

Abstract
The Schlemm’s canal (SC) is a circular, lymphatic-like vessel located at the limbus of the eye that participates in the regulation of aqueous humor drainage to control intraocular pressure (IOP). Circumferential flow of aqueous humor within the SC lumen generates shear stress, which regulates SC cell behaviour. Using biochemical analysis and real-time live cell imaging techniques, we have investigated the activation of autophagy in SC cells by shear stress. We report, for the first time, the primary cilium (PC)-dependent activation of autophagy in SC cells in response to shear stress. Moreover, we identified PC-dependent shear stress-induced autophagy to be positively regulated by phosphorylation of SMAD2 in its linker and C-terminal regions. Additionally, SMAD2/3 signaling was found to transcriptionally activate LC3B, ATG5 and ATG7 in SC cells. Intriguingly, concomitant to SMAD2-dependent activation of autophagy, we also report here the activation of mTOR pathway, a classical autophagy inhibitor, in SC cells by shear stress. mTOR activation was found to also be dependent on the PC. Moreover, pharmacological inhibition of class I PI3K increased phosphorylation of SMAD2 at the linker and activated autophagy. Together, our data indicates an interplay between PI3K and SMAD2/3 signaling pathways in the regulation of PC-dependent shear stress-induced autophagy in SC cells.
Keywords: autophagy, glaucoma, primary cilia, Schlemm’s canal, shear stress, SMAD2/3, mTOR
Introduction
The Schlemm’s canal (SC) is a circular, unique lymphatic-like vessel structure located in the anterior segment of the eye. The SC, together with the trabecular meshwork (TM) form the conventional outflow pathway, the main tissue regulating intraocular pressure (IOP) [1]. IOP is defined as the fluid pressure inside the eye. Elevation in IOP leads to ocular hypertension (OHT) and predisposes to the development of glaucoma, the leading cause of permanent blindness worldwide. The TM/SC maintains fluid homeostasis by draining aqueous humor (AH) from the interior eye into the systemic circulation. In the SC, the circumferential flow of AH inside the vessel exerts shear stress to the endothelial cells lining the inner wall. Shear stress is intensified with increasing IOP, with the collapse of SC lumen. Higher shear stress is also locally observed in the regions proximal to the collector channels, downstream of the SC, where AH exits the eye to join the systemic circulation [2–4]. SC cells respond to shear stress by rearranging the actin architecture [5] and production of nitric oxide (NO) [6]. NO has been shown to increase AH outflow and lower IOP in both mouse and human eyes, suggesting a role of shear stress-induced responses in maintaining IOP homeostasis [7] [8].
Activation of autophagy (macroautophagy) in response to shear stress or fluid flow have been described in a number of cell types, including other endothelial cells (ECs) [9–23]. Autophagy is a catabolic process for degradation or recycle of intracellular components, which is emerging as a critical cellular pathway to maintain cellular and tissue homeostasis [24,25]. In vascular ECs, shear stress induces autophagy together with an increase in NOS3/eNOS (nitric oxide synthase 3) expression [23] and importantly, autophagy inhibition impaired the NO production in response to shear stress [23,26], suggesting that autophagy regulates NO production to maintain vascular tissue homeostasis. In addition, autophagy also functions as a regulator of the antioxidant and anti-inflammatory homeostasis to protect vascular ECs in response to shear stress [21,27].
Prior studies conducted in our laboratory have reported the activation of autophagy in TM cells with mechanical stretch and have shown autophagy to be an important component of the physiological response regulating IOP homeostasis [28–31]. Furthermore, we identified the primary cilium (PC) as the mechanosensor, and a reciprocal activation between AKT-SMAD2/3 as the signaling pathway regulating stretch-induced autophagy in TM cells. Similar to our findings in stretched TM cells, the PC has been implicated in the activation of autophagy with shear stress [32–34]. The PC is a sensory organelle that projects from the apical surface in most eukaryotic cells, which functions as a transducer of external chemical or mechanical stimuli, including fluid flow and fluid flow-induced shear stress [35,36], into an intracellular response. The PC membrane is enriched with receptors, ion channels and signaling components that coordinate a number of signal transduction pathways. In kidney epithelial cells, PC-mediated autophagy is activated in response to fluid flow, mediated by Ca2+ influx generated by PKD2 (polysystin 2, transient receptor cation channel) in the PC. [32,37]. Activation of autophagy was dependent on the mobilization of phosphatidylinositol-3 phosphate (PI3P) to the basal body of the PC by PIK3C2A (phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 alpha), a class II PI3K [38]. PC-dependent autophagy has also been shown to trigger lipophagy to fuel mitochondria biogenesis in response to fluid flow-induced shear stress in proximal tubule kidney epithelial cells [33].
In this study, we investigated the activation of autophagy in response to shear stress in SC cells. We report here that autophagy is activated in response to shear stress, and it is regulated by PC-mediated SMAD2/3 signalling pathway in SC cells. We postulate that autophagy constitutes one of the physiological cellular mechanisms used by SC cells to regulate IOP by i.e increasing permeability to AH outflow. We further postulate that malfunction of autophagy, as reported in outflow pathway cells in glaucoma [39,40] 41], could jeopardize such cellular mechanism, increasing resistance and contribute to IOP elevation.
Results
Autophagy is activated by shear stress in SC cells.
Autophagy activation is a cellular response to mechanical stretch and high pressure in TM cells [28–31]. We wanted to examine whether SC cells also activate autophagy in response to shear stress. For this, we evaluated the expression levels of LC3 and SQSTM1 by western blot (WB) in primary cultures of human SC cells subjected to laminar flow (1 or 10 dyn/cm2 for 24 h). SC cells are predicted to experience a shear stress of ~1–28 dyn/cm2 based on the proximity to the collector channels, with a time-average of 11 dyn/cm2 at elevated IOP [5]. As seen in Fig.1A, the protein levels of LC3-II and SQSTM1 significantly increased by approximately 2-fold (p<0.05, n=6) upon flow application (10 dyn/cm2 for 24 h) to SC cells. A shear stress of 10 dyn/cm2 was chosen hereafter since we previously showed that strength to induce NO production by SC cells and to lower outflow resistance [6]. Interestingly, 10 dyn/cm2 shear stress did not significantly increase LC3-II levels in cultured primary human TM cells (Fig.S1A).
Figure 1.
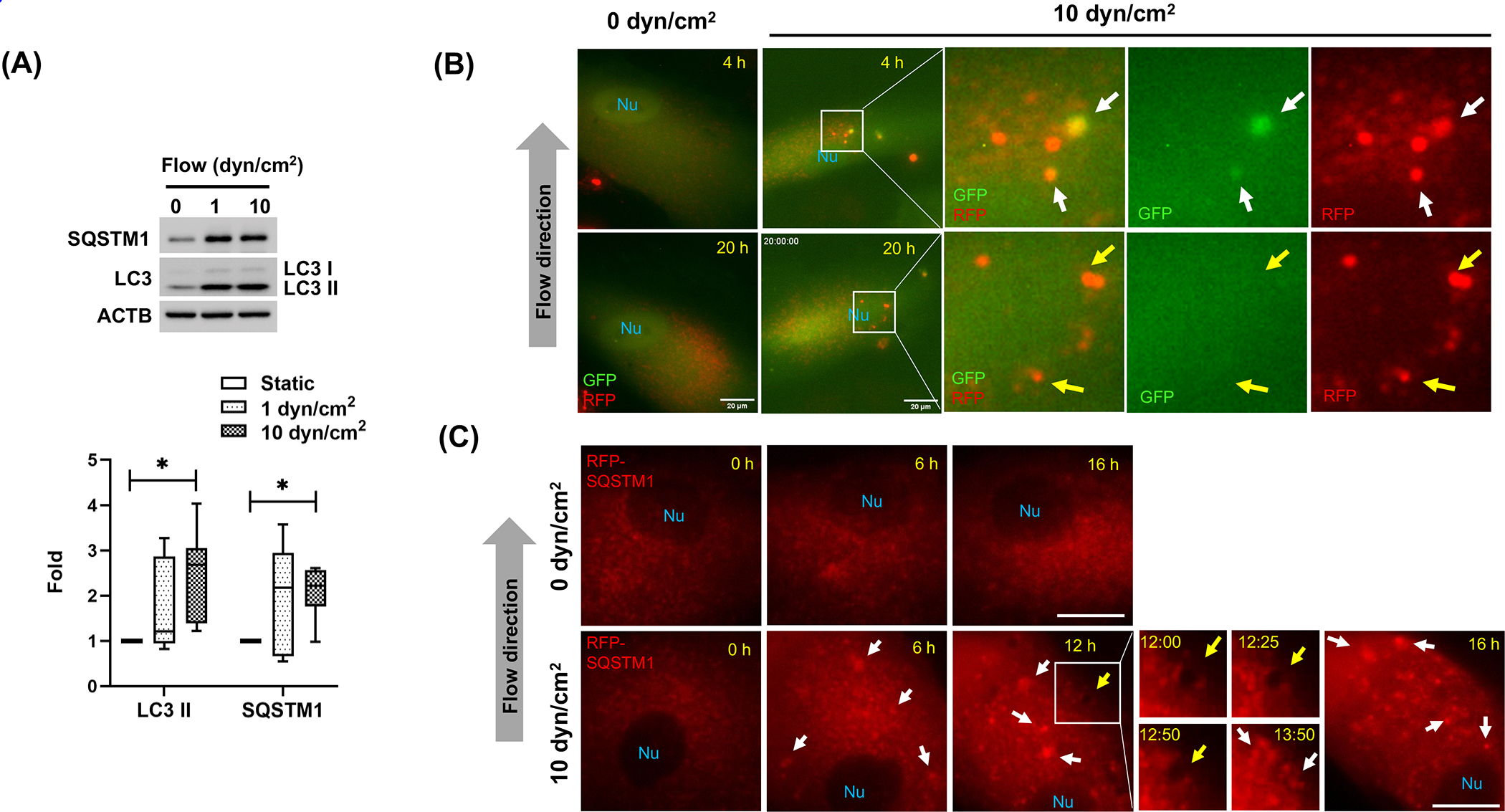
Autophagy is activated by shear stress in human primary SC cells. (A) Western blot analysis for protein expression levels of LC3 and SQSTM1 in human SC cells subjected to different flow rates (1 or 10 dyn/cm2) for 24 h. Band densities were quantified by Image Lab™ touch software, normalized with ACTB and fold changes calculated and graphed. Max to min and mean values are shown in interleaved box and whiskers plot (n=6). *, p<0.05, (One-way ANOVA with Tukey’s post hoc test). (B-C) Live cell imaging of tfLC3- or RFP-SQSTM1-transduced human SC cells in the presence or absence of fluidic flow (0 or 10 dyn/cm2). Time-lapse live cell images were acquired with CELENA® X High Content Imaging System equipped with the ibidi stage top incubation system and processed by Fiji software. White arrowhead and yellow arrows represent autophagosome and autolysosomes, respectively (B). SQSTM1 aggregates and autophagic-like vacuole are indicated by white and yellow arrows, respectively (C). Scale bars: 20 μm. Nu: nucleus.
To further investigate autophagy activation upon shear stress, we monitored autophagic flux by real time live cell imaging analysis, using the tandem fluorescence LC3 (tfLC3) reporter. For this, we transduced SC cell with a recombinant adenovirus expressing tfLC3 (AdtfLC3, [40]). At 72 h post-transduction, cells were subjected to shear stress (10 dyn/cm2), and GFP and RFP signals were traced up to 20 h using the CELENA® X realtime imaging system. Timelapse imaging analysis showed presence of autophagosomes (yellow puncta, white arrows) maturing into autolysosomes (red puncta, yellow arrows) in cells under fluid flow (Fig.1B and Video S1), suggesting that autophagosome maturation was not affected by shear stress. Corroborating that, autophagy flux analysis using BafA1 showed higher LC3-II levels in stressed BafA1-treated cells, compared to non-treated ones (Fig. S1B).
SQSTM1 is a well-known autophagy adaptor protein, which is degraded by autophagy and serve as a marker for autophagy flux [42]. SC cells were transduced with an adenovirus expressing RFP-SQSTM1 and RFP puncta was monitored real time following shear stress application (10 dyn/cm2). As shown in Fig.1C, video S2, RFP dots aggregated and accumulated in cells subjected to shear stress (white arrows in Fig.1 C). Interestingly, the aggregated RFP dots were continuously removed by vacuole-like structures (yellow arrows in Fig.1 C). Co-expression of GFP-LC3 and RFP-SQSTM1 showed that those vacuoles represent autophagic vacuoles (Video S3). Together, these results clearly demonstrate that the increased LC3-II levels in SC cells under shear stress result from autophagy activation.
PC regulate shear stress-induced autophagy in SC cells.
We recently showed that PC is a mechanosensor for cyclic mechanical stress (CMS)-induced autophagy in human TM cells [31]. PC has been reported to mediate shear stress-induced autophagy in kidney tubular epithelial cells [32]; therefore, we investigated whether PC also mediate shear stress-induced autophagy in SC cells. We first confirmed the presence of PC in SC cells since this has not been yet examined in the literature to date. PC was visualized by immunocytochemistry in fixed cells, using PC markers (acetylated-TUBA4A and IFT88, Fig.2A), or by time-lapse live cell imaging using 5HT6-mcherry, as previously described [31] (Fig.2B and Video S4). Both methodologies clearly demonstrate the presence of PC in SC cells. Under steady conditions, PC in SC cells showed to be highly dynamic, changing its size, shape and orientation, and connecting with PC with adjacent cells (Fig. 2C, Video S5). PC dynamism increased in SC cells subjected to flow (1–10 dyn/cm2). PC were seen to reorganize, retract and branch (Fig. 2D and Video S6). Interestingly, this type of remodeling of PC morphology has been reported to reflect changes in PC signaling [43,44].
