


Abstract
Although localized haploid phasing can be achieved using long read genome sequencing without parental data, reliable chromosome-scale phasing remains a great challenge. Given that sperm is a natural haploid cell, single-sperm genome sequencing can provide a chromosome-wide phase signal. Due to the limitation of read length, current short-read-based single-sperm genome sequencing methods can only achieve SNP haplotyping and come with difficulties in detecting and haplotyping structural variations (SVs) in complex genomic regions. To overcome these limitations, we developed a long-read-based single-sperm genome sequencing method and a corresponding data analysis pipeline that can accurately identify crossover events and chromosomal level aneuploidies in single sperm and efficiently detect SVs within individual sperm cells. Importantly, without parental genome information, our method can accurately conduct de novo phasing of heterozygous SVs as well as SNPs from male individuals at the whole chromosome scale. The accuracy for phasing of SVs was as high as 98.59% using 100 single sperm cells, and the accuracy for phasing of SNPs was as high as 99.95%. Additionally, our method reliably enabled deduction of the repeat expansions of haplotype-resolved STRs/VNTRs in single sperm cells. Our method provides a new opportunity for studying haplotype-related genetics in mammals.
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Gametes are natural haploid cells that carry genetic information that can be inherited by future generations, making gametes ideal for genetic research. The first single-sperm genome sequencing technology was developed a decade ago (1,2), and since then, continuous improvements in sequencing methods have provided new insights into single-gamete studies. Different single-sperm sequencing methods possess unique features, such as high-throughput single sperm genome sequencing methods utilizing droplet or combinatorial indexing (SCI) strategies on short-read sequencing platforms (3,4) or high genome coverage methods achieved by high-depth sequencing of a single sperm (5). Although these methods differ, shared biological issues are addressed using single sperm cells and are concentrated in three areas: the study of meiosis and crossovers, genomic instability in sperm, and phasing of the haplotypes.
Sequencing a single gamete using short-read technologies has made it easy and accurate to study meiotic phenotypes without the need for genomic segment sharing information among relatives or analysis of linkage-disequilibrium patterns in populations (1–5). Additionally, long-read sequencing techniques, such as the Oxford Nanopore Technologies (ONT), Pacific Biosciences (PacBio) or Linked-Read sequencing, have also been developed for bulk sperm genome sequencing and are mainly used to improve the detection resolution of crossovers (6–10). These existing sequencing methods have provided great technical support for meiosis research.
Currently, there have been many studies investigating the association between single nucleotide variations (SNVs) and CNVs in sperm cells using short-read technologies and their potential relationships with diseases (11–14). However, the genomic differences between individuals caused by SVs have been estimated to be 3–10 times higher than those caused by SNVs (15–17). In meiosis, homologous pairs of chromosomes undergo physical exchanges of genetic material through homologous recombination and crossing over, which requires DNA double-strand break and repair processes (15). These repair events can result in genomic changes ranging from SNVs to structural and copy number variations, some of which can have significant consequences in offspring. However, studies of SVs in sperm have been limited because short-read technologies have difficulty in detecting genetic changes in complex genomic regions and have a high false-positive rate in detecting SVs (18). In fact, the study of SVs is valuable, especially for repeat-type SVs (the sequences of SVs contain repetitive elements). Repetitive elements (REs) are widespread in the genome and can generally be grouped into two types: tandem repeats and transposable elements (19), both of which have a major impact on global genome stability and have been shown to play significant roles in regulating gene expression, RNA splicing, and DNA methylation (20,21). Importantly, more than 30 Mendelian disorders are caused by tandem repeat expansions via a range of molecular mechanisms (22). The absence of reliable studies of SVs in the genome of gametes may result in missed opportunities to study infertility or genetic diseases.
For the study of haplotype phasing, previous research has shown that localized phasing can be achieved using long reads, ultralong reads, or linked reads sequencing, even without parental data. However, these methods are limited in their ability to extend the phased block to the entire chromosome (from dozens to hundreds of megabases long) without additional data such as Hi-C or Bionano data (23–26), indicating that reliable chromosome-scale phasing remains challenging. The ability to determine chromosome-scale haplotypes would be valuable in studying cis-interactions between regulatory element variants on the same chromosome (long-range promoter-enhancer interactions) (27,28) and understanding the relationships between variants of different genomic elements on the two alleles. For example, there were two SVs more than 1 Mb apart in the unphased genome, with one located in an enhancer element and the other located in the exons of the gene it regulates. After chromosome-scale phasing, we were able to understand whether these two variants were colocated on the same copy of a chromosome (in cis) or whether each of the parental chromosomes harbored a particular variant (in trans) (Supplementary Figure 1A–C). In addition, Strand-seq is a short-read-based method that preserves the structural contiguity of individual chromosomes in every single cell and is considered the gold standard for chromosome-wide haplotype phasing (29–31). However, Strand-seq requires culturing cells in BrdU for one round of cell division to label the nascent DNA strands, and only SNVs and large inversions (>5 kb) can be phased with Strand-seq data alone.
To address these challenges, we developed a long-read-based single-sperm genome sequencing method and a corresponding data analysis pipeline. We sequenced sperm cells from F1 hybrid mouse B6D2F1/Crl [BDF1] (female Mus musculus C57BL/6NCrl [B6] × male Mus musculus DBA/2NCrl [DBA]), and we designed 24 types of Tn5 enzymes (Tn5 enzyme is a transposon compound with a ‘cut and paste’ function) with different barcodes. In this way, high amplification efficiency could be achieved by combining different barcodes of subsequent amplification primers for single sperm genome sequencing (Figure 1A). This approach provided us with high-quality single sperm genome sequencing data to infer crossover events (Figure 1B). By leveraging long read sequencing, we were able to accurately detect structural variations (SVs) in single sperm cells (Figure 1C). More importantly, long-read-based single-sperm genome sequencing enabled chromosome-wide phasing and measurement of haplotype-specific DNA features (Figure 1D). Long reads also enabled us to identify the expansions of haplotype-resolved tandem repeats (STRs/VNTRs) in single sperm cells (Figure 1E).
Figure 1.

Long-read-based single sperm genome sequencing workflow. (A) Schematic view of the experimental process for long-read-based single sperm genome sequencing using the Oxford Nanopore Technologies (ONT) platform. (B) High-quality single sperm genome sequencing data enable the identification of crossover events in a single sperm. (C) Insertions and deletions from a single sperm can be efficiently detected. (D) Single-sperm long-read sequencing enables chromosome-wide haplotype phasing, including structural variations (SVs) and single nucleotide polymorphisms (SNPs). (E) Estimation of the expansions of haplotype-resolved STRs/VNTRs in a single sperm cell.
MATERIALS AND METHODS
Preparation and lysis of mouse sperm cells
All animal experiments were performed according to the guidelines of the Institutional Animal Care and the Ethics Committee of the Peking University (Beijing, China), the research license number is LSC-TangFC-4. We collected the cauda epididymis from an 8-week-old male B6D2F1/Crl. Next, we poked the cauda epididymis gently with a needle and placed them in 1.5 ml Eppendorf tubes. Then, we add pre-warmed Human Tubal Fluid (HTF; EasyCheck, cat#M1130) slowly up to 1 ml and mature sperm cells were incubated in a CO2 incubator at 37°C for 30 min to allow individual mature sperm cells to swim out. To reduce somatic contamination, sperm in the fluid upstream could be transferred for several more times. Finally, 400 μl of 1 ml of liquid containing high-purity sperm was gently pipetted into a tube and heated directly at 55°C for 10 min to allow for better staining of DAPI. After heating, centrifugation was performed at 2000 × g, and the precipitate obtained was resuspended with 0.1% BSA to obtain a single sperm suspension. For FACS sorting, DAPI was used in single sperm cell suspension and we selected the haploid cell group which represents single sperm cells. Before sorting, we checked whether the haploid cell group was sperm cells through microscope. And then single sperm cells were sorted into 96-well plates by FACS, each well contain 2.5 μl cell lysis buffer consisting of 0.375 μl 0.1M DTT, 0.25 μl 100mM Tris-EDTA, 0.125 μl Qiagen protease (20 mg/ml), 0.075 μl 10% Triton X-100, 0.05 μl 1 M KCl and 1.625 μl H2O. And then cell lysis was incubated at 50°C for 3 h to digest proteins and then at 70°C for 30 min to inactivate the protease.
