

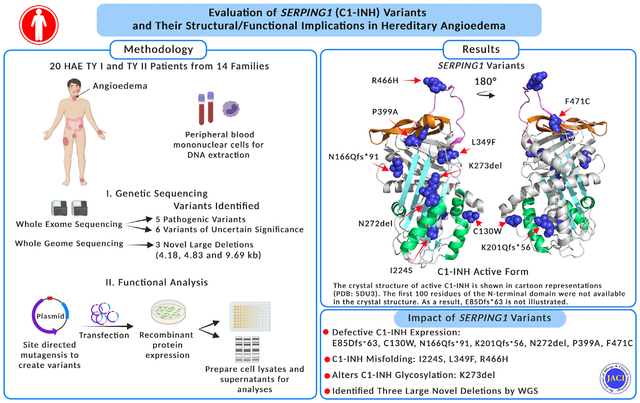
Abstract
Background:
The pathogenesis of hereditary angioedema (HAE) type I and type II is linked to defective C1 esterase inhibitor (C1-INH) encoded by the SERPING1 gene. There are substantial variabilities in the clinical presentations of patients with HAE that are not directly correlated to the serum levels of C1-INH. The impact of SERPING1 variants on C1-INH expression, structure, and function is incompletely understood.
Objective:
To investigate the influence of SERPING1 variants on the C1-INH expression, structure, and function of 20 patients with HAE from 14 families with no prior genetic diagnosis.
Methods:
Patients underwent whole-exome sequencing (WES). If no variants were identified, whole-genome sequencing (WGS) was performed. Except for the frameshift and large deletions, each C1-INH variant was recombinantly produced and, if synthesized and secreted, was subjected to structural, oligosaccharide, and functional analyses.
Results:
We identified 11 heterozygous variants in the SERPING1 gene, of which 5 were classified as pathogenic (E85Dfs*63, N166Qfs*91, K201Qfs*56, P399A, and R466H) and 6 as variants of uncertain significance (C130W, I224S, N272del, K273del, L349F, and F471C). Three large heterozygous deletions were discovered through WGS. Our data indicate that C130W, N272del, P399A, and F471C are poorly synthesized, I224S prevents proper C1-INH folding, and K273del impairs C1-INH function by adding an additional oligosaccharide. Further evaluation suggests that compound variant P399A/L349F contributes to a more severe clinical phenotype.
Conclusions:
Our combined approach of WES and WGS uncovered SERPING1 gene alternations in each patient. The recombinant protein production followed by systematic antigenic, structural, and functional assessment facilitates the identification of underlying pathogenic mechanisms in HAE.
Keywords: Hereditary angioedema, SERPING1 gene, C1 esterase inhibitor, mechanism, genetic variant
GRAPHICAL ABSTRACT
Hereditary angioedema (HAE) is a rare autosomal-dominant disorder with recurrent yet highly unpredictable episodes of soft tissue swelling.1 More than half the patients with HAE experience laryngeal attacks, of which approximately 50% are life-threatening because of asphyxiation.2 The worldwide prevalence of HAE is estimated to be between 1:10,000 and 1:50,000.3 The clinicopathologic events in HAE are caused by variants in the SERPING1 gene that encode the C1 esterase inhibitor (C1-INH).4
C1-INH is a heavily glycosylated protein with an estimated relative molecular weight (Mr) of 105,000, and it contains 7 N-linked and 14 O-linked oligosaccharides (Fig 1).5 It belongs to the serine protease inhibitor superfamily, with a highly conserved serpin domain comprising 9 α-helices and 3 β-sheets. The reactive site P1-P1’ (R466–T467) is located in a flexible reaction center loop (RCL), which involves protease recognition and mediates conformational transformation (Fig 2).6–8 C1-INH regulates complement, kinin, and contact system activation. A decrease in the function of the C1-INH leads to overproduction of bradykinin, which, in turn, causes increased vascular permeability and tissue swelling.9
FIG 1.
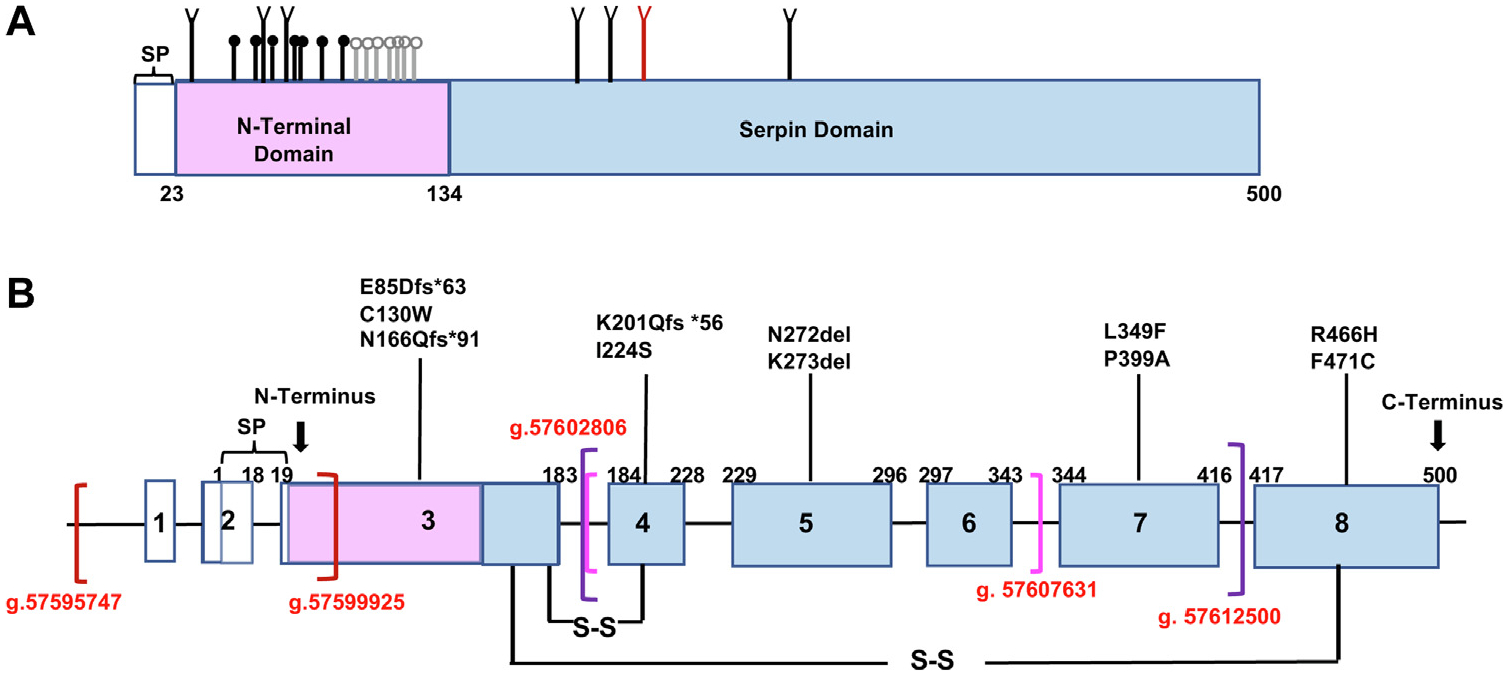
C1-INH point mutation spectrum in this cohort. A, Schematic representation of protein domains for C1-INH. SP contains 22 residues and is included in the numbering. The N-terminal domain includes residues 23–134 (pink). The C-terminal serpin domain consists of residues 135–500 (blue). N-glycosylation (n = 7) (branched symbols); verified O-glycosylation sites (n = 7) (black circles); potential O-glycosylation (n = 7) (gray circles); N272-glycosylation site (red) (slightly modified from Barnum and Schein5). B, Schematic representation of the C1-INH locus showing exon distribution (numbered boxes), untranslated regions (white boxes), protein-coding regions (blue boxes), and introns (black lines connecting boxes). Translation starts at exon 2. SP, Signal peptide; S-S, disulfide bonds. Brackets represent 3 large deletions in red, magenta, and purple, respectively.
FIG 2.
