

Abstract
Sleep loss is associated with cognitive decline in the aging population and is a risk factor for Alzheimer’s disease (AD). Considering the crucial role of immunomodulating genes such as that encoding the triggering receptor expressed on myeloid cells type 2 (TREM2) in removing pathogenic amyloid plaque aggregates and regulating neurodegeneration in the brain, our aim was to investigate whether and how changes in sleep influence microglial function in mouse models. We chronically sleep-deprived mice expressing the humanized TREM2 common variant or the loss-of-function R47H AD-associated risk variant as well as mice without TREM2 expression. Mice were either wildtype without parenchymal amyloid-β (Aβ) plaques or the 5xFAD mouse model of cerebral amyloidosis with parenchymal Aβ plaques. In 5xFAD mice, sleep deprivation not only enhanced TREM2-dependent Aβ plaque deposition compared to 5xFAD mice with normal sleeping patterns, but also induced microglial reactivity that was independent of the presence of Aβ plaques in the brain. We investigated lysosomal morphology using transmission electron microscopy and found lysosomal abnormalities, particularly in mice without Aβ plaques, and also observed lysosomal maturation impairments in a TREM2-dependent manner in both microglia and neurons, suggesting that changes in sleep modified neuro-immune crosstalk. Unbiased transcriptome and proteome profiling provided mechanistic insights into functional pathways triggered by sleep deprivation that were unique to TREM2 and Aβ pathology and that converged on metabolic dyshomeostasis. Our findings highlight that sleep deprivation directly impacts microglial reactivity, for which TREM2 is required, by altering their metabolic ability to cope with the energy demands of prolonged wakefulness, leading to further Aβ deposition. Our study underlines the importance of sleep modulation as a promising future therapeutic approach to prevent the chronic inflammatory effects of sleep loss on increasing AD risk.
One sentence summary
Sleep deprivation exacerbates microglial reactivity and amyloid-β deposition in a TREM2-dependent fashion.
Accessible Summary:
Sleep loss is associated with cognitive decline in older adults, and is likely a risk factor for Alzheimer’s disease (AD). However, the underlying mechanisms are unclear. Here we investigated the effects of sleep loss on microglia, the primary immune cells in the brain, and found that sleep loss over time worsens microglial ability to remove amyloid aggregates in the brain that are a key hallmark of Alzheimer’s disease. Our findings suggest that sleep loss could increase the risk of dementia such as Alzheimer’s disease due to compromised immune function.
Introduction
Irregular sleep patterns have been linked to a greater likelihood of developing dementia associated with neurodegenerative disorders (1), though it is not clear whether these sleep changes contribute to the disease or simply reflect early symptoms. Sleep loss has been shown to influence memory consolidation (2), waste clearance in the brain (3, 4), as well as changes in glial morphology indicative of inflammation (5–7). Additionally, our lab showed that acute sleep deprivation in mice and in humans results in rapid increases, over hours, in both brain interstitial fluid and cerebrospinal fluid (CSF), of two proteins that make up key pathological hallmarks of the most common neurodegenerative dementia – Alzheimer’s disease (AD), in a neuronal activity-dependent fashion (8, 9). Further, chronic sleep deprivation in mice increases Aβ plaque and tau tangle pathology in mouse brains (8, 9). This strongly suggests that sleep loss earlier in life could trigger pathomechanisms in the brain that accelerate neurodegenerative disease onset and progression. Several studies have shown a direct correlation between sleep loss, in particular non-rapid eye movement (NREM) sleep, and increased CSF Aβ in cognitively healthy adults (1, 10–14), supporting the hypothesis that sleep loss over time increases AD risk in part due to changes in Aβ production and release (8, 13, 15), as well as changes in Aβ clearance (3, 4). Sleep in AD is compromised by prolonged wakefulness, decreased REM and NREM sleep, all of which correlate with the degree of cognitive impairment (15–17). In line with this, Aβ deposition in the preclinical stage of AD appears to be associated with worse sleep quality with reports of shorter sleep duration concomitant with greater Aβ burden (16–18). These findings suggest that disrupted sleep could lead to a cumulative effect on cellular metabolism that progressively disrupts disease modifying functions, including promoting the buildup of certain cellular substrates and decreasing their proteolytic degradation. Understanding the pathological consequences of early changes in sleep could therefore not only map the altered risk for developing AD, but also could help in identification of effective therapeutic interventions early during disease progression.
Research investigating the cellular substrates for sleep/wake or cell-specific consequences of sleep loss have traditionally focused on neurons (8, 19, 20). Yet, increasing evidence points to the role for glial cells, especially astrocytes (5, 21, 22), as a key component of the sleep/wake cycle, though much less is known about the role of microglia. Besides Aβ plaques, microgliosis is a well-known overarching pathological phenomenon characteristic of several neurodegenerative diseases including AD and plays a crucial role in modulating early AD pathogenesis (23, 24). Microglial reactivity is an important response to brain injury or protein aggregates such as Aβ plaques, which elicits a neuroinflammatory response by promoting an increase of cytokines, complement proteins, interleukins, and chemokines presumably in an attempt to recover from the diseased state (23, 24). However, prolonged periods of uncontrolled microglial reactivity results in chronic neuroinflammation that can promote neuronal dysfunction and neurodegeneration (25). To this end, acute and chronic sleep deprivation has been shown to induce systemic inflammation with increased production of IL-1β, IL-6 and TNFα (26, 27). Also, direct injection of TNFα and IL-6 in mice directly influences the duration of NREM sleep. These studies suggest that changes in sleep and inflammatory processes may interact with each other in a bidirectional manner, which highlights the need for further research into the genetic relationship between microglia and sleep.
Variants in several immunomodulating genes expressed in microglia including triggering receptor expressed on myeloid cells type 2 (TREM2) were recently identified to increase the risk of developing AD. For example, the AD associated R47H loss-of-function mutation impairs microglial chemotaxis and clustering around plaques (28–31), suggesting that loss of TREM2 function disrupts the immune-surveillance ability of microglia to identify and remove pathogenic threats. It is not yet known whether and how microglia alter their transcriptional and functional profile similar to that observed with AD pathology (32–35) in response to changes in sleep. Therefore, we investigated whether chronic sleep deprivation influenced microglial function in both wild-type mice and amyloid plaque-depositing 5xFAD mice, and further, if TREM2, as a key immunomodulating component in AD pathology, was required.
Results
Increased Aβ plaque deposition upon sleep deprivation in mice is TREM2 dependent
Several studies have reported increased amyloid accumulation with differing degrees of sleep loss. However, the effects of immunoregulating genes on sleep/wake states and whether these genes affect Aβ plaque deposition in response to sleep loss is not known. To investigate the effects of chronic sleep deprivation on Aβ plaque pathology, we used 5xFAD mice expressing the humanized TREM2 common variant (5xFAD/T2CV), loss-of-function R47H variant (5xFAD/T2R47H), or TREM2 knockout mice (5xFAD/T2KO) (28). Sleep deprivation experiments were started at 2.5 months of age (Fig. 1A) once Aβ plaques started to appear in the brain (36), and lasted over six weeks after which mice were euthanized to study changes on early plaque deposition in the brain. We verified that the mice were indeed sleep deprived as we observed a decrease in the percentage of sleep (p=0.026) (Fig. 1B) as well as sleep bout length (p<0.0001) (Fig. 1C) compared to normally sleeping 5xFAD mice. Further analyses revealed reduced percentage of sleep specifically during the light phase compared to the dark phase, with a similar pattern noted in sleep bout length in sleep-deprived mice, regardless of genotype (Fig. S1A–D). In line with this, sleep-deprived mice displayed an increase in NREM sleep with a concomitant decrease in time spent awake measured by electroencephalography immediately after sleep deprivation (Fig. S1E) (p=0.001), a sign of sleep rebound following a substantial period of sleep loss. To eliminate stress as a potential confounding factor, we measured plasma corticosterone and found no changes in sleep-deprived mice compared to normally sleeping control mice, suggesting no major change in stress as a result of our sleep deprivation method (Fig. S1F).
Figure 1. Sleep deprivation exacerbates amyloid plaque deposition in 5xFAD mice in a TREM2-dependent manner.
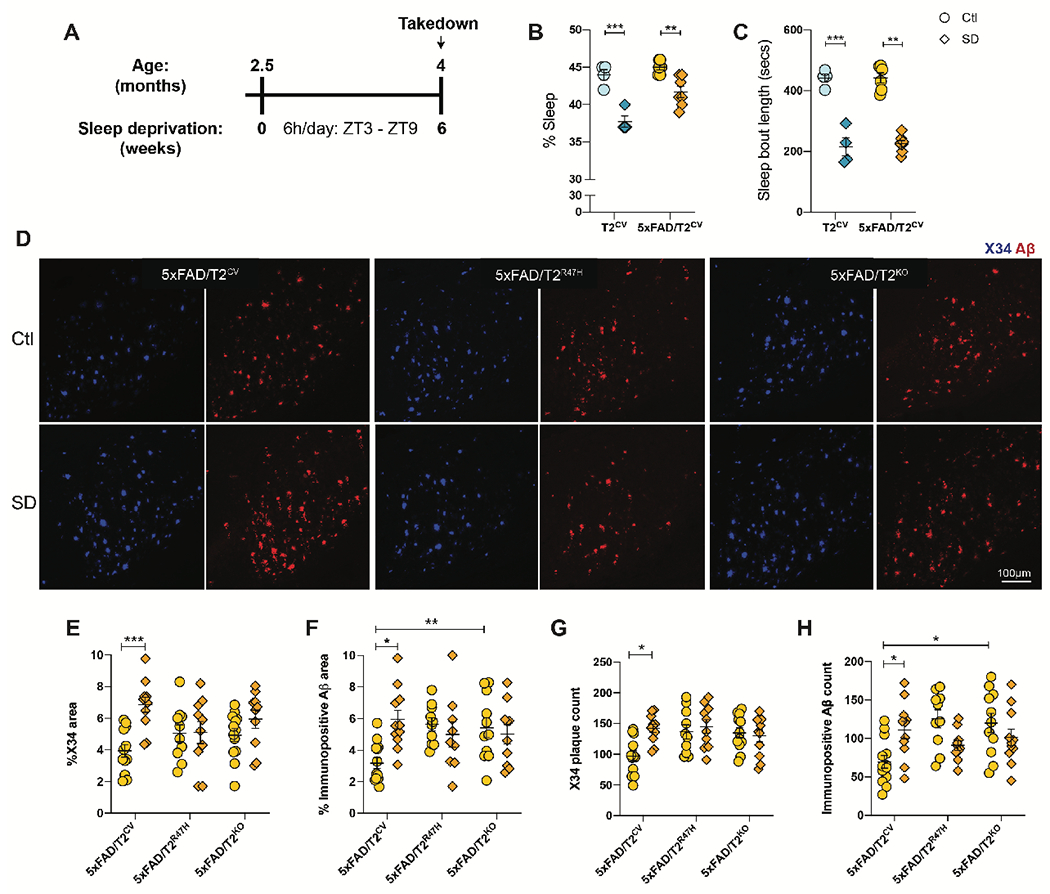
(A) Design of sleep deprivation study. (B, C) Shown is the percentage sleep (B) and sleep bout length recorded during sleep deprivation (SD) compared to normally sleeping control (Ctl) mice expressing TREM2 common variant (T2CV). Data were recorded over a 24h time period. (D) Representative confocal images of immunohistochemically stained fibrillar Aβ plaques (X34) and immunopositive plaques (6E10) in the subiculum of 5xFAD/T2CV, 5xFAD/T2R47H and 5xFAD/T2KO mice that were either sleep deprived or allowed normal sleep as control (Ctl). (E,F,G,H) Quantifications of immunohistochemical images shown in panel D; n=10-14 mice/genotype. Data represent mean ± S.E.M. Two-way ANOVA, Tukey’s multiple comparison test; *P<0.05; **P<0.01; ***P<0.001. ZT, Zeitgeber time (where ZT 0 is the start of the light phase and ZT 12 is the start of the dark phase).
Consistent with previous reports (8, 9, 37), we found that chronic sleep deprivation increased fibrillar Aβ plaques that stained with the small molecule X34 (p=0.002) as well as overall immunopositive Aβ plaque burden (p=0.006) (Fig. 1D–H) by approximately two-fold in the subiculum of the mouse brain compared to normally sleeping control mice. Though not statistically significant, we observed a slight increase in X34+ fibrillar plaques in 5xFAD/T2KO mice after sleep deprivation compared to 5xFAD/T2KO normally sleeping controls (p=0.715). This could potentially reflect a plateau on earlier saturation of amyloid deposition (30) and therefore a lack of further effect of sleep deprivation in these mice. Interestingly, an increase in immunopositive Aβ plaque accumulation was only present in sleep deprived 5xFAD/T2CV mice and was not found in the loss of TREM2 function 5xFAD/T2R47H mice or 5xFAD/T2KO mice. Nonetheless, Aβ accumulation was higher in 5xFAD/T2KO versus 5xFAD/T2CV mice (p=0.047). Given that the 5xFAD mouse model predominantly develops plaques in the subiculum at this age (38), the strongest effect of sleep deprivation-induced plaque deposition was most prominent in the subiculum, followed by the thalamic nuclei and cortical regions where a similar change in plaque deposition was observed though to a lesser extent (Fig. S2). Altogether, these findings suggest that normal TREM2 function is important for modulating Aβ deposition in the 5xFAD mouse model and, importantly, TREM2 is required for the effects of sleep deprivation on amyloid deposition.