Tagmentation of the genomes of single sperm cells
7.5 μl tagmentation mixture including 2 μl 5 × TAPS_PEG8K (50 mM TAPS–NaOH (or KOH), pH 8.3, 25 mM MgCl2, 40% PEG8K), and 1 μl 0.2 ng/μl one of 24 types of adaptor conjunct Tn5 enzyme (Vazyme, Cat#S601-01) were added into each sperm cell nuclei lysate. The tagmentation reaction was performed on 55°C for 10 min, 2.5 μl 0.2% SDS was added and then the reaction stood at room temperature for 5 min to deactivate of Tn5 enzyme. After tagmentation, 24 single cells with different Tn5 barcodes were pooled together, and then purified with 0.8 volume of Ampure XP beads (Beckman, Cat. A63882) and finally used 50 μl H2O to elute.
Amplification of single sperm cell genome for nanopore sequencing
These purified genomic DNAs of sperm cells were then used for strand displacement and amplification. Amplification buffer mixture including 2 μl 1.25 U/μl Tks Gflex DNA Polymerase (TAKARA, Cat# R060B), 50 μl 2× Gflex PCR Buffer,13 μl H2O and 10 μl 1 μM I5-nano PCR primer which containing 24 bp cell barcode, added 75 μl amplification buffer mixture for each 25 μl purified genomic DNAs. Then amplification reaction was performed as follows: 72°C 3 min, 98°C 1 min and then 20–22 cycles of 98°C for 15 s, 60°C for 30 s, and finally 68°C for 5min.The amplified genomic DNA (gDNA) products were then purified with 0.5× AMPure XP beads twice. For further library construction based on Nanopore platform, we pooled 240–960 single sperm cells together, and the total amount needed was about 1–2 ug. Products were sequenced on Oxford Nanopore PromethION 48 (Oxford Nanopore Technologies; R9.4.1).
Bulk genomic DNA extraction from mouse tissues
Kidney tissues of B6D2F1/Crl♂[BDF1],C57BL/6NCrl♀[B6] and DBA/2NCrl♂[DBA] were cut into small pieces, and mouse genomic DNA (gDNA) was extracted using the QIAGEN DNeasy Blood and Tissue Kit (QIAGEN, cat# 69504) following the manufacturer's Quick-StartProtocol for the part that samples derived from tissues. Then the extracted mouse gDNA was quantified using the Equalbit 1 × dsDNA HS Assay Kit (Vazyme, cat#EQ121). C57BL/6NCrl♀and DBA/2NCrl♂ gDNA were used to construct bulk third-generation sequencing library based on Nanopore platform, and B6D2F1/Crl♂, C57BL/6NCrl♀and DBA/2NCrl♂ gDNA were used for validation of structure variations.
Validation of structure variations in mouse cells
We detected the multiple types of structure variation events between C57BL/6NCrl♀ and DBA/2NCrl♂ mouse for SV phasing. And we selected deletion and insertion event candidates for PCR validation, including SV types defined as simple repeat, LINE1, ERV and complex SVs, such as combinations of these types. Validation-PCR primers were designed by NCBI-Primer designing tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and the pair of PCR primers need to flank the detected breakpoints. For PCR amplification, we used Tks Gflex DNA Polymerase and the details of SV validation primers and parameters of PCR were shown Supplemental Table 4. All PCR amplicons were analyzed on 1% agarose gel, running at 100 V, for 40 min.
Sequencing and data pre-processing
We totally generated six single-sperm ONT sequencing libraries, and each library contains 240–960 single sperm cells. For ONT single-sperm reads from BDF1, we used nanoplexer v0.1 (https://github.com/hanyue36/nanoplexer) and cutadapt v3.4 (https://github.com/marcelm/cutadapt) to demultiplex and trim barcoded ONT reads for each sperm (cell barcode contained 24 bp Tn5 barcodes and 24bp PCR primer barcodes). Cleaned ONT reads were then mapped to mm10 mouse reference genome using minimap2 v2.24 (https://github.com/lh3/minimap2) with parameters ‘–MD -ax map-ont –secondary = no’, and only reads with mapping quality >20 and length >1 kb were used for subsequent analysis.
For ONT bulk reads from B6 and DBA mouse, reads were directly mapped to mm10 mouse reference genome using minimap2 with parameters ‘–MD -ax map-ont –secondary = no’, and only reads with mapping quality >20 and length greater than 1 kb were used for subsequent analysis.
For Illumina bulk reads from B6 and DBA mouse, we used fastp v0.23.2 (https://github.com/OpenGene/fastp) to remove low-quality bases. Then bwa mem -M v0.7.17 (https://github.com/lh3/bwa) was used to map clean reads to mm10 mouse reference genome, and only reads with mapping quality greater than 30 were used for subsequent analysis.
Single nucleotide variation (SNV) calling
We totally used ∼30× Illumina sequencing data for SNP calling. After removing the duplicates, GATK4 HaplotypeCaller v4.1.5.0 (https://github.com/broadinstitute/gatk) was applied to call SNPs from B6 and DBA data. GATK4 VariantFiltration was used to identify high quality homozygous SNPs with parameters ‘GQ > 30; QD > 2.0; QUAL > 30’. Finally, the identified homSNPs were further filtered by requiring as known mouse SNPs in the dbSNP database. Then bcftools (https://github.com/samtools/bcftools) was used to identify the specific SNPs between B6 and DBA, which was 4 664 507 in total, and will be used as the gold standard for heterozygous SNPs of BDF1. For each single sperm, we only examined the HetSNPs loci from the previous step to determine the genotype of these loci in single sperm. Then we used genotype function of whatshap (https://github.com/whatshap/whatshap) to generate chromosomal phase in single sperm from BDF1, and only homozygous SNPs in single sperm will be retained.
Identification of sperm cell doublets
Errors in chromosome segregation during meiosis can result in chromosomes with different (homologous) haplotypes, which only occurred on very small number of chromosomes in a cell, but if happening on the vast majority of chromosomes, it will be defined as having more than one cell with the same barcode. We counted the number of different parental haplotype transitions in each sperm and normalized the genome coverage (as a score) to infer whether this sperm was a doublets cell. We ranked each sperm by the score, and using the KneeLocator function from kneed python package (https://github.com/arvkevi/kneed) to determine a reasonable threshold to filter doublets cells. Finally, only single sperms with genome coverage >1% were used for subsequent analysis.
Inferring of aneuploidy for single sperm cells
To identify single sperm chromosomal aneuploidies, we normalized the sequencing depth and chromosome length for each cell, correcting for differences in sequencing depth and chromosome length between cells. The values were then normalized to one by multiplying by a constant. We identified chromosomes with values less than 0.03 as whole chromosome losses. Chromosomes with values greater than 2 and exhibiting different (homologous) haplotypes were identified as chromosome gains. The copy number results were visualized using the R package ggplot2 v.3.3.5 (https://github.com/tidyverse/ggplot2).
To weigh the stability of the aneuploidy detection signal after normalized sequencing depth, and chromosome length, we used Control-FREEC v11.5 (https://github.com/BoevaLab/FREEC) to calculate chromosome copy number variation with 500 kb windows. We set the following parameters: ploidy = 1, breakPointThreshold = 0.8, and window = 500 000. The copy number results were visualized using the R package IdeoViz v.1.26.0 (https://github.com/shraddhapai/IdeoViz).
