

Significance
Fungal pathogens cause severe diseases in humans, animals, and plants. To develop new defense strategies, we need to understand how infection is precisely regulated at the DNA and RNA levels. However, knowledge on RNA regulation in fungal pathogens is currently scarce. Here, we adopt a powerful in vivo RNA labeling technique for fungi. Thereby, we uncover how a key RNA-binding protein (RBP) orchestrates the polar growth of infectious hyphae by determining the exact stability of mRNAs encoding regulators of membrane trafficking. Thus, we disclose a different regulatory concept for infection: a single RBP determines the precise timing of expression for numerous regulatory proteins at the level of mRNA stability. This opens up the opportunity to use RBPs as novel fungicide targets.
Keywords: GTPase, membrane trafficking, mRNA stability, polar growth, fungal pathogen
Abstract
Fungal pathogens depend on sophisticated gene expression programs for successful infection. A crucial component is RNA regulation mediated by RNA-binding proteins (RBPs). However, little is known about the spatiotemporal RNA control mechanisms during fungal pathogenicity. Here, we discover that the RBP Khd4 defines a distinct mRNA regulon to orchestrate membrane trafficking during pathogenic development of Ustilago maydis. By establishing hyperTRIBE for fungal RBPs, we generated a comprehensive transcriptome-wide map of Khd4 interactions in vivo. We identify a defined set of target mRNAs enriched for regulatory proteins involved, e.g., in GTPase signaling. Khd4 controls the stability of target mRNAs via its cognate regulatory element AUACCC present in their 3′ untranslated regions. Studying individual examples reveals a unique link between Khd4 and vacuole maturation. Thus, we uncover a distinct role for an RNA stability factor defining a specific mRNA regulon for membrane trafficking during pathogenicity.
Fungal pathogens are ubiquitous and pose a serious threat to public health, agriculture, and wildlife. Fungal infections are estimated to cause more than 1.5 million deaths per year (1) and account for a 30% reduction in crop yield worldwide (2). A widespread virulence strategy of these pathogens is their capacity for morphological plasticity, i.e., the alternation between yeast and hyphal growth forms (3, 4). In particular, hyphae formation is a criterion for successful host invasion in many pathogenic fungi (5). For example, the human pathogen Candida albicans requires the morphological transition from yeast-like to hyphal growth to invade host tissue and selectively escape the host immune system (6, 7). In the plant pathogen Ustilago maydis, the switch from yeast to hyphal form is tightly linked to mating and is essential for plant entry and infection (8).
The polarized growth of fungal hyphae is intimately associated with intracellular membrane trafficking. Vesicle transport, for example, mediates the delivery of new plasma membranes and cell wall material to growth poles and therefore determines the shape and growth direction of hyphal cells (9). Equally important is the recycling of excess plasma membrane components via endocytosis. Furthermore, the balance between endocytosis and exocytosis forms the basis for dynamic hyphal growth (10). The transport of cargo between different cellular compartments is robustly controlled by distinct regulatory proteins. In particular, small GTPases such as Rab, Rho, Arf, and Arl, and their interaction partners serve as master regulators of vesicle trafficking (11).
The link between membrane trafficking and infectious hyphal growth is well-studied in the plant pathogen U. maydis causing corn smut disease (12). To maintain its morphology and pathogenicity, this phytopathogen inordinately depends on endocytosis mediated by the bidirectional shuttling of early endosomes (13). Besides their role in endocytosis, the shuttling early endosomes also offer a transportation platform for organelles such as peroxisomes as well as mRNAs and associated polysomes throughout the hyphal cells (12, 14). For such a complex process to be coordinated, robust spatiotemporal control of the associated proteins is essential. At the posttranscriptional level, it was found that the RNA-binding protein (RBP) Rrm4 transports mRNAs of morphological regulators, such as all four septin-encoding mRNAs, along fungal hyphae, determining the local translation and the spatial distribution of the translation products (15, 16).
Previously, we discovered that a second RBP, Khd4, is crucial for morphogenesis and pathogenesis in U. maydis. The loss of Khd4 resulted in cytokinesis defects in yeast-like cells, aberrant morphology of hyphae, and impaired virulence (Fig. 1A and SI Appendix, Fig. S1A; 17). Khd4 is a multi-KH domain-containing RBP, with KH domains 3 and 4 being crucial for RNA binding. Based on binding studies in the heterologous yeast three-hybrid assay, Khd4 recognizes the RNA motif AUACCC (A-Adenine, U-Uracil, C-Cytosine), and its KH domains 3 and 4 are necessary and sufficient for binding. Mutations in KH domains 3 and 4 severely impact the morphology and virulence of U. maydis, suggesting that the RNA binding potential of Khd4 is responsible for its function (17). However, it is currently unclear how Khd4 tunes the morphological transition and pathogenic development. Here, we perform a transcriptome-wide identification of in vivo target mRNAs in infectious hyphae. We demonstrate that Khd4 regulates the expression of a distinct set of regulatory proteins involved in membrane trafficking and reveal a unique link to posttranscriptional control of vacuole biogenesis.
Fig. 1.
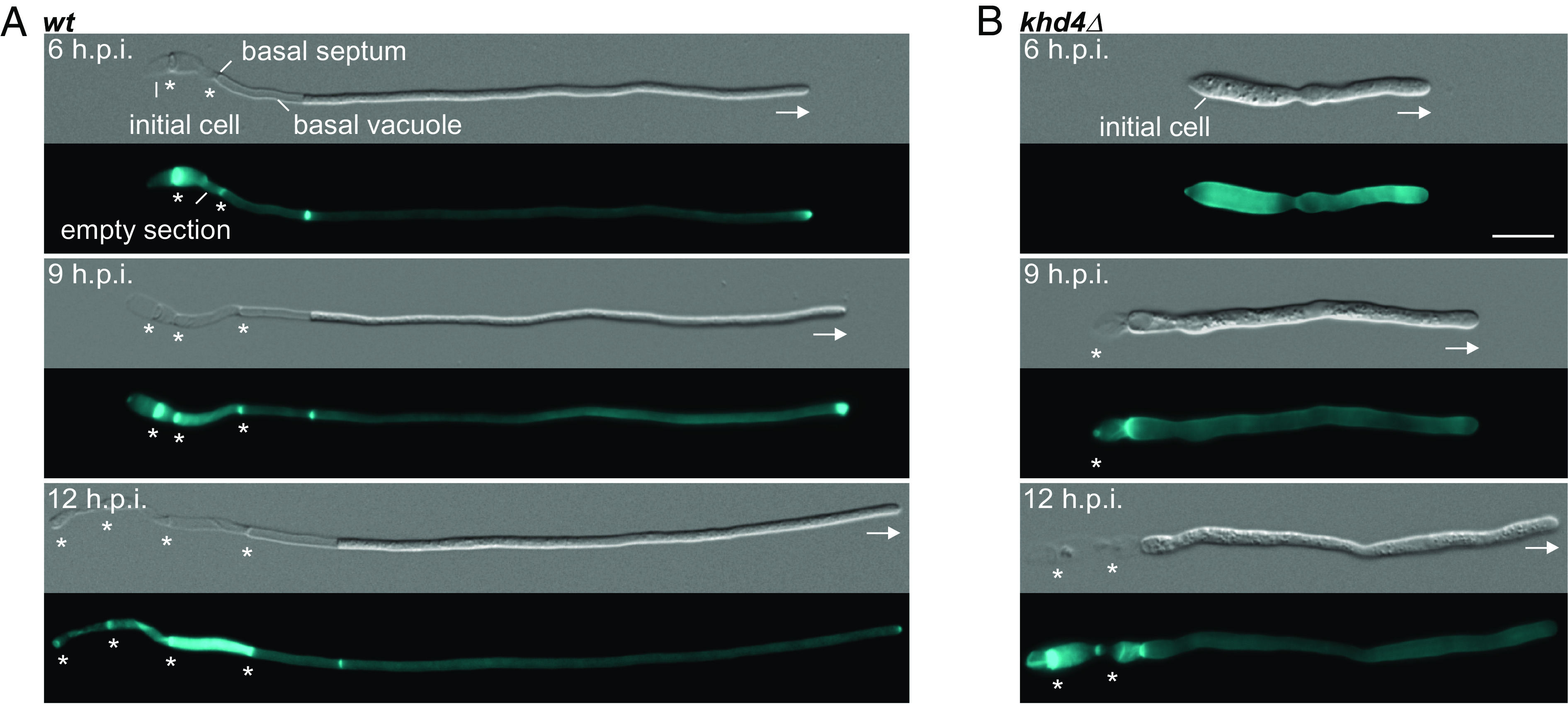
Khd4 is important for hyphal growth. Differential interference contrast (DIC, Top panels) and Calcofluor white staining (Bottom panels; CFW, 18) of (A) wild-type (wt) and (B) khd4Δ hypha grown for 6, 9, and 12 hours post induction (h.p.i.). All strains are derivatives of AB33. Basal septa and growth directions are marked by asterisks and arrows, respectively (scale bar: 10 µm.).
Results
Loss of Khd4 Causes Defects in Polar Growth of Infectious Hyphae.
To study the role of Khd4 during pathogenic development, we focused on the formation of infectious hyphae. For this, we conducted a time-course analysis to determine the appropriate induction time needed for hyphae establishment. As the genetic background, we used the laboratory strain AB33, which expresses the heteromeric master transcription factor (bE/bW) for hyphal formation under the control of the nitrate-inducible Pnar1 promoter. By switching the nitrogen source, hyphal growth can be induced synchronously in a highly reproducible manner (Fig. 1A; 19). The resulting hyphae grow unipolarly by tip expansion up to a defined length of about 100 µm. This maximal length is maintained over time by the insertion of regularly spaced septa, generating characteristic cytoplasm-free sections consisting of cell wall remnants at the basal pole (Fig. 1A; 16, 20). Wildtype hyphae with basal septa were already evident at six hours post induction (6 h.p.i.; Fig. 1A). In contrast, the deletion of khd4 resulted in delayed hyphal development (Fig. 1 A and B and SI Appendix, Fig. S1 B–E). Compared to wildtype hyphae, the khd4Δ hyphae were shorter, thicker, and aggregated at 6 h.p.i. (Fig. 1 A and B and SI Appendix, Fig. S1 A and F). However, prolonging the induction time caused a gradual recovery of hyphal length, reduced the diameter, and increased the basal septa insertion in khd4Δ cells (Fig. 1B and SI Appendix, Fig. S1 B–E). A longer period of hyphal induction also diminished hyphal aggregation (SI Appendix, Fig. S1F). Although khd4∆ hyphae can attain comparable lengths to wildtype, it is known that the virulence of khd4∆ strains is severely reduced, highlighting the critical role of Khd4-mediated RNA regulation also during later stages of pathogenic development (17). In essence, the loss of RBP Khd4 disturbs the morphological dynamics during the formation of infectious hyphae. Based on these results, we determined 9 h.p.i. as the optimal time point for further analysis.
Establishing HyperTRIBE for Fungal RBPs.