Exacerbated microglial reactivity after sleep deprivation is TREM2 dependent
As chemotaxis and plaque association of microglia is impaired with loss of TREM2 function (28, 30, 39), we next investigated whether sleep deprivation influences microglial function by examining changes in reactive and homeostatic microglial markers (Fig. 2). We found a strong reduction in homeostatic microglial populations expressing P2RY12 (Fig. 2A, D) and TMEM119 (Fig. 2A, E) after sleep deprivation, but only in T2CV (p=0.0001 and p=0.0028, respectively) and 5xFAD/T2CV (p=0.039 and p=0.038, respectively) groups. Compared to the wild-type (WT) mice that do not otherwise accumulate Aβ plaques, 5xFAD groups presented greater coverage of ionized calcium-binding adaptor molecule 1 (IBA1)-positive microglia in the subiculum (Fig. 2B, C). Clustering of IBA1+ microglia around Aβ plaques was more prominent in sleep-deprived 5xFAD/T2CV mice compared to normally sleeping 5xFAD/T2CV controls, which was not observed in the loss-of-function 5xFAD/T2R47H or 5xFAD/T2KO mice (Fig. 2C, I, Fig. S2). This suggested a TREM2-dependent effect of sleep deprivation-induced microglial reactivity. We observed similar TREM2-dependent changes after sleep deprivation in X34+ plaque deposition, IBA1+ microglia and CD68+ lysosomes in 5xFAD/T2CV mice, in both the thalamic nuclei and cortex, however to a lesser extent than the subiculum (Fig. S2). We also assessed whether sleep deprivation affected Cd33 and Bmal1 expression, both of which play an important role in glial immune responses and cellular metabolism, with the latter directly impacting circadian microglial responses. We found no changes across different genotypes or sleep conditions (Fig. S2L, M). The percentage area of IBA1 staining in the subiculum did not show TREM2 genotype-dependent changes in the 5xFAD groups under control conditions, likely due to their young age, however, a larger percentage of IBA1+ microglia was found closely clustering around Aβ plaques in 5xFAD/T2CV mice compared to 5xFAD/T2KO mice (p=0.044) (Fig. 2I). Differences in microglial reactivity were further supported by changes in microglial morphology following sleep deprivation noted by a reduced number of branches per cell and increased cell soma size in the cortex where substantially fewer Aβ plaques were present compared to the subiculum at this age (Fig. S2). In line with this, microglial lysosomal CD68 immunostaining (p<0.0001) as well as the reactive microglial marker Clec7a (p=0.045) were increased in sleep-deprived 5xFAD/T2CV mice (Fig. 2B, G, F, H), suggesting that sleep deprivation induced a less functional microglial state. These changes in microglial markers, homeostatic or reactive, were not exclusive to mice depositing plaques as these changes were also present in T2CV mice (Fig. 2 and fig. S2). Similar to the 5xFAD groups, changes in microglia reactivity were only observed in the presence of functional TREM2 in WT mice after sleep deprivation, indicating TREM2 dependency of microglial responses to changes in sleep (Fig. 2 and Fig. S2). Overall, these findings suggest that TREM2 is required to promote a dysfunctional microglial phenotype in response to chronic sleep deprivation, in the presence or absence of Aβ plaques.
Figure 2. Sleep deprivation alters microglial reactivity according to TREM2 expression.
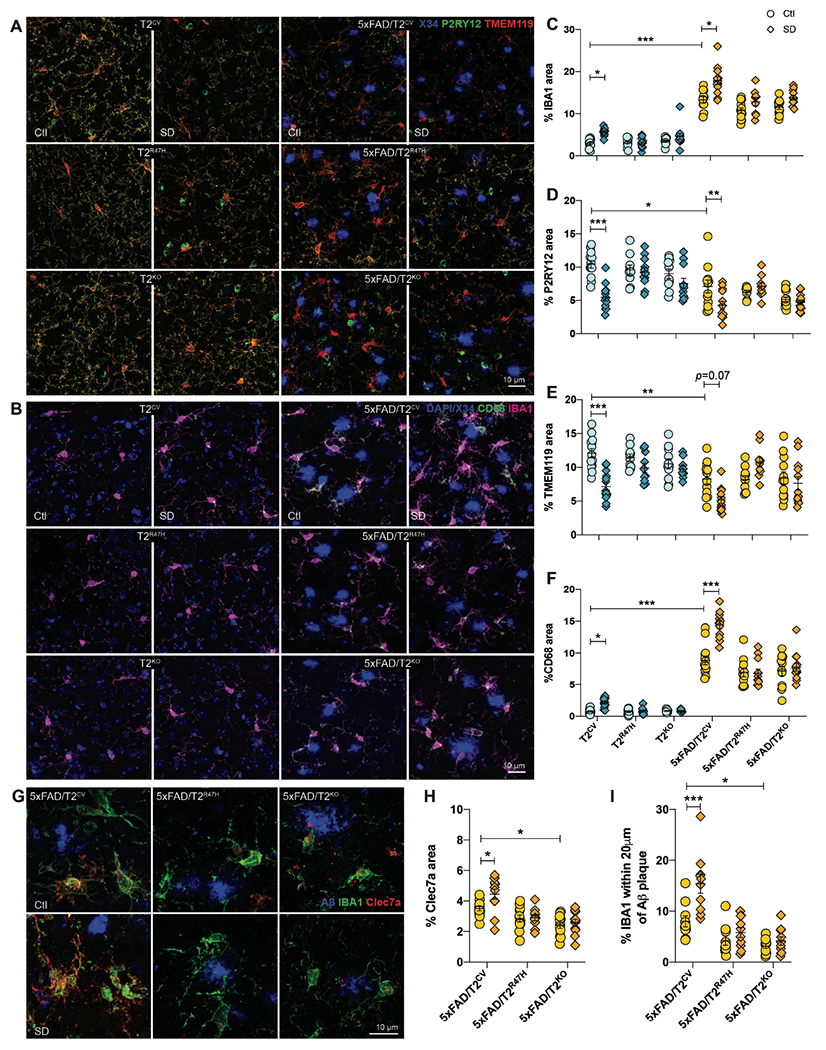
(A) Shown are representative confocal images of the subiculum immunostained with X34, P2RY12 or TMEM119, which detect fibrillar Aβ plaques and homeostatic microglia, respectively. (B) Shown are representative confocal images of the subiculum immunostained with X34 and costained with CD68+ phagolysosomes within microglia (green) and IBA1+ microglia (magenta), with DAPI nuclear stain (blue). (C) The percentage of subiculum area stained positive for IBA1 is shown (including microglia close to or far away from amyloid plaques) quantified from panel B. (D) The percentage of subiculum area stained positive for P2RY12 quantified from panel A. (E) The percentage of subiculum area stained positive for TMEM119 is shown. (F) The percentage of subiculum area stained positive for CD68 quantified from panel A is shown. (G) Representative confocal images of subiculum immunopositive for Aβ (6E10 antibody, blue), IBA1 (green) and Clec7a (red). (H) The percentage of the subiculum area stained positive for Clec7a. (I) The percentage of IBA1+ microglia within 20μm of Aβ plaques in the subiculum were quantified. n=10-14 mice/genotype. SD, Sleep deprived; Ctl, normally sleeping control mice. Data represent mean ± S.E.M. Two-way ANOVA, Sidak’s multiple comparison test; *P<0.05; **P<0.01; ***P<0.001.
3D reconstructed images of CD68 and IBA1 stained microglia around Aβ plaques in the subiculum showed that despite the increased microglial reactivity surrounding plaques after sleep deprivation in 5xFAD/T2CV mice, these microglia engulfed fibrillar Aβ plaques yet accumulated this material in their lysosomes (p=0.008) (Fig. 3A, B). This supports our hypothesis that sleep deprivation induces a dysfunctional microglial state that is inefficient in degrading or removing Aβ plaques in the brain despite recruiting larger numbers of reactive microglia to the plaques. In addition, a similar reactive microglial state was observed even in the absence of plaques (Fig. 2 and fig. S2). Therefore, we further investigated whether microglia in sleep-deprived versus normally sleeping control mice functionally differed in their ability to cluster around or degrade Aβ. We first sleep deprived T2CV and 5xFAD/T2CV mice for six weeks, followed by intracerebrally injecting 488 dye-labeled Aβ fibrils, and then we euthanized the mice two days after injection to compare the effects of microglia function acutely after changes in sleep (Fig. 3C). Prior to injecting the Aβ fibrils, we verified their nature by transmission electron microscopy, both pre- and post-fluorophore labelling (Fig. 3D, E). We found that IBA1+ microglia heavily clustered around exogenously inoculated Aβ488 fibrils in both T2CV (p=0.020) and 5xFAD/T2CV mice (p=0.054), with a greater amount of labelled fibrils within microglia in sleep-deprived mice compared with normally sleeping control mice (Fig. 3F, G). These findings suggest that when faced with a challenge, such as Aβ fibrils, microglia function in sleep-deprived mouse brain becomes compromised as they were not only hyperreactive compared to those in the control mice, but also they were acutely unable to efficiently degrade the engulfed material. To further assess this hypothesis, we next examined lysosomes.
Figure 3. Sleep deprivation results in an increase in Aβ within microglia.
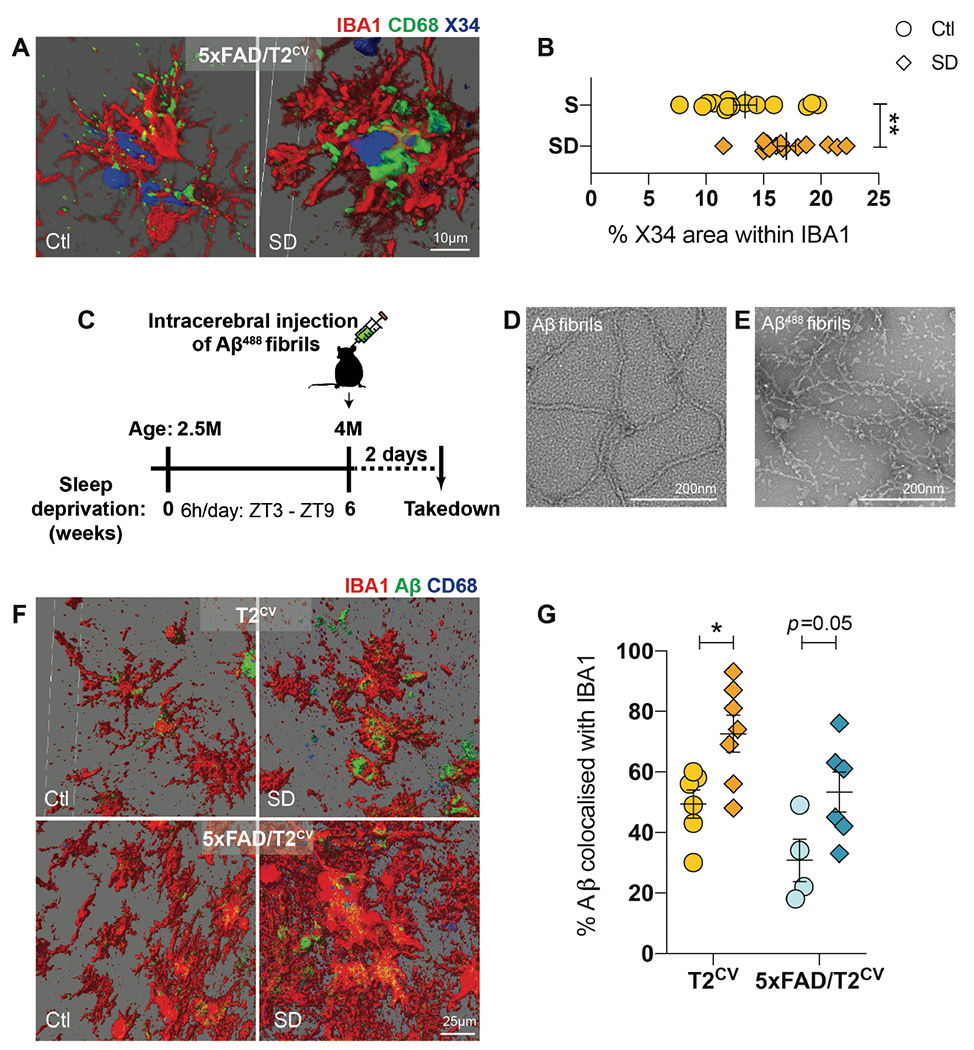
(A) 3D reconstructed images of X34, CD68 and IBA1 immunostained subiculum from normally sleeping control (Ctl) 5xFAD/T2CV mice compared to sleep deprived (SD) 5xFAD/T2CV mice. (B) Quantification of the percentage of X34-positive (blue) area within CD68+ stained microglia (green). n=10-14 mice/genotype. Unpaired t-test. (C) Study design for intracerebral inoculation of 488-labelled Aβ fibrils into the subiculum of sleep deprived T2CV and 5xFAD/T2CV mice versus normally sleeping controls. (D,E) Representative transmission electron microscopy images of unlabeled (D) and 488-labelled (E) Aβ fibrils. (F) Shown are 3D reconstructed images of 488-labelled Aβ (green), CD68 (blue) and IBA1 (red) immunostaining of subiculum from T2CV control mice (n=6-7 mice/group) and 5xFAD/T2CV mice (n=4-6/group). (G) Shown is the percentage of 488-labelled Aβ fibrils within IBA1+ microglia quantified from panel F. Data represent mean ± S.E.M. Two-way ANOVA, Sidak’s multiple comparison test; *P<0.05; **P<0.01; ***P<0.001. ZT, zeitgeiber time.