Identification of crossover events in single sperm cells
Because of sequencing and PCR errors, the continuity of phased blocks will be greatly compromised, we used a Hidden Markov Model (HMM) to identify the most likely sequence of haplotypes. For each chromosome in each sperm, we build the following HMM model:
Observations
In VCF file from whatshap, each variant location has three possible diploid states 0/0, 1/0 and 1/1 (where 0 represents the reference and 1 represents the alternative allele), and the locations that were called as heterozygous 1/0 were removed. Then, by comparing the genotypes of this result with the gold standard of phased HetSNPs from Illumina data, this variant location could be phased with two ‘observations’ : 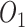 (same genotype as B6) and
(same genotype as B6) and  (same genotype as DBA).
(same genotype as DBA).
States
The observation values can be affected by sequencing and PCR errors, since crossovers cannot occur at a high frequency. We need to infer the most likely hidden state based on the observed value, and the hidden state is the 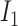 (B6 chromosome) and
(B6 chromosome) and 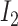 (DBA chromosome).
(DBA chromosome).
Emission probabilities
In VCF file from whatshap, which generates a posterior log-likelihood for each variant, representing the likelihood of each genotype (0/0, 1/0 and 1/1), and we used the value of 0/0, and 1/1 to construct the emission matrix. If the log-likelihoods from whatshap at a particular site are 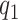 (same genotype as B6) and
(same genotype as B6) and 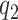 (same genotype as DBA), the emission probabilities are:
(same genotype as DBA), the emission probabilities are:
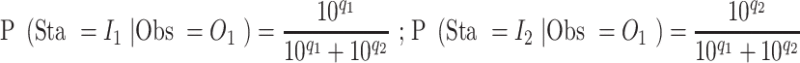 |
Transition probabilities
By relating the probability of state transition to the distance between two HetSNPs, the impact of ONT sequencing errors could be reduced (5). For sites i and j, transition from 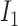 to
to 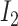 reflects a crossover event from B6 at site i to DBA at site j in the parental hybrid mouse. The transition probability of a change in state between sites i and j is set to be
reflects a crossover event from B6 at site i to DBA at site j in the parental hybrid mouse. The transition probability of a change in state between sites i and j is set to be 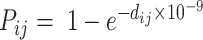 , where
, where 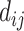 is the distance between the sites i and j (in bp).
is the distance between the sites i and j (in bp).
Initial probabilities
The initial state distribution is 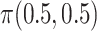 .
.
We used the Viterbi algorithm to infer the most likely sequence of states in the Hidden Markov Model, and only the phased blocks with the length greater than 500 kb and containing more than 100 SNPs will be used to identify crossover events. We observe an excess of multiple transitions in a region of chromosome 4 between ∼143 to 149 Mb. For this region we required the length of phased blocks >1 Mb and contained >200 SNPs. We observe a long region in which SNPs are so sparsely distributed (chr10: 21–82 Mb), and the crossovers across this region will not be recorded.
SV detection from single sperm and benchmarking
We used CuteSV v2.0.1 (https://github.com/tjiangHIT/cuteSV) to identify insertions and deletions (≥50 bp) from single sperm sequencing data with parameters ‘–max_cluster_bias_INS 100 –diff_ratio_merging_INS 0.3 –max_cluster_bias_DEL 100 –diff_ratio_merging_DEL 0.3 –min_support 1 –genotype’. And all SVs that were called heterozygous were removed, since sperm sample is expected to be haploid. We have phased each chromosome of each cell based on the genotype of SNPs, next the SVs called from chromosomes without detected crossover events were used to evaluate the accuracy of SV calling from single-sperm data. We used SURVIVOR v1.0.7 (https://github.com/fritzsedlazeck/SURVIVOR) to merge single-sperm SV sets for the sake of calculation with parameters ‘500${how many cells are needed to support SV} 1 -1 -1 -1’.
For B6 and DBA bulk ONT data, CuteSV was used to identify insertions and deletions (≥50 bp) with parameters ‘–max_cluster_bias_INS 100 –diff_ratio_merging_INS 0.3 –max_cluster_bias_DEL 100 –diff_ratio_merging_DEL 0.3 –min_support 2 –genotype’. And all SVs that were called heterozygous were removed, since the genome of B6 and DBA were expected as homozygous, and those HomSVs will be treated as ground truth. We used precision = TP/(TP + FP), recall = TP/(TP + FN), and F1 = 2 × precision × recall/(precision + recall) to quantify the performance of insertion and deletion detection from single sperm cells.
chromosome-wide haplotype phasing
HetSV phasing
We only need the first 100 cells covering the most HetSVs to complete HetSVs phasing. The different SVs between B6 and DBA was taken as gold truth for BDF1 HetSVs set. We take the intersection of single-sperm SVs and this HetSVs set, the sperm was considered to have an overlap SV only if the size and position are close to the SV from HetSVs set. Finally, a cell-by-HetSV sparse binary matrix was created to accomplish SV phasing (1 means the cell had identified this SV, 0 means it didn’t, Null means no coverage). Due to the HetSVs were sparsely distributed across the genome, and the fact that multi-cell supported SVs have higher accuracy, the SV errors from single sperm need to be corrected before phasing. We did the following preprocessing:
HMM for correction of genotyping errors
In cell-by-HetSVs sparse binary matrix, we compare the column of cell i with the cell j to create a vector of bool value indicating whether those two cells have the same SV. Theoretically, the ‘same’ or ‘different’ value had high continuity. However, due to the accuracy of SV identified in a sperm was not enough, if there is an error in genotyping, the continuity will be affected. We used HMM to detect those genotyping errors. In the HMM, there are two observations ‘a’ and ‘b’ indicating either same or different. Two hidden states ‘A’ and ‘B’ indicating the same or different genotypes. Emission probabilities:  . The definition of Transition probabilities refers to the above, and it needs to be associated with the distance of two HetSVs. The initial probabilities of the two states are 0.5. Then, viterbi algorithm can be used to determine the most likely sequence of the hidden states. The locus markers with genotyping errors are determined where there are conflicts between the observed and the inferred states. We did a pairwise comparison of 100 cells, and if a locus from a cell was marked as error with more than four times, it needed to switch to another genotype. This procedure was repeated for several times to correct most of the genotyping errors.
. The definition of Transition probabilities refers to the above, and it needs to be associated with the distance of two HetSVs. The initial probabilities of the two states are 0.5. Then, viterbi algorithm can be used to determine the most likely sequence of the hidden states. The locus markers with genotyping errors are determined where there are conflicts between the observed and the inferred states. We did a pairwise comparison of 100 cells, and if a locus from a cell was marked as error with more than four times, it needed to switch to another genotype. This procedure was repeated for several times to correct most of the genotyping errors.
The corrected genotyping of all cells was used to impute the dropout of cell-by-HetSVs sparse binary matrix. The function ‘imputationFun1’ from Hapi (https://github.com/Jialab-UCR/Hapi) was used for the imputation of missing genotypes. And then the SV linkage information was used to phase HetSVs with Hapi software. The phasing results will be compared with the HomSVs from B6 and DBA to calculate the accuracy of phasing.
HetSNP phasing
The method of phasing HetSNPs was similar to that of phasing HetSVs. To improve the accuracy of phasing, we selected the first 400 sperm cells with the highest coverage for subsequent calculations. Firstly, we utilized Hidden Markov Model (HMM) to correct genotyping errors, and the HMM parameters were identical to those used for HetSV phasing. Next, we used the function ‘imputationFun1’ from Hapi (https://github.com/Jialab-UCR/Hapi) to impute missing genotypes. Finally, we employed linkage information from SNPs to phase HetSNPs with the Hapi software. The phasing results were then compared with HomSNPs from B6 and DBA to assess the accuracy of phasing results.
The annotation of successfully phased SVs
SVs longer than 100 bp and could be correctly phased will be used for annotation, which was 36 271 in total. Since these SVs could be identified in both B6 or DBA bulk ONT sequencing data and BDF1 single sperm ONT sequencing data, we used the sequences of SVs from bulk data for repeat annotation. For inserted sequences, the insertion of novel sequences from cuteSV were used; for deleted sequences, the sequence between breakpoints relating to the reference were used. Then, we used RepeatMasker v4.1.2 (https://github.com/rmhubley/RepeatMasker) and TRF v4.09.1 (https://github.com/Benson-Genomics-Lab/TRF) with default parameters to identify repeats in each sequence, and we considered sequences as covered by repetitive elements if repeat(s) occupied >80% of the sequences, which was 25 664 in total.