Mutations in the RNA-binding domains mimic the loss-of-function phenotype of khd4 deletion (17). Identifying mRNA targets of Khd4 is therefore crucial to dissect its function. Hence, we adapted hyperTRIBE, a technique well suited for the investigation of proteins with high molecular weight or low expression that are difficult to purify (21–23). HyperTRIBE exploits the RNA editing activity of the catalytic domain of ADAR (Adenosine Deaminase Acting on RNA) from Drosophila melanogaster carrying the mutation E488Q to increase editing (24; designated Ada). We fused the codon-optimized catalytic Ada domain with the green fluorescent protein (enhanced version, designated Gfp, Clontech) to the C terminus of the RBP of interest (Fig. 2 A and B). Expression of the fusion protein is controlled by the arabinose-inducible Pcrg1 promoter that is repressed by glucose (Fig. 2 A and B; 19, 25). The resulting construct was inserted ectopically at the ips locus (26) in the deletion mutant of the RBP of interest. To assess RBP–Ada–Gfp functionality, we compared it to a control strain expressing RBP–Gfp (Fig. 2B).
Fig. 2.
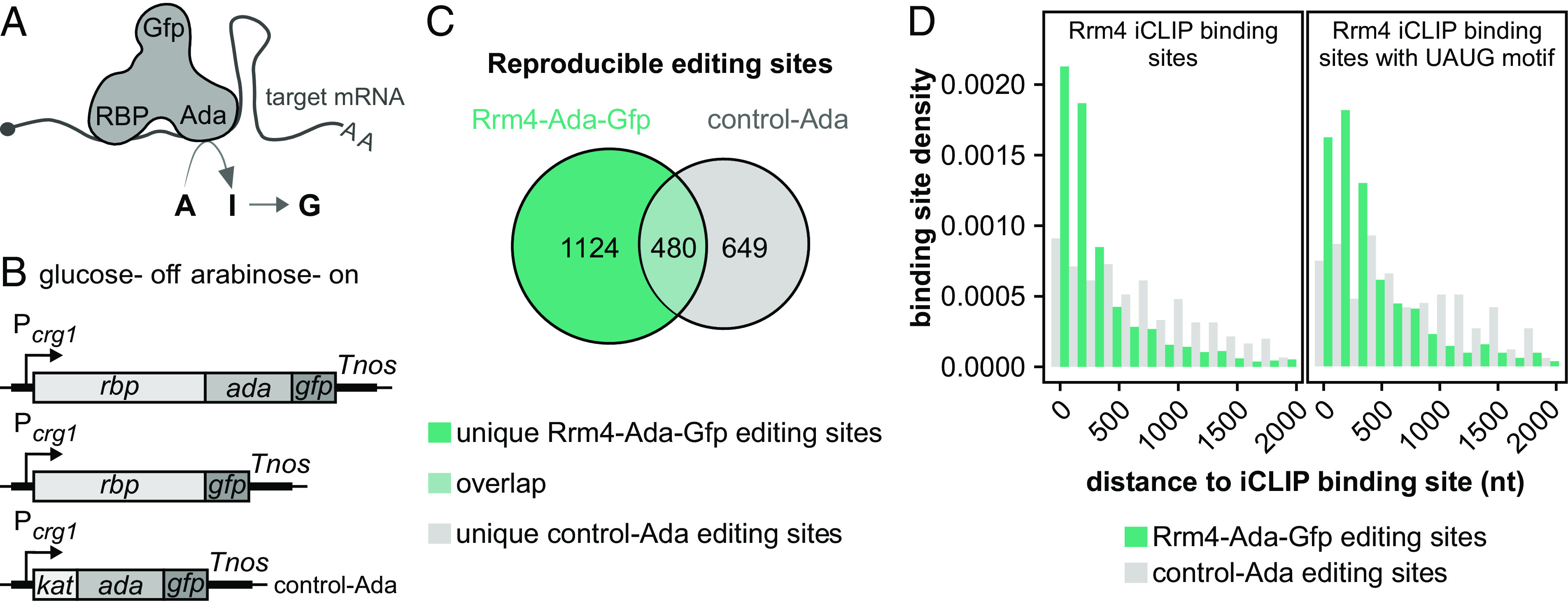
HyperTRIBE identifies bona fide targets of Rrm4. (A) Schematics of the hyperTRIBE technique. The binding of the RBP–Ada–Gfp fusion protein to target mRNA results in the editing of adjacent adenosine (A) to inosine (I) which is converted to guanine (G; Top) (Ada—hyperactive version of Drosophila ADAR catalytic domain; Gfp—green fluorescent protein; AA—poly(A) tail, 5′ cap structure is depicted by a gray circle). (B) HyperTRIBE constructs with arabinose inducible Pcrg1 promoter (on), which is repressed by glucose (off). Fusion proteins of RBP, Ada, Gfp, and/or Kat (Kat—mKate2, red fluorescent protein; Tnos—transcription terminator). (C) Overlap of reproducible editing events from Rrm4–Ada–Gfp (sea green) and control–Ada (gray). The number of reproducible editing events within each category is stated inside. (D) Histogram displaying the distance of editing events to the nearest Rrm4 iCLIP binding sites (Left) or the nearest UAUG-containing Rrm4 iCLIP binding sites (Right) in edited transcripts specific to Rrm4–Ada–Gfp (sea green) compared to control–Ada (gray).
For a proof-of-principle study, we chose Rrm4, as its cognate mRNA targets have been extensively investigated at the transcriptome-wide level using iCLIP (individual-nucleotide resolution UV Cross-Linking and ImmunoPrecipitation; 27). A detailed description of the experimental setup is given in the SI Appendix, Figs. S2–S4 and Text. In brief, the expression of the Rrm4–Ada–Gfp protein by arabinose induction rescued the rrm4∆ phenotype (SI Appendix, Fig. S2 A–F), indicating that the fusion protein is functional. As a control for background editing, we used the strain expressing Ada–Gfp protein with N-terminal mKate2 fusion (a monomeric form of the red fluorescent protein, designated Kat; 28) in the wildtype background (control–Ada; Fig. 2B). Control–Ada was expressed at higher levels than Rrm4–Ada–Gfp, localized uniformly throughout hyphae and did not interfere with hyphal growth (SI Appendix, Figs. S2D and S3 A–C). To avoid potential artifacts caused by overexpression of RBPs, we did not attempt to express the RBP–Ada–Gfp levels similar to control–Ada.
We sequenced mRNAs of hyphae expressing the hyperTRIBE constructs that were induced under promoter-on conditions (arabinose) for 6 hours. The A-to-G editing events in the sequencing data were detected using the published hyperTRIBE pipeline (23). Editing events were deemed reproducible when detected in two independent experimental replicates (SI Appendix, Fig. S4A). Notably, Rrm4–Ada–Gfp expression led to twice as many reproducible editing sites as control–Ada (Fig. 2C and SI Appendix, Fig. S4 A–D). In our system, the control–Ada construct harboring only the catalytic domain of ADAR shows substantial background activity. This is different from the initial reports on mammalian and Drosophila hyperTRIBE systems but fits with observations for the more recently established plant hyperTRIBE system (26, 29). Importantly, the increased background activity of control–Ada can be clearly separated from the editing events for the RBP of interest (see below).
Comparing the hyperTRIBE data with Rrm4-bound transcripts from iCLIP data showed significant overlap, whereby, ~84% of the transcripts with unique Rrm4–Ada–Gfp editing sites had also been detected as Rrm4-bound in the published iCLIP data, demonstrating a higher enrichment compared to the control–Ada (SI Appendix, Fig. S4 E–G).
Interestingly, compared to control–Ada editing sites, the majority of editing sites on mRNAs unique to Rrm4–Ada–Gfp were predominantly located near the actual Rrm4 binding sites [Fig. 2D; median distance = 159 nucleotides (nt)]. Additionally, only the Rrm4–Ada–Gfp editing sites demonstrated enrichment in proximity to the UAUG-containing binding sites, a motif recognized by the third RRM domain of Rrm4 (Fig. 2D; median distance = 252 nt). In summary, we conclude that the specificity of Ada in hyperTRIBE is largely determined by the attached RBP. Our pilot study has thus laid the foundation for using hyperTRIBE for in vivo detection of Khd4 target mRNAs.
Khd4 Interacts with mRNAs Enriched for Its Binding Motif AUACCC.
Following the same experimental strategy (Fig. 2B and SI Appendix, Fig. S5 A and B), we first verified that Khd4–Ada–Gfp rescued the mutant khd4∆ phenotype with respect to hyphal morphology, length, and thickness (SI Appendix, Fig. S5 C–E). In line with Rrm4–Ada–Gfp, the levels of Khd4–Ada–Gfp were also lower compared to control–Ada (SI Appendix, Fig. S5 F and G). To assess the specificity of Khd4–Ada–Gfp editing, we designed a reporter mRNA, containing six tandem copies of the previously identified Khd4 binding motif AUACCC (17) in the 3′ untranslated region (UTR) of the Kat coding sequence. This synthetic mRNA was constitutively expressed using the Ptef promoter (SI Appendix, Fig. S5H). We harvested hyphae (6 h.p.i.) grown in the promoter-on condition and sequenced kat_(auaccc)6 mRNA using transcript-specific primers (SI Appendix, Materials and Methods). Strikingly, we observed two reproducible A-to-G editing events in the reporter mRNA only after the expression of Khd4–Ada–Gfp (SI Appendix, Fig. S5H). Thus, the Khd4–Ada–Gfp fusion protein is fully functional, and the Ada domain can edit Khd4 mRNA targets with high specificity. Notably, this experiment also demonstrated for the first time that the AUACCC motif indeed serves as an in vivo Khd4 binding motif in U. maydis.
To detect Khd4 targets, reproducible editing events were identified from two replicate experiments for Khd4–Ada–Gfp (Fig. 3A and SI Appendix, Fig. S6 A and B). In contrast to Rrm4–Ada–Gfp, editing events for Khd4–Ada–Gfp were substantially lower; however, the reproducibility was higher than for those obtained with Rrm4–Ada–Gfp or control–Ada (SI Appendix, Figs. S4 C and D and S6 B–D). Comparing reproducible editing events from Khd4–Ada–Gfp and control–Ada showed that the majority of editing sites were unique to either Khd4–Ada–Gfp (n = 269) or the control–Ada (n = 1,021), with 108 editing sites overlapping between both sets (n = 108; Fig. 3B). Importantly, de novo motif enrichment analysis using XSTREME identified only the motif AUACCC near Khd4–Ada–Gfp editing sites as highly enriched (Fig. 3C). About 81% of the transcripts with unique Khd4–Ada–Gfp editing events contained the AUACCC motif (SI Appendix, Fig. S7A). In contrast, the control motifs, including the AGAUCU motif, a point-mutated version of AUACCC that demonstrated no binding by Khd4 in the yeast three-hybrid assay (17), the antisense motif GGGUAU, and the scrambled sequence ACACUC were not significantly enriched compared to the entire transcriptome (SI Appendix, Fig. S7A). Moreover, neither of these motifs showed specific enrichment in transcripts unique to the control–Ada (SI Appendix, Figs. S6E and S7A). Thus, Khd4–Ada–Gfp recognizes mRNAs via the AUACCC motif at the transcriptome-wide level in vivo.
Fig. 3.
