

ABSTRACT
Antibody-cytokine fusions targeted against tumor-associated antigens (TAAs) are promising cancer immunotherapy agents, with many such molecules currently undergoing clinical trials. However, due to the limited number of tumor-specific targets, on-target off-tumor effects can lead to systemic toxicity. Additionally, targeted cytokines can be scavenged by cytokine receptors on peripheral cells, decreasing tumor penetration. This study aims at overcoming these issues by engineering a platform for targeted conditionally active type I cytokines. Building on our previously reported PACE (Prodrug-Activating Chain Exchange) platform, we split the type I cytokine interleukin-4 (IL-4) to create two inactive IL-4 prodrugs, and fused these split IL-4 counterparts to the C-termini of antibody-like molecules that undergo proximity-induced chain exchange. In doing so, we developed IL-4 prodrugs that preferentially reconstitute into active IL-4 on target cells. We demonstrate that pre-assembled split IL-4 (without additional inactivation) retains activity and present two different strategies of splitting and inactivating IL-4. Using an IL-4 responsive cell-line, we show that IL-4 prodrugs are targeted to TAAs on target cells and regain activity upon chain exchange, primarily in a cis-activation setting. Furthermore, we demonstrate that split IL-4 complementation is also possible in a trans-activation setting, which opens up the possibility for activation of immune cells in the tumor vicinity. We demonstrate that targeted on-cell prodrug conversion is more efficient than nonspecific activation in-solution. Due to the structural similarity between IL-4 and other type I cytokines relevant in cancer immunotherapy such as IL-2, IL-15, and IL-21, cytokine-PACE may be expanded to develop a variety of targeted conditionally active cytokines for cancer immunotherapy.
KEYWORDS: antibody engineering, antibody-cytokine fusions, cancer immunotherapy, conditional activation
Introduction
Cytokines are small proteins that regulate the survival, proliferation, differentiation, and effector functions of immune cells. They act as short-distance paracrine and autocrine signaling molecules, achieving their effects by binding to specific receptors on target cells.1 Cytokines are classified into families based on shared structural features. The four-helix bundle family binds to class I cytokine receptors and consists of, among others, the cytokines interleukin (IL)-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-11, IL-13, IL-15, IL-21, leukemia inhibitory factor (LIF), oncostatin-M (OSM), cardiotrophin-1 (CT-1), ciliary neurotrophic factor (CNTF), and granulocyte-macrophage-colony-stimulating factor (GM-CSF).2 These cytokines are also referred to as type I cytokines.3
Cytokines have been used for the treatment of rheumatoid arthritis, asthma, multiple sclerosis, viral infections, and various forms of cancer.4 The use of cytokines in cancer immunotherapy (CIT) has emerged as a particularly promising approach. Of particular relevance are the cytokines IL-2, IL-12, IL-15, and IL-21, which are under investigation in ongoing clinical trials.5,6 The US Food and Drug Administration has approved two cytokines for cancer therapy – IL-2 for the treatment of metastatic melanoma and renal cell carcinoma and IFN-α for the treatment of hairy cell leukemia, melanoma, follicular non-Hodgkin lymphoma, and AIDS-related Kaposi’s sarcoma.5,6 Due to their rapid clearance, cytokines need to be administered at high doses, which can often be accompanied by undesired side effects including hypotension, capillary leak syndrome, nausea and vomiting, neutropenia, leukopenia, and depression.5 To overcome the drawbacks of systemic high-dose cytokine CIT, a targeted approach is critical. Targeting can be accomplished by fusing the cytokine to an antibody or antibody-derived molecule directed against a tumor-associated antigen (TAA), leading to its specific accumulation in the tumor microenvironment. To date, many antibody-cytokine fusions have entered clinical trials and show promising results, particularly those containing IL-2, IL-12, and tumor necrosis factor.7 However, while TAAs can be overexpressed in tumors, they are often also expressed at lower levels in healthy tissue, which can lead to on-target off-tumor toxicity.8 Furthermore, antibody-cytokine fusions can be scavenged by cytokine receptors found on circulating immune cells or in normal tissues, reducing their tumor penetration.7,9
We recently developed PACE (Prodrug-Activating Chain Exchange) as a method that enables the assembly of active entities from antibody prodrugs on target cells.10 Chain exchange is driven by opposing charges introduced into the CH3 domains.11 When the prodrugs are in close proximity, such as via targeted accumulation on the cell surface, spontaneous heavy chain exchange results in the reconstitution of a TriFab with three binding sites. After chain exchange, the “dummy” chains dissociate from the cell, as they contain no relevant binding entity (Figure S1). The PACE technique is designed to overcome on-target, off-tumor effects by initiating prodrug conversion only when the TAA concentration on the cell surface reaches a critical concentration.10
In this study, we show that chain exchange driven by inter-chain interface modulation, the underlying principle of PACE, can be expanded to cover not only prodrug-antibodies, but also prodrug-cytokines. In this manner, we aimed to address on-target, off-tumor liabilities of antibody-cytokine fusions. To provide proof-of-concept for the cytokine-PACE approach, we generated a targeted conditionally active IL-4. To do so, we split this type I cytokine into two inactive prodrugs and fused them to antibody-like PACE entities, which preferentially recombine to generate active IL-4 upon accumulation on target cells. The split cytokine approach has previously been applied to increase the therapeutic window and efficacy of IL-12 and IL-2.12,13 This approach is especially well suited for PACE because it relies on the target density of TAAs for discrimination of tumor cells over healthy cells, in contrast to other environment-dependent conditional activation approaches such as cleavage of a masking domain by tumor-specific proteases,14–17 targeted accumulation of attenuated cytokines,18–20 and pH-dependent cytokine engineering21,22. Our cytokine-PACE platform has the potential to be applied to additional relevant cytokines in immunotherapy approaches.
Results
Split IL-4 antibody fusions
To create a generalized platform for conditionally active targeted cytokines, we chose IL-4 as a model type I cytokine. IL-4 promotes T-cell differentiation to the Th2 lineage, B-cell isotype switching to IgE, B-cell antibody production, and proliferation of eosinophils and basophils.23 IL-4 dysregulation has been linked to asthma and atopic dermatitis, and blocking IL-4 signaling has shown success in treating these conditions.24,25 Importantly, IL-4 shares a similar structure to IL-2, IL-15, and IL-21, which are promising candidates of CIT.5,6
Human IL-4 consists of four alpha helices (denoted ABCD, Figure 1) connected in an “up-up-down-down” manner.26,27 To create IL-4 cytokine-PACE prodrugs, we designed two split versions of IL-4. For the 3 + 1 split approach, we first removed the disulfide bridge between helices A and D by introducing C3S and C127S mutations. We then artificially split IL-4 directly downstream of helix A. The N-terminal portion of 3 + 1 split IL-4 (denoted as helix A) contains residues 1–21 while the C-terminal portion (helices BCD) contains residues 22–129 (Figure 1a). In the 2 + 2 split approach, we circularly permutated IL-4 in a similar manner as previously described,28 by connecting the C- and N-termini of IL-4 with a flexible 7-residue Gly-Ser linker. We set the new N- and C-termini at V101 and P100, respectively (denoted as DABC, Figure 1b). One split IL-4 reactant (DA) consists of amino acids 101–129 and amino acids 1–21 in WT IL-4, connected by a 7 amino acid Gly-Ser linker. The other split IL-4 pair (BC) consists of amino acids 22–100.
Figure 1.
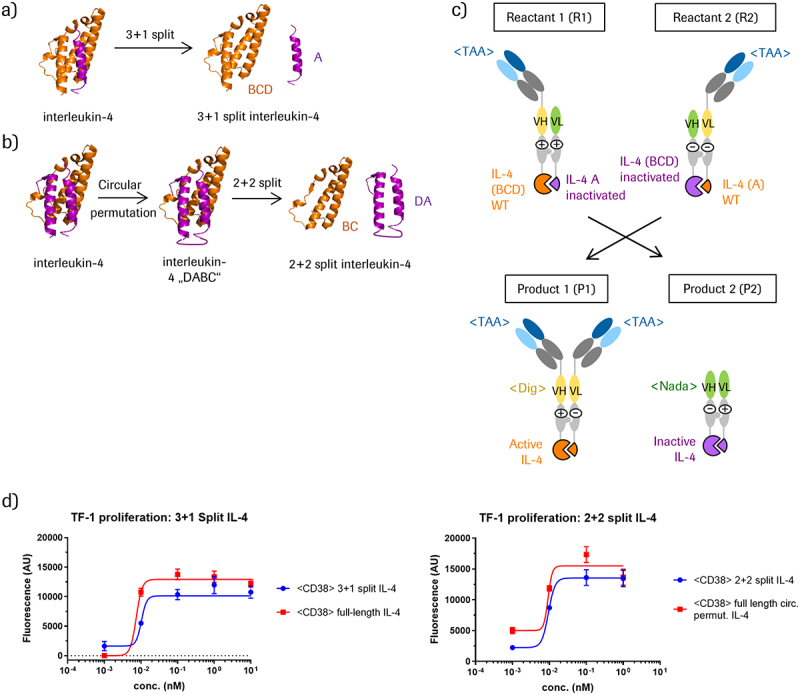
Design of split IL-4 modules and cytokine-PACE formats.