Abnormal lysosomal protein accumulation after sleep deprivation in microglia and neurons
An increase in CD68+ microglia in the brain typically indicates enhanced microglial phagocytosis due to amplified lysosomal activity, for example, as a response to amyloid plaques. Considering we observed a sleep deprivation-induced increase in amyloid plaque burden as well as an increase in CD68+ microglia with increased fibrillar Aβ in these cells, we hypothesized that microglial-mediated Aβ degradation was likely to be impaired and unable to cope with the cellular demands caused by sleep deprivation-provoked metabolic stress. To address this hypothesis, we investigated lysosomal morphology using transmission electron microscopy and found that indeed chronic sleep deprivation leads to enlarged lysosomes, but only in the absence of amyloid plaques (Fig. 4A–C). As expected, lysosomal area was substantially larger in 5xFAD/T2KO mice compared with 5xFAD/T2CV mice (p=0.0001), indicative of metabolic dysfunction due to a lack of TREM2 expression (40). However, no additional changes in lysosomal morphology were found in sleep-deprived 5xFAD groups. In contrast, sleep deprived T2CV mice not only displayed deformed and distended lysosomal morphology as measured by lysosomal area (Fig. 4B), but also contained a greater number of lysosomes with electron-dense inclusions compared with normally sleeping T2CV or T2KO controls (Fig. 4C). Again, the sleep deprivation-induced changes in lysosomes were observed only in T2CV but not in T2KO mice, suggesting a TREM2-dependent effect. We further validated these changes in lysosomal activity by immunoblotting for several proteins important in lysosomal proteolytic functions including but not limited to fusion with autophagosomes, cargo trafficking, as well as hydrolase activity-dependent digestion (Fig. 4D–K). In line with the transmission electron microscopy data, we found a TREM2-dependent effect of sleep deprivation on lysosomal proteins, with a larger effect in the absence of amyloid plaques in T2CV mice. Microgliosis is often accompanied by an elevated expression of lysosomal cathepsins (41). Therefore, we hypothesized that the hydrolase activity of cathepsins B and D, subtypes of hydrolases located within the lysosomal lumen, would concomitantly increase with microglial reactivity in sleep-deprived T2CV groups. However, we found that the heightened microglial response in sleep-deprived mice had an opposite effect. Cathepsins B and D were overall reduced in sleep-deprived T2CV (p<0.0001 and p<0.0001, respectively) and 5xFAD/T2CV mice (p=0.002 and p=0.0003, respectively) (Fig. 4F, G), despite the increase in transcription factor EB (TFEB), particularly in the T2CV mice (p=0.002) (Fig. 4J). This suggests that TFEB could compensate for the compromised lysosomal function triggered by sleep deprivation. Both cathepsins B and D showed altered maturation (Fig. 4D–G), suggesting impaired enzymatic activity in the ability of lysosomes to digest cargo. In line with this, we investigated whether the changes in lysosomal morphology as well as proteases affected the formation of autophagosomes (Fig. S3). Indeed, sleep-deprived T2CV mice displayed a greater number of incompletely fused autophagosomes-lysosomes (p=0.06) (Fig. S3A, C) noted by partially-fused double membrane bilayer consistent with autophagosomes that are docked with lysosomes (Fig. S3B), similar to that previously reported (42). Further, as TFEB is essential for autophagic processes and clearance of damaged lysosomes (43), we additionally probed for LC3b and found it to be elevated particularly in the sleep-deprived T2CV mice compared with normally sleeping controls (p=0.038) or other TREM2 genotypes (Fig. 4D, E, K). These findings provide further evidence that sleep deprivation provokes lysosomal dysfunction in the inflamed brain, especially in the absence of amyloid plaque pathology.
Figure 4. Lysosomal impairments after sleep deprivation are dependent on TREM2 expression.
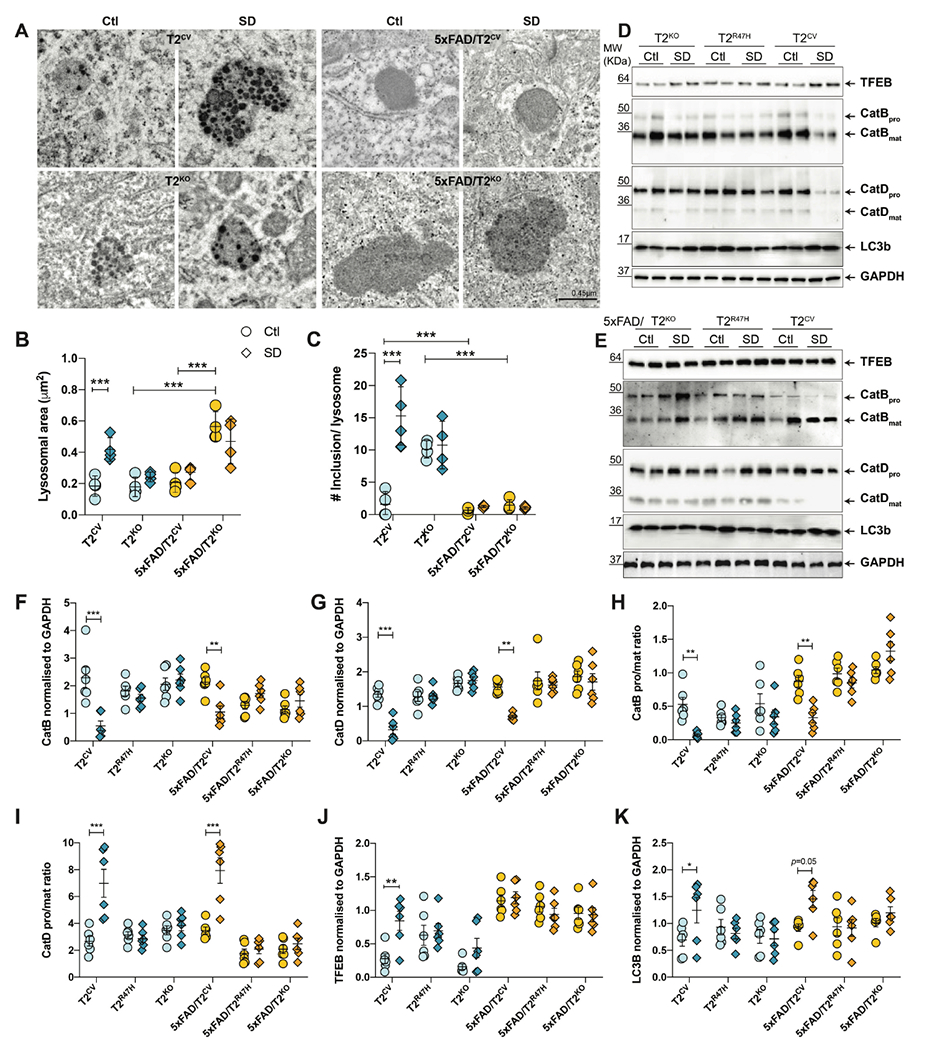
(A) Representative transmission electron microscopy images of lysosomes in subiculum from T2CV, 5xFAD/T2CV, T2KO and 5xFAD/T2KO mice that were sleep deprived (SD) or normally sleeping controls (Ctrl). (B, C) Quantification of lysosomal area (B) and number of electron dense inclusions per lysosome (C) from transmission electron microscopy images in panel A. Two-way ANOVA, Tukey’s multiple comparison test; ***P<0.001. (D) Immunoblotting of hippocampal lysates for TFEB, cathepsins B and D, LC3b, with GAPDH as loading control, for T2CV, T2R47H and T2KO groups that were sleep deprived (SD) or normally sleeping (Ctl). (E) Immunoblotting of hippocampal lysates for TFEB, cathepsins B and D, LC3b, with GAPDH as loading control, for 5xFAD/T2CV, 5xFAD/T2R47H and 5xFAD/T2KO groups that were sleep deprived (SD) or normally sleeping (Ctl). (F,G) Quantification of immunoblots for (F) cathepsin B and (G) cathepsin D normalized to GAPDH. (H,I) Quantification of immunoblots for the pro form to mature form ratio of (H) cathepsin B and (I) cathepsin D. (J, K) Quantification of immunoblots for TFEB (J) and LC3b (K) normalized to GAPDH. (n=6/genotype). All data represent mean ± S.E.M. Two-way ANOVA, Sidak’s multiple comparison test; *P<0.05; **P<0.01; ***P<0.001.
As we observed a TREM2-dependent effect on changes in lysosomal morphology and proteins after sleep deprivation, we aimed to identify whether these changes were exclusive to microglia. We co-stained lysosomal markers that are abundant in microglia under physiological conditions (44) such as lysosome associated membrane protein 1 (LAMP1) as well as cathepsins B and D, together with IBA1, glial fibrillary acidic protein (GFAP), and neuronal nuclear marker (NeuN) to examine co-localization in microglia, astrocytes, and neurons, respectively (Fig. 5). Compared to normally sleeping controls, sleep-deprived T2CV mice displayed large LAMP1+ puncta within NeuN+ neurons as well as within IBA1+ and GFAP+ microglia and astroglia, respectively (Fig. 5A–D). A similar change was not present in sleep-deprived T2R47H or T2KO mice, supporting the hypothesis that functional TREM2 is required for lysosomes to respond to sleep deprivation-triggered metabolic stress. Whereas LAMP1 co-localization was increased in all three cell types after sleep deprivation in T2CV mice, it was morphologically most evident in neurons with an overall increase in co-localization by approximately 50% compared with normally sleeping control mice (Fig. 5A, D). In line with our results in Fig. 4, changes in LAMP1 immunoreactivity after sleep deprivation were more prominent in T2CV mice compared with 5xFAD groups. 5xFAD mice showed no changes in LAMP1 co-localization after sleep deprivation regardless of TREM2 genotype, suggesting that LAMP1+ lysosomes respond differentially to sleep deprivation-dependent metabolic stress in the presence or absence of Aβ plaques. Consistent with these results, a reduction in cathepsins B and D were found only in the T2CV mice after sleep deprivation compared with normally sleeping controls of other genotypes (Fig. 5E–I). The decrease in co-localization was once again more prominent in neurons and microglia with the largest effect in the sleep deprivation-dependent reduction in cathepsin D in neurons by ~ 45% (p=0.026) (Fig. 5H, I). Cathepsin B was reduced overall in both microglia (p=0.0035) and neurons (p=0.007) in sleep-deprived compared to normally sleeping T2CV mice (Fig. 5F, G). These data suggest that in the absence of Aβ plaques, sleep deprivation acts as a primary metabolic stressor impairing microglial lysosomal functions, which may also result in additional burden on neurons that are unable to process and excrete substrates, especially in the presence of dysfunctional microglia.
Figure 5. Cell-specific lysosomal protein changes after sleep deprivation.
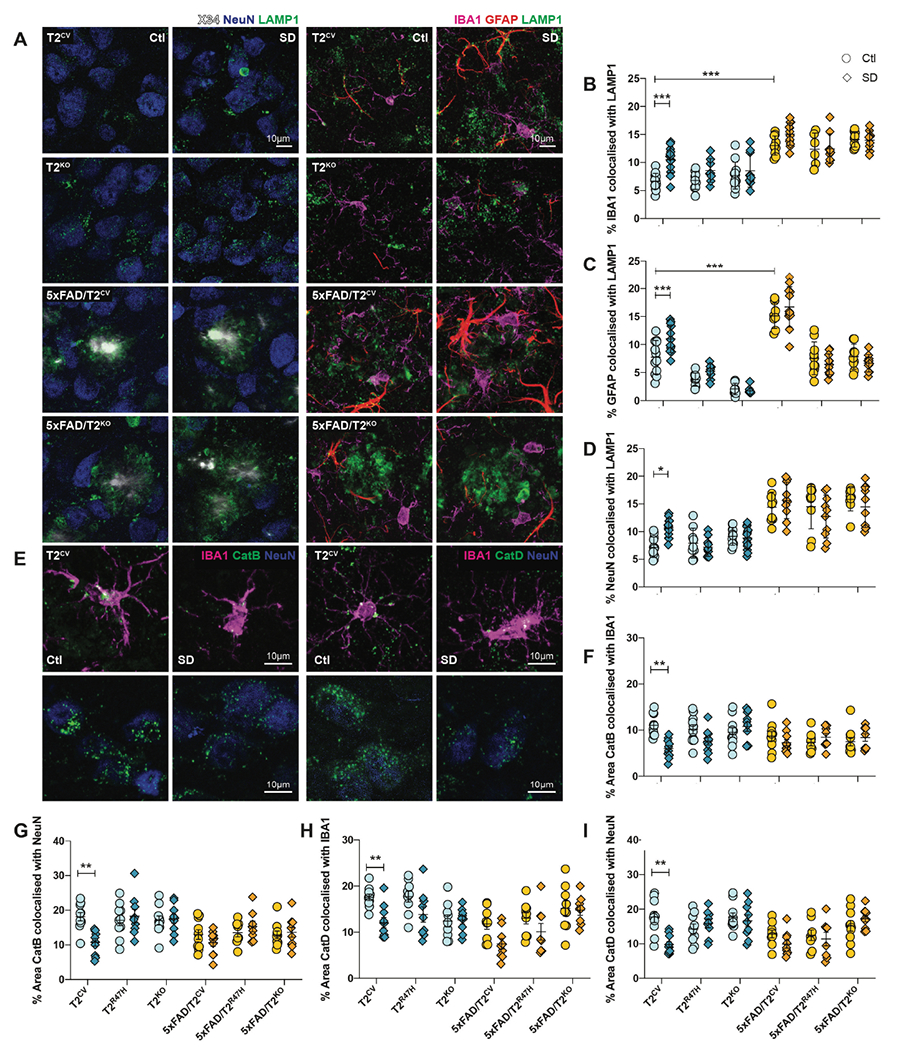
(A) Left panels show representative confocal images of X34, NeuN and LAMP1 immunostaining of subiculum from T2CV, 5xFAD/T2CV, T2KO, and 5xFAD/T2KO mice that were sleep deprived (SD) or normally sleeping controls (Ctl). Right panels show representative confocal images of IBA1, GFAP and LAMP1 immunostaining of subiculum from T2CV, 5xFAD/T2CV, T2KO, and 5xFAD/T2KO mice that were sleep deprived (SD) or normally sleeping controls (Ctl). (B) Quantification of the percentage of subiculum stained for colocalized IBA1 and LAMP1. (C) Quantification of the percentage of subiculum stained for colocalized GFAP and LAMP1. (D) Quantification of the percentage of subiculum stained for colocalized NeuN and LAMP1. (E) Left panels show representative confocal images of subiculum from T2CV mice costained with IBA1 (magenta), cathepsin B (CatB, green), and NeuN (blue). Right panels show representative confocal images of subiculum from sleep deprived or normally sleeping T2CV mice costained with IBA1 (magenta), cathepsin D (CatD, green), and NeuN (blue). (F, G) Quantification of the percentage of subiculum in which cathepsin B colocalizes with IBA1 (F) or NeuN (G). (H,I) Quantification of the percentage of subiculum in which cathepsin D colocalizes with IBA1 (H) or NeuN (I). n=9-11 mice/genotype. Data represent mean ± S.E.M. Two-way ANOVA, Tukey’s multiple comparison test; *P<0.05; **P<0.01; ***P<0.001.