Figure 2.
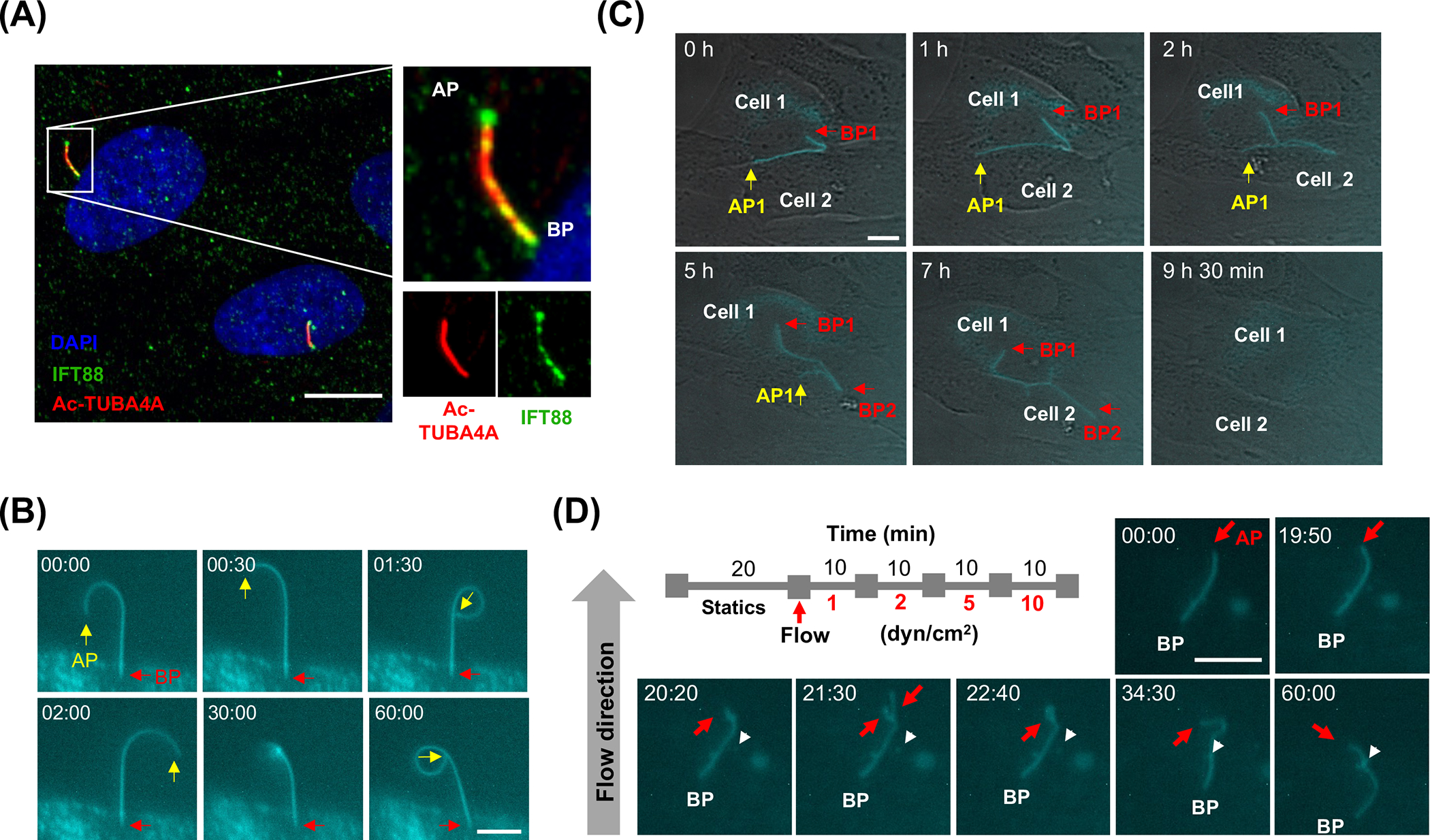
PC are present and respond to shear stress in SC cells. (A) Representative immunocytochemical analysis of PC in SC cells. Acetylated TUBA4A (red fluorescence) and IFT88 (green fluorescence) antibodies were used to identify the PC. DAPI was used to stain nuclei. Images were acquired with confocal microscope and processed by using Fiji software. BP and AP represent basal and apical process, respectively. (B) Time-lapse live cell imaging of PC in SC cells transfected with p5HT6–mCherry. Images were acquired with CELENA® X live cell imaging system and processed by using Fiji software. BP and AP are indicated by red and yellow arrows, respectively. Captured time (min: sec) is represented in upper left of each picture. (C) Live cell imaging of PC dynamics. Note that the two cells are connected by forming PC network and the PC is disappeared in certain cell stages. (D) Response of PC upon fluid flow. AP is indicated by red arrows. Arrowheads represent a vesicle containing 5HT6–mCherry. Captured time (min: sec) is represented in upper left of each picture. Scale bars: 10 μm.
Next, we investigated whether PC mediate shear stress-induced autophagy in SC cells. For this, we chemically removed PC by treating the cells with chloral hydrate (CH, 2 mM for 3 days), as conducted in our previous study [31]. CH treatment completely removed PC without affecting SC cell viability (Fig.S2). Cells were then switched to fresh media to remove and prevent any potential effect of CH and subjected to shear stress (10 dyn/cm2) for 24 h. Autophagy was evaluated by monitoring LC3-II expression levels in whole cell lysates by WB analysis. As shown in Fig. 3A, deciliation significantly prevented the increase in LC3-II levels by shear stress in SC cells, compared to their non-treated control (CNT vs deciliated: 2.36±0.58 vs 0.78 ±0.62, n=4, p<0.05). We additionally investigated the recruitment of ATG16L to the basal body of the PC, which is considered the hallmark for PC-dependent autophagy [45,46]. ATG16L was found at the basal body of the PC, co-localizing with γ-TUB under shear stress and control conditions (Fig. 3B, C). Together these results strongly demonstrate that PC regulates autophagy activation in response to shear stress in SC cells.
Figure 3.
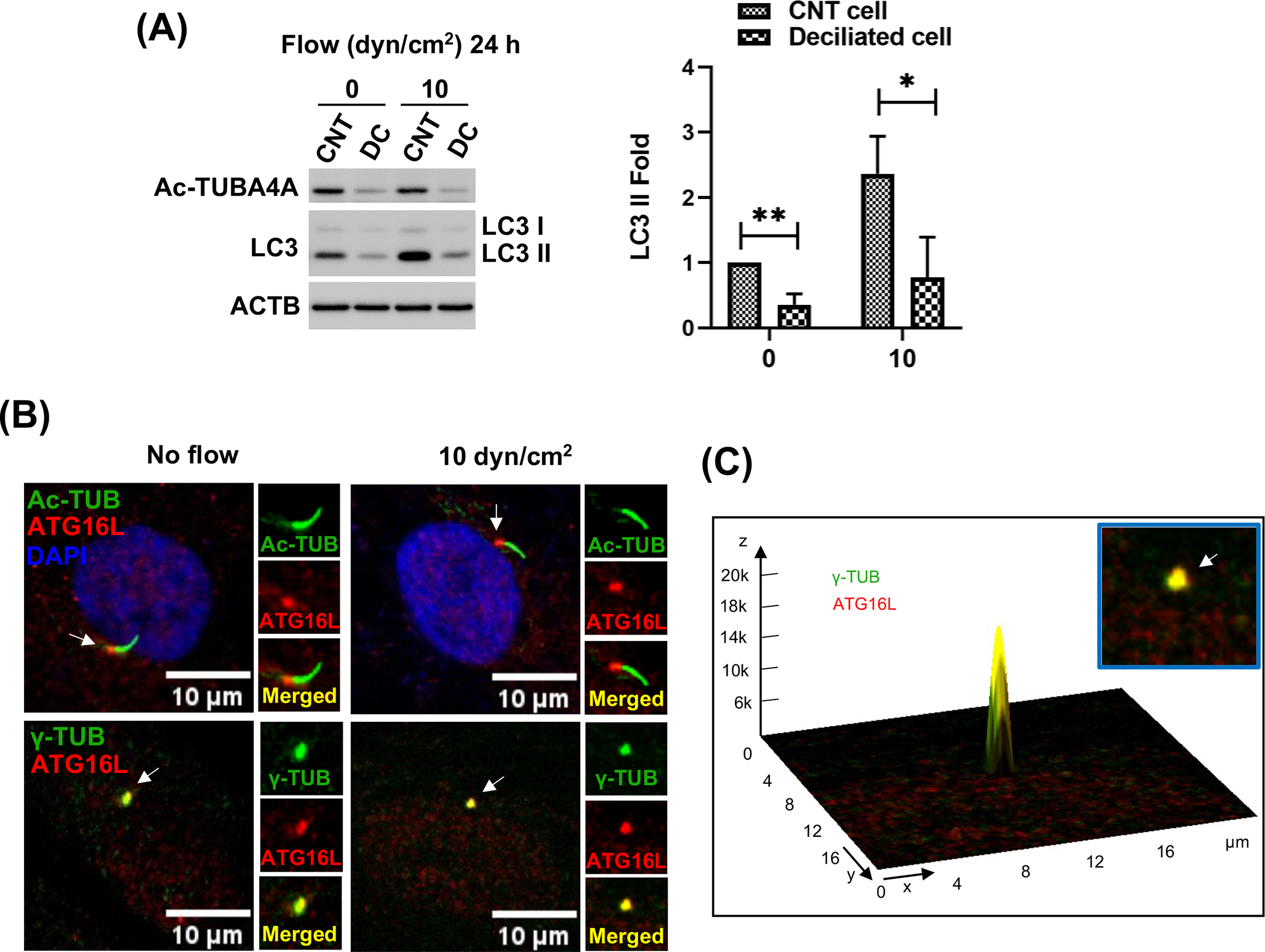
PC regulate shear stress-induced autophagy in SC cells. (A) PC were disrupted by treating SC cells with 2 mM of CH for 3 days. CH was removed by switching to fresh media and SC cells were then subjected to shear stress (10 dyn/cm2) for 24 h. Protein levels of LC3-II, and acetylated TUBA4A (PC marker) were evaluated by WB. Band intensities were quantified by Image Lab™ touch software and normalized with ACTB. Data are shown as the mean ± S.D. (n=4). *, p<0.05; **, p<0.01 (Student’s t-test). CNT: control, DC: deciliated. (B) Representative immunostaining of ATG16L (red) with acetylated TUBA4A (green, upper panels) and ɣ-tubulin (green, lower panels), a maker for the basal body of PC, in SC cells in the absence or presence of shear stress for 24 h. (C) Interactive 3D surface plot analysis visualizing the colocalization of ATG16L with ɣ-tubulin (white arrows).
Shear stress-induced autophagy in SC cells is not directly regulated by PKD2 or PIK3C2A
Ca2+ signalling regulated by PKD2 [32,37] and mobilization of phosphatidylinositol-3, phosphate (PIP3) by PIK3C2a [38] have been reported to regulate and activate PC-dependent autophagy upon exposure of kidney tubular epithelial cells (KTECs) to shear stress. Therefore, we first examined whether similar mechanism controls shear stress-induced autophagy in SC cells. As seen in Fig.S3, mRNA (Fig.S3A) and protein levels (Fig. S3B) of PKD2 were significantly increased upon fluid flow; however, silencing PKD2 expression did not reduce the increase in LC3-II levels by shear stress (Fig. S3B), indicating that PKD2-mediated Ca2+ influx is not responsible for the induction of autophagy in response to shear stress in SC cells. Similarly, in contrast to the findings in KTECs, we did not observe upregulation of PIK3C2A by shear stress (10 dyn/cm2, 24 h) in SC cells (Fig. S4). Moreover, PIK3C2A knockdown did not reduce the increase in LC3-II induced by shear stress (Fig. S4). Together, these results suggest that SC cells use an alternative signalling pathway different from that used by KTECs.
SMAD2 and AKT activity is regulated by PC in response to shear stress in SC cells.
Our laboratory recently identified the reciprocal activation of AKT and SMAD2/3 to regulate PC-dependent CMS-induced autophagy in TM cells [31]. Shear stress has been shown to stimulate the phosphorylation of AKT in cultured human umbilical vein endothelial cells (HUVECs) [47]; therefore, we questioned whether a PC-regulated AKT-SMAD2/3 crosstalk could mediate the activation of autophagy in response to shear stress in SC cells. First, we tested whether shear stress stimulates AKT and SMAD2/3 phosphorylation in SC cells. For this, we applied shear stress (10 dyn/cm2) to SC cells and evaluated pAKT (S473) and pSMAD2/3 at C-terminal region (S465,467/S423,425; pSMAD2/3C) at early times post stress (1 or 3 h). As seen in Fig 4A, shear stress significantly increased both pAKT (8.18 ± 3.53-fold, p<0.001, n=4) and pSMAD2C (4.80 ± 1.80-fold, p<0.01, n=3) levels at 1h, gradually decreasing afterwards. We additionally examined noncanonical phosphorylation of SMAD2 at the linker region (pSMAD2L) since phosphorylation at this site has been recently reported with shear stress in HUVECs [48]. In agreement, pSMAD2L levels were found to be significantly increased in stressed SC cells (4.86 ± 1.93-fold, p<0.001, n=3) and showed a similar decreasing pattern as pAKT and pSMAD2C (Fig.4A). Intriguingly, we consistently found an additional upper band of SMAD2/3 (Fig.4A. question mark) whose apparent MW size is different from that of pSMAD2C and L, suggesting the possibility that shear stress might induce another yet non-identified posttranslational modification of SMAD2/3 in SC cells.