The identification of haplotype-resolved STR/VNTR expansions
RepeatHMM (https://github.com/WGLab/RepeatHMM) is a novel computational tool to accurately estimate expansion counts from PacBio and Oxford Nanopore data, and we used this software to identify the expansion or contraction of haplotype-resolved STRs/VNTRs. First, we only examined the annotated loci of STRs/VNTRs in mm10 reference downloading in http://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/. Second, RepeatHMM was used to estimate the expansion counts of those loci from bam files with parameters ‘–MinSup 4 –FlankLength 100 –SeqTech Nanopore –repeatName all –RepeatTime 3’. Third, the loci with different length between B6 and DBA allele >100 bp, and the different repeat counts >5 were retained. In order to make the annotation more accurate, we only keep the loci where RepeatHMM estimated as allele specific STRs/VNTRs expansions and the HetSVs annotated as tandem.
The visualization of haplotype-resolved STR/VNTR expansions
To produce a sequence composition view, we require (1). The loci had the same length of STR/VNTR expansions with HetSVs. (2) There were no other SVs or repeats within 500 bp of the STR/VNTR flanks. First, we estimate the locations of the STR/VNTR region from mm10 reference annotation, and then the flanking sequences (500 bp) and expansion sequences of SVs from B6 and DBA allele were used as haplotype-resolved sequences for following analysis. Second, jellyfish v0.9.0 (https://github.com/gmarcais/Jellyfish) was used to establish the abundance of each available k-mer, where the length of k-mer was the same as the length of STR/VNTR unit. Third, the k-mers are ordered by decreasing order of abundance, and each k-mer sequence is paired with a specific color based on their frequencies.
RESULTS
Obtaining 1573 high-quality single-sperm genome sequencing data on the ONT platform
In our long-read-based single-sperm genome sequencing method, cells with different Tn5 barcodes were pooled together for amplification, which may have caused potential intercellular contamination between single sperm cells. Such contamination could lead to the appearance of heterozygosity or non-haploid regions in a sperm cell genome, thereby affecting the accuracy of identifying sperm aneuploidy and crossover locations. To demonstrate the reliability of our single-sperm genome sequencing method, we conducted a species-mixing experiment by mixing mouse embryonic stem cells (mESCs) and human lymphoblast cells (HG002) in the same sequencing library. Specifically, an equal number of cells from these two cell lines were pooled together before single-cell genome amplification. The cross-contamination rate was determined by the ratio of read alignment to the false reference genome. In this experiment, the cross-contamination rate was <2%, and we unambiguously recovered the information for 72 human cells and 72 mouse cells from the read mapping results, indicating nearly no cross-contamination among these 144 loaded cells (Figure 2A). These results validate the feasibility of our method and demonstrate the accuracy of our subsequent analyses.
Figure 2.

Quality of our long-read-based single sperm genome sequencing data. (A) Performance of the species-mixing experiment. Scatterplot shows the numbers of reads aligned to human (hg38) or mouse (mm10) reference. Each orange dot represents a mouse cell, and each blue dot represents a human cell. (B) Genome sequencing depths and coverages of 1573 single sperm cells. (C) Distribution of average read length from each sperm. (D) A heatmap showing the whole-chromosome losses and gains in 1573 single sperm, including 29 whole-chromosome loss events in 23 sperm cells, with 15 on autosomes and 14 on sex chromosomes, as well as 4 gain events in 4 individual sperm cells (the chromosome that was identified as lost is shown in purple, and gain is shown in orange). Additionally, shown are 20 normal sperm cells carrying the X (10) or Y (10) chromosome. (E) Examples of complex aneuploidy on chromosome 3 in H_F1_500_cell_v2_sc14708. The normal single sperm chromosome (chromosome 2) is haploid, whereas we identified a chromosome-arm-level gain event on chromosome 3. DNA copy number was estimated by normalizing the read coverage in 500 kb bins. The SNP track shows SNP positions on the chromosome (blue: haplotype 1; red: haplotype 2).
Using our newly developed sequencing technology, we obtained the sequences of a total of 1573 sperm cells from F1 hybrid mouse B6D2F1/Crl [BDF1] (filtering out samples containing multiple sperm cells or genome coverage less than 1%) (Supplementary Figure 2). Haploid genome sequencing depths ranged from ∼0.02× to ∼1.37× (median sequencing depth of ∼0.1×), and the genome coverages ranged from ∼1% to ∼25.5% (median genome coverage of ∼4.9%) (Figure 2B). The numbers of sperm cells carrying the X (846) or Y (713) chromosome were comparable, as expected. The average read length from each sperm was approximately 5.5 kb. (Figure 2C and Supplementary Table 1).
Aberrant meiotic divisions might cause whole chromosomal aneuploidies (whole chromosome gains or losses). Aneuploidies in sperm can be inherited by the embryo after fertilization, leading to congenital disorders, infertility, and pregnancy loss (15). In our haploid dataset, we identified whole chromosome loss and gain events by normalizing the read density across the 21 mouse chromosomes (19 autosomes plus X & Y chromosomes). To ensure accuracy, we performed manual inspection, where chromosomes with copy number increases and different (homologous) haplotypes were considered to have undergone chromosome gains (see Methods). Finally, among the 1573 sperm cells analyzed, we identified 29 whole-chromosome loss events in 23 sperm cells, with 15 on autosomes and 14 on sex chromosomes. The frequency of whole-chromosome losses was 1.5% per sperm cell (Figure 2D and Supplementary Figure 3). Additionally, we identified 4 autosome gain events in 4 individual sperm cells (Figure 2D).
Chromosome 3 appears to be more susceptible to aneuploidies. Specifically, we observed two instances of autosome gain and five instances of whole-chromosome loss occurring on chromosome 3. For example, in a particular sperm cell, the normal single sperm chromosome (chromosome 2) was haploid and exhibited a low frequency of crossover events (maintaining the same genotype for a long chromosome region), as expected. However, we identified a complex aneuploidy event on chromosome 3, with a significant increase in copy number in a large region of the long arm, and the phased blocks also showed a high frequency of ‘exchange’ in this area. This was likely caused by chromosome segregation errors (Figure 2E).
Chromosome-wide HetSNP phasing and detection of crossovers
Due to the higher error rate of ONT sequencing compared to short read technologies, we utilized bulk Illumina data from the parental genomes of the donor mice (B6 and DBA) to aid in the phasing of heterozygous single nucleotide polymorphisms (HetSNPs). We identified ∼4.6 million potential HetSNPs in the F1 hybrid mouse by comparing the homozygous SNPs between the parental genomes. For each single sperm cell from ONT sequencing, only ‘homozygous’ SNPs that overlapped with the set of HetSNPs from the Illumina data were retained and used for phasing. Ultimately, each single sperm cell covered an average of ∼0.24 million HetSNP sites (Supplementary Figure 4). Next, we performed haplotype phasing on these ∼4.6 million heterozygous SNPs. We first used a hidden Markov model (HMM) to correct genotyping errors and phased them at the chromosomal scale by comparing the SNP linkage information (see Methods) (Supplementary Figure 5). Using this approach, we were able to phase ∼ 4.58 million (98.15%) HetSNPs into two sets of haplotypes, and by comparing them with the known parental information, we calculated a phasing accuracy rate of 99.95% (Table 1).
Table 1.
