


Abstract
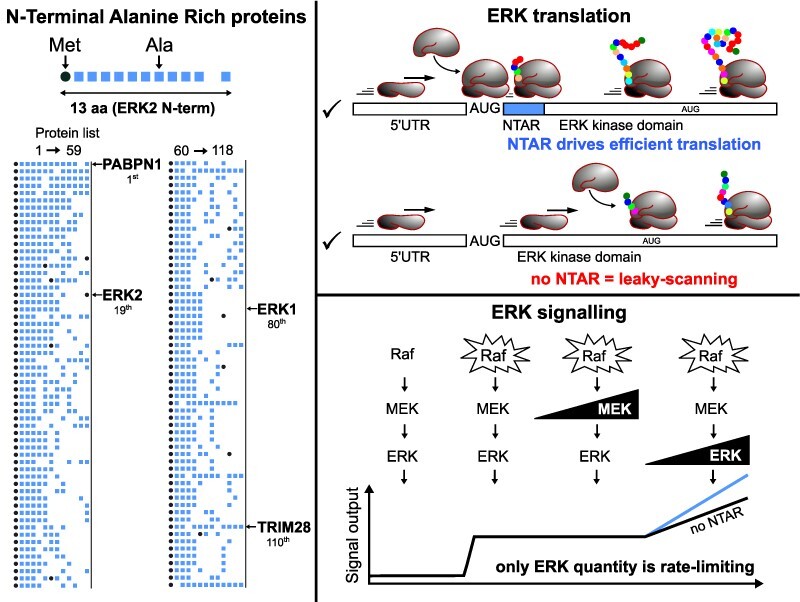
We report the discovery of N-terminal alanine-rich sequences, which we term NTARs, that act in concert with their native 5′-untranslated regions to promote selection of the proper start codon. NTARs also facilitate efficient translation initiation while limiting the production of non-functional polypeptides through leaky scanning. We first identified NTARs in the ERK1/2 kinases, which are among the most important signaling molecules in mammals. Analysis of the human proteome reveals that hundreds of proteins possess NTARs, with housekeeping proteins showing a particularly high prevalence. Our data indicate that several of these NTARs act in a manner similar to those found in the ERKs and suggest a mechanism involving some or all of the following features: alanine richness, codon rarity, a repeated amino acid stretch and a nearby second AUG. These features may help slow down the leading ribosome, causing trailing pre-initiation complexes (PICs) to pause near the native AUG, thereby facilitating accurate translation initiation. Amplification of erk genes is frequently observed in cancer, and we show that NTAR-dependent ERK protein levels are a rate-limiting step for signal output. Thus, NTAR-mediated control of translation may reflect a cellular need to precisely control translation of key transcripts such as potential oncogenes. By preventing translation in alternative reading frames, NTAR sequences may be useful in synthetic biology applications, e.g. translation from RNA vaccines.
Graphical Abstract
Graphical Abstract.
INTRODUCTION
Activation of the ERK pathway is required for a variety of cell processes, including proliferation and differentiation (1,2), and plays a role in cell survival and death [reviewed in (3)]. How this single signaling pathway confers different effects in similar cell types remains a matter of debate (4).
ERK kinase activity can be increased up to 1000-fold upon double phosphorylation by MEK (5). Nearly all studies have focused on ERK activation as the primary mechanism through which its downstream effects are manifested. However, decreased ERK quantity can also modulate downstream signaling, as has been shown with altered wing development in Drosophila when deficient splicing reduces dERK quantity (6). In mice, combinations of ERK1 and/or ERK2 knockouts trigger increasingly deleterious phenotypes due to decreased total ERK quantity [reviewed in (7)]. In humans, children lacking one erk2 allele display minor neurological disorders (8). These studies suggest that a threshold of ERK quantity may be necessary for regulation of kinase activity by MEK and phosphatases. Increased ERK quantity resulting from erk2 gene amplification, which can confer resistance to chemotherapy (9), was demonstrated to occur frequently in lung cancers (10); according to the datasets available in the Tissue Cancer Genome Atlas (TCGA), both erk1 and erk2 are amplified in many cancers including bladder and non-small cell lung cancer (NSCLC) (11). With the goal of further clarifying the role of ERK quantity in regulating downstream signaling, we first assessed pathway activation using a classical reporter of ERK activity, the GAL4–ELK/gal4–luc system (12). In addition, we studied the phosphorylation pattern of an ELK chimera (13), because the progressive multi-phosphorylation of transcription factors constitutes a molecular switch for cell fate decisions driven by ERK (14). Here, we show that the quantity of ERK, but not the quantity of MEK, the kinase immediately upstream, impacts signaling. This observation led us to explore the mechanisms regulating ERK quantity. We focused our attention on the N-terminus (Nt) of ERK1/2, which contain an unusual alanine-rich tract. In the context of erk1 or erk2 promoters/5′-untranslated region (UTR), we discovered that ERK Nts drive elevated protein synthesis of functional ERKs by allowing precise and efficient start codon selection.
A short RNA sequence flanking the initiation codon, termed the Kozak sequence (15), drives start codon selection in mammals. Some mRNAs harbor several AUGs in their 5′-UTRs, and switching start codon selection from the native AUG [i.e. the one that sets the main open reading frame (ORF)] to an upstream AUG (uAUG) is known to regulate biological responses such as resistance to amino acid starvation (16,17). Alternatively, the native AUG can be skipped and downstream AUGs may be selected to start translation, a process termed ‘leaky scanning’ (18). We show that in the absence of its own Nt-coding sequence, translation of erk mRNA led to production of shorter, non-functional polypeptides via leaky scanning, starting at downstream AUGs. The action of ERK Nts is restricted to specific 5′-UTRs, suggesting that various rules govern start codon selection.
Several hundreds to a few thousand human proteins display N-terminal alanine-rich sequences (NTARs) similar to those of ERK1/2. We further demonstrated that the NTARs of PABPN1, TRIM28, MECP2 and NIPA1 are required for the high expression of a functional reporter protein in the context of their proximal promoter/5′ UTR, similar to what we found for ERK1/2.
Studying the mechanism by which NTARs regulate translation may provide tools to understand physio-pathological conditions such as polyalanine expansion disorders. In this family of diseases, protein synthesis is altered by the presence of alanine repeats that are orders of magnitude greater than those found in ERK1/2 (19). Hence, studying the much shorter ERK Nts may help to decipher the molecular processes at play in these diseases. The specific regulation of ERK quantity by NTAR sequences may offer new perspectives to fight cancer because nearly 40% of all human cancers display overactivation of the ERK pathway, including ERK overexpression (10). It might also be helpful in developing therapies against RASopathies, a family of diseases originating from germline mutations of proteins that occupy different positions along the ERK signaling pathway (20).
MATERIALS AND METHODS
Proximal promoters and Nt sequences were assembled by Golden Gate cloning which requires entry plasmids with kanamycin selection and assembly plasmids with ampicillin selection (21).
Entry plasmids for Golden Gate cloning
The entry vector was derived from the pCRII-topo plasmid (Thermo Fisher, Invitrogen). The BsaI site in the ampicillin gene of the pCRII-topo plasmid was inactivated by point mutation (new sequence agcgtgggtcAcgcggt), then it was cut by XmnI and ScaI restriction enzymes (REs) and religated to remove ampR (the plasmid remained KanaR). The following sequence was cloned between the two BstXI sites: gaattcAggcctgatttaaatattcccgggatatcacgtggaattcaagctt, introducing a stretch of restriction sites for blunt REs (StuI, SwaI, SspI, SmaI, EcoRV and PmlI) between two EcoRI sites for diagnostic purposes. The resulting plasmid was named Empal-Blunt (manuscript in preparation).
Fragments of promoter regions were amplified by polymerase chain reaction (PCR) with primers containing BsaI sites at their extremities, such that upon cutting by BsaI, a cohesive fragment is released with non-palindromic extremities. High fidelity enzymes produced blunt-end PCR products that were cloned directly into the plasmid Empal-BluntkanaR after mixing the purified PCR fragment with the Empal-Blunt plasmid in the presence of a blunt-end RE that does not cut the fragment. Incubation was done with cycles of temperature, from the optimal temperature for ligation to the optimal temperature for RE digestion (Supplementary Table S2, tab5). Minipreps from white colonies were sequenced to ensure the integrity of the expected fragments bordered by BsaI sites. PCR was performed using the KOD hot-start enzyme [Merck Millipore Toyobo ref 71086- (4)], the Q5 hot-start enzyme (New England Biolabs, M0493S) or the Phusion hot-start enzyme (Finnzymes F-540L). For high GC-rich sequences, 3% dimethylsulfoxide (DMSO) was added. Typical reactions are listed in Supplementary Table S2.
Assembly plasmids for Golden Gate cloning
Assembly plasmids harboring either firefly luciferase (ffLuc) with a vPEST sequence (luc2P) or the catalytic domain of mouse ERK1 were derived from pGL4 from Promega bought from Switchgear (pSGG-Basic-3′GAPDH).
pSGG-GG-luc2P and pSGG-GG-lucHA
pSGG-Basic-3′GAPDH harbors the RPLP10 promoter upstream of Luc2P (ffLuc with PEST sequences) with the 3′-UTR of human glyceraldehyde phosphate dehydrogenase (GAPDH) downstream. Firstly, the BsaI site in 3′GAPDH was mutated into the BsmBI site by overlap extension PCR with external primers PL-09–58 and PL-09–59 and mutagenesis primers PL-12–58 and PL-12–59 (sequences of primers in Supplementary Table S2, tab2). The resulting plasmid is called pSGG-Basic. Secondly, a fragment comprising the RPLP10 promoter downstream from KpnI, up to the BsrGI inside luc2P, was replaced by a PCR fragment with primers PL-09–57 and PL-12–39 encompassing the Nt sequence of luc2P (into KpnI and BsrGI); the resulting plasmid has no promoter upstream of luc2P but instead it has two BsaI sites (GGTCTC) in opposite orientation: accgaGAGACCtgtacaGGTCTCggaag. This vector, named pSGG-GG-Luc2P, allows cloning of fragments into the non-palindromic sites accg and gaag upon cutting by BsaI RE. On the 3′ cohesive end, gaa is the second codon of luc2P (glutamic acid).
For immunoblotting, the PEST sequence of luc2P was replaced by the hemagglutinin (HA) tag. A PCR fragment from luc2P as template was obtained with primers PL-16-11 and PL-16-27 (harboring the sequence of the HA tag). This fragment cut by BsaI was inserted into the AgeI and XbaI sites of plasmid pSGG-GG-luc2P to generate the plasmid pSGG-GG-lucHA (the same BsaI RE sites upstream of ffLuc for cloning).
pSGG-mERK1
A PCR product containing the mouse ERK1 coding sequence (primers PL-16-28 and PL-16-29; BsmBI external cloning sites) was inserted between the KpnI and XbaI sites of plasmid pSGG-Basic-3′GAPDH. The sequences of promoter RPLP10 and luc2P were replaced by mERK1 with two upstream BsaI sites in opposite orientation: accgaGAGACCtgtacaGGTCTCggaag. On the 3′ cohesive end of this sequence, the gaa codon corresponds to glutamic acid of mERK1 at sequence EVVKG. The glutamic acid was chosen to be able to use the same cloning strategy for pSGG-GG-luc2P and pSGG-GG-ERK1.
Assembly strategy
Golden Gate assembly was performed in a single tube; several fragment-bearing plasmids (in Empal-Blunt, KanaR) were mixed with pSGG-GG-luc2P (AmpR) in the presence of BsaI and T4 DNA ligase. When needed, annealed oligonucleotides were added at a final concentration of 40 ng each. In a thermocycler, incubation cycles switched from the best temperature for the RE (37°C or 55°C) to the best temperature for T4 ligase (16°C) overnight. A typical reaction is presented in Supplementary Table S2, tab 5. This file also includes all the primers used to clone the proximal promoters or the 5′-UTRs and oligonucleotides annealed to generate all constructs. Golden Gate cloning allowed us to assemble up to four fragments at once with high efficiency. Transformation was performed in NEB-stable bacteria (cat. # C3040). AmpR colonies were screened and subsequently sequenced.
Existing plasmids and ERK1 truncations
For Figure 1, Chinese hamster HA-MEK1-WT and HA-MEK1-SSDD (S218D-S222-D) were described previously (22). HA-ERK-WT plasmid harbors Chinese hamster wild-type ERK1 in the same plasmid backbone (pECE) as HA-MEK1-WT (23). RAF1-WT and RAF1-CAAX were described previously (24). For other figures, mouse ERK1 was used, derived from mouse HA-mERK1 (25). pGAL4-ELK and 5×-gal4-ffLuciferase were cited previously (26). For immunoblotting, Myc-tag was added to GAL4(1–146)–linker(PVEL)–hELK1-307–428) by PCR with primers PL-19-56 and PL-19-57, and the fragment generated by BsaI cutting was assembled with the cytomegalovirus (CMV) promoter fragment (PCR primers PL-18-69 and PL-18-12) into pSGG-GG-luc2P cut by BsaI/XbaI. The CMV promoter was amplified from plasmid pCRHL. The final vector is called pSGG-GAL4-ELK-myc (ELK-myc).
Figure 1.