Figure 4.
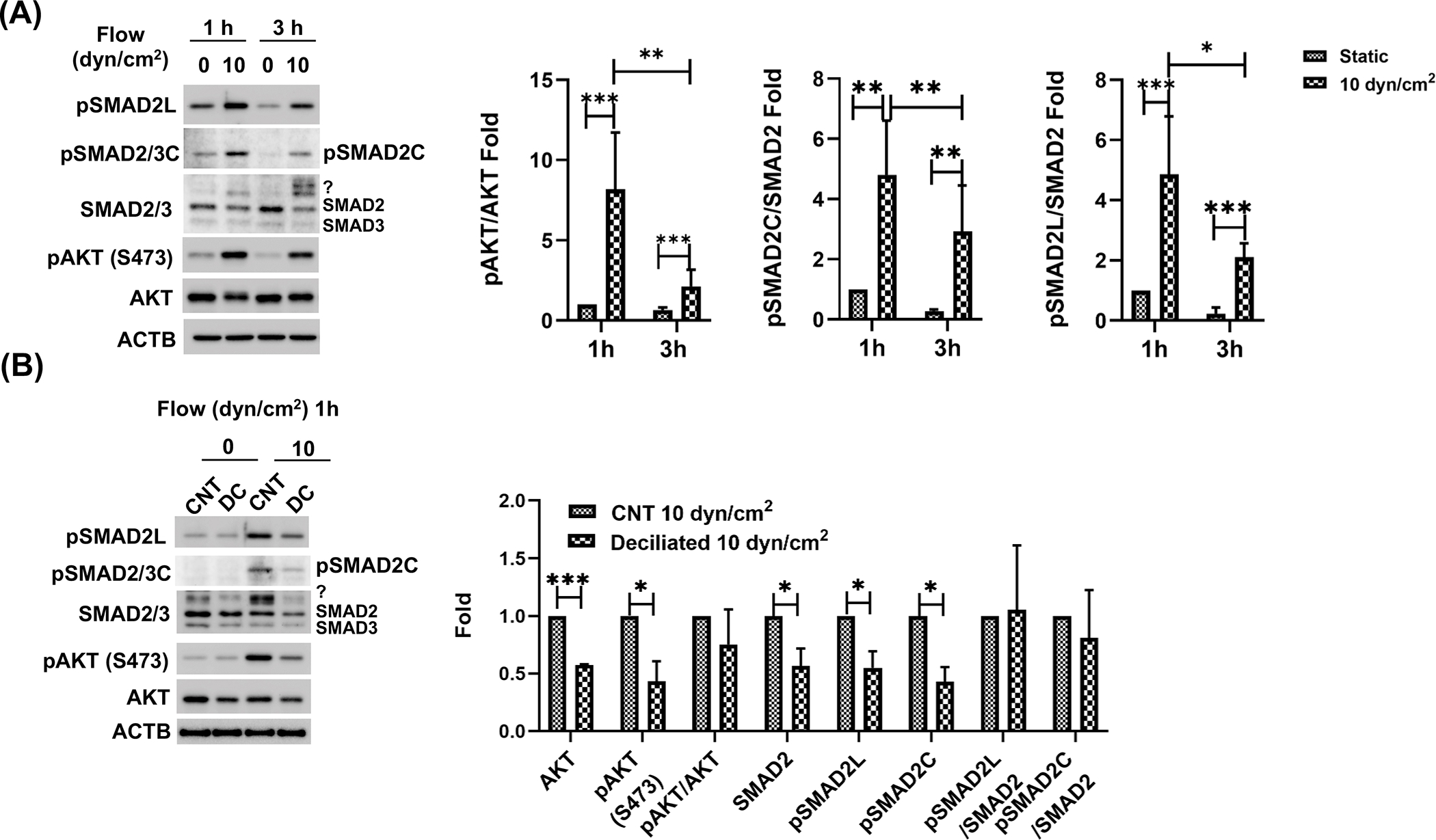
SMAD2 and AKT activity is regulated by PC in response to shear stress in SC cells. (A) Effect of shear stress on SMAD2/3 and AKT. SC cells were subjected to shear stress (10 dyn/cm2) for 1 or 3h. SMAD2/3 and AKT non-phosphorylated and phosphorylated protein levels (pAKT, pSMAD2/3C, pSMAD2L) were evaluated by WB. pSMAD2L and C represent phosphorylation at the linker regions (S245, 250 and 255) and c-terminal regions (S465,467) of SMAD2, respectively. Question mark represents additional unidentified upper bands detected by SMAD2/3 antibody in SC cells under shear stress. SMAD2 and AKT phosphorylated versus total protein ratio were calculated and represented in the right panel. Data are shown as the mean ± S.D. (n =3). *, p<0.05; **, p<0.01; ***, p<0.001 (Two-way ANOVA with Tukey’s post hoc). (B) Effect of deciliation on SMAD2/3 and AKT non-phosphorylated and phosphorylated protein levels. PC was chemically disrupted in SC cells with 2mM CH treatment for 3 days and subjected to shear stress (10 dyn/cm2) for 1 h. SMAD2/3 and AKT non-phosphorylated and phosphorylated protein levels were evaluated by WB. Data are shown as the mean ± S.D. (n=3). *, p<0.05; ***, p<0.001 (Student’s t-test). CNT: control, DC: deciliated.
To test whether PC regulate the activity of AKT and/or SMAD2/3, we chemically removed PC, as described above, and examined the levels of pAKT, pSMAD2C, and pSMAD2L after shear stress (10 dyn/cm2, 1h). As observed in Fig.4B, we found diminished absolute expression levels of pAKT, pSMAD2C and pSMAD2L (0.43 ± 0.18, 0.43 ± 0.13 and 0.55 ± 0.15-fold, respectively p<0.05, n=3), as well as those of the non-phosphorylated forms (AKT: 0.58 ± 0.01, p<0.001 and SMAD2/3:0.56 ± 0.16-fold, respectively, n=3). Interestingly, the upper band of SMAD2/3 also decreased by deciliation (question mark in SMAD2/3 WB image in Fig.4B). These results clearly demonstrate that PC regulates the AKT and SMAD2/3 signaling in response to shear stress.
SMAD2/3 directly regulates the PC-mediated autophagy in response to shear stress in SC cells.
Next, we tested whether SMAD2/3 and AKT1 regulate PC-dependent shear stress-induced autophagy in SC cells. For this, we knocked down SMAD2/3 (Fig. 5A, B) or AKT1 (Fig. 5C) in SC cells using specific siRNAs and subjected the cells to shear stress (10 dyn/cm2) for 24 h; LC3 II levels were evaluated. SMAD2/3 knock-down significantly reduced the increase in LC3-II levels by shear stress (Fig. 5A, siCNT-SS vs siSMAD2/3-SS; 1.79 ± 0.31 vs 1.41 ± 0.28-fold, p<0.05, n=6). However, in contrast to our findings in mechanically stretched TM cells, silencing SMAD2/3 did not affect pAKT/AKT ratio in SC cells, under control or shear stress conditions (Fig.5 B). Similarly, silencing AKT1 did not significantly alter pSMAD2L or SMAD2/3 expression in SC either (Fig. 5C). Moreover, AKT1 knockdown had no effect on shear stress-induced LC3-II. Together, these results indicate that shear stress-induced autophagy is regulated by SMAD2/3 without direct crosstalk with AKT1.
Figure 5.
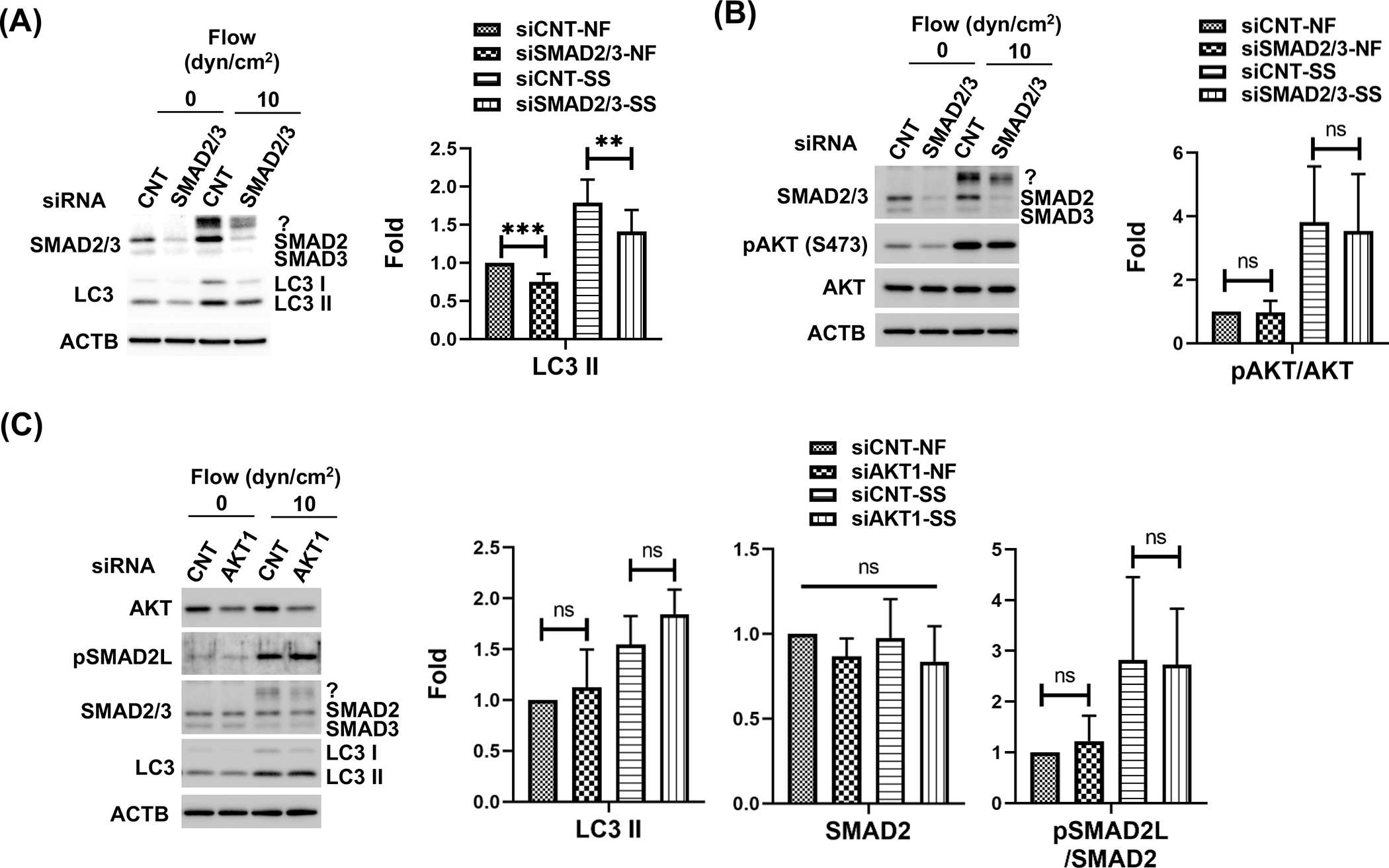
SMAD2/3, not AKT1 directly participates the PC-mediated autophagy in response to shear stress in SC cells. (A) Effect of SMAD2/3 knockdown on shear stress-induced autophagy. (B) Effect of SMAD2/3 knockdown on AKT activation. (C) AKT1 knockdown effect on LC3 and SMAD2/3 protein level. Protein expression levels were analysed by WB. Fold changes were calculated and represented at each section. Data are shown as the mean ± S.D. (n=3). *, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant (One-way ANOVA with Tukey’s post hoc test).
SMAD2/3 transcriptionally regulates LC3 in response to shear stress in SC cells.
Phosphorylation of SMAD2/3C triggers nuclear localization of SMAD2/3, where it regulates the transcription of its target genes [49]. We investigated whether SMAD2/3 phosphorylation in response to shear stress regulates transcriptional activation of autophagy genes. To test this, we first examined whether shear stress stimulates SMAD2/3 nuclear localization in SC cells, using immunocytochemistry analysis. SC cells subjected to shear stress (10 dyn/cm2, 1h) were immunostained with pSMAD2/3C antibody and observed by confocal microscope. Higher overall immunoreactivity of pSMAD2/3 (red fluorescence) and nuclear localization of pSMAD2/3 (white arrows, Fig.6A) were found in the stressed cells (Fig.6A). Next, we quantified the transcriptional levels of LC3B, ATG5 and ATG7 in SC cells exposed to flow (1 or 10 dyn/cm2, 24 h). As seen in Fig.6B, realtime PCR analysis revealed that shear stress (10 dyn/cm2) significantly increases the mRNA levels of LC3B, ATG5 and ATG7 (128 ± 14, 180 ± 63 and 222 ± 90%, p<0.01, 0.05 and 0.05, respectively, n=3). Moreover, these were reduced in cells with knockdown expression of SMAD2/3 (Fig.6C). As an aside, there was high variability in the expression levels of ATG5/7 in response to shear stress among different SC cell strains. Changes in LC3B were more robust regardless the strain and it is only upregulated in shear stress conditions. Together, these results clearly indicate that SMAD2/3 activates transcription of LC3B.