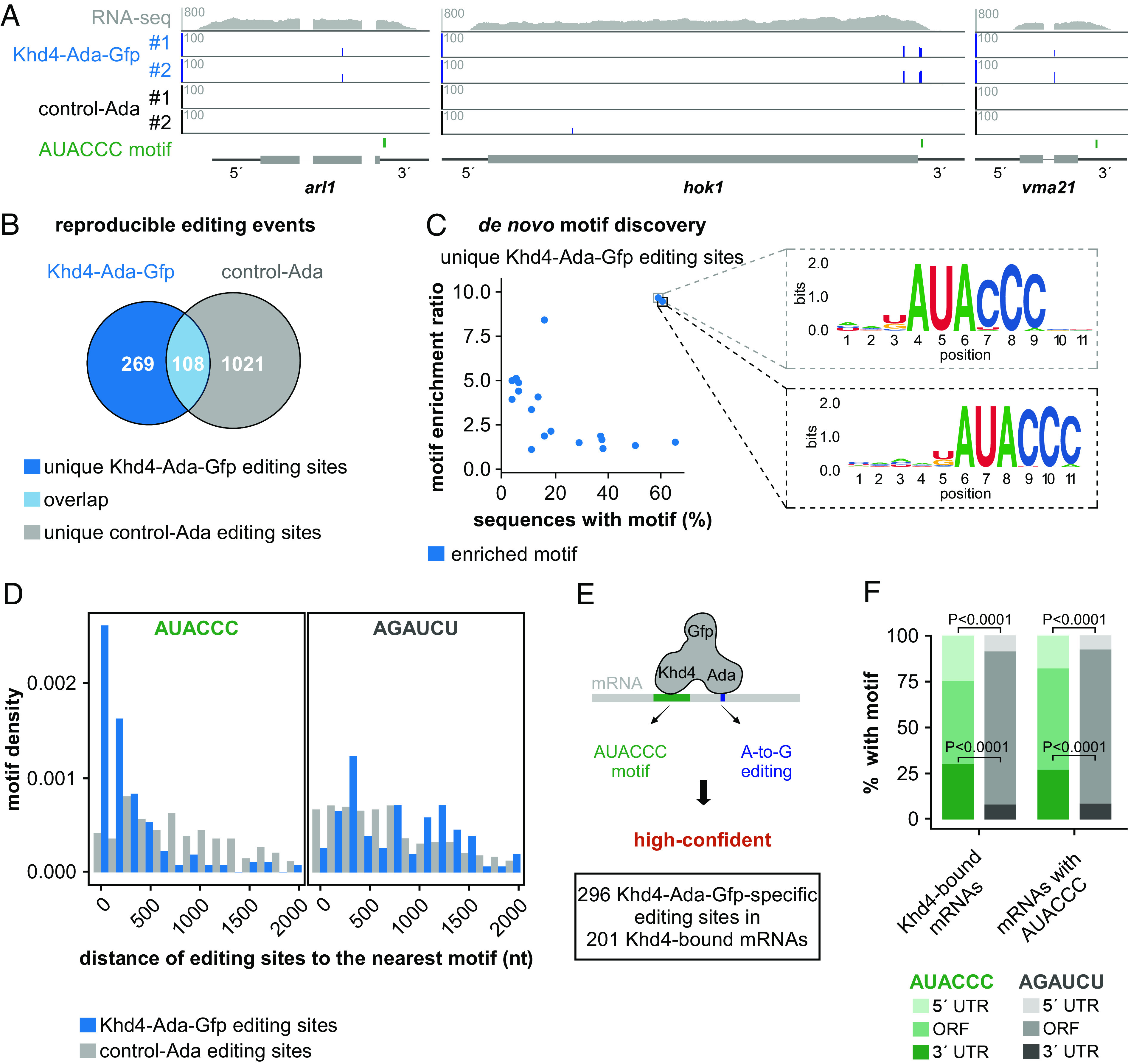
HyperTRIBE identifies highly specific targets of Khd4. (A) HyperTRIBE data of Khd4 on selected target transcripts (arl1, UMAG_10313; hok1, UMAG_11790; vma21, UMAG_11418). Tracks showing RNA-seq read coverage (Top) followed by editing tracks of Khd4–Ada–Gfp and control–Ada (#1-replicate 1, #2-replicate 2). The bottom track displays the position of the AUACCC motif. The gene model with the exon/intron structure below was extended by 300 nt on either side to include 5′ and 3′ UTRs (5′ and 3′, respectively). (B) Overlap of reproducible editing sites from the Khd4–Ada–Gfp (blue) and control–Ada (gray). The overlapping region is depicted in light blue color. The numbers of reproducible editing sites in each category are indicated. (C) De novo motif discovery analysis on sequences carrying unique Khd4–Ada–Gfp editing sites. Scatter plot compares the relative enrichment ratio of enriched motifs (blue; relative to the background control) with the percentage of each motif in the tested sequences. The enlarged region (indicated by dotted box) presents the sequence logo of motifs exhibiting both high enrichment and overrepresentation in Khd4–Ada–Gfp-edited sequences. For motif enrichment, a 501-nt window centered on Khd4–Ada–Gfp editing sites was used. (D) Histogram showing the distribution of editing sites from the nearest AUACCC (Left) or AGAUCU (Right; a point mutated version of AUACCC) motifs in transcripts specific to Khd4–Ada–Gfp (blue) compared to control–Ada (gray). (E). Schematic depiction of high-confident, Khd4-bound mRNA selection using hyperTRIBE. High-confident target mRNAs are those that contain Khd4–Ada–Gfp-specific editing sites (blue; A-to-G editing) and at least one AUACCC motif (green). (F) Stacked bar graph describing the percentage of AUACCC (green) or AGAUCU (gray) motifs per transcripts region (5′ UTR, ORF and 3′ UTR) for Khd4-bound mRNAs and all AUACCC-containing mRNAs. Statistical analysis was carried out using Fisher’s exact test.
Similar to Rrm4–Ada–Gfp, most of the Khd4–Ada–Gfp-specific editing sites were found within 500 nt from the nearest AUACCC motif (median distance = 120 nt) and progressively declined with increasing distance (Fig. 3D). In contrast, the background editing sites from control–Ada were not enriched toward the closest AUACCC motif (median distance = 863 nt) nor did the control motifs (AGAUCU, GGGUAU, and ACACUC) show any spatial enrichment toward either type of editing sites (Fig. 3D and SI Appendix, Fig. S7 B–D).
The transcripts overlapping between Khd4–Ada–Gfp and control–Ada were nonetheless significantly enriched for the AUACCC motif (n = 60 mRNAs with 66 editing sites), indicating Khd4-dependent editing, and were hence included as Khd4–Ada–Gfp targets in order to avoid false negatives (Fig. 3E and SI Appendix, Fig. S7A). Based on these results, we defined a high-confident set of 201 Khd4-bound mRNA targets that harbored Khd4–Ada–Gfp editing sites (n = 296) and contained at least one AUACCC motif (Fig. 3E). Transcripts carrying Khd4–Ada–Gfp-specific editing sites but lacking the AUACCC motif (n = 74 mRNAs with 81 editing sites) did not show enrichment for other motifs and were not investigated further (SI Appendix, Fig. S8 A–C).
Examining the positional distribution of the AUACCC motifs in Khd4-bound mRNAs and other AUACCC-containing transcripts revealed a predominant occurrence in the 5′ and 3′ UTRs (Fig. 3F). The strong enrichment of AUACCC motifs in these regions was even more apparent when compared to the control motif AGAUCU, which was almost exclusively found in the open reading frames (ORFs), reflecting that ORFs are generally much longer than UTRs in U. maydis (Fig. 3F). Within the 3′ UTR, the AUACCC motifs preferentially occurred toward the beginning (SI Appendix, Fig. S8D). Hence, our data suggest that AUACCC motifs present in the 3′ UTR constitute the major binding sites for Khd4 in the identified target mRNAs.
To evaluate the biological roles of Khd4-bound mRNAs, we performed gene ontology (GO) enrichment analysis using the R package gProfiler2 (Ensembl annotation; 30). We observed a significant overrepresentation of processes related to small GTPase-mediated signal transduction processes, such as Ras and Rho protein signaling and GTPase regulator activity (SI Appendix, Figs. S8E and S9A). The Khd4-bound mRNAs associated with these GO terms encode well-studied membrane-trafficking regulators such as the small GTPase Arl1 that participates in the trans-Golgi network and secretion, the Ras GTPase activating protein Sar1, and the Rho guanine nucleotide exchange factor (GEF) Rom2 that functions in actin cytoskeleton regulation (SI Appendix, Fig. S9A; 31–34). In addition, by manually analyzing the Khd4-bound mRNAs for encoded functions and subcellular localization, we found that a distinct subset of target mRNAs was involved in the endomembrane system such as endoplasmic reticulum, Golgi apparatus, and vacuoles, with the most carrying the AUACCC motif in their 3′ UTR (SI Appendix, Fig. S9B). Hence, Khd4 might be involved in the posttranscriptional control of membrane-trafficking regulators. Khd4 appears to coordinate the regulation of these functionally related mRNAs into an mRNA regulon, which enables robust and dynamic control over membrane trafficking. In summary, we find that Khd4–Ada–Gfp interacts specifically with a distinct set of target mRNAs in vivo via the AUACCC binding motif, preferentially in the 3′ UTR. Intriguingly, the identified target mRNAs frequently encode regulatory proteins involved in membrane trafficking.
Loss of Khd4 Increases the Abundance of mRNAs with AUACCC Motif in Their 3′ UTR.
Our previous microarray analysis in yeast cells showed that the AUACCC motif was enriched in the 3′ UTR of the transcripts that exhibited an increase in mRNA abundance in the absence of Khd4 (17). This suggests an impact on mRNA stability. Since our hyperTRIBE data showed that Khd4 binds only a subset of AUACCC-containing mRNAs in hyphae, we tested for a differential mRNA abundance between wildtype and khd4∆ hyphae using RNA sequencing (RNA-seq; khd4Δ hyphae vs. wt hyphae; 9 h.p.i. see Fig. 1 A and B and SI Appendix, Fig. S1 B–F).
Differential expression analysis revealed that about one-fifth of all transcripts showed significant changes in mRNA abundance following khd4 deletion (1,273 out of 6,765 protein-coding genes; >1.5 fold change; P < 0.05; Benjamini–Hochberg correction, >10 average normalized reads), most of which had increased mRNA levels (Fig. 4A). Loss of khd4 caused a significant increase in the expression levels of Khd4-bound mRNA targets compared to all transcripts, confirming that the hyperTRIBE-identified targets are indeed regulated by Khd4. Of note, this trend could not be observed when analyzing all AUACCC-containing mRNAs (Fig. 4 B and C), underlining that the knowledge of binding motifs alone is not sufficient to dissect the regulatory function of Khd4.
Fig. 4.