(a) Separation of helix A from the helices BCD generates 3+1 split IL-4 components. (b) 2+2 split IL-4 components are derived from a circular permutated IL-4 in the DABC composition that are separated between A and B to result in DA and BC fragments.(c) Cytokine-PACE-mediated conversion of targeted 3+1 split IL-4 prodrugs into an active IL-4-containing TriFab. Reconstitution of the digoxigenin binder <Dig> in the TriFab stem region can be utilized to monitor CH3-triggered exchange reactions independent of IL-4 activity assays. The Nada VH/VL domains serve as non-binding “dummy” VH/VL domains, which stabilize the split TriFab. (d) Split IL-4 fusions retain activity compared to full-length IL-4 and circular permutated IL-4 fusions. Activity was assessed by IL-4-dependent proliferation of TF-1 cells treated with CD38-targeting TriFabs fused with either full-length IL-4, circular permutated IL-4, pre-assembled 3+1 split IL-4, or pre-assembled 2+2 split IL-4.
Split entities lacking corresponding counterparts may contain exposed hydrophobic interfaces, which generate biophysical challenges such as expression problems, aggregation, and instability.29 To avoid these problems, and to show that split IL-4 entities retain activity, we fused complementary split IL-4 portions with a flexible Gly-Ser linker to the C-termini of antibody-like TriFabs (Figure 1c, molecule P1).30,31 These molecules were produced in transiently transfected HEK293 cells and were purified to homogeneity from cell culture supernatants (see Figure S2 for protein analytics). Expression yields of the pre-assembled split IL-4 TriFab fusions ranged between 1–5 mg/L for the 3 + 1 splits and 5–20 mg/L for the 2 + 2 splits. The chain composition was correct and the molecules were mostly monomeric, with a low percentage of aggregates (Figure S2). Furthermore, they had favorable thermal stability profiles (Table S2). We next tested whether the activity of the pre-assembled split IL-4 variants containing complementary parts fused to the two CH3 domains (A+BCD or DA+BC) is preserved. Accordingly, we measured proliferation of IL-4-responsive TF-1 cells in the presence of educts containing 3 + 1 split IL-4, 2 + 2 split IL-4, or intact IL-4. Figure 1d shows that split IL-4 variants retained signaling activity, with activities being similar to that of the non-split, full-length IL-4. Thus, split IL-4, when fused via a flexible linker to the CH3 domain, assembles into the correct structure, resulting in binding and activation of the IL-4 receptor (see Figure S3 for binding data).
Antibody fusions harboring split IL-4 prodrugs
The PACE reaction generates active TriFab entities from inactive prodrugs by chain exchange (Figure S1).10 To expand the antibody prodrug approach toward cytokine prodrugs, the first step was to inactivate the cytokine signaling capability of each of the split IL-4 educts. To enable reconstitution of active IL-4 via chain exchange, inactivation of the two complementary educts needed to be achieved by mutating one or the other side of the split IL-4 entities (Figure 1c). To generate such inactive split entities (termed dummy chains), we directed our efforts toward abrogating the interaction of IL-4 with the IL-4 receptor α chain (IL-4 Rα). IL-4 binds to IL-4 Rα with a high affinity, while its affinity for its other receptor chain, the common γ-chain (γc), is relatively low.32,33 Previous structural and biochemical studies of the IL-4/IL-4 Rα interface identified helices A and C in IL-4 as the main interaction sites with IL-4 Rα (Figure 2a).32–35 Both 3 + 1 and 2 + 2 split IL-4 contain helix A in one reactant and helix C in the other. Guided by rational design based on the crystal structure of the IL-4 ternary complex,34 we generated cytokine-PACE constructs (Figure 1c, Figure S2) and screened them for mutations in helices A and C that inactivate IL-4 signaling. We identified mutations in helix A (T6D E9A) and in helix C (R81E R88Q) that completely abrogate IL-4 activity, as measured by proliferation of IL-4-responsive TF-1 cells (Figure 2b).36 Analysis of binding kinetics by surface plasmon resonance (SPR) showed no detectable binding of reactants containing T6D E9A or R81E R88Q mutations to IL-4 Rα (Figure S3, Table S3). Split IL-4 reactants without inactivating mutations bound to IL-4 Rα, albeit with a lower affinity than TriFabs that contain full-length (non-split) IL-4 (Figure S3). We therefore placed these mutations into the dummy chain of each reactant, thereby creating IL-4 prodrug antibody fusions. Expression yields of the split IL-4 prodrug educts ranged from 0.5–12 mg/L for 3 + 1 splits and 1–10 mg/L for 2 + 2 splits. The 2 + 2 split IL-4 educts showed correct chain composition and were primarily monomeric, with a minimal percentage of aggregates (Figure S2). The majority of 3 + 1 split IL-4 educts also showed correct chain composition, were mainly monomeric, and had a low percentage of aggregates, with the exception of one HER2-targeted construct, which had a higher percentage of aggregates (Figure S2). Thermal stability profiles of most split IL-4 constructs were favorable, with constructs containing the inactivated IL-4 (A) dummy helix showing lower thermal stability (Table S2).
Figure 2.
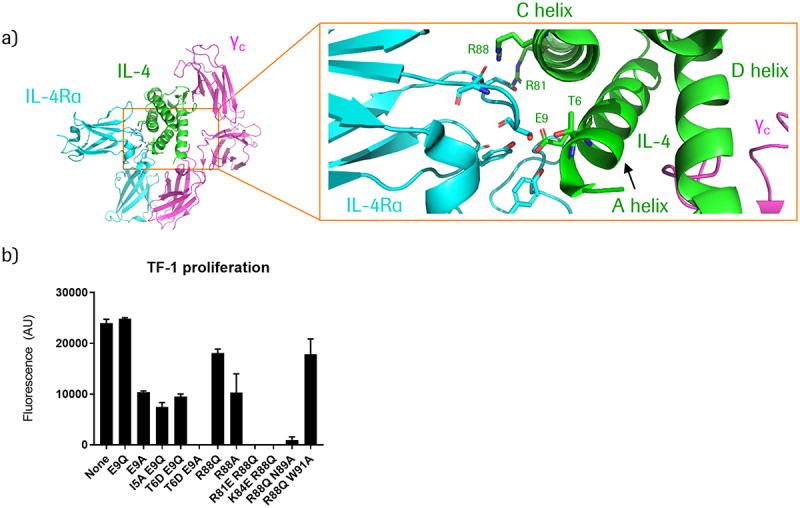
Inactivation of targeted split IL-4 generates prodrug modules.
(a) Structural depiction of the IL-4/IL-4Rα/common gamma chain (γC) ternary complex, PDB ID 3BPN.34 Key residues in IL-4 that interact with IL-4Rα are indicated in green. (b) Effect of IL-4 mutations (generated in the context of CD38-targeting 3+1 split IL-4 PACE educts) on IL-4 signaling activity, as measured by proliferation of TF-1 cells treated with 100 nM of the respective molecules. TF-1 cells express CD38 – see following section for more details.