Sleep deprivation triggers distinct transcriptional pathways in the presence or absence of Aβ plaques
We observed a reactive microglial-like phenotype in both T2CV and 5xFAD/T2CV mice after sleep deprivation (Figs. 2 and S2), although the lysosomal phenotype was most prominent in T2CV mice (Figs. 4 and 5). Therefore, we aimed to investigate the mechanism in what set the two groups apart in their response to sleep deprivation. Additionally, as we mainly observed TREM2-dependent effects on sleep deprivation, we aimed to acquire an unbiased understanding of the transcriptional pathways involved in the presence or absence of functional TREM2. RNA sequencing of bulk hippocampal tissue revealed a distinct transcriptional signature in sleep-deprived compared to normally sleeping controls, with changes in lysosomes being the only common functional pathway between T2CV and 5xFAD/T2CV mice (Fig. 6). We found that gene expression changes were not only specific to genotypes, but also to changes in sleep (Fig. 6A). Strikingly, gene ontology pathways in normally sleeping versus sleep-deprived T2CV and 5xFAD/T2CV mice were distinct with regard to the presence or absence of Aβ plaques (Fig. 6B–E). Sleep deprivation in T2CV mice led to changes in gene expression mainly associated with lysosomes, such as Atp6v0a1, Npc1, Ctsd, LaptmS, Cd68 and Lamp1, as well as microglia and the interleukin-2 signaling pathway including but not limited to P2ry12, Aif1, Slc2a9, (Clcn7, Tmem119, Sec22c and Rab29. We additionally found several genes involved in autophagy such as Atg16l, Nlrp6, Lgals8, and Becn1, which further suggested sleep deprivation-induced impairment of lysosomal degradation and accumulation of cargo (Fig 6C–G). Comparing the different sleep states of 5xFAD/T2CV mice showed more modest changes in a set of gene pathways unique to Aβ pathology such as the syndecan-1 pathway, collagen biosynthesis and extracellular metabolism as well as insulin-like growth factors such as Col8a1, Col8a2, Col3a1, Fmod, Igfr1, Igfr2, Irs4, Ctnnb1 and Cdh1 among others (Fig. 6C–G). Interestingly, sleep-deprived 5xFAD/T2CV mice also showed an upregulation of microglial and macrophage genes, though unique to Aβ plaque pathology, such as Clec7a, Clec10a, Ccl4, Cst7, Mgl2 and Itgae. (Fig. 6C–G). Even though most of the gene expression changes due to sleep deprivation in T2CV and 5xFAD/T2CV groups were unique, the only common entities were lysosomal and microglial genes. To obtain a better understanding of whether the observed effects were primarily TREM2 genotype-dependent rather than due to sleep deprivation, we compared the transcriptomes of sleep-deprived 5xFAD/T2CV versus normally sleeping 5xFAD/T2CV controls to normally sleeping 5xFAD/T2CV versus 5xFAD/T2KO mice (Fig. 6B). We found nine genes overlapped between the two groups including Bhmt, Cd68, Clec7a, Col8a1, Col8a2, Igf2, Irs4, Mpzl2, and Slc47a1 (Fig. 6B, F, G). Thus, while sleep deprivation-dependent gene expression changes were not exclusively TREM2-dependent, the effects of sleep deprivation on inflammation were largely present in mice with TREM2 expression suggesting that TREM2 is required to mediate a response to sleep deprivation.
Figure 6. Sleep deprivation triggers transcriptional pathways depending on TREM2 and Aβ plaques .
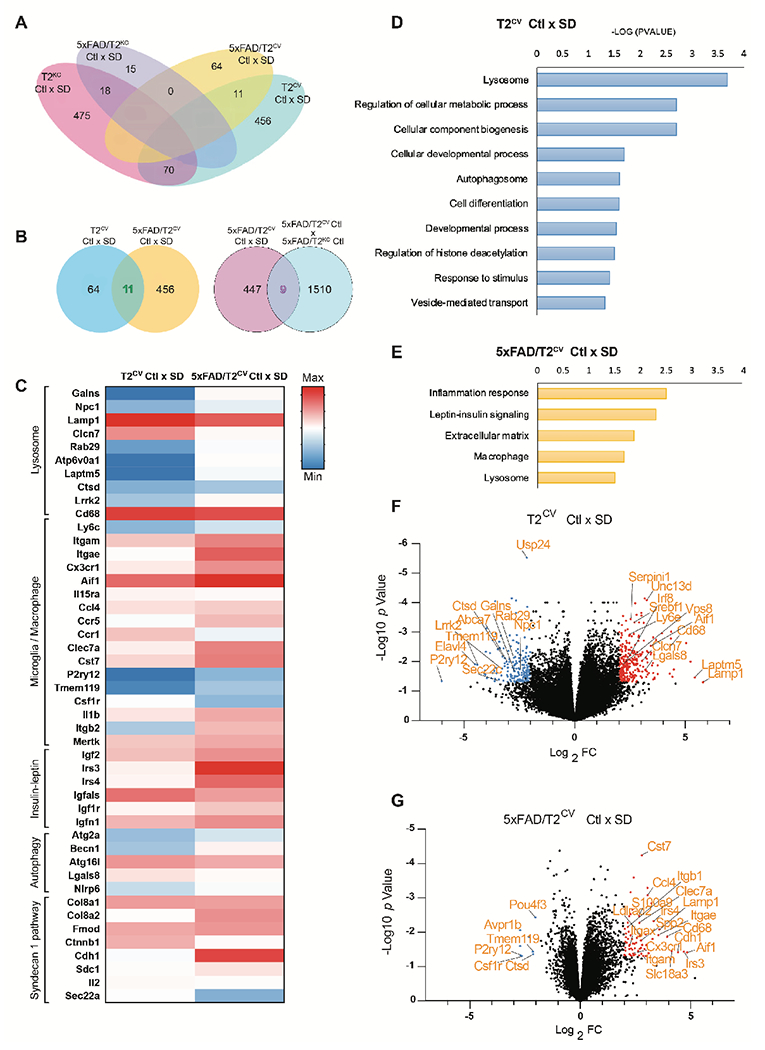
(A) Venn diagram illustrates the number of genes mutually inclusive or exclusive in the four different mouse genotypes: T2CV, 5xFAD/T2CV, T2KO, and 5xFAD/T2KO. (B) Venn diagrams compare the number of genes mutually inclusive or exclusive for sleep deprived (SD) T2CV mice and 5xFAD/T2CV mice, and their normally sleeping controls (Ctl), as well as for sleep deprived 5xFAD/T2CV mice compared to normally sleeping 5xFAD mice as control. (C) Heat map of RNA sequences that were upregulated or downregulated in bulk hippocampal tissue from T2CV and 5xFAD/T2CV mice, either sleep deprived (SD) or normally sleeping controls (Ctl), grouped according to gene ontology pathways. (D, E) Shown are gene ontology pathways affected by sleep deprivation in T2CV mice (D) and 5xFAD/T2CV mice (E). (F, G) Volcano plot of gene expression changes in T2CV mice (F) and 5xFAD/T2CV mice (G) that were sleep deprived (SD) or allowed sleep normally (Ctl). Significantly downregulated genes are indicated by blue dots and upregulated genes by red dots. n=10/genotype. Significance for volcano plots was marked at p<0.05 with a 2-fold change in Log2.
Sleep deprivation evokes changes in the proteome
Despite the unique gene expression changes after sleep deprivation, both T2CV and 5xFAD/T2CV mice displayed a similar inflammatory profile in the brain. Therefore, we further investigated whether sleep loss caused global changes in the proteome by analyzing cerebrospinal fluid (CSF) using unbiased mass-spectrometry to identify proteolytic processes unique, or similar to changes linked to sleep and TREM2 genotype, in the presence or absence of Aβ plaques. Interestingly, sleep loss predominantly influenced metabolic processes in all groups regardless of TREM2 expression or plaque pathology (Fig. 7). However, in line with our previous results, the specific proteins involved in the metabolic processes were unique to TREM2 and Aβ plaques. For example, sleep loss-induced changes in proteins involved in the endolysosomal pathway, the inflammatory response to stimuli as well as the TGFβ signaling pathway—such as Eef1a2, Rab5b, Ctsd, Il6st, Cxcl16, Cd207, Megf10, Ppp2cb, Fbn1, Mapk3, and Rock1—were present in the absence of Aβ plaques (Fig. 7A, B). In contrast, sleep-deprived 5xFAD/T2CV mice displayed changes in metabolic processes that additionally influenced glycolysis, ATPase binding, and the insulin signaling pathway, such as Hk2, Pygl, Fbpl, Atp6v1e1, Atp1a4, Aldh1b1 and Apoc3 (Fig. 7C, D). Sleep deprivation-induced proteome changes in the CSF in mice without TREM2 mostly affected metabolic pathways associated with autophagy, GTPase activity and ubiquitin-mediated proteolysis, to a larger extent in T2KO mice compared to 5xFAD/T2KO mice. Examples of this include Mtor, Atg4b, Usp26, Rab1, Rab18, Ube2e1, and Mtif2 (Fig. 7D–G). We additionally compared sleep-deprived T2CV groups and separately, sleep-deprived T2KO groups to obtain a better understanding of the contribution of Aβ pathology, by removing sleep deprivation as the sole contributing factor to any effects observed within these comparisons (Fig. S4). Each comparison shared differential regulation of specific proteins across genotypes and sleep conditions including, Sox5, Fgl2, A1Bg, Enpp6, Neug, Clca, Nrp1, Lmnb1, among others (Figs. 7 and S4). Interestingly, we still observed changes in metabolic dysfunction while comparing sleep-deprived T2CV to sleep-deprived 5xFAD/T2CV mice, suggesting that Aβ pathology further contributed to metabolic dyshomeostasis even when the sleep deprivation-induced changes in each individual genotype already showed metabolic impairments. In contrast, metabolic dysfunction was not a primary contributor to the proteome changes observed in sleep-deprived T2KO mice versus sleep-deprived 5xFAD/T2KO mice. This suggested that not only is TREM2 required to instigate proteomic changes in the sleep-deprived brain, but also the presence of Aβ pathology can be detrimental in further exacerbating metabolic dysfunction. However, in the absence of TREM2, Aβ pathology did not further aggravate the dysfunction already triggered by sleep deprivation. Altogether, these data suggest that even though additional genetic factors such as TREM2 or APP may compensate, or impact specific pathways triggered by sleep deprivation, sleep loss eventually results in metabolic dysfunction.
Figure 7. Sleep deprivation induces metabolic dysregulation in the mouse CSF proteome.
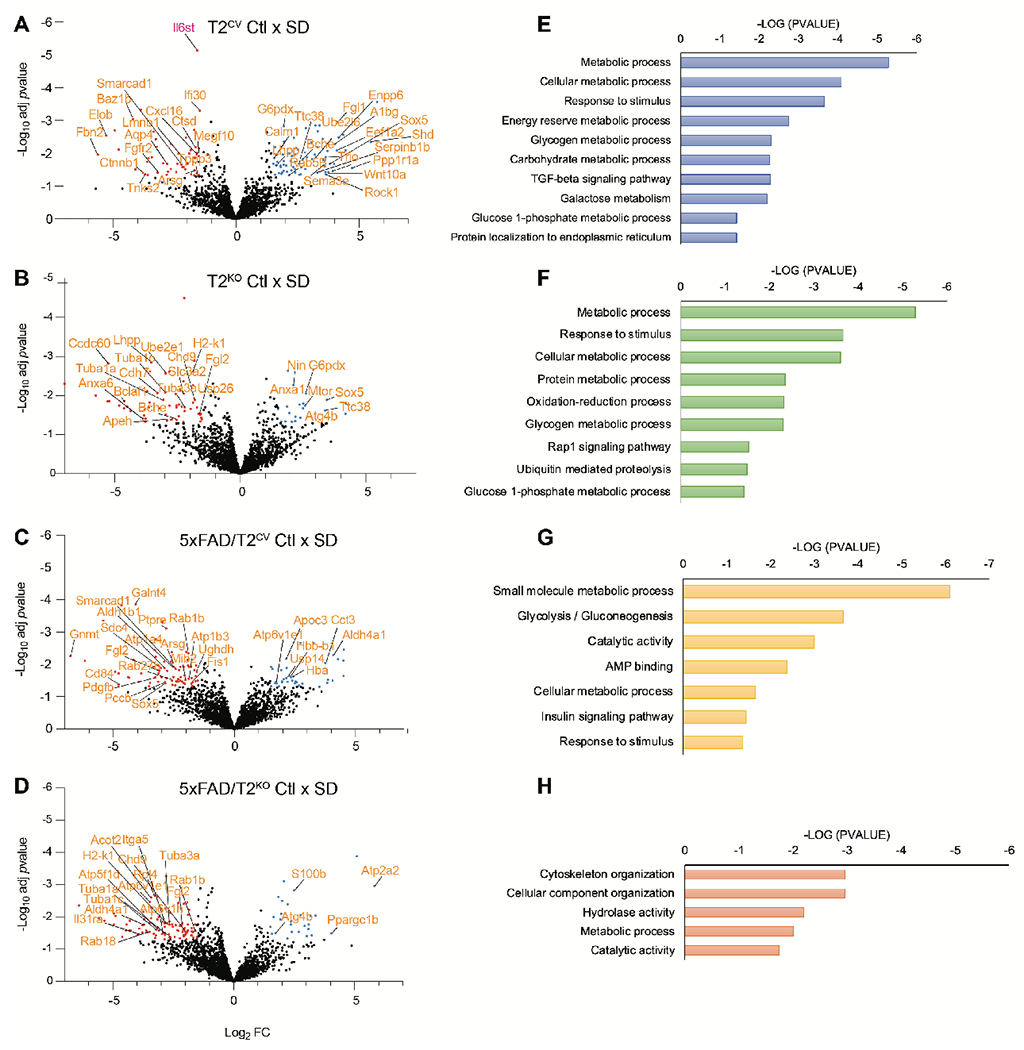
(A-D) Shown are volcano plots of the CSF proteomes for T2CV mice (A), 5xFAD/T2CV mice (B), T2KO mice (C) and 5xFAD/T2KO mice (D), that were either sleep deprived (SD) or allowed to sleep normally as a control (Ctl). Significantly downregulated proteins are indicated by blue dots and upregulated proteins are indicated by red dots. (E-H) Gene ontology functional pathways are shown for CSF proteomes for T2CV mice (E), 5xFAD/T2CV mice (F), T2KO mice (G) and 5xFAD/T2KO mice (H). n=9-11 mice/genotype. Significance for volcano plots was marked at p<0.05 with a 1.5-fold change in Log2.
