

The ATP8A2 (ATPase phospholipid transporting 8A2) gene, located in chromosome 13q12.13, belongs to a family of phospholipid transporting ATPases, which are involved in the transport of aminophospholipids. It is involved in lipid flipping, aiding in the asymmetry in membrane lipids, being highly expressed in brain, cerebellum, retina, and key for inner ear morphogenesis. Variants in this gene have been previously associated with cerebellar ataxia, mental retardation, and disequilibrium syndrome 4 (CAMRQ4) phenotype (OMIM: 615268). 1
An 8‐year‐old girl was referred to our service because of psychomotor delay and abnormal movements. Born to first‐degree consanguineous parents after an uneventful gestation, shortly after birth, global hypotonia and visual impairment were noticed. She did not acquire developmental milestones and was unable to sit or produce meaningful speech. Family history was unremarkable. The patient had no history of hearing loss or epileptic events.
On neurological examination, she had no expressive or receptive language; misaligned teeth, bilateral ptosis, ophthalmoplegia (Fig. 1) (characterized mainly by horizontal gaze impairment), and bilateral optic disc pallor were of note. Visual contact was absent, and pupils reacted poorly to light. She displayed hypotonia and muscular atrophy involving neck, trunk, and four limbs. Continuous hyperkinetic movements, characterized as chorea, at times choreodystonic, affected her neck, upper, and lower limbs (Video 1). Screening for inborn errors of metabolism and electroneuromyography performed at the age of one were unremarkable. Brain magnetic resonance imaging did not show significant abnormalities.
FIG. 1.
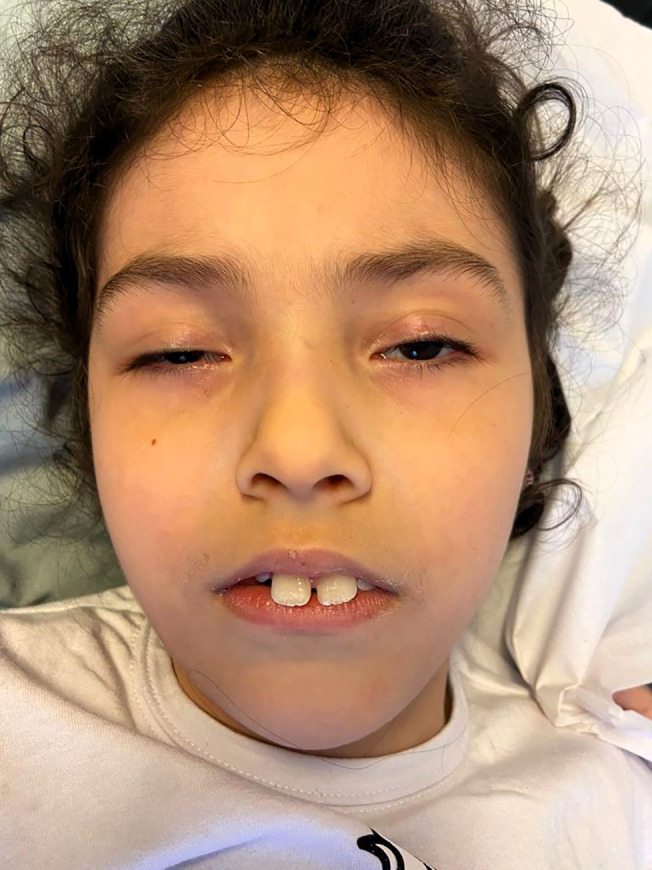
Bilateral semi‐ptosis and teeth misalignment.
Video 1.
Choreo‐dystonia and hypotonia.
A whole exome sequencing (WES) was performed, disclosing a homozygous frameshift variant, NM_016529.6(ATP8A2):c.1761dup;p.(Arg588fs), on the ATP8A2 gene. This variant causes a premature stop codon, and the resulting protein is predicted to undergo nonsense mediated decay (NMD). Therefore, a loss‐of‐function effect is expected. It is rare on population databases (gnomAD allele frequency of 0.000029). An entry on ClinVar was registered (Variation ID: 522073); however, no reports on literature of this variant were found. Therefore, it has been classified as pathogenic. 2
The first description of CAMRQ4 phenotype—notable for quadrupedal gait—was published in 2006 3 and the identification of the causative gene in 2013. 1 Several new cases and numerous pathogenic ATP8A2 variants have been reported with varying degrees of cerebellar ataxia and developmental delay severity and even isolated mild ataxia. 4 Underlying molecular mechanisms are heterogeneous and range from point mutations 4 to chromosome translocations. 5
Loss‐of‐function biallelic variants were associated with a severer phenotype characterized, in 2016, by encephalopathy, intellectual disability, central hypotonia, psychomotor delay, chorea, and optic atrophy, akin to mitochondrial diseases, without broader systemic involvement. A subsequent study from 2018, described 11 individuals with ATP8A2 mutations, figuring hearing loss and cerebellar ataxia with or without cerebellar atrophy in some patients. 1
Heidari et al 4 proposed an ATP8A2 phenotype expansion to include dystonia, below average head circumference, mild optic atrophy, developmental delay, and teeth abnormalities.
This paper describes a new pathogenic variant in the ATP8A2 gene, disclosing a complex phenotype, which can mimicry a mitochondrial disease minus multisystemic impairment. In patients presenting with developmental delay, optic atrophy, bilateral ptosis, movement disorders, and cerebellar ataxia, ATP8A2 mutations must feature in differential diagnosis.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution. (2) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
L.A.C.: 1A, 1B, 1C, 2A.
V.L.B.: 1A, 1B, 1C, 2A, 2B.
T.Y.T.S.: 1B, 1C, 2A, 2B.
J.L.P.: 1A, 1B, 2A.
O.G.P.B.: 1A, 1B, 1C, 2A, 2B.
Disclosures
Ethical Compliance Statement: Written consent for publication of videos, images, and case report was obtained from the patient's family. Patient signed a standard institutional consent form, which includes publication in medical journals. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: No specific funding was received for this work and the authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: The authors declare that there are no additional disclosures to report.
Supporting information
Text S1. Variant classification – According to ACMG 2015.
Relevant disclosures and conflict of interest are listed at the end of this article.
References
- 1. McMillan HJ, Telegrafi A, Singleton A, et al. Recessive mutations in ATP8A2 cause severe hypotonia, cognitive impairment, hyperkinetic movement disorders and progressive optic atrophy. Orphanet J Rare Dis 2018;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uner T. A new syndrome with quadrupedal gait, primitive speech, and severe mental retardation as a live model for human evolution. Int J Neurosci 2006;116(3):361–369. [DOI] [PubMed] [Google Scholar]
- 4. Heidari E, Harrison AN, Jafarinia E, Almadani N, Molday RS, Garshasbi M. Novel variants in critical domains of ATP8A2 and expansion of clinical spectrum. Hum Mutat 2021;42:491–497. [DOI] [PubMed] [Google Scholar]
- 5. Cacciagli P, Haddad MR, Mignon‐Ravix C, et al. Disruption of the ATP8A2 gene in a patient with a t(10;13) de novo balanced translocation and a severe neurological phenotype. Eur J Hum Genet 2010;18:1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Text S1. Variant classification – According to ACMG 2015.