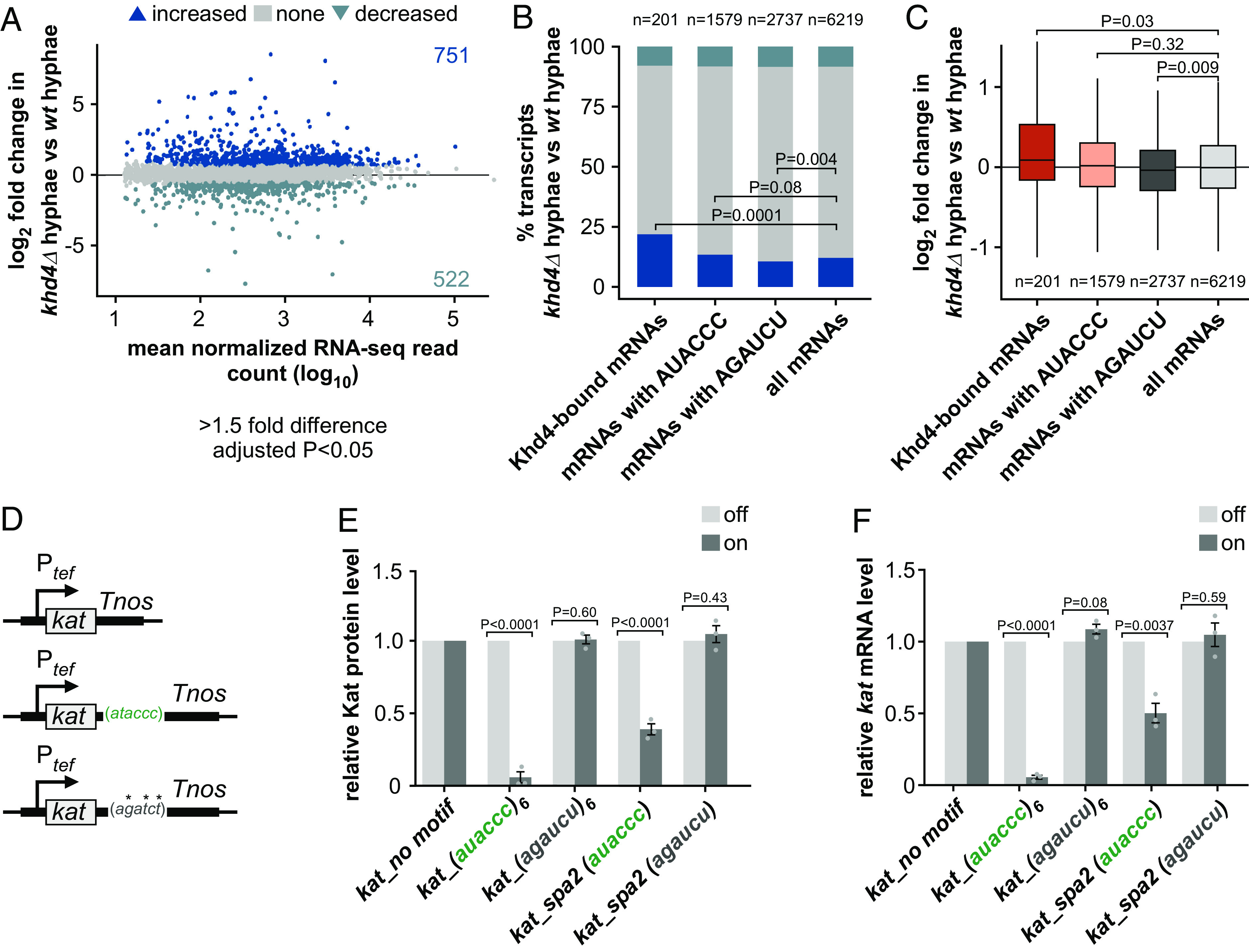
khd4 deletion increases the abundance of transcripts with 3′ UTR AUACCC. (A) Difference in mRNA abundance (khd4Δ hyphae vs. wt hyphae; wt-wildtype) in hyphal cells following khd4 deletion. Shown is the log2-transformed fold change plotted against the log10-transformed mean normalized RNA-seq read counts. Blue dots represent values of transcripts with a significant change in their mRNA abundance (>1.5-fold change, P < 0.05, Benjamini–Hochberg correction, >10 average normalized reads), and gray dots represent remaining transcripts. Numbers inside the plots indicate genes with significantly increased or decreased mRNA abundance. (B) Stacked bar graph of the percentage of genes with differential mRNA abundance in hyphal cells after khd4 deletion for Khd4-bound mRNAs, all mRNAs with AUACCC motif, all mRNAs with AGAUCU motif, and all expressed mRNAs in hyphae (>10 average normalized reads). The dark and light blue bars represent the percentage of transcripts that are significantly increased and decreased in their mRNA abundance, respectively (>1.5-fold change, P < 0.05, Benjamini–Hochberg correction). The percentage of remaining transcripts is depicted by a gray bar. Statistical analysis of the percentage of transcripts with increased expression levels was performed using Fischer’s exact test. The number of transcripts in each set is represented at the Top. (C) Box plot of changes in mRNA expression (log2-transformed fold change, khd4Δ hyphae vs. wt hyphae) for Khd4-bound mRNAs (red), all mRNAs with AUACCC motif (salmon), mRNAs with AGAUCU motifs (dark gray), and all expressed mRNAs (light gray) in hyphal cells. Statistical significance between the mean mRNA expression level (log2-transformed fold change) was calculated using the Student’s t test. The number of transcripts in each set is given at the Bottom. (D) Schematics of reporter constructs for testing the binding effect of Khd4 on the 3′ UTR AUACCC. The constitutively active Ptef promoter mediates the expression of the Kat fluorescent reporter in the genetic background of arabinose-inducible Khd4–Gfp. Reporter constructs carrying no binding motif (Top), the AUACCC binding motif (center), or the AGAUCU binding motif (Bottom) in their 3′ UTR are depicted chronologically. Tnos—transcription terminator. *—mutated nucleotides. (E) Shown are the relative fluorescence level of Kat proteins in on (dark gray; Khd4-induced) compared to off (light gray; Khd4-repressed) conditions in strains expressing kat mRNA tethered to different 3′ UTR regions (n = 3 independent experiments; error bars: SEM; auaccc6 or agaucu6—3′ UTR consisting of six repeats of the respective motifs; kat_spa2 (auaccc)—36 nt-long endogenous 3′ UTR sequence of spa2 gene that contains a single AUACCC motif; kat_spa2 (agaucu)—36 nt-long endogenous 3′ UTR sequence of spa2 gene with mutated AUACCC motif). (F) The relative kat transcript level in on conditions (dark gray) compared to the transcript levels under off conditions (light gray), measured via RT-qPCR. Statistical significance between on and off conditions in E and F was calculated using multiple unpaired Student’s t tests. (n = 3 independent experiments; error bars: SEM).
Interestingly, for the Khd4-bound mRNA targets as well as for all AUACCC-containing mRNAs with increased mRNA levels in khd4∆ hyphae, the AUACCC motif was primarily enriched in their 3′ UTR. (SI Appendix, Fig. S10A). This indicates that AUACCC located in the 3′ UTR affects mRNA stability. Essentially, these results strengthen our hypothesis that the interaction of Khd4 with the 3′ UTR AUACCC determines mRNA stability.
AUACCC in the 3′ UTR Functions as a Khd4-Dependent mRNA Stability Element.
To test whether Khd4 binding causes increased mRNA turnover, we applied our Kat reporter system to quantify the regulatory potential of AUACCC motifs in the 3′ UTR. We inserted either six tandem repeats of AUACCC or an endogenous sequence of equal length carrying a single AUACCC motif from the 3′ UTR of spa2 mRNA (UMAG_04468), encoding a polarisome protein, into the 3′ UTR of the kat ORF (Materials and methods; 35, 36). The spa2 3′ UTR with mutated binding motif, AGAUCU, and the Kat construct without any motifs served as controls (Fig. 4D). All constructs were generated in the strain expressing arabinose-inducible Khd4–Gfp as mentioned before. The expression of the reporter constructs was controlled by the constitutively active Ptef promoter to exclude differences in mRNA amounts due to transcriptional regulation.
Measuring the fluorescence intensity of Kat under the promoter-on condition revealed that the presence of a single AUACCC binding motif in the 3′ UTR was sufficient to significantly reduce the amount of kat reporter mRNA. Concurrently, turning off the Khd4 expression rescued the Kat fluorescence level (Fig. 4E and SI Appendix, Fig. S10 B and C), validating that the regulatory potential of the AUACCC motif is dependent on Khd4. Indeed, quantifying the reporter transcript level manifested the same expression pattern, implying that the decrease in mKate2 fluorescence is caused at the mRNA level by Khd4 binding to the 3′ UTR (Fig. 4F). At both protein and mRNA levels, the presence of six AUACCC repeats in the 3′ UTR caused a stronger reduction, indicating a dose-dependent response. This effect was lost when mutating the binding site (Fig. 4 E and F and SI Appendix, Fig. S10 B and C). Thus, the AUACCC motif in the 3′ UTR functions as a cis-regulatory mRNA instability element in a Khd4-dependent manner.
Khd4 Tightly Regulates the Expression of Distinct Targets Involved in Membrane Trafficking.
In addition to rapid clearance, intrinsically unstable mRNAs reach steady-state levels faster than stable mRNAs after transcriptional induction. Therefore, regulatory proteins are often encoded by such unstable mRNAs permitting faster response time (see below, 37). Since Khd4 targets encode regulatory proteins involved in membrane trafficking, we wondered whether the Khd4-mediated mRNA instability controls the amount of protein produced during hyphal morphogenesis. Such mRNA targets should be expressed upon hyphal induction and carry the regulatory element AUACCC in their 3′ UTR for increased turnover.
To identify hyphal-specific genes, we sequenced mRNAs from yeast cells and compared them with our hyphal transcriptome (9 h.p.i.). This analysis detected more than 3,500 genes with differential mRNA abundance as a response to hyphae formation (Fig. 5A). Studying the Khd4-bound mRNAs identified by hyperTRIBE revealed that 61% of them showed a significant differential mRNA abundance during morphogenesis (122 out of 201 transcripts), with most exhibiting increased mRNA amounts in hyphae compared to yeast cells, suggesting a hyphal-specific transcriptional induction (77 out of 122 transcripts, 63%; Fig. 5 B and C). Interestingly, 52% of these hyphal-specific targets also showed increased mRNA abundance in the absence of khd4 (40 out of 77 transcripts), with half containing the AUACCC motif in their 3′ UTR (25 out of 40 transcripts, 63%). In contrast, the minor fraction of hyphal-specific targets with decreased levels after khd4 deletion showed enrichment of the motif in the 5′ UTR (17 out of 37 transcripts, 46%), suggesting a role of Khd4 in translation (SI Appendix, Fig. S11A). Nevertheless, we noted that most of the Khd4-bound mRNA targets responded to khd4 deletion with increased mRNA levels and harbored the AUACCC motif in their 3′ UTR (Fig. 5C).
Fig. 5.