Accuracy of haplotype phasing of SNPs and SVs
| Type of genomic variants | SVs | SNPs | |
|---|---|---|---|
| No. of genomic variants from parental genomes | C57BL/6NCrl | 5901 | 8837 |
| DBA/2NCrl | 57 116 | 4 665 972 | |
| No.of heterozygous genomic variants in the F1 hybrid | 54 712 | 4 664 507 | |
| No. of genomic variants phased in single sperms | 51 624 (94.36%) | 4 578 324 (98.15%) | |
| No. of phased genomic variants consistent with family trio | 50 899 (98.59%) | 4 576 131 (99.95%) | |
| The length of Phased blocks | Whole chromosome | Whole chromosome |
Total number of detected and phased SVs (insertions and deletions) and SNPs with respect to the mm10 reference genome. The mm10 reference genome is derived from the widely used C57BL/6J strain, while the C57BL/6NCrl strain we used here is a substrain of C57BL, which is slightly different from C57BL/6J.
To measure the uniformity of HetSNP distribution, we divided the genome into windows of 500 kb and calculated the number of HetSNPs in each window covered by every single sperm cell (Figure 3A). We found that the HetSNPs were unevenly distributed across the genome, with some loci having a high density of HetSNPs (chr1:153.0–175.5 Mb; chr8:29.5 Mb- 41.5 Mb; chr14:86.0–99.5 Mb) and others having sparse HetSNPs (chr8:41.5–56.0 Mb; chr9:3.0–31.0 Mb; chr10: 19.5–82.0 Mb) (Figure 3A). Unevenly distributed HetSNPs may affect the resolution of crossover sites.
Figure 3.

Detection of crossover position and crossover interference. (A) Distribution of HetSNPs. The heatmap from CMplot visualizes the number of HetSNPs called from bulk Illumina data (1 Mb window). The dot point visualizes the median HetSNPs in 1573 single sperm cells (500 kb window). (B) A HMM was used to identify the most likely crossover position. Dots represent observations (red is B6 genotype and blue is DBA genotype), which could be affected by sequencing and PCR errors. Lines represent hidden states (red is B6, blue is DBA and gray is undetermined). We used the transition of hidden states to infer the position of crossovers. (C) Crossover distribution map of the 19 chromosomes from H_F1_2304_cell_v6_sc10111. The normalized coverage depth that reflects chromosome copy number is shown on the right side. Blue lines represent the SNPs from the DBA allele; red lines represent the SNPs from the B6 allele. (D) Distribution of the number of crossovers identified per sperm cell (only cells that 19 autosomes all fit our criteria were used). (E) Average number of crossovers (±SE) called per chromosome per sperm cell. (F) Crossover interference. The base-pair distances between two crossover events (the chromosomes with only two crossovers were used). The red line represents the distribution curve fitted from the experimental data. The black lines show randomly generated distance fitting lines, while the dotted line represents the median of the random distributions.
We utilized a hidden Markov model (HMM) to identify the most likely crossover positions (Figure 3B). The key parameters for this algorithm in single-sperm ONT sequencing data were the transition probabilities between haplotypes. By adjusting the probability of state transition and relating it to the distances between two HetSNPs, we could minimize the impact of ONT sequencing errors and unevenly distributed HetSNPs (see Materials and Methods). Using this approach, we could accurately identify the location of crossovers and the genotype of each region of the genome in individual sperm cells. Figure 3C visualizes the locations of crossovers identified in a sperm cell. Additionally, we have presented the crossover locations in four single sperm cells with different genomic coverage (Supplementary Figure 6). Finally, we identified 17 445 autosomal crossover events in 1573 sperm samples, with a median resolution of crossover sites of 945 kb (Supplementary Figure 7 and Supplementary Table 2). The recombination rates ranged from 4 to 27 crossovers per sperm cell (with an average of 12 crossovers per cell) (Figure 3D), which is consistent with the findings of previous studies (5). The average number of crossovers (±SE) called per chromosome in a single sperm cell is shown in Figure 3E. Interestingly, we found that chromosome 3 had a lower frequency of crossovers compared to other chromosomes of similar size, while exhibiting a higher percentage of aneuploidy. This suggests that crossovers may protect against meiosis nondisjunction of the chromosomes on which they occur (4).
In addition, we investigated crossover interference by analyzing chromosomes with only two crossovers (1476 chromosomes). We calculated the base-pair distances between two crossover events and compared their length distribution to that expected from random simulation. Our analysis showed that the median observed distance between crossovers was approximately 122 Mb on chromosome 1, which was 63% of the length of chromosome 1. This distance was much larger than the expected distance of 53 Mb based on random simulation. This result was consistent with the phenomenon of repulsion of crossovers in close proximity on the same chromosome, which is known as crossover interference (Figure 3F and Supplementary Figure 8).
Detection of insertion and deletion events in single sperm cells
Long-read sequencing is advantageous for accurately identifying structural variations, and our method enabled the identification of genome-wide structural variations at the resolution of the single sperm cell for the first time. Next, we aimed to identify insertions and deletions in the single sperm genome, as they represent the second most common type of genomic variations (32). To evaluate the feasibility of SV detection from long-read-based single-sperm genome sequencing data, we sequenced the parental homozygous genome on the bulk ONT platform as a gold standard (see Methods), and we identified 57 116 and 5901 SVs within the DBA/2NCrl and C57BL/6NCrl genomes, respectively. The Mus musculus reference genome (mm10) was derived from the widely used C57BL/6J strain, while the C57BL/6NCrl strain we used here is a substrain of C57BL, which is slightly different from C57BL/6J. Therefore, a very small number of SNPs and SVs were identified in its genome when compared to the mm10 reference genome (Table 1). The number of SVs we identified was comparable to those reported in previous studies using long-read sequencing platforms (33,34).
We only used reads that were mapped to chromosomes without detected crossover events to identify SVs (>50 bp). We used three metrics: precision, recall and F1-score to evaluate the accuracy of SV detection. As shown in Figure 4A, the precision of SV detection increased with the number of supporting cells. The precision with >6 supported cells improved to 90%, while SVs supported in three cells had the highest F1-score (78%). This accuracy is consistent with results from long-read-based K562 single-cell genome sequencing data (35).
Figure 4.

Detection and phasing of heterozygous insertions and deletions. (A) Precision, recall, and F1-score were evaluated for our sequencing method in detecting SVs with varying numbers of supporting sperm cells. (B) Distribution of successful phasing or unsuccessful phasing HetSVs across the genome. The red lines represent heterozygous SV sites where phasing was unsuccessful, while the green lines represent sites where phasing was successful. (C) Length distribution of successfully phased HetSVs. The red line represents the length distribution of deletion events relative to the reference genome, while the blue line represents the length distribution of insertion events relative to the reference genome.
To further demonstrate the accuracy of our SV detection method, we examined the alignment of single sperm genome sequencing reads around the Spata6 and Wrn genes and identified a 1118 bp deletion event and a 976 bp insertion event in DBA mice compared to B6 mice, respectively (Supplementary Figure 9). These SVs contain many repeat sequences and are difficult to identify in short-read data but were accurately detected in our long-read-based single-sperm genome sequencing data. For example, some single sperm cells had a 6449 bp deletion and a 571 bp insertion near an LTR repeat element on chr1 and chr4, respectively, while others did not. Therefore, by determining the presence of these structural variations in this region, the genotypes of these single sperm cells in this local region could be clearly distinguished, and their genotypes could be determined as either B6 or DBA by comparison with bulk genome sequencing data from the parents (Supplementary Figure 10).
Chromosome-wide HetSV phasing with single-sperm genome sequencing data
Subsequently, we used these data to complete chromosome-wide HetSV phasing. We developed an innovative SV phasing pipeline that can achieve entire-chromosome scale SV phasing using only 100 single sperm cells (see Methods) (Supplementary Figure 5). We identified a total of 54 712 HetSVs (specific SVs from the B6 and DBA cells) from parental bulk ONT data, of which 94.36% were phased, with a phasing accuracy rate of 98.59% (Table 1). The accuracy of SV phasing was found to be slightly different across different chromosomes (Figure 4B), with the accuracy of SV phasing on chromosome 10 being the best and the ratio of the number and total length of correctly phased SV being lower on chromosome 19 (Supplementary Figure 11a, b). The size distribution of successfully phased SVs displayed an ∼190 bp peak and a 6 kb peak, which could be explained by the dominance of two types of repeat elements: SINEs and LINEs (34) (see below) (Figure 4C). We compared the genomic features overlapping with successfully phased HetSVs to understand their distribution and found that half of the HetSVs intersected with the distal intergenic regions (Supplementary Figure 11c), suggesting that they rarely affect gene expression directly within promoter or gene body regions (36).