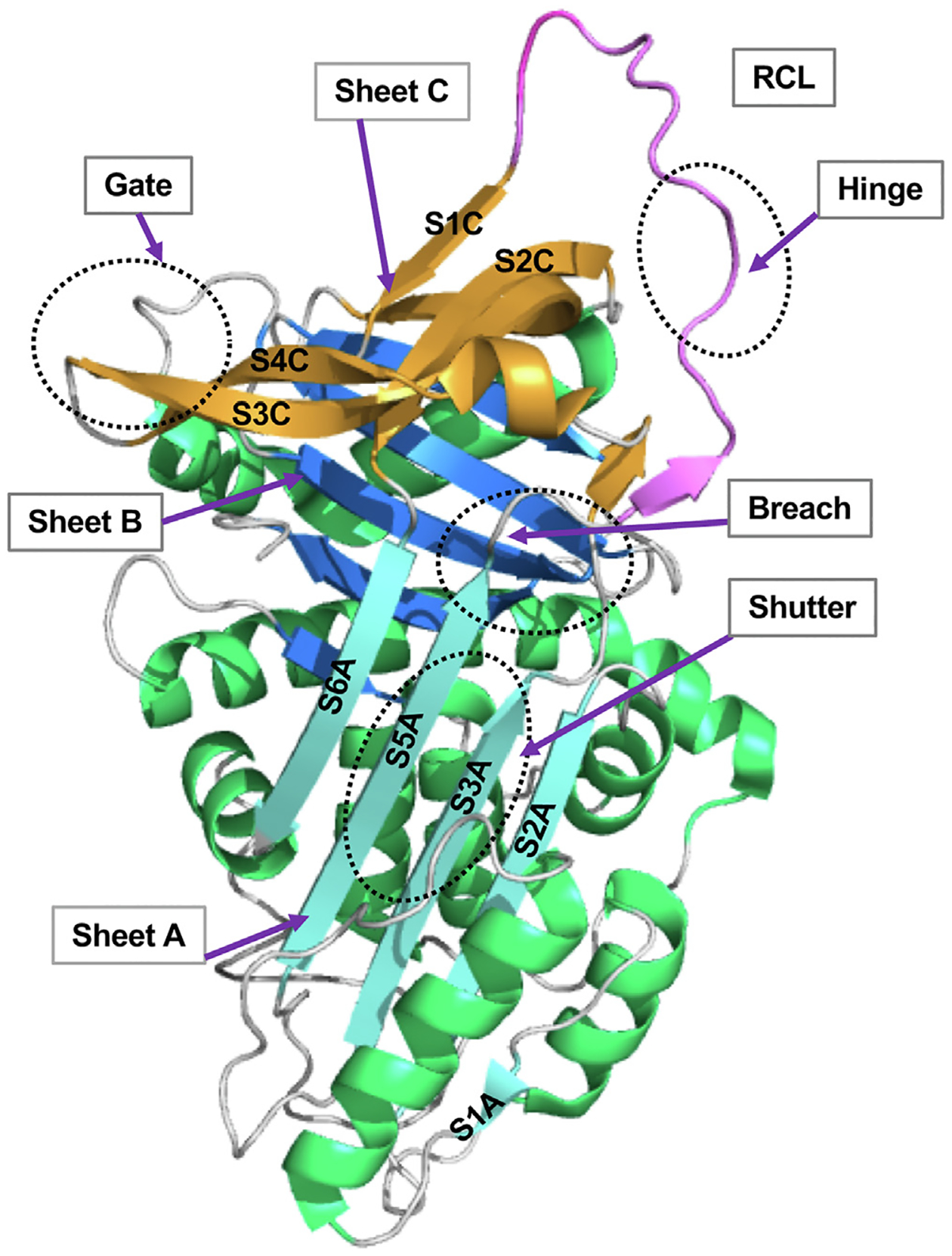
The overall structure of the key domains in C1-INH. Regions involved in the serpin function are labeled (PDB: 5DU3).6,7 The RCL is in pink. The P15–P9 portion of the RCL, the hinge domain, is highly conserved and facilitates the insertion of RCL into the β-sheets A (SA).6 The breach region lies on the top of SA, the initial insertion site of RCL. Composed of strands S3A and S5A, the shutter domain is located in the center of SA, which opens and accepts the RCL insertion. The gate region consists of strands 3 and 4 from β-sheets C (S3C and S4C).6–8 PDB, Protein Data Bank; SA, β-sheets A.
The SERPING1 gene is located on chromosome 11 (11q12.q13.1) and is approximately 17 kilobase pairs (kb) in length (Fig 1).10 More than 700 variants have been published in the SERPING1 gene, including single-nucleotide variants (SNVs), small insertions or deletions, large deletions, and duplications.11 The challenge in providing a definitive genetic diagnosis of HAE is that mutations are not easily detected in up to 10% of cases using conventional Sanger gene sequencing (SGS) panels. In addition to sequencing all SERPING1 coding regions, long-range PCR and multiplex ligation-dependent probe amplification are required to detect deletions, insertions, and copy number variations in the SERPING1 gene, which can be time-consuming and labor-intensive.12,13 Compared with SGS, targeted next-generation sequencing (NGS) is an efficient platform for the molecular diagnosis of HAE with a low average cost, which can be used as the first tool for HAE genotyping. However, it requires a large number of already diagnosed and genotyped HAE samples to validate, which consequently limits its application in small-sized single-center studies such as ours (see this article’s Methods section in the Online Repository at www.jacionline.org for additional rationale for using whole-exome sequencing [WES]).
HAE is routinely categorized on the basis of C1-INH laboratory findings, namely, low plasma antigenic levels (type I) or normal antigen but with a dysfunctional protein (type II). However, these 2 forms of HAE are clinically indistinguishable. In addition, there is substantial variability in the clinical manifestations in both type I and type II. Furthermore, the frequency and severity of angioedema (AE) attacks are not directly correlated to the serum level of C1-INH.14 Patients with HAE maintain 1 functional wild-type (WT) SERPING1 allele. Therefore, approximately 50% of the average C1-INH value would be expected. However, serum antigen levels of C1-INH in patients with HAE are usually 5% to 30% of the normal values.1 The molecular mechanism(s) leading to a decreased level of C1-INH is (are) not well understood. So far, there are no generally accepted processes to explain the decrease in circulating C1-INH.15–17
Furthermore, an increasing number of HAE cases are being reported with normal antigenic and functional C1-INH serum levels (HAE-nC1-INH). Rare pathogenic mutations of factor XII, plasminogen, angiopoietin 1, kininogen 1, myoferlin, and heparan sulfate glucosamine 3-O-sulfotransferase 6 have been reported in HAE-nC1-INH cases.18 Despite these encouraging discoveries, most HAE-nC1-INH cases fall into the AE of unknown cause category.
We present the results of a clinical cohort with HAE from a single tertiary referral center where the patients had not had genetic tests performed. We carried out WES to determine the genetic alterations in the SERPING1 gene. Whole-genome sequencing (WGS) was used for patients in whom no sequence alterations were detected. We then focused on characterizing rare SERPING1 variants identified in this cohort and examined their influence on protein folding, glycosylation, and C1-INH function. The long-term goal of this project is to explore the use of genomic sequencing to assess whether it could enhance our understanding of the pathological consequences of SERPING1 variants and facilitate treatment decisions.
METHODS
Study population
We studied 20 patients from 14 families with HAE who were followed in the Division of Allergy and Immunology at Washington University School of Medicine (WUSM). The diagnosis of HAE with impaired C1-INH activity was based on clinical symptoms and laboratory findings that meet the guidelines.19 This study, initiated in October 2021, was approved by the Institutional Review Board of WUSM, and informed written consent was obtained from each participant. Their clinical characteristics and complement levels are presented in Table I.
TABLE I.
Immunologic and clinical data from the cohort with HAE at WUSM
| Patient no. | HAE phenotype | Age (y) | Sex | Onset (y) | Location of edema* | Frequency of edema† | Duration of attacks (d) | C1-INH antigen (19–37 mg/dL) | C1-INH function‡ (%) | C4 (mg/dL) |
|---|---|---|---|---|---|---|---|---|---|---|
| 101 | Type I | 48 | F | 8 | E, F, L | 3/y | 2 | <3 | 0 | <4§ |
| 201 | Type I | 55 | M | 18 | A, E, F, L | 12/y | 2 | 8 | <10 | 3‖ |
| 301 | Type I | 73 | F | 12 | A, E, F, L | 15/y | 1 | 6 | 48 | 6§ |
| 401 | Type I | 58 | F | 16 | A, E, F, L | 8/wk | 2 | 4 | 0 | 2‖ |
| 501 | Type I | 49 | F | 13 | A, E, F, L | 3/wk | 3 | 10 | 43 | 3‖ |
| 502 | Type I | 44 | M | 8 | A, E, F, G, L | 4/wk | 1 | 7 | 42 | 2‖ |
| 503 | Type I | 8 | M | 8 | E, G | 2/y | 1 | 12 | 60 | 12‖ |
| 504 | Type I | 25 | F | 15 | A, E, F, G, L | 3/wk | 1 | 8 | 52 | <4‖ |
| 601 | Type I | 30 | M | 21 | A, E, F | 2/wk | 1 | 9 | 63 | 10§ |
| 701 | Type I | 40 | F | 33 | A, E, G, L | 2/y | 0.5 | 9 | 45 | 6‖ |
| 702 | Type I | 14 | M | 14 | A, E | 3/mo | 0.5 | 5 | 38 | 3¶ |
| 801# | Type I | 11 | F | 8 | A, E, F, L | 3/mo | 1 | 7 | 27 | 8§ |
| 901 | Type I | 30 | F | 1.5 | A, E, F, G, L | 3/wk | 1 | 4 | 56 | 10‖ |
| 902 | Type I | 30 | F | 1 | A, E, F, L | 1–2/wk | 2 | 11 | 38 | 3‖ |
| 903 | Type I | 68 | M | 10 | A, E, F, G, L | 2–3/mo | 1 | 14 | 85 | 16‖ |
| 1001 | Type I | 25 | M | 13 | A, E, F, G, L | 2–3/mo | 2 | 5 | 34 | 3‖ |
| 1101 | Type I | 26 | M | 14 | A, E, F, L | 1–2/wk | 1 | 8 | 68 | 6‖ |
| 1201 | Type II | 35 | F | 18 | A, E, F | 1/mo | 2 | 29 | 18 | NA |
| 1301 | Type II | 26 | M | 14 | A, E | 3–4/mo | 1 | 33 | 16 | 16** |
| 1401 | Type II | 17 | M | 0.5 | A, E, F, L | 2–3/wk | 2 | 24 | 24 | 2‖ |
All study candidates are White.