Figure 6.
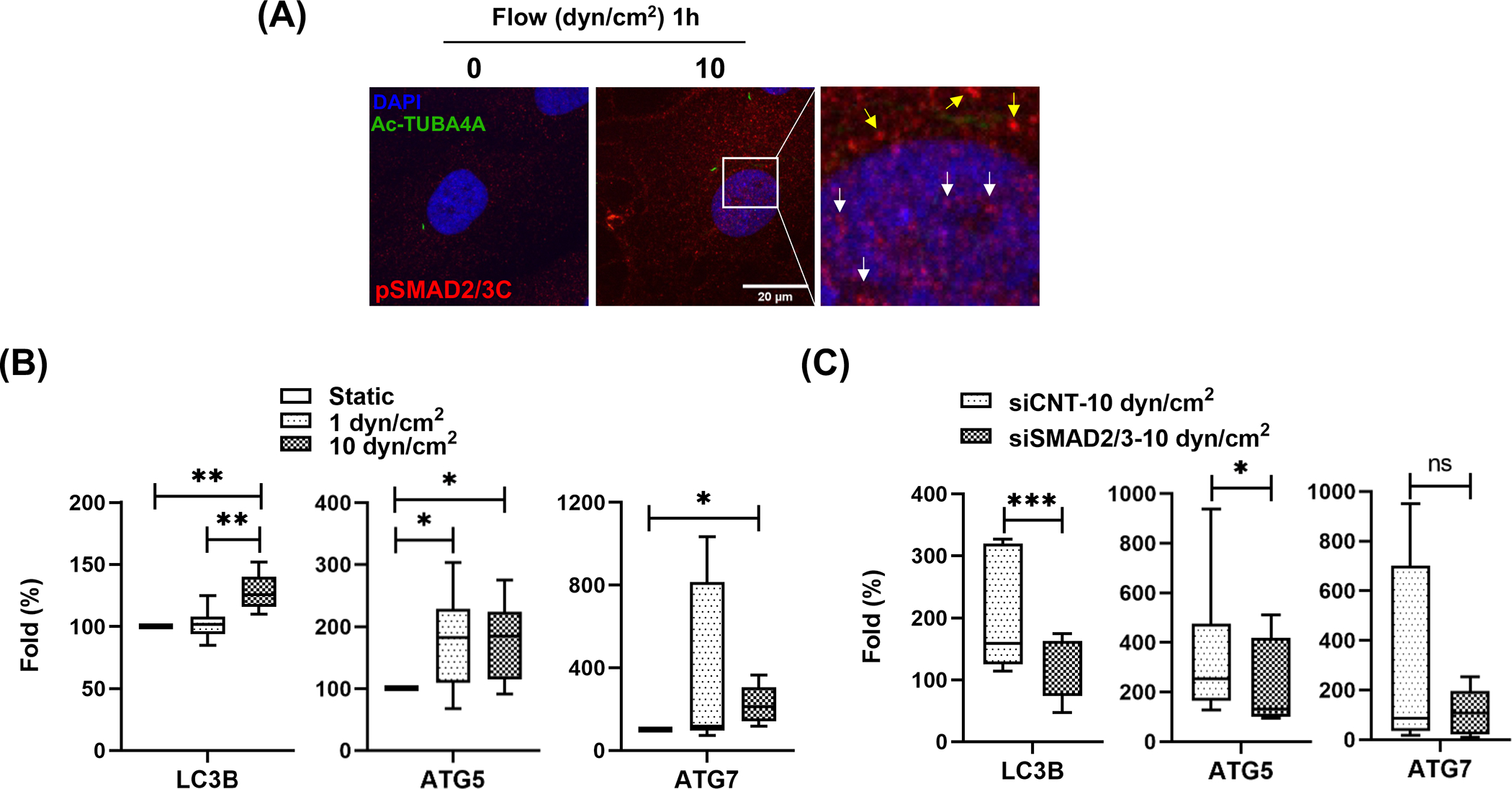
SMAD2/3 regulates transcription of LC3 in response to shear stress in SC cells. (A) Immunocytochemistry analysis of pSMAD2/3C localization. One hour after shear stress (10 dyn/cm2), SC cells were immunostained with pSMAD2/3C (red fluorescence) and acetylated TUBA4A (green fluorescence) antibodies. DAPI was used to stain nuclei. Images were acquired with confocal microscope and processed by using Fiji software. The fluorescent signals stained with pSMAD2/3C in nucleus and cytosol are indicated by white and yellow arrows in inbox, respectively. (B) Quantitative real-time PCR analysis for examining expression levels of ATG genes in SC cells subjected to fluidic flow (1 or 10 dyn/cm2) for 24 h. The mRNA expression level of LC3B, ATG5 and 7 were normalized by that of B2M. For calculating fold changes, the mRNA expressions of each gene in controls cells (0 dyn/cm2) were set to a value of 100%. Data are shown as interleaved box and whiskers plot (n =3). *, p<0.05; **, p<0.01 (One-way ANOVA with Tukey’s post hoc test). (C) SMAD2/3 knockdown effect on the mRNA level of the ATG genes in SC cells subjected to shear stress (10 dyn/cm2). Data are shown as interleaved box and whiskers plot (n =3). *, p<0.05; ***, p<0.001; ns, not significant (Student’s t-test).
Inhibition of PI3K/MTOR activates autophagy and increase pSMAD2L and C expression levels in SC cells.
Class I PI3Ks has been reported to regulate pSMAD2L levels in flow-exposed human aortic endothelial cells [50]. Since our data showed changes in pSMAD2L levels with shear stress (Fig.4A), as well as in deciliated SC cells (Fig. 4B), we questioned whether SMAD2/3 could regulate PC-dependent activation of autophagy under shear stress through the action of class I PI3Ks on pSMAD2L. First, we examined the effects of pharmacological inhibition of Class I PI3Ks on shear stress-induced activation of autophagy on SC cells. For this, SC cells transfected with the autophagosome reporter GFP-LC3 were treated with PI-103, a dual class I PI3K and MTOR inhibitor (10 μM [51]). GFP fluorescence signal was monitored up to 24 h by time-lapse live cell imaging. GFP-LC3 puncta (Fig.7A, red arrows) together with autophagic vacuoles (Fig.7A, white arrows) were started to form within 1 or 2 h upon PI-103 treatment and the formations are continued up to 24 h. Furthermore, the GFP-LC3 puncta were dynamically recruited to and processed in the autophagic vacuoles (Fig.7A, inbox and Video S7), indicating autophagy activation by PI3K/MTOR in SC cells.
Figure 7.
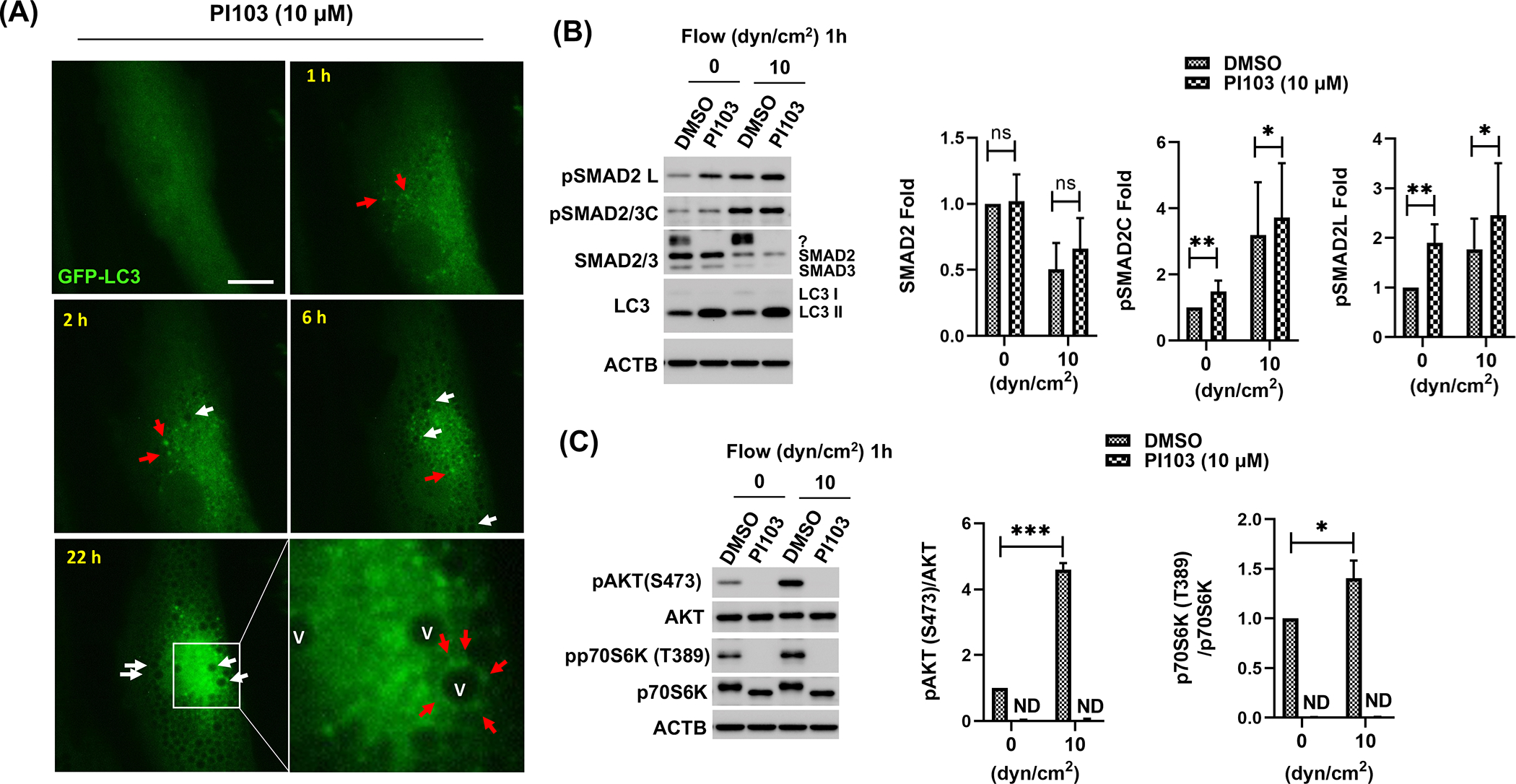
Class I PI3K inhibition triggers autophagy with increasing pSMAD2 L and C level in SC cells. (A) Autophagy activation induced by PI-103 treatment. SC cells expressing GFP-LC3 were treated with PI-103 (10 μM); cell morphology and GFP fluorescent signals were monitored for 24 h. LC3 puncta and autophagic vacuoles are indicated by red and white arrows, respectively. Captured time is represented in upper left of each picture. Scale bars: 10 μm. (B and C) Effect of PI-103 treatment on posttranslational modification of SMAD2 (B), AKT and mTOR signaling (C). ACTB was used for normalization. Data are shown as the mean ± S.D (n=3). *, p<0.05; **, p<0.01; ns, not significant (Student’s t-test).
Next, we investigated SMAD2 phosphorylation in response to shear stress in SC cells in the presence of PI-103. SC cells were pre-treated for 2h with PI-103 (10 μM) and exposed to shear stress (10 dyn/cm2) for 1 h. Levels of pSMAD2L and pSMAD2C were quantified by WB (Fig.7B). WB analysis showed a significant increase in the levels of pSMAD2L with PI-103 treatment in both static (1.90 ± 0.38, p<0.01, n=6) and shear stress conditions (CNT vs PI-103; 1.77 ± 0.62 vs 2.46 ± 1.05, p<0.05, n=6). Higher levels were also observed, although to a lesser degree, in pSMAD2C under both static (1.48 ± 0.33, p<0.01, n=6) and shear stress (CNT vs PI-103; 3.20 ± 1.60 vs 3.72 ± 1.65, p<0.05, n=6). No changes in total SMAD2 were observed with PI-103 treatment (Fig.7B). Intriguingly, the unidentified upper band of SMAD2/3 induced by shear stress completely disappeared upon PI-103 treatment (Fig.7B, question mark). Confirming GFP-LC3 results, higher LC3-II levels were observed in PI-103- treated cells. Together, these results suggest that the activity of SMAD2, a key player mediating PC-dependent shear stress-induced autophagy, is regulated by PI3K-MTOR signaling pathway SC cells.
Shear stress activates MTOR signaling in SC mediated by PC.
PI3K/AKT/MTOR is a well-known pathway regulating autophagy activation. We wanted to investigate the status of mTOR signaling in SC cells under shear stress. For this, we monitored phosphorylation of the downstream effector of MTOR, pp70S6K (T389), in SC cells under shear stress (10 dyn/cm2) for 1h (Fig. 7C) or 24h (Fig. S5). WB analysis showed significantly higher levels of pp70S6K [1.40 ± 0.18, p<0.05, n=4] in stressed cells. mTOR activation was not affected by silencing SMAD2/3 or AKT1 expression (Fig. S5). However, phosphorylation of p70S6K, as well as that of AKT was completely blocked upon PI-103 treatment (Fig. 7C). In view of these results, we investigated whether PC mediates mTOR activation in response to flow. Phosphorylation of p70S6K (T389) and p70S6K levels were evaluated in deciliated SC cells exposed to shear stress (10 dyn/cm2, 24h). As seen in Fig. 8, PC disruption decreased the protein levels of pp70S6K (T389) and p70S6K with fluid flow. Together, our results indicate that PC regulates mTOR signaling in response to shear stress-induced fluid flow in SC cells
Figure 8.