Chain-exchange-mediated cis-activation of CD38-targeted split IL-4 prodrugs
In the PACE reaction, reactants undergo spontaneous chain exchange when located in close spatial proximity, such as when bound to the cell surface.10 In a similar manner, cytokine-PACE should lead to accumulation of reactants on target cells, which would lead to chain exchange and reconstitution of active IL-4. To investigate the cis-activation of split IL-4 prodrugs on the surface of target-expressing cells (IL-4 receptor activation on the targeted cell), we generated prodrugs with Fab arms directed against CD38, a transmembrane glycoprotein upregulated in multiple myeloma.37,38 We used the IL-4 responsive TF-1 cell line as the target cell line and monitored its proliferation as a readout for IL-4 signaling. The TF-1 cell line is an erythroleukemia-derived cell line that expresses a high level of CD38 (approximately 30,000 molecules per cell, see Fig. S6, Table S6 for quantification) and proliferates in the presence of IL-4.36 As a non-binding control, we generated prodrugs directed against HER2, which is not expressed on TF-1 cells (Figure S6). We added the split IL-4 prodrugs to TF-1 cells and monitored proliferation as a readout of IL-4 signaling. The results of these assays are shown in Figure 3 and reveal that treatment of TF-1 cells with CD38-targeted complementary split IL-4 prodrugs induces proliferation, indicative of IL-4 receptor engagement (Figure 3a, d). We then asked whether chain exchange of prodrugs into active IL-4 occurs in a targeted manner preferentially on the surface of target cells, or also in-solution. We hypothesized that proximity-induced chain exchange occurs between the heavy chains of the prodrugs upon accumulation on tumor cells, resulting in IL-4 activation. Since chain-exchange-mediated activation and IL-4 receptor signaling of the product occurs on the same cell, this mode of action represents cis-activation. The exchange and ability to cis-activate is dependent on antigen density, accessibility, and possibly also geometry. Ideally, activation should occur predominately on target cells and not in-solution. Consequently, we compared the addition of CD38-targeting prodrugs with non-targeted (HER2) prodrugs to TF-1 cells, and assessed IL-4 activity. We observed that the addition of either prodrug alone does not lead to TF-1 proliferation, confirming that the individual prodrugs are indeed inactive (Figure 3a, b, d, e). Inactivity of individual targeted prodrugs was observed for all 2 + 2 as well as 3 + 1 split IL-4 formats, except for one HER2-targeted 3 + 1 prodrug, which retained some residual IL-4 signaling (Figure 3e). Interestingly, this same HER2-targeted prodrug showed no activity in isolation in a HEK-Blue IL-4 assay, which also monitors cis-activation (Figures 4a and S4e-f, see following section). A comparison of TF-1 cell proliferation between CD38 (targeted) and HER2 (non-targeted) prodrug combinations revealed more efficient TF-1 proliferation with targeted CD38 prodrugs than with the non-targeted HER2 prodrugs (Figure 3c, f). This demonstrates CD38-mediated targeting and activation of prodrugs on the surface of target cells. However, addition of non-targeted (HER2) prodrugs also resulted in TF-1 proliferation, albeit to a lesser degree. This indicates that there is notable in-solution reconstitution of IL-4 activity (Figure 3c, f). Comparison of CD38 and HER2 prodrug combinations yielded a 6.7-fold difference in EC50 values between targeting (CD38) and non-targeting (HER2) 2 + 2 split IL-4 prodrugs (0.54 nM vs 3.6 nM), and a 3.7-fold difference for 3 + 1 split IL-4 prodrugs (2.4 nM vs 8.8 nM). Thus, although we observed targeted cis-activation of split IL-4 prodrugs, the therapeutic window between on-cell and in-solution activation may benefit from further optimization.
Figure 3.
CD38-targeted IL-4 prodrugs elicit IL-4 cis-signaling and mediate proliferation of CD38-expressing, HER2-negative TF-1 cells.
(a) Proliferation of TF-1 cells after treatment with CD38-targeting prodrugs containing 2+2 split IL-4 or pre-assembled split IL-4. b) Proliferation of TF-1 cells after treatment with non-targeted (HER2-directed) prodrugs containing 2+2 split IL-4 or pre-assembled split IL-4. c) Comparison of TF-1 cell proliferation after treatment with either CD38- or HER2-targeting prodrugs containing 2+2 split IL-4. d) Proliferation of TF-1 cells after treatment with CD38-targeting prodrugs containing 3+1 split IL-4 or pre-assembled split IL-4. e) Proliferation of TF-1 cells after treatment with non-targeted (HER2-directed) prodrugs containing 3+1 split IL-4 or pre-assembled split IL-4. f) Comparison of TF-1 cell proliferation after treatment with either CD38 or HER2-targeting prodrugs containing 3+1 split IL-4.
Figure 3 consists of six panels showing dose-response curves from TF-1 proliferation experiments with various targeted split IL-4 prodrugs and corresponding non-targeted or pre-assembled controls. Panel A shows that individual CD38-targeted 2+2 split IL-4 prodrugs are inactive, while the respective prodrug combination is active. Panel B shows that individual HER2-targeted 2+2 split IL-4 prodrugs, which serve as non-targeted controls on TF-1 cells, are inactive. The respective prodrug combination is active, however with lower potency that the targeted combination in panel A. Panel C shows dose response curves for targeted (CD38) and non-targeted (HER2) prodrug combinations, with the targeted combination being more active than the non-targeted combination. Panels D, E, and F show the same experiments performed in panels A-C, but with 3+1 split IL-4 prodrugs. The dose-response curves indicate inactivity of all prodrugs with the exception of non-targeted (HER2) IL-4 BCD, which shows some residual activity. In panel F, greater activity is observed for targeted (CD38) compared to non-targeted (HER2) prodrug combinations.
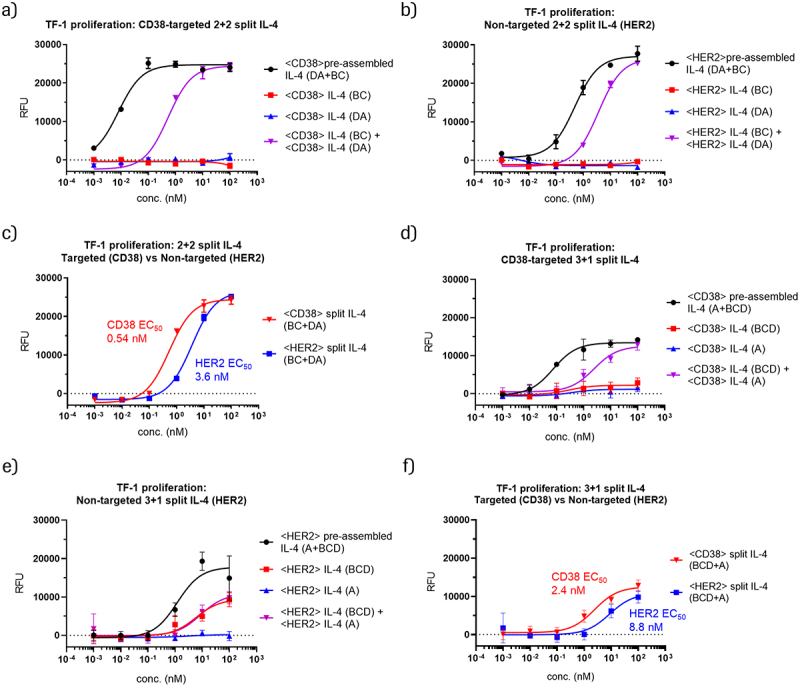
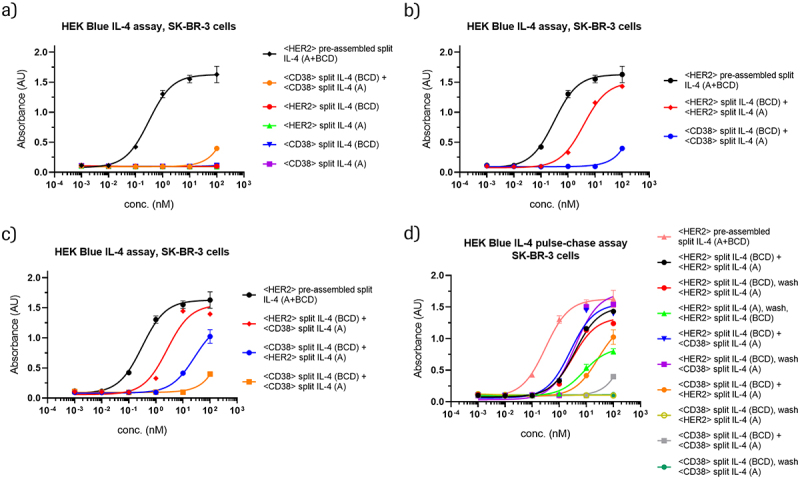
Figure 4.
HER2-binding IL-4 prodrugs assemble on HER2-expressing, CD38-negative SK-BR-3 cells and elicit IL-4 cis- and trans-signaling.
HEK-Blue IL-4 reporter assays reveal activation of HER2-targeted prodrugs, which become activated on HER2-expressing SK-BR-3 cells. For more details about differentiation between cis- and trans-activation in these assays, see Figures S4-S5. Data in panels a-d come from the same experiment. Selected curves are represented in multiple plots for comparison purposes. (a) Individual educts or the combination of non-targeting educts (CD38-targeted) elicit no or only very little IL-4 signaling compared to a pre-assembled HER2-targeted control. (b) The combination of HER2-targeting educts leads to prodrug conversion and IL-4 signaling with a low nM EC50, and approximately two orders of magnitude greater activity compared to non-targeted (CD38) control educts. (c) The combination of one HER2-targeting educt with a complementary non-targeting educt (CD38) can also trigger prodrug conversion and IL-4 signaling. In this setting, relative efficacy depends on the format and composition of the entity that harbors the cell-surface binder. (d) In pulse-chase experiments, one prodrug is added to target cells, followed by a wash step to remove unbound educt before addition of the second prodrug. IL-4 signaling observed upon subsequent addition of complementary non-targeting educts indicates on-cell assembly with the first prodrug, as free prodrugs are removed during the wash step. The fact that removing the first prodrug from the medium does not have a major impact on activity indicates that most of the activation occurs on target cells during the setting of simultaneous addition.