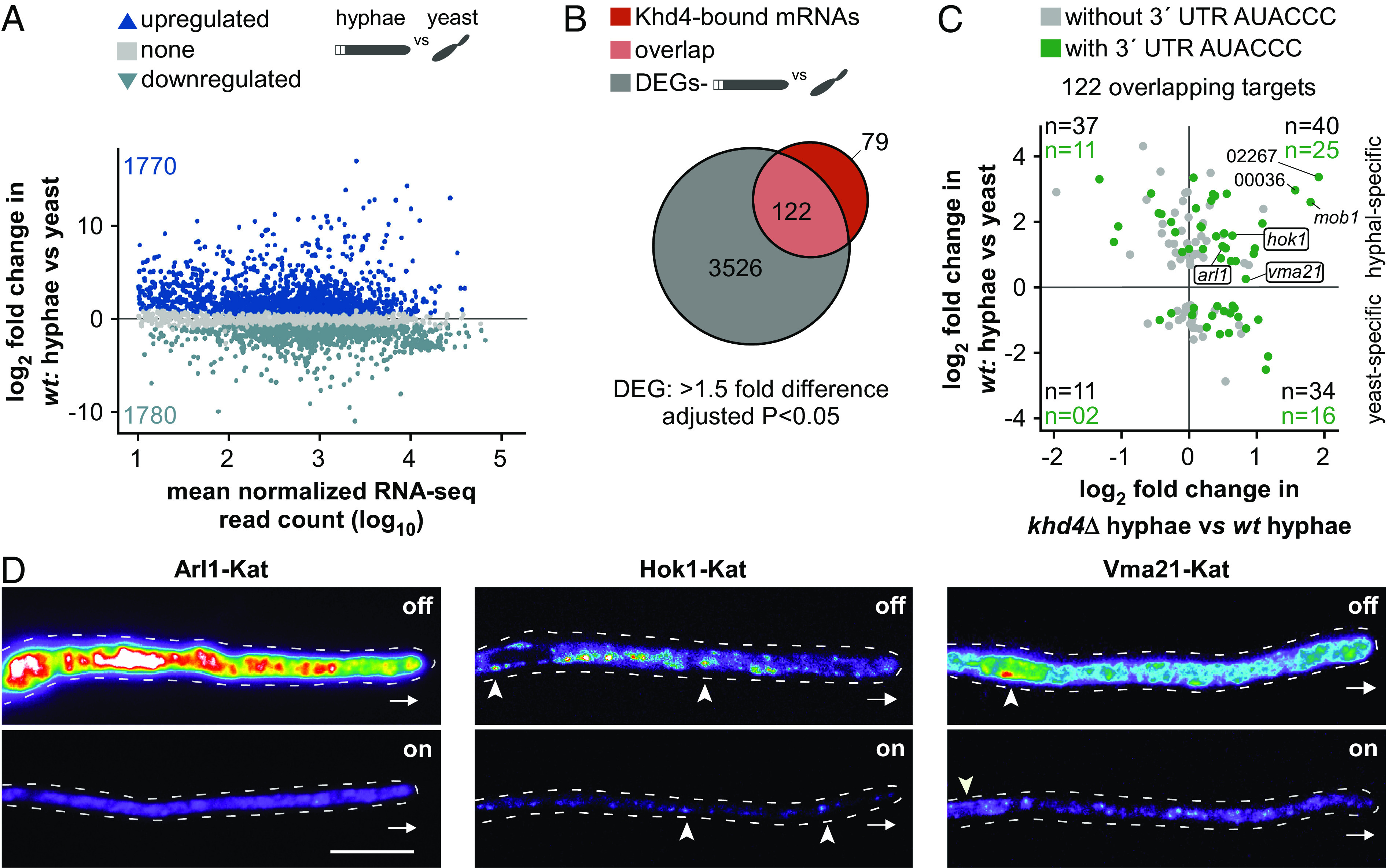
Khd4 regulates subcellular protein levels of membrane-trafficking regulators. (A) MA plot showing the differential mRNA abundance in wt cells upon hyphal induction (wt: hyphae vs. yeast). Shown is the log2-transformed fold change plotted against the log10-transformed mean normalized read counts. Blue dots represent values of transcripts with a significant change in their mRNA abundance (>1.5-fold change, P < 0.05, Benjamini–Hochberg correction, >10 average normalized reads), and gray dots represent remaining transcripts. Numbers inside the plot indicate upregulated and downregulated genes. Silhouette images of elongated hyphae and dividing yeast are shown. (B) Overlap (light red) between the differentially expressed genes (DEGs) in wildtype cells (gray) following the switch from yeast to hyphae (wt: hyphae vs. yeast; >1.5-fold difference, P < 0.05, Benjamini–Hochberg correction, >10 average normalized reads; gray) and Khd4-bound mRNAs (red). (C) Scatterplot comparing mRNA expression levels (log2-transformed fold change) of the overlapping high-confident Khd4-bound mRNAs from B in wildtype cells (wt: hyphae vs. yeast), and hyphal cells following khd4 deletion (khd4Δ hyphae vs. wt hyphae). Transcripts with a 3′ UTR AUACCC motif are indicated by green dots. The numbers in black and green within each quadrant indicate the total number of target transcripts and the count of targets with 3′ UTR AUACCC, respectively. We manually included vma21 in our analysis since it showed a significant increase in mRNA abundance when khd4 is absent, despite a marginal increase in expression during hyphal growth. In addition, 02267,00036 are shortened IDs representing UMAG_02267 and UMAG_00036, respectively. (D) Fluorescent micrographs in false color (black/blue—low intensity, red/white—high intensity) of hyphae (6 h.p.i.) constitutively expressing target genes (arl1—left, hok1—center, vma21—right) under Khd4-repressed off (Top) and Khd4-induced on (Bottom) conditions. Arrowheads and arrow marks indicate the corresponding target protein’s location (Hok1 on early endosomes; Vma21 in the perinuclear region) and the growth direction, respectively (scale bar: 10 µm.).
To validate whether Khd4 determines the steady-state levels of the encoded proteins of its target mRNAs in vivo, we selected the target transcripts arl1, hok1, and vma21, encoding proteins functioning in small GTPase signaling, early endosome transport, and vacuolar biogenesis, respectively (UMAG_10313 and UMAG_11418 exhibit high-sequence identity with the respective Arl1 and Vma21 orthologs of other fungi; SI Appendix, Fig. S11 B and C). In pathogenic fungi, the highly conserved Arf GTPase Arl1 participates in hyphal growth and virulence by regulating the endosomal trans-Golgi network and secretion (31, 38, 39). Hok1 regulates the bidirectional movement of early endosomes by recruiting motor proteins Kin3 and split dynein Dyn1/2 during long-distance transport (40). Vma21 is required for the precise assembly of the multisubunit V-ATPase complex in the vacuole of the budding yeast Saccharomyces cerevisiae (41). The three mRNAs encoding Arl1, Hok1, and Vma21 harbored 3′ UTR AUACCC motifs and showed increased expression levels during hyphal growth. Interestingly, loss of Khd4 enhanced this increase even further (Fig. 5C and SI Appendix, Fig. S11D).
We ectopically expressed the target transcripts using the constitutively active Ptef promoter in the background of the arabinose-inducible Khd4–Gfp strain. To detect the abundance of the encoded proteins and their subcellular localization, the ORFs of the target transcripts were tagged with Kat, followed by the insertion of the corresponding endogenous 3′ UTR (SI Appendix, Fig. S11E). Indeed, as seen in our RNA-seq analysis, the absence of Khd4 resulted in an increased amount of encoded protein in all three cases (Fig. 5D and SI Appendix, Fig. S11 F and G). These results reinforce that Khd4 regulates the expression of these target mRNAs at the level of mRNA stability. Despite the same constitutively active promoter and insertion at the same ectopic locus, each target mRNA exhibited a different protein expression level under promoter-on and -off conditions (Fig. 5D and SI Appendix, Fig. S11G), suggesting the presence of an additional layer of regulation at the posttranscriptional or posttranslational level.
Based on these results, we hypothesize that Khd4 destabilizes mRNAs encoding regulatory proteins and thereby functions as a key variable for achieving faster steady-state levels of these transcripts during hyphal development (Fig. 5C). Using a single-compartment model, we simulated the impact of mRNA stability on gene expression (37, 42). Our analysis confirmed that unstable mRNAs attain faster steady-state levels, enabling quick response times. Thus, intrinsically unstable mRNA is indispensable for rapid on-off kinetics for faster cellular responses (SI Appendix, Fig. S12A).
Building upon these findings, we used the mathematical model to investigate the dynamics of mob1 mRNA, a Khd4 target encoding a kinase regulator in the presence and absence of Khd4. Using the measured mob1 expression levels (SI Appendix, Fig. S12B) and an estimated mRNA half-life (t1/2) of 8 min (as determined in S. cerevisiae; 43), we simulated mob1 induction kinetics during hyphal induction (SI Appendix, Fig. S12C). Accordingly, the loss of Khd4 resulted in the stabilization of mob1 mRNA to a half-life of t1/2 = 22 min, which was sufficient to explain the elevated initial and new steady-state levels, without changing the synthesis rate. Furthermore, this stabilization caused a significant delay in mob1 induction kinetics, reaching the new steady state only after 2 h of morphogenesis. While termination of mob1 synthesis rapidly cleared the mRNA in wildtype cells, stabilized mob1 took considerably longer to return to its original steady-state levels (SI Appendix, Fig. S12C).
Thus, our model showcases that the absence of Khd4 causes the stabilization of mob1 mRNA, resulting in delayed induction kinetics and an overshooting of absolute expression levels. These aspects will likely have adverse effects since regulatory proteins should not accumulate excessively in the cell. We, therefore, propose that the continuous availability of Khd4 during hyphal morphogenesis (confirmed at the mRNA, protein, or subcellular localization level; SI Appendix, Fig. S12 D–H) is crucial to ensure the rapid response kinetics of its target mRNAs.
Loss of Khd4 Causes Dysregulation of Membrane-Trafficking Factors in khd4∆ Cells.
As Khd4 plays a vital role in controlling the precise expression levels of membrane-trafficking regulators, we wondered if loss of Khd4 would cause compensatory effects through altered gene expression. Akin to the wildtype cells, we determined the hyphal-specific transcripts in khd4∆ cells by evaluating our RNA-seq data of khd4∆ hyphae and yeast cells (9 h.p.i.; khd4∆: hyphae vs. yeast; see Materials and methods) and detected ~3,500 genes with differential mRNA abundance in response to hyphae formation in the khd4∆ cells (Fig. 6A; >1.5-fold change, P < 0.05, Benjamini–Hochberg correction, >10 average normalized reads). Comparing genes with differential mRNA abundance revealed that the majority were regulated similarly in both wildtype and khd4Δ cells, suggesting that the hyphal growth program itself is not abolished in the absence of Khd4 (Pearson correlation coefficient R = 0.86, P < 2.26e-16; Fig. 6B and SI Appendix, Fig. S13A). However, we identified about 2,000 genes exhibiting exclusive regulation in either wildtype or khd4Δ cells (Fig. 6B; 818 or 927 genes specifically regulated in the wildtype or khd4Δ cells, respectively).
Fig. 6.