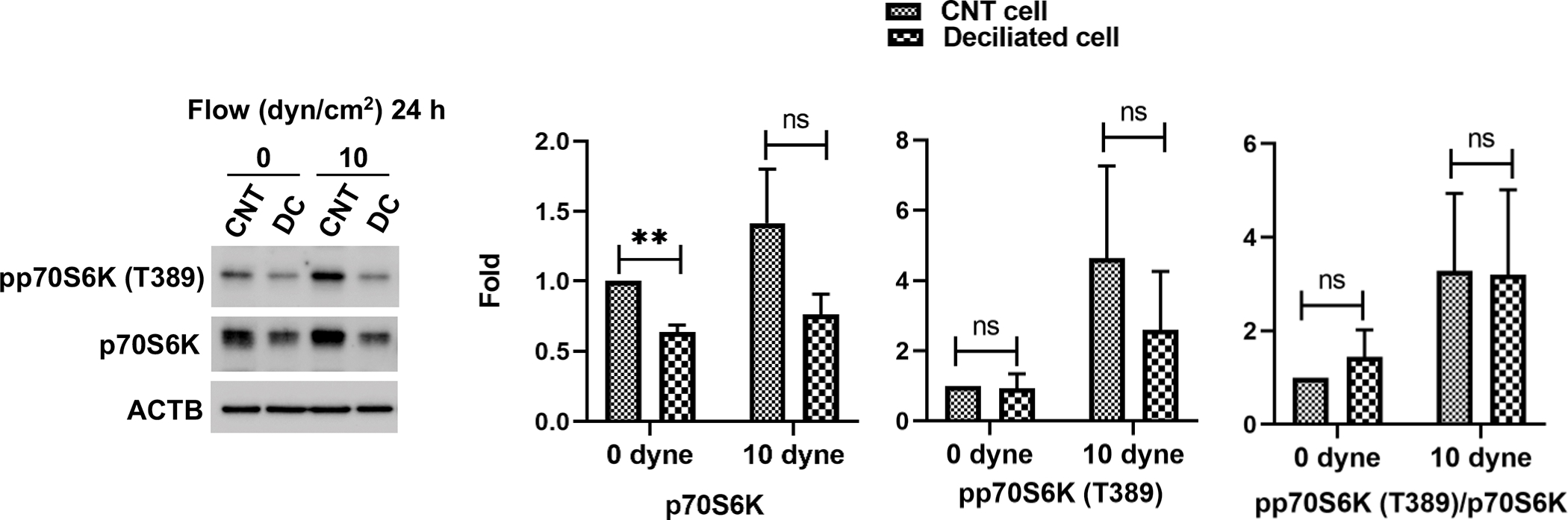
PC mediates mTOR activation in response to fluid flow. PC were disrupted by treating SC cells with 2 mM of CH for 3 days. CH was removed by switching to fresh media and SC cells were then subjected to shear stress (10 dyn/cm2) for 24 h. Protein levels of pp70S6K (T389) and p70S6K were evaluated by WB. Band intensities were quantified by Image Lab™ touch software and normalized with ACTB. Data are shown as the mean ± S.D. (n=3). **, p<0.01; ns, not significant (Student’s t-test).
MTOR does not mediate pSMAD2L in SC cells with shear stress.
As mentioned above, PI-103, is a dual class I PI3K and MTOR inhibitor. PI3K/mTORC2 has been reported to negatively regulate SMAD2/3 activity by increasing the ubiquitin-mediated degradation of SMAD2/3 via modulating the phosphorylation status of SMAD2/3 at the linker region [60]. Since our data implicated PC in the regulation of mTOR signaling with shear stress, we questioned whether MTOR could mediated pSMAD2L. For this, we quantified by WB levels of pSMAD2L in SC under shear stress (10 dyn/cm2, 1 h) in the presence of the mTOR inhibitor rapamycin (1 μM). As seen in Figure 9, no significant effect on basal (1.29 ± 0.40-fold, ns, n=3) or stress-induced pSMAD2L levels (Vehicle: 5.24 ± 3.58; Rap: 7.18 ± 5.23-fold, ns, n=3) were observed with mTOR inhibition.
Figure 9.
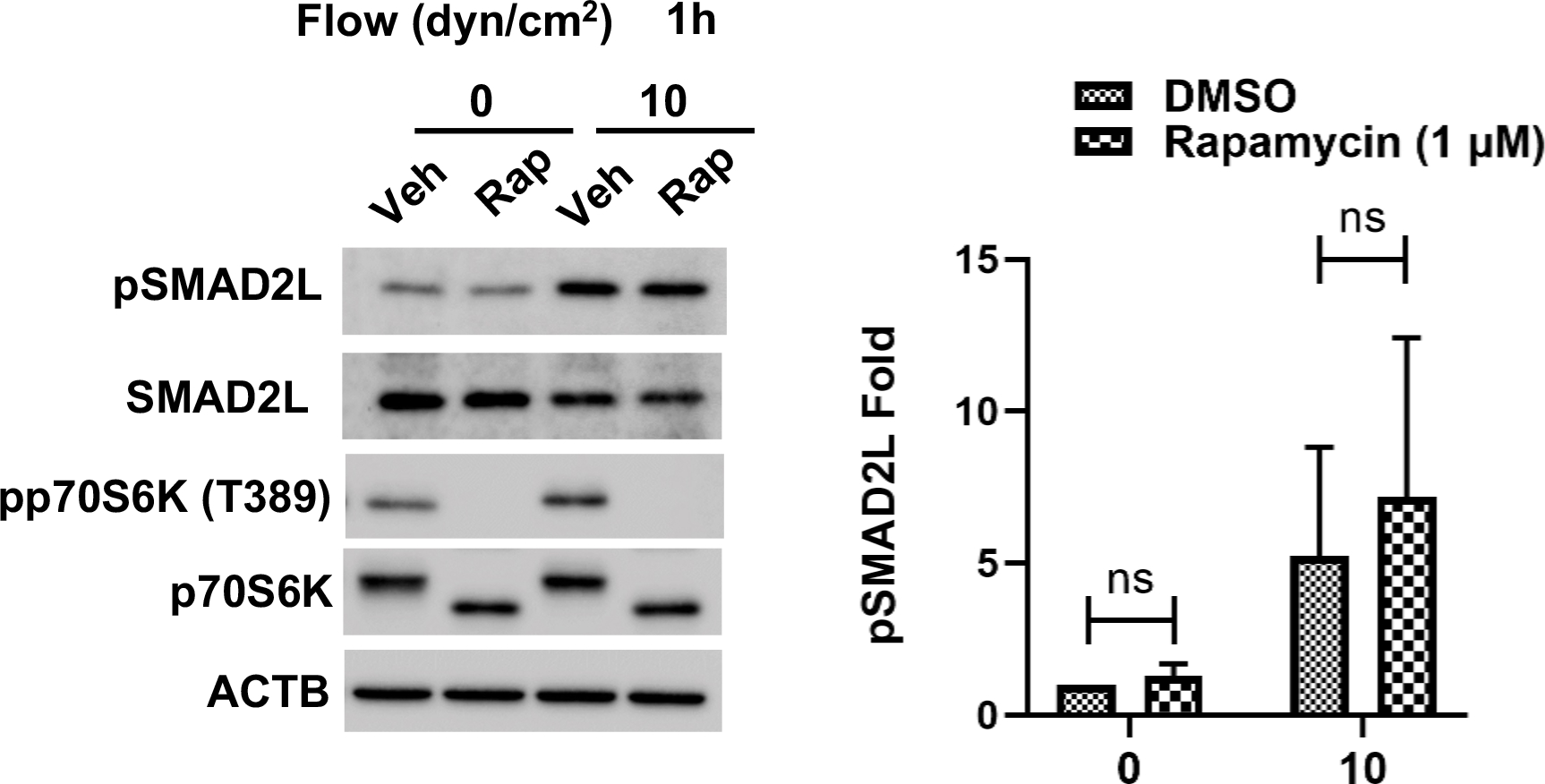
MTOR inhibition does not induce SMAD2 linker phosphorylation in SC cells. Western blot analysis showing protein expression levels of pSMAD2L in human SC cells treated and rapamycin. SC cells were treated with an MTOR inhibitor (rapamycin; 1 μM) and subjected to shear stress (10 dyn/cm2) for 1 h. Protein levels of pSMAD2L and pp70S6K were measured by western blot analysis. ACTB was used for normalization. Data are shown as the mean ± S.D (n=3). *, p<0.05; ns, not significant (Student’s t-test).
Discussion
In this study, we report for the first time the PC-dependent activation of autophagy in SC cells in response to flow-induced shear stress. Moreover, we identified phosphorylation of SMAD2 in its linker and c-terminal regions as a critical positive regulator of the event. SMAD2/3 signaling was also found to transcriptionally activate LC3B, ATG5 and ATG7. Concomitant to SMAD2-dependent activation of autophagy, we observed the PC-dependent activation of mTOR pathway in SC cells by shear stress. Pharmacological inhibition of class I PI3K, but not of mTOR, activated autophagy and increased phosphorylation of SMAD2 at the linker. Together, this data indicates an interplay between PI3K and SMAD2/3 signaling pathways regulating PC-dependent shear stress-induced autophagy in SC cells
Autophagy activation in response to fluid flow has been described in other cell types, in particular vascular ECs and KTECs [14,32,34,52–54]. In line with the findings in these cell types, we observed increased LC3-II levels in SC when subjected to physiological fluid flow forces. Time-lapse live imaging using tfLC3 also showed formation of autophagosomes and their transport to the perinuclear region maturing into autolysosomes. Similarly, RFP-SQSTM1 puncta formation and their subsequent removal by GFP-LC3-labelled vacuoles were observed in SC cells under shear stress. Activation of autophagy with shear stress was prevented by chemically disrupting PC. Moreover, co-localization of ATG16L1 at the basal of the PC - a hallmark of PC-dependent autophagy - was observed. However, in our setting, ATG16L1 at the PC was also noticed in SC cells in the absence of applied fluid flow. ATG16L1 has been recently shown to participate in ciliogenesis by regulating the turnover of phosphoinositides at the PC [55]. In this regard, PC in SC cells were found to be highly dynamic, changing in size and forming physical contacts with PC from adjacent cells. Higher dynamism, including branching and retraction was observed under fluidic conditions. This type of phenomenon has been described in some, but not all, cell types, and it is believed to reflect changes in PC signaling [43,44], suggesting that PC might sense the extracellular space not just as a passive antennae, but also through direct contact [56].
The signaling pathway regulating PC-dependent shear stress-induced autophagy in SC cells differed from that reported in KTECs [32,37]. First, our data showed the activation of AKT/mTOR pathway - a classical autophagy inhibitor - with fluid flow in SC cells. Interestingly, AKT/mTOR activation was found to also be mediated by PC; therefore, indicating that autophagy must occur in an mTOR-independent manner. Silencing of PKD2 or PIK3C2A, which have been implicated in PC-dependent autophagy in KTECs and PTEKECs, respectively, did not decrease fluid flow-induced LC3-II levels. In contrast, our data identified SMAD2/3 signaling as a critical component regulating shear stress-induced autophagy in SC cells. SMAD2/3 was previously reported by our group to participate in the activation of autophagy in TM cells in response to mechanical stretch [31]. Unlike in TM cells, in which stretch-induced activation of autophagy was regulated by a reciprocal cross-talk between AKT and non-canonical SMAD2/3, SMAD2/3 seems to directly regulate shear stress-induced autophagy in SCs, and no cross-talk between both pathways was observed.
SMAD2/3, a generally known mediator for TGF-β signalling pathway, is composed of 3 common structures: N-terminal domain (MAD homology 1), the proline rich linker region and a carboxyterminal (MAD homology 2) [49,57]. Interestingly, flow-induced shear stress phosphorylated SMAD2 in both, its C-terminal and linker regions, which was dependent on the presence of PC. Phosphorylation at C-terminal is normally associated to canonical TGFβ signaling, occurring upon binding of TGF-β to TGFβ receptor-I. pSMAD2/3C enters the nucleus as a complex with SMAD4 and regulates transcriptional activation of its target genes [57,58]. SMAD2 linker phosphorylation has been reported to suppress SMAD2/3 transcriptional activity by preventing its nuclear localization [59]. Also, high shear stress (25 dyn/cm2) prevented SMAD2/3 nuclear translocation through phosphorylation at the linker domain in HUVECs [48]. However, contrary to these reports, we observed immunoreactivity of SMAD2/3C in the nucleus of SC cells subjected to flow. This data supports emerging evidence suggesting that SMAD linker region phosphorylation not only is a modulator of canonical TGF-β, but it is a signaling pathway on its own, activated by alternative signals other than TGF and driving the expression of different target genes [49]. Interestingly, unlike in mechanical stretch [31], shear stress promotes the SMAD2/3-mediated transcriptional activation of LC3B, as well as that of ATG5 and ATG7, although, ATG7 did not reach statistical significance due to the individual variation among the different cell strains. Whether this is regulated by phosphorylation at the linker region and/or C-terminal needs further investigation.