To assess the potential impacts of SVs on phenotypes, previous studies tested 281 246 SVs from thirteen classic and four wild-derived inbred strains of mice (37). These SVs were tested in association with 100 phenotypes measured in over 2000 HS mice (38). The study identified 12 high-confidence quantitative trait loci (QTLs) where the SVs overlapped a gene or flanking region ( 2 kb up and downstream) and the effect size was in the top 5% of the distribution (37). Among these 12 SVs, we identified 5 successfully phased HetSVs using our long-read-based single-sperm genome sequencing data that overlapped with them. Supplementary Table 3 provides the details of these SVs, including their exact lengths and the putative phenotypes that these SVs were associated with, such as T-cell phenotype (39) (in the promoter of the H2-Ea-pa gene) and cellular hemoglobin (37) (in the exon of the Trim30b gene). By accurately phasing these SVs, we could infer phenotypic differences between DBA and B6 (Supplementary Figure 12 and Supplementary Figure 13).
Sequence features of haplotype-specific insertions and deletions
A significant fraction of SVs were considered to be repeat-type SVs. To analyze the repeat features of these SVs, we used RepeatMasker software (http://www.repeatmasker.org; see Methods) and focused only on correctly phased HetSVs longer than 100 bp. We considered sequences as being covered by repetitive elements if repeat(s) occupied >80% of the sequences (40). Finally, among the 36 271 successfully phased HetSVs (length >100 bp), 25 664 were involved in repetitive elements. Among these, 29.3% were covered by a single LINE, 17.0% by a single SINE, 16.2% by a single LTR, 12.4% by a tandem repeat, 24.9% by multiple repeats and 0.2% by other repeats (Figure 5A). The proportion of different types of repetitive elements contained in heterozygous SVs is similar across different autosomes (Supplementary Figure 14a), and the length distribution of the single repeats was consistent with the findings of previous studies (34) (Supplementary Figure 14b). We further analyzed the combination of multiple repeats in the same SV and found that the combination with tandem repeats was predominant, with the combination of one LINE and tandem repeat being the most frequent (Figure 5B).
Figure 5.

DNA sequence features of haplotype-specific insertion and deletion events. (A) Classification of the repeat(s) in 25 664 successfully phased HetSVs (>100 bp). We considered sequences as covered by repeat(s) if repeat(s) occupied ≥80% of the sequences. (B) Composition of 6398 multirepeats. Families with counts ≤ 50 were classified as other. (C) Examples of HetSVs covered by multiple repeats. A 732 bp deletion of DBA haplotype containing three subfamilies of SINEs (including B1, B2 and B4). (D) 7159 bp deletion of DBA haplotype, including L1 (LINE) and ERVK (LTR). (E) PCR validation of 2 HetSV events covered by multiple repeats, identified in B6, DBA and BDF1. (F) Examples of haplotype-specific tandem repeat expansions. The sequences from each haplotype were colored according to their k-mer abundance. A B6-specific STR expansion is located in the intronic region of the Ccnf gene and composed of 19/149 (DBA/B6) repeats of GAAA. (G) A DBA-specific STR expansion is located in the distal intergenic region of chromosome 7 and composed of 191/71 (DBA/B6) repeats of CTAT.
Additionally, we examined reads that were aligned around certain DBA deleted alleles, which were successfully phased in single-sperm reads, and compared our annotations with those from the UCSC mm10 reference genome to verify the accuracy of our annotations (Supplementary Figure 15 and Supplementary Figure 16). For instance, we identified a 732 bp deletion in the Trpm1 gene that contained three SINEs, and the annotation from the mm10 reference genome indicated that this region contained three subfamilies of SINEs, as expected, including B1, B2, and B4 (Figure 5C and E). We also identified a 7159 bp deletion on chromosome 1, which contained an LTR and a LINE (Figure 5D and E). By comparing our annotations with those from the mm10 reference genome, we were able to demonstrate the validity of our annotation to some extent. Furthermore, to verify our results, we used genomic DNA (gDNA) from parental (B6 and DBA) and offspring (BDF1) mice as PCR templates for SV validation, selecting 7 SVs containing multiple repeats as candidates. All 7 selected events were validated with the expected sizes (Supplementary Figure 17).
The estimation of haplotype-specific tandem repeat expansions
As a highly abundant type of repetitive elements, tandem repeats are important for forensic testing, evolution, and genetic disease research. Therefore, we aimed to identify expansions or reductions in haplotype-resolved STRs/VNTRs (STRs: repeating units of DNA of 1 to 6 bp; VNTRs: repeating units of DNA of >6 bp) in single sperm cells. We identified 3190 tandem repeats that were specifically expanded or reduced between haplotypes. To avoid artifacts from ONT sequencing or the base-calling process, it is not appropriate to de novo identify repeat units from base-called ONT data (41). We only identified expansions or reductions of annotated tandem repeat loci from the mm10 reference genome, where the type of repeat unit had been previously annotated. Using RepeatHMM software (42), we estimated the approximate locations of the repeat region in each ONT read and used an HMM to estimate repeat counts. We then used the k-mer frequency information to visualize the structure and sequence composition of these repeat loci across the B6 and DBA haplotypes (Supplementary Figure 18). For example, we identified a 520 bp HetSV in the intronic region of the Ccnf gene that contained 149 GAAA repeats in the B6 allele and 19 GAAA repeats in the DBA allele (Figure 5F). We also detected 191 CTAT repeats in the distal intergenic region of chromosome 7 in the DBA allele and 71 CTAT repeats in the B6 allele (Figure 5G).
To ensure the accuracy of the repeat-type HetSVs that we detected and successfully phased, we selected 70 HetSVs for SV validations. These HetSVs were covered by different types of repeats, including SINEs, LTRs, LINEs, and STRs, as well as combinations of multiple types of repeats. We used gDNAs from B6, DBA, and BDF1 as PCR templates for SV validation. As a result, 15 out of 18 SINE-associated SV events (83%) (Supplementary Figure 19), 14 out of 15 (93%) LTR-associated SV events (Supplementary Figure 20), 15 out of 18 (83%) LINE-associated SV events (Supplementary Figure 21) and 12 out of 12 (100%) STR-associated SV events (Supplementary Figure 22) could be successfully amplified with the expected size (Supplementary Table 4). These results demonstrate the reliability and accuracy of the structural variations identified in single sperm cells and the haplotype phasing of HetSVs.
DISCUSSION AND CONCLUSIONS
The development of long-read genome sequencing has greatly promoted the study of haplotype analysis. In cases where parental information is available, haplotypes can be easily resolved using the trio binning method (43). In the absence of parental information, additional data are needed to achieve chromosome-scale haplotype phasing, such as chromosome sorting (44), Strand-seq (45) and Hi-C (46), which can link variants over a much longer range, delivering chromosome-scale phase blocks. However, these methods are time-consuming and expensive (43). In comparison, direct sequencing of individual gamete genomes is a more efficient way to perform haplotype analysis, but currently, all single gamete sequencing technologies can only be completed using short-read sequencing platforms, limiting single gamete sequencing to haplotype phasing of HetSNPs.
We developed a long-read-based single-sperm genome sequencing method and a corresponding data analysis pipeline. This approach represents a significant improvement over short-read single-sperm sequencing, which can only partially detect SV breakpoints. Additionally, our innovative SNP and SV phasing pipeline allows us to accurately phase SNPs and SVs at a whole chromosome scale, which could aid in the study of genomic imprinting and parent-of-origin effects. Notably, our method enables us to classify repeat-type SVs and identify repeat expansions and reductions at a haplotype-specific resolution, which has important implications for the study of genetics and related diseases.