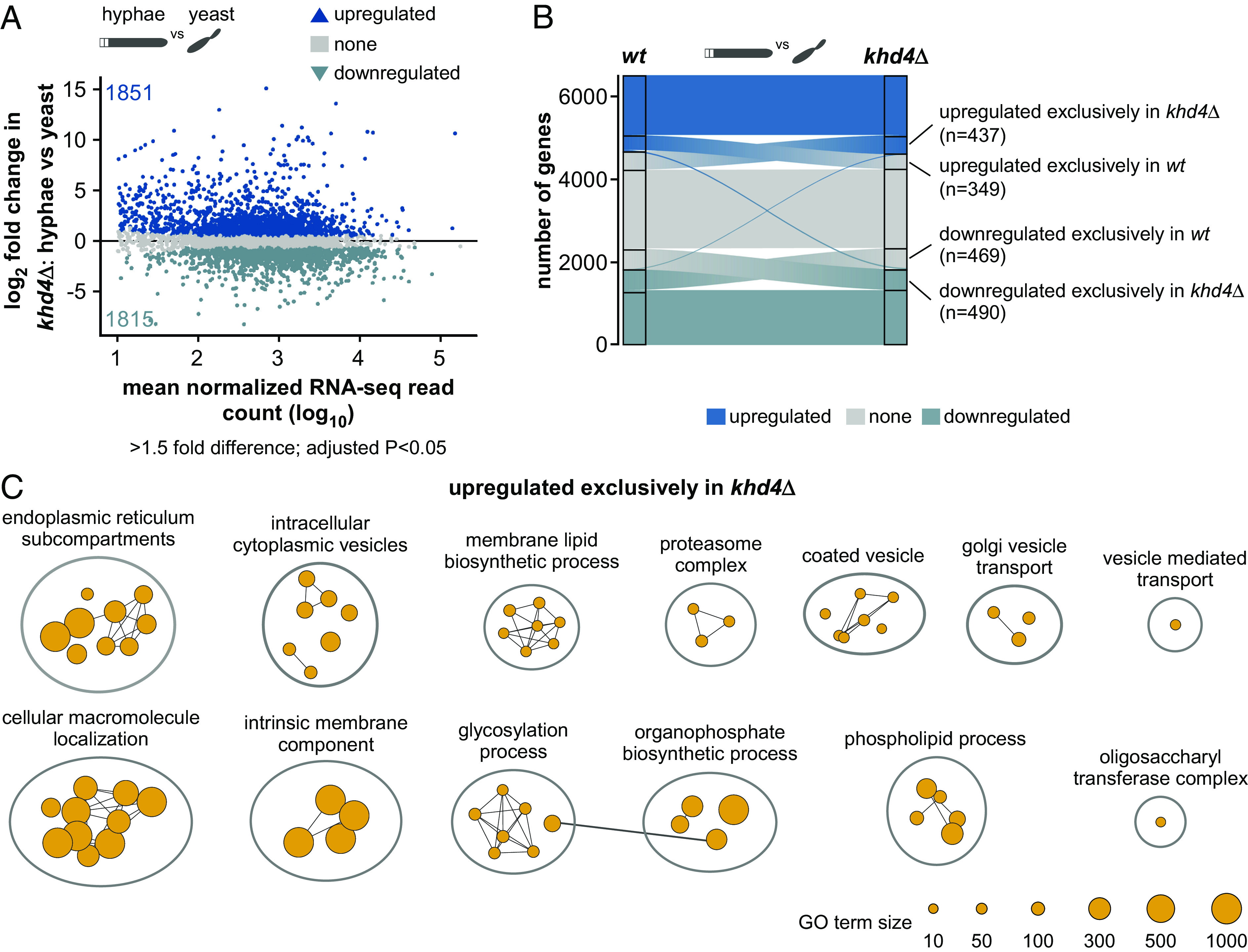
Loss of Khd4 causes dysregulation of membrane trafficking. (A) MA plot showing the differential mRNA abundance in khd4∆ cells (khd4∆: hyphae vs. yeast) following hyphae formation. Log2-transformed fold change values of genes are plotted against the log10-transformed mean normalized read counts. Blue dots represent transcripts with a significant change in their mRNA abundance (>1.5-fold change, P < 0.05, Benjamini-Hochberg correction, >10 average normalized reads), and gray dots represent remaining transcripts. Numbers inside the plots indicate the upregulated and downregulated genes. Silhouette images of elongated hyphae and dividing yeast cells are shown on Top. (B). Alluvial plot showing the distribution of upregulated (up; dark blue), downregulated (down; light blue), and nonregulated (none; gray) genes in wt and khd4∆ cells following the switch from yeast to hyphae. The labeled regions represent genes that are exclusively upregulated (n = 349 in wt, n = 437 in khd4∆) or downregulated (n = 469 in wt, n = 490 in khd4∆) in either wt or khd4∆ cells. Importantly, these genes were regulated exclusively in wt or khd4∆ cells but not in the corresponding regions of the other set. Two slender blue lines depict differentially expressed genes exhibiting opposite regulation between wt and khd4∆ cells. The central flow in the middle of the alluvial plot depicts the gene transition between different categories. The width of the flow accurately represents the number of genes undergoing this transition. (C) GO terms that are significantly (P < 0.05, multiple-testing correction with g: SCS algorithm) overrepresented in genes upregulated exclusively in khd4∆ cells after hyphal induction (orange; >1.5-fold change, P < 0.05, Benjamini–Hochberg corrections, >10 average normalized reads). The reference GO term sizes are given below.
To distinguish the effect of khd4 deletion in hyphal cells, we performed a GO term analysis on upregulated genes that are specific either to wildtype or khd4Δ cells (n = 349, upregulated exclusively in wildtype cells; n = 437, upregulated exclusively in khd4Δ cells; Fig. 6 B and C and SI Appendix, Fig. S13B). Interestingly, in contrast to the wildtype cells, the genes upregulated exclusively in khd4∆ cells were mainly enriched for functional categories associated with the endomembrane system such as endoplasmic reticulum, Golgi vesicle transport, cytoplasmic vesicles, and cellular macromolecule localization. We also found enriched terms related to ER-Golgi metabolisms such as glycosylation, carbohydrate, and phospholipid metabolic processes (44; Fig. 6C and SI Appendix, Fig. S13B), indicating that in comparison to wildtype cells, the genes encoding membrane-trafficking components are dysregulated during hyphal growth in the khd4Δ cells. Thus, loss of Khd4 causes specific alterations in the amounts of mRNAs encoding membrane-trafficking components, supporting the notion that Khd4 is an important regulatory protein to orchestrate membrane trafficking during hyphal growth.
Loss of Khd4 Causes Defects in Vacuole Formation and Localization.
To evaluate the impact of khd4 deletion on membrane trafficking in more detail, we examined the endocytic pathway leading to vacuole biogenesis in hyphal cells. As a control for defective membrane trafficking, we used cells lacking Did2, an ESCRT-III regulator involved in endosome maturation and identity (endosomal sorting complex required for transport; 45). Studying the endocytic pathway with FM4-64 uptake assays revealed that loss of Khd4 heavily delayed endocytosis. In time-resolved microscopy of wildtype hyphae, FM4-64 first stained the plasma membrane followed by bidirectionally shuttling endosomes and finally the vacuoles. After 25 min of staining, most of the FM4-64 signals in the wildtype hyphal cells were static (49%), while about 13% of the signals were found on processively moving early endosomes (SI Appendix, Fig. S13 C and D). By contrast, the loss of Khd4 caused a reduction in both static (36%) and processive signals (4%) and a significant increase (60%) in diffusively moving signals (SI Appendix, Fig. S13 C and D). A similar defect was observed for cells lacking Did2, leading to a deficiency in vacuolar protein sorting. Also, both khd4Δ and did2Δ cells showed a strong reduction in the intensity and distance between static signals throughout the hyphae (SI Appendix, Fig. S13 C and D).
To analyze vacuole morphology more closely, we stained vacuoles with CMAC (7-amino-4-chloromethylcoumarin; 46). In general, vacuoles were evenly distributed in wildtype hyphae (Fig. 7 A and B). However, in khd4Δ and did2Δ hyphae, the vacuoles were smaller, less defined, and found scattered throughout the hyphal cells (Fig. 7 A and B). Interestingly, in about 80% of khd4Δ hyphae, the vacuoles were positioned aberrantly toward the cell cortex, neatly outlining the hyphal cells (Fig. 7 C and D), whereas less than 10% of the wildtype cells and ~40% of the did2Δ cells contained cortically positioned vacuoles (Fig. 7 C and D). In summary, loss of Khd4 results in defective vacuole formation that correlates with aberrant polar growth of infectious hyphae and discloses a unique link between RNA biology and vacuole formation.
Fig. 7.
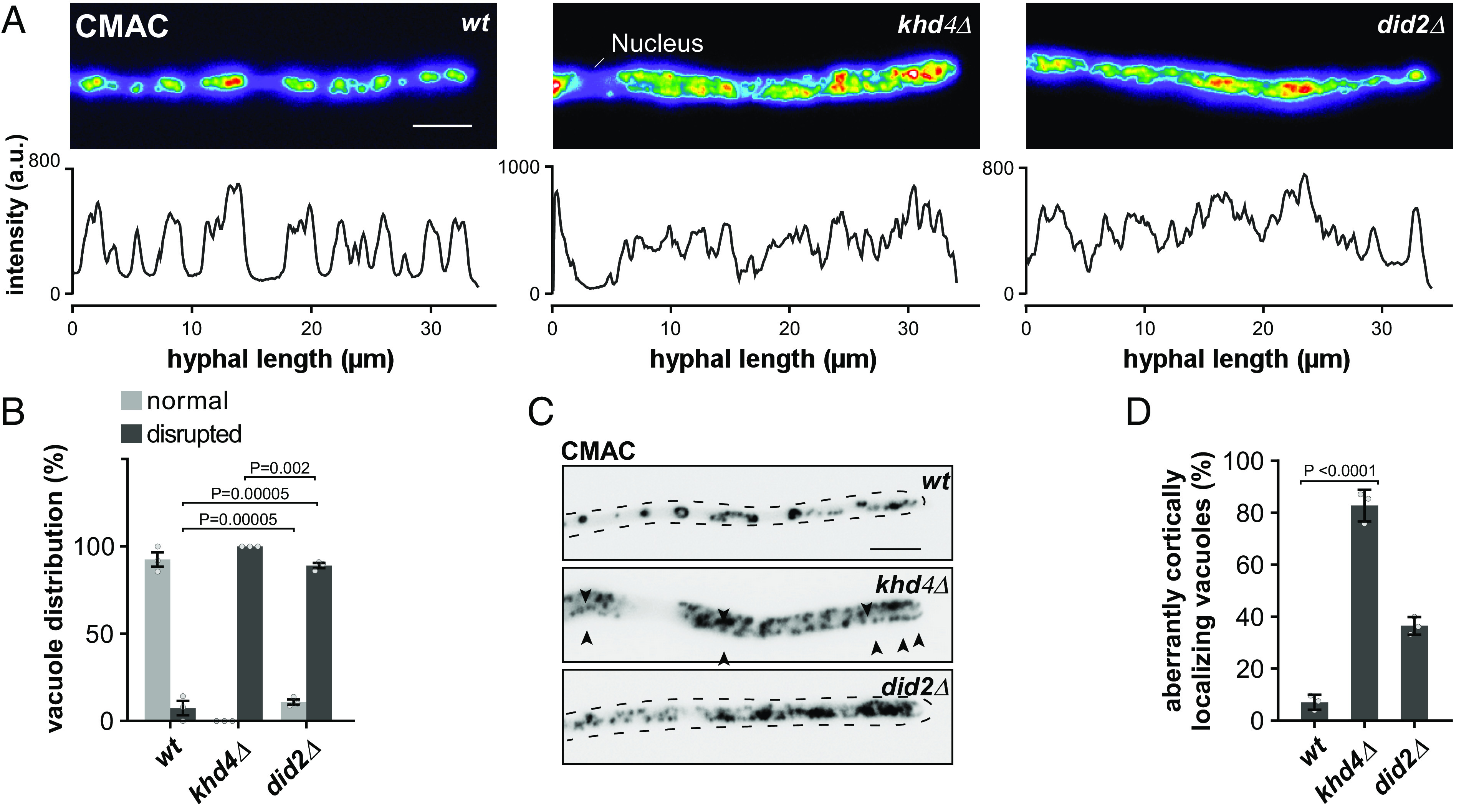
Loss of Khd4 severely affects vacuole formation and localization. (A) False colored maximum projections of z-stacks acquired from hyphal cells (AB33 derivatives; 10 h.p.i.) stained with the vacuolar stain CMAC (Top). The line graphs below depicts the fluorescent intensity profile of the respective hyphal cells. a.u.—arbitrary units (scale bar: 10 µm.). (B) Bar graph depicting the percentage of vacuole distribution (normal—light gray, disrupted—dark gray) in hyphal cells from AB33 derivatives (>25 hyphal cells per strain; n = 3, gray dots represent the mean value of independent experiments; error bars: SEM; unpaired Student’s t test). (C) Inverted micrographs of hyphal cells (10 h.p.i.) stained with CMAC dye to visualize the vacuole. (Scale bar: 5 µm.) (D) Percentage of hyphal cells containing aberrant cortical localizing vacuoles. The mean values from n = 3 independent experiments are shown as gray dots (>25 hyphal cells were analyzed; error bar: SEM; unpaired Student’s t test). Quantification in B and D involved manual assessment through visual inspection.