Figure 4 consists of four panels that show dose-response curves from HEK-Blue IL-4 reporter assays with HER2-expressing SK-BR-3 cells. Data from targeted and non-targeted 3 + 1 split IL-4 prodrugs, prodrug combinations, and pre-assembled controls are shown. In panel A, targeted (HER2) and non-targeted (CD38) prodrugs alone show no activity, and the non-targeted prodrug combination shows minimal activity only at the highest concentration. Panel B shows that the targeted prodrug combination (HER2) is markedly more active than the otherwise identical non-targeted combination. Panel C shows that combinations of targeted (HER2) and non-targeted (CD38) prodrugs also elicit activity, with the targeted IL-4 (BCD) plus non-targeted IL-4 (A) combination being more active than the targeted IL-4 (A) plus non-targeted IL-4 (BCD) combination. Panel D shows the results of pulse-chase experiments for different targeted and non-targeted 3 + 1 split IL-4 prodrug combinations, revealing the highest activities when targeted IL-4 (BCD) prodrugs (HER2) are applied before the wash step, combined with either a targeted (HER2) or non-targeted (CD38) prodrug after the wash.
HER2-targeted, chain-exchange-mediated cis- and trans-activation of split IL-4 prodrugs
Antibody-cytokine fusions are not only restricted to triggering cognate receptors on the same cells to which they bind (cis-activation), they can also activate receptors on neighboring cells in spatial proximity (trans-activation). Such trans-activation of cytokine receptors is the mechanism by which microenvironment-targeted cytokines elicit therapeutic activities.9,39 To assess if the cytokine-PACE approach also enables trans-activation of IL-4 receptors, HER2 or CD38- binding split IL-4 prodrugs in the 3 + 1 format were applied to SK-BR-3 cells, which express a very high level of HER2 and no detectable CD38 (quantification in Figure S6). In parallel, the 3 + 1 split IL-4 prodrugs were also applied to SK-OV-3, NCI-H1650, and MCF-7 cells, which also lack detectable CD38 but express differing levels of HER2 (quantification in Figure S6). An additional control was also performed with no target cells. The potency of IL-4 receptor trans-activation was assessed by co-culturing the SK-BR-3 cells with HEK-Blue IL-4 reporter cells (Invivogen), which express very low levels of HER2 and no detectable CD38 (Figure S6).
Figure 4 shows that split IL-4 prodrugs that are targeted to high HER2-expressing SK-BR-3 cells elicit IL-4 receptor activation. The individual prodrugs by themselves do not lead to IL-4 receptor activation above background levels (Figure 4a), confirming inactivation of the educts also in this assay setting. The combination of complementary prodrugs in a non-targeting setting (CD38 binders) leads to minimal IL-4 signaling only at the highest concentration tested (Figure 4a). In contrast, IL-4 signaling was observed at relatively low concentrations with HER2-targeted prodrugs on SK-BR-3 cells, with an EC50 value of 3.8 nM (Figure 4b). The potency of HER2-targeted split IL-4 prodrugs is just 12-fold lower compared to the pre-assembled fully active split IL-4 control, which does not require chain-exchange-mediated activation. This shows that accumulation of split IL-4 prodrugs on cells that express a high density of target antigens leads to efficient effector cell activation. In addition, we observe almost no IL-4 signaling when both prodrugs are non-targeting (Figure 4b).
Interestingly, we also observed IL-4 receptor activation upon co-incubation of the reporter cells with target cells that express lower densities of HER2 (Figure S4a-d, Table S4a). This activity was HER2-specific because CD38-targeted prodrug combinations generated only minimal activity at the highest concentration tested. The observed EC50 for SK-BR-3 and SK-OV-3 cells, which both express a very high level of HER2 (approximately 600,000 copies per cell, see Figure S6), was two-to-three fold lower than that observed for cells with lower HER2 levels, indicating that the accumulation on SK-BR-3 or SK-OV-3 cells may activate effector cells in a trans setting. The fact that HEK-Blue IL-4 reporter cells also express HER2, albeit at rather low levels (approximately 3000 copies per cell, Figure S6), may explain the relative lack of receptor-density-mediated dependence in these experiments. Cis-activation may have a lower receptor threshold than trans-activation, and would thus be present at a background level in these experiments. Indeed, applying the 3 + 1 split IL-4 prodrugs to HEK-Blue IL-4 cells alone resulted in IL-4 activity, but at an EC50 value 6–7 fold greater than that observed in the presence of SK-BR-3 cells (Figure S4e-f, Table S4b). This result indicates that there is indeed background cis-activation of HEK-Blue IL-4 cells. Additional trans-activation likely accounts for the increased potency observed in the presence of target cells. We therefore conclude that 3 + 1 split IL-4 PACE prodrugs can elicit trans-activation of effector cells. Interestingly, the targeting window of cis-activation on HEK-Blue IL-4 cells for the 3 + 1 split format (targeted vs. non-targeted setting) was notably greater than that of the TF-1 cis-activation assay (Figure S4f vs Figure 3f). One possible explanation is that the internalization kinetics of CD38-bound complexes may differ from those of HER2-bound complexes. In contrast to the 3 + 1 split IL-4 format, the 2 + 2 split IL-4 format did not show a notable activity difference in the HEK-Blue IL-4 assay between high HER2-expressing SK-BR-3 cells, medium-expressing NCI-H1650 cells, and no target cells (Fig. S5, Table S5). As such, we could not demonstrate trans-activation for the 2 + 2 split format.
In another experimental setting, SK-BR-3 cells were treated with one IL-4 prodrug targeting HER2 in combination with a complementary IL-4 prodrug containing a non-targeting binder (CD38). Figure 4c indicates successful IL-4 receptor activation (as a combination of cis- and trans-activation) also under these “semi-targeted” conditions. These results indicate that IL-4 prodrug activation requires only one educt to be bound to the cell surface, which could be due to productive chain exchange or another mechanism. Interestingly, we observed significantly more potent and efficient IL-4 activation when the prodrug containing WT IL-4 (BCD) was bound to the cell surface as compared to conditions where the prodrug containing WT IL-4 (A) was bound to the cell surface (Figure 4c).
Pulse-chase experiments indicate that the chain exchange reaction appears to proceed predominantly upon accumulation of one educt on the surface of target cells. Treatment of the target cells with the targeting prodrug (HER2) followed by a wash step and subsequent addition of the targeting (HER2) or non-targeting prodrug (CD38) generates the same IL-4 receptor activation as with simultaneous application of both, where undesired in-solution exchange may additionally occur (Figure 4d). In contrast, initial exposure to a non-targeting educt (CD38) followed by a wash step and subsequent treatment with the targeting counterpart (HER2) does not result in signals above background levels (Figure 4d). Overall, these results demonstrate that cytokine-PACE with 3 + 1 split IL-4 can be applied in a cis-activation setting as well as a trans-activation setting, provided the target cells contain a high antigen density.
Discussion
Cytokine immunotherapy has emerged as a promising modality in cancer therapy in recent years.5,6 The previous experience of undesired toxicity of systemic cytokine therapy has driven efforts to create engineered targeted cytokines, among them antibody-cytokine fusions.7 Although antibody-cytokine fusions have improved upon the targeting, pharmacokinetics, and dosing of cytokines as therapeutics, they nevertheless have some unresolved issues. One major issue with tumor-targeting approaches such as antibody-cytokine fusions is that TAAs are often not exclusively expressed on tumor cells, but are also expressed in healthy tissue at low levels.8 This can lead to potential on-target off-tumor-related toxicity. A second problem is that antibody-cytokine fusions can bind to cytokine receptors on various cell types in circulation, which can lead to decreased tumor penetration and additional off-target toxicity issues.7 Such specificity issues are not unique to cytokine immunotherapy; they are major challenges for many tumor-targeting cancer therapies, including T-cell bispecific antibodies (TCBs), chimeric-antigen receptor T cells, and antibody-drug conjugates.8,40,41
To overcome these challenges, the development of conditionally active targeted immunotherapies is essential. Notable conditional activation approaches in the TCB field include cleavage of a masking moiety by tumor-specific proteases,42–44 splitting of a T-cell binding Fv to create two inactive prodrugs which assemble preferentially on tumor cells,45 or generating inactive chain exchange-enabled prodrugs that re-associate into functional entities on tumor cells.10 In a similar manner, conditionally active cytokine prodrugs are currently being developed to address the aforementioned problems of conventional antibody-cytokine fusions. Some of the approaches undertaken to date include targeted reconstitution of split cytokines,12,13 cleavage of a masking domain by tumor-specific proteases,14–17 attenuation of targeted cytokines,18–20 and engineering of pH-dependent cytokines.21,22 Cytokines including, IL-2, IL-15, and IL-12, which have shown great promise in cancer immunotherapy, are among the primary candidates being developed as conditionally active prodrugs.