ERK quantity shapes signal output. (A) Analysis of ERK activity (expressed in relative luciferase units, RLU) in extracts of HeLa cells transiently transfected with the GAL4-ELK/gal4-Luc system (ELK-GAL4/Luc), along with increasing amounts of WT or constitutive active forms of either RAF1 or MEK (RAF1-CAAX or MEK-SSDD) or WT-ERK (n = 3). (B) RLU from extracts of HeLa cells transfected with ELK-GAL4/Luc, RAF1-CAAX and increasing quantities of either WT-ERK or WT-MEK as in (A) (n = 3). (C) Western blot experiment from extracts of HeLa cells transiently transfected with myc-tagged GAL4-ELK in the presence of RAF1-CAAX and increasing amounts of either HA-tagged-ERK or HA-tagged-MEK. HA-tagged protein expression was visualized with an anti-HA antibody (second panel). Phosphorylation of either ERK or GAL4-ELK was assessed using specific anti-phospho antibodies (third and fourth panels). Arrows on the fourth panel indicate the phosphorylated GAL4-ELK forms. HSP90 expression was used as a loading control (first panel).
Plasmids expressing catalytic mouse ERK or truncations of mouse ERK1 behind the CMV promoter were generated into a psgg vector, with an ACCG cohesive site in 5′ (BsaI) and a CTAG cohesive site in 5′ (XbaI). The CMV promoter was amplified with primers PL-18-08 and PL-19-69. The ERK fragments were generated by PL-16-29 in 3′, producing a CTAG site by BsmBI cutting. In 5′, the catalytic mERK1 was amplified with primer PL-19-69; the second in-frame AUG with PL-19-70; the third AUG in-frame with PL-19-71; and the fourth AUG in-frame with PL-19-72, all producing an ACCG cohesive site by BsmBI cutting. Note that catalytic mouse ERK1 starts here with the sequence MGEVEVVK. Two fragments were assembled at once in the vector cut by XbaI and BsaI.
Proximal promoters
The sequences of the promoters used in this study are presented in Supplementary Figure S9.
Mouse erk2 promoter
A genomic fragment encompassing the mouse erk2 (m-erk2) promoter was amplified with primers PL-09-47 and PL-09-48. The m-erk2 promoter has three BsaI sites, hence the assembly was carried out by overlap PCR between the promoter and luc2P. For assembly with the Nt moiety, a fragment of the erk2 promoter was amplified by primers PL-09-62 and PL-09-63, and the luc2P fragment was amplified by primers PL-09-64 and PL-09-57. External primers were then used to generate an assembled fragment that was cloned into the plasmid pSGG-Basic in the KpnI and BsrG1 RE sites. For assembly without the Nt, a fragment of the erk2 promoter was amplified with primers PL-09-62 and PL-09-65, the luc2P fragment was amplified with primers PL-09-66 and PL-09-57, and the same strategy was used with external primers.
Mouse erk1 promoter
A genomic fragment encompassing the m-erk1 promoter was amplified with primers PL-09-49 and PL-09-50. The m-erk1 proximal promoter was initially assembled with ffLuc by overlap PCR; the external primer on erk1 is PL-09-54 and on luc2P it is PL-09-57. For assembly in the presence of the ERK1 Nt, the internal reverse primer was PL-09-55 on erk1 and the internal sense primer on luc2P was PL-09-56. For assembly in the absence of the ERK1 Nt, the internal reverse primer of erk1 was PL-09-60 and the internal sense primer on luc2P was PL-09-61. To compare the Kozak sequences in the presence or absence of the Nt, refer to the alignment in Supplementary Figure S8. The proximal promoter of mouse erk1 harbors two BsaI sites that were subsequently mutated to facilitate assembly of all the modified sequences: –163 G to C and –536 A to T. No difference was observed with promE1WT versus promE1BsaImut when comparing the presence or absence of the ERK1 Nt sequence (not shown). Therefore, all results presented here are with the m-erk1 promoter with the two point mutations on BsaI sites. For Figures 2B and 3A, the m-erk1 promoter (fragment amplified with primers PL-13-02 and PL-13-63) was assembled with fragment Nt-ERK1 (obtained with primers PL-12-40 and PL-12-41) or with Nt-ERK2 (annealed with primers PL-12-42 and PL-12-43).
Figure 2.

Effect of ERK NTARs on ERK signaling, activity and expression. (A) Nucleotide and amino acid composition of mouse and human ERK1 and ERK2 Nt domains upstream of the glutamic acid residue common to both ERKs (highlighted in yellow). Downstream of the initiating methionine (in blue), alanine stretches occur and their corresponding GCG codons are highlighted in red (full nucleotide sequence in Supplementary Figure S10). (B) Measurement of ERK activity using the GAL4-ELK/Luc system (RLU) in HeLa cells transfected with an empty plasmid or increasing quantities of ERK1 under the control of its own proximal promoter, in the absence (white bars) or presence of its Nt moiety (gray bars). Except for the first empty plasmid condition, cells were all co-transfected with 100 ng of constitutive active RAF1-CAAX construct (n = 3; *P < 0.05 or **P < 0.01 bilateral Welch's t-test, representative of three experiments). (C) Western blot analysis from extracts of HeLa cells transfected with active RAF1-CAAX and myc-tagged Gal4-ELK (ELK-myc), together with HA-MEK plasmid (lane 1) or decreasing amounts of HA-tagged ERK1, either with its Nt (NtE1-ERK1-HA, lanes 6–8) or without (ERK1catalyt-HA, the catalytic moiety of mouse ERK1 lacking the first 26 amino acids, lanes 2–5). The top panel shows expression of HSP90 as a loading control, the second panel shows expression of ERK1-HA and HA-MEK, the third panel reveals phosphorylated ERKs (ectopic and endogenous), and the fourth and fifth panels show the profile of total myc-tagged GAL4-ELK and the multi-phosphorylated GAL4-ELK profile. (D) Western blot analysis from extracts of HEK293 cells transfected with various HA-tagged ERK constructs depicted with their predicted molecular weights above the blots. One day post-transfection, cells were treated overnight with 100 nM bortezomib to decrease proteasome-mediated protein degradation, then cells were stimulated for 15 min with 0.1 M orthovanadate prior to lysis to increase detection of phosphorylated ERK. The first four constructs were expressed under the erk1 promoter (lanes 1–4). ERK1 was expressed from either a mutated AUG (GUG, lane 1) or a normal AUG initiation codon (lanes 2–4). Nt domains of ERK1 or ERK2 were fused to catalytic ERK1 (lanes 3 and 4). Truncated ERK1 forms were expressed under the CMV promoter (lanes 5–8). Of note, lanes 5–8 were transfected with 18-fold less plasmid than lanes 1–4 (CMV versus ERK1 promoter). Lane 5 shows catalytic ERK1, whereas lanes 6–8 show polypeptides starting at the second, third or fourth in-frame AUG codons. Red arrows indicate the full-length ERK1-HA proteins. In lanes 1–4, smaller polypeptides expressed under the erk1 promoter are indicted by decreasing sizes (#1 to #5) on the left of the upper blot. Detection of HA-tagged proteins was performed using an anti-HA antibody (upper panel), whereas phosphorylated ERK (ectopic and endogenous) was assessed using a specific phospho-ERK antibody (lower panel). (E) Western blot analysis of HeLa cells transfected and processed as in (D). The first three lanes show expression of ERK1-HA whose catalytic domain is WT while the last three lanes show expression of ERK1-HA whose catalytic domain has the second in-frame AUG mutated to GUG. In lanes 1 and 4, ERK1 lacks its first AUG, while in lanes 3 and 6 ERK1 contains the Nt domain. Arrows indicate the functional ERK1-HA proteins. The lower molecular weight polypeptides are indicated on the left (#1 to #3).
Figure 3.

Firefly luciferase (ffLuc) as a reporter to study ERK Nt action on start codon selection. (A) Normalized ffLuc activity (RLU) from extracts of NIH3T3 cells transfected with plasmids containing the erk1 promoter and the 5′-UTR (prom E1), in the absence (lane 1) or presence of ERK1 or ERK2 Nt moieties fused to ffLuc (lanes 2 and 3). Transfection of plasmids with the erk2 promoter and 5′-UTR (prom E2) in the absence (lane 4) or presence of ERK2 Nt fused to ffLuc (lane 5) (n = 3, representative of >3 experiments). (B) Western blot analysis of extracts from HeLa cells and HEK293 cells transfected with various constructs, all in the context of the erk1 promoter and 5′-UTR. The presence of Nt domains (red), the position of the HA tag (green) and the predicted molecular weight of full-length proteins are indicated. Detection of HA-tagged luciferase was performed using an anti-HA antibody. The red arrow corresponds to ffLuc-HA at the expected size (lane 1); asterisks (*) indicate leaky scanning bands. (C) Western blot analysis from HEK293 cells transfected and processed as in (B). The first three lanes express WT-ffLuc-HA while the last three lanes express ffLuc-HA with its second in-frame AUG mutated to GUG. In lanes 1 and 4, ffLuc lacks the first AUG while in lanes 3 and 6 the ERK1 Nt domain was fused to ffLuc. Small molecular weight polypeptides expressing the HA-tag are indicated on the left (#1 and #2). Arrows indicate functional ffLuc-HA at the expected size.
Human hstk promoter
The hstk minimal promoter was reconstituted from the pRL-TK plasmid sequence (Promega). Upstream primer PL-13-27 on the thymidine kinase (TK) promoter was combined with a long reverse primer PL-15-69 for PCR. Primer PL-15-69 reconstitutes the human hstk minimal promoter as it is present in sequence JQ673480.1 of GenBank [human herpesvirus 1 (HSV) strain KOS, complete genome]. For luciferase assays, this WT hstk promoter fragment was amplified with PL-13-27 and PL-15-69 primers to insert the promoter alone upstream of luc2P, or with PL-13-27 and PL-15-70 to be assembled with Nt-coding sequences, such as annealed primer PL-16-06/07 to add the HSTK Nt or annealed primers PL-12-42/43 to add the ERK2 Nt.
Human kras promoter
The plasmid containing the human kras proximal promoter was cloned from genomic DNA from human Beas-2b cells with primers PL-15-73 and PL-15-74. To shunt exon0, kras promoter fragments upstream of exon0 were amplified with primers that reconstitute the native ATG after exon0: PL-16-00 and PL-16-01, PL-16-00 and PL-16-02, or PL-16-00 and primer PL-16-08. The fragment PL-16-00/PL-16-01 contains the kras promoter alone to be cloned into pSGG-GG-luc2P. The fragment PL-16-00/PL-16-02 allows cloning of the annealed primers PL-12-42/43 to assemble the ERK2 Nt after the AUG. The fragment PL-16-00/PL-16-08 includes the promoter kras with its native Nt moiety (size of Nt-ERK2).
Note: for final assembly, it was necessary to re-assemble the promoter after the first Golden Gate cloning step into the plasmid pSGG-GG-luc2P because the kras promoter has three BsaI RE sites (–649, –794 and –891). As the cohesive ends of these three BsaI sites are non-palindromic, they can assemble in the right order. Therefore, after Golden Gate assembly, BsaI was inactivated by 20 min at 80°C, then T4 ligase and ATP (0.5 mM) were added for 10 min at 16°C and 30 min at 25°C to ligate promoter fragments. The full-length h-kras minimal promoter was readily obtained.
Mouse trim28 promoter
The proximal promoter of trim28 was amplified from mouse genomic DNA with primers PL-19-41 and PL-19-43 (size 1965 bp). Then the proximal promoter was amplified with 5′ primer PL-19–48 and with 3′ primers PL-19-45 (proximal promoter alone 886 bp), PL-19-46 (promoter with 180 nucleotides of coding sequence) or PL-19-47 (promoter with 342 nucleotides of coding sequence).
Human nipa1 promoter
The proximal promoter of nipa1 was amplified from human genomic DNA from normal human foreskin fibroblasts with primers PL-22-98 and PL-22-100 (size 1550 bp). Then the proximal promoter was amplified with 5′ primer PL-22-101 and 3′ primers PL-22-103 (proximal promoter alone), PL-22-102 (promoter with 81 nucleotides of coding sequence) or PL-22-104 (promoter with ATGG cohesive site). With the fragments PL-22-101/PL-22-104, a fragment lacking the first two amino acids of the Nt was assembled (PL-22-105 and PL-22-102). As an inadvertent BsaI site was present inside the promoter, after inactivation of the Golden Gate enzymes, ATP and ligase were added for 1 h to close back the plasmids prior to transformation of bacteria.
Human mecp2 promoter
The proximal promoter of mecp2 was amplified from human genomic DNA from normal human foreskin fibroblasts with primers PL-22-07 and PL-22-10 (size 2324 bp). Then the proximal promoter was amplified with 5′ primer PL-22-13 and with 3′ primers PL-22-15 (proximal promoter alone 969 bp), PL-22-21 (promoter with 63 nucleotides of coding sequence) or PL-22-22 (promoter for adding annealed primers). To this last fragment, primers PL-22-23/PL-22-26 were annealed to change codon sequences to GCG, or primers PL-22-24/PL-22-27 to change codon sequences to GCA-GCT.
Specific strategies for individual figures
Site-directed mutagenesis of the second AUG of mERK1 and ffLuc
The second AUGs were converted to GUG by the method of Q5 Site-Directed Mutagenesis from NEB. The primers were designed by the NEB-tool changer (primers PL-22-68 and PL-22-69 for ffLuc and primers PL-22-70 and PL-22-71 for mERK1; sequences in Supplementary Table S2), For each protein, site-directed mutagenesis was performed only on the GG vector (pSGG-mERK1 and pSGG-GG-lucHA); after sequence verification, in these vectors assembly of the erk1 promoter with its 5′-UTR was generated by Golden Gate cloning with the fragments PL-13-02/PL-13-59 and PL-13-58/PL-19-100, PL-13-58/PL-13-74 or PL-13-58/PL-12-41 to generate proteins without their first AUG, without or with the ERK1 Nt. The mutagenesis was performed from 1 μl of the Q5 PCR performed according to the manufacturer’s conditions, then 20 U of DpnI, 400 U of T4 DNA ligase and 10 U of T4 polynucleotide kinase were added in a final volume of 20 μl with T4 DNA ligase buffer, and incubated at 10 min cycles from 37°C to 25°C overnight prior to transformation into competent bacteria.