Discussion
The morphogenesis of fungal pathogens is intensively regulated at the level of transcription. Key transcription factors, for example, the heteromeric master regulator bE/bW in U. maydis and Efg1 in C. albicans, control the dimorphic switch from yeast to hyphae as part of the underlying pathogenic program (47, 48). The precise expression output is, however, determined at the level of RNA regulation. Yet, little is known about how transcriptional and posttranscriptional mechanisms cooperate during pathogenic development. Here, we report that in U. maydis, the RBP Khd4 is a vital regulator of the polar growth of infectious hyphae. Mapping the RNA binding of Khd4 uncovers a distinct set of target mRNAs encoding regulatory proteins involved in membrane trafficking. We find that Khd4 is required to maintain the accurate levels of these target mRNAs by regulating their mRNA stability, thereby functioning at the core of the hyphal gene expression program.
A key technical advance of our study is the successful application of the RNA editing-based hyperTRIBE method (23) to identify direct mRNA targets of RBPs in infectious hyphae of U. maydis. To this end, we conduct a pilot study with the well-known RBP Rrm4 and a detailed analysis of Khd4. In both cases, unique target transcripts are identified, with editing sites consistently enriched near the RBP binding sites. This feature serves as a critical quality criterion for the obtained dataset, enabling the discovery of unknown binding motifs for other RBPs in the future. In the case of Khd4, de novo motif discovery analysis successfully recovers the AUACCC binding motif as a top candidate, which is found in over 80% of the edited transcripts. This implies that, despite the increased background editing observed in our control experiments, specific hyperTRIBE editing is guided by the tested RBP in the fungal system. Importantly, using this in vivo binding information rather than relying only on motif content enables the identification of Khd4-regulated RNA networks (see below).
RBPs determine RNA regulation such as localization, translation, and stability by interaction with their cognate cis-regulatory elements. We discover that Khd4 functions at the level of mRNA stability by controlling the abundance of target mRNAs when the regulatory motif AUACCC is present in their 3′ UTR. Khd4 thereby regulates the spatiotemporal gene expression in hyphal cells by mediating mRNA turnover. In other systems, several RBPs are thought to mediate mRNA decay by directly recruiting the Ccr4–Not deadenylation complex. For instance, in D. melanogaster, the RBP Bicaudal-C interacts with the NOT3/5 subunit of the Ccr4–Not complex via its KH domain (49, 50). We hypothesize that Khd4 recruits the evolutionarily conserved Ccr4–Not complex for mRNA-specific poly(A) tail shortening (51, 52). Studies on the roles of such cis and trans-acting factors in fungal pathogens are currently scarce. However, similar functions are known from the mammalian RBPs TTP and CELF1, which cause rapid mRNA turnover by binding to the AU-rich and GU-rich mRNA decay elements in the 3′ UTR of mRNAs, respectively (53, 54).
Interestingly, we observe that targets of the mRNA stability factor Khd4 encode numerous regulatory proteins such as GTPases. Consistently, regulatory proteins such as cytokines and transcriptional factors are commonly encoded by unstable mRNAs in other systems (55, 56). This permits the transient expression of these proteins, which otherwise would cause undesirable side effects from prolonged exposure. Furthermore, unstable mRNAs facilitate the rapid attainment of steady-state levels following transcriptional induction (SI Appendix, Fig. S12A; 37, 42, 57, 58). Consistently, we observe that Khd4-bound mRNAs are transcriptionally induced during the transition from yeast to hyphal cells, suggesting that mRNA destabilization is crucial for the precise expression of regulatory proteins during hyphal morphogenesis. Consequently, the loss of Khd4 changes the underlying regulatory dynamics, reflected in hyphae with a delayed polar growth program (SI Appendix, Fig. S12 A–C).
Our findings implicate Khd4 as a regulator of membrane trafficking. By binding to AUACCC motifs in the 3′ UTR, Khd4 destabilizes a distinct subset of mRNAs encoding membrane-trafficking regulators, dictating their exact subcellular protein levels. This coordinated regulation is consistent with the concept that RBPs coregulate functionally related mRNAs, also referred to as mRNA regulons (59). Thus, we hypothesize that Khd4 defines a specific mRNA regulon for the coordinated regulation of membrane trafficking. Consistently, in the absence of khd4, the dynamic control of membrane trafficking is disturbed, as evidenced by aberrant vacuole biogenesis in fungal hyphae. It is already known that fungal RBPs such as Ssd1 (60–62), Puf4 (63), She3 (64), and Rrm4 (12, 16) play vital roles in the morphology and virulence of fungal pathogens. However, the concept of a defined mRNA regulon for coordinating a specific biological process is predominantly described in higher eukaryotes. For example, by recognizing 3′ UTR AU-rich elements and stem-loop structures, RBPs such as TTP and Roquin promote the degradation of mRNA regulons that encode inflammatory regulators, such as cytokines, modulating immune responses (55, 65).
Our study unveils a unique link between RNA regulation and vacuole maturation. Correct vacuole biogenesis plays an instrumental role in the virulence of fungal pathogens (66–69). Notably, appressoria-like infection structures in phytopathogens are strongly related to the development of vacuole-dependent basal septa (20), a process that is severely disturbed in khd4Δ hyphae. Therefore, Khd4-mediated control of the membrane-trafficking mRNA regulon might also impact early infection processes such as plant entry.
Strikingly, the Khd4-bound mRNAs are enriched for regulatory proteins involved in small GTPase signaling. Small GTPases participate in a wide range of biological processes (11, 70, 71). For example, Arl1, an Arf-like small GTPase, regulates secretion in C. albicans (31). The small GTPases Rac1 and Cdc42 control the morphology and pathogenicity of U. maydis (72). Our findings reveal that Khd4 is essential for the exact levels of Arl1 in hyphal cells. While most studies on small GTPase regulation focus on the GTP hydrolysis cycle and posttranslational modifications (73, 74), our study discloses a regulation at the level of the mRNA amounts. Khd4 also determines the precise subcellular levels of mRNAs encoding other endomembrane regulators, such as Hok1 and Vma21, which act in endosomal motor function (40) and V-ATPase assembly in the vacuole, respectively (75). Thus, RNA regulation appears to play a major role in the membrane-trafficking pathway during infectious hyphal growth.
In essence, we find that the RBP Khd4 functions as a key mRNA stability factor orchestrating membrane trafficking during pathogenic development. Identifying direct target mRNAs has been highly rewarding in resolving the underlying RNA regulation and disclosing new links to cell biological processes such as vacuole maturation. Our study thereby demonstrates the importance of RBPs during fungal pathogenicity and will serve as a blueprint to study the role of RBPs in other fungal/host pathosystems. Noteworthily, hyperTRIBE is very well suited to identify target mRNAs when the pathogen is in intimate contact with the host. This will be central to developing our understanding of the role of RNA biology in fungal virulence and creating new avenues of research into antifungal therapeutic strategies.
Materials and Methods
All strains and plasmids were generated following standard methods and incubation procedures (76). The mRNA targets of RBPs were identified using HyperTRIBE. De novo motif discovery analysis was conducted using the XSTREME tool from the MEME suite. Differential gene expression analysis comparing RNA-seq data was performed using DESeq2. The GO term enrichment analysis was carried out using the R package gProfiler2 and visualized with the Cytoscape plugin, EnrichmentMap (77, 78). Relative fluorescence levels were measured using the Infinite M200 plate reader (Tecan Group Ltd., Männedorf, Switzerland). RT-qPCR analysis was performed using the exp(-∆Ct) method in Stratagene Mx3000P (Agilent Technologies). Western blotting analysis, microscopy, and image processing were performed as described previously (79). For detailed methods, see SI Appendix, Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Acknowledgments
We would like to sincerely thank Dr. Andreas Brachmann, Dr. Kerstin Schipper, and Dr. Seomun Kwon for valuable scientific inputs; Thorsten Wachtmeister for NGS support; U. Gengenbacher and S. Krüger provided tremendous technical assistance; Melina Klostermann, Nina Kim Stoffel for the support with bioinformatics and biochemical analyses, respectively. The work was funded by grants from the Deutsche Forschungsgemeinschaft (German Research Foundation; https://www.dfg.de/) to M.F. (Germany’s Excellence Strategy EXC-2048/1—Project ID 39068111; CRC1535/1-A03—Project ID 458090666, FOR2333-TP03 FE 448/12-1), K.Z. (FOR2333-TP02 ZA 881/3-1), and A.M. (CRC1535/1-B02—Project ID 458090666) as well as by the graduate school Molecules of Infection-III to M.F. and S.S. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
S.S., C.H., K.K., A.M., K.Z., and M.F. designed research; S.S., P.P., and A.M. performed research; S.S., C.H., P.P., K.K., A.M., K.Z., and M.F. analyzed data; and S.S., K.Z., and M.F. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Kathi Zarnack, Email: kathi.zarnack@bmls.de.
Michael Feldbrügge, Email: feldbrue@hhu.de.
Data, Materials, and Software Availability
All sequencing data are available in the NCBI Gene Expression Omnibus under the SuperSeries accession number GSE224487 and are accessible to the public (80).