Here we have developed cytokine-PACE as a prodrug approach to reconstitute split versions of type I cytokines on target cells (Figure 1). This novel approach combines 1) splitting of cytokines for inactivation with 2) the addition of stabilizing dummy chains to reduce aggregation propensity of exposed core regions with high hydrophobicity, and 3) antibody CH3-domain-driven chain exchange for targeted re-assembly of active cytokines. IL-4, a representative type I cytokine, was split and mutated to create two inactive IL-4 prodrugs. IL-4 can be split into two different topologies and fused to the C-terminus of antibody-like TriFab molecules. Pre-assembled split IL-4 retains activity (Figure 1d) and can be inactivated through mutations in each split half to generate inactive prodrugs (Figure 2b). The inactivation of split IL-4 prodrugs persists even upon TAA-mediated accumulation on target cells (Figures 3, 4 and S4-S5), and thereby relies on chain exchange for reactivation. The expression yields of split IL-4 PACE prodrugs and the pre-assembled split IL-4 products were lower compared to PACE molecules lacking split IL-410 (ranging from 4-fold to 40-fold lower). Nevertheless, expression yields of split IL-4 PACE molecules are in the range previously reported for other antibody-cytokine fusions.46–48 Almost all split IL-4 PACE molecules were primarily monomeric with a low percentage of aggregates, as determined by analytical size-exclusion chromatography (SEC) (Figure S2). We also investigated the thermal stability of split IL-4 PACE molecules using nanoDSF (Table S2). Most split IL-4 PACE molecules showed favorable thermal stability, with many of the constructs having melting temperatures (Tm) and aggregation temperatures (Tagg) in the range of IgGs.49 Molecules containing an inactivated IL-4 (A) dummy helix showed an overall lower thermal stability. Split IL-4 prodrugs recovered their activity and triggered IL-4 receptor activation in a cis setting (Figures 3 and 4), and in a trans setting for 3 + 1 split IL-4 (Figures 4 and S4). The reconstitution of IL-4 activity was driven through chain exchange, triggered by charge mutations in the CH3 domains of the cytokine-PACE prodrugs. We also demonstrated that the IL-4 prodrugs are targeted to TAA-expressing cells and that chain exchange most likely occurs on the cell surface (Figure 4d and below).
A key feature for improving immunotherapies with cytokine prodrugs is a sufficient window between desired activation on the target cell and undesired systemic activation. In cell culture assays, this is reflected by differences between targeted (on-cell) and non-targeted (in-solution) conversion of prodrugs into active cytokines. The window of 3 + 1 split IL-4 PACE entities reflected by the activities of targeted vs non-targeted prodrugs is estimated to be between 20-to-100-fold in HEK-Blue IL-4 reporter assays (Figures 4b, S4), yet is < 10-fold in TF-1 cis-activation assays, which monitor proliferation of IL-4 dependent TF-1 cells (Figure 3). The relatively larger window between targeted and non-targeted activation of 3 + 1 split IL-4 observed in HEK-Blue IL-4 assays indicates that on-cell activation is more efficient than in-solution activation, and we find no clear evidence for excessive in-solution prodrug conversion in these assays (Figures 4, S4). The relatively smaller window observed for the 3 + 1 split IL-4 format in TF-1 cis-activation assays may thus not be the consequence of excessive in-solution activation (Figure 3). It is instead possible that the tethering of IL-4 fusions to CD38 limits access to the IL-4 receptor on the same cell surface. Such accessibility constraints may be addressed in the future by optimizing the formats to improve cis-activation of cytokine receptors.11,50
The 3 + 1 split IL-4 format is the preferred format in regard to trans-activation and minimization of in-solution activation. Indeed, we observed an increased potency in the presence of target cells expressing a high HER2 density in HEK-Blue IL-4 assays (Figure S4). This indicates additional HER2-targeted trans-activation on top of the HER2-targeted cis-activation observed in this assay. We were surprised to find that, although trans-activation was dependent on target antigen density, the dependence was not as pronounced as expected. We observed roughly a two-fold increased potency on very high-HER2 expressing cell lines (approximately 600,000 molecules per cell) compared to medium HER2-expressing cell lines (approximately 10,000–30,000 molecules per cell) (Figures S4, S6). However, the background cis-activation observed in the HEK-Blue IL-4 assays (Figure S4e-f) may have led to an underestimation of trans-activation. Cis-activation observed in HEK-Blue IL-4 assays was indeed target-specific, as the combination of CD38-targeting prodrugs did not lead to notable IL-4 activity (Figure S4e-f). Given that HEK-Blue IL-4 cells express only approximately 3000 copies of HER2 per cell (Figure S6), the threshold for IL-4 prodrug activation is notably lower than we expected. Cis-activation is predicted to have a lower threshold than trans-activation, since a relatively small amount of productive chain exchange on the surface of IL-4 receptor-expressing cells may result in a sufficiently high local concentration of active IL-4 on these cells.
For the 2 + 2 split format, we observed similar activity in HEK-Blue IL-4 assays with no target cells compared to assays in the presence of SK-BR-3 cells (Figure S5a-c). As such, we cannot infer any level of trans-activation for this format. Additionally, we observe greater levels of non-targeted activation (with the combination of CD38-targeting prodrugs) for the 2 + 2 format compared to the 3 + 1 format (Figure S5 compared to Figure S4). This may contribute to a higher background level of IL-4 activation in the absence of target cells for the 2 + 2 format.
One interesting observation is that combining one targeting prodrug with a complementary non-targeting prodrug also results in IL-4 signaling in HEK-Blue IL-4 reporter assays (Figure 4c, d). Pulse-chase experiments (addition of prodrug 1 – wash – addition of prodrug 2) show that this mode of activation can take place on the surface of target cells even though one of the prodrugs is not targeted to a TAA. Thus, the IL-4 activation observed in our assays likely represents ‘hybrid-activation’ events, namely the sums of bi-targeted and mono-targeted prodrug conversion on cell surfaces. The chain-exchange-enabled, cell-bound IL-4 prodrug may capture the complementary (and otherwise non-targeted) IL-4 prodrug from the solution to complete the exchange reaction. Such a mechanism may also explain why cell-bound IL-4 (BCD) 3-helix entities, which are expected to be more structured and thus likely a better capture module, are more effective in capturing a complementary non-targeted single IL-4 (A) helix than vice versa. The efficiencies of capture and on-cell exchange of non-targeted prodrugs might be dependent on the formats or composition of the split entities. As such, modulation of formats and interfaces (such as in refs. 11 and 50) may provide additional ways to regulate the contributions of nonspecific or hybrid activation, and to minimize premature activation. Importantly, the pulse-chase experiments in Figure 4d also show that cytokine-PACE is amenable to pre-targeting as another option to minimize systemic, non-targeted prodrug activation. Initial application of one prodrug would result in its accumulation on target cell surfaces, while the complementary second prodrug can subsequently be supplied after a defined time window (mimicking addition after the wash step in cell culture experiments). This may result in a reduced probability for the second prodrug to encounter exchange-enabled entities in the circulation, and would likely result in increased partner educt levels at desired target sites.
Development of two-component, conditionally active prodrug therapies may be associated with higher manufacturing costs than the development of a single, constitutively active molecule. However, the potential higher cost is warranted if the prodrugs are safer than the parental molecule, while maintaining efficacy. Such conditionally active targeted cytokines are in high demand in cancer immunotherapy. We conclude that cytokine-PACE provides a foundation for further conditionally active targeted cytokines. In particular, other type I cytokines such as IL-2, IL-15, and IL-21, which are promising candidates for cancer immunotherapy and share similar structures to IL-4, may be amenable to cytokine-PACE.
Materials and methods
Protein design
To create targeted split IL-4 PACE constructs, we used the following target binders: <CD38> from Daratumumab51 and <HER2> clone 4D5–8.52 We used VH/VL domains of the following binders in the stem region of the TriFab: <Dig> Digoxigenin,53 <Nada> Nadaceptin.10 Split IL-4 prodrugs were designed using the PACE format as previously described.10 Heterodimerization of heavy chains was achieved using knob-into-hole mutations in the CH3 domains (knob: T366W, hole: T366S, L368A, Y407V).54 IL-4 derivatives were fused to the C-terminus of PACE educts with a flexible 3×G4S (Gly-Ser) linker. Control TriFabs were designed as previously described.10,31 The details regarding the design of the split IL-4 variants are described in the main text. Residues in IL-4 chosen as candidates for weakening or interfering with the IL-4 R interaction were defined based on structural information of the IL-4/IL-4 Rα/common gamma chain (γC) ternary complex, PDB ID 3BPN.34 PyMOL (Schroedinger, Inc.) was used to visualize the effects of proposed mutations and to prepare the structural representations in Figure 2.
Production of IL-4 TriFab fusions, split IL-4 derivatives, and IL-4 prodrugs
All antibody-fusion proteins were expressed in transiently transfected HEK293 cells (Freestyle or Expi293F™), as previously described.10 The desired proteins were purified from supernatants by affinity chromatography and subsequent cation exchange chromatography (CEX) and/or SEC. Affinity chromatography was performed on an ÄKTAexplorer with a HiTrap KappaSelect column (Cytiva, 17545812) to capture the desired proteins, as TriFabs and PACE prodrugs lack CH2 domains and hence do not bind Protein A. In order to separate the desired product from side products of a similar molecular weight, a subsequent CEX step was performed using a HiTrap SP HP column (Cytiva, 17115201). For CEX, the protein was diluted in 20 mM Histidine pH 6.0 and was loaded onto the column with no salt. Elution was performed in 20 mM Histidine pH 6.0 with a linear gradient of NaCl. SEC was subsequently performed on a HiLoad 16/600 Superdex 200 column (Cytiva, 28989335) or a Superdex 200 Increase 10/300 GL column (Cytiva, 28990944) to isolate monomeric proteins of desired composition. The fractions of interest were subjected to SDS-PAGE, and fractions with the correct polypeptide chain composition were pooled, concentrated, sterile-filtered (0.22 µm). The final protein concentrations were calculated from absorbance values at 280 nm, measured on a NanoDrop™ 2000 instrument (Thermo Fisher). Examples of chain composition and purity of the prepared split IL-4 proteins applied as prodrugs or controls are provided in Figure S2.