A, abdomen; E, extremities, F, face; G, genitals; L, larynx.
Frequency of edema before HAE treatment.C4 antigen levels were measured at 4 institutions with slightly different normal ranges.
C1-INH function: >67%, normal; 41%–67%, equivocal; <41%, abnormal.
Normal level of C4: 12–54.
Normal level of C4: 10–40.
Normal level of C4: 14–59.
Except for patient 801, all other patients have a family history of HAE.
Normal level of C4: 17–42.
Sequencing
Genomic DNA was isolated from peripheral blood samples using the Qiagen DNA blood kit (Qiagen, Hilden, Germany). WES was performed by the Genome Technology Access Center at WUSM using the IDT Exome (Integrated DNATechnologies, Coralville, Iowa) with 150 × coverage. WGS was performed with 30 × coverage. WES and WGS sequencing data were analyzed using a Dynamic Read Analysis for GENomics Bio-IT processor (Illumina, San Diego, Calif) and compared with the GRCh38 reference genome.20 Duplicate reads, SNVs, structural variants, and copy number variants were called for the genomic samples. SERPING1 reference sequence NM_000062.3 was used for variant calling. A variant was confirmed using PCR, with primers flanking each candidate mutation designed using Primer3. PCR products were purified with gel extraction (Qiagen) and then sequenced.
Preparation and expression of variants
The SERPING1 expression pcDNA3.1 vector (GeneScript, Piscataway, NJ) was used to construct the C1-INH variants. The variants were produced using the QuikChange XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, Calif). Each SERPING1 cDNA clone was sequenced. The variants were transfected into human embryonic kidney 293T cells where Dulbecco modified Eagle medium was replaced with OptiMEM (Invitrogen, Grand Island, NY). Transient transfections were performed using the Xfect reagent (Takara Bio, Mountain View, Calif). Supernatants were collected after 48 hours, centrifuged to remove debris, concentrated 40 ×, and stored in aliquots at −80 ° C.21
C1-INH quantification and functional analysis
The quantity of each recombinant C1-INH variant protein was determined using a C1-INH ELISA kit (Abcam, Cambridge, Mass) asper the manufacturer’s protocol.22 Purified C1-INH, recombinant C1-INH, and L349F were mixed with biotinylated C1s and then applied to the streptavidin-coated plate. Active C1-INH molecules were detected through binding with biotinylated C1s.23
Western blotting
Supernatants were analyzed under reducing conditions using 4% to 20% SDS-PAGE, transferred to nitrocellulose, probed with 1:1000 mouse C1-INH mAb (MA5–37889) (Abcam), recognizing the N-terminus of C1-INH between 22 aa and 100 aa, and then followed by a 1:5000 horseradish peroxidase goat antimouse IgG (Invitrogen, Carlsbad, Calif) (Fig 3).
FIG 3.
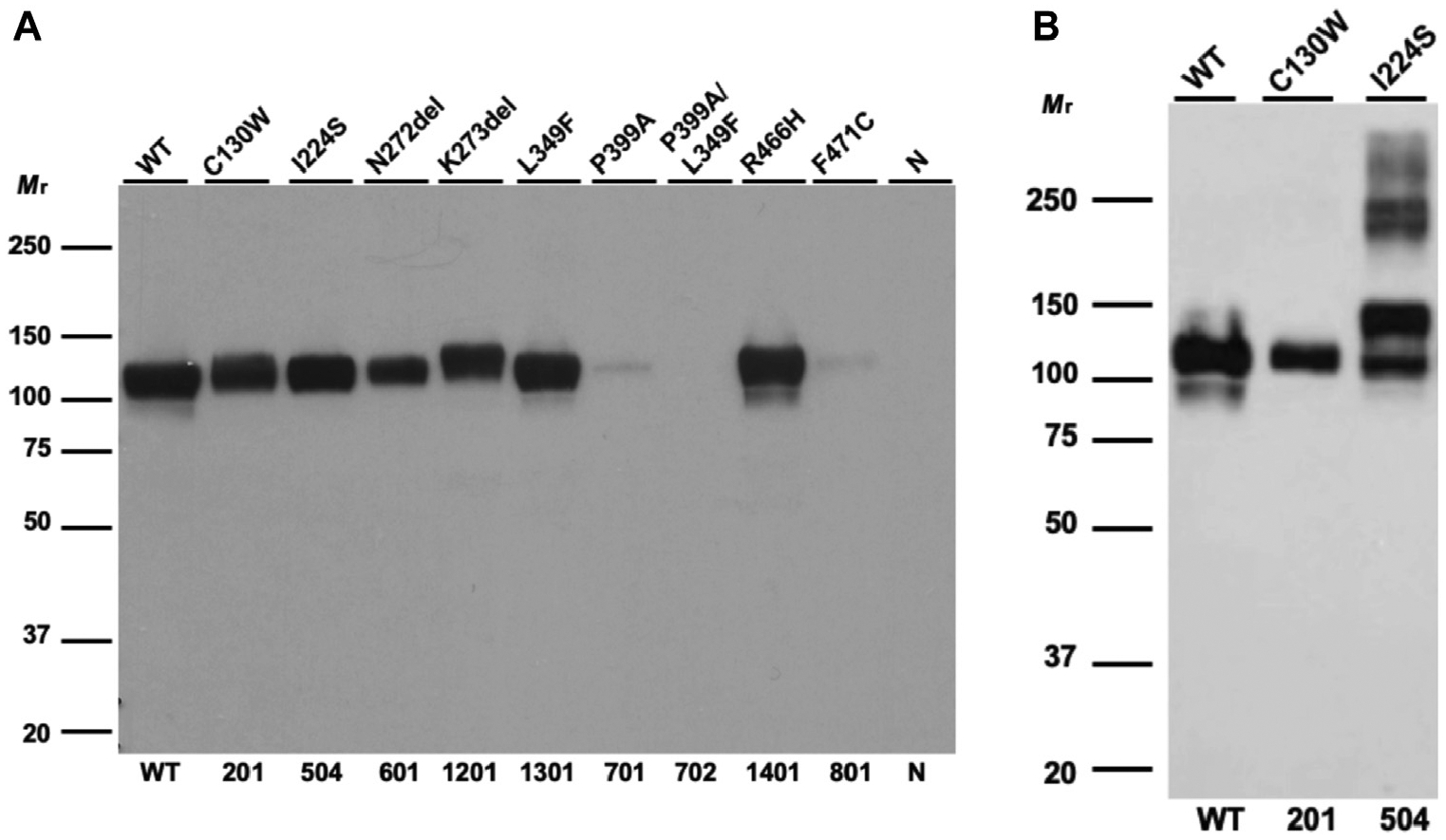
Recombinant expression of C1-INH variants. A, WB analysis of supernatants from transfected WT C1-INH and variant constructs in reducing conditions. Recombinant expressions of P399A, P399A/L349F, and F471C are markedly decreased (~90% compared with that of WT). The expressions of C130W and N272del are decreased by approximately 50% compared with that of WT. K273del and L349F have normal secretion compared with that of WT (Table II). B, WB analysis of supernatants from transfected WT and variant constructs in nonreducing conditions. I224S is secreted and forms oligomers. This experiment was repeated 4 times with similar results.
Molecular modeling
PyMOL (The PyMOL Molecular Graphics System, Version 2.1; Schrödinger, LLC, New York, NY) was used to visualize and analyze protein structures.
Statistical analyses
Statistical analyses were performed using Prism 9 (GraphPad, San Diego, Calif). Comparisons between 2 groups were assessed using paired t test (nonparametric). Comparisons among groups were performed using a 1-way ANOVA with Dunnett multiple comparison test (P <.05 was considered as significant).
More detailed methodologies including DNA library preparation, sequencing, variant classification, and molecular modeling are outlined in this article’s Methods section in the Online Repository.
RESULTS
Eleven variants were initially identified in 15 study candidates, including 6 SNVs, 2 nonframeshift (fs) deletions, and 3 fs variants (Fig 1 and Table II). Of the 6 SNVs, 1 (F471C) was novel and 5 (C130W, I224S, L349F, P399A, and R466H) were rare variants with a minor allele frequency (MAF) of less than 0.01%. Two insertions and deletions (N272del and K273del) have been reported.24,25 Among the 3 fs variants, 2 (N166Qfs*91 and E85Dfs*63) were novel, whereas K201Qfs*56 had been noted.26 These fs variations likely result in premature termination of C1-INH protein synthesis.27 No causative mutation was initially identified in 5 study candidates from 3 unrelated HAE families. However, when WGS was performed, we identified 3 large-size deletions in the SERPING1 gene, each not having been reported previously (Fig 1 and Table II).
TABLE II.