Bicistronic vector to assemble erk1 5′-UTRs of various sizes
To determine the size of the erk1 5′-UTR that drives start codon choice with the ERK1 Nt, a plasmid pSGG-renilla-luciferase-GG-ffLuc was constructed. PCR was performed from plasmid pCRHL (27) to obtain the CMV promoter upstream of Renilla luciferase (R-Luc). To remove the BsaI site at the end of this CMV promoter, a fragment of the CMV promoter was amplified by primers PL-17-36 and PL-17-80 to be joined with the fragment of R-Luc which was amplified with primers PL-17-81 and PL-17-88. These fragments, cut by BsmBI, were inserted into the plasmid pSGG-GG-luc2P between KpnI and BsaI sites. The final plasmid had two BsaI sites between R-Luc and the ffLuc, with the BsaI cohesive extremities: ACCG and GAAG, with GAA being the second codon of ffLuc. The control FGF-1A internal ribosome entry site (IRES) fragment (441 bp) with only the erk1 Kozak context was generated by PCR from the vector pCRF-1AL with the primers Pl-17-42 and PL-17-91. The FGF-1A IRES fragment to be assembled with fragments of the erk1 5′-UTR was generated by PCR from the vector pCRF-1AL with the primers Pl-17-42 and PL-17-44. Fragments with erk1 5′-UTRs of increasing size were generated with 5′ primers (sense): PL-17-89; PL-17-109; PL-17-108; PL-13-85 and PL-13-58; the 3′ primer (reverse) was either PL-13-16 to generate fragments without the Nt or PL-12-41 to add the ERK1 Nt sequence.
Assembly of various Kozak sequences
To assemble the different Kozak sequences between the erk1 proximal promoter and the luc2P coding sequence, in the presence or absence of the ERK2 Nt moiety, the erk1 promoter was truncated in 3′. This fragment was generated by PCR with primers PL-13-02 and PL-14-120. To evaluate Kozak contexts in the absence of the ERK2 Nt, the following annealed primers were assembled with the promoter fragment: PL-15-24/25; PL-14-118/119; PL-15-12/13; PL-15-14/15; and PL-15-16/17 (order of Figure 4B). To evaluate Kozak contexts in the presence of the ERK2 Nt sequence, the following annealed primers were assembled with the promoter's fragment: PL-15-26/27; PL-15-01/02; PL-15-06/07; PL-15-08/09; and PL-15–10/11 (order of Figure 4B).
Figure 4.

Leaky scanning in the presence of two AUGs, in a controlled or native context. (A) RLU measured from NIH3T3 cells transfected with ffLuc constructs under control of the erk1 promoter and the 5′-UTR (pE1) with its native AUG in a G+4 context, separated from a second AUG in the same reading frame by a 69 bp spacer. This second AUG is in the context of a poor Kozak sequence (boxed in red) or the canonical Kozak sequence (boxed in blue), both Kozak sequences being in a G+4 context. In lanes 1 and 4, the spacer is located immediately downstream of the first AUG; in lanes 2 and 5, the first AUG is fused to the HSTK Nt; and in lanes 3 and 6, the first AUG is fused to the ERK1 Nt (n = 3, representative of three experiments). (B) as in (A), except that for lanes 1–3 the spacer is 70 bp long, while in lanes 4–6 the spacer is 71 bp long. Changes in spacer length led ffLuc to be coded in alternative reading frames relative to the first AUG (n = 3, representative of three experiments). (C) Western blot analysis from lysates of HeLa cells transfected with HA-tagged ffLuc constructs under the erk1 promoter and 5′-UTR, and harboring or not a second potential start site downstream of the ERK2 Nt. This site is an AUG in lanes 4 and 7 (native position in human ERK2) or its near-cognate codons GUG or GUC in lanes 5 and 6. Control constructs were the kras promoter with the KRAS Nt fused to ffLuc in lane 8, prom E1 with the E1 Nt fused to ffLuc in lane 2, and prom E1 upstream of ffLuc in lane 1. HA-MEK was co-transfected to normalize the transfection efficiency. Detection of HA-tagged proteins was performed using an anti-HA antibody. Presence of low molecular weight proteins due to leaky scanning is indicated (upper panel). The graph represents the percentage of leaky scanning products relative to full-length forms.
Experiments with two AUGs in a defined environment
In Figure 4A and B, two AUGs, within defined contexts, are separated by spacers. We chose to use the Nt of ffLuc as a template for spacers. All four spacers were obtained by PCR from the plasmid pSGG-GG-luc2P, then they were cloned into the ACCG and GAAG sites of this vector cut by BsaI. In 5′, the four PCR products possess the Golden Gate sites of the pSGG-GG-luc2P vector. As a result of inserting the PCR products into the vector (cohesive ends generated by BsmBI cutting), the N-terminus of ffLuc is now duplicated, the copy/spacer being placed between the BsaI Golden Gate sites and a second AUG in the reading frame of the full-length ffLuc protein. The four PCR spacers differ in the environment of this second AUG. All previous cloning strategies can be performed in the ACCG/GAAG sites by BsaI. In 5′, all spacers were amplified by the forward primer PL-22-108. The spacer with a canonical Kozak sequence (GCCGCCACC-ATG) or a poor Kozak sequence (CTTATATTA-ATG) upstream of the second AUG were amplified in 3′ by the primers PL-22-111 and PL-22-112, respectively. These two spacers introduce 69 nucleotides between the two AUGs (in final constructions lacking th eERK Nt). The 3′ primers PL-22-109 and PL-22-113 produced spacers with the second AUG in the second or third reading frames, respectively. In those cases, there are 70 or 71 nucleotides between the two AUGs (in final constructions lacking the ERK Nt). Please note that the Kozak contexts of these AUGs in alternative frames (with respect to the first AUG) are nearly close to the canonical sequences (only 1G at –9 is missing): CCGCCACC-ATG. All eight AUGs have Kozak contexts with a G+4.
Changing the nucleotide sequence of the ERK2 Nt moiety
For Figure 6C, several PCR fragments of the mouse erk1 promoter were used for Golden Gate assembly as presented in Supplementary Table S2. Promoter erk1 amplified from primers PL-13-02 and PL-14-120 was assembled with annealed primers PL-15-57/58 or PL-15-59/60 to generate promoter erk1 with one or two alanines, respectively, in the Nt fused to luc2P. Promoter erk1 amplified from primers PL-13-02 and PL-13-63 was assembled with annealed primers PL-15-32/33 or PL-15-30/31 to generate promoter erk1 with three alanines in the Nt fused to luc2P, or with the six alanine codons switched to GCC codons in the Nt. Promoter erk1 amplified from primers PL-13-02 and PL-13-11 was assembled with annealed primers PL-13-42/43, PL-13-46/47, PL-13-64/65, PL-14-01/02 or PL-13-19/20 to generate promoter erk1 with an Nt with only six alanines (absence of glycine–proline–glutamic acid after the stretch of alanines), an Nt shifted by one nucleotide, by two nucleotides or by three nucleotides, or with the codons of the ERK2 Nt switched to codons GCA/T.
Figure 6.

Analysis of ERK Nt features that contribute to start codon selection. (A) RLU from NIH3T3 cells transfected with ffLuc constructs containing the erk1 promoter and 5′-UTR (pE1) without (line 1) or with Nt sequences, either the full-length ERK2 Nt (line 5) or shorter Nt sequences with only one, two or three alanine residues instead of six (lines 2–4) (n = 6, ns = not significant, ***P < 0.001 bilateral Welch's t-test, representative of three experiments). (B) RLU measurement as in (A) of constructs with ffLuc fused or not to the ERK2 Nt (control conditions, lines 2 and 1), or with an Nt frameshifted by one, two or three nucleotides (lines 3–5). The reading frame was recovered downstream of the ERK2 Nt as indicated. Corresponding protein coding sequences are shown on the right (n = 3, representative of three experiments). (C) RLU as in (A), control conditions as in (B) (lines 1 and 2). Distinct alanine codon composition of the ERK2 Nt is tested in lines 3 and 4, as indicated by sequence alignments with modified nucleotides in red (n = 6, ***P < 0.001 bilateral Welch's t-test, representative of three experiments).
Adding a second AUG 3′ adjacent to the Nt sequence
For Figure 4C, erk1 promoter was amplified with primers PL-13-02 and PL-13-63; this fragment was assembled into pSGG-GG-lucHA with annealed primers PL-20-19/20 with a second ATG (after the ERK2 Nt sequence), or with primers PL-20-21/22 or PL-20-23/24 with GTG or GTC after the ERK2 Nt sequence instead of ATG.
Cell culture and transfection
NIH3T3 mouse fibroblasts, HeLa and HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX medium (GIBCO #31966–021) supplemented with 7% fetal calf serum (FCS; Dutscher #S005I30003) and penicillin/streptomycin (GIBCO #15140–122) [100 IU (50 μg)/ml] in a humidified atmosphere containing 5% CO2 in air at 37°C. Cells were seeded in 100, 60 or 30 ml diameter cell dishes for western blot or RNA extraction experiments, and 24-well plates for luciferase experiments.
Transfections were performed using the home-optimized PEI method by using polyethylenimine ‘Max’ (Polysciences, Inc., #24765) as the transfection agent in a 1 mg/ml solution (pH 7.0) in water. PEI was complexed to the different plasmids with a ratio of 2.5 μl of PEI for 1 μg of plasmid DNA. Formation of PEI–DNA complexes occurred by incubating PEI and plasmid DNA in 100 mM NaCl for 30 min at room temperature [the final volume constituting 1/10 (v/v) of the total cell medium with FCS]. The PEI–DNA precipitate was deposited drop by drop onto the cell culture medium. The medium was changed 16–24 h later. For 24-well plates, the amount of plasmid DNA per well was 0.5 μg. The DNA amount and volume of the transfection mix were adapted according to the surface of the culture dish.
Luciferase assays
For luciferase experiments, cells were seeded in 24-well dishes and transfected the next day. A 0.5 μg aliquot of total plasmid DNA was transfected, including the ffLuc construction and the plasmid pRL-TK (Promega) encoding the R-Luc which was co-transfected in a ratio of a 1/10 of the total DNA to control the variability of transfection efficiency in the reporter assays. The medium with transfection mix was changed to fresh medium 16–24 h after transfection. At 48 h after transfection, soluble extracts were harvested in 50 μl of Promega 1× passive lysis buffer (# E1941); lysis was carried out for 30 min under constant shaking. A single volume of 20 μl was assayed for R-Luc and ffLuc according to the published protocol (28). Transfections were performed in triplicate, and different plasmid preparations of the same construct were assayed. Luminescent counts were read using opaque 96-well plates in the dual injector luminometer Solaris from Robion (Germany).
Use of the GAL4-ELK1-myc/5×-gal4-luc system to report ERK activity
Transfection conditions for luciferase measurement
The GAL4-ELK-myc/5×-gal4-luc system was used to quantify ERK signaling. ffLuc activity was measured to report increased transcriptional activity on the 5×-gal4 promoter when the chimera GAL4-ELK was phosphorylated by ERK on its ELK moiety containing the ERK phosphorylation sites. HeLa cells were seeded in 24-well plates. Each triplicate of wells was transfected by a total of 100 ng of pSGG-GAL4-ELK-myc, 160 ng of plasmid-5×-gal4-luc, 30 ng of Renilla pRL-TK and 50 ng of the Raf1-CAAX plasmid; finally pSGG-promE1-ERK1catal or pSGG-promE1-ERK1full (with the ERK1 Nt) were transfected in increasing quantities (0.3, 0.5, 0.7 and 1.1 μg), compensated with empty psgg-vector. Transfection was performed using PEI as described before with a 2.5 ratio of PEI/plasmid (μl/μg). At 48 h after transfection, cells were lysed for simultaneous ffLuc and R-Luc activity assays.
Transfection conditions for western blot analysis
To analyze the GAL4-ELK phosphorylation profile by western blot, HeLa cells seeded in 6 cm diameter plates were transfected with 1 μg of Raf1-CAAX and 10 μg of GAL4-ELK-myc plasmids in the presence of increasing quantities of pECE-HA-ERK or pECE-HA-MEK (from 0.15 μg to 4 μg as indicated in Figure 1C). Transfection was performed with PEI as described previously, At 16 h post-transfection, medium was rinsed and replaced by fresh medium and 24 h later the cells were lysed in 1.5× Laemmli sample buffer and immunoblotting analysis was performed as described below.
Immunoblotting
Cells were lysed in Laemmli sample buffer prior to boiling. Proteins were separated by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE) [10% acrylamide-bis acrylamide (29:1) gels] loaded with 15–40 μg of protein per lane depending on gel size. Proteins were transferred onto BioTrace Nt nitrocellulose membranes from PALL (#66435). Membranes were blocked by incubating with 2.5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) containing 0.12% cold-skin fish gelatin and 0.1% casein (all from Sigma). Antibodies were incubated in PBS containing 0.1% cold-skin fish gelatin and 0.08% casein. The HA constructs were visualized by the monoclonal antibody HA11 1:1000 (Eurogentec #16B12). Phosphorylated ERK1/2 was detected with the mouse monoclonal anti-phospho-ERK1/2 antibody 1:3000 (Sigma #M8159); identical results were obtained with the rabbit monoclonal anti-phospho ERK 1:1000 (Cell Signaling Technologies #4370). Total ERK was detected with the rabbit monoclonal anti-ERK 1:2000 (Cell Signaling #4695) or the rabbit anti-rat ERK1 1:4000 (Fisher 1019–9152). MEK was detected with a home-made antibody (1:2000) described previously (29). The ffLuc protein was detected using the rabbit polyclonal antibody anti-ffLuc 1:1000 (MBL #PM016), the phospho-GAL4-ELK protein was visualized with the rabbit polyclonal antibody 1:1000 (Cell Signaling Technologies #9181S), the total GAL4-ELK protein was detected with the anti-myc antibody clone 4A6 1:3000 (Merck Millipore # 05–724) and the rabbit anti-HSP90 antibody was from Cell Signaling Technologies (#4877S). Secondary antibodies were IR dyes anti-rabbit 800CW (1:7000) and anti-mouse 680RD (1:7000) from LI-COR. The imager used is LI-COR Odyssey for detecting bands and quantification; all images are presented with gamma = 1 during image processing.