Cell culture and reagents
TF-1 (ATCC CRL-2003), SK-BR-3 (ATCC HTB-30), SK-OV-3 (ATCC HTB-77), NCI-H1650 (ATCC CRL-5883), and MCF-7 cells (ATCC HTB-22) were cultivated in Roswell Park Memorial Institute Medium 1640 (RPMI-1640, PAN Biotech), supplemented with 2 mM glutamine and 10% fetal calf serum (FCS; Gibco, 10500–064). Human GM-CSF (Thermo Fisher Scientific) was added to a final concentration of 2 ng/ml to TF-1 cells for sub-culturing. HEK-Blue IL-4/IL-13 SEAP reporter cells (Invivogen, hkb-il413) were cultivated in Dulbecco’s Modified Eagle Medium, high glucose (DMEM-HG, Sigma-Aldrich, D5796), supplemented with 10% FCS, 100 µg/ml Zeocin, and 10 µg/ml Blastidicin. All cells were cultured in an incubator at 37°C with 5% CO2. Adherent cells were detached with Accutase (Sigma-Aldrich, A6964) during sub-culturing. Cells were counted using a Vi-CELL XR cell counter (Beckman Coulter).
Flow cytometry
Expression of CD38 and HER2 on TF-1 and SK-BR-3 cells was assessed by flow cytometry (Figure S6A). Briefly, 4 × 105 total cells were resuspended in phosphate-buffered saline (PBS) with 2% FCS and incubated with 5 µl of either phycoerythrin (PE)-labeled anti-human CD38 antibody (clone HB-7, BioLegend, 356604), PE-labeled anti-human HER2 antibody (clone 24D2, BioLegend, 324406) or isotype control (PE mouse IgG1, k isotype control, clone MOPC-21, BioLegend, 400112). The cells were incubated for 30 min on ice in the dark, washed twice with ice-cold PBS, resuspended in cold PBS, and analyzed on a FACS Canto II instrument (BD Biosciences). The data was analyzed and visualized using FlowJo software (BD Biosciences).
Surface plasmon resonance
In order to capture His-tagged IL-4R alpha, an anti-histidine antibody (GE Healthcare, 28-9980-56) was immobilized at high density (>10 000 RU) on a CM5 sensor. Human recombinant His-tagged IL-4R alpha (Abcam, ab167726) was applied at a concentration of 5 nM for 45 s (capture level was approximately 55 RU). TriFabs containing C-terminally fused IL-4 variants were analyzed by single cycle kinetics at a concentration of 7.4 nM to 600 nM with an association time of 120 s and a dissociation time of 900 s, at a flow rate of 50 µl/min. The measurements were performed in HBS-P+ pH 7.4 running buffer (GE Healthcare, Br-1008–27) at 25°C. The kinetic properties were analyzed using T200 evaluation software (Cytiva) using a 1:1 Langmuir binding model.
TF-1 proliferation assay
Cis-activation of split IL-4 prodrugs was evaluated using a TF-1 proliferation assay. TF-1 cells were washed three times with PBS to remove residual GM-CSF and were resuspended in RPMI-1640 media with supplements. The washed TF-1 cells were seeded out in flat-bottom 96-well plates at 2 × 104 cells per well. The cells were treated with serial dilutions of cytokine-PACE molecules and were subsequently incubated for 120 h at 37°C and 5% CO2. Each prodrug was added sequentially to the cells to prevent premature in-solution chain exchange. Proliferation of TF-1 cells was measured by the Resazurin assay kit (Abcam, 129732). In this assay, the non-fluorescent molecule resazurin is added to the cells and is converted by mitochondria to the fluorescent molecule resofurin. The fluorescence of resofurin correlates directly with the number of living cells, serving as a readout for cell proliferation.55 The working resazurin solution was prepared according to the manufacturer’s instructions and was added directly to the cells. The plates were incubated for 4 h in a 37°C incubator and fluorescence was measured (excitation 550 nm, emission 590 nm) in a plate reader (Tecan Infinite F200 Pro). The data were analyzed with GraphPad Prism software and were fit with a three parameter, non-linear regression.
HEK-Blue IL-4 cis- and trans-activation assays
Cis- and trans-activation of split IL-4 prodrugs was analyzed using a co-culture assay of target cells (SK-BR-3, SK-OV-3, NCI-H1650, or MCF-7) with IL-4 reporter cells (HEK-Blue IL-4). Briefly, target cells and HEK Blue IL-4 cells were detached with Accutase, resuspended in DMEM-HG, counted, and pipetted into a 96-well plate. Each well contained 6 × 104 target cells and 7.5 × 104 HEK-Blue IL-4 cells. Serial dilutions of split IL-4 PACE molecules were added to the wells and the plates were incubated for 40 h at 37°C, 5% CO2. Each prodrug was added sequentially to the plate to prevent premature in-solution chain exchange. Following incubation, 20 µl of supernatant was transferred to clear 96-well flat bottom plates and was mixed with 180 µl of freshly prepared QUANTI-Blue™ solution (Invivogen, rep-qbs2), as per manufacturer’s instructions. Upon IL-4 R signaling, secreted alkaline phosphatase (SEAP) reporter activity is measured by an increase in the absorbance of QUANTI-Blue at 640 nm, which serves as a readout for IL-4 activity. After QUANTI-Blue addition, the plates were incubated for 30 min at 37°C and absorbance at 640 nm was measured using a Tecan Infinite F200 Pro plate reader. The data were analyzed with GraphPad Prism software and were fit with a three-parameter, non-linear regression. In pulse-chase experiments, the same protocol was performed, with the exception that the supernatant was removed 1 h after incubation with the first prodrug (at 37°C). The cells were then washed three times with DMEM-HG, and the second split IL-4 prodrug was added to the wells (with a subsequent total incubation time of 40 h, as above). For experiments investigating the background cis-activation of HEK-Blue IL-4 cells (Figures S4f, S5c), the same protocol was performed, but without the addition of target cells.
HER2 and CD38 quantification on various cell lines
The average HER2 and CD38 copy numbers were quantified for each cell line using the QIFIKIT (Agilent, K0078), according to the manufacturer’s instructions. Briefly, 3 × 105 total cells were transferred into round-bottom 96-well plates and were resuspended in ice-cold QIFI buffer (PBS + 0.1% BSA). Saturating concentrations of unlabeled mouse monoclonal antibodies against HER2, CD38, or an isotype control were added to the cells. The following antibodies were used for the primary incubation: unlabeled anti-human HER2 (clone 24D2, BioLegend, 324402), unlabeled anti-human CD38 (clone HB-7, BioLegend, 356602), and unlabeled isotype control (mouse IgG1, k isotype control, clone MOPC-21, BioLegend, 400102). The cells were incubated with primary antibodies for 45 min at 4°C. During the primary antibody incubation, the setup beads and calibration beads were washed with 1 ml of ice-cold FACS buffer, centrifuged at 300 × g for 5 min, and the supernatant was aspirated. This was repeated two more times. The plate containing the cells was centrifuged at 300 × g for 5 min at 4°C, the supernatant was aspirated, and the cells were washed three times with 200 µl of ice-cold FACS buffer. The cells were then incubated with saturating concentrations of the supplied fluorescein isothiocyanate (FITC)-conjugated secondary antibody, as per the manufacturer’s instructions. The cells were incubated with the secondary antibody for 45 min at 4°C, after which they were washed three times as above. In parallel, the setup beads and the calibration beads were treated with the FITC-conjugated secondary antibody in the same manner. The mean fluorescence intensity (MFI) of the calibration beads was measured in the FITC channel of a FACS Canto II instrument (BD Biosciences). The MFI of the cells was then measured in the FITC channel using the same instrument settings. Quantification of mean receptor counts for each cell line (specific antibody-binding capacity) was calculated as per the manufacturer’s instructions, using the calibration curve shown in Fig. S6b. The results of the quantification are shown in Table S6.