Pathogenetic data on the cohort with HAE at WUSM
| Patient no. | Mutation | Protein | Region | ACMG/AMP classification | Recombinant secretion (μg/mL), mean ± SEM* | P value for recombinant variant† | ||
|---|---|---|---|---|---|---|---|---|
| Location | gDNA numbering | cDNA numbering | ||||||
| 101 | Exon 3 | g.57600081delA | c.255del | p.E85Dfs*63 | N-terminus | Pathogenic‡ | nd | nd |
| 201 | Exon 3 | g.57600217C>G | c.390C>G | p.C130W | N-terminus | VUS | 18.0 ± 2.3 | § |
| 301 | Exon 3 | g.57600321insA | c.494_495insA | p.N166Qfs*91 | s6B | Pathogenic‡ | nd | nd |
| 401 | Exon 4 | g.57602080dupC | c.600dup | p.K201Qfs*56‖ | Loop after Helix C | Pathogenic | nd | nd |
| 501, 502, 503, 504 | Exon 4 | g.57602155T>G | c.671T>G | p.I224S | s2A strand | VUS | 32.2 ± 2.8 | § |
| 601 | Exon 5 | g.57606134_57606136delCAA | c.816_818del | p.N272del | Helix F/s3A | VUS | 16.0 ± 1.6 | § |
| 701 | Exon 7 | g.57611882C>G | c.1195C>G | p.P399A¶ | s2C | Likely pathogenic | 0.2 ± 0.2 | § |
| 702 | Exon 7 | g.57611732C>T | c.1045C>T | p.L349F¶# | Distal hinge (s2B) | VUS | 34.7 ± 2.3 | NS |
| Exon 7 | g.57611882C>G | c.1195C>G | p.P399A¶ | s2C | Likely pathogenic | 0.2 ± 0.2 | § | |
| 801 | Exon 8 | g.57614490T>G | c.1412T>G | p.F471C | s4B | VUS‡ | ND | nd |
| 901, 902, 903 | Exon 1–2 | g. 57595747_ 57599925del | Large deletion 4.18 kb | nd | nd | Pathogenic | nd | nd |
| 1001 | Exon 5–6 | g. 57602806_ 57607631del | Large deletion 4.83 kb | nd | nd | Pathogenic | nd | nd |
| 1101 | Exon 5–7 | g.57602806_57612500del | Large deletion 9.69 kb | nd | nd | VUS | nd | nd |
| 1201 | Exon 5 | g.57606141_57606143delAAG | c.818_820del | p.K273del | Loop after Helix F | VUS | 37.6 ± 0.9 | NS |
| 1301 | Exon 5 | g.57606141_57606143delAAG | c.818_820del | p.K273del | Loop after Helix F | VUS | 37.6 ± 0.9 | NS |
| 1401 | Exon 8 | g.57614475G>A | c.1397G>A | p.R466H‖ | RCL-P1 | Pathogenic | 44.7 ± 3.3 | NS |
ACMG/AMP, American College of Medical Genetics/Association of Molecular Pathology; ND, not detected; NS, no significant difference; nd, not done.
WT recombinant C1-INH concentration, 36 ± 3.1 μg/mL (7 experiments with 95% CI ranging from 29.1 to 44.1 μg/mL).
P values were computed by using 2-sided independent-sample t tests and comparing the results with those for the WT.
Novel mutation.
P < .0001.
Two submitters reported in ClinVar.
One single submitter in ClinVar.
Variant c.1045C>T (p.L349A) has an MAF of 0.007% on gnomAD. The MAF information is not reported for other variants.
To further ascertain the pathological consequences of the variants, we investigated the 6 missense SERPING1 variants. Antigenic, functional, and structural analyses were carried out.
Family 5, a classic HAE type I kindred
Patient 504.
This 22-year-old woman was admitted to her local hospital for laryngeal edema. She had an episode of upper respiratory tract infection that started 2 days before admission. Treatment with antihistamines, epinephrine, and fresh frozen plasma did not lead to improvement. She was intubated for airway protection and transferred to our center. Her symptoms improved within approximately 4 hours after starting on human C1-INH concentrate.
A heterozygous variant I224S in the SERPING1 gene was identified. I224S was previously reported in a large cohort with HAE from Spain.28 Genomic information on this variant is, however, absent in the Genome Aggregation Database (gnomAD). Variant I224 is located in S2A near the shutter region, facilitating the RCL insertion (Fig 4, A). The hydrophobic I bearing a long side chain enhances the stability of the hydrophobic core of β-sheets A (SA) in C1-INH. Serine is a hydrophilic residue that prefers to be in an α-helix structure than in a β-strand. The change of I224 to S could disrupt the stability of SA (Fig 2). Recombinant expression of I224S was comparable with that of WT. However, there was a significant degree of oligomerization of this variant (Fig 3, B), providing a potential explanation for this patient’s low degree of C1-INH antigen (Table II). These data suggest that variant I224S, albeit secreted in normal quantities, is likely misfolded, leading to protein aggregation and a reduced intravascular half-life.
FIG 4.
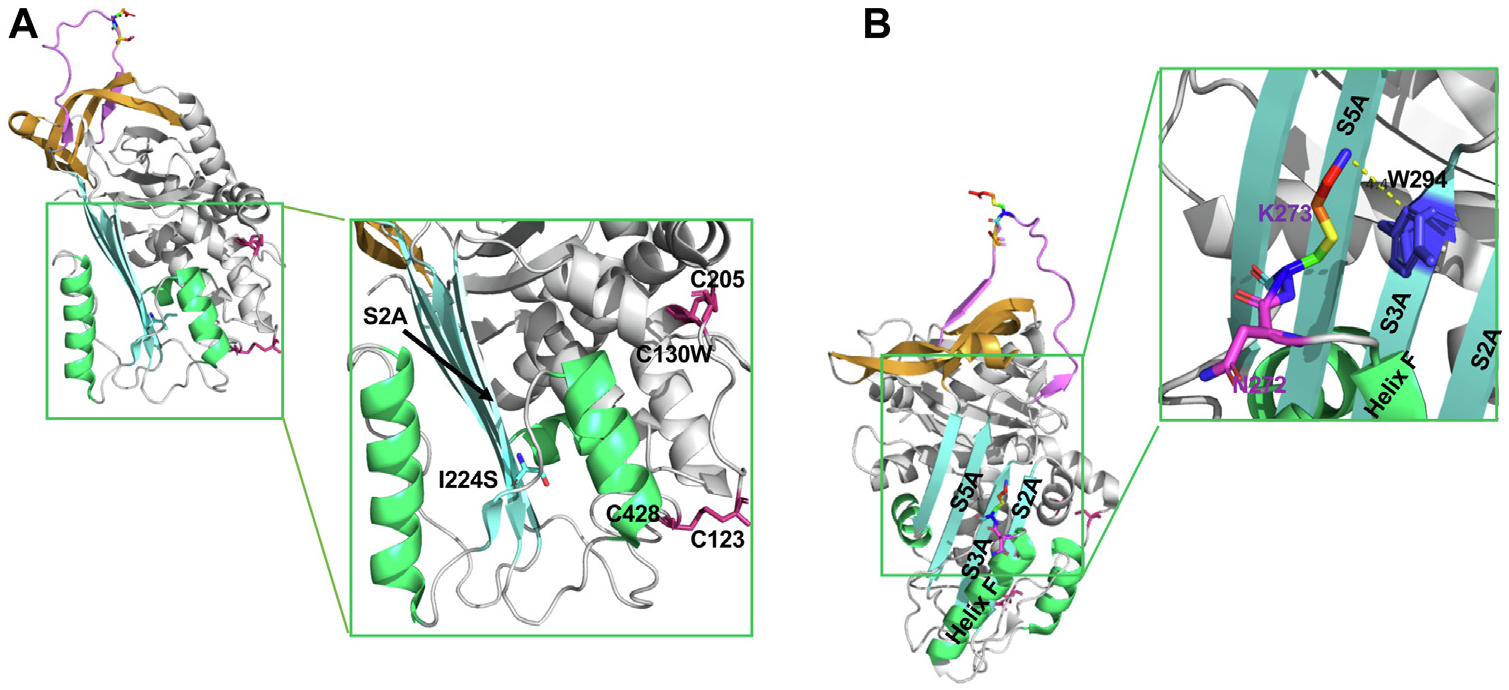
Structural analysis of I224S, N272del, and K273del. A, Structural analysis of I224S. The “reoriented” structure of active C1-INH is shown in a cartoon representation (PDB: 5DU3).7 Isoleucine has a large hydrophobic side chain, which enhances the stability of the hydrophobic core of β-strands in C1-INH. The change of I224 to S destabilizes the hydrophobic core of the antiparallel β-strands (SA) in C1-INH. The 2 disulfide bonds are shown in pink. B, Structural analysis of N272del and K273del. N272 is one of the N-glycosylation sites. The deletion of N272 will likely disrupt the N-linked glycosylation site and impair protein folding. K273 is located in the loop right after helix F. The side chain of K273 forms the stabilizing cation-π interactions with the aromatic side chain from W294. The deletion of K273 will disrupt this interaction and affect the conformation of helix F. K273del previously reported resulting in a new N-glycosylation site in C1-INH.25 The N272 residue is shown in pink and K273 is shown in rainbow. PDB, Protein Data Bank; SA, β-sheets A.
Genetic neighbors, N272del and K273del
Patient 601.