Quantitative PCR
NIH3T3 fibroblasts plated in 60 mm plates were transfected with plasmids and PEI mix as described previously. A 6 μg aliquot of various pSGG-GG-luc2P-plasmids was transfected as indicated in the legend of Supplementary Figure S3A. RNAs were extracted using TRIzol reagent (Invitrogen) and the RNeasy mini kit from QIAGEN (ref #74106). RNA (1.5 μg) was treated with the RNase-free DNase I set from QIAGEN (ref #79254) for 15 min according to the manufacturer's instructions. This step removed any trace of DNA, including plasmid DNA. Reverse transcription was performed with the Omniscript kit from QIAGEN (ref # 205111) according to the manufacturer's instructions. Luciferase cDNA expression levels were quantified by real-time PCR using SYBR-Green PCR Master Mix RT-SN2X-03+ (with ROX) from Eurogentec (Belgium). The oligonucleotides to amplify mouse luc2P firefly luciferase transcript were luc2P-forward (CTGTTCATCGGTGTGGCTGT) and luc2P-reverse (GCGCTCGTTGTAGATGTCGTT). Luc2P levels were normalized using the mouse erk2 cDNA expression level. Mouse-erk2 was amplified with mERK2-forward primer (GGAGCAGTATTATGACCCAAGTGA) and mERK2-reverse primer (TCGTCCAACTCCATGTCAAACT). Results are represented as the average of five independent experiments.
Statistical analysis
Statistical tests were conducted in Excel with the Analysis ToolPak. Welch's t-test was chosen to compare two independent groups due to suspected differences in variances linked to sample size. Details on sample sizes and significance can be found in the figure legends. All experiments have been reproduced at least three times with similar results. Independent plasmid preparations confirmed luciferase measurements after transient transfection.
Gene classification and protein sorting
Human gene sequences were obtained from Biomart Ensembl Release 104. The protein coding sequence and 12 nucleotides upstream were exported and converted to single-lane Fasta format. Each sequence was segmented into the Kozak sequence (9 nucleotides) and the first 39 coding nucleotides. Coding sequences were translated and the Swissprot number was concatenated with its translated coding sequence (13 amino acids). Duplicates were removed, dropping entries from 99K to ∼23K. This strategy allows entries with the same accession number but different Nt-coding sequences to be scrutinized. The sum of alanine residues and glycine residues among the sequence of 13 amino acids was calculated. The frequency of each of the four alanine codons was determined among the first six amino acid positions. This size was chosen because it is the size of the ERK2 alanine stretch that was demonstrated to provide full effect. Ranking of proteins was determined arbitrarily according to the following rules: 30 points given if the second amino acid is alanine; 25 points per alanine in the third or fourth position; 3 points per alanine in the fifth to sixth position; 2 points per alanine in the seventh to ninth position; and 1 point per alanine in the 10th to 13th position. To this number, the total number of alanine residues among the first 13 amino acids is added. The first protein in the list has 11 alanines out of 13; beyond the protein ranked in position 15 370, no alanine residues are present in the Nt (overall, 7923 proteins in the list have no alanine in their first 13 amino acids). From this list, families of proteins were sorted, such as the 85 proteins harboring a canonical Kozak sequence (Supplementary Table S1), or proteins harboring at least a stretch of six consecutive Nt alanines (Supplementary Figure S5B). For searching the presence of housekeeping genes (HKGs) in groups of 500 proteins, the list of HKGs was downloaded from the protein atlas web site in 2021:
https://www.proteinatlas.org/humanproteome/tissue/housekeeping and searching was operated by the Excel formula VLOOKUP. A similar procedure was done with the list of HKGs in the supplementary materials of the publication of Eisenberg and Levanon (30).
RESULTS
Both ERK activity and quantity play crucial roles in ERK signaling
We studied the relevance of ERK quantity on the ERK cascade signal output using the GAL4-ELK/gal4-luc system in which the production of ffLuc increases when the chimeric GAL4-ELK protein is phosphorylated by ERK. Then, we evaluated the consequences of increasing the quantity of WT or constitutively active kinases.
Using transfected HeLa cells, we confirmed that either a constitutively active RAF1 mutant, i.e. RAF1-CAAX, or a constitutively active MEK mutant, i.e. MEK-SSDD, strongly increased ffLuc activity in a dose-dependent manner compared with their respective WT forms or with WT-ERK (Figure 1A).
In cells with constitutively active RAF1-CAAX, increasing amounts of WT-ERK further induced a dose-dependent increase in ffLuc activity, which was not seen with increasing quantities of WT-MEK (Figure 1B). This result strongly suggests that under persistent activation of the ERK pathway, only ERK quantity is rate limiting. To confirm this observation, phosphorylation of a chimera of ELK, a direct ERK substrate, was measured by immunoblotting. Basal myc-tagged ELK phosphorylation was fully inhibited in the presence of the MEK inhibitor PD184352, whereas in the presence of RAF1-CAAX the multi-phosphorylation profile of ELK shifted to higher molecular weight bands (Supplementary Figure S1). Under this basal activation by RAF1-CAAX, increasing amounts of WT-ERK led to an increase in phospho-ERK levels as well as the increase in ELK phosphorylation (Figure 1C; Supplementary Figure S1). The ELK expression pattern mostly mimics the ELK multi-phosphorylation profile. Thus, in the context of constitutive activation of the ERK pathway, increasing ERK expression not only increases the intensity of ELK activation but also changes its multi-phosphorylation profile. In contrast, increasing expression of MEK induced neither ERK nor ELK phosphorylation (Figure 1C).
The ERK Nt impacts ERK signaling
ERK1 and ERK2 are both ubiquitously expressed and readily detected by immunoblotting in cell lines and tissues. Furthermore, quantitative proteomic analysis indicates that ERKs are among the most expressed kinases. For example, in NIH3T3 cells, only CDK1 and a few metabolic kinases are more highly expressed than ERK2, while ERK1 is expressed in the range of MEK1 or MEK2 (ERK2 being the 375th most expressed protein in this cell model) (31). In a panel of 32 human tissues, the quantity of ERK quantity is than that of MEK in 90% of multiple samples (32). To better understand the basis of elevated ERK1/2 expression, we studied erk promoters and the role of ERK Nt sequences that are highly G/C rich similarly to both 5′-UTRs. It is easy to distinguish the ERK Nt moieties from the catalytic domain because, after their first shared glutamic acid (E12 of ERK2, E29 of ERK1), human ERK1 and ERK2 are 83% identical (33). Although the ERK1 Nt is twice as long as the ERK2 Nt (29 versus 12 amino acids), both include a stretch of AlaGCG codons immediately downstream of the initiating methionine (Meti) (Figure 2A).
To investigate a putative role for ERK Nts in regulating ERK signaling, the GAL4-ELK/gal4-luc reporter system was transfected along with increasing quantities of an ERK1-encoding construct, containing its proximal promoter in the absence or presence of its Nt (Figure 2B). In the presence of RAF1-CAAX alone, ffLuc activity was stimulated 20-fold compared with the basal level, while increasing quantities of ERK1 further induced ERK signaling in a dose-dependent manner. However, in the absence of the ERK1 Nt, signaling output was decreased (Figure 2B). We then investigated whether the absence of the Nt could alter ERK1 kinase activity. Because there is a direct correlation between ERK activity and the extent of ELK multi-phosphorylation (Supplementary Figure S1; Figure 1C), we co-transfected HeLa cells with ELK and increasing quantities of ERK1 with or without its Nt. As shown in Figure 2C, when similar amounts of full-ERK1 (with the Nt) or catalytic-ERK1 (without theNt) were expressed (lane 4 versus lane 7), the activation levels of ELK were similar, either when measured by an anti-phospho-ELK antibody or by observing the molecular weight shift of multi-phosphorylated myc-ELK. Therefore, the ERK1 Nt domain does not play a role in ERK-specific kinase activity.
The ERK Nt increases ERK quantity and reduces the synthesis of smaller polypeptides
Cells were transiently transfected to assess the ability of the ERK Nt domains to regulate ERK quantity, catalytic-ERK1, under the control of the native erk1 promoter and fused or not to the ERK1 or ERK2 Nt (Figure 2D). As shown by the phospho-ERK and ERK-HA profiles (Figure 2D, first four lanes), ERK-HA proteins displayed an increased molecular weight when the Nt moieties were present (+29 amino acids for ERK1 and +12 amino acids for ERK2, red arrows of Figure 2D, upper panel, lanes 3 and 4 versus lane 2). Mutation of the native start codon to GUG led to lack of ERK1 expression (lane 1). Phosphorylated (functional) HA-tagged ERK proteins with Nt extensions from either ERK1 or ERK2 were ∼4-fold more abundant than those lacking the Nt (red arrows of Figure 2D, lower panel, lanes 3 and 4 versus 2).
Initial detection of transfected ERK-HA by immunoblot revealed bands of lower molecular weight expressed at variable levels. Here, to maximize detection of these bands, cells were treated overnight with the proteasome inhibitor bortezomib. A major low molecular weight band was expressed at ∼43 kDa (Figure 2D, upper panel, #1). These lower molecular weight bands (from #1 to #5) are unlikely to correspond to degradation products, firstly because they are less abundant under conditions of strong expression of full-size HA-tagged ERK proteins (upper panel, lanes 3 and 4 versus lanes 1 and 2), and secondly because they are more abundant when catalytic-ERK1 is not expressed due to a mutated AUG initiation codon (lane 1). The ERK1 Nt appeared more efficient than the ERK2 Nt in reducing expression of the lower molecular weight bands (lane 3 versus lane 4). We therefore hypothesized that they correspond to products translated by leaky scanning from in-frame downstream AUGs. Therefore, we sought to compare the size of these low molecular weight proteins with polypeptides produced from either the second, third or fourth downstream in-frame AUG codons (sequences in Supplementary Figure S2A). The truncated forms of the catalytic ERK-HA were expressed at higher levels using the CMV promoter (lane 5 versus lane 2) because smaller quantities of plasmids were transfected. The main bands produced from the second, third or fourth AUG (Figure 2D, lanes 6, 7 and 8) corresponded to the sizes of the putative leaky scanning products (#1, #2 and #3 bands). Only proteins that included the catalytic moiety of ERK1 were phosphorylated and thus activated by MEK (lanes 2–5, lower panel).
To confirm that the smaller polypeptides observed in the absence of the Nt moiety corresponded to leaky scanning products, we deleted the second in-frame AUG codon of ERK1 by mutating A166 to G (M66V). As shown in Figure 2E, in the absence of the first AUG or in the absence of the Nt moiety, a band of smaller molecular weight was observed (Figure 2E, control lanes 1 and 2, band #1). When the second AUG of ERK1 was mutated (Figure 2E, lanes 4 and 5), this band disappeared, while expression of lower molecular weight bands slightly increased (#2 and #3). In the presence of the ERK1 Nt, only the full-length protein was expressed, independently of the status of the second in-frame AUG (WT or mutant, Figure 2E, lanes 3 and 6, respectively). Taken together, these data suggest that the presence of Nt domains ensures the synthesis of full-length functional ERK proteins while inhibiting the expression of shorter polypeptides from downstream AUGs through leaky scanning.
Firefly luciferase reporter gene recapitulates the action of ERK-Nts
We next tested whether the ERK Nt downstream of the ERK1 promoter and 5′-UTR prevented leaky scanning to additional proteins such as the reporter ffLuc. We transfected NIH3T3 cells with constructs that fuse the ERK1 Nt or the ERK2 Nt to ffLuc. Compared with the absence of Nt, such constructs demonstrated increased luciferase activity by ∼10-fold (Figure 3A, upper panel). Even though the mouse erk2 proximal promoter drove ffLuc basal expression >100-fold higher than the erk1 promoter, fusion of the ERK2 Nt moiety increased ffLuc activity by ∼2.5-fold (Figure 3A, lower panel). These observations were reproduced in other mammalian cell lines, including mouse neuro2a, B16 melanoma and C2C12 (data not shown), and human cell lines HEK293 and HeLa. Fusions that included ERK Nt domains did not alter ffLuc mRNA level as determined by quantitative reverse transcription–PCR (RT–qPCR) in NIH3T3 cells (Supplementary Figure S3A), suggesting that the ERK Nt acts post-transcriptionally.
To assess whether the ERK Nt improves expression of full-length ffLuc at the expense of smaller polypeptides, as was observed with ERK1 protein expression itself, we analyzed ffLuc expression by immunoblotting. To maximize the signal, we replaced the C-terminal PEST sequence of ffLuc with the HA-tag and transfected HeLa and HEK293 cells (Figure 3B). In constructs containing the erk1 promoter, in the absence of the ERK Nt sequence, full-length ffLuc was weakly expressed at the expected size of 61.8 kDa (lane 1, red arrow), while there was greater intensity for two bands of ∼58 and 55 kDa, sizes corresponding to the expected sizes of polypeptides generated from the second and third in-frame AUG codons of ffLuc (sequences in Supplementary Figure S2B). When the ERK1 or ERK2 Nt was present (lanes 2 and 3), intense bands appeared with sizes consistent with full-length ffLuc, while expression of lower molecular weight bands markedly decreased. Compared with the ERK2 Nt, the ERK1 Nt more efficiently reduced expression of the low molecular weight forms (Figure 3B, lane 2 versus 3), as was also observed for ERK1 protein (Figure 2D).
