

Abstract
Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycemia in infancy. CHI is a challenging disease to diagnose and manage. Moreover, complicating the course of the disease with another metabolic disease, in this case maple syrup urine disease (MSUD), adds more challenges to the already complex management. We report a term neonate who developed symptomatic, non-ketotic hypoglycemia with a blood glucose (BG) level of 1.9 mmol/L at 21-hours of life. A critical sample at that time showed high serum insulin and C-peptide levels confirming the diagnosis of CHI. Tandem mass spectrometry done at the same time was suggestive of MSUD which was confirmed by high performance liquid chromatography. The diagnosis of both conditions was subsequently confirmed by molecular genetic testing. His hypoglycemia was managed with high glucose infusion with medical therapy for CHI and branched chain amino acids (BCAA) restricted medical formula. At the age of four months, a near-total pancreatectomy was done, due to the failure of conventional therapy. Throughout his complicated course, he required meticulous monitoring of his BG and modified plasma amino acid profile aiming to maintain the BG at ≥3.9 mmol/L and levels of the three BCAAs at the disease therapeutic targets for his age. The patient is currently 29 months old and has normal growth and development. This patient is perhaps the only known case of the co-occurrence of CHI with MSUD. Both hypoglycemia and leucine encephalopathy can result in death or permanent neurological damage. The management of CHI and MSUD in combination is very challenging.
Keywords: Hypoglycemia, congenital hyperinsulinemia, maple syrup urine disease, ABCC8 mutation, BCKDHA mutation
What is already known on this topic?
Glucose homeostasis is maintained through multiple pathway interactions. Congenital hyperinsulinism is the most common cause of persistent hypoglycemia in infancy. Maple syrup urine disease is a rare cause of neonatal hypoglycemia caused by impairment of branched-chain alpha-keto acid dehydrogenase complex.
What this study adds?
This patient is perhaps the only known case of the co-occurrence of congenital hyperinsulinism with maple syrup urine disease. This case report describes the challenging clinical management of both conditions in combination.
Introduction
Neonatal hypoglycemia is a medical emergency with multiple possible underlying causes. Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycemia in infancy. CHI comprises a group of heterogenic genetic disorders with the common finding of recurrent episodes of hyperinsulinemic hypoglycemia due to inappropriate secretion of insulin by the pancreatic β-cells (1). The most common causes of CHI are mutations in ABCC8 (OMIM #600509) and KCNJ11 (OMIM #600937). Recessive mutations in these genes cause diffuse hyperinsulinism that is a severe, drug-resistant form, which may require resection of the pancreas. In this case, 18F-fluoro-L-DOPA positron emission tomography (PET) scan is not indicated. Dominant mutations in these genes have been associated with diazoxide-responsive disease (1,2).
Maple syrup urine disease (MSUD) is a rare cause of neonatal hypoglycemia. It is an autosomal recessive disease affecting the metabolism of branched chain amino acids (BCAA). The genetic defects result in impairment of branched-chain alpha-keto acid dehydrogenase complex. This complex is the rate-limiting step in breaking down the BCAA; leucine, isoleucine, and valine.
The combination of those two diseases makes the management very challenging, as there are multiple interactions between insulin and BCAA. Leucine is the most potent insulin stimulus among BCAA. It increases insulin secretion indirectly by inhibition of KATP channel activity via its intermediate metabolite; α-ketoisocaproate. Moreover, it regulates insulin release by acting on glutamate dehydrogenase. Additional mechanisms include triggering calcium oscillations in pancreatic β-cells and stimulation of the mTOR signaling pathway. The mTOR pathway mediates nutrient sensing and regulates protein synthesis in the pancreas. Inhibition of mTOR by rapamycin impairs glucose-induced insulin secretion in the pancreatic β-cells which might explain the risk for new-onset diabetes in organ transplanted patients (3,4).In contrast, insulin has a pronounced leucine lowering effect, hence it is used in the treatment of MSUD catabolic status (5).
In this case report, we present a child with CHI and MSUD who underwent near-total pancreatectomy. The main objective is to describe the management challenges in pre- and post-pancreatectomy status.
Case Report
A currently 29-month-old boy was born to a double first-cousin parent with no significant family history. His mother was 30 years old, G4P3 with no significant medical comorbidity. The pregnancy was uneventful. He was born at a peripheral hospital at 37 weeks of gestation via lower segment caesarian section (LSCS) as his mother had previous LSCS. His APGAR scores were 9 and 10 at 5 and 10 minutes, respectively. His birth weight was 3.46 kg (46th percentile), head circumference 35 cm (35th percentile), and length 51 cm (61st percentile). Breastfeeding was started immediately following delivery. At 21 hours of life, the baby was noted to be dusky, not breathing and he was resuscitated with bag-mask ventilation and transferred to the neonatal intensive care unit. He was found to be hypoglycemic, with a blood glucose (BG) of 1.9 mmol/L. He received four boluses of 10% dextrose and a critical blood sample, taken during hypoglycemia, showed high insulin, 58.7 mIU/L (2.6-24.9 mIU/L) and C-peptide of >1160 ng/mL (0.5-2 ng/mL). The urine was negative for ketones. The baby required nasal cannula oxygen 2 L/min for the first two days of life and then gradually weaned to room air.