Biochemical quality control of molecules: SDS-PAGE and analytical SEC
Molecule purity and chain composition was characterized by non-reducing and reducing SDS-PAGE. Monomeric content and aggregation was analyzed by analytical SEC. For non-reducing SDS-PAGE, approximately 2 µg of each protein was prepared in LDS sample buffer (NuPAGE, Invitrogen) and the proteins were separated on a 4–12% Bis-Tris gel (NuPAGE, Invitrogen). For reducing SDS-PAGE, the sample buffer contained 1X NuPAGE sample reducing agent (Invitrogen). The proteins were visualized by colloidal Coomassie staining with ReadyBlue™ protein gel stain (Sigma-Aldrich). The molecular weight (MW) was estimated by comparison of bands with a protein standard (Precision Plus Protein Dual Color Standard, Bio-Rad). For analytical SEC, 20 µg of each protein was loaded onto a BioSuite 250, 5 µm HR SEC column (Waters) using an UltiMate 3000 UHPLC system (Thermo Fisher Scientific). The data was analyzed using Chromeleon CDS software (Thermo Fisher Scientific).
Thermal unfolding analysis
Prior to analysis, all samples were diluted in PBS, resulting in protein concentrations ranging from 0.4–1.0 mg/ml. Thermal unfolding and aggregation analysis was determined on a Prometheus Panta instrument (NanoTemper Technologies, Munich, Germany). The measurements were performed with a heating ramp of 1.5 K/min from 20°C to 95°C, 45% excitation power, and 100% DLS laser power, using standard capillary chips (NanoTemper Technologies, Munich, Germany, Cat # PR-AC002). Data analysis and calculations were carried out using the PR.Panta Analysis software (NanoTemper Technologies, Munich, Germany).
Supplementary Material
Acknowledgments
We thank our colleagues at Roche for their support, insight and discussions regarding the concept and the manuscript. VV, CB, and SD were supported by the Roche Postdoctoral Fellowship (RPF).
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Abbreviations
- ABC:
Antibody-binding capacity (from QIFIKIT)
- BSA:
Bovine serum albumin
- CEX:
Cation exchange chromatography
- CIT:
Cancer immunotherapy
- Dig:
Digoxigenin
- DLS:
Dynamic light scattering
- FACS:
Fluorescence-activated cell sorting
- FCS:
Fetal calf serum
- FITC:
Fluorescein isothiocyanate
- IL-4:
Interleukin-4
- IL-4 Rα:
Interleukin-4 receptor-alpha
- IL-4 R:
Interleukin-4 receptor
- MFI:
Mean fluorescence intensity
- nanoDSF:
Nano differential scanning fluorimetry
- PACE:
Prodrug-activating chain exchange
- PBS:
Phosphate-buffered saline
- PE:
Phycoerythrin
- SDS-PAGE:
Sodium dodecyl-sulfate-polyacrylamide gel electrophoresis
- SEAP:
Secreted alkaline phosphatase
- SEC:
Size-exclusion chromatography
- SPR:
Surface plasmon resonance
- TAA:
Tumor-associated antigen
- Tagg:
Aggregation temperature
- TCB:
T cell bispecific antibody
- Tm:
Melting temperature
- γc:
Common gamma chain
Disclosure statement
All authors are either employed by Roche, or were employed by Roche, as indicated. Roche has an interest in targeted immunotherapies. VV, CB, SD, GG, KM, & UB are co-inventors of patent applications that cover targeted or conditional immunotherapies related to this study. UB owns Roche stocks.
Author contributions
VV, CB, SD, and UB devised the concept. UB supervised the project and provided guidance and mentoring. GG performed structural modeling and aided in designing split IL-4 formats. VV, CB, MR, SD, and KM designed, purified, and characterized constructs, and performed experiments. VV, CB, MR, SD, and UB coordinated and planned experiments, and analyzed/interpreted data. CS designed, performed and analyzed SPR experiments. AP and TM designed, performed and analyzed thermal stability experiments. VV, CB, and UB wrote the manuscript. All coauthors reviewed and edited the manuscript.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2245111.
References
- 1.Leonard WJ, Lin J-X, O’Shea JJ.. The γc family of cytokines: basic biology to therapeutic ramifications. Immunity. 2019;50(4):832–12. doi: 10.1016/j.immuni.2019.03.028. [DOI] [PubMed] [Google Scholar]
- 2.Wang X, Lupardus P, LaPorte SL, Garcia KC. Structural biology of shared cytokine receptors. Annu Rev Immunol. 2009;27(1):29–60. doi: 10.1146/annurev.immunol.24.021605.090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulay J-L, O’Shea JJ, Paul WE. Molecular phylogeny within type I cytokines and their cognate receptors. Immunity. 2003;19(2):159–63. doi: 10.1016/S1074-7613(03)00211-5. [DOI] [PubMed] [Google Scholar]
- 4.Feldmann M. Many cytokines are very useful therapeutic targets in disease. J Clin Invest. 2008;118(11):3533–36. doi: 10.1172/JCI37346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waldmann TA. Cytokines in Cancer Immunotherapy. Csh Perspect Biol. 2018;10(12):a028472. doi: 10.1101/cshperspect.a028472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, Rodríguez-Ruiz ME, Ponz-Sarvise M, Castañón E, Melero I. Cytokines in clinical cancer immunotherapy. Brit J Cancer. 2019;120(1):6–15. doi: 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutmacher C, Neri D. Antibody-cytokine fusion proteins: Biopharmaceuticals with immunomodulatory properties for cancer therapy. Adv Drug Deliver Rev. 2018;141:67–91. doi: 10.1016/j.addr.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 8.Ellerman D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods. 2019;154:102–17. doi: 10.1016/j.ymeth.2018.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6(3):00–00. doi: 10.1080/2162402X.2016.1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickopf S, Buldun C, Vasic V, Georges G, Hage C, Mayer K, Forster M, Wessels U, Stubenrauch K-G, Benz J, et al. Prodrug-Activating Chain Exchange (PACE) converts targeted prodrug derivatives to functional bi- or multispecific antibodies. Biol Chem. 2022;403(5–6):495–508. doi: 10.1515/hsz-2021-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dengl S, Mayer K, Bormann F, Duerr H, Hoffmann E, Nussbaum B, Tischler M, Wagner M, Kuglstatter A, Leibrock L, et al. Format chain exchange (FORCE) for high-throughput generation of bispecific antibodies in combinatorial binder-format matrices. Nat Commun. 2020;11(1):4974. doi: 10.1038/s41467-020-18477-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venetz D, Koovely D, Weder B, Neri D. Targeted reconstitution of cytokine activity upon antigen binding using split cytokine antibody fusion proteins*. J Biol Chem. 2016;291(35):18139–47. doi: 10.1074/jbc.M116.737734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quijano-Rubio A, Bhuiyan AM, Yang H, Leung I, Bello E, Ali LR, Zhangxu K, Perkins J, Chun J-H, Wang W, et al. A split, conditionally active mimetic of IL-2 reduces the toxicity of systemic cytokine therapy. Nat Biotechnol. 2022;41(4):532–40. doi: 10.1038/s41587-022-01510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nirschl CJ, Brodkin HR, Hicklin DJ, Ismail N, Morris K, Seidel-Dugan C, Steiner P, Steuert Z, Sullivan JM, Tyagi E, et al. Discovery of a conditionally activated interleukin-2 that promotes anti-tumor immunity and induces tumor regression. Cancer Immunol Res. 2022;10(5):581–96. doi: 10.1158/2326-6066.CIR-21-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu EJ, Cao X, Moon B, Bae J, Sun Z, Liu Z, Fu Y-X. A cytokine receptor-masked IL2 prodrug selectively activates tumor-infiltrating lymphocytes for potent antitumor therapy. Nat Commun. 2021;12(1):2768. doi: 10.1038/s41467-021-22980-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao X, Liang Y, Hu Z, Li H, Yang J, Hsu EJ, Zhu J, Zhou J, Fu Y-X. Next generation of tumor-activating type I IFN enhances anti-tumor immune responses to overcome therapy resistance. Nat Commun. 2021;12(1):5866. doi: 10.1038/s41467-021-26112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo J, Liang Y, Xue D, Shen J, Cai Y, Zhu J, Fu Y-X, Peng H. Tumor-conditional IL-15 pro-cytokine reactivates anti-tumor immunity with limited toxicity. Cell Res. 2021;31(11):1190–98. doi: 10.1038/s41422-021-00543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcin G, Paul F, Staufenbiel M, Bordat Y, der HJ, Wilmes S, Cartron G, Apparailly F, Koker SD, Piehler J, et al. High efficiency cell-specific targeting of cytokine activity. Nat Commun. 2014;5(1):3016. doi: 10.1038/ncomms4016. [DOI] [PubMed] [Google Scholar]
- 19.Deak LC, Nicolini V, Hashimoto M, Karagianni M, Schwalie PC, Lauener L, Varypataki EM, Richard M, Bommer E, Sam J, et al. PD-1-cis IL-2R agonism yields better effectors from stem-like CD8+ T cells. Nature. 2022;610(7930):161–72. doi: 10.1038/s41586-022-05192-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Carrascosa LC, Yeung YA, Chu M-H, Yang W, Djuretic I, Pappas DC, Zeytounian J, Ge Z, de RV, et al. An engineered IL15 cytokine mutein fused to an Anti-PD-1 improves intratumoral T-Cell function and antitumor immunity. Cancer Immunol Res. 2021;9(10):1141–57. doi: 10.1158/2326-6066.CIR-21-0058. [DOI] [PubMed] [Google Scholar]