Discussion
We show that chronic sleep deprivation triggers microglial reactivity in the brain independent of Aβ plaques, the mechanism of which is largely influenced by TREM2. As sleep loss increases parenchymal Aβ deposition (9, 45), we initially hypothesized that this increase may partially be owed to early inflammatory responses by microglia. To test this hypothesis, we compared the effects of sleep deprivation in mice with or without TREM2 expression, and those expressing the AD-associated TREM2 R47H variant to additionally compare both loss and partial loss of TREM2 on microglial function (28, 30). Using the humanized TREM2 mouse model additionally served as an essential genetic tool to investigate the bi-directional relationship between microglial function and sleep/wake behavior. Indeed, we confirmed that chronic sleep deprivation exacerbated Aβ deposition in 5xFAD transgenic mouse model of cerebral amyloidosis compared to normally sleeping control mice that were matched for age, sex, and genotype. We found that microglia were further reactive upon sleep deprivation, with a similar microglial and macrophage gene expression profile to that previously reported in response to amyloid plaques (34, 35). However, microglia were activated to a greater extent in sleep-deprived mice compared to normally sleeping control mice with Aβ plaque deposition. Whereas part of this sleep deprivation-induced change in microglial reactivity might be explained by concomitant increased Aβ plaque accumulation, arguing against this is that we found a similar exaggeration in microglial reactivity in sleep-deprived mice without Aβ plaques. This suggested that chronic sleep deprivation alone triggered pathological pathways that evoked a microglial response akin to that observed in the injured or diseased brain.
Our findings demonstrate that the inflammatory response from chronic sleep deprivation in mice is dependent on two additional factors, (i) the baseline reactivity of microglia, and (ii) variation in genes expressed in microglia that may further dictate whether microglia are functionally equipped to respond to an additional stressor such as chronic sleep deprivation. Under physiological conditions, microglia function as alert sentinels of the nervous system clearing debris, looking for signs of infiltration by infectious agents, and mediating the inflammatory and repair response to brain injuries. Increased microglial responses to Aβ plaque accumulation triggers microglia to transcriptionally switch to a disease-associated state that essentially allows the immune cells to rapidly react based on what is required of them from their immediate vicinity. Given that TREM2 is critical in this switch to orchestrate multiple microglial functions, we hypothesized that loss of TREM2 would influence sleep/wake behavior. Whereas we did not observe any direct effects of the TREM2 genotype on sleep/wake states in either the 5xFAD mice or non-transgenic mice (Fig. S5), microglial responses to sleep deprivation largely depended on the TREM2 genotype. This was supported by the lack of changes in microglial markers such as IBA1, P2RY12, TMEM119 or Clec7a in mice expressing the loss-of-function R47H variant or no TREM2. This is likely due to loss of TREM2 leading to microglia existing in more of a static and metabolically impaired state, thereby unable to respond to challenges (40, 46), including those initiated by chronic sleep deprivation.
Sleep is a period of low metabolic demand and is considered a restorative brain state that relieves the metabolic cost of wakefulness. This is supported by our findings demonstrating that chronic sleep deprivation leads to metabolic dysfunction with reduced lysosomal maturation, impairment of cathepsin proteases, as well as decreased autophagic degradation. Interestingly, these changes were mainly present in non-plaque depositing T2CV mice, suggesting that microglial responses to sleep deprivation vary in the presence or absence of Aβ plaques and may reflect differences in their baseline reactivity. It is plausible that microglia could be stimulated and primed in response to early changes in soluble or oligomeric Aβ in the brain, which heightens microglial reactivity, resulting in long-lasting changes in glial responses further down the line. This may explain why sleep-deprived 5xFAD/T2CV mice adapt a transcriptional profile similar to that observed in plaque depositing mice but with even greater changes in gene expression. This is further supported by our data showing that despite increased IBA1 and CD68 staining in microglial lysosomes potentially indicating elevated lysosomal activity, sleep-deprived 5xFAD/T2CV mice displayed increased, undigested fibrillar Aβ plaque remains within CD68+ lysosomes, suggesting that microglia may amplify their response in an attempt to phagocytose progressive accumulation of Aβ plaques, yet fail to degrade the material due to the increasing cellular metabolic demand. Consistent with this notion, sleep-deprived 5xFAD/T2CV mice had similar amounts of Aβ plaques compared to normally sleeping 5xFAD/T2KO mice, further suggesting that increased Aβ plaque deposition after sleep deprivation may be a consequence of dysfunctional microglia.
Microglia are crucial to the maintenance of neural networks by breaking down substrates that neurons are unable to process and excrete, as well as forming frequent contacts with neuronal synapses to monitor and regulate firing activity (47). Prolonged wakefulness from chronic sleep deprivation leads to increased neuronal activity, which in itself is known to acutely change interstitial release of soluble Aβ and tau (8, 48). This may additionally contribute to the elevated Aβ plaque accumulation observed in this study, and also seen in previous reports from our lab (9, 37). Increased neuronal activity from chronic sleep deprivation likely results in toxic excitatory neuronal inputs that dysfunctional microglia are unable to modulate due to an impaired negative-feedback mechanism, which may therefore additionally overburden neurons. Concomitantly, the effects of chronic inflammatory signaling by aberrant neuroglial contacts may further contribute to dysregulated synaptic firing and early neuronal dysfunction. Our findings support the hypothesis that chronic sleep deprivation amplifies these combined negative effects by metabolically overwhelming microglia and neurons, which may therefore increase the brain’s vulnerability to insult.
Interestingly, we found that chronic sleep deprivation not only led to TREM2-dependent cellular responses, but also Aβ plaque-specific changes. Sleep deprivation led to transcriptional changes that were only observed in 5xFAD/T2CV mice, in the leptin-insulin signaling pathways, syndecan 1 pathway, regulation of extracellular matrix, as well as glycolysis and insulin signaling pathway in the CSF proteome. Of note, these pathways play a key role in Aβ plaque accumulation by modulating amyloid aggregation, uptake and degradation, as well as clearance and may therefore mechanistically contribute to chronic sleep deprivation induced by Aβ plaque deposition. For example, syndecans and extracellular matrix proteins have been implicated in endocytic uptake and clearance of Aβ through interacting with proteoglycans such as heparan sulfate (49). Additionally, impaired glucose metabolism and reduced glycolytic flux are prominent in the early stages of AD and correlate closely with the severity of disease (50). In line with this, insulin resistance has been shown to reduce Aβ accumulation (51, 52), additionally supporting our findings that expression of insulin receptors and insulin-like growth factor increases in sleep-deprived 5xFAD mice together with enhanced Aβ plaque accumulation. Thus, increased neuronal activity due to chronic sleep deprivation may also account for glycolytic abnormalities that contribute to greater Aβ release, which in turn may lead to a vicious feedback loop in which progressively accruing Aβ deposits further influence glucose metabolism, insulin and leptin signaling as well as neuronal activity (53). This suggests that chronic sleep deprivation earlier in life could contribute considerably to the glycolytic impairment associated with AD that ultimately disrupts metabolic homeostasis, leading to abnormal protein deposition and cognitive decline.
Disrupted sleep is a common feature of aging with decreased sleep, particularly NREM sleep, associated with cognitive impairment that is typical of many neurodegenerative diseases. Based on previous reports and our current findings, it is tempting to speculate that sleep loss over time leads to increasing risk of developing dementia due to a dysfunction of intracellular protein degradation leading to or in response to a damaging inflammatory cascade that is unable to be subdued with simultaneous changes in sleep and inflammation. Individuals who may have a genetic predisposition to developing early or late onset AD, may additionally be influenced by changes in sleep/wake patterns and are more likely to show accelerated progression to AD pathology. Thus, sleep loss over time is detrimental regardless of one’s genetic predisposition. These findings raise the possibility that sleep loss may contribute to cognitive impairment not only by influencing protein aggregation, but also through an immune mechanism. Considering the critical role of immunomodulating genes such as TREM2 in dictating the risk of developing AD as well as the role of microglia in modulating neuronal activity (47), we anticipated changes in sleep/wake behavior with differences in TREM2 genotype. However, we observed no TREM2-dependent differences in either non-transgenic WT mice nor plaque depositing 5xFAD mice, prior to or post Aβ plaque onset. It is plausible that changes in sleep/wake cycles are more prominent later in aging and with substantially greater Aβ plaque burden in aged mice. Future studies should explore whether lysosomal storage disorders with elevated microglial reactivity influence the sleep/wake cycle.
Sleep dysregulation has been shown to increase CSF concentrations of tau and Aβ, which are associated with an increased risk of AD. Here, we show that regardless of a predisposition to developing Aβ plaques, sleep loss impairs multiple cellular processes that lead to metabolic dysfunction, that in part includes a TREM2-dependent role in microglia. A multitude of metabolic pathways being influenced by changes in sleep suggest that this may further increase the risk of developing different diseases; for example, glucose and glycogen dysregulation may lead to increased risk of developing diabetes, obesity, insulin resistance, as well as additional neuroendocrine impairments that may further cascade the likelihood and severity of developing co-pathologies, which is often observed in many neurodegenerative diseases including AD. We observed sleep loss-induced changes in inflammation and metabolic dysfunction in young mice, further suggesting that sleep loss may kindle metabolic dyshomeostasis early in life that progressively exacerbates the cumulative hazard of aging-related disorders (54). Future studies should explore the effects of sleep loss in aging mouse models and in humans as well as other non-plaque depositing models that are also relevant to neurodegenerative disease pathology.
Out study has limitations. The study relies on animal models, which may not fully represent the complex nature of sleep loss in humans over time and its influence on neurodegenerative disease. As it is currently not feasible to experimentally study the effects of chronic sleep loss on microglia reactivity in humans, we used genetic models with humanized APP and TREM2 to obtain a better understanding of their functional response to sleep deprivation. Further, the study focuses on the effects of chronic sleep deprivation on microglial reactivity and Aβ plaque accumulation in the short term. Long-term follow-up studies are needed to determine the persistence of these effects and their potential implications for the progression of neurodegenerative diseases such as Alzheimer’s disease. Additionally, our study offers novel hypothesis-generating insights into how sleep deprivation influences microglial reactivity in mice with or without plaques, though further studies are needed to elucidate the precise mechanisms by which sleep deprivation affects microglial function and Aβ plaque accumulation. We identified TREM2 as an important regulator of microglial response to sleep deprivation, however we cannot rule out TREM2-independent effects of sleep deprivation on microglia function as evidenced by the metabolic dysfunction observed in T2KO mice and 5xFAD/T2KO mice groups.
Taken together, our findings demonstrate that chronic sleep loss plays a critical role in modulating microglial inflammatory and metabolic responses, as well as Aβ plaque burden in the brain. This study raises important questions about charting early changes in disease progression and suggests an interaction between sleep loss, Aβ, TREM2, and microglial reactivity. Further mechanistic studies can assess whether augmentation of microglial function can prevent the impairment seen in these and other cells due to sleep loss. Prospective human studies will be required to test whether improving sleep quality by treating underlying sleep disorders can reduce microglial reactivity, long-term risk of amyloid deposition, and progression to cognitive impairment observed in AD. Understanding how sleep regulates these complex neuroglial interactions will open new avenues regarding which molecular pathways may be most amenable to early therapeutic interventions targeting both sleep disorders as well as neurodegenerative diseases.
Materials and Methods
Study design
The major goal of this study was to investigate how sleep deprivation influences neuroinflammation in the presence or absence of amyloid plaques in the mouse brain and to determine mechanisms that underlie the effect of sleep loss on increasing AD risk. To bridge this knowledge gap, we used a 5xFAD amyloid depositing mouse model of cerebral amyloidosis as well as non-transgenic wildtype control mice and sleep deprived them over 6 weeks starting at 2.5 months of age, to determine the effects of chronic sleep deprivation on early amyloid deposition and associated neuroinflammation in the brain. To additionally examine effects of changes in sleep on microglial function, we crossed these mice so that they expressed the humanized common TREM2 variant, the AD-associated R47H variant or no TREM2 expression. ARRIVE guidelines were followed throughout the study design. Sample size was determined based on power calculation and previous findings (28). A power of 0.8 at an alpha <0.05 was estimated by N=6 mice per group. Mice were allocated randomly after genotyping, with blinded identity to the experimenter through pseudonymization. All analyses were performed blinded by two investigators, except for the western blot experiments. Potential outliers were statistically identified using ROUT analysis (Q=1%) on Graphpad Prism. To ensure reliable replication, experimental method and analysis were designed to include multiple technical as well as biological replicates and to reduce as much subjective quantification as possible. For example, sleep/wake analysis was performed using a machine learning approach in Matlab (MathWorks). All immunohistochemical stainings were performed on at least 2 brain sections per mouse with 10-14 biological replicates, which allowed for at least 2-3 images per region of interest per animal with all images quantified by standard Fiji (ImageJ) protocols. Quantification for mouse brain tissue was excluded if tissue folds were present or the quality of the staining was poor. Reproducibility has been indicated either in the figure legends or shown in quantifications as each animal presented as an individual biological replica as an average of 2-3 technical replicates. Using these mice, we compared the effects of sleep deprivation to normally sleeping control mice using the right hemisphere of the brain for immunohistochemical analyses, and the flash frozen subdissected left hemisphere to biochemically analyze brain region dependent effects of sleep loss.