Supporting Information
References
- 1.Brown G. D., Denning D. W., Levitz S. M., Tackling human fungal infections. Science 336, 647 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Fisher M. C., et al. , Threats posed by the fungal kingdom to humans, wildlife, and agriculture. mBio 11, e00449-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gow N. A., Brown A. J., Odds F. C., Fungal morphogenesis and host invasion. Curr. Opin. Microbiol. 5, 366–371 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Min K., Neiman A. M., Konopka J. B., Fungal pathogens: Shape-shifting invaders. Trends Microbiol. 28, 922–933 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brand A., Gow N. A., Mechanisms of hypha orientation of fungi. Curr. Opin. Microbiol. 12, 350–357 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sudbery P. E., Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 9, 737–748 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Erwig L. P., Gow N. A. R., Interactions of fungal pathogens with phagocytes. Nat. Rev. Microbiol. 14, 163–176 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Lanver D., et al. , Ustilago maydis effectors and their impact on virulence. Nat. Rev. Microbiol. 15, 409–421 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Riquelme M., et al. , Fungal morphogenesis, from the polarized growth of hyphae to complex reproduction and infection structures. Microbiol. Mol. Biol. Rev. 82, e00068-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Commer B., Shaw B. D., Current views on endocytosis in filamentous fungi. Mycology 12, 1–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dautt-Castro M., Rosendo-Vargas M., Casas-Flores S., The small GTPases in fungal signaling conservation and function. Cells 10, 1039 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Müntjes K., Devan S. K., Reichert A. S., Feldbrügge M., Linking transport and translation of mRNAs with endosomes and mitochondria. EMBO Rep. 22, e52445 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wedlich-Söldner R., Bölker M., Kahmann R., Steinberg G., A putative endosomal t-SNARE links exo- and endocytosis in the phytopathogenic fungus Ustilago maydis. EMBO J. 19, 1974–1986 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinberg G., Endocytosis and early endosome motility in filamentous fungi. Curr. Opin. Microbiol. 20, 10–18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.König J., et al. , The fungal RNA-binding protein Rrm4 mediates long-distance transport of ubi1 and rho3 mRNAs. EMBO J. 28, 1855–1866 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baumann S., König J., Koepke J., Feldbrügge M., Endosomal transport of septin mRNA and protein indicates local translation on endosomes and is required for correct septin filamentation. EMBO Rep. 15, 94–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vollmeister E., et al. , Tandem KH domains of Khd4 recognize AUACCC and are essential for regulation of morphology as well as pathogenicity in Ustilago maydis. RNA 15, 2206–2218 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ram A. F., Klis F. M., Identification of fungal cell wall mutants using susceptibility assays based on Calcofluor white and Congo red. Nat. Protoc. 1, 2253–2256 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Brachmann A., Weinzierl G., Kämper J., Kahmann R., Identification of genes in the bW/bE regulatory cascade in Ustilago maydis. Mol. Microbiol. 42, 1047–1063 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Freitag J., et al. , Septation of infectious hyphae is critical for appressoria formation and virulence in the smut fungus Ustilago maydis. PLoS Pathog. 7, e1002044 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McMahon A. C., et al. , TRIBE: Hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell 165, 742–753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu W., Rahman R., Rosbash M., Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. RNA 24, 173–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahman R., Xu W., Jin H., Rosbash M., Identification of RNA-binding protein targets with HyperTRIBE. Nat. Protoc. 13, 1829–1849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuttan A., Bass B. L., Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U.S.A. 109, E3295–E3304 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bottin A., Kämper J., Kahmann R., Isolation of a carbon source-regulated gene from Ustilago maydis. Mol. Gen. Genet. 253, 342–352 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Loubradou G., Brachmann A., Feldbrügge M., Kahmann R., A homologue of the transcriptional repressor Ssn6p antagonizes cAMP signalling in Ustilago maydis. Mol. Microbiol. 40, 719–730 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Olgeiser L., et al. , The key protein of endosomal mRNP transport Rrm4 binds translational landmark sites of cargo mRNAs. EMBO Rep. 20, e46588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Müntjes K., et al. , Establishing polycistronic expression in the model microorganism Ustilago maydis. Front Microbiol. 11, 1384 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Göhre V., Vollmeister E., Bölker M., Feldbrügge M., Microtubule-dependent membrane dynamics in Ustilago maydis: Trafficking and function of Rab5a-positive endosomes. Commun. Integr. Biol. 5, 485–490 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raudvere U., et al. , g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Labbaoui H., et al. , Role of Arf GTPases in fungal morphogenesis and virulence. PLoS Pathog. 13, e1006205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Just W. W., Peränen J., Small GTPases in peroxisome dynamics. Biochim. Biophys. Acta 1863, 1006–1013 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Berepiki A., Lichius A., Read N. D., Actin organization and dynamics in filamentous fungi. Nat. Rev. Microbiol. 9, 876–887 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Manning B. D., Padmanabha R., Snyder M., The Rho-GEF Rom2p localizes to sites of polarized cell growth and participates in cytoskeletal functions in Saccharomyces cerevisiae. Mol. Biol. Cell 8, 1829–1844 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng P., et al. , Spitzenkörper assembly mechanisms reveal conserved features of fungal and metazoan polarity scaffolds. Nat. Commun. 11, 2830 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carbó N., Pérez-Martín J., Spa2 is required for morphogenesis but it is dispensable for pathogenicity in the phytopathogenic fungus Ustilago maydis. Fungal Genet. Biol. 45, 1315–1327 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Hargrove J. L., Schmidt F. H., The role of mRNA and protein stability in gene expression. FASEB J. 3, 2360–2370 (1989). [DOI] [PubMed] [Google Scholar]
- 38.Patiño-Medina J. A., et al. , Role of Arf-like proteins (Arl1 and Arl2) of Mucor circinelloides in virulence and antifungal susceptibility. Fungal Genet. Biol. 129, 40–51 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Yu C. J., Lee F. J., Multiple activities of Arl1 GTPase in the trans-Golgi network. J. Cell Sci. 130, 1691–1699 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Bielska E., et al. , Hook is an adapter that coordinates kinesin-3 and dynein cargo attachment on early endosomes. J. Cell Biol. 204, 989–1007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finnigan G. C., Ryan M., Stevens T. H., A genome-wide enhancer screen implicates sphingolipid composition in vacuolar ATPase function in Saccharomyces cerevisiae. Genetics 187, 771–783 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White E. J., Brewer G., Wilson G. M., Post-transcriptional control of gene expression by AUF1: Mechanisms, physiological targets, and regulation. Biochim. Biophys. Acta 1829, 680–688 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller C., et al. , Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hebert D. N., Garman S. C., Molinari M., The glycan code of the endoplasmic reticulum: Asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 15, 364–370 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Haag C., Pohlmann T., Feldbrügge M., The ESCRT regulator Did2 maintains the balance between long-distance endosomal transport and endocytic trafficking. PLoS Genet. 13, e1006734 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haugland R. P., Detecting enzymatic activity in cells using fluorogenic substrates. Biotech. Histochem. 70, 243–251 (1995). [DOI] [PubMed] [Google Scholar]
- 47.Heimel K., et al. , The transcription factor Rbf1 is the master regulator for b-mating type controlled pathogenic development in Ustilago maydis. PLoS Pathog. 6, e1001035 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumamoto C. A., Vinces M. D., Contributions of hyphae and hypha-co-regulated genes to Candida albicans virulence. Cell Microbiol. 7, 1546–1554 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Chicoine J., et al. , Bicaudal-C recruits CCR4-NOT deadenylase to target mRNAs and regulates oogenesis, cytoskeletal organization, and its own expression. Dev. Cell 13, 691–704 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Minegishi K., et al. , Fluid flow-induced left-right asymmetric decay of Dand5 mRNA in the mouse embryo requires a Bicc1-Ccr4 RNA degradation complex. Nat. Commun. 12, 4071 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller J. E., Reese J. C., Ccr4-not complex: The control freak of eukaryotic cells. Crit. Rev. Biochem. Mol. Biol. 47, 315–333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collart M. A., Panasenko O. O., Nikolaev S. I., The Not3/5 subunit of the Ccr4-Not complex: A central regulator of gene expression that integrates signals between the cytoplasm and the nucleus in eukaryotic cells. Cell. Signal. 25, 743–751 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Barreau C., Paillard L., Osborne H. B., AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 33, 7138–7150 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vlasova-St Louis I., Bohjanen P. R., Coordinate regulation of mRNA decay networks by GU-rich elements and CELF1. Curr. Opin. Genet. Dev. 21, 444–451 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anderson P., Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol. 10, 24–35 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Thompson M. K., Ceccarelli A., Ish-Horowicz D., Davis I., Dynamically regulated transcription factors are encoded by highly unstable mRNAs in the Drosophila larval brain. RNA 29, 1020–1032 (2023), 10.1261/rna.079552.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bruce S. R., Wilkinson M. F., “Messenger RNA stability” in Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine (Springer Berlin Heidelberg, Berlin, Heidelberg, 2006), pp. 1065–1065, 10.1007/3-540-29623-9_2230. [DOI] [Google Scholar]
- 58.Moore A. E., Chenette D. M., Larkin L. C., Schneider R. J., Physiological networks and disease functions of RNA-binding protein AUF1. Wiley Interdiscip. Rev. RNA 5, 549–564 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Keene J. D., RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Bayne R. A., et al. , Yeast Ssd1 is a non-enzymatic member of the RNase II family with an alternative RNA recognition site. Nucleic Acids Res. 50, 2923–2937 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thammahong A., Dhingra S., Bultman K. M., Kerkaert J. D., Cramer R. A., An Ssd1 homolog impacts trehalose and chitin biosynthesis and contributes to virulence in Aspergillus fumigatus. mSphere 4, e00244-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gank K. D., et al. , SSD1 is integral to host defense peptide resistance in Candida albicans. Eukaryot. Cell 7, 1318–1327 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalem M. C., Subbiah H., Leipheimer J., Glazier V. E., Panepinto J. C., Puf4 mediates post-transcriptional regulation of cell wall biosynthesis and caspofungin resistance in Cryptococcus neoformans. mBio 12, e03225-20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elson S. L., Noble S. M., Solis N. V., Filler S. G., Johnson A. D., An RNA transport system in Candida albicans regulates hyphal morphology and invasive growth. PLoS Genet. 5, e1000664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diaz-Muñoz M. D., Osma-Garcia I. C., The RNA regulatory programs that govern lymphocyte development and function. Wiley Interdiscip. Rev. RNA 13, e1683 (2022). [DOI] [PubMed] [Google Scholar]
- 66.Palmer G. E., Kelly M. N., Sturtevant J. E., The Candida albicans vacuole is required for differentiation and efficient macrophage killing. Eukaryot. Cell 4, 1677–1686 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu G., et al. , Vam6/Vps39/TRAP1-domain proteins influence vacuolar morphology, iron acquisition and virulence in Cryptococcus neoformans. Cell Microbiol. 23, e13400 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu X., Hu G., Panepinto J., Williamson P. R., Role of a VPS41 homologue in starvation response, intracellular survival and virulence of Cryptococcus neoformans. Mol. Microbiol. 61, 1132–1146 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Li B., et al. , The FgVps39-FgVam7-FgSso1 complex mediates vesicle trafficking and is important for the development and virulence of Fusarium graminearum. Mol. Plant Microbe Interact. 30, 410–422 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Rojas A. M., Fuentes G., Rausell A., Valencia A., The Ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 196, 189–201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arkowitz R. A., Bassilana M., Regulation of hyphal morphogenesis by Ras and Rho small GTPases. Fungal Biol. Rev. 29, 7–19 (2015). [Google Scholar]
- 72.Mahlert M., Leveleki L., Hlubek A., Sandrock B., Bölker M., Rac1 and Cdc42 regulate hyphal growth and cytokinesis in the dimorphic fungus Ustilago maydis. Mol. Microbiol. 59, 567–578 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Bos J. L., Rehmann H., Wittinghofer A., GEFs and GAPs: Critical elements in the control of small G proteins. Cell 129, 865–877 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Ahearn I. M., Haigis K., Bar-Sagi D., Philips M. R., Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 13, 39–51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Malkus P., Graham L. A., Stevens T. H., Schekman R., Role of Vma21p in assembly and transport of the yeast vacuolar ATPase. Mol. Biol. Cell 15, 5075–5091 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brachmann A., König J., Julius C., Feldbrügge M., A reverse genetic approach for generating gene replacement mutants in Ustilago maydis. Mol. Genet. Genomics 272, 216–226 (2004). [DOI] [PubMed] [Google Scholar]
- 77.Kolberg L., Raudvere U., Kuzmin I., Vilo J., Peterson H., gprofiler2–an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Res. 9, ELIXIR-709 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Merico D., Isserlin R., Stueker O., Emili A., Bader G. D., Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS One 5, e13984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Devan S.-K., et al. , A MademoiseLLE domain binding platform links the key RNA transporter to endosomes. PLoS Genet. 18, e1010269 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sankaranarayanan S., Zarnack K., Feldbrügge M., The mRNA stability factor Khd4 defines a specific RNA regulon for membrane trafficking in the pathogen Ustilago maydis. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE224487. Deposited 3 February 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Data Availability Statement
All sequencing data are available in the NCBI Gene Expression Omnibus under the SuperSeries accession number GSE224487 and are accessible to the public (80).