This 30-year-old man developed his first AE episode at age 24 years. Since then, he has experienced swelling of limbs and/or stomach approximately 2 times a week. During each swelling episode, he took icatibant, leading to symptom control within about an hour. Subsequently, lanadelumab was started for prophylactic management and the frequency of his AE episodes decreased to approximately 2 times a year.
Patient 1201.
This 36-year-old woman with a history of HAE type II became symptomatic at age 18 years. She developed recurrent extremity and/or stomach swelling, usually occurring a few days before her period. Icatibant was inadequate to control her acute symptoms. After subcutaneous (SC) administration of ecallantide, she developed flushing, dyspnea, and loss of consciousness within a few minutes and has been avoiding it since (see Table E1 in this article’s Online Repository at www.jacionline.org). Since starting C1-INH concentrate SC for prophylactic treatment in November 2019, her AE flares have markedly improved, with the frequency of attacks reduced to approximately 5 times a year.
Patient 1301.
This 31-year-old man presented with episodes of stomach pain or testicular swelling weekly since his early teens. He recalled an attack of stomach pain when he was 14 years old, which was thought to be secondary to appendicitis. An operation was performed, and a normal appendix was removed. At age 28 years, the patient was diagnosed with HAE type II. Icatibant was initially used weekly to manage acute AE attacks. He was then started on lanadelumab, with a remarkable improvement in symptoms considering that he has had no further episodes since its initiation in December 2019.
A heterozygous variant N272del in the SERPING1 gene was identified in patient 601, whereas patients 1201 and 1301 harbored a heterozygous variant—K273del, a genetic neighbor of N272del. There is no available genomic information for these 2 variants in gnomAD; therefore, they were classified as variants of uncertain significance (VUS). N272 is an N-linked glycosylation site (Fig 4, B). The consensus sequence for an N-glycosylation site is NXS/T/C, where “X” can be any amino acid except proline.29,30 The protein sequence for the N272 glycosylation site is NN(272)KIS. The deletion of N272 likely disrupts the recognition site, thereby altering protein folding and function. K273 is located in the loop connecting helix F and S3A. The deletion of K273 decreases the flexibility of the helix F/S3A loop, which is critical for the RCL insertion during the transition from an active to a latent state (Fig 5). Recombinant expression of variant N272del shows decreased secretion, whereas the expression of K273del was normal compared with that of WT (Table II). The variants N272del and K273del had Mr’s of 105,000 and 110,000, respectively, on Western blotting (WB), indicating a possible additional N-glycosylation site on K273del (see Fig E1 in this article’s Online Repository at www.jacionline.org). To confirm this observation, we conducted an analysis of the variants N272del and K273del. After the treatment with glycosidases, recombinant proteins N272del, K273del, and WT were equivalent in size by WB (see Fig E1).
FIG 5.
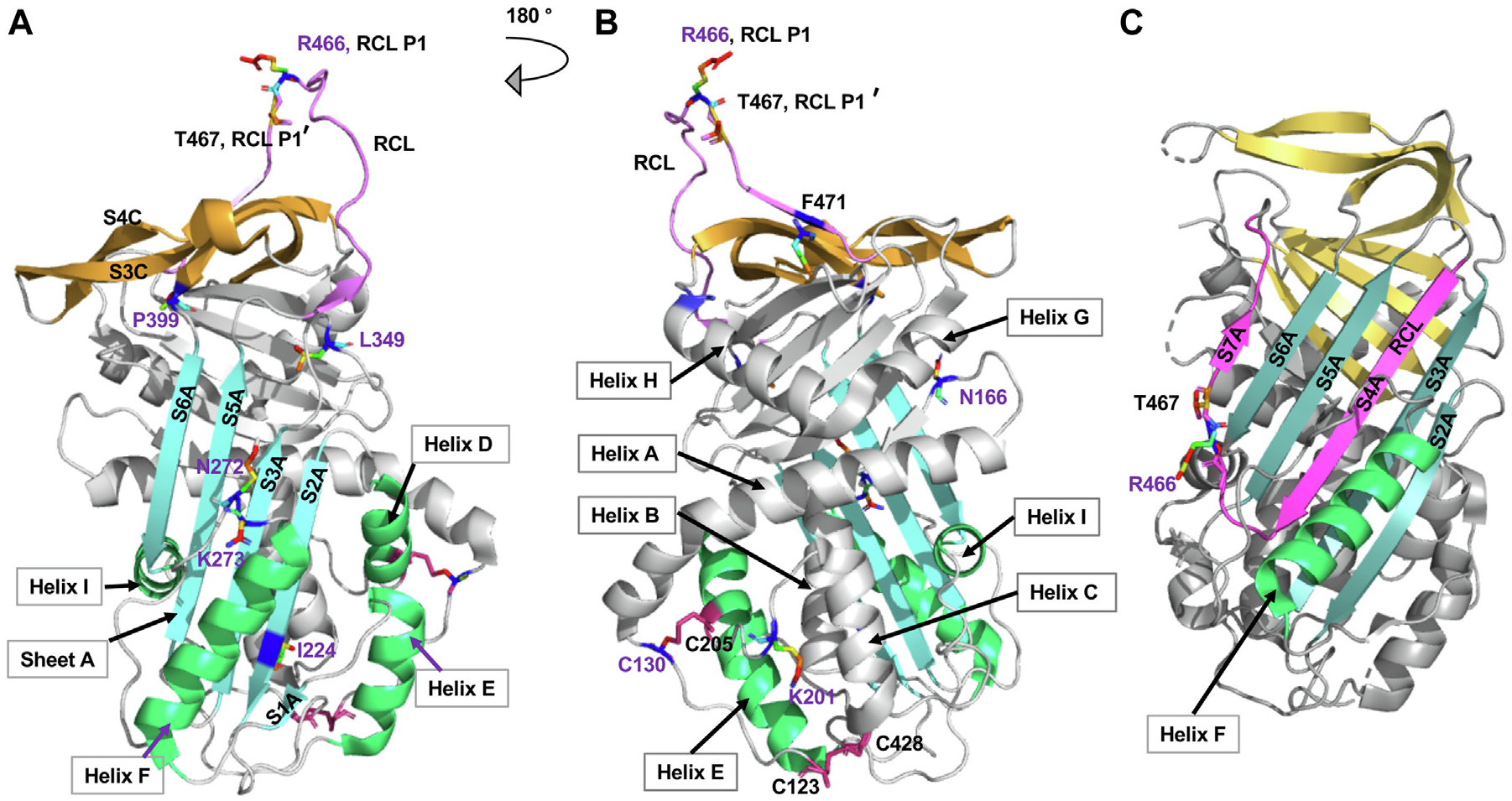
Structure of active and latent forms of C1-INH. A and B, The structure of active C1-INH is shown in a cartoon representation in 2 different orientations (PDB: 5DU3).7 The RCL is in pink. The P1 R466 and P19 T467’ residues are displayed in rainbow stick and are responsible for trapping the target protease. Strands of central SA are in cyan. Helix F1 is the polyanion binding site. The disulfide bridges are labeled and colored in red. The variants identified in our cohort are labeled in purple. C, The structure of the latent form of C1-INH is shown in a cartoon representation (PDB: 2OAY).8 The RCL is incorporated into sheet A, forming S4A. PDB, Protein Data Bank; SA, β-sheets A.
Our interpretation is that the N-glycosylation site N272 is critical for C1-INH protein folding and function. Deletion of N272 results in decreased C1-INH antigen level and function, whereas deletion of the adjacent K273 results in only reduced C1-INH function (Table II).
Family 7, carrying 2 SERPING1 variants
Patient 701 (proband, mother).
This 40-year-old woman began to experience AE symptoms at age 33 years. Swelling occurred on her hand, arm, and/or abdomen approximately 2 times a year (Table I). Icatibant was used as needed to control her symptoms, which commonly took approximately 12 hours to fully resolve.
Patient 702 (son).
This 14-year-old boy experienced hand, arm, and/or abdominal swelling since age 7 years. The episodes occur approximately 3 times a month. He uses icatibant on an as-needed basis, with symptom relief within approximately 6 hours.
The proband is heterozygous for the P399A variant in SERPING1. The son, however, has heterozygous SERPING1 variants, L349F and P399A. L349F has an MAF of 0.007% in gnomAD and is classified as VUS because of a lack of functional data (Table II). P399A has been reported in ClinVar and classified as likely pathogenic.4 SGS of patient 702 confirmed that L349F and P399A are located on different alleles (see Fig E2 in this article’s Online Repository at www.jacionline.org).
The substitution of L349 to F introduces an aromatic side chain that likely generates clashes between L349 and W299 (Fig 6, A and B).31–33 Because W299 directly links to S3A, the steric clashes could perturb the conformation of S3A and thereby disrupt the RCL insertion into the shutter region. The P399 is at the end of the gate domain as it connects to the center shutter domain through S6A. The rigid pyrrolidine ring on P399 constrains the main chain dihedral angle and restricts the conformation of the preceding S6A (Fig 6, A). Substituting P with A would destabilize the protein and thereby lead to faster degradation.34 L349F, P399A, and the combined variant P399A/L349F were expressed recombinantly. The results demonstrated that the L349F variant did not alter C1-INH protein secretion, whereas the secretion of P399A and P399A/L349F was abolished (Fig 3, A; see Fig E3 in this article’s Online Repository at www.jacionline.org).
FIG 6.