To confirm that the shorter polypeptides are due to leaky scanning, we mutated the second in-frame AUG of ffLuc (A88 to G, corresponding to a ffLuc M30V protein mutation). In the absence of the second AUG (Figure 3C, lanes 4 and 5), the 58 kDa band (#1) disappeared while the 55 kDa band (#2) increased in intensity. This result demonstrates that translation starts at an AUG further downstream when the second in-frame AUG is mutated. When the ERK1 Nt is fused to ffLuc, mutation of the second in-frame AUG leads to weak expression of the 55 kDa polypeptide (Figure 3C, lane 6), while when the ERK1 Nt is fused to the WT-ffLuc the bands at 58 and 55 kDa are not expressed significantly (Figure 3C, lane 3). This suggests a potential interaction between the first two AUGs, leading to a preference for translation from the first one. These data support the idea that the ERK Nts can optimize start codon selection, leading to increased expression of full-length proteins, and thereby improve translational fidelity through the prevention of leaky scanning.
When tested in the settings of two nearby AUGs, the ERK Nt reduces leaky scanning in all reading frames
To further support our hypothesis that start codon selection in erk1 is inefficient in the absence of the ERK Nt domain, we tested whether a nearby downstream AUG in a favorable Kozak context would be recognized by the scanning pre-initiation complex (PIC). We inserted a sequence of 69 nucleotides flanked by two AUGs upstream of the ffLuc coding sequence, the first AUG being the native erk1 AUG with its native Kozak context. When the second AUG was in an unfavorable Kozak context (CUUAUAUUA-AUG-G, termed ‘Kozak poor’), ffLuc activity appeared low in the absence of the Nt (Figure 4A, lane 1) and ffLuc remained low in the presence of the control HSTK Nt (lane 2). However, in the presence of the ERK1 Nt upstream of the first AUG, ffLuc activity increased almost 5-fold (lane 3). When the second AUG was in a favorable context (GCCGCCGCC-AUG-G, termed ‘Kozak canonical’), ffLuc activity increased even in the absence of the Nt (Figure 4A, compare lanes 4 and 1). Indeed, there was no significant difference in ffLuc activity in the presence or the absence of the ERK1 Nt on the first AUG (compare lanes 4 and 6). In the presence of the HSTK Nt downstream of the first AUG, a second AUG in the good Kozak context also increased ffLuc activity (compare lanes 2 and 5). Overall, the results in Figure 4A suggest that when the ERK Nt is removed, the native mouse erk1 AUG itself is not favored for start codon selection and the PIC scans the mRNA until a more favorable AUG is encountered.
In Figure 4A, both nearby AUGs were in the same reading frame, whereas in Figure 4B we sought to test whether the presence of the ERK Nt at the first AUG could also influence AUG choice downstream, in alternative reading frames. Here, the second AUG is either in the second reading frame relative to erk1 AUG (lanes 1–3), or in the third reading frame (lanes 4–6). It is important to note that only the use of this second AUG in alternative reading frames will allow expression of the functional ffLuc. In the absence of the Nt at the first AUG, ffLuc expression was elevated when the second AUG was in the second reading frame (Figure 4B, lane 1) or in the third reading frame (lane 4). In the presence of the HSTK Nt, expression of ffLuc remained elevated in alternative reading frames, although slightly less than in the absence of the Nt, particularly in the third reading frame (lane 5). However, in the presence of the ERK1 Nt, expression of ffLuc was maximally reduced in both alternative reading frames (Figure 4B, lanes 3 and 6). These results confirm that the presence of the ERK1 Nt enforces start codon selection and dictates the reading frame, such that expression from alternative reading frames is markedly reduced, even when the downstream AUGs are in a very favorable context (canonical Kozak). Note that all AUGs examined here have a G+4 context. Thus, although the ERK1 Kozak context has an optimal composition with a purine at –3 and a G at +4 (15), leaky scanning occurs in the absence of the Nt in the same reading frame (Figure 4A) or in alternative reading frames (Figure 4B).
Human ERKs have a second nearby AUG to further reduce leaky scanning
A natural in-frame AUG codon exists at the end of mouse ERK2, human ERK2 and human ERK1 Nt sequences (Figure 2A). We investigated whether this second AUG participated in ERK start codon selection and as a consequence further reduced leaky scanning. An AUG, or the control codons GUG or GUC, was inserted between the ERK2 Nt and ffLuc in the context of the erk1 promoter, because this promoter/Nt combination allows better visualization of the leaky scanning products (Figures 2D and 3B). In Figure 4C, immunoblot analysis indicates that all constructs produced the functional ffLuc protein (upper band) along with some leaky scanning products at 58 and 55 kDa (see quantification in Figure 4C, lower panel). Leaky scanning was maximized in the absence of the ERK1 Nt (lane 1) but diminished in the presence of the ERK1 Nt and ERK2 Nt (lanes 2 and 3). In the ERK2 Nt constructs, insertion of GUC or the near-cognate GUG codon after the Nt did not affect luciferase production (lanes 5 and 6 versus 3), whereas insertion of an AUG codon at the same position reduced leaky scanning (different plasmid preparations of lanes 4 and 7 versus 3). Leaky scanning observed in the context of the erk1 promoter is also observed with the human KRAS Nt with its native promoter (lane 8). Considering that ERK polypeptides resulting from leaky scanning are not phosphorylated by MEK (Figure 2D), these results suggest that the presence of two in-frame AUGs in close proximity increases synthesis of fully functional ERKs.
Features of the erk1 5′-UTR that confer ERK Nt action
We studied the specificity of the ERK Nt by using proximal promoters from two other genes, HSV-TK and human KRAS. For these promoters of strengths similar to that of mouse erk1, fusion of their respective Nt moieties to ffLuc had no impact on ffLuc activity (Figure 5A). The ERK2 Nt induced a modest but significant increase of activity in the context of both the HSV-TK promoter and the KRAS promoter (Figure 5A). These results demonstrate that the maximal increase of ffLuc expression is specific to the combination of erk promoters/5′-UTR and ERK Nts.
Figure 5.

Analysis of 5′-UTR features that cooperate with the ERK Nt to determine start codon selection. (A) RLU from extracts of NIH3T3 cells transfected with plasmids bearing HSV-TK (orange) or KRAS (gray) promoters and the 5′-UTR. ffLuc was expressed directly behind promoters (white bars) or fused to their homologous Nt domain (orange and gray) or ERK2 Nt moiety (blue) (n = 6, ns = not significant, **P < 0.01 or ***P < 0.001 bilateral Welch's t-test, representative of three experiments). (B) NIH3T3 cells transfected with bicistronic constructs depicted above the graph. The CMV promoter expresses one mRNA, with R-Luc normalizing ffLuc (RLU). The second cistron, ffLuc, is fused (yellow bars) or not (white bars) to the ERK1 Nt. Upstream of the erk1 Kozak AUG sequence, fragments of the mouse erk1 5′-UTR are cloned with the indicated sizes (0–177 nucleotides, deletions from the 5′ side) (n = 3, ns = not significant, *P < 0.05 or **P < 0.01 bilateral Welch's t-test, representative of three experiments). (C) RLU from NIH3T3 cells transfected with plasmids harboring distinct Kozak sequences in the context of promoter erk1 and its 5′-UTR (pE1). ffLuc is fused (yellow bars) or not (white bars) to the ERK1 Nt, and fold induction is indicated on the graph near the yellow bars. Lines 1 and 2 contain erk1 Kozak and lines 3 and 4 the canonical Kozak sequence. From lines 5–10, nucleotides of Kozaks were swapped as indicated. Guanine nucleotides shared by erk1 and the canonical Kozak context are in black, nucleotides specific to the erk1 Kozak context in blue and those specific to the canonical Kozak context in red (n = 3, representative of three experiments).
We explored whether specific domains within the erk 5′-UTR were required for efficient translation of full-length protein. Recognizing that deletions of the 5′-UTR may remove promoter sequences, progressive shortening of the erk1 5′-UTR was analyzed between two cistrons, transcribed as a single mRNA from an upstream CMV promoter. Ribosome entry for the second cistron was provided by FGF-1A IRES, a weak cellular IRES (34). This weak cellular IRES avoids caveats of strong viral IRES, such as driving AUG choice without scanning in some cases (35). For these constructs, the AUG of the second cistron (ffLuc) was in the context of the erk1 Kozak sequence (Figure 5B). When the ERK1 Nt was fused to ffLuc, increasing the length of the erk1 5′-UTR progressively increased ffLuc activity (Figure 5B), while in the absence of the Nt, increasing the length of the erk1 5′-UTR had a progressively inhibitory effect. With very short 5′-UTR sequences (9–20 nucleotides, including the erk1 Kozak context), no significant differences were observed in the presence or absence of the ERK1 Nt, whereas maximal ffLuc activity was observed in the presence of the ERK1 Nt with the complete 5′-UTR sequence of mouse erk1 (186 nucleotides, including the erk1 Kozak sequence 36). These results show that the entire 5′-UTR is needed for the full response.
To study the specificity of the erk1 Kozak sequence for ERK1 Nt action, we swapped its nucleotides with those of the canonical Kozak sequence, in the presence or absence of the ERK1 Nt fused to ffLuc. Importantly, all constructs were in a G+4 context, from AUG–GCG (Met–Ala) with the Nt or from AUG–GAA (Met–Glu) in the absence of the Nt. In the absence of the ERK1 Nt (Figure 5C, white bars), when the erk1 Kozak sequence was replaced by the canonical Kozak sequence, ffLuc activity was increased by 4-fold (line 1 versus line 3). This indicates that, compared with the erk1 Kozak sequence, the canonical Kozak sequence more efficiently drives start codon selection. However, when the ERK1 Nt is fused upstream of the reporter gene (yellow bars), the canonical Kozak sequence was associated with reduced ffLuc activity (lane 2 versus lane 4). As the G nucleotides at positions –3, –6 and –9 are conserved in the erk1 Kozak sequences and the canonical sequence, we swapped other positions by pairs, creating hybrid Kozak sequences (Figure 5C). In the absence of the ERK1 Nt (white bars), any changes to the canonical Kozak sequence within the six nucleotides closest to AUG reduced luciferase activity (lanes 7 and 9); however, changes further upstream had no effect. In the presence of the ERK1 Nt, the intact erk1 Kozak sequence was the most efficient for driving ffLuc activity (lane 2 versus lanes 4, 6, 8 and 10). These results suggest that either the erk1 Kozak context is required for full action of the ERK1 Nt, or the canonical Kozak sequence is incompatible with the ERK1 Nt sequence.
Features of the ERK Nt that enhance start codon selection
To characterize the Nt domain requirements for this function, and particularly the role of the stretch of AlaGCG, we first modified the ERK2 Nt nucleotide sequence in order to progressively decrease the number of alanine residues and evaluated the impact on ffLuc activity. Figure 6A shows that the presence of all six alanine residues induced the highest ffLuc output (16-fold), whereas all other combinations were no more than half as efficient, the lowest effect being with two alanine residues. Moreover, removing the last three amino acids of the ERK2 Nt (GPE) had no impact (Supplementary Figure S3B). Taken together, these results demonstrate that the number of alanine residues in the Nt is of high importance.
To test whether the Nt nucleotide sequence is required to be located immediately downstream of the initiating AUG, we frameshifted the position of the ERK2 Nt progressively while maintaining the ffLuc coding frame downstream (Figure 6B). Frameshifting was carried out with G or C nucleotides to adhere to the GC richness of the ERK2 Nt (96%, Figure 6C upper sequence). When the start codon and the ERK2 Nt were positioned in their native state, ffLuc activity increased by 13-fold compared with ffLuc without the ERK2 Nt (Figure 6B, lane 1 versus 2). The insertion of one, two or three nucleotides between the start codon and the Nt abolished this response (lanes 3–5), demonstrating that the position of the Nt is essential to mediate full expression of functional ffLuc. Hence, replacing all the alanine (GCG) by glycine (GGC) in the Nt (lane 3), or inserting a single glycine before the ERK Nt (lane 5), markedly reduced luciferase activity, although these glycine codons are 100% GC rich. These findings indicate that either the position of the Nt, immediately downstream of the AUG, or the stretch of alanine residues itself is required, particularly an alanine residue next to Meti.
Alanine can be translated from four different codons, but in both the ERK1 and the ERK2 Nt, the rarest mammalian alanine codon (GCG) is used. We therefore tested various combinations of alternative alanine codons in the Nt to determine if the nucleotide sequence rather than the amino acid sequence was the root cause of the Nt effect. As shown before, WT-ERK2-Nt, which contains only AlaGCG codons, increased luciferase activity by 18-fold compared with constructs lacking the Nt (Figure 6C, lane 1 versus 2). Upon switching to AlaGCC codons, the response was lower (10-fold induction, lane 3 versus 1), and luciferase activity was even further reduced when all the codons were maximally A/U rich (4-fold induction, lane 4 versus 1), demonstrating that codon composition is crucial. Considering that the amino acid sequences of these proteins are identical (Figure 6C, lower panel), our results also suggest that the marked differences in response cannot be attributed to changes in protein half-life. Hence, the optimal alanine codon composition of the ERK2 Nt to mediate its function on protein expression is a stretch of six GCG codons.