On examination, he had no dysmorphic features; his anterior fontanelle was level and there was no organomegaly. He was noted to have absent sucking and rooting reflexes with normal Moro’s reflex. No abnormal body odor was noted and there were no abnormal movements. Feeding was started with 30 mL expressed breast milk plus glucose powder via nasogastric tube. The BG level was very labile between the days two and nine of life, needed a glucose infusion rate (GIR) of 15.6 mg/kg/minute to maintain his BG at a safe level. On day ten of life, he was started on octreotide 14 mcg/kg/day divided every 4 hours (as diazoxide was not available). Three days later, diazoxide (15 mg/kg/day) in three divided doses was added with hydrochlorothiazide 3.5 mg twice a day. On day 12 of life, the tandem mass spectrometry which was done as part of the critical sample at 21hours of life was reported. It showed a high serum (leucine and isoleucine) and valine suggesting the diagnosis of MSUD. Following this result, he was kept nil per mouth on a high glucose infusion and shifted to our hospital for further management. MSUD diagnosis was further confirmed by plasma amino acid analysis by HPLC with the presence of allo-isoleucine at 101 µmol/L, and elevated levels of the three BCAA: Leucine 724 µmol/L (45.0-160.0 µmol/L), isoleucine 240 µmol/L (28.0-95.0 µmol/L), and valine 390 µmol/L (60.0-294.0 µmol/L). The patient was started on oral feeding formula consisting of MSUD Anamix, Similac 1, and maxijul with adjustment of BCAA supply based on regular profiling of plasma BCAA levels; MSUD profile was performed three times weekly, on average. Along with enteral feeding, he continued to require intravenous dextrose infusion giving a GIR of 12.5 mg/kg/min to maintain his glucose. Medical therapy started initially was continued with adjustment of doses reaching the maximum dose of diazoxide (20 mg/kg/day). The dose of octreotide was built up reaching a maximum of 35 mcg/kg/day. Despite that, he kept requiring high GIR and had recurrent episodes of hypoglycemia associated with attempts of weaning the dextrose infusion requiring subcutaneous injections of glucagon. Diazoxide was stopped on day 20 of life due to a lack of clinically significant response.
Brain magnetic resonance imaging was performed at 24 days of life and showed an abnormal white matter signal with areas of restricted diffusion in the brainstem, cerebral peduncles, and cortical spinal tract features consisting with leucine encephalopathy.
CHI was genetically confirmed through the finding of a known pathogenic homozygous variant c.3748C>T, p.Arg1250, in the ABCC8 gene. A homozygous variant in the BCKDHA a (c.1087, p.Arg363Trp) was identified supporting the diagnosis of MSUD that was confirmed biochemically.
His management course was complicated with central venous line (CVL) related issues, including central line-related infections, cardiac arrest due to CVL migration-related pericardial effusion, and the need for multiple CVL insertions. As the patient continued to need high GIR with the maximum dose of octreotide to maintain his BG, he was transferred to another local center for near-total pancreatectomy at the age of 4 months. Pre-operatively, PET-scan was not done as the patient had been genetically confirmed to have diffuse disease. This was subsequently also confirmed on histopathology which showed diffuse hyperplasia of beta islet cells. Post-operatively, he had a stormy course; with central line-related septicemia, venous thrombosis, and difficulty establishing enteral feeding. In the immediate postoperative days, he had persistent hyperglycemia requiring IV insulin infusion initially, and then shifted to insulin infusion pump at a basal rate only.
At the age of 5 months, he was discharged home on an insulin pump and BCAA-restricted formula via nasogastric tube. Initially, he was only requiring basal rate insulin, later he required bolus insulin for feeds on follow-up.
Three months after the discharge, the patient presented with frequent episodes of hypoglycemia, hence insulin was stopped. Subsequently, he continued to have recurrent episodes of hypoglycemia so he was restarted on diazoxide with hydrochlorothiazide for which he showed a good response. Parents stopped the medication on their own after one month for a claim of episodes of hypotension in the peripheral hospital. The child continued to have hypoglycemic episodes on a daily basis, especially at night. Thus, uncooked cornstarch was added to bedtime formula once daily. With that, he continued to have recurrent hypoglycemia so a gastrostomy tube was inserted and continuous night feeding via feeding pump was started. Afterward, the glycemic control was much better.