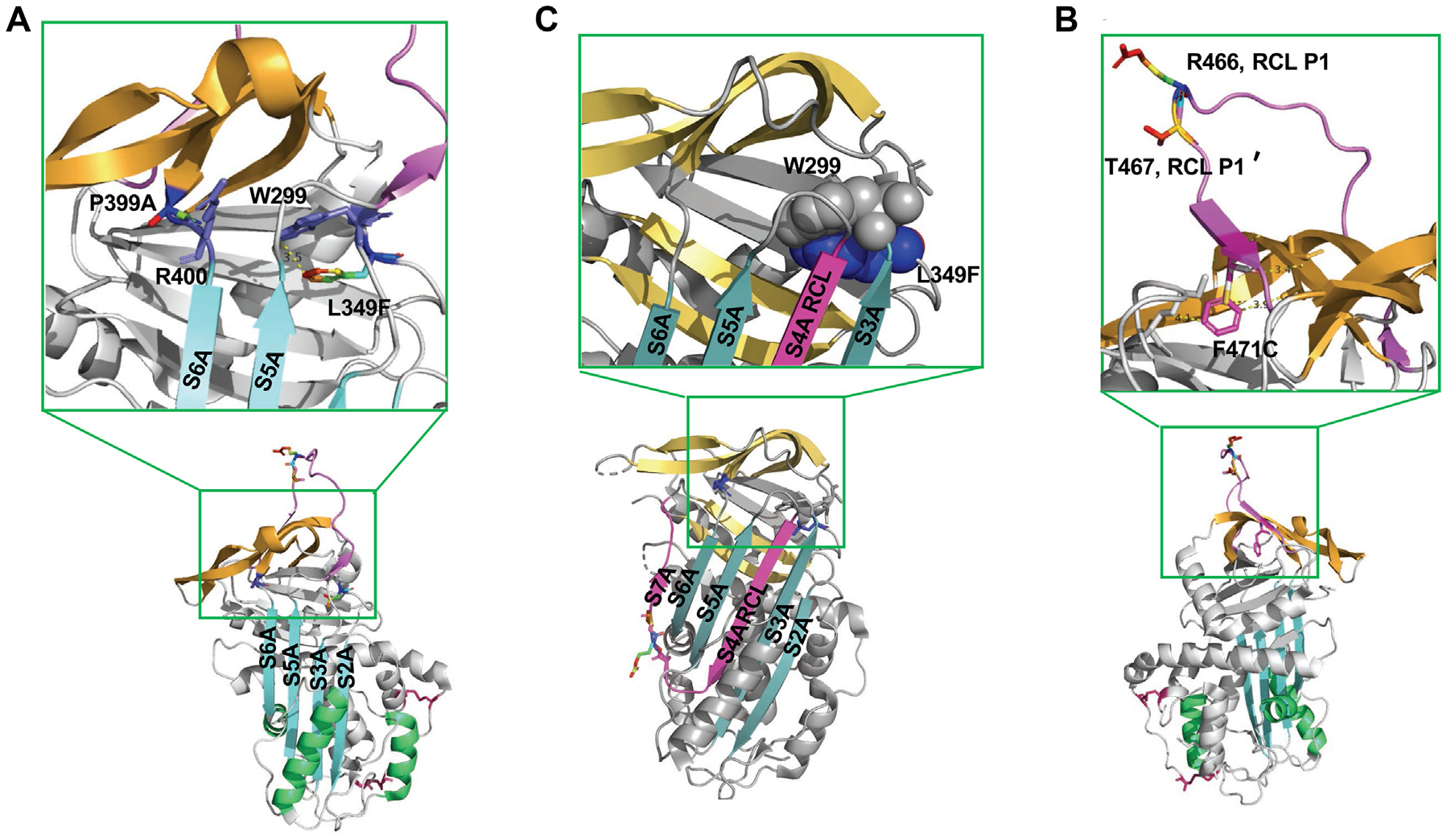
Structural analysis of L349F, P399A, and F471C. A and B, L349 is located in the loop in front of the strands of β-sheet B (PDB: 5DU3 and 2OAY).7,31 The substitution of aromatic amino acid F to L349 may cause side-chain clashes with the W299, which could further alter the conformation of S3A and affect the RCL insertion. The P399 is located in a tight turn connecting to the center shutter S6A. The alternation of P to A will disrupt the loop formation and further affect the orientation of R400. C, Structural analysis of F471C. F471 is located at the end of the RCL (PDB: 5DU3).7 It is a highly conserved residue in serpins.32 F is a hydrophobic aromatic amino acid in the protein hydrophobic core. C belongs to the group of polar amino acids. It often resides at the surface of proteins. The introduction of a polar side chain from F471C disrupts the RCL insertion.33 PDB, Protein Data Bank.
Notably, the proband has less severe disease than her son on the basis of the late-onset age and less frequent AE attacks. Unusually, the son carries 2 SERPING1 variants. One is inherited from his mother, whereas the source of the other remains unknown because the unaffected father’s DNA sample is not available for confirmation. We reasoned that carrying 2 defective alleles leads to a more severe AE phenotype in the son.
Family 8, a sporadic heterozygous SERPING1 variant
Patient 801.
An 8-year-old girl presented with intermittent swelling of her arms and episodes of abdominal pain. Complement studies were consistent with HAE type I (Table I). Ecallantide was started, but she developed an anaphylactoid reaction (see Table E1). She was unable to tolerate lanadelumab because of headaches and insomnia and had frequent (~1–2 times a week) breakthrough attacks while on berotralstat. However, she tolerated the C1-INH SC formula and obtained marked improvement with a decreased frequency of AE episodes from almost weekly to approximately 6 to 8 times a year. Her parents have normal C1-INH antigen and function.
A novel heterozygous variant, F471C, in the SERPING1 gene was identified. It has not been reported in gnomAD previously and is classified as a VUS because of its rarity and lack of functional data. F471 is located distal to the hinge region. It facilitates the mobility of RCL and is critical to loop insertion (Fig 6, C).35 It is highly conserved among serine proteases.32,33 The aromatic side chain from F471 is buried in the hydrophobic protein core, whereas C is a polar amino acid that usually resides on the surface of proteins. The F471C alteration will likely affect the packing of the RCL and lead to degradation. Consistent with this appraisal, WB barely detected secretion of the recombinantly produced F471C (Fig 3). Our protein expression analyses established that the SERPING1 variant F471C is likely deleterious.
Cases that underwent WGS
Patient 901 (family 9).
This 27-year-old woman was initially diagnosed with HAE type I at 18 months. From the age of 3 until age 14 years, she had extremely frequent attacks (~3–4 times a week). At age 15 years, she was placed on danazol, which significantly reduced attacks to approximately 1 to 2 times a year. At age 25 years, danazol was tapered off because of hyperlipidemia, and berotralstat was initiated for long-term prophylactic therapy. She had AE attacks approximately 4 times a week while on berotralstat. In 2019, lanadelumab was administered, with decreased AE episodes to approximately once a month.
Patient 1001.
This 25-year-old man was diagnosed with HAE type I at age 13 years after which he took danazol daily for HAE control. He still experienced extremity swelling approximately 2 to 3 times a month and abdominal pain or throat swelling approximately 2 times a year. After discontinuing danazol at age 18 years, he started to have AE attacks weekly. He was hospitalized at age 19 years for throat swelling that required intubation. Since the initiation of lanadelumab at age 22 years, his AE episodes have decreased from weekly to approximately 6 times a year.
Patient 1101.
For this 26-year-old man, AE attacks occurred primarily as abdominal pain or extremity swelling, approximately 6 times a year. Initially, his AE symptoms were controlled on an as-needed icatibant. At age 19 years, he began experiencing AE episodes weekly. At age 25 years, he started on lanadelumab biweekly as prophylactic therapy. Since then, his AE episodes decreased drastically, from almost weekly to approximately 2 times a year. During his follow-up visit, he asked whether his baby would have HAE because he anticipated becoming a father in about 2 weeks.
WES revealed no causative variant in these individuals. However, WGS demonstrated that patient 901, her twin sister (patient 902), and their father (patient 903) all carried a 4.18-kb deletion spanning exons 1 and 2 in the SERPING1 gene. Patient 1001 had a 4.83-kb deletion spanning exons 5 and 6, whereas patient 1101 harbored a 9.69-kb deletion affecting exons 5, 6, and 7 (Table II and Fig 1, B). These large deletions are located in deep intronic regions and cover coding areas. Detailed genetic information on these deletions is lacking in gnomAD and the Database of Genomic Variants. Also, of note, among the 5 individuals who underwent WGS, 4 are of childbearing age. The knowledge of large deletions uncovered by WGS will directly contribute to choosing the appropriate genetic testing methods to facilitate an early diagnosis.
DISCUSSION
In this study, we have characterized genetic, protein structural, and functional information for a cohort with HAE with no prior genetic diagnosis. We highlighted lessons learned from establishing the genetic diagnosis by (1) ascertaining the potential pathogenic role of a novel variant; (2) accessing the mechanism of variation on protein structure that leads to a decreased C1-INH level; (3) evaluating the impact of a glycosylation site on CI-INH laboratory phenotype (type I vs type II); (4) defining the role of compound variants in the HAE phenotype; and (5) exploring the use of WGS to detect large deletions not discovered by a conventional sequencing platform. In addition, de novo variants are reported to account for 20% to 25% of the probands,26,36,37 whereas only 1 proband (5%) in our cohort carried a de novo mutation. Its secretion was abolished, demonstrating a deleterious effect of this novel variant.