All animal procedures and protocols were approved by the Animal Studies Committee at Washington University School of Medicine.
Animals
5xFAD/T2CV and 5xFAD/T2R47H mice were generated (28) on mouse T2KO background as previously described (55). Both male and female animals were used and sacrificed at 4 months of age. Age- and sex- and T2 genotype matched littermates negative for the APP and presenilin transgenes were used as non-transgenic wild-type controls. All mice used in this study were backcrossed onto C57BL/6 background for at least ten generations and bred as well as housed in specific pathogen-free conditions. All mice were housed under the same 12h light/dark cycle, ambient room temperature as well as with food and water available ad libitum.
Sleep deprivation
Mice were placed in automated sleep fragmentation chambers (LaFayette Instruments; model 80390) that include a swipe bar, which was set to move every 30secs for six hours per day, starting ZT3 to ZT9 over six weeks. The duration of sleep deprivation was based on a combination of previous findings (9) and from existing literature on the time course of chronic inflammation (23). Mice were monitored daily for signs of stress.
Corticosterone ELISA
Plasma corticosterone was quantified by a colorimetric competitive enzyme immunoassay kit (Enzo, ADI-900-097) following the manufacturer’s protocol.
Measurement and analysis of sleep/wake states
Sleep/wake behavior in mice were monitored using two independent methods, electroencephalography (EEG) and PiezoSleep mouse behavioral tracking system (SignalSolutions). For EEG experiments, animals were anesthetized with isofluorane 0.5-3%. Any signs of pain were assessed by toe pinching before an incision was made. Mice were then surgically implanted with screw electrodes in the skull for EEG and stainless wire electrodes in the nuchal muscle for electromyography (EMG). After a midline vertical incision to expose the skull, forceps and 3% hydrogen peroxide were used to remove any connective tissue and dry the skull for electrode placement. Burr holes for the frontal reference electrodes were made (anterior +0.5 mm, lateral ±0.5 mm; bregma) using a micro drill with a 0.9 mm tip and screws were secured in the skull. Two bilateral active recording electrodes were placed over the parietal cortex (posterior −2.5 mm, lateral ±1.5; bregma) and a ground screw secured over the cerebellum (posterior −6.2 mm, lateral ±0.5; bregma), using the same technique as the reference electrode. The exposed skull, screws, and all wires were covered in a layer of dental cement (SNAP, Parkell) with the pin header secured to the head for subsequent recording. The skin was sutured around the exposed dental cement/pin header and tissue glue (Vetbond, 3M) used to close the remainder of the incision. After the procedure, mice were placed in a warmed chamber to fully recover from anesthesia and individually housed in monitoring cages with fresh bedding, water, food and Carprofen (orally; ¼ tab of 5g tab; ad libitum) supplement. After recovery from surgery over three days, mice were habituated in the recording cage for two weeks, followed by undisturbed EEG/EMG recording performed for two consecutive days in freely moving mice. Bilateral cortical EEG signals were acquired by a P511K A.C. Preamplifier (Grass-Telefactor Instruments, Warwick, RI USA), digitized with a BIOPAC MP150, recorded digitally using the BIOPAC’s AcqKnowlege software with a sampling rate of 250Hz, and converted into (.edf) format for analysis. EEG was processed in MATLAB (MathWorks) through a band-pass filter of 1-30 Hz to remove DC offset and high-frequency noise. EEG/EMG recordings were manually scored in 10-second epochs for wake, NREM and REM sleep to create a calibration file containing mixture-z scoring variables specific for the recording subject. The calibration file was imported into AccuSleep, a machine learning-based, automated sleep scoring program in MATLAB, to complete the remainder of the scoring (56).
We additionally used PiezoSleep mouse behavioral tracking system (Signal Solutions, LLC, Lexington, KY, USA), as described previously described (57). The non-invasive method includes a thin dielectric piezo sensor pad that generates a voltage signal in response to changes in real-time fluctuations in pressure on its surface. Mice were individually housed with the piezo pad underneath fresh bedding, with fresh water and food available ad libitum and recorded without disturbance over a period of six days to determine TREM2 dependent changes in sleep/wake behavior. To validate and compare sleep/wake behavior between sleep deprived and normally sleeping mice, individually housed mice were recorded over 24h either undergoing sleep deprivation as mentioned above or while normally sleeping. Sleep assessment was compared between genotype and age-matched mice that either were normally sleeping or had been sleep deprived for four consecutive weeks. Recording started at ZT1 and stopped at ZT1 the following day to obtain a representation of the animal’s sleep/wake cycle before, during and after sleep deprivation compared to controls. Data was acquired using SleepStats software (Signal Solutions, LLC, Lexington, KY, USA).
Preparation of 488-labelled Aβ42 fibrils and stereotactic intracerebral injections
Human Aβ42 peptide was purchased from GenScript (Cat# 107761-42-2). Monomeric Aβ42 was prepared in DMSO following previous literature (58) and conjugated with Alexa Fluor™ 488 NHS Ester (Succinimidyl Ester) (Thermo Fisher Scientific, Cat# A20000) at molar ratio Aβ42:dye=1:5 in sodium bicarbonate buffer, pH 8.5 at room temperature overnight. Unconjugated dye was removed after 24h 3.5KDa cutoff dialysis in H2O at 4°C. Following this, Aβ42-488 peptide was dried with speed vacuum at room temperature overnight. Unlabeled Aβ42 fibrils and Aβ42-488 fibrils were prepared following the same protocol (58) and dialyzed to PBS for three days. Samples were imaged with negative staining using JEOL 1400 Plus transmission electron microscope (JEOL USA Inc., Peabody, MA). Final products were prepared into 10uM aliquots and stored in −80°C until further use for intracerebral injections.
Following sleep deprivation for six weeks, mice were anaesthesized with isofluorane and immobilized in a stereotaxic frame (Kopf instruments). A 0.5-1.0cm long incision was made in the shaved and sterilized skin to expose the bregma. Prior to intracerebral injection, Aβ42-488 fibril aliquots were thawed on ice and sonicated with amplitude 50% for 30s. Aβ42-488 fibrils were injected into the subiculum at specific stereotaxic coordinates (anterior/posterior: −2.5 mm; medial/lateral: −0.4 mm; dorsal/ventral: −1.8 mm). A total amount of 2.5uL of the 10uM stock was injected using a Hamilton syringe (Hamilton; syringe: 80265–1702RNR; needle: 7803–07). The needle was retained for additional 5 mins before it was slowly withdrawn. The incision was cleaned with povidone-iodine antiseptic solution and sutured. Mice were allowed to recover on a 37°C heating pad and monitored for any signs of stress. Food and water were provided ad libitum. Two days following the injection, mice were euthanized and brains were harvested for immunohistochemical analyses.
Cerebrospinal fluid and tissue collection
All mice were perfused between ZT3 and ZT7, at a time window when mice were sleep deprived, to avoid circadian influence on transcriptional fluctuation of microglial gene expression. Prior to transcardiac perfusion, mice were anesthetized with pentobarbital (100 mg/kg, intraperitoneal). Cerebrospinal spinal fluid (CSF) was collected from animals from the cisterna magna through the dura matter as previously described (59). After CSF sampling, blood was collected from the heart before transcardiac perfusion, which was centrifuged at 5000xg for 5mins at 4°C to obtain plasma. Mice were transcardially perfused with ice-cold phosphate-buffered saline containing 0.3% heparin. One hemibrain was dissected, snap-frozen and stored at −80°C for biochemical analyses. The other hemibrain was immerse-fixed for 24h in 4% paraformaldehyde following by cryoprotection in 30% sucrose for 48h and frozen at −80°C until tissue samples were sectioned for immunohistochemical analyses.
Immunofluorescence staining
30μm microtome-cut sagittal sections were incubated in X34 for one hour at room temperature in 60% PBS/40% EtOH mix; pH was adjusted with 1N NaOH. Following this, sections were washed briefly with 60% PBS/40% EtOH before blocking in 5% donkey or goat serum for 1h at room temperature. Primary antibodies were diluted in blocking buffer and incubated at 4°C overnight with slow agitation unless mentioned otherwise. Primary antibodies were used as follows: IBA1 (1:500; 019-19741, Fujifilm or NB100-1028, Novus Biologicals), CD68 (1:100; FA-11, BioRad), P2RY12 (1:100 at room temperature, S16007D, BioLegend), TMEM119 (1:500; E3E1O, Cell Signaling Technology), Clec7a (1:50 at room temperature; mabg-mdect, InvivoGen), GFAP (1:2000; 2E1.E9 Alexa Flour 488-conjugated, BioLegend), 6E10 (1:1000; Alexa Flour 647-conjugated, BioLegend), NeuN (1:2000; ab104224, Abcam), LAMP1/CD107a (1:500; AF4320, Novus Biologicals), CathepsinB (1:50; AF965, R&D Systems), Cathepsin D (1:50; AF1029, R&D Systems). The next day, sections were washed and incubated with secondary antibody diluted in blocking buffer, followed by a 4′,6-Diamidin-2-phenylindol (DAPI, 5μg/mL) where appropriate before mounting sections onto slides (Prolong™ Gold Antifade reagent, Thermo Fisher Scientific).
Confocal imaging and analyses
Images were acquired using a Leica Stellaris 5 confocal microscope and the Leica Application Suite X software (4.2.1.23810). Laser and detector settings were maintained constant for the acquisition of each immunostaining. For all analyses, at least two images were taken per brain region and slide using 20x (Apo CS 10x/0.40 dry), 40x (Apo CS 40.0x 1.25) and 63x (Apo CS 63.0x 1.4 Oil) differential interference contrast objective, respectively, at 2048 × 2048pixel resolution, with a 15μm z-step thickness. High-resolution stacks imaged by confocal microscopy were 3D reconstructed using the Leica Application Suite X software. Image analyses was performed using Fiji (ImageJ). For the feasibility of the quantification, all layers from a single image stack were projected on a single slice (Stack\Z projection). Next, the plaques per microglia were segmented using automatic thresholding methods in Fiji and presented as % area covered by selected stain. Plaques were segmented and counted using Analyze particles function.
Transmission electron microscopy
Mice were transcardially perfused with 37°C warmed Ringer’s solution containing xylocaine and heparin for 2 min at 37°C, followed by perfusion with a fixative including 2.5% glutaraldehyde, 2% paraformaldehyde and 0.15M cacodylate buffer pH 7.4 with 2mM calcium chloride (CaCl2) at room temperature. Brain tissue was allowed to fix in the fixative solution overnight with gentle agitation at 4°C. Post-fixation, samples of mouse brain were cut into 100μm thick sections using a vibratome (Leica VT1200S, Vienna, Austria). Sections containing relevant regions of interest were rinsed in 0.15M cacodylate buffer containing 2 mM CaCl2 3 times for 10 min each followed by a secondary fixation in 2% osmium tetroxide in 0.15M cacodylate buffer containing 2mM CaCl2 for 2h in the dark. The samples were then rinsed 3 times in ultrapure water for 10 minutes each and en bloc stained with 2% aqueous uranyl acetate overnight at 4°C in the dark. After another 4 washes in ultrapure water, the samples were dehydrated in a graded ethanol series (30%, 50%, 70%, 90%, 100% x3) for 10 minutes each step. Once dehydrated, samples were infiltrated with LX112 resin (Electron Microscopy Sciences, Hatfield, PA), flat embedded and polymerized at 60°C for 48 hours. Post curing, specific regions of interest were excised and mounted on a blank epoxy stub for sectioning. 70nm sections were then cut, post-stained with 2% aqueous uranyl acetate and Sato’s lead. Samples were imaged on a JEOL 1400 Plus transmission electron microscope (JEOL USA Inc., Peabody, MA) operating at an accelerating voltage of 120kV using a NanoSprint15-MkII 16-megapixel sCMOS camera (Advanced Microscopy Techniques, Woburn, MA). Images were acquired by two investigators and were analyzed using Fiji software (ImageJ).
Protein extraction
Frozen mouse hippocampal tissue was weighed and homogenized in a bullet blender homogenizer (Next Advance) using beaded tubes with 200μl RAB buffer pH 7.0 (100mM MES, 1mM EGTA, 0.5mM MgSO4, 750mM NaCl, 20mM NaF, 1mM Na3VO4) supplemented with 1x protease inhibitor (cOmplete™, Roche) and 1x phosphatase inhibitor (PhosSTOP, Roche). This homogenate was centrifuged for five min at 4°C at 5000×g to pellet RAB insoluble material and the supernatant was ultracentrifuged for 20 minutes at 50’000×g with an MLA-130 rotor in an Optima MAX-XP ultracentrifuge (Beckman Coulter) to obtain the RAB extracts. From the remaining cellular pellet, proteins were extracted with RIPA buffer pH 8.0 (150mM NaCl, 50mM TRIS, 0.5% deoxycholic acid, 1% Triton-X 100, 0.1% sodium deoxycholate, 5mM EDTA, 20mM NaF, 1mM Na3VO4) supplemented with protease and phosphatase inhibitors. After a five min clearing for RIPA insoluble material at 5000×g at 4°C, the supernatant was again ultracentrifuged for 30 minutes at 50’000×g to obtain the RIPA soluble protein fraction. The RIPA insoluble pellet was dissolved with ice-cold 70% formic acid (FA) and sonicated for one minute at 30% amplitude in short pulses at room temperature using a sonicator (Model FB120, Fisher Scientific), followed by a final ultracentrifugation for 20 minutes at 50’000×g at 4°C. Protein concentrations were measured for RIPA fractions using a BCA assay (Pierce). All samples were aliquoted and frozen at −80°C until use.