The child was admitted twice at the age of six and eight months respectively with hyperglycemia associated with significant ketosis mainly due to feeding intolerance. These episodes need to be identified as either diabetic ketoacidosis or metabolic decompensation related to his MSUD. With further investigations doing a blood gas the child was not acidotic hence the ketosis was most likely related to metabolic decompensations.
As his BCKDHA variant was novel the patient was given a trial of thiamin for two months with no marked reduction in leucine as would be expected in patients with thiamin-responsive MSUD.
Upon the last visit at 29 months, the patient was not on any medication but was still on continuous overnight feeding through a gastrostomy tube. Overall, his BG readings were within normal. His assessment revealed that his development was appropriate for his age with a normal neurological examination. His growth parameters were all acceptable for his age; with a height of 87 cm (15th percentile), weight: 11.8 kg (12th percentile), and head circumference of 48 cm (21st percentile).
Discussion
Non-ketotic hypoglycemia is a rare neonatal presentation, associated with disorders of fructose or galactose metabolism, hyperinsulinism, fatty acid oxidation, and growth hormone deficiency. MSUD, which is an aminoacidopathy, is usually marked by early encephalopathy, sweet odor, and ketosis. In a country where there is no newborn screening for MSUD, the diagnosis of MSUD with non-ketotic hypoglycemia presentation may be missed. As reported by Haymond et al. (6), lethal hypoglycemia in MSUD is rare and it’s due to a defect in gluconeogenesis. Given the high insulin level in the critical sampling, and the absence of ketone and sweet-smell, the initial hypoglycemia in the presented patient was likely mainly secondary to CHI.
As previously reported, the homozygous variant in the ABCC8 gene in the presented patient is associated with a diffuse and severe CHI that is diazoxide unresponsive and required near-total pancreatectomy (7). Postoperatively, the hypoglycemia was less severe, as shown in Figure 1, which may explain the better response to diazoxide as reported before (8).
Figure 1.
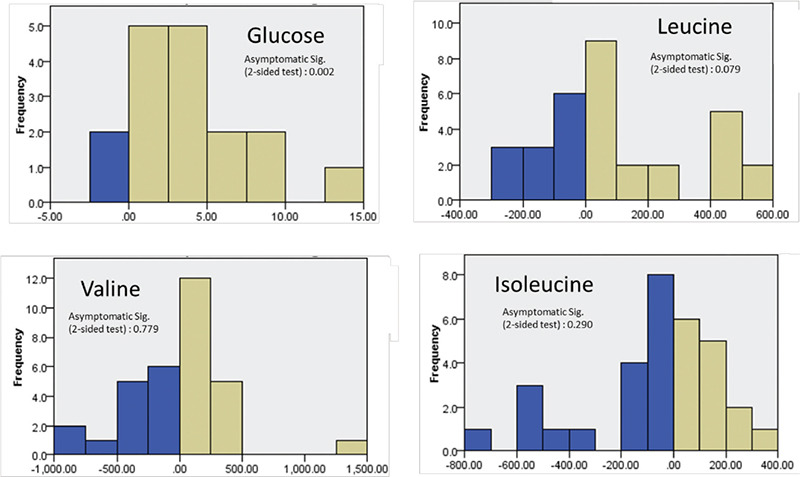
Wilcoxon signed-ranking test related samples for glucose, leucine, valine and isoleucine level. The test conducted on the serum level that was taken four months before the pancreatectomy and four months after the procedure. It showed statistically significant difference on glucose level before and after the procedure. On the other hand, there was no statistically significant difference on the BCAA level. This might indicate indirectly that hyperinsulinemia has no major rule in BCAA status in pre and post pancreatectomy
BCAA: branched chain amino acids
In the pre-pancreatectomy period, the leucine level was fluctuating and difficult to predict compared to the post-procedure period. This is thought to be partially explained by concordant hyperinsulinemia. The relatively mild MSUD course so far, in which leucine maximally reached 500 umol/L under stress conditions (surgery), was thought to be a beneficial effect of hyperinsulinemia. However, in the post-procedure period, leucine levels remain reasonably controlled. This was supported by Wilcoxon signed-ranking test which does not show a statistically significant difference in the leucine level during the pre- and post-operative period (shown in Figure 1). We hypothesize that the mild course of MSUD is most likely related to the genetic variant effect rather than CHI concurrence.
As this patient showed no response to conventional medical therapy, the use of sirolimus, an mTOR inhibitor was discussed. However, given currently limited data of its success and the complexity of our case, near-total pancreatectomy was the preferable option (9). The discussion of an early liver transplant was raised but due to uncertainty of the MSUD status post pancreatectomy, this option was delayed.