Assessing the expression of recombinant C1-INH variants provided key data for multiple variants. For example, it demonstrated that I224S, a mutation located in S2A in proximity to the shutter region (Fig 5, A), results in C1-INH forming oligomers extracellularly. A similar finding was observed in a hinge region variant, A458T, likely resulting in the polymerization of C1-INH in the patient’s serum.38 These observations provide a potential mechanism to explain a decrease in circulating C1-INH.
C1-INH is a heavily glycosylated protein but the exact role of “glycosylation” relative to the function of C1-INH remains unclear (Fig 1).39 N272 is an N-glycosylation site whose deletion resulted in a decreased protein level both in vitro and in vivo. The genetic neighbor, K273del, caused a decreased C1-INH function, specifically a decreased binding affinity for the C1s substrate (Table II), but there was no impact on the protein antigen level. K273del created an additional N-glycosylation site (next to the N272), which could hinder the C1-INH transformation from the active to the latent state. These observations support the hypothesis that the N272-glycosylation site is crucial for protein stability and activity.40
Defining the role of compound variants in the SERPING1 gene has been challenging.30 In our cohort, compound variant P399A/L349F was detected in patient 703 in a trans configuration. Interestingly, a more severe phenotype was observed in patient 703 compared with that in his mother (patient 702) who harbored a single deleterious variant P399A. Another compound variant (A21V/L349F) was reported in a kindred where it was associated with HAE type I phenotype by 2 daughters, but the heterozygous parents were asymptomatic with normal C1-INH function and expression.30 On the basis of these observations, we conclude that L349F is likely pathogenic.
HAE diagnosis of newborns is established by the same complement testing as adults. However, unlike in adults, the levels of C4 and C1-INH in newborns are often decreased because of an immature complement system.41 Therefore, the offspring of a patient with HAE is considered affected until proven otherwise, and repeat complement testing is recommended.41,42 If a parental variant is known, genetic testing can be performed to facilitate an early diagnosis. The ability to offer WGS allowed us to discover large deletions that WES did not detect. This genetic information enables clinicians to choose the appropriate methods to offer an early diagnosis for newborns. Notably, all the patients presenting with large deletions have responded well to lanadelumab (see Table E1). We speculate that these patients have functional C1-INH synthesized from the normal SERPING1 alleles. Blocking the activation of the kallikrein-kinin cascade results in a decreased consumption of C1-INH,43 which likely contributes to their excellent response to lanadelumab.
The combined approach of WES and WGS uncovered all SERPING1 gene alterations in our cohort. Characterization of the recombinant protein production followed by systematic antigenic and structural assessment enables the discovery of underlying pathogenic mechanisms in HAE. Putative limitations to our analyses include the following: (1) the cost of WGS is 2-fold compared with that of WES and is not covered by standard medical insurance; (2) the time interval between analyzing WES and WGS data was about 3 to 6 months in our study; and (3) the clinical observations are drawn from a limited number of study candidates. We shall continue to collect further data to confirm our observations. To further investigate therapeutic responses, we plan on analyzing the function of C1-INH variants in complement, contact, and coagulation systems.23 As outlined, we anticipate that our approach to defining the disease will allow for further informative insights into its pathophysiology and treatment.
METHODS
Rationale for the genetic sequencing platform used
When we started the study in October 2020, we sought to use NGS but soon realized that we would need to wait to apply this approach. The reasons were the following: (1) To build the platform, we needed to design unique probes to cover the entire 17-kb SERPING1 gene. The coverage rate of the SERPING1 gene was only about 80%.E1 (2) Also, validating the NGS platform for SERPING1 genotyping became problematic. We would require a large number of samples to validate the platform. Because we were a single-study center and none of our patients had a genetic diagnosis, it was challenging to validate the NGS platform. (3) After obtaining the total cost of designing specific probe sets, including the SERPING1 gene and other genes associated with HAE (usually starting with 96 count/set/gene), we found that the total cost was higher than that for performing WES. For these reasons, we decided to apply WES as the first genetic sequencing tool in our study candidates, including HAE type I, HAE type II, and HAE-nC1-INH.
WES and WGS library preparation, sequencing, data processing, and variant calling
DNA libraries were constructed using a SciCloneG3 NGS instrument (Perkin-Elmer Inc, Waltham, Mass) and KAPA Hyper PCR-free Library Prep Kits (KAPA Biosystems, Wilmington, Mass). Genomic DNA of 600 ng was fragmented using a Covaris LE220 (Covaris, Woburn, Mass) targeting 350-bp inserts. The libraries were size-selected using a paramagnetic bead cleanup after fragmentation. Sequence libraries were pooled at an equimolar ratio yielding up to 5 μg per library pool before the hybrid capture. Library pools were hybridized with the Gen Exome Research Panel v1.0 reagent (IDT Technologies, Coralville, Iowa) according to manufacturer protocols.
The concentration of each library pool was determined using qPCR (KAPA Biosystems) and diluted to 2.5 μM. The libraries were sequenced on the NovaSeq6000 Instrument (Illumina, San Diego, Calif), generating 2 × 150 paired-end reads. Illumina’s bcl2fastq2 software was used to demultiplex individual libraries and generate FASTQ files.
The sequence reads were analyzed using a Dynamic Read Analysis for GENomics Bio-IT processor (Illumina) using the latest software available at the time of processing (v3.7–v3.9). Reads were mapped to human genome reference GRCh38 and output in Compressed Reference-oriented Alignment Map (CRAM) format, duplicate reads were marked, and SNV and structural variants were called. Copy number variants were also called for the genomic samples.
All variants are reported according to the Human Genome Variation Society nomenclature and classified on the basis of the guidelines established by the joint consensus of the American College of Medical Genetics and the Association of Molecular Pathology. On the basis of these standards, variants are classified as follows: level 1, pathogenic; level 2, likely pathogenic; level 3, VUS; level 4, likely benign; and level 5, benign.
Supplementary Material
Key messages.
This study investigated the influence of SERPING1 gene variants on the C1-INH expression, structure, and function.
Through this work, we sought to enhance our understanding of the pathological consequences of SERPING1 variants in HAE and facilitate treatment approaches.
Disclosure of potential conflict of interest:
H. J. Wedner reports receiving research funding from BioMarin, KalVista, Pharvaris, and Takeda; consulting fees from CSL, Takeda, BioCryst, and BioMarin; and speaking fees from Genentech, GlaxoSmithKline, CSL, Takeda, and BioCryst. The rest of the authors declare that they have no relevant conflicts of interest.
This study has been supported by the Division of Allergy and Immunology in the Department of Medicine at the Washington University School of Medicine, the National Institutes of Health under award R35 GM136352 (J.P.A.), and the Washington University Institute of Clinical and Translational Sciences, which is, in part, supported by the National Institutes of Health/National Center for Advancing Translational Sciences (CTSA grant no. UL1TR002345). This publication is solely the responsibility of the authors and does not necessarily represent the official view of the National Center for Research Resources or the National Institutes of Health.
We thank Dr Jonathan Sheehan and M. Kathryn (Kathy) Liszewski (WUSM) for their helpful suggestions related to structural analysis, experimental design, and manuscript preparation. We also thank the participants of the HAE study without whose collaboration this study could not have been accomplished. We thank the clinical research team in the Division of Allergy and Immunology at WUSM who were responsible for patient recruitment, samples, and data handling. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. This center is partially supported by NCI Cancer Center Support Grant P30CA91842 to the Siteman Cancer Center and by ICTS/CTSA (grant no. UL1TR002345) from the National Center for Research Resources, a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research.