Immunoblotting
Hippocampal RIPA lysates were used for TFEB (1:2000; A17106C, BioLegend), Cathepsin B (1:100; ab214428, Abcam), Cathepsin D (1:100; ab6313, Abcam), LC3b (1:1000; ABC232, EMD Millipore), GAPDH (1:1000; 60004-1-Ig, Proteintech) western blots. 10ug total protein per sample was loaded in 4-20% precast polyacrylamide gels (BioRad) under denaturing conditions and subjected to SDS-PAGE. After separation, proteins were transferred onto nitrocellulose membranes (Trans-Blot Turbo, BioRad) and blocked in 5% milk or 5% albumin in TRIS buffered saline with 0.1% triton-x 100 for one hour at room temperature. Blots were incubated in primary antibodies as listed above at 4°C with gentle agitation. Blots were then washed and incubated in corresponding HRP-conjugated secondary antibodies for one hour at room temperature and visualized using enhanced chemiluminescence technique (Pierce).
RNA extraction
Frozen hippocampal tissue was weighed and homogenized in RNAase-free beaded tubes (REDE, Next Advance) in chloroform with TRIzol™. Samples were centrifuged for 15 minutes at 12’000xg at 4°C and the aqueous upper supernatant transferred for RNA isolation with the RNeasy Mini Kit (Qiagen) following manufacturer’s instructions. RNA quality was controlled using Bioanalyzer prior to Next Generation Sequencing by Clontech SMARTer.
Realtime quantitative PCR
RNA was reverse transcribed to cDNA using the high-capacity RNA-to-cDNA kit (Applied Biosystems, #4387406) following the manufacturer’s instructions. Quantitative PCR was performed with Taqman primers (Cd33 ID: Mm00491152_m1, and Bmal1 ID:Mm00500223_m1 Thermo Fisher Scientific) and Taqman universal PCR master mix (Applied Biosystems, #4304437) using the QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems). Data were normalized to the geometric mean values of β-actin (ID: Mm01205647_g1) and Gapdh (ID: Mm99999915_g1).
RNA sequencing and analyses
Samples were prepared according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina NovaSeq 6000. Basecalls and demultiplexing were performed with Illumina’s bcl2fastq software and a custom python demultiplexing program with a maximum of one mismatch in the indexing read. RNA-seq reads were then aligned to the Ensembl release 76 primary assembly with STAR version 2.7.9a (Doblin et al). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 2.0.3. Isoform expression of known Ensembl transcripts were quantified with Salmon version 1.5.2. Sequencing performance was assessed for the total number of aligned reads, total number of uniquely aligned reads, and features detected. The ribosomal fraction, known junction saturation, and read distribution over known gene models were quantified with RSeQC version 4.0. All gene counts were then imported into the R/Bioconductor package EdgeR and TMM normalization size factors were calculated to adjust for samples for differences in library size. Ribosomal genes and genes not expressed in the smallest group size minus one sample greater than one count-per-million were excluded from further analysis. The TMM size factors and the matrix of counts were then imported into the R/Bioconductor package Limma. Weighted likelihoods based on the observed mean-variance relationship of every gene and sample were then calculated for all samples with the voomWithQualityWeights function and were fitted using a Limma generalized linear model with additional unknown latent effects as determined by surrogate variable analysis (SVA). The performance of all genes was assessed with plots of the residual standard deviation of every gene to their average log-count with a robustly fitted trend line of the residuals. Differential expression analysis was then performed to analyze for differences between conditions and the results were filtered for only those genes with Benjamini-Hochberg false-discovery rate adjusted p-values less than or equal to 0.05.
For each contrast extracted with Limma, global perturbations in known Gene Ontology (GO) terms, MSigDb, and KEGG pathways were detected using the R/Bioconductor package GAGE9 to test for changes in expression of the reported log2 fold-changes reported by Limma in each term versus the background log2 fold-changes of all genes found outside the respective term. The R/Bioconductor package heatmap3 was used to display heatmaps across groups of samples for each GO or MSigDb term with a Benjamini-Hochberg false-discovery rate adjusted p-value less than or equal to 0.05. Perturbed KEGG pathways where the observed log2 fold-changes of genes within the term were significantly perturbed in a single-direction versus background or in any direction compared to other genes within a given term with p-values less than or equal to 0.05 were rendered as annotated KEGG graphs with the R/Bioconductor package Pathview.
To find the most critical genes, the Limma voomWithQualityWeights transformed log2 counts-per-million expression data was then analyzed via weighted gene correlation network analysis with the R/Bioconductor package WGCNA. Briefly, all genes were correlated across each other by Pearson correlations and clustered by expression similarity into unsigned modules using a power threshold empirically determined from the data. An eigengene was then created for each de novo cluster and its expression profile was then correlated across all coefficients of the model matrix. Because these clusters of genes were created by expression profile rather than known functional similarity, the clustered modules were given the names of random colors where grey is the only module that has any pre-existing definition of containing genes that do not cluster well with others. These de-novo clustered genes were then tested for functional enrichment of known GO terms with hypergeometric tests available in the R/Bioconductor package clusterProfiler. Significant terms with Benjamini-Hochberg adjusted p-values less than 0.05 were then collapsed by similarity into clusterProfiler category network plots to display the most significant terms for each module of hub genes to interpolate the function of each significant module. The information for all clustered genes for each module were then combined with their respective statistical significance results from Limma to determine whether those features were also found to be significantly differentially expressed.
Peptide preparation for LC-MS analysis
Peptides were prepared from mouse CSF using a modified filter aided sample preparation method (60). 8μL of mouse CSF was dried then solubilized in SDS buffer (4% SDS, 100 mM Tris-HCl pH8.0) containing 0.2% sodium deoxycholate and reduced with 100 mM DTT at 95°C for 10 min. Reduced samples were mixed with 400μL of 100 mM Tris-HCL buffer, pH 8.5 containing 8M urea (UA buffer) and transferred to the top chamber of a 30K filter unit (Millipore, MRCF0R030). The samples were spun in a microcentrifuge (Eppendorf, Model No. 5424) at 10,000rcf for 10 min followed by an addition of 400μL of UA buffer and centrifugation (10,000rcf for 10 min). The flow through was discarded and the proteins were alkylated by addition of 100 μL of 50mM Iodoacetamide in UA buffer to the top chamber of the filter unit. Samples were gyrated at at 550 rpm (Thermomixer, ThermoFisher Scientific) for 30 min at room temperature in the dark. The filter units were spun at 10,000rcf for 10 min and the flow through discarded. Two UA buffer centrifugation cycles were used to remove excess iodoacetamide. The urea buffer was exchanged into digestion buffer (50mM ammonium bicarbonate buffer, pH 8.0) using two centrifugation cycles with of 200 μL the digestion buffer. The filters were transferred to a new collection tube and 80 μL of LysC (1 μAU) in 100 mM ammonium bicarbonate buffer (pH 8) was added. Samples were incubated for 2h at 37°C followed by the addition of trypsin (1:20) and incubation overnight at 37°C. Following day, filter units were centrifuged at 14,000rcf for 15 min to collect the peptides in the flow through. The filters were washed with 50 μL of 100 mM ammonium bicarbonate buffer and the wash was collected with the peptides. The peptides were acidified with 1% trifluoroacetic acid (TFA) final concentration and were desalted using two micro-tips (porous graphite carbon, BIOMETNT3CAR) (Glygen) on a Beckman robot (Biomek NX) (61). The peptides were eluted with 60% acetonitrile (MeCN) in 0.1% TFA and dried in a Speed-Vac (Model No. Savant DNA 120 concentrator, Thermo Scientific), after adding TFA to 5%. The peptides were dissolved in 20 μL of 1% MeCN in water. An aliquot (10%) was removed for quantification using the Pierce Quantitative Fluorometric Peptide Assay kit (#23290, Thermo Scientific). The remaining peptides were transferred to autosampler vials (#200046, Sun-Sri), spiked with 50fmol of six heavy isotope-labeled mucin peptides, dried and stored at −80°C for LC-MS analysis.
LC-MS analysis
The peptides were analyzed using a nano-Elute chromatograph coupled online to a hybrid trapped ion mobility-quadrupole time of flight mass spectrometer (timsTOF Pro, Bruker Daltonics, Bremen Germany) with a modified nano-electrospray source (CaptiveSpray, Bruker Daltonics). The mass spectrometer was operated in PASEF mode (62). Samples in 1% (vol/vol) aqueous FA were loaded (2 μL) onto a 75 μm i.d. × 25 cm Aurora Series column with CSI emitter (Ionopticks) on a Bruker nano-ELUTE (Bruker Daltonics). The column temperature was set to 50°C. The column was equilibrated using constant pressure (800 bar) with 8 column volumes of solvent A (0.1% (vol/vol) aqueous FA). Sample loading was performed at constant pressure (800bar) at a volume of 1 x sample pick-up volume plus 2 μL. The peptides were eluted using the one column separation mode with a flow rate of 300 nL/min and using solvents A and B (0.1% (vol/vol) FA/MeCN): solvent A containing 2%B increased to 17% B over 60 min, to 25% B over 30 min, to 37% B over 10 min, to 80% B over 10 min and constant 80% B for 10 min. Mass spectra of peptide precursors (MS1) and fragmentation mass spectra of peptides selected in narrow mass range (2Da) from MS1 spectra were recorded from m/z 100 to 1700. Suitable precursor ions for PASEF-MS/MS were selected in real time from TIMS-MS survey scans by a PASEF scheduling algorithm (62). A polygon filter was applied to the m/z and ion mobility plane to select features most likely representing peptide precursors rather than singly charged background ions. Quadrupole isolation width was set to 2 Th for m/z < 700 and 3 Th for m/z > 700, and the collision energy was ramped stepwise as a function of increasing ion mobility: 52 eV for 0–19% of the ramp time; 47 eV from 19–38%; 42 eV from 38–57%; 37 eV from 57–76%; and 32 eV for the remainder. The TIMS elution voltage was calibrated linearly using the Agilent ESI-L Tuning Mix (m/z 622, 922, 1222).
Identification of and Quantification of Proteins
MS data were searched against Mascot search engine (ver 2.8.0.1). Mascot was set up to search against a concatenated UniProt (ver March 2020) database mouse (17,033 entries) and common contaminant proteins (cRAP, version 1.0 Jan. 1st, 2012; 116 entries), assuming the digestion enzyme was trypsin/P with a maximum of 4 missed cleavages allowed. The searches were performed with a fragment ion mass tolerance of 20 ppm and a parent ion tolerance of 20 ppm. Carbamidomethylation of cysteine was specified in Mascot as a fixed modification. Deamidation of asparagine, formation of pyro-glutamic acid from N-terminal glutamine, acetylation of protein N-terminus, oxidation of methionine, and pyro-carbamidomethylation of N-terminal cysteine were specified as variable modifications. PSMs were filtered at 1% false-discovery rate (FDR) by searching against a reversed database.
Processing, quality assurance and analysis of LC-MS data were performed with proteoQ (version 1.5.0.0, https://github.com/qzhang503/proteoQ), software developed with the tidyverse approach (63) tidyverse: Easily Install and Load the ‘Tidyverse’. R package version 1.3.1. https://CRAN.R-project.org/package=tidyverse) with open-source software for statistical computing and graphics, R (R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/) and RStudio (RStudio Team (2016). RStudio: Integrated Development for R. RStudio, Inc., Boston, MA http://www.rstudio.com/). Precursor intensities were converted to logarithmic ratios (base 2), relative to the average precursor intensity across all samples. Within each sample, Dixon’s outlier removals were carried out recursively for peptides with greater than two identifying PSM’s. The median of the ratios of PSM that could be assigned to the same peptide was first taken to represent the ratios of the incumbent peptide. The median of the ratios of peptides was then taken to represent the ratios of the inferred protein. To align protein ratios across samples, likelihood functions were first estimated for the log-ratios of proteins using finite mixture modeling, assuming two-component Gaussian mixtures (64). Ratio distributions were then aligned so that the maximum likelihood of log-ratios was centered at zero for each sample. Scaling normalization was performed to standardize the log-ratios of proteins across all samples. To reduce the influence of outliers from either log-ratios or reporter-ion intensities, the values between the 5th and 95th percentile of log-ratios and 5th and 95th percentile of intensity were used in the calculations of standard deviations.
Metric multidimensional scaling (MDS) and Principal component analysis (PCA) of protein log2-ratios was performed with the base R function stats:cmdscale and stats:prcomp, respectively.
Linear modelings were performed using the contrast fit approach in limma (65) to assess the statistical significance in protein abundance differences between indicated groups of contrasts. Adjustments of p-values for multiple comparison were performed with Benjamini-Hochberg (BH) correction.