The patient course in the post pancreatectomy period has followed the expected route in which 40-60% of patients developed persistent hypoglycemia (8). He had an initial period of persistent hyperglycemia followed by persistent hypoglycemia that is now responding to overnight feeding.
The outcome for this patient remains unpredictable. The post-pancreatectomy glucose level has become significantly high which might indicate an early sign of diabetes. There are still multiple challenging aspects in his management, like the long-term risk of developing diabetes and its interaction with MSUD, the need for a liver transplant for his MSUD control, and the overall quality of life. We intend to continue reporting the progress of this patient in the future, given the possibly unique nature of his conditions.
Acknowledgments
We would like to acknowledge our dietitian, neonatal intensive care unit team at sultan Qaboos University hospital and the pediatric surgery, metabolic and gastroenterology teams at the Royal Hospital for their incredible role in the management of our patient.
Footnotes
Ethics
Informed Consent: Informed consent for publishing this case was obtained from the patient’s parents.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Azza Al Shidhani, Abdulhamid Al Hinai, Khalid Al Thihli, Hilal Al Mandhari, Saif Al Yaarubi, Irfan Ullah, Nadia Al-Hashmi, Fathiya Al Murshedi, Concept: Azza Al Shidhani, Data Collection or Processing: Azza Al Shidhani, Abdulhamid Al Hinai, Analysis or Interpretation: Azza Al Shidhani, Abdulhamid Al Hinai, Khalid Al Thihli, Hilal Al Mandhari, Saif Al Yaarubi, Irfan Ullah, Nadia Al-Hashmi, Fathiya Al Murshedi, Literature Search: Azza Al Shidhani, Abdulhamid Al Hinai, Writing: Azza Al Shidhani, Abdulhamid Al Hinai.
Conflict of Interest: None declared.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, Brunelle F, Fournet JC, Robert JJ, Aigrain Y, Bellanné-Chantelot C, de Lonlay P. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis. 2011;6:63. doi: 10.1186/1750-1172-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.James C, Kapoor RR, Ismail D, Hussain K. The genetic basis of congenital hyperinsulinism. J Med Genet. 2009;46:289–299. doi: 10.1136/jmg.2008.064337. [DOI] [PubMed] [Google Scholar]
- 3.Gran P, Cameron-Smith D. The actions of exogenous leucine on mTOR signalling and amino acid transporters in human myotubes. BMC Physiol. 2011;11:10. doi: 10.1186/1472-6793-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J, Dolinger M, Ritaccio G, Mazurkiewicz J, Conti D, Zhu X, Huang Y. Leucine stimulates insulin secretion via down-regulation of surface expression of adrenergic α2A receptor through the mTOR (mammalian target of rapamycin) pathway: implication in new-onset diabetes in renal transplantation. J Biol Chem. 2012;287:24795–24806. doi: 10.1074/jbc.M112.344259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wendel U, Langenbeck U, Lombeck I, Bremer HJ. Maple syrup urine disease--therapeutic use of insulin in catabolic states. Eur J Pediatr. 1982;139:172–175. doi: 10.1007/BF01377350. [DOI] [PubMed] [Google Scholar]
- 6.Haymond MW, Karl IE, Feigin RD, DeVivo D, Pagliara AS. Hypoglycemia and maple syrup urine disease: defective gluconeogenesis. Pediatr Res. 1973;7:500–508. doi: 10.1203/00006450-197305000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Al-Badi MK, Al-Azkawi HS, Al-Yahyaei MS, Mula-Abed WA, Al-Senani AM. Clinical characteristics and phenotype-genotype review of 25 Omani children with congenital hyperinsulinism in infancy. A one-decade single-center experience. Saudi Med J. 2019;40:669–674. doi: 10.15537/smj.2019.7.24291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arya VB, Senniappan S, Demirbilek H, Alam S, Flanagan SE, Ellard S, Hussain K. Pancreatic endocrine and exocrine function in children following near-total pancreatectomy for diffuse congenital hyperinsulinism. PLoS One. 2014;9:e98054. doi: 10.1371/journal.pone.0098054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szymanowski M, Estebanez MS, Padidela R, Han B, Mosinska K, Stevens A, Damaj L, Pihan-Le Bars F, Lascouts E, Reynaud R, Ferreira C, Bansept C, de Lonlay P, Saint-Martin C, Dunne MJ, Banerjee I, Arnoux JB. mTOR Inhibitors for the Treatment of Severe Congenital Hyperinsulinism: Perspectives on Limited Therapeutic Success. J Clin Endocrinol Metab. 2016;101:4719–4729. doi: 10.1210/jc.2016-2711. [DOI] [PubMed] [Google Scholar]