Many human proteins possess ERK-like Nt moieties
ERK1 and ERK2 are encoded by two separate genes that both require closely related NTARs for precise and efficient translation initiation. To identify other proteins with ERK-like Nt domains, we screened for alanine-rich sequences specifically located at the N-termini of human proteins. Arbitrarily, we limited our quest to a 13 amino acid long sequence, corresponding to the size of the ERK2 NTARs up to the second AUG. Out of 23 325 human proteins analyzed, 21% start with Met–Ala, alanine being by far the most frequent amino acid in the second position (Supplementary Figure S4A). To rank NTAR proteins, we gave increasing weight to alanine residues near Meti because we showed that a glycine after AUG is detrimental, and considered the total number of alanine residues because an increased number correlates with increased effectiveness of the ERK2 Nt. We identified PABPN1 as a leading candidate, with 10 consecutive alanine residues after Meti (Figure 7; full list in Supplementary Table S1). Mutations within the PABPN1 gene that extend the alanine stretch are associated with oculopharyngeal muscular dystrophy (OPMD 37).
Figure 7.

Hundreds of proteins display repeated alanine codons at their Nt sequences. A list of the first 100 NTAR-containing proteins ranked in Supplementary Table S1. Each column indicates the protein rank, its name, the Nt sequence, the frequency of alanine codons among the first six amino acids as well as the percentage of Ala plus Gly residues among the first 12 amino acids. Methionine residues are highlighted in red; the color code for the frequencies of Ala codons is indicated below the list. The six proteins that have been demonstrated to possess functional NTARs in the context of their own promoters are boxed in red: PABPN1 #1; ERK2 (MAPK1) #19; MECP2 #20; ERK1 (MAPK3) #80; TRIM28 #110; and NIPA1 #529. At the bottom of the list, nine lower ranked proteins are presented to illustrate the extent of the NTAR selection list.
Each of the first 100 proteins on our list has at least three alanine residues immediately after Meti, and an overall alanine content of 55% within the first 12 amino acids downstream of Meti (versus 10% average among all human proteins). In addition to alanine, glycine residues are over-represented in our top 100 NTAR proteins (Supplementary Figure S4B). However, glycine is not observed adjacent to Meti in the first 448 proteins of the NTAR list, and only in 1% of the first 1000 NTAR proteins, while it is observed adjacent to Meti in 7.6% of all human proteins (Supplementary Figure S4A). Selected proteins ranked lower on the list are presented in Figure 7 to demonstrate the breadth of ERK-like NTAR proteins. Some lack alanine immediately adjacent to Meti but have other features consistent with NTARs, such as NIPA1, ranked in 529th position, with 10 alanine residues within the first 12 amino acids (MGTAAAAAAAAAA), and PRKACA, ranked 1997th, that has four consecutive AlaGCC codons (MGNAAAAKKGSEQ).
To confirm that the ERK Nt translational regulation also applies to other NTAR proteins, we examined the Nt sequence of several proteins. The proximal promoter/5′-UTRs of PABPN1 (1.5 kb, ranked 1 in the list), TRIM28 (886 bp, ranked 110), MECP2 (966 bp, ranked 20) and NIPA1 (1444 bp, ranked 529) were cloned in the absence or presence of their Nt sequences upstream of the ffLuc reporter. Like the ERK2 Nt, the PABPN1 Nt has six AlaGCG codons located immediately downstream of Meti, and four additional alanine codons follow (sequence shown in Supplementary Figure S10). The PABPN1 Nt increased luciferase activity 3-fold (manuscript in preparation).
Unlike the Nt of ERKs, the TRIM28 Nt has only one AlaGCG directly downstream of Meti, and it also does not harbor stretches of repeated alanine codons. However, TRIM28 protein has an alanine-rich Nt with 14 alanine residues out of the first 19 residues (bottom of Figure 8A), with overall 50% of alanine in the first 48 amino acids downstream of Meti. Fusion of the TRIM28 Nt to ffLuc increased ffLuc expression by ∼4-fold (Figure 8A). A similar boosting effect was obtained when fusing the entire sequence upstream of the second AUG to ffLuc (114 amino acids named ‘long-Nt’).
Figure 8.

The TRIM28, MECP2 and NIPA1 NTARs function in a manner similar to the ERK NTARs in the induction of reporter gene expression. (A) RLU from cell extracts of NIH3T3 cells transfected with constructs harboring the TRIM28 promoter (pTRIM28) without or with its short or long Nt domains fused to ffLuc (sequences are shown beneath the graph with AlaGCG codons in red) (n = 3, representative of three experiments). (B) RLU from cell extracts of NIH3T3 cells transfected with constructs harboring the MECP2 promoter (pMECP2) without or with its Nt domain fused to ffLuc (lanes 1 and 2). In lane 3, alanine codons were mutated to GCG, and in lane 4, alanine codons were mutated to GCA/GCU (sequence of the MECP2 Nt is shown beneath the graph, with AlaGCC codons in green) (n = 3, representative of three experiments). (C) RLU from cell extracts of NIH3T3 cells transfected with constructs harboring the NIPA1 promoter (pNIPA1) without or with its Nt domain fused to ffLuc (lanes 1 and 2). In lane 3, the first two amino acids (G–T) are deleted and in lane 4, two AlaGCG of the stretch are deleted (sequence of the NIPA1 Nt is shown beneath the graph with AlaGCG codons in red) (n = 3, representative of three experiments).
Unlike the ERK2 Nt with its six AlaGCG, the Nt of MECP2 displays a row of six AlaGCC (followed by one AlaGCG, sequence at the bottom of Figure 8B and in Supplementary Figure S10). The Nt of MECP2 increased ffLuc expression 5-fold (Figure 8B, lanes 1 versus 2) which was significantly reduced when codons were switched to AlaGCG (lane 3), and induction was nearly abolished when codons were switched to AlaGCA/GCU (lane 4). Therefore, when comparing the Nt of ERK2 (Figure 6C) with the Nt of MECP2 (Figure 8B), alanine codons rich in G/C are favored in both cases, but the most effective codon (GCG versus GCC) depends on the promoter/5′-UTR upstream of AUG.
Unlike the Nt of ERKs, the NIPA1 Nt has a very long stretch of 13 alanine residues that is separated from Meti by a glycine and threonine residues, and, unlike the Nt of ERKs, the NIPA1 stretch of eight AlaGCG codons is located seven residues downstream from Meti (sequence at the bottom of Figure 8C). Despite these differences, the WT sequence of the NIPA1 Nt increased ffLuc expression 3-fold (Figure 8C, lanes 1 versus 2). Because insertion of a single glycine between Meti and the six alanines of the ERK2 Nt nearly abolished completely the action of the ERK2 Nt (Figure 6B), we decided to test whether the two non-alanine residues could hamper the strength of the alanine stretch of NIPA1. Indeed, removal of glycine and threonine adjacent to Meti increased ffLuc expression 10-fold (lane 3), i.e. >3-fold higher than the WT NIPA1 sequence. This increased effect is not due to the mere shortening of the NIPA1 Nt because even with removal of two AlaGCG codons ffLuc levels were increased as efficiently as with the WT sequence (lane 4). These observations confirm that NTARs seem to function better when alanine residues are located adjacent to Meti.
Hundreds of human proteins display alanine-rich sequences at their N-termini (Supplementary Table S1). For ERK1, ERK2, PABPN1, TRIM28, MECP2 and NIPA1, their respective NTARs increase expression of functional ffLuc in the context of their own promoter/5′-UTR, which is not the case for proteins without an alanine-rich Nt such as KRAS (Figure 3D). Collectively, alanine richness seems to be the common feature of NTAR proteins; however, for each gene, specific NTAR features may additionally contribute to proper start codon selection in the context of their proximal promoters. In summary, our work shows that NTAR sequences play a key role in translation initiation by favoring the proper start codon, thereby increasing the amounts of an important subset of human proteins.
DISCUSSION
Although the N-terminal domains of mammalian ERK1 and ERK2 differ in length and composition, the kinase cores share 83% identical amino acids after human ERK2Glu-12/ERK1Glu-29 (33). The nucleotide sequences encoding the ERK Nt are highly GC rich, and the protein sequences include a stretch of 5–6 AlaGCG codons immediately downstream from the initiating Meti, the NTARs (Figure 2A). In this work, we demonstrated that the ERK1 NTAR has no role in kinase catalytic activity (Figure 2C), which we assumed for two reasons: first, invertebrate ERK proteins lack these Nt repeated amino acids, and second, the crystal structures of the catalytic domains of mammalian ERK proteins do not include the Nt amino acids (e.g. Protein Data Bank entries 2ZOQ or 4QTB).
We have also shown that ERK NTARs play no role in transcription (Supplementary Figure S3A). However, we found that in the context of their own proximal promoter/5′-UTR, ERK NTAR moieties are associated with increased synthesis of the functional protein (Figure 2D, E). They ensure start codon selection and reduce leaky scanning through interactions involving the entire 5′-UTR (Figures 4 and 5). The nature of the smaller polypeptides translated in the absence of ERK NTARs was suspected to originate from leaky scanning because their sizes matched those of artificial proteins starting at the successive in-frame AUGs. Leaky scanning was confirmed by site-directed mutagenesis of the second AUG, thereby ruling out other mechanisms such as degradation. Synthetic constructs revealed that the ERK1 NTAR also enforces its reading frame, thereby avoiding synthesis of peptides from alternative frames (Figure 4B). The co-evolution of ERK1 and ERK2 NTARs indicates that there is a strong evolutionary pressure to select these peculiar sequences, therefore they must play an important role. Indeed, NTAR moieties could have arisen to decrease the production of toxic proteins translated via alternative reading frames (Figure 4B), or to increase the quantity of functional proteins (Figure 2D, E). Considering that protein synthesis requires more energy than any other metabolic process (38), NTARs should benefit the cell by tipping the balance in favor of functional protein synthesis at the expense of leaky scanning products. This could be particularly relevant when considering that NTAR-containing proteins are highly enriched among housekeeping proteins (Supplementary Figure S7).
It is extremely puzzling that both erk1 and erk2 require NTARs because the native AUGs of these two genes fulfill the known criteria for efficient start codon choice. On one hand, the Kozak sequences of both erk1 and erk2 are purportedly ‘good’ contexts because they harbor a purine in –3 and a G in +4 (+1 being the A of AUG). On the other hand, in mouse, the erk2 5′-UTR has no uAUG while the erk1 5′-UTR has only one uAUG which cannot be functional because it is adjacent to a stop codon [the sizes of mouse 5′-UTRs were determined by primer extension to be 186 nucleotides for erk1 and 223 nucleotides for erk2 (36,39), and, at these sizes, human sequences have no uAUGs]. Why then does ERK translation not start at the first AUG, which is in a ‘good’ context, in the absence of NTARs? By definition, NTARs are downstream of the AUG because they code for proteins, and so far the only downstream feature known to increase AUG choice is a stem–loop placed exactly 14 nucleotides downstream of the AUG according to M. Kozak (40). Because a single mRNA molecule is scanned simultaneously by several ribosomes, it was proposed that slowing down the elongating ribosome by the stem–loop makes it collide with the trailing scanning PICs and pause them close to the AUG codon to facilitate initiation. Hence, here we have searched for features of ERK NTARs that could be involved in slowing down the nascent elongating ribosome.
First, we have shown that the ERK2 NTAR sequence harbors a second in-frame AUG that reduces leaky scanning significantly (Figure 4C). Indeed, previous in silico analyses have found that a second AUG is often present in mRNAs with a suboptimal Kozak context at their native start codon (41). This fits with the observation that the erk1 Kozak sequence inefficiently flags the start codon in the absence of NTARs (Figure 5C). Ribosome assembly on AUG takes longer than the time required for a PIC to scan a codon (42). In the case of erk2 mRNA, the distance between the two AUGs is the same size as RNA fragments protected from nuclease treatment by a single ribosome, the so-called mRNA channel (43). Therefore, this second AUG is strategically located downstream of the first AUG at the distance of the RNA channel because a ribosome on this second AUG would temporarily roadblock an incoming PIC exactly on the first AUG. As with human ERK1 and ERK2, many NTAR proteins (11% of those in Figure 7) have a second nearby in-frame AUG.
Second, it was proposed that rare codons slow the progression of the elongating ribosome because it has been shown that clusters of rare codons slow down translation in order to allow time for proper folding of protein domains (44). In addition, in silico analysis revealed that the Nt of human proteins is enriched in rare codons, mainly for AlaGCG, ProCGG and SerUCG (45,46), and among the first 100 proteins of Figure 7, we showed that 51% of all four alanine codons are the rarest AlaGCG. Indeed, as shown in Figure 6C, the strength of ERK2 NTARs decreased when the six rare AlaGCG codons were replaced by six frequent AlaGCC codons. Therefore, codon rarity could very well participate to slow the nascent elongating ribosome and, as such, favor start codon choice for the trailing PICs. Alternatively, the codon specificity may reflect the requirement for secondary structures, possibly by pairing NTARs with 5′-UTR sequences. In favor of a structural role for NTARs, the ERK2 NTAR acts specifically in the context of erk promoters because it is ineffective downstream of the Kras proximal promoter (Figure 5A). However, when modifying alanine codon composition, changing the third base of the codon from G to C was less detrimental than changing from G to A/U in ERK2 (Figure 6C). This argues against the formation of secondary structures, because a change from G to C disrupts secondary structures more than G to A, or even more than G to U which can generate wobble pairings (47). Due to the duality of codon/nucleotide sequence, it is difficult to unambiguously demonstrate that a stretch of rare codons acts by forming a secondary structure rather than as a consequence of the rare codons themselves. Therefore, we cannot dismiss completely a role for secondary structures, but we favor the idea that codon rarity in the ERK2 NTAR slows down the leading ribosome. The balance was tipped towards codon rarity by observing that there are twice as many AlaGCG than AlaGCC codons in NTAR proteins (51% of all alanine codons versus 23% respectively, among the first 100 NTARs, Supplementary Table S1). Therefore, although both codons are 100% GC rich, which should allow formation of secondary structures of similar strength, the rarest codon is highly preferred (codon frequencies of 0.11 for AlaGCG versus 0.40 for AlaGCC, in mammals).
