nanotechnology. ACS Nano. 2017;11:1142–1164. doi: 10.1021/acsnano.6b05737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghimire C., Wang H., Li H., Vieweger M., Xu C., Guo P. RNA Nanoparticles as Rubber for Compelling Vessel Extravasation to Enhance Tumor Targeting and for Fast Renal Excretion to Reduce Toxicity. ACS Nano. 2020;14:13180–13191. doi: 10.1021/acsnano.0c04863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X., Bhullar A.S., Binzel D.W., Guo P. The dynamic, motile and deformative properties of RNA nanoparticles facilitate the third milestone of drug development. Adv. Drug Deliv. Rev. 2022;186:114316. doi: 10.1016/j.addr.2022.114316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shu Y., Pi F., Sharma A., Rajabi M., Haque F., Shu D., Leggas M., Evers B.M., Guo P. Stable RNA nanoparticles as potential new generation drugs for cancer therapy. Adv. Drug Deliv. Rev. 2014;66:74–89. doi: 10.1016/j.addr.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta R., Dastane A.M., Forozan F., Riley-Portuguez A., Chung F., Lopategui J., Marchevsky A.M. Evaluation of EGFR abnormalities in patients with pulmonary adenocarcinoma: the need to test neoplasms with more than one method. Mod. Pathol. 2009;22:128–133. doi: 10.1038/modpathol.2008.182. [DOI] [PubMed] [Google Scholar]
- 34.Shu D., Li H., Shu Y., Xiong G., Carson W.E., 3rd, Haque F., Xu R., Guo P. Systemic Delivery of Anti-miRNA for Suppression of Triple Negative Breast Cancer Utilizing RNA Nanotechnology. ACS Nano. 2015;9:9731–9740. doi: 10.1021/acsnano.5b02471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fleming J.B., Shen G.L., Holloway S.E., Davis M., Brekken R.A. Molecular consequences of silencing mutant K-ras in pancreatic cancer cells: justification for K-ras-directed therapy. Mol. Cancer Res. 2005;3:413–423. doi: 10.1158/1541-7786.MCR-04-0206. [DOI] [PubMed] [Google Scholar]
- 36.Pecot C.V., Wu S.Y., Bellister S., Filant J., Rupaimoole R., Hisamatsu T., Bhattacharya R., Maharaj A., Azam S., Rodriguez-Aguayo C., et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol. Cancer Therapeut. 2014;13:2876–2885. doi: 10.1158/1535-7163.MCT-14-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuan T.L., Fellmann C., Lee C.S., Ritchie C.D., Thapar V., Lee L.C., Hsu D.J., Grace D., Carver J.O., Zuber J., et al. Development of siRNA payloads to target KRAS-mutant cancer. Cancer Discov. 2014;4:1182–1197. doi: 10.1158/2159-8290.CD-13-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xue W., Dahlman J.E., Tammela T., Khan O.F., Sood S., Dave A., Cai W., Chirino L.M., Yang G.R., Bronson R., et al. Small RNA combination therapy for lung cancer. Proc. Natl. Acad. Sci. USA. 2014;111:E3553–E3561. doi: 10.1073/pnas.1412686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zorde Khvalevsky E., Gabai R., Rachmut I.H., Horwitz E., Brunschwig Z., Orbach A., Shemi A., Golan T., Domb A.J., Yavin E., et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA. 2013;110:20723–20728. doi: 10.1073/pnas.1314307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross S.J., Revenko A.S., Hanson L.L., Ellston R., Staniszewska A., Whalley N., Pandey S.K., Revill M., Rooney C., Buckett L.K., et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017;9:eaal5253. doi: 10.1126/scitranslmed.aal5253. [DOI] [PubMed] [Google Scholar]
- 41.Afonin K.A., Bindewald E., Yaghoubian A.J., Voss N., Jacovetty E., Shapiro B.A., Jaeger L. In vitro assembly of cubic RNA-based scaffolds designed in silico. Nat. Nanotechnol. 2010;5:676–682. doi: 10.1038/nnano.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J.B., Hong J., Bonner D.K., Poon Z., Hammond P.T. Self-assembled RNA interference microsponges for efficient siRNA delivery. Nat. Mater. 2012;11:316–322. doi: 10.1038/nmat3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z., Yang L., Wang H., Binzel D.W., Williams T.M., Guo P. Non-Small-Cell Lung Cancer Regression by siRNA Delivered Through Exosomes That Display EGFR RNA Aptamer. Nucleic Acid Therapeut. 2021;31:364–374. doi: 10.1089/nat.2021.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Awad M.M., Liu S., Rybkin I.I., Arbour K.C., Dilly J., Zhu V.W., Johnson M.L., Heist R.S., Patil T., Riely G.J., et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021;384:2382–2393. doi: 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inamura K., Ninomiya H., Ishikawa Y., Matsubara O. Is the epidermal growth factor receptor status in lung cancers reflected in clinicopathologic features? Arch. Pathol. Lab Med. 2010;134:66–72. doi: 10.1043/2008-0586-RAR1.1. [DOI] [PubMed] [Google Scholar]
- 46.Agustoni F., Suda K., Yu H., Ren S., Rivard C.J., Ellison K., Caldwell C., Jr., Rozeboom L., Brovsky K., Hirsch F.R. EGFR-directed monoclonal antibodies in combination with chemotherapy for treatment of non-small-cell lung cancer: an updated review of clinical trials and new perspectives in biomarkers analysis. Cancer Treat Rev. 2019;72:15–27. doi: 10.1016/j.ctrv.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 47.DeNicola G.M., Karreth F.A., Humpton T.J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K.H., Yeo C.J., Calhoun E.S., et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pietrantonio F., Vernieri C., Siravegna G., Mennitto A., Berenato R., Perrone F., Gloghini A., Tamborini E., Lonardi S., Morano F., et al. Heterogeneity of Acquired Resistance to Anti-EGFR Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017;23:2414–2422. doi: 10.1158/1078-0432.CCR-16-1863. [DOI] [PubMed] [Google Scholar]
- 49.Gazdar A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shu D., Shu Y., Haque F., Abdelmawla S., Guo P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011;6:658–667. doi: 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shu Y., Haque F., Shu D., Li W., Zhu Z., Kotb M., Lyubchenko Y., Guo P. Fabrication of 14 different RNA nanoparticles for specific tumor targeting without accumulation in normal organs. RNA. 2013;19:767–777. doi: 10.1261/rna.037002.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shu Y., Shu D., Haque F., Guo P. Fabrication of pRNA nanoparticles to deliver therapeutic RNAs and bioactive compounds into tumor cells. Nat. Protoc. 2013;8:1635–1659. doi: 10.1038/nprot.2013.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sunaga N., Shames D.S., Girard L., Peyton M., Larsen J.E., Imai H., Soh J., Sato M., Yanagitani N., Kaira K., et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol. Cancer Therapeut. 2011;10:336–346. doi: 10.1158/1535-7163.MCT-10-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang L., Shen C., Pettit C.J., Li T., Hu A.J., Miller E.D., Zhang J., Lin S.H., Williams T.M. Wee1 Kinase Inhibitor AZD1775 Effectively Sensitizes Esophageal Cancer to Radiotherapy. Clin. Cancer Res. 2020;26:3740–3750. doi: 10.1158/1078-0432.CCR-19-3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information. Additional data related to this paper may be requested from the authors.