Statistical analysis
Graphpad Prism 8.0 was used to perform all statistical analyses. Data are presented as mean ± SEM unless otherwise stated. Data were checked for normality using the Shapiro-Wilk method, D’Agostino and Pearson normality test as well as KS normality test. Statistical significance between groups with normally distributed data was calculated by one-way ANOVA followed by Dunnett’s post hoc test for group-wise comparisons unless otherwise stated. For statistical analysis of more than one group, two-way ANOVA followed by Tukey’s or Sidak’s post hoc test for group-wise comparisons were used based on number of groups and power involved. P less than 0.05 was considered significant: *p< 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Material
Acknowledgements
We thank Marco Colonna for gifting the 5xFAD mouse line crossed with humanized TREM2 variants. We would like to thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help on genomic analysis, and the Proteomics Shared Resource for assistance on data acquisition, processing, and analysis of proteomics data. The expert technical assistance of Yiling Mi and Rose Connors is gratefully acknowledged. The proteomic experiments were performed at the Washington University Proteomics Shared Resource (WU-PSR) directed by Reid Townsend. We would like to thank Sihui Song for fruitful discussions on the manuscript. Finally, we would like to thank Adam Turner for providing assistance with data organization and analysis.
Funding
This work was supported by NIH grant P01NS074969 (D.M.H.), the JPB Foundation (D.M.H.), Alzheimer’s Association Research Fellowship AARF-21-850865 (S.P.), NIH grant P50 HD103525 to the Washington University Intellectual and Developmental Disabilities Research Center (M.W) and 5K08NS10929222 (E.L). The WU-PSR is supported in part by the Washington University Institute of Clinical and Translational Sciences (NCATS UL1 TR000448), the Mass Spectrometry Research Resource (NIGMS P41 GM103422) and the Siteman Comprehensive Cancer Center Support Grant (NCI P30 CA091842). Transmission electron micrograph images were acquired using an AMT Nanosprint15-MkII sCMOS camera, which was purchased with support from the Office of Research Infrastructure Programs (ORIP), a part of the NIH Office of the Director under grant OD032186.3.
Footnotes
Competing interests
DMH is as an inventor on a patent licensed by Washington University to C2N Diagnostics on the therapeutic use of anti-tau antibodies, US patent 9,834,596. DMH co-founded and is on the scientific advisory board of C2N Diagnostics. DMH is on the scientific advisory board of Denali, Genentech, and Cajal Neuroscience and consults for Alector and Asteroid
Data and materials availability
All data associated with this study are present in the paper or the Supplementary Materials. The microarray dataset generated in this study is available in the NCBI Gene Expression Omnibus (accession # GSE225360), and the proteomics dataset is available in the ProteomeXchange (accession # PXD040242).
References and Notes
- 1.Sabia S, Fayosse A, Dumurgier J, van Hees VT, Paquet C, Sommerlad A, Kivimäki M, Dugravot A, Singh-Manoux A, Association of sleep duration in middle and old age with incidence of dementia. Nat. Commun 12, 2289 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prince T-M, Abel T, The impact of sleep loss on hippocampal function. Learn. Mem 20, 558–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M, Sleep Drives Metabolite Clearance from the Adult Brain. Science. 342, 373–377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achariyar TM, Li B, Peng W, Verghese PB, Shi Y, McConnell E, Benraiss A, Kasper T, Song W, Takano T, Holtzman DM, Nedergaard M, Deane R, Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol. Neurodegener 11, 74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellesi M, de Vivo L, Chini M, Gilli F, Tononi G, Cirelli C, Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. J. Neurosci 37, 5263–5273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaneshwaran K, Olah M, Tasaki S, Yu L, Bradshaw EM, Schneider JA, Buchman AS, Bennett DA, De Jager PL, Lim ASP, Sleep fragmentation, microglial aging, and cognitive impairment in adults with and without Alzheimer’s dementia. Sci. Adv 5, eaax7331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie Y, Ba L, Wang M, Deng S-Y, Chen S-M, Huang L-F, Zhang M, Wang W, Ding F-F, Chronic sleep fragmentation shares similar pathogenesis with neurodegenerative diseases: Endosome-autophagosome-lysosome pathway dysfunction and microglia-mediated neuroinflammation. CNS Neurosci. Ther 26, 215–227 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM, Lucey BP, Holtzman DM, The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 363, 880–884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang J-E, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM, Amyloid-β Dynamics Are Regulated by Orexin and the Sleep-Wake Cycle. Science. 326, 1005–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ju Y-ES, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, Fagan AM, Mignot E, Zempel JM, Claassen JAHR, Holtzman DM, Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain 140, 2104–2111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen D-W, Wang J, Zhang L-L, Wang Y-J, Gao C-Y, Cerebrospinal Fluid Amyloid-β Levels are Increased in Patients with Insomnia. J. Alzheimer’s Dis 61, 645–651 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Sharma RA, Varga AW, Bubu OM, Pirraglia E, Kam K, Parekh A, Wohlleber M, Miller MD, Andrade A, Lewis C, Tweardy S, Buj M, Yau PL, Sadda R, Mosconi L, Li Y, Butler T, Glodzik L, Fieremans E, Babb JS, Blennow K, Zetterberg H, Lu SE, Badia SG, Romero S, Rosenzweig I, Gosselin N, Jean-Louis G, Rapoport DM, de Leon MJ, Ayappa I, Osorio RS, Obstructive Sleep Apnea Severity Affects Amyloid Burden in Cognitively Normal Elderly. A Longitudinal Study. Am. J. Respir. Crit. Care Med 197, 933–943 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucey BP, Hicks TJ, McLeland JS, Toedebusch CD, Boyd J, Elbert DL, Patterson BW, Baty J, Morris JC, Ovod V, Mawuenyega KG, Bateman RJ, Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol 83, 197–204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sprecher KE, Koscik RL, Carlsson CM, Zetterberg H, Blennow K, Okonkwo OC, Sager MA, Asthana S, Johnson SC, Benca RM, Bendlin BB, Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 89, 445–453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, Ancoli-Israel S, Jagust WJ, Walker MP, β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat. Neurosci 18, 1051–1057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ju Y-ES, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, Morris JC, Holtzman DM, Sleep Quality and Preclinical Alzheimer Disease. JAMA Neurol. 70, 587 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, Fagan AM, McCue L, Xiong C, Morris JC, Benzinger TLS, Holtzman DM, Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Transl. Med 11 (2019), doi: 10.1126/scitranslmed.aau6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, Zhou Y, Wong DF, Ferrucci L, Resnick SM, Self-reported Sleep and β-Amyloid Deposition in Community-Dwelling Older Adults. JAMA Neurol. 70, 1537–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mander BA, Local Sleep and Alzheimer’s Disease Pathophysiology. Front. Neurosci 14, 525970 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucey BP, It’s complicated: The relationship between sleep and Alzheimer’s disease in humans. Neurobiol. Dis 144, 105031 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingiosi AM, Frank MG, Goodnight, astrocyte: waking up to astroglial mechanisms in sleep. FEBS J. (2022), doi: 10.1111/febs.16424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haydon PG, Astrocytes and the Modulation of Sleep, doi: 10.1016/j.conb.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ransohoff RM, How neuroinflammation contributes to neurodegeneration. Science 353, 777–783 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Long JM, Holtzman DM, Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM, Serrano JR, Hoyle R, Holtzman DM, Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med 216, 2546–2561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullington JM, Simpson NS, Meier-Ewert HK, Haack M, Sleep loss and inflammation. Best Pract. Res. Clin. Endocrinol. Metab 24, 775–84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Everson CA, Clinical assessment of blood leukocytes, serum cytokines, and serum immunoglobulins as responses to sleep deprivation in laboratory rats. Am. J. Physiol. Regul. Integr. Comp. Physiol 289, R1054–63 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M, Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med 215, 745–760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ulrich JD, Ulland TK, Colonna M, Holtzman DM, Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 94, 237–248 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong M, Ghasemigharagoz A, Katzmarski N, Krasemann S, Lichtenthaler SF, Müller SA, Colombo A, Monasor LS, Tahirovic S, Herms J, Willem M, Pettkus N, Butovsky O, Bartenstein P, Edbauer D, Rominger A, Ertiirk A, Grathwohl SA, Neher JJ, Holtzman DM, Meyer-Luehmann M, Haass C, Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. 22, 191–204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leyns CEG, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VMY, Ulrich JD, Holtzman DM, TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci 22, 1217–1222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M, Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang SS, Ebbert MTW, Baker KE, Cook C, Wang X, Sens JP, Kocher J-P, Petrucelli L, Fryer JD, Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med 215, 2235–2245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O, The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I, A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.el7 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R, Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM, Disruption of the Sleep-Wake Cycle and Diurnal Fluctuation of β-Amyloid in Mice with Alzheimer’s Disease Pathology. Sci. Transl. Med 4 (2012), doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O, Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 33, 196.e29–196.e40 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM, Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener 9, 20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ulland TK, Song WM, Huang SC-C, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M, TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 170, 649–663.el3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakanishi H, Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regen. Res 15, 25–29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bustos V, Pulina MV, Bispo A, Lam A, Flajolet M, Gorelick FS, Greengard P, Phosphorylated Presenilin 1 decreases β-amyloid by facilitating autophagosome–lysosome fusion. Proc. Natl. Acad. Sci 114, 7148–7153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura S, Shigeyama S, Minami S, Shima T, Akayama S, Matsuda T, Esposito A, Napolitano G, Kuma A, Namba-Hamano T, Nakamura J, Yamamoto K, Sasai M, Tokumura A, Miyamoto M, Oe Y, Fujita T, Terawaki S, Takahashi A, Hamasaki M, Yamamoto M, Okada Y, Komatsu M, Nagai T, Takabatake Y, Xu H, Isaka Y, Ballabio A, Yoshimori T, LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat. Cell Biol. 2020 2210 22, 1252–1263 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ, An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–47 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roh JH, Jiang H, Finn MB, Stewart FR, Mahan TE, Cirrito JR, Heda A, Snider BJ, Li M, Yanagisawa M, de Lecea L, Holtzman DM, Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J. Exp. Med 211, 2487–2496 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, Lucas A, Baskaran S, Haddick PCG, Lenser M, Earr TK, Shi J, Dugas JC, Andreone BJ, Logan T, Solanoy HO, Chen H, Srivastava A, Poda SB, Sanchez PE, Watts RJ, Sandmann T, Astarita G, Lewcock JW, Monroe KM, Di Paolo G, TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 105, 837–854.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, Hwang P, Chan AT, Graves SM, Uweru JO, Ledderose C, Kutlu MG, Wheeler MA, Kahan A, Ishikawa M, Wang YC, Loh YHE, Jiang JX, Surmeier DJ, Robson SC, Junger WG, Sebra R, Calipari ES, Kenny PJ, Eyo UB, Colonna M, Quintana FJ, Wake H, Gradinaru V, Schaefer A, Negative feedback control of neuronal activity by microglia. Nat. 2020 5867829 586, 417–423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee J-M, Holtzman DM, Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci 14, 750–756 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu C-C, Zhao N, Yamaguchi Y, Cirrito JR, Kanekiyo T, Holtzman DM, Bu G, Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med 8, 332ra44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, Chia CW, Egan JM, Ferrucci L, Troncoso J, Levey AI, Lah J, Seyfried NT, Legido-Quigley C, O’Brien R, Thambisetty M, Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 14, 318–329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freude S, Hettich MM, Schumann C, Stöhr O, Koch L, Köhler C, Udelhoven M, Leeser U, Müller M, Kubota N, Kadowaki T, Krone W, Schroder H, Brüning JC, Schubert M, Neuronal IGF-1 resistance reduces Aβ accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 23, 3315–3324 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Stöhr O, Schilbach L, Moll L, Hettich MM, Freude S, Wunderlich FT, Ernst M, Zemva J, Brüning JC, Krone W, Udelhoven M, Schubert M, Insulin receptor signaling mediates APP processing and β-amyloid accumulation without altering survival in a transgenic mouse model of Alzheimer’s disease. Age (Omaha). 35, 83–101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stanley M, Macauley SL, Holtzman DM, Changes in insulin and insulin signaling in Alzheimer’s disease: cause or consequence? J. Exp. Med 213, 1375–1385 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hahn EA, Wang H-X, Andel R, Fratiglioni L, A Change in Sleep Pattern May Predict Alzheimer Disease. Am. J. Geriatr. Psychiatry 22, 1262–1271 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M, TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160 (2015), doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barger Z, Frye CG, Liu D, Dan Y, Bouchard KE, Robust, automated sleep scoring by a compact neural network with distributional shift correction. PLoS One 14, e0224642 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yaghouby F, Donohue KD, O’Hara BF, Sunderam S, Noninvasive dissection of mouse sleep using a piezoelectric motion sensor. J. Neurosci. Methods 259, 90–100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stine WB, Jungbauer L, Yu C, Ladu MJ, Preparing Synthetic Aβ in Different Aggregation States. Methods Mol. Biol 670, 13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu L, Duff K, A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J. Vis. Exp (2008), doi: 10.3791/960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Erde J, Loo RRO, Loo JA, Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res 13, 1885–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Z-W, Fuchs K, Sieghart W, Townsend RR, Evers AS, Deep amino acid sequencing of native brain GABAA receptors using high-resolution mass spectrometry. Mol. Cell. Proteomics 11, M111.011445 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meier F, Brunner A-D, Koch S, Koch H, Lubeck M, Krause M, Goedecke N, Decker J, Kosinski T, Park MA, Bache N, Hoerning O, Cox J, RÄther O, Mann M, Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteomics 17, 2534–2545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wickham H, Averick M, Bryan J, Chang W, D’ L, Mcgowan A, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Lin Pedersen T, Miller E, Bache SM, Müller K, Ooms J, Robinson D, Seidel DP, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H, RStudio 2 cynkra 3 Redbubble 4 Erasmus University Rotterdam 5 Flatiron Health 6 Department of Integrative Biology. J. Open Source Softw 4, 1686 (2019). [Google Scholar]
- 64.Benaglia T, Chauveau D, Hunter DR, Young DS, Mixtools: An R package for analyzing finite mixture models. J. Stat. Softw 32, 1–29 (2009). [Google Scholar]
- 65.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The microarray dataset generated in this study is available in the NCBI Gene Expression Omnibus (accession # GSE225360), and the proteomics dataset is available in the ProteomeXchange (accession # PXD040242).