A third feature of the ERK NTARs that could slow down the initiating ribosome is the repetition of 5–6 identical codons, which is suggestive of tRNA channeling, a process in which a limited set of tRNAs are constantly reloaded in the vicinity of the ribosome upon association of aminoacyl-tRNA synthetases with the ribosome (48,49). When studied as doublets, or when ordered along the mRNA, correlated codons were shown to drive faster translation (49). However, in NTAR sequences, the codons are often repeated in rows, requiring constant reloading of the same tRNA, which will be limited by local concentration effects. Importantly, the strength of action of the ERK2 NTARs markedly increased with six alanines in a row instead of three (Figure 6A). Among other NTARs, codon repetition is observed for other amino acids, bolstering a role for tRNA channeling to contribute to start codon choice. For example, the NTAR of human MECP2 contains six consecutive AlaGCC codons, five consecutive GlyGGA codons and three consecutive GluGAG codons.
The fourth and, so far, final feature is the alanine richness 3′ adjacent to Meti for both the ERK1 and ERK2 NTARs. This feature might also contribute to slowing the pace of the elongating ribosome. Alanine richness can be highly elevated among NTAR proteins, as indicated in Figure 7 and Supplementary Table S1. The Translation Initiator of Short 5′-UTR (TISU) that drives scanning-independent translation (50) also favors two consecutive alanine residues after Meti with the consensus sequence AUG–GCG–GCN, coding for Met–Ala–Ala (51). Furthermore, among highly expressed proteins, alanine has been shown to be the most common amino acid immediately following the Meti across a broad range of species (52). Considering that selection of the start codon in bacteria is driven by a direct interaction between rRNA and mRNA via the Shine–Dalgarno sequence, alanine preference may suggest a biophysical advantage during translation. In our case, this advantage is experimentally confirmed with the replacement of alanine by glycine residues, the two most common amino acids present in NTARs (Figure 7; Supplementry Figure S4B). The replacement by seven glycines in a row, or just the insertion of a single glycine between the Meti and the stretch of six alanines, was sufficient to markedly reduce start codon selection (Figure 6B). Similarly, in the case of NIPA1 protein, removal of a glycine and a threonine at the second and third positions resulted in the alanine stretch being adjacent to Meti, which significantly increased the efficacy of the NTARs (Figure 8C). The detrimental effect of a glycine at the second position may be a general phenomenon among NTAR proteins. For NIPA1, the first four alanine codons that were juxtaposed with the initiating Meti via mutation were neither rare nor repeated codons, suggesting a role for alanine per se. Glycine is not observed adjacent to Meti in the first 448 proteins of the list of NTARs, and Met–Gly is observed in only 1% of the first 1000 NTAR proteins, while it is observed adjacent to Meti in 7.6% of all human proteins (Supplementary Figure S4A), reinforcing this notion. Of note, only seven human proteins start with Met–Gly–Gly–Gly and 81 start with Met–Gly–Gly (for comparison, 280 human proteins start with Met–Ala–Ala–Ala and 1080 start with Met–Ala–Ala; see Supplementary Table S1). Moreover, inserting a glycine upstream of the NTARs did not alter the GC-richness or the G+4 rule of erk1 Kozak, suggesting a pivotal role for alanine residues during translation itself.
At this point, one wonders about the biophysical advantage of alanine versus glycine. It has been shown that repeats of large and charged amino acids located at the Nt of proteins block emergence of the nascent peptidyl chain through the ribosomal exit tunnel (even destabilizing the ribosome) (53). Therefore, because alanine and glycine are the two smallest amino acids, their elevated presence in NTARs (Figure 7; Supplementary Figure S4B) might help to nudge the nascent amino acid chain through the ribosomal exit tunnel. With its methyl side chain, alanine is slightly larger than glycine and neither is charged. Hydropathy measurements have indicated that alanine is weakly hydrophobic whereas glycine is weakly hydrophilic (54,55). Interestingly, hydrophobic stretches of amino acids have been estimated to cross the ribosomal exit tunnel more slowly than hydrophilic ones (56). Therefore, by being hydrophobic, a stretch of alanine residues may slow the emergence of the nascent peptide from the ribosomal exit tunnel. In turn, this may retard departure of the ribosome, increasing its probability of colliding with an incoming PIC, improving start codon selection by increasing its dwell time on the AUG/Kozak sequence.
Overall, the ERK2 NTAR accumulates the four features described above, which could converge to slow the departure of the initiating ribosome. First, the ERK2 NTAR displays a second AUG in phase; second, the ERK2 NTAR is rich in rare codons; third, the ERK2 NTAR possesses a stretch of six repeated codons; and fourth, the ERK2 NTAR is highly alanine rich. In humans and great apes, ERK1 also possesses these four features. This model predicts that ribosome density ought to be higher around an AUG when the initiating ribosome is slowed down. Indeed, this has been observed in the vicinity of AUGs in general (43). However, it is unlikely that NTARs would increase ribosome density at AUGs at unusually high levels for two reasons. First, we observed that the ERK2 NTAR does not form a ‘stand-alone structure’ because it is ineffective in the context of the Kras promoter/5′-UTR (Figure 5A). Second, ribosome stalling by NTARs must be limited to avoid triggering the ribosome quality control mechanism (RQC) that leads to decay of the mRNA (57). NTARs may simply be required to establish normal ribosome dwell time at an initiating AUG in the context of the 5′-UTRs of highly expressed/peculiar HKGs (Supplementary Figure S7).
As indicated by their name, NTARs are N-terminal sequences rich in alanine residues, and therefore possess at least one feature that seems to boost start codon selection. NTAR proteins other than ERKs may possess only a subset of the four features present in ERK NTARs that seem to foster start codon selection. For example, the MECP2 mRNA does not possess rare AlaGCG codons, but has three stretches of repeated codons as presented above (sequences in Supplementary Figure S10). The sequence encoding the TRIM28 NTARs does not include a long stretch of repeated codons, nor does it have groups of rare codons; however, 14 out of its 19 first amino acids are alanine, as are 24 out of its first 50 residues. The NIPA1 NTAR does not have an alanine next to Meti, but it possesses a stretch of 13 alanines, 8 of which are tandemly repeated rare AlaGCG codons.
Mirroring the NTARs, the Kozak sequence on the other side of the AUG plays a major role in start codon selection. It is widely believed that the most effective context to promote accurate start codon selection is the canonical Kozak sequence (GCC)GCCG/ACC-AUG-G (15). In our study, we were initially surprised to find that very few mRNAs with canonical Kozak sequences have alanine residues in their Nt, and none of them has NTARs (Supplementary Table S1). This is unexpected because both the canonical and erk1 Kozak sequences possess the specific nucleotides that interact with the scanning PIC, notably a purine at the –3 position that interacts with eIF2-α and a G at +4 that interacts with eIF1A (58). Therefore, we sought to evaluate the relative strength of the canonical Kozak sequence and its compatibility with NTARs, in the context of the erk1 proximal promoter. In the absence of an NTAR, the canonical Kozak context is up to 4-fold more efficient than the erk1 Kozak context, which confirms the validity of previous claims (Figure 5C). However, in the presence of an NTAR, the canonical Kozak context became inhibitory. Swapping nucleotides between the erk1 and the canonical Kozak sequence confirmed these conclusions by providing intermediate results (Figure 5C). Replacing the erk1 Kozak sequence with the canonical one could disrupt a secondary structure formed between the NTAR sequence and the erk1 Kozak sequence. However, we do not favor this possibility because NTAR proteins do not share a common Kozak sequence (Supplementary Figure S5A). Even proteins with the same N-terminal stretch of six alanines do not share a common Kozak sequence (Supplementary Figure S5B). We favor the idea of an incompatibility of ERK NTARs with a canonical Kozak sequence because they form a very stable stem–loop, and similar stem–loops formed at AUGs were shown to reduce translation initiation (59). Indeed, with a minimal free energy (MFE) of –23.6 kcal/mol, this stem–loop is much more stable than all other combinations between Kozak sequences and the NTARs tested (Supplementary Figure S6A). Furthermore, human 5′-UTRs with canonical Kozak sequences form stem–loops of much lower strength in the context of their native Nt-coding sequences (average MFE of –7.14 kcal/mol; Supplementary Figure S6B).
It has been shown that the canonical Kozak context is seldom encountered in the vicinity of vertebrate AUGs (60), while the boundaries of the NTAR protein family remain to be established. For example, among all members of the RAS–MEK–ERK signaling cascade, only ERK1 and ERK2 rely on NTAR moieties for efficient translation. In parallel, we have shown that the quantity of ERK, but not MEK, influences signaling output (Figure 1C). This is surprising because ERKs are among the most abundant signaling kinases and are more highly expressed than MEK (31,32). The co-evolution of the ERK NTARs is stunning. On the one hand, the second nearby methionine is present in vertebrate ERK2s from amphibians to mammals but is seldom found in teleost fish and is absent in cartilaginous fishes. On the other hand, the second nearby methionine is present in ERK1 of humans and great apes but not in ERK1 of other vertebrates, including tarsiers, a closely related primate. The convergent evolution leading to an in-frame second methionine in both human ERK1 and ERK2, and evolution towards stretches of the rarest alanine codons for both proteins, suggest the importance of producing precisely full-length functional ERKs in humans, which may in turn have implications for signaling.
Increasing evidence indicates that the proteome is greatly shaped through translational regulation. For example, the choice of start codon between AUGs of upstream ORFs versus the AUG of the major ORF has been shown to regulate the expression of many proteins. This mechanism is at play for ATF4 expression during the integrated stress response, for the balance of eIF1/eIF5 expression [reviewed in (18)] and for the regulation of Hox gene expression (61). Having shown that the presence of NTARs strongly increases expression of the main ORFs of ERK1/2, we propose that the mechanism of NTARs is not subject to short-term regulation. Furthermore, most NTAR-containing proteins are encoded by HKGs (Supplementary Figure S7), whose expression is usually not subject to rapid regulation because they are constitutively and ubiquitously expressed to drive basic cellular functions. In fact, harmful pathophysiological consequences arise when Kozak sequences or NTAR-encoding sequences are altered. For example, mutation of the BRCA1 Kozak sequence contributes to cancer progression (62), while increasing the number of alanines in the PABPN1 NTARs causes OPMD (37).
For cell fate decisions, ERK proteins have attracted considerable attention because the duration of their activation drives the progressive multi-phosphorylation of specific substrates, particularly of immediate early genes such as FOS (63). Here, we reveal for the first time a direct link between ERK quantity and the extent of substrate multi-phosphorylation (Figure 1C). ERK expression might be elevated and tightly regulated because ERK interacts with all its partners via the same docking sites, either the CDS (common docking site) or the FRS (F-site recruitment) [reviewed in (7)]. Indeed, on the activator side, a balanced expression of MEK has been shown to be required to establish normal cytoplasmic localization of micro-injected ERK (64). On the substrate side, expression of short-lived partners was shown to sustain ERK nuclear localization (65).
Unregulated activation of the ERK pathway occurs in many cancers, and congenital disorders designated RASopathies are caused by mutations of ERK signaling cascade members (20). Furthermore, amplification of erk genes has been found in several cancers (10,11), and some cases of resistance to EGFR tyrosine kinase inhibitors have been linked to erk2 gene amplification (9). Understanding the role of ERK Nt sequences may facilitate the development of new strategies to improve the treatment outcomes for these diseases. Because ERK1/2 are among the hundreds of human NTAR proteins, we propose that the NTAR mechanism represents a general system controlling start codon choice, leading to enhanced translation initiation for a substantial fraction of human proteins, particularly those involved in housekeeping functions. A better understanding of NTARs could have implications for synthetic biology, corrective gene therapy and even efficient antigen production by mRNA vaccines.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to Dr A.C Prats for providing the bicistronic vectors pCRHL and pRCF1AL. We thank Olivier Croce and Olivier Casile for their generous help with converting the multi-line fasta formatted files into single line format. We thank Drs Nathalie Yazbeck and P. Brest for help with cloning of the hkras proximal promoter. We thank Marie-Angela Domdom for technical help and Dr Steve Olsen for careful reading of the manuscript.
Notes
Present address: Mylène Egensperger, EVOTEC, Göttingen, Germany.
Contributor Information
Roser Buscà, Université Côte d’Azur (UCA), CNRS UMR 7284 and INSERM U 1081, Institute for Research on Cancer and Aging Nice (IRCAN), 28 Avenue de Valombrose, 06107 Nice, France; Centre Antoine Lacassagne, Nice, France.
Cercina Onesto, Université Côte d’Azur (UCA), CNRS UMR 7284 and INSERM U 1081, Institute for Research on Cancer and Aging Nice (IRCAN), 28 Avenue de Valombrose, 06107 Nice, France; Centre Antoine Lacassagne, Nice, France; Polytech’Nice Sophia, Bioengineering Department, Sophia-Antipolis, France.
Mylène Egensperger, Université Côte d’Azur (UCA), CNRS UMR 7284 and INSERM U 1081, Institute for Research on Cancer and Aging Nice (IRCAN), 28 Avenue de Valombrose, 06107 Nice, France; Centre Antoine Lacassagne, Nice, France.
Jacques Pouysségur, Université Côte d’Azur (UCA), CNRS UMR 7284 and INSERM U 1081, Institute for Research on Cancer and Aging Nice (IRCAN), 28 Avenue de Valombrose, 06107 Nice, France; Centre Antoine Lacassagne, Nice, France; Centre Scientifique de Monaco, Biomedical Department, Principality of Monaco.