Abbreviations used
- AE
Angioedema
- C1-INH
C1 esterase inhibitor
- fs
Frameshift
- gnomAD
Genome Aggregation Database
- HAE
Hereditary angioedema kb: Kilobase pairs
- MAF
Minor allele frequency
- NGS
Next-generation sequencing
- RCL
Reaction center loop
- SC
Subcutaneous
- SGS
Sanger gene sequencing
- SNV
Single-nucleotide variant
- VUS
Variant of uncertain significance
- WB
Western blotting
- WES
Whole-exome sequencing
- WGS
Whole-genome sequencing
- WT
Wild type
- WUSM
Washington University School of Medicine
REFERENCES
- 1.Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008;359: 1027–36. [DOI] [PubMed] [Google Scholar]
- 2.Hofman ZL, Relan A, Zeerleder S, Drouet C, Zuraw B, Hack CE. Angioedema attacks in patients with hereditary angioedema: local manifestations of a systemic activation process. J Allergy Clin Immunol 2016;138:359–66. [DOI] [PubMed] [Google Scholar]
- 3.Altman KA, Naimi DR. Hereditary angioedema: a brief review of new developments. Curr Med Res Opin 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 4.Bissler JJ, Aulak KS, Donaldson VH, Rosen FS, Cicardi M, Harrison RA, et al. Molecular defects in hereditary angioneurotic edema. Proc Assoc Am Physicians 1997;109:164–73. [PubMed] [Google Scholar]
- 5.Barnum SR, Schein TN. The complement factsbook. 2nd ed. London (UK): Elsevier/Academic Press; 2018. [Google Scholar]
- 6.Khan MS, Singh P, Azhar A, Naseem A, Rashid Q, Kabir MA, et al. Serpin inhibition mechanism: a delicate balance between native metastable state and polymerization. J Amino Acids 2011;2011:606797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dijk M, Holkers J, Voskamp P, Giannetti BM, Waterreus WJ, van Veen HA, et al. How dextran sulfate affects C1-inhibitor activity: a model for polysaccharide potentiation. Structure 2016;24:2182–9. [DOI] [PubMed] [Google Scholar]
- 8.Beinrohr L, Harmat V, Dobo J, Lorincz Z, Gal P, Zavodszky P. C1 inhibitor serpin domain structure reveals the likely mechanism of heparin potentiation and conformational disease. J Biol Chem 2007;282:21100–9. [DOI] [PubMed] [Google Scholar]
- 9.Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet 2000;356:213–7. [DOI] [PubMed] [Google Scholar]
- 10.Carter PE, Duponchel C, Tosi M, Fothergill JE. Complete nucleotide sequence of the gene for human C1 inhibitor with an unusually high density of Alu elements. Eur J Biochem 1991;197:301–8. [DOI] [PubMed] [Google Scholar]
- 11.Steiner UC, Keller M, Schmid P, Cichon S, Wuillemin WA. Mutational spectrum of the SERPING1 gene in Swiss patients with hereditary angioedema. Clin Exp Immunol 2017;188:430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bock SC, Skriver K, Nielsen E, Thogersen HC, Wiman B, Donaldson VH, et al. Human C1 inhibitor: primary structure, cDNA cloning, and chromosomal localization. Biochemistry 1986;25:4292–301. [DOI] [PubMed] [Google Scholar]
- 13.Nicolicht P, Faria DOS, Martins-Silva L, Maia LSM, Moreno AS, Arruda LK, et al. Gene mapping strategy for Alu elements rearrangements: detection of new large deletions in the SERPING1 gene causing hereditary angioedema in Brazilian families. Gene 2019;685:179–85. [DOI] [PubMed] [Google Scholar]
- 14.Nzeako UC, Frigas E, Tremaine WJ. Hereditary angioedema: a broad review for clinicians. Arch Intern Med 2001;161:2417–29. [DOI] [PubMed] [Google Scholar]
- 15.Konings J,Cugno M,Suffritti C, Ten Cate H,Cicardi M,Govers-Riemslag JW. Ongoing contact activation in patients with hereditary angioedema. PLoS One 2013;8:e74043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haslund D, Ryo LB, Seidelin Majidi S, Rose I, Skipper KA, Fryland T, et al. Dominant-negative SERPING1 variants cause intracellular retention of C1 inhibitor in hereditary angioedema. J Clin Invest 2019;129:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caccia S, Suffritti C, Carzaniga T, Berardelli R, Berra S, Martorana V, et al. Intermittent C1-inhibitor deficiency associated with recessive inheritance: functional and structural insight. Sci Rep 2018;8:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santacroce R, D’Andrea G, Maffione AB, Margaglione M, d’Apolito M. The genetics of hereditary angioedema: a review. J Clin Med 2021;10:2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygoren-Pursun E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2021 revision and update. Allergy 2022;77:1961–90. [DOI] [PubMed] [Google Scholar]
- 20.Tran CG, Borbon LC, Mudd JL, Abusada E, AghaAmiri S, Ghosh SC, et al. Establishment of novel neuroendocrine carcinoma patient-derived xenograft models for receptor peptide-targeted therapy. Cancers (Basel) 2022;14:1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren Z, Perkins SJ, Love-Gregory L, Atkinson JP, Java A. Clinicopathologic implications of complement genetic variants in kidney transplantation. Front Med (Lausanne) 2021;8:775280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin J, Robeson H, Fagan P, Orloff MS. Association of PARP1-specific polymorphisms and haplotypes with non-small cell lung cancer subtypes. PLoS One 2020;15:e0243509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajdacsi E, Jandrasics Z, Veszeli N, Mako V, Koncz A, Gulyas D, et al. Patterns of C1-inhibitor/plasma serine protease complexes in healthy humans and in hereditary angioedema patients. Front Immunol 2020;11:794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loli-Ausejo D, Lopez-Lera A, Drouet C, Lluncor M, Phillips-Angles E, Pedrosa M, et al. In search of an association between genotype and phenotype in hereditary angioedema due to C1-INH deficiency. Clin Rev Allergy Immunol 2021;61:1–14. [DOI] [PubMed] [Google Scholar]
- 25.Parad RB, Kramer J, Strunk RC, Rosen FS, Davis AE III. Dysfunctional C1 inhibitor Ta: deletion of Lys-251 results in acquisition of an N-glycosylation site. Proc Natl Acad Sci U S A 1990;87:6786–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Speletas M, Szilagyi A, Psarros F, Moldovan D, Magerl M, Kompoti M, et al. Hereditary angioedema: molecular and clinical differences among European populations. J Allergy Clin Immunol 2015;135:570–3. [DOI] [PubMed] [Google Scholar]
- 27.Szabo E, Csuka D, Andrasi N, Varga L, Farkas H, Szilagyi A. Overview of SERPING1 variations identified in Hungarian patients with hereditary angioedema. Front Allergy 2022;3:836465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roche O, Blanch A, Duponchel C, Fontan G, Tosi M, Lopez-Trascasa M. Hereditary angioedema: the mutation spectrum of SERPING1/C1NH in a large Spanish cohort. Hum Mutat 2005;26:135–44. [DOI] [PubMed] [Google Scholar]
- 29.Rout PK, Verma M. Post translational modifications of milk proteins in geographically diverse goat breeds. Sci Rep 2021;11:5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ponard D, Gaboriaud C, Charignon D, Ghannam A, Wagenaar-Bos IGA, Roem D, et al. SERPING1 mutation update: mutation spectrum and C1 inhibitor phenotypes. Hum Mutat 2020;41:38–57. [DOI] [PubMed] [Google Scholar]
- 31.Bork K, Frank J, Grundt B, Schlattmann P, Nussberger J, Kreuz W. Treatment of acute edema attacks in hereditary angioedema with a bradykinin receptor-2 antagonist (icatibant). J Allergy Clin Immunol 2007;119:1497–503. [DOI] [PubMed] [Google Scholar]
- 32.Li M, Takahashi D, Kanost MR. Peptides based on the reactive center loop of Manduca sexta serpin-3 block its protease inhibitory function. Sci Rep 2020;10:11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanrattana W, Maas C, de Maat S. SERPINs—from trap to treatment. Front Med (Lausanne) 2019;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bajaj K, Madhusudhan MS, Adkar BV, Chakrabarti P, Ramakrishnan C, Sali A, et al. Stereochemical criteria for prediction of the effects of proline mutations on protein stability. PLoS Comput Biol 2007;3:e241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hopkins PC, Carrell RW, Stone SR. Effects of mutations in the hinge region of ser pins. Biochemistry 1993;32:7650–7. [DOI] [PubMed] [Google Scholar]
- 36.Germenis AE, Speletas M. Genetics of hereditary angioedema revisited. Clin Rev Allergy Immunol 2016;51:170–82. [DOI] [PubMed] [Google Scholar]
- 37.Pappalardo E, Cicardi M, Duponchel C, Carugati A, Choquet S, Agostoni A, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol 2000;106:1147–54. [DOI] [PubMed] [Google Scholar]
- 38.Aulak KS, Eldering E, Hack CE, Lubbers YP, Harrison RA, Mast A, et al. A hinge region mutation in C1-inhibitor (Ala436→Thr) results in nonsubstrate-like behavior and in polymerization of the molecule. J Biol Chem 1993;268:18088–94. [PubMed] [Google Scholar]
- 39.Stavenhagen K, Kayili HM, Holst S, Koeleman CAM, Engel R, Wouters D, et al. N- and O-glycosylation analysis of human C1-inhibitor reveals extensive mucin-type O-glycosylation. Mol Cell Proteomics 2018;17:1225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bos IG, Hack CE, Abrahams JP. Structural and functional aspects of C1-inhibitor. Immunobiology 2002;205:518–33. [DOI] [PubMed] [Google Scholar]
- 41.Andrasi N, Balla Z, Visy B, Szilagyi A, Csuka D, Varga L, et al. Diagnosing pediatric patients with hereditary C1-inhibitor deficiency—experience from the Hungarian angioedema center of reference and excellence. Front Allergy 2022;3: 860355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farkas H Pediatric hereditary angioedema due to C1-inhibitor deficiency. Allergy Asthma Clin Immunol 2010;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ravindran S, Grys TE, Welch RA, Schapira M, Patston PA. Inhibition of plasma kallikrein by C1-inhibitor: role of endothelial cells and the amino-terminal domain of C1-inhibitor. Thromb Haemost 2004;92:1277–83. [DOI] [PubMed] [Google Scholar]
- E1.Loules G, Zamanakou M, Parsopoulou F, Vatsiou S, Psarros F, Csuka D, et al. Targeted next-generation sequencing for the molecular diagnosis of hereditary angioedema due to C1-inhibitor deficiency. Gene 2018;667:76–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.