Our results identified inhibition of Class I PI3K as a critical upstream signal regulating pSMAD2/3 linker phosphorylation and autophagy activation in SC cells in response to shear stress. Class I PI3Ks has been also reported to regulate pSMAD2L levels in flow-exposed human aortic endothelial cells [50]. In addition to promoting pSMAD2/3 linker phosphorylation, Class I PI3K inhibition completely blocked AKT and mTOR activation with fluid flow. It is plausible, then that mTOR inhibition in PI103-treated cells contributes to the observed increased autophagy levels. Interestingly, PI3K/mTORC2 has been reported to negatively regulate SMAD2/3 activity by increasing the ubiquitin-mediated degradation of SMAD2/3 via modulating the phosphorylation status of SMAD2/3 at the linker region [60]. Although no changes in pSMAD2L levels were observed in cells treated with rapamycin, we do not discount a similar cross-talk between PI3K and mTORC1. Very intriguingly, our data consistently showed an upper band immunoreacting with SMAD2/3 antibody in SC cells subjected to shear stress, which was not recognized by any of the antibodies targeting the phosphorylated forms. Moreover, this band disappeared in DC cells as well as in cells treated with PI103, suggesting that this SMAD2/3 posttranslational modification is also regulated by class I PI3K and/or mTOR. We speculate that this upper band represents polyubiquitinated SMAD2/3, targeting SMAD2/3 for degradation, as reported in [60]. That would certainly explain the lower levels of SMAD2/3 observed after longer shear stress application or in DC SC cells. We further postulate this can represent a molecular mechanism negatively regulating the activation of autophagy by SMAD2/3 in SC cells under fluid flow. An illustrative summary of our finding sis shown in Fig. 10.
Figure 10.
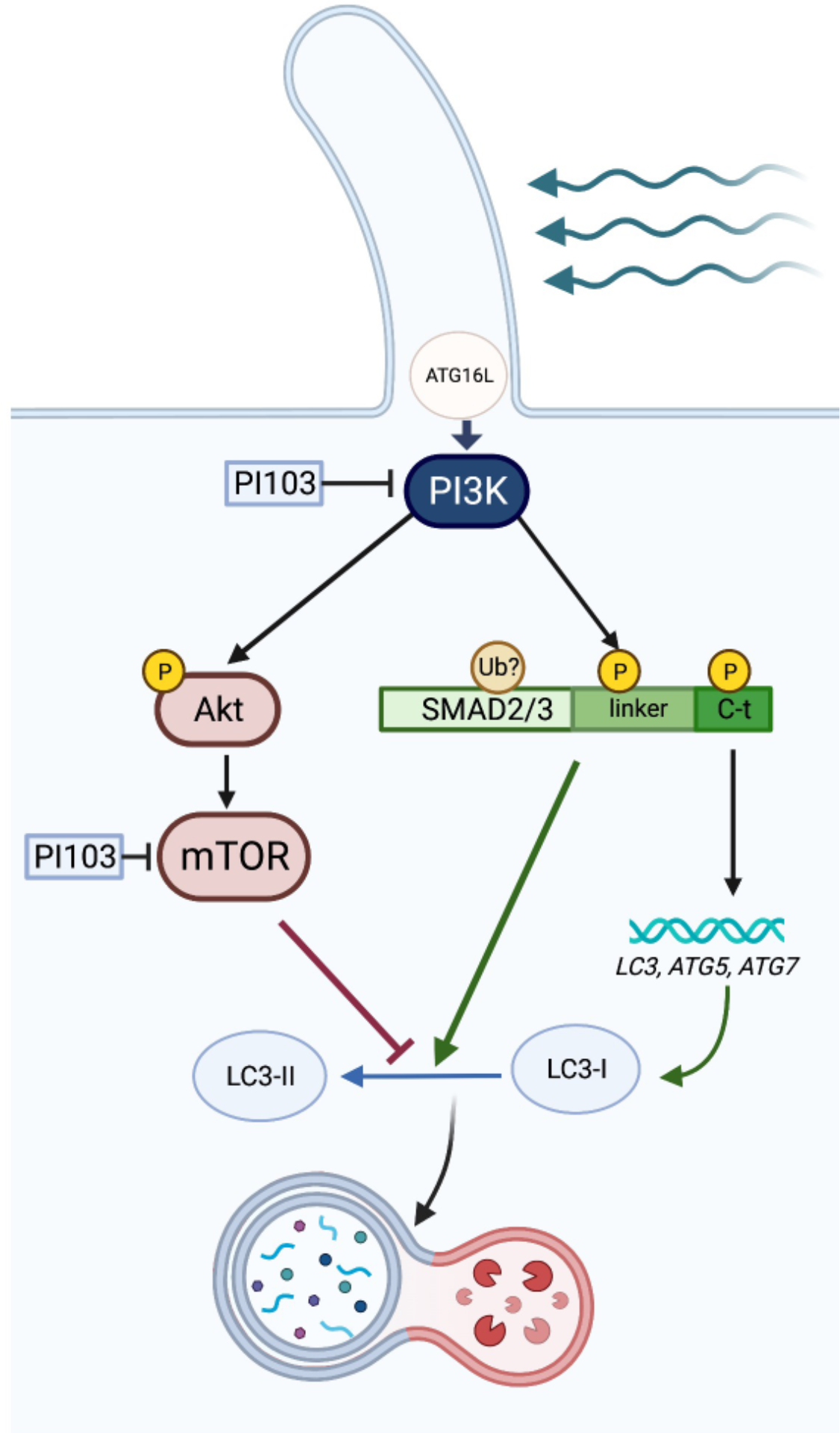
Illustrative summary of the key molecular events involved in flow-induced shear stress-induced autophagy in SC cells Flow-induced shear stress triggers PC-dependent activation of autophagy in SC cell, which is regulated by phosphorylation at the C-terminal and linker region of SMAD2/3. Class I PI3K acts was identified as a critical upstream signal, regulating the phosphorylation of SMAD2/3 linker and subsequent autophagy activation in response to shear stress. Activation of SMAD2/3 signaling further induces the transcriptional upregulation of autophagy genes
Together, our data strongly demonstrate that flow-induced shear stress triggers PC-dependent activation of autophagy in SC cell, and that this is regulated by phosphorylation at the C-terminal and linker region of SMAD2/3. Moreover, our results further suggest a role of class I PI3Kin regulating SMAD2/3 phosphorylation. Future studies will be directed at elucidating the detailed molecular mechanisms by which PC controls SMAD2/3 for the regulation of shear stress-induced autophagy and the physiological roles of PC-dependent flow-induced autophagy in SC cells and IOP homeostasis. Although still not investigated in SC cells, TM cells isolated from eyes with glaucoma showed constitutive activation of mTOR signaling and dysregulated basal and stress-induced autophagy [39]. Similar dysregulation of autophagy in SC cells in response to fluid flow might compromise the physiological response and promote or contribution to elevated IOP.
Materials and methods
Reagents.
Chemicals and other materials were obtained from the following sources: Chloral hydrate (CH, C8383), PI-103 (528100), rapamycin (R8781–200UL, Sigma), DMSO (D2650), goat serum (G9023), HEPES (H0887), bovine serum albumin (BSA; A7906), Trion X100 (Tx1568–1), β-mecaptoethanol (M6250) from MilliporeSigma; phosphate-buffered saline (PBS,10X; Corning, 46-013-CM); Dulbecco’s modified eagle medium (DMEM; 11885084), 100 × penicillin–streptomycin (Pen/Strep; 15240062), fetal bovine serum (FBS; 10082147), 100 × non-essential amino acids (11140050), gentamycin (15750060), Hank’s Balanced Salt Solution (HBSS; 14025092), Lipofectamine® RNAiMAX Reagent (13778075), DAPI (62248), Fluoromount-G™ (00-4958-02), super signal west femto chemiluminescent substrates (34096), SuperScript® First-strand kit (11904–018) from Thermo Fisher Scientific; ECL western blotting detection reagent (RPN2106) from GE Healthcare; 4x Laemmli sample buffer (161–0747), protein size marker (161–0373), protein assay dye reagent concentrate (5000006), Blotting-Grade Blocker non-fat dry milk (1706404), Tween 20 (1706531) and polyvinylidene fluoride (PVDF) membrane for protein blotting (1620177), SsoFast™ EvaGreen® Supermix (172–5204) from Bio-Rad; RNeasy kit (74106) from Qiagen.
Isolation and maintenance of primary human SC cells.
Primary human SC cells were isolated by using the cannulation method from human donor eyes, cultured and characterized as previously described [61,62]. Briefly, SC cell strains (passages 4–8) isolated from donors (SC66, -71, -75, -77, -82 and -87, corresponding to ages of no information, 44, 10, 23, 56 and 62 years old at time of death, respectively) were used in experiments. SC cells were cultured in DMEM supplemented with 10% FBS, non-essential amino acids, Pen/Strep and gentamycin (50 μg/mL) at 37 °C with 5% CO2. The protocols involving the use of human tissue were consistent with the tenets of the Declaration of Helsinki.
Flow-induced shear stress.
Laminar flow was applied to confluent SC cells using an ibidi pump system (ibidi USA, Inc., Fitchburg, WI, USA) as described previously [7] with minor modification. Briefly, human primary SC cells (1× 105 or 2× 104 cells/one channel of slide) were seeded onto ibidi μ-slides I 0.6 Luer (80186) or VI 0.4 (80606) in 150 or 30 μl volume of media, respectively, and cultured at 37 °C with 5% CO2. SC cells were allowed to settle for 3 to 7 days before a constant shear was applied. The computer-controlled ibidi pump system was set up for each flow (1~10 dyn/cm2) according to manufacturer’s instruction, and a fluidic flow were applied to the cells for the indicated time in each experiment. Control cells were cultured under the same conditions, but no fluidic flow was applied.
Transfection or transduction of plasmids, adenovirus and siRNA.
5HT6-mCherry plasmid [63] (kindly provided by Dr. Takanari Inoue, Johns Hopkins University School of Medicine was transfected using the Ca2+ phosphate methods described previously [31] with minor modifications. Primary human SC cells were plated at 80% confluence on 6-well plates in 2 mL of the growth media. Five micrograms of the plasmid were dissolved in 167 μl of 0.3 M CaCl2. The DNA/CaCl2 mixtures were then added dropwise to equal volume of 2xHBS (280 mM NaCl, 1.5 mM Na2HPO4, 50 mM HEPES) and mixed gently by repeated pipetting. DNA/CaCl2/HBS suspensions were immediately added to the cells in a dropwise fashion while gently swirling the plate, and cells were incubated at 37°C, 5% CO2 for 16 h. The DNA-calcium phosphate precipitates were then removed by incubating the cells in a medium pre-equilibrated in a 10% CO2 for approximately 4 h and replaced with original growth medium. The cells were incubated at 37°C, 5% CO2 for the further experiments. Adenoviruses that express tfLC3 [40], RFP-SQSTM1 [30] and GFP-LC3 [64] were transduced to SC cells using the Ca2+ phosphate method described previously [65] and above. Adenoviruses (100 pfu) were used for producing adenovirus/CaCl2 mixtures. For siRNA transfection, 1 day after SC cells (1× 105 cells/slide) were seed onto ibidi μ-slides I 0.6 Luer, a total of either 5 pmol of siRNA against SMAD2/3 (siSMAD2/3, sc-37238), AKT1 (siAKT1, sc-29195), PKD2 (siPKD2, sc-40863), PIK3C2A (siPIK3C2A, sc-61340) or 5 pmol of non-targeting siRNA (siNC, sc-37007) were transfected using Lipofectamine® RNAiMAX Reagent, according to the manufacturer’s instructions. All siRNAs were obtained from Santa Cruz Biotechnology.
Live cell time lapse imaging.
Live cell images were captured using the CELENA® X High Content Imaging System (Logos biosystems, Annandale, VA, USA), equipped with high power LED filters coupled with the ibidi perfusion system equipped with a CO2 and temperature-controlled stage top incubation system (ibidi USA, Inc., Fitchburg, WI, USA). To monitor autophagic flux and dynamics, the cells expressing tfLC3 or RFP-SQSTM1 were plated at 100% confluence onto μ-Slide VI 0.4 in 30 μl volume, incubated for additional 72 h and subjected to fluidic flow (10 dyn/cm2) for 24 h. GFP and/or RFP signal were traced with every 5 min up to 24 h. To observe PC, 5HT6-mCherry-expressing SC cells grown on μ-Slide VI 0.4 were observed directly after the media change during indicated time. To monitor the response of PC upon shear stress, the cell expressing 5HT6-mCherry were subjected to fluidic flow (1,2,5 or10 dyn/cm2 for each 10 min) using growth medium.. Fiji [66] was used for image processing.
Immunocytochemical analyses.
Immunostaining was performed as described previously [30,31] with minor modification. Briefly, SC cells grown on type IV collagen-coated flexible membranes mounted into 24well culture plate (BioFlex culture plates, Flexcell International Corp, Burlington, NC, USA) or ibidi μ-slides VI 0.4 were fixed with 4% paraformaldehyde/PBS for 15 min at room temperature. After washing three times with PBS, the cells were incubated in blocking solution (5% goat serum, 2% BSA, 0.1% Trion X100 in PBS) at room temperature for 30 min. Cells were then incubated for 1 h or overnight at room temperature or 4°C, respectively, with primary antibodies diluted in the blocking solution, washed several times with PBS, and incubated for 2 h at room temperature with Alexa Fluor 488 or 594 dye-conjugated goat anti-mouse or rabbit IgG antibodies (Invitrogen, A-11032 or A-11037) diluted 1:1000 in blocking solution without serum. The cells were subsequently washed with PBS, counterstained with DAPI (1 μg/ml) and mounted with Fluoromount-G™ onto micro cover glass (VWR, 48393 059). Images were acquired on a Nikon C2si confocal microscope (Nikon Instruments Inc., Melville, NY, USA) with a × 100 or 60/1.4 numerical aperture objective. Confocal z-stacks (0.2 μm step size) were generated containing the entire cell depth. Final images were processed with Fiji. Primary antibodies and dilutions used in this study were: anti-acetylated TUBA4A (1:2000: MilliporeSigma, T7451), anti-IFT88 (1:1000, Proteintech, 13967-1-AP), anti-pSMAD2 (S465/467)/3(S423/425), 1:500, Cell signaling Technology, 8828S). anti-ATG16L (1:1000, MBL, PM040) and anti-γ-tubulin (1:500, MilliporeSigma, T6557)
Whole cell lysate preparation and western blot analysis.