Based on our experiments, we found that pooling ∼480 single sperm cells together per sequencing run ensured uniform genome coverage and low duplication rates for each cell. The ONT platform produced ∼110 Gb of data per run, and the cost per individual sperm was ∼$3. Using our method, we were able to achieve SV phasing at the scale of the entire chromosome using only 100 single sperm cells, with a theoretical cost of ∼$300 to achieve whole genome haplotyping. However, it should be noted that the error rate of ONT sequencing was higher, and that to improve haplotyping accuracy, somatic bulk Illumina sequencing data were needed. Additionally, in this study, we only completed haplotyping of SVs longer than 50 bp. For haplotype phasing of indels, the PacBio high-fidelity (HiFi) sequencing platform may be more appropriate.
Compared to short-read-based single-sperm genome sequencing technologies that apply the drop-seq strategy (4), our method can achieve similar genome coverage at the same sequencing depth. Specifically, our method can achieve a genome coverage of approximately 2% for a single sperm cell when sequenced at ∼0.1 Gb, which is comparable to the coverage reported for drop-seq at approximately 1.5%. (Supplementary Figure 23a). However, the average sequencing depth per cell is ∼0.15 Gb or even lower, resulting in suboptimal coverage uniformity for individual sperm cells. Detecting CNVs at such a low sequencing depth is challenging, especially for long-read sequencing, which poses limitations for detecting CNVs ranging from 100 kb to several tens of megabases in sperm cells, while remaining accurate in detecting the loss or gain of an entire chromosome. We selected some cells with high sequencing depth to calculate their CNV values. These sperm cells exhibited haploid features and could be clearly separated into those carrying the X or Y chromosome. Additionally, a cell with a deletion on chromosome 3 was also clearly identified (Supplementary Figure 23b). This result suggests that with increased sequencing depth, our method can still achieve CNV detection at a high resolution.
There are several potential applications of our method in genomic research. First, similar to the Strand-seq technique, our approach can be combined with bulk long read sequencing to achieve highly accurate haplotype assembly. Alternatively, by sequencing individual sperm cells more deeply and in greater numbers, our method can also enable direct de novo haplotype assembly (47). Second, our method can be applied to the study of human single sperm cells, providing opportunities to investigate infertility, genetic diseases, and the patterns of clonal mosaic mutations in male gonads through the identification of SVs in the genome of infertile men. Third, the accuracy of the ONT sequencing platform we used in detecting de novo tandem repeat units and counts is limited. Therefore, in this study, we only identified expansions of known STR loci. In future studies, to detect de novo STR expansion in sperm, the PacBio HiFi sequencing platform could be used for sequencing, and our method is also applicable to the PacBio HiFi platform (35).
Through our study, we have demonstrated the versatility of our method in characterizing genome diversity in single sperm cells, thereby enabling new avenues of exploration into the genetic roles of SVs or repeat expansions from germ cells in research on evolution, infertility, and genetic diseases. Furthermore, the long-read genome sequencing of a single gamete offers a convenient and highly accurate method for performing whole chromosome-scale haplotyping. Overall, our method represents a valuable tool for investigating the complex genetic processes that occur in germ cells and has important implications for the field of reproductive and genomic medicine.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Beijing Advanced Innovation Centre for Genomics and Changping Laboratory for support, and part of the analysis was performed on the High Performance Computing Platform of the Center for Life Sciences(Peking University).
Authors’ contributions: F.T. conceived the project and supervised the overall experiments. H.X. was in charge of the bioinformatics analysis with the help of Y.G., and Y.G. developed the method to identify aneuploidy. W.L. was in charge of the experimental part and developed the experimental protocol for Nanopore platform. W.L. performed SVs validation with the help of X.S.. K.C. was involved in the FACS of sperm cells and the design of experimental protocols. H.X., W.L., Y.G. and F.T. wrote the manuscript with help from all authors.
Contributor Information
Haoling Xie, Biomedical Pioneering Innovation Center, School of Life Sciences, Peking University, Beijing 100871, China; Beijing Advanced Innovation Center for Genomics (ICG), Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, Beijing 100871, China; Changping Laboratory, Changping Laboratory, Yard 28, Science Park Road, Changping District, Beijing 102206, China.
Wen Li, Biomedical Pioneering Innovation Center, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China; Beijing Advanced Innovation Center for Genomics (ICG), Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, Beijing 100871, China.
Yuqing Guo, Biomedical Pioneering Innovation Center, School of Life Sciences, Peking University, Beijing 100871, China; Beijing Advanced Innovation Center for Genomics (ICG), Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, Beijing 100871, China.
Xinjie Su, Biomedical Pioneering Innovation Center, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China; Beijing Advanced Innovation Center for Genomics (ICG), Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, Beijing 100871, China.
Kexuan Chen, Biomedical Pioneering Innovation Center, School of Life Sciences, Peking University, Beijing 100871, China; Beijing Advanced Innovation Center for Genomics (ICG), Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, Beijing 100871, China.
Lu Wen, Biomedical Pioneering Innovation Center, School of Life Sciences, Peking University, Beijing 100871, China; Beijing Advanced Innovation Center for Genomics (ICG), Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, Beijing 100871, China.
Fuchou Tang, Biomedical Pioneering Innovation Center, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China; Beijing Advanced Innovation Center for Genomics (ICG), Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, Beijing 100871, China; Changping Laboratory, Changping Laboratory, Yard 28, Science Park Road, Changping District, Beijing 102206, China.
Data Availability
The analysis code is deposited in github (https://github.com/hlxie/Single-sperm-ONT-sequencing) and Zenodo (10.5281/zenodo.8008766). The sperm data have been deposited in the Sequence Read Archive (SRA) under BioSample accession: PRJNA893252.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work is supported by the National Natural Science Foundation of China [32288102].
Conflict of interest statement
None declared.