- 21.Sarkar CA, Lowenhaupt K, Horan T, Boone TC, Tidor B, Lauffenburger DA. Rational cytokine design for increased lifetime and enhanced potency using pH-activated “histidine switching”. Nat Biotechnol. 2002;20(9):908–13. doi: 10.1038/nbt725. [DOI] [PubMed] [Google Scholar]
- 22.Gaggero S, Martinez-Fabregas J, Cozzani A, Fyfe PK, Leprohon M, Yang J, Thomasen FE, Winkelmann H, Magnez R, Conti AG, et al. IL-2 is inactivated by the acidic pH environment of tumors enabling engineering of a pH-selective mutein. Sci Immunol. 2022;7(78):eade5686. doi: 10.1126/sciimmunol.ade5686. [DOI] [PubMed] [Google Scholar]
- 23.Heeb LEM, Egholm C, Boyman O. Evolution and function of interleukin-4 receptor signaling in adaptive immunity and neutrophils. Genes Immun. 2020;21(3):143–49. doi: 10.1038/s41435-020-0095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Junttila IS. Tuning the cytokine responses: An update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol. 2018;9:888. doi: 10.3389/fimmu.2018.00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paul WE. History of interleukin-4. Cytokine. 2015;75(1):3–7. doi: 10.1016/j.cyto.2015.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wlodaver A, Pavlovsky A, Gustchina A. Crystal structure of human recombinant interleukin‐4 at 2.25 Å resolution. FEBS Lett. 1992;309(1):59–64. doi: 10.1016/0014-5793(92)80739-4. [DOI] [PubMed] [Google Scholar]
- 27.Walter MR, Cook WJ, Zhao BG, Cameron RP, Ealick SE, Walter RL, Reichert P, Nagabhushan TL, Trotta PP, Bugg CE. Crystal structure of recombinant human interleukin-4. J Biol Chem. 1992;267(28):20371–76. doi: 10.1016/S0021-9258(19)88711-2. [DOI] [PubMed] [Google Scholar]
- 28.Kreitman RJ, Puri RK, Pastan I. A circularly permuted recombinant interleukin 4 toxin with increased activity. Proc Natl Acad Sci. 1994;91(15):6889–93. doi: 10.1073/pnas.91.15.6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dolberg TB, Meger AT, Boucher JD, Corcoran WK, Schauer EE, Prybutok AN, Raman S, Leonard JN. Computation-guided optimization of split protein systems. Nat Chem Biol. 2021;17(5):531–39. doi: 10.1038/s41589-020-00729-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayer K, Baumann A-L, Grote M, Seeber S, Kettenberger H, Breuer S, Killian T, Schäfer W, Brinkmann U. TriFabs—Trivalent IgG-Shaped bispecific antibody derivatives: design, generation, characterization and application for targeted payload delivery. Int J Mol Sci. 2015;16(11):27497–507. doi: 10.3390/ijms161126037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickopf S, Lauer ME, Ringler P, Spick C, Kern P, Brinkmann U. Highly flexible, IgG-shaped, trivalent antibodies effectively target tumor cells and induce T cell-mediated killing. Biol Chem. 2019;400(3):343–50. doi: 10.1515/hsz-2018-0338. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Shen B-J, Sebald W. A mixed-charge pair in human interleukin 4 dominates high-affinity interaction with the receptor α chain. Proc Natl Acad Sci. 1997;94:1657–62. doi: 10.1073/pnas.94.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruse N, Shen BJ, Arnold S, Tony HP, Müller T, Sebald W. Two distinct functional sites of human interleukin 4 are identified by variants impaired in either receptor binding or receptor activation. Embo J. 1993;12(13):5121–29. doi: 10.1002/j.1460-2075.1993.tb06207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM, Keegan AD, Garcia KC. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;132(2):259–72. doi: 10.1016/j.cell.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hage T, Sebald W, Reinemer P. Crystal structure of the interleukin-4/Receptor α chain complex reveals a mosaic binding interface. Cell. 1999;97(2):271–81. doi: 10.1016/S0092-8674(00)80736-9. [DOI] [PubMed] [Google Scholar]
- 36.Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T, Miyagawa K, Piao Y, Miyazono K, Urabe A, Takaku F. Establishment and characterization of a unique human cell line that proliferates dependently on GM‐CSF, IL‐3, or erythropoietin. J Cell Physiol. 1989;140(2):323–34. doi: 10.1002/jcp.1041400219. [DOI] [PubMed] [Google Scholar]
- 37.Nooka AK, Kaufman JL, Hofmeister CC, Joseph NS, Heffner TL, Gupta VA, Sullivan HC, Neish AS, Dhodapkar MV, Lonial S. Daratumumab in multiple myeloma. Cancer. 2019;125(14):2364–82. doi: 10.1002/cncr.32065. [DOI] [PubMed] [Google Scholar]
- 38.Hogan KA, Chini CCS, Chini EN. The multi-faceted ecto-enzyme CD38: roles in immunomodulation, cancer, aging, and metabolic diseases. Front Immunol. 2019;10:1187. doi: 10.3389/fimmu.2019.01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waldhauer I, Gonzalez-Nicolini V, Freimoser-Grundschober A, Nayak TK, Fahrni L, Hosse RJ, Gerrits D, Geven EJW, Sam J, Lang S, et al. Simlukafusp alfa (FAP-IL2v) immunocytokine is a versatile combination partner for cancer immunotherapy. MAbs. 2021;13(1):1913791. doi: 10.1080/19420862.2021.1913791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327–44. doi: 10.1038/s41571-021-00470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geiger M, Stubenrauch K-G, Sam J, Richter WF, Jordan G, Eckmann J, Hage C, Nicolini V, Freimoser-Grundschober A, Ritter M, et al. Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat Commun. 2020;11(1):3196. doi: 10.1038/s41467-020-16838-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boustany LM, LaPorte SL, Wong L, White C, Vinod V, Shen J, Yu W, Koditek D, Winter MB, Moore S, et al. A probody T cell–engaging bispecific antibody targeting EGFR and CD3 inhibits colon cancer growth with limited toxicity. Cancer Res. 2022;82(22):4288–98. doi: 10.1158/0008-5472.CAN-21-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panchal A, Seto P, Wall R, Hillier BJ, Zhu Y, Krakow J, Datt A, Pongo E, Bagheri A, Chen T-H, et al. COBRA™: a highly potent conditionally active T cell engager engineered for the treatment of solid tumors. MAbs. 2020;12(1):1792130. doi: 10.1080/19420862.2020.1792130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banaszek A, Bumm TGP, Nowotny B, Geis M, Jacob K, Wölfl M, Trebing J, Kucka K, Kouhestani D, Gogishvili T, et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat Commun. 2019;10(1):5387. doi: 10.1038/s41467-019-13196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kermer V, Baum V, Hornig N, Kontermann RE, Müller D. An antibody fusion protein for cancer immunotherapy mimicking IL-15 trans-presentation at the tumor site. Mol Cancer Ther. 2012;11(6):1279–88. doi: 10.1158/1535-7163.MCT-12-0019. [DOI] [PubMed] [Google Scholar]
- 47.Nitto CD, Neri D, Weiss T, Weller M, Luca RD. Design and characterization of novel antibody-cytokine fusion proteins based on interleukin-21. Antibodies. 2022;11(1):19. doi: 10.3390/antib11010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hess C, Neri D. Tumor-targeting properties of novel immunocytokines based on murine IL1β and IL6. Protein Eng Des Sel. 2014;27(6):207–13. doi: 10.1093/protein/gzu013. [DOI] [PubMed] [Google Scholar]
- 49.Nemergut M, Žoldák G, Schaefer JV, Kast F, Miškovský P, Plückthun A, Sedlák E. Analysis of IgG kinetic stability by differential scanning calorimetry, probe fluorescence and light scattering. Protein Sci. 2017;26(11):2229–39. doi: 10.1002/pro.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dickopf S, Georges GJ, Brinkmann U. Format and geometries matter: structure-based design defines the functionality of bispecific antibodies. Comput Struct Biotechnol J. 2020;18:1221–27. doi: 10.1016/j.csbj.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, Oomen LA, Peipp M, Valerius T, Slootstra JW, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. The Journal Of Immunology. 2011;186(3):1840–48. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 52.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci. 1992;89(10):4285–89. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Metz S, Haas AK, Daub K, Croasdale R, Stracke J, Lau W, Georges G, Josel H-P, Dziadek S, Hopfner K-P, et al. Bispecific digoxigenin-binding antibodies for targeted payload delivery. Proc Natl Acad Sci. 2011;108(20):8194–99. doi: 10.1073/pnas.1018565108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ridgway JBB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng Des Sel. 1996;9(7):617–21. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 55.Czekanska EM. Mammalian Cell Viability. In: Stoddart, M editor. Assessment of Cell Proliferation with Resazurin-Based Fluorescent Dye. Humana Press; 2011;740:27–32. doi: 10.1007/978-1-61779-108-6_5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.