Whole cell lysates preparation and western blot analysis are performed as described previously [7,31] with minor modification. After shear stress application, the cells on the ibidi μ-slides I 0.6 Luer were washed with PBS (3 times). After complete removal of the PBS from the slide, 130~150 μl of 1x Laemmli sample buffer containing 2.5 % β-mecaptoethanol were directly added to the slide. The whole cell lysates were collected and boiled for 10 min. Same volume of the lysates was loaded. Proteins were separated by SDS-PAGE (7.5–15%) and electrotransferred to PVDF membrane. Non-specific sites were blocked with 5% non-fat dry milk in PBS-T (0.1% Tween-20) for 1 h. Membranes were then incubated with primary antibodies overnight at 4°C, washed in PBS-T (3 times), and incubated with peroxidase-conjugated donkey anti-mouse or rabbit IgG (1:5,000; Jackson ImmunoResearch Inc.,715-035-151 or -152) for 2 h at room temperature. Signal was developed using enhanced chemiluminescence substrate system. The images were captured and quantified by using ChemiDoc™ Touching Image system with Image Lab™ touch software (Bio-Rad, Hercules, CA, USA). The following primary antibodies and dilutions were used: anti-LC3B antibody (1:3,000; Cell Signaling Technology, 3868S), anti-SQSTM1 antibody (1:5,000; MilliporeSigma, P0067), anti-acetylated TUBA4A (1:2000: MilliporeSigma, T7451), anti-SMAD2/3 antibody (1:2000, Cell Signaling, 3102S), anti-phospho-SMAD2/3 antibody (S465,467/S423,425; 1:2000, Cell signaling Technology, 8828S), anti-phospho-SMAD2 antibody (S245/250/255; 1:2000, Cell signaling Technology, 3104S), anti-AKT antibody (1:2000, Cell signaling Technology, 9272S), anti-phospho-AKT antibody (S473; 1:2000, Cell signaling Technology, 4060S), anti-PKD2 antibody (1:2,000; Proteintech, 19126-1-AP), anti-PIK3C2A (1:1,000, Santa Cruz Biotechnology, SC-365290) and anti-ACTB antibody (1:1,000; Santa Cruz Biotechnology, SC-69879). Band densities were obtained with Image Lab™ software and normalized with that of ACTB.
Total RNA isolation and quantitative real-time PCR analysis.
RNA isolation and realtime PCR were performed as described previously [67,68]. In brief, total RNAs were isolated using RNeasy kit, according to the manufacturer’s protocol and RNA concentration and quality were measured by using DS-11+ spectrophotometer (DeNovix, Inc, Wilmington, DE). The total RNAs (0.5–1 μg) were used to synthesize cDNA using SuperScript® First-strand kit with oligo(dT) primer. Real-time PCRs were performed using SsoFast™ EvaGreen® Supermix in the Bio-Rad CFX 96™ system (Bio-Rad, Hercules, CA). The following PCR parameters were used: 95 °C for 5 min, followed by 40 cycles of 95 °C for 5 s, 55 °C for 5 s, and 72 °C for 5 s. Output data were obtained as Ct values by using the CFX 96™ system software and the differential mRNA expression of each gene among samples was calculated using the comparative Ct method. B2M mRNA, an internal control, was amplified along with the target genes, and the Ct value of B2M was used to normalize the expression of target genes. The sequences of the primers used for the amplifications were: LC3B (F: AGCAGCATCCAACCAAAATC, R: CTGTGTCCGTTCACCAACAG), ATG5 (F: AAAGATGTGCTTCGAGATGTGT, R: CACTTTGTCAGTTACCAACGTCA), ATG7 (F: GGATGAAGCTCCCAAGGACAT R: CCAGCAGAGTCACCATTGTAGTA), PKD2 (F: CGAGCCAAACTGAAGAGGAG, R: ACTGATCTGGGAAGCTGCAT), B2M (F: AGGCTATCCAGCGTACTCCA, R: TCAATGTCGGATGGATGAA).
Statistical analyses.
All experimental procedures were repeated at least three times in independent experiments with at least three different primary cultured SC cells. Unless otherwise stated, “n” in text and figure legends represents biological replicates including technical replication with different cells strains. Data were presented as the mean ± S.D. or interleaved box and whiskers plot with max to min and mean values. Statistical analysis was done in GraphPad Prism software using Student’s t-test between samples or, one- or two-way ANOVA with Tukey’s post hoc test among groups. p < 0.05 was considered to be statistically significant.
Supplementary Material
Figure S1. (A) Shear stress does not induce autophagy activation in human TM cells. Western blot analysis for protein expression levels of LC3 and SQSTM1 in human TM cells subjected to shear stress (10 dyn/cm2) for 1 or 24 h. Band densities were quantified by Image Lab™ touch software, normalized with ACTB and fold changes calculated and graphed. Data are shown as the mean ± S.D. (n=4). *, p<0.05; ns, not significant (Student’s t-test). CNT: control (0 dyn/cm2). (B) Autophagy flux analysis in SC cells subjected to shear stress. Human SC cells were subjected to fluid flow (10 dyn/cm2) for 24 h, the last two hours under the presence of bafilomycin (BafA1, 10 nM). LC3-II levels were evaluated by western blot analysis. Band densities were quantified by Image Lab™ touch software, normalized with ACTB and fold changes calculated and graphed.
Figure S2. Chemical disruption of PC in SC cells. SC cells were treated with different concentrations of CH (0–8 mM) for 3 days and then stained with acetylated TUBA4A (red fluorescence) antibody to detect PC. DAPI was used to stain nuclei. Transmission and fluorescent images were acquired with CELENA X and confocal microscope, respectively, and processed by Fiji software. Note that PC were still detected in SC cells treated with 1 mM CH. A 2mM CH was chosen for subsequent experiments, as that concentration efficiently removes PC without compromising cell morphology or viability.
Figure S3. PKD2-mediated calcium signaling in PC does not mediates shear stress-induced autophagy in SC cells. (A) Effect of fluidic flow on PKD2 mRNA expression. Data are shown as interleaved box and whiskers plot (n =3). *, p<0.05 (One-way ANOVA with Tukey’s post hoc test). (B) Effect of silencing PKD2 on LC3-II levels. ACTB was used as a loading control. Graphs shows fold changes in LC3-II levels calculated from the densitometric analysis of the bands. Data are shown as the mean ± S.D (n=3). ns, not significant (Student’s t-test).
Figure S4. PIK3C2A does not regulate shear stress-induced autophagy in SC cells. PIK3C2A expression levels upon shear stress and effect of PIK3C2A knockdown on LC3 II levels. ACTB was used for normalization. Data are shown as the mean ± S.D (n=3). *, p<0.05; ns, not significant (Student’s t-test).
Figure S5. SMAD2/3 and AKT1 does not affect MTOR activation. (A-C) Protein levels of pp70S6K and p70S6K were evaluated by WB in SMAD2/3 (A) or AKT1 (B) knocked-down SC cells in the absence or presence of shear stress (10 dyn/cm2) for 24 h, and fold changes were calculated and represented at each section (C). Data are shown as the mean ± S.D. (n=3). *, p<0.05; ns, not significant (Student’s t-test).
Video S1. Realtime time-lapse imaging of tfLC3 in human SC cells.
Video S2. Realtime time-lapse imaging of human SC cells expressing RFP-SQSTM1 demonstrating that degradation of RFP-SQSTM1 aggregates by autophagosome upon shear stress in human SC cells.
Video S3. Realtime time-lapse imaging demonstrating that autophagy vacuole like structure is autophagic vacuoles which contain. GFP-LC3 and RFP-SQSTM1
Video S4. PC dynamics in human SC cells.
Video S5. Cell communication via PC dynamics in human SC cells.
Video S6. Reorganization of PC membrane is a mechanotransduction mechanism against shear stress in SC cells.
Video S7. PI-103 treatment activates autophagy in SC cells.
Acknowledgments
We thank Dr. Takanari Inoue at Johns Hopkins University School of Medicine for kindly providing 5HT6-mCherry plasmid; Dr. Xiao-Ming Yin at Indiana University and Dr. Wen-Xing Ding at University of Kansas Medical Center for permission to use and providing AdGFP-LC3, respectively.
Funding
Funding was provided by National Institute of Health, Eye Institute (EY026885, EY027733, EY005722, EY022359, EY028608), Glaucoma Research Foundation (2022 Shaffer Grant), BrightFocus Foundation (G2022010S) and Unrestricted Research to Prevent Blindness Grant.
Abbreviation:
- ACTB
actin beta
- AKT1/PKB
AKT serine/threonine kinase 1
- ATG
autophagy-related
- BSA
bovine serum albumin
- B2M
beta-2 microgloblin
- CH
chloral hydrate
- CMS
cyclic mechanical stretch
- CNT
control
- DAPI
4′,6-diamidino-2-phenylindole
- DC
deciliated
- ECs
endothelial cells
- ECM
extracellular matrix
- GFP
green fluorescent protein
- HBSS
Hanks balanced salt solution
- HUVECs
human umbilical vein endothelial cells
- IFT
intraflagellar transport
- IOP
intraocular pressure
- KTECs
kidney tubular and epithelial cells
- MAP1LC3/LC3
microtubule associated protein 1 light chain 3
- NO
nitric oxide
- NOS3/eNOS
nitric oxide synthase 3
- PBS
phosphate-buffered saline
- PC
primary cilium (or cilia)
- PI3K
phosphatidylinositol 3-kinase
- PIK3C2A
phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 alpha
- PIP
phosphatidylinositol-phosphate
- PKD2
polycystin 2 transient receptor cation channel
- pSMAD2L
phosphorylated SMAD2 at linker region
- pSMAD2/3C
phosphorylated SMAD2/3 at c-terminal regions
- PVDF
polyvinylidene fluoride
- RFP
red fluorescent protein
- siCNT
Scrambled siRNA used as control
- SC
Schlemm’s canal
- SMAD
SMAD family member
- SQSTM1/p62
sequestosome 1
- TGF
transforming growth factor
- TM
trabecular meshwork
- tfLC3
tandem fluorescence LC3
- TUBA4A
tubulin alpha 4a
- WB
western blot
Footnotes
Disclosure statement
We declare no conflict of interest, and we have no manuscripts in the press or in the submission containing the research presented here.