Gilles Pagès, Université Côte d’Azur (UCA), CNRS UMR 7284 and INSERM U 1081, Institute for Research on Cancer and Aging Nice (IRCAN), 28 Avenue de Valombrose, 06107 Nice, France; Centre Antoine Lacassagne, Nice, France; Centre Scientifique de Monaco, Biomedical Department, Principality of Monaco.
Philippe Lenormand, Université Côte d’Azur (UCA), CNRS UMR 7284 and INSERM U 1081, Institute for Research on Cancer and Aging Nice (IRCAN), 28 Avenue de Valombrose, 06107 Nice, France; Centre Antoine Lacassagne, Nice, France.
Data Availability
The data underlying this article are available in the article and in its online supplementary data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
CNRS running fund for the laboratory.
Conflict of interest statement. None declared.
REFERENCES
- 1. Meloche S., Seuwen K., Pagès G., Pouysségur J.. Biphasic and synergistic activation of p44mapk (ERK1) by growth factors: correlation between late phase activation and mitogenicity. Mol. Endocrinol. 1992; 6:845–854. [DOI] [PubMed] [Google Scholar]
- 2. Marshall C.J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995; 80:179–185. [DOI] [PubMed] [Google Scholar]
- 3. Lavoie H., Gagnon J., Therrien M.. ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020; 21:607–632. [DOI] [PubMed] [Google Scholar]
- 4. Patel A.L., Shvartsman S.Y.. Outstanding questions in developmental ERK signaling. Development. 2018; 145:dev143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Canagarajah B.J., Khokhlatchev A., Cobb M.H., Goldsmith E.J.. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997; 90:859–869. [DOI] [PubMed] [Google Scholar]
- 6. Ashton-Beaucage D., Udell C.M., Lavoie H., Baril C., Lefrancois M., Chagnon P., Gendron P., Caron-Lizotte O., Bonneil E., Thibault P.et al.. The exon junction complex controls the splicing of MAPK and other long intron-containing transcripts in Drosophila. Cell. 2010; 143:251–262. [DOI] [PubMed] [Google Scholar]
- 7. Buscà R., Pouysségur J., Lenormand P.. ERK1 and ERK2 Map kinases: specific roles or functional redundancy?. Front. Cell Dev. Biol. 2016; 4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Samuels I.S., Karlo J.C., Faruzzi A.N., Pickering K., Herrup K., Sweatt J.D., Saitta S.C., Landreth G.E.. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. 2008; 28:6983–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ercan D., Xu C., Yanagita M., Monast C.S., Pratilas C.A., Montero J., Butaney M., Shimamura T., Sholl L., Ivanova E.V.et al.. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012; 2:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campbell J.D., Alexandrov A., Kim J., Wala J., Berger A.H., Pedamallu C.S., Shukla S.A., Guo G., Brooks A.N., Murray B.A.et al.. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016; 48:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salaroglio I.C., Mungo E., Gazzano E., Kopecka J., Riganti C.. ERK is a pivotal player of chemo-immune-resistance in cancer. Int. J. Mol. Sci. 2019; 20:2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Janknecht R., Zinck R., Ernst W.H., Nordheim A.. Functional dissection of the transcription factor Elk-1. Oncogene. 1994; 9:1273–1278. [PubMed] [Google Scholar]
- 13. Mylona A., Theillet F.X., Foster C., Cheng T.M., Miralles F., Bates P.A., Selenko P., Treisman R.. Opposing effects of Elk-1 multisite phosphorylation shape its response to ERK activation. Science. 2016; 354:233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murphy L.O., Smith S., Chen R.H., Fingar D.C., Blenis J.. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002; 4:556–564. [DOI] [PubMed] [Google Scholar]
- 15. Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986; 44:283–292. [DOI] [PubMed] [Google Scholar]
- 16. Lu P.D., Harding H.P., Ron D.. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004; 167:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loughran G., Sachs M.S., Atkins J.F., Ivanov I.P.. Stringency of start codon selection modulates autoregulation of translation initiation factor eIF5. Nucleic Acids Res. 2012; 40:2898–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hinnebusch A.G., Ivanov I.P., Sonenberg N.. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016; 352:1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Messaed C., Rouleau G.A.. Molecular mechanisms underlying polyalanine diseases. Neurobiol. Dis. 2009; 34:397–405. [DOI] [PubMed] [Google Scholar]
- 20. Tajan M., Paccoud R., Branka S., Edouard T., Yart A.. The RASopathy family: consequences of germline activation of the RAS/MAPK pathway. Endocr. Rev. 2018; 39:676–700. [DOI] [PubMed] [Google Scholar]
- 21. Engler C., Kandzia R., Marillonnet S.. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008; 3:e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pagès G., Brunet A., L’Allemain G., Pouysségur J.. Constitutive mutant and putative regulatory serine phosphorylation site of mammalian MAP kinase kinase (MEK1). EMBO J. 1994; 13:3003–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meloche S., Pagès G., Pouysségur J.. Functional expression and growth factor activation of an epitope-tagged p44 mitogen-activated protein kinase, p44mapk. Mol. Biol. Cell. 1992; 3:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buscà R., Abbe P., Mantoux F., Aberdam E., Peyssonnaux C., Eychene A., Ortonne J.P., Ballotti R.. Ras mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000; 19:2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lefloch R., Pouyssegur J., Lenormand P.. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol. Cell. Biol. 2008; 28:511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Knockaert M., Lenormand P., Gray N., Schultz P., Pouyssegur J., Meijer L.. p42/p44 MAPKs are intracellular targets of the CDK inhibitor purvalanol. Oncogene. 2002; 21:6413–6424. [DOI] [PubMed] [Google Scholar]
- 27. Créancier L., Morello D., Mercier P., Prats A.C.. Fibroblast growth factor 2 internal ribosome entry site (IRES) activity ex vivo and in transgenic mice reveals a stringent tissue-specific regulation. J. Cell Biol. 2000; 150:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hampf M., Gossen M.. A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal. Biochem. 2006; 356:94–99. [DOI] [PubMed] [Google Scholar]
- 29. Lenormand P., Sardet C., Pagès G., L’Allemain G., Brunet A., Pouysségur J.. Growth factors induce nuclear translocation of MAP kinases (p42maPk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell Biol. 1993; 122:1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eisenberg E., Levanon E.Y.. Human housekeeping genes, revisited. Trends Genet. 2013; 29:569–574. [DOI] [PubMed] [Google Scholar]
- 31. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M.. Global quantification of mammalian gene expression control. Nature. 2011; 473:337–342. [DOI] [PubMed] [Google Scholar]
- 32. Jiang L., Wang M., Lin S., Jian R., Li X., Chan J., Dong G., Fang H., Robinson A.E., Aguet F.et al.. A quantitative proteome map of the human body. Cell. 2020; 183:269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buscà R., Christen R., Lovern M., Clifford A.M., Yue J.X., Goss G.G., Pouyssegur J., Lenormand P.. ERK1 and ERK2 present functional redundancy in tetrapods despite higher evolution rate of ERK1. BMC Evol. Biol. 2015; 15:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martineau Y., Le Bec C., Monbrun L., Allo V., Chiu I.-M., Danos O., Moine H., Prats H., Prats A.-C.. Internal ribosome entry site structural motifs conserved among mammalian fibroblast growth factor 1 alternatively spliced mRNAs. Mol. Cell. Biol. 2004; 24:7622–7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mailliot J., Martin F.. Viral internal ribosomal entry sites: four classes for one goal. Wiley Interdiscip. Rev. RNA. 2018; 9:e1458. [DOI] [PubMed] [Google Scholar]
- 36. Pages G., Stanley E.R., Le Gall M., Brunet A., Pouyssegur J.. The mouse p44 mitogen-activated protein kinase (extracellular signal-regulated kinase 1) gene. Genomic organization and structure of the 5′-flanking regulatory region. J. Biol. Chem. 1995; 270:26986–26992. [DOI] [PubMed] [Google Scholar]
- 37. Brais B., Bouchard J.P., Xie Y.G., Rochefort D.L., Chretien N., Tome F.M., Lafreniere R.G., Rommens J.M., Uyama E., Nohira O.et al.. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998; 18:164–167. [DOI] [PubMed] [Google Scholar]
- 38. Wieser W., Krumschnabel G.. Hierarchies of ATP-consuming processes: direct compared with indirect measurements, and comparative aspects. Biochem. J. 2001; 355:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugiura N., Suga T., Ozeki Y., Mamiya G., Takishima K.. The mouse extracellular signal-regulated kinase 2 gene. Gene structure and characterization of the promoter. J. Biol. Chem. 1997; 272:21575–21581. [DOI] [PubMed] [Google Scholar]
- 40. Kozak M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc. Natl Acad. Sci. USA. 1990; 87:8301–8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kochetov A.V. AUG codons at the beginning of protein coding sequences are frequent in eukaryotic mRNAs with a suboptimal start codon context. Bioinformatics. 2005; 21:837–840. [DOI] [PubMed] [Google Scholar]
- 42. Hinnebusch A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014; 83:779–812. [DOI] [PubMed] [Google Scholar]
- 43. Ingolia N.T., Lareau L.F., Weissman J.S.. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011; 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Komar A.A. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 2009; 34:16–24. [DOI] [PubMed] [Google Scholar]
- 45. Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y.. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010; 141:344–354. [DOI] [PubMed] [Google Scholar]
- 46. Park J.-H., Kwon M., Yamaguchi Y., Firestein B.L., Park J.-Y., Yun J., Yang J.-O., Inouye M.. Preferential use of minor codons in the translation initiation region of human genes. Hum. Genet. 2017; 136:67–74. [DOI] [PubMed] [Google Scholar]
- 47. Varani G., McClain W.H.. The G × U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000; 1:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stapulionis R., Deutscher M.P.. A channeled tRNA cycle during mammalian protein synthesis. Proc. Natl Acad. Sci. USA. 1995; 92:7158–7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cannarozzi G., Schraudolph N.N., Faty M., von Rohr P., Friberg M.T., Roth A.C., Gonnet P., Gonnet G., Barral Y.. A role for codon order in translation dynamics. Cell. 2010; 141:355–367. [DOI] [PubMed] [Google Scholar]
- 50. Sinvani H., Haimov O., Svitkin Y., Sonenberg N., Tamarkin-Ben-Harush A., Viollet B., Dikstein R.. Translational tolerance of mitochondrial genes to metabolic energy stress involves TISU and eIF1-eIF4GI cooperation in start codon selection. Cell Metab. 2015; 21:479–492. [DOI] [PubMed] [Google Scholar]
- 51. Elfakess R., Dikstein R.. A translation initiation element specific to mRNAs with very short 5′UTR that also regulates transcription. PLoS One. 2008; 3:e3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tats A., Remm M., Tenson T.. Highly expressed proteins have an increased frequency of alanine in the second amino acid position. BMC Genomics. 2006; 7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ito Y., Chadani Y., Niwa T., Yamakawa A., Machida K., Imataka H., Taguchi H.. Nascent peptide-induced translation discontinuation in eukaryotes impacts biased amino acid usage in proteomes. Nat. Commun. 2022; 13:7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kyte J., Doolittle R.F.. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982; 157:105–132. [DOI] [PubMed] [Google Scholar]
- 55. Di Rienzo L., Miotto M., Bò L., Ruocco G., Raimondo D., Milanetti E.. Characterizing hydropathy of amino acid side chain in a protein environment by investigating the structural changes of water molecules network. Front. Mol. Biosci. 2021; 8:626837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bui P.T., Hoang T.X.. Hydrophobic and electrostatic interactions modulate protein escape at the ribosomal exit tunnel. Biophys. J. 2021; 120:4798–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Filbeck S., Cerullo F., Pfeffer S., Joazeiro C.A.P.. Ribosome-associated quality-control mechanisms from bacteria to humans. Mol. Cell. 2022; 82:1451–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hussain T., Llácer J.L., Fernández I.S., Munoz A., Martin-Marcos P., Savva C.G., Lorsch J.R., Hinnebusch A.G., Ramakrishnan V.. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell. 2014; 159:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kozak M. Influences of mRNA secondary structure on initiation by eukaryotic ribosomes. Proc. Natl Acad. Sci. USA. 1986; 83:2850–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakagawa S., Niimura Y., Gojobori T., Tanaka H., Miura K.. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res. 2008; 36:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ivanov I.P., Saba J.A., Fan C.-M., Wang J., Firth A.E., Cao C., Green R., Dever T.E.. Evolutionarily conserved inhibitory uORFs sensitize Hox mRNA translation to start codon selection stringency. Proc. Natl Acad. Sci. USA. 2022; 119:e2117226119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Signori E., Bagni C., Papa S., Primerano B., Rinaldi M., Amaldi F., Fazio V.M.. A somatic mutation in the 5′UTR of BRCA1 gene in sporadic breast cancer causes down-modulation of translation efficiency. Oncogene. 2001; 20:4596–4600. [DOI] [PubMed] [Google Scholar]
- 63. Murphy L.O., Blenis J.. MAPK signal specificity: the right place at the right time. Trends Biochem. Sci. 2006; 31:268–275. [DOI] [PubMed] [Google Scholar]
- 64. Adachi M., Fukuda M., Nishida E.. Nuclear export of MAP kinase (ERK) involves a MAP kinase kinase (MEK)-dependent active transport mechanism (published erratum appears in J Cell Biol 2000 May 1;149(3):754). J. Cell Biol. 2000; 148:849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Volmat V., Camps M., Arkinstall S., Pouyssegur J., Lenormand P.. The nucleus, a site for signal termination by sequestration and inactivation of p42/p44 MAP kinases. J. Cell Sci. 2001; 114:3433–3443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary data.