REFERENCES
- 1. Wang J., Fan H.C., Behr B., Quake S.R.. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012; 150:402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu S., Zong C., Fan W., Yang M., Li J., Chapman A.R., Zhu P., Hu X., Xu L., Yan L.et al.. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science. 2012; 338:1627–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yin Y., Jiang Y., Lam K.W.G., Berletch J.B., Disteche C.M., Noble W.S., Steemers F.J., Camerini-Otero R.D., Adey A.C., Shendure J.. High-throughput single-cell sequencing with linear amplification. Mol. Cell. 2019; 76:676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bell A.D., Mello C.J., Nemesh J., Brumbaugh S.A., Wysoker A., McCarroll S.A.. Insights into variation in meiosis from 31,228 human sperm genomes. Nature. 2020; 583:259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hinch A.G., Zhang G., Becker P.W., Moralli D., Hinch R., Davies B., Bowden R., Donnelly P.. Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science. 2019; 363:eaau8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun H., Rowan B.A., Flood P.J., Brandt R., Fuss J., Hancock A.M., Michelmore R.W., Huettel B., Schneeberger K.. Linked-read sequencing of gametes allows efficient genome-wide analysis of meiotic recombination. Nat. Commun. 2019; 10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dréau A., Venu V., Avdievich E., Gaspar L., Jones F.C.. Genome-wide recombination map construction from single individuals using linked-read sequencing. Nat. Commun. 2019; 10:4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Z., Xie L., Tang X., Zhang Z.. NanoCross: a pipeline that detecting recombinant crossover using ONT sequencing data. Genomics. 2022; 114:110499. [DOI] [PubMed] [Google Scholar]
- 9. Chen Z., Xie L., Tang X., Zhang Z.. Recombination map construction method using ONT sequence. MethodsX. 2023; 10:101969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. López-Nandam E.H., Albright R., Hanson E.A., Sheets E.A., Palumbi S.R.. Mutations in coral soma and sperm imply lifelong stem cell renewal and cell lineage selection. Proc. R. Soc. B Biol. Sci. 2023; 290:20221766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dai C., Zhang Z., Shan G., Chu L.T., Huang Z., Moskovstev S., Librach C., Jarvi K., Sun Y.. Advances in sperm analysis: techniques, discoveries and applications. Nat. Rev. Urol. 2021; 18:447–467. [DOI] [PubMed] [Google Scholar]
- 12. Tang S., Wang X., Li W., Yang X., Li Z., Liu W., Li C., Zhu Z., Wang L., Wang J.et al.. Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm Flagella. Am. J. Hum. Genet. 2017; 100:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coutton C., Martinez G., Kherraf Z.E., Amiri-Yekta A., Boguenet M., Saut A., He X., Zhang F., Cristou-Kent M., Escoffier J.et al.. Bi-allelic mutations in ARMC2 lead to severe astheno-teratozoospermia due to sperm Flagellum malformations in humans and mice. Am. J. Hum. Genet. 2019; 104:331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagaoka S.I., Hassold T.J., Hunt P.A.. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012; 13:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shukla V., Høffding M.K., Hoffmann E.R.. Genome diversity and instability in human germ cells and preimplantation embryos. Semin. Cell Dev. Biol. 2021; 113:132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Collins R.L., Brand H., Karczewski K.J., Zhao X., Alföldi J., Francioli L.C., Khera A.V., Lowther C., Gauthier L.D., Wang H.et al.. A structural variation reference for medical and population genetics. Nature. 2020; 581:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kosugi S., Momozawa Y., Liu X., Terao C., Kubo M., Kamatani Y.. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019; 20:8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lyu R., Tsui V., McCarthy D.J., Crismani W.. Personalized genome structure via single gamete sequencing. Genome Biol. 2021; 22:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Padeken J., Zeller P., Gasser S.M.. Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev. 2015; 31:12–19. [DOI] [PubMed] [Google Scholar]
- 20. Anwar S.L., Wulaningsih W., Lehmann U.. Transposable elements in human cancer: causes and consequences of deregulation. Int. J. Mol. Sci. 2017; 18:974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sulovari A., Li R., Audano P.A., Porubsky D., Vollger M.R., Logsdon G.A., Warren W.C., Pollen A.A., Chaisson M.J.P., Eichler E.E.. Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:23243–23253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saini S., Mitra I., Mousavi N., Fotsing S.F., Gymrek M.. A reference haplotype panel for genome-wide imputation of short tandem repeats. Nat. Commun. 2018; 9:4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cretu Stancu M., Van Roosmalen M.J., Renkens I., Nieboer M.M., Middelkamp S., De Ligt J., Pregno G., Giachino D., Mandrile G., Espejo Valle-Inclan J.et al.. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017; 8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jarvis E.D., Formenti G., Rhie A., Guarracino A., Yang C., Tracey A., Thibaud-nissen F., Vollger M.R., Porubsky D., Cheng H.. Automated assembly of high-quality diploid human reference genomes. Nature. 2022; 611:519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kronenberg Z.N., Rhie A., Koren S., Concepcion G.T., Peluso P., Munson K.M., Porubsky D., Kuhn K., Mueller K.A., Low W.Y.et al.. Extended haplotype-phasing of long-read de novo genome assemblies using Hi-C. Nat. Commun. 2021; 12:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng H., Jarvis E.D., Fedrigo O., Koepfli K.P., Urban L., Gemmell N.J., Li H.. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 2022; 40:1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garg S. Computational methods for chromosome-scale haplotype reconstruction. Genome Biol. 2021; 22:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tewhey R., Bansal V., Torkamani A., Topol E.J., Schork N.J.. The importance of phase information for human genomics. Nat. Rev. Genet. 2011; 12:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Falconer E., Hills M., Naumann U., Poon S.S.S., Chavez E.A., Sanders A.D., Zhao Y., Hirst M., Lansdorp P.M.. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods. 2012; 9:1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanders A.D., Falconer E., Hills M., Spierings D.C.J., Lansdorp P.M.. Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc. 2017; 12:1151–1176. [DOI] [PubMed] [Google Scholar]
- 31. Hanlon V.C.T., Chan D.D., Hamadeh Z., Wang Y., Mattsson C.A., Spierings D.C.J., Coope R.J.N., Lansdorp P.M.. Construction of Strand-seq libraries in open nanoliter arrays. Cell Rep. Methods. 2022; 2:100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong J.H., Shigemizu D., Yoshii Y., Akiyama S., Tanaka A., Nakagawa H., Narumiya S., Fujimoto A.. Identification of intermediate-sized deletions and inference of their impact on gene expression in a human population. Genome Med. 2019; 11:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arslan A., Fang Z., Wang M., Tan Y., Cheng Z., Chen X., Guan Y., Pisani L.J., Yoo B., Bejerano G.et al.. Analysis of structural variation among inbred mouse strains. BMC Genomics. 2023; 24:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferraj A., Audano P.A., Balachandran P., Eichler E.E., Reinholdt L.G., Beck C.R., Ferraj A., Audano P.A., Balachandran P., Czechanski A.et al.. Resource Resolution of structural variation in diverse mouse genomes reveals chromatin remodeling due to transposable elements Resolution of structural variation in diverse mouse genomes reveals chromatin remodeling due to transposable elements. Cell Genomics. 2023; 3:100291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fan X., Yang C., Li W., Bai X., Zhou X., Xie H., Wen L., Tang F.. SMOOTH-seq: single-cell genome sequencing of human cells on a third-generation sequencing platform. Genome Biol. 2021; 22:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chiang C., Scott A.J., Davis J.R., Tsang E.K., Li X., Kim Y., Hadzic T., Damani F.N., Ganel L., Montgomery S.B.et al.. The impact of structural variation on human gene expression. Nat. Genet. 2017; 49:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yalcin B., Wong K., Agam A., Goodson M., Keane T.M., Gan X., Nellåker C., Goodstadt L., Nicod J., Bhomra A.et al.. Sequence-based characterization of structural variation in the mouse genome. Nature. 2011; 477:326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valdar W., Solberg L.C., Gauguier D., Burnett S., Klenerman P., Cookson W.O., Taylor M.S., Rawlins J.N.P., Mott R., Flint J.. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat. Genet. 2006; 38:879–887. [DOI] [PubMed] [Google Scholar]
- 39. Yalcin B., Nicod J., Bhomra A., Davidson S., Cleak J., Farinelli L., Østerås M., Whitley A., Yuan W., Gan X.et al.. Commercially available outbred mice for genome-wide association studies. PLoS Genet. 2010; 6:e1001085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fujimoto A., Wong J.H., Yoshii Y., Akiyama S., Tanaka A., Yagi H., Shigemizu D., Nakagawa H., Mizokami M., Shimada M.. Whole-genome sequencing with long reads reveals complex structure and origin of structural variation in human genetic variations and somatic mutations in cancer. Genome Med. 2021; 13:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tan K.-T., Slevin M.K., Meyerson M., Li H.. Identifying and correcting repeat-calling errors in nanopore sequencing of telomeres. Genome Biol. 2022; 23:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Q., Zhang P., Wang D., Gu W., Wang K. Interrogating the ‘unsequenceable’ genomic trinucleotide repeat disorders by long-read sequencing. Genome Med. 2017; 9:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koren S., Rhie A., Walenz B.P., Dilthey A.T., Bickhart D.M., Kingan S.B., Hiendleder S., Williams J.L., Smith T.P.L., Phillippy A.M.. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 2018; 36:1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang H., Chen X., Hung W.. Completely phased genome sequencing through chromosome sorting. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Falconer E., Lansdorp P.M.. Strand-seq: a unifying tool for studies of chromosome segregation. Semin. Cell Dev. Biol. 2013; 24:643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Selvaraj S., Dixon J.R., Bansal V., Ren B.. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat. Biotechnol. 2013; 31:1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xie H., Li W., Hu Y., Yang C., Lu J., Guo Y., Wen L., Tang F.. De novo assembly of human genome at single-cell levels. Nucleic Acids Res. 2022; 50:7479–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The analysis code is deposited in github (https://github.com/hlxie/Single-sperm-ONT-sequencing) and Zenodo (10.5281/zenodo.8008766). The sperm data have been deposited in the Sequence Read Archive (SRA) under BioSample accession: PRJNA893252.