References
- 1.Kizhatil K, Ryan M, Marchant JK, et al. Schlemm’s canal is a unique vessel with a combination of blood vascular and lymphatic phenotypes that forms by a novel developmental process. PLoS Biol. 2014. Jul;12(7):e1001912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamm ER, Braunger BM, Fuchshofer R. Intraocular Pressure and the Mechanisms Involved in Resistance of the Aqueous Humor Flow in the Trabecular Meshwork Outflow Pathways. Prog Mol Biol Transl Sci. 2015;134:301–14. [DOI] [PubMed] [Google Scholar]
- 3.Carreon T, van der Merwe E, Fellman RL, et al. Aqueous outflow - A continuum from trabecular meshwork to episcleral veins. Prog Retin Eye Res. 2017. Mar;57:108–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stamer WD, Braakman ST, Zhou EH, et al. Biomechanics of Schlemm’s canal endothelium and intraocular pressure reduction. Prog Retin Eye Res. 2015. Jan;44:86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ethier CR, Read AT, Chan D. Biomechanics of Schlemm’s canal endothelial cells: influence on F-actin architecture. Biophys J. 2004. Oct;87(4):2828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashpole NE, Overby DR, Ethier CR, et al. Shear stress-triggered nitric oxide release from Schlemm’s canal cells. Invest Ophthalmol Vis Sci. 2014. Nov 13;55(12):8067–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDonnell F, Perkumas KM, Ashpole NE, et al. Shear Stress in Schlemm’s Canal as a Sensor of Intraocular Pressure. Sci Rep. 2020. Apr 2;10(1):5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reina-Torres E, De Ieso ML, Pasquale LR, et al. The vital role for nitric oxide in intraocular pressure homeostasis. Prog Retin Eye Res. 2021. Jul;83:100922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan Z, Guo D, Tao R, et al. Fluid shear stress induces cell migration via RhoA-YAP1-autophagy pathway in liver cancer stem cells. Cell Adh Migr. 2022. Dec;16(1):94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng Q, Pu L, Qi M, et al. Laminar shear stress inhibits inflammation by activating autophagy in human aortic endothelial cells through HMGB1 nuclear translocation. Commun Biol. 2022. May 6;5(1):425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das J, Maiti TK. Fluid shear stress influences invasiveness of HeLa cells through the induction of autophagy. Clin Exp Metastasis. 2022. Jun;39(3):495–504. [DOI] [PubMed] [Google Scholar]
- 12.Chen S, Qin L, Wu X, et al. Moderate Fluid Shear Stress Regulates Heme Oxygenase-1 Expression to Promote Autophagy and ECM Homeostasis in the Nucleus Pulposus Cells. Front Cell Dev Biol. 2020;8:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan Z, Su G, Gao W, et al. Fluid shear stress induces cell migration and invasion via activating autophagy in HepG2 cells. Cell Adh Migr. 2019. Dec;13(1):152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazari MM, Orhon I, Codogno P, et al. Monitoring of Autophagy and Cell Volume Regulation in Kidney Epithelial Cells in Response to Fluid Shear Stress. Methods Mol Biol. 2019;1880:331–340. [DOI] [PubMed] [Google Scholar]
- 15.Zhang JX, Qu XL, Chu P, et al. Low shear stress induces vascular eNOS uncoupling via autophagy-mediated eNOS phosphorylation. Biochim Biophys Acta Mol Cell Res. 2018. May;1865(5):709–720. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Zhang Y, Feng T, et al. Fluid Shear Stress Promotes Autophagy in Hepatocellular Carcinoma Cells. Int J Biol Sci. 2018;14(10):1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun L, Zhao M, Liu A, et al. Shear Stress Induces Phenotypic Modulation of Vascular Smooth Muscle Cells via AMPK/mTOR/ULK1-Mediated Autophagy. Cell Mol Neurobiol. 2018. Mar;38(2):541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bharath LP, Cho JM, Park SK, et al. Endothelial Cell Autophagy Maintains Shear Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler Thromb Vasc Biol. 2017. Sep;37(9):1646–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao P, Zhao H, Mo W, et al. Laminar Shear Stress Promotes Vascular Endothelial Cell Autophagy Through Upregulation with Rab4. DNA Cell Biol. 2016. Mar;35(3):118–23. [DOI] [PubMed] [Google Scholar]
- 20.Yang Q, Li X, Li R, et al. Low Shear Stress Inhibited Endothelial Cell Autophagy Through TET2 Downregulation. Ann Biomed Eng. 2016. Jul;44(7):2218–27. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Bi X, Chen T, et al. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015. Jul 16;6(7):e1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding Z, Liu S, Deng X, et al. Hemodynamic shear stress modulates endothelial cell autophagy: Role of LOX-1. Int J Cardiol. 2015. Apr 1;184:86–95. [DOI] [PubMed] [Google Scholar]
- 23.Guo F, Li X, Peng J, et al. Autophagy regulates vascular endothelial cell eNOS and ET-1 expression induced by laminar shear stress in an ex vivo perfused system. Ann Biomed Eng. 2014. Sep;42(9):1978–88. [DOI] [PubMed] [Google Scholar]
- 24.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015. Aug;16(8):461–72. [DOI] [PubMed] [Google Scholar]
- 25.Liton PB. The autophagic lysosomal system in outflow pathway physiology and pathophysiology. Exp Eye Res. 2016. Mar;144:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bharath LP, Mueller R, Li Y, et al. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can J Physiol Pharmacol. 2014. Jul;92(7):605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu H, Fan Y, Qiao C, et al. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci Signal. 2017. Jan 31;10(464). [DOI] [PubMed] [Google Scholar]
- 28.Porter KM, Jeyabalan N, Liton PB. MTOR-independent induction of autophagy in trabecular meshwork cells subjected to biaxial stretch. Biochim Biophys Acta. 2014. Jun;1843(6):1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirt J, Liton PB. Autophagy and mechanotransduction in outflow pathway cells. Exp Eye Res. 2017. May;158:146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shim MS, Nettesheim A, Hirt J, et al. The autophagic protein LC3 translocates to the nucleus and localizes in the nucleolus associated to NUFIP1 in response to cyclic mechanical stress. Autophagy. 2020. Jul;16(7):1248–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shim MS, Nettesheim A, Dixon A, et al. Primary cilia and the reciprocal activation of AKT and SMAD2/3 regulate stretch-induced autophagy in trabecular meshwork cells. Proc Natl Acad Sci U S A. 2021. Mar 30;118(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orhon I, Dupont N, Zaidan M, et al. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat Cell Biol. 2016. Jun;18(6):657–67. [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Lopez N, Singh R. Shear stress turns on the primary cilium and lipophagy. Nat Cell Biol. 2020. Sep;22(9):1029–1030. [DOI] [PubMed] [Google Scholar]
- 34.Miceli C, Roccio F, Penalva-Mousset L, et al. Fluid flow-induced shear stress controls the metabolism of proximal tubule kidney epithelial cells through primary cilium-dependent lipophagy and mitochondria biogenesis. Autophagy. 2020. Dec;16(12):2287–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Praetorius HA. The primary cilium as sensor of fluid flow: new building blocks to the model. A review in the theme: cell signaling: proteins, pathways and mechanisms. Am J Physiol Cell Physiol. 2015. Feb 1;308(3):C198–208. [DOI] [PubMed] [Google Scholar]
- 36.Nauli SM, Jin X, AbouAlaiwi WA, et al. Non-motile primary cilia as fluid shear stress mechanosensors. Methods Enzymol. 2013;525:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003. Feb;33(2):129–37. [DOI] [PubMed] [Google Scholar]
- 38.Boukhalfa A, Nascimbeni AC, Ramel D, et al. PI3KC2alpha-dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat Commun. 2020. Jan 15;11(1):294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porter K, Hirt J, Stamer WD, et al. Autophagic dysregulation in glaucomatous trabecular meshwork cells. Biochim Biophys Acta. 2015. Mar;1852(3):379–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porter K, Nallathambi J, Lin Y, et al. Lysosomal basification and decreased autophagic flux in oxidatively stressed trabecular meshwork cells: implications for glaucoma pathogenesis. Autophagy. 2013. Apr;9(4):581–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pulliero A, Seydel A, Camoirano A, et al. Oxidative damage and autophagy in the human trabecular meshwork as related with ageing. PLoS One. 2014;9(6):e98106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007. Aug 17;282(33):24131–45. [DOI] [PubMed] [Google Scholar]
- 43.Toro-Tapia G, Das RM. Primary cilium remodeling mediates a cell signaling switch in differentiating neurons. Sci Adv. 2020. May;6(21):eabb0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higginbotham H, Guo J, Yokota Y, et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nature Neuroscience. 2013;16(8):1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ávalos Y, Peña-Oyarzun D, Budini M, et al. New Roles of the Primary Cilium in Autophagy. BioMed Research International. 2017;2017:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orhon I, Dupont N, Codogno P. Primary cilium and autophagy: The avengers of cell-size regulation. Autophagy. 2016. Nov;12(11):2258–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dimmeler S, Fleming I, Fisslthaler B, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999. Jun 10;399(6736):601–5. [DOI] [PubMed] [Google Scholar]
- 48.Deng H, Min E, Baeyens N, et al. Activation of Smad2/3 signaling by low fluid shear stress mediates artery inward remodeling. Proc Natl Acad Sci U S A. 2021. Sep 14;118(37). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamato D, Do BH, Osman N, et al. Smad linker region phosphorylation is a signalling pathway in its own right and not only a modulator of canonical TGF-beta signalling. Cell Mol Life Sci. 2020. Jan;77(2):243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shepherd RD, Kos SM, Rinker KD. Flow-dependent Smad2 phosphorylation and TGIF nuclear localization in human aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2011. Jul;301(1):H98–h107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan QW, Knight ZA, Goldenberg DD, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006. May;9(5):341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim SW, Ehrman J, Ahn MR, et al. Shear stress induces noncanonical autophagy in intestinal epithelial monolayers. Mol Biol Cell. 2017. Nov 1;28(22):3043–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zemirli N, Boukhalfa A, Dupont N, et al. The primary cilium protein folliculin is part of the autophagy signaling pathway to regulate epithelial cell size in response to fluid flow. Cell Stress. 2019. Feb 25;3(3):100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Claude-Taupin A, Codogno P, Dupont N. Links between autophagy and tissue mechanics. J Cell Sci. 2021. Sep 1;134(17). [DOI] [PubMed] [Google Scholar]
- 55.Boukhalfa A, Roccio F, Dupont N, et al. The autophagy protein ATG16L1 cooperates with IFT20 and INPP5E to regulate the turnover of phosphoinositides at the primary cilium. Cell Rep. 2021. Apr 27;35(4):109045. [DOI] [PubMed] [Google Scholar]
- 56.Ott C, Elia N, Jeong SY, et al. Primary cilia utilize glycoprotein-dependent adhesion mechanisms to stabilize long-lasting cilia-cilia contacts. Cilia. 2012. Apr 25;1(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005. Dec 1;19(23):2783–810. [DOI] [PubMed] [Google Scholar]
- 58.Wordinger RJ, Sharma T, Clark AF. The role of TGF-beta2 and bone morphogenetic proteins in the trabecular meshwork and glaucoma. J Ocul Pharmacol Ther. 2014. Mar-Apr;30(2–3):154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kretzschmar M, Doody J, Timokhina I, et al. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. 1999. Apr 1;13(7):804–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu JSL, Ramasamy TS, Murphy N, et al. PI3K/mTORC2 regulates TGF-β/Activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nature Communications. 2015;6(1):7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stamer WD, Roberts BC, Howell DN, et al. Isolation, culture, and characterization of endothelial cells from Schlemm’s canal. Invest Ophthalmol Vis Sci. 1998. Sep;39(10):1804–12. [PubMed] [Google Scholar]
- 62.Perkumas KM, Stamer WD. Protein markers and differentiation in culture for Schlemm’s canal endothelial cells. Exp Eye Res. 2012. Mar;96(1):82–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hong SR, Wang CL, Huang YS, et al. Spatiotemporal manipulation of ciliary glutamylation reveals its roles in intraciliary trafficking and Hedgehog signaling. Nat Commun. 2018. Apr 30;9(1):1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ni HM, Bockus A, Wozniak AL, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011. Feb;7(2):188–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fasbender A, Lee JH, Walters RW, et al. Incorporation of adenovirus in calcium phosphate precipitates enhances gene transfer to airway epithelia in vitro and in vivo. J Clin Invest. 1998. Jul 1;102(1):184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012. Jun 28;9(7):676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shim MS, Kim KY, Bu JH, et al. Elevated intracellular cAMP exacerbates vulnerability to oxidative stress in optic nerve head astrocytes. Cell Death Dis. 2018. Feb 19;9(3):285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nettesheim A, Shim MS, Dixon A, et al. Cathepsin B Localizes in the Caveolae and Participates in the Proteolytic Cascade in Trabecular Meshwork Cells. Potential New Drug Target for the Treatment of Glaucoma. Journal of Clinical Medicine. 2020;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) Shear stress does not induce autophagy activation in human TM cells. Western blot analysis for protein expression levels of LC3 and SQSTM1 in human TM cells subjected to shear stress (10 dyn/cm2) for 1 or 24 h. Band densities were quantified by Image Lab™ touch software, normalized with ACTB and fold changes calculated and graphed. Data are shown as the mean ± S.D. (n=4). *, p<0.05; ns, not significant (Student’s t-test). CNT: control (0 dyn/cm2). (B) Autophagy flux analysis in SC cells subjected to shear stress. Human SC cells were subjected to fluid flow (10 dyn/cm2) for 24 h, the last two hours under the presence of bafilomycin (BafA1, 10 nM). LC3-II levels were evaluated by western blot analysis. Band densities were quantified by Image Lab™ touch software, normalized with ACTB and fold changes calculated and graphed.
Figure S2. Chemical disruption of PC in SC cells. SC cells were treated with different concentrations of CH (0–8 mM) for 3 days and then stained with acetylated TUBA4A (red fluorescence) antibody to detect PC. DAPI was used to stain nuclei. Transmission and fluorescent images were acquired with CELENA X and confocal microscope, respectively, and processed by Fiji software. Note that PC were still detected in SC cells treated with 1 mM CH. A 2mM CH was chosen for subsequent experiments, as that concentration efficiently removes PC without compromising cell morphology or viability.
Figure S3. PKD2-mediated calcium signaling in PC does not mediates shear stress-induced autophagy in SC cells. (A) Effect of fluidic flow on PKD2 mRNA expression. Data are shown as interleaved box and whiskers plot (n =3). *, p<0.05 (One-way ANOVA with Tukey’s post hoc test). (B) Effect of silencing PKD2 on LC3-II levels. ACTB was used as a loading control. Graphs shows fold changes in LC3-II levels calculated from the densitometric analysis of the bands. Data are shown as the mean ± S.D (n=3). ns, not significant (Student’s t-test).
Figure S4. PIK3C2A does not regulate shear stress-induced autophagy in SC cells. PIK3C2A expression levels upon shear stress and effect of PIK3C2A knockdown on LC3 II levels. ACTB was used for normalization. Data are shown as the mean ± S.D (n=3). *, p<0.05; ns, not significant (Student’s t-test).
Figure S5. SMAD2/3 and AKT1 does not affect MTOR activation. (A-C) Protein levels of pp70S6K and p70S6K were evaluated by WB in SMAD2/3 (A) or AKT1 (B) knocked-down SC cells in the absence or presence of shear stress (10 dyn/cm2) for 24 h, and fold changes were calculated and represented at each section (C). Data are shown as the mean ± S.D. (n=3). *, p<0.05; ns, not significant (Student’s t-test).
Video S1. Realtime time-lapse imaging of tfLC3 in human SC cells.
Video S2. Realtime time-lapse imaging of human SC cells expressing RFP-SQSTM1 demonstrating that degradation of RFP-SQSTM1 aggregates by autophagosome upon shear stress in human SC cells.
Video S3. Realtime time-lapse imaging demonstrating that autophagy vacuole like structure is autophagic vacuoles which contain. GFP-LC3 and RFP-SQSTM1
Video S4. PC dynamics in human SC cells.
Video S5. Cell communication via PC dynamics in human SC cells.
Video S6. Reorganization of PC membrane is a mechanotransduction mechanism against shear stress in SC cells.
Video S7. PI-103 treatment activates autophagy in SC cells.