

Abstract
Mitochondria are responsible for ATP production but are also known as regulators of cell death, and mitochondrial matrix Ca2+ is a key modulator of both ATP production and cell death. Although mitochondrial Ca2+ uptake and efflux have been studied for over 50 years, it is only in the past decade that the proteins responsible for mitochondrial Ca2+ uptake and efflux have been identified. The identification of the mitochondrial Ca2+ uniporter (MCU) led to an explosion of studies identifying regulators of the MCU. The levels of these regulators vary in a tissue and disease specific manner providing new insight into how mitochondrial Ca2+ is regulated. This review will focus on the proteins responsible for mitochondrial transport and what we have learned from mouse studies with genetic alterations in these proteins.
Keywords: mitochondria, Ca2+, sodium, energetics
2. Introduction
Mitochondrial Ca2+ uptake and efflux pathways were defined decades ago and the consequences of physiological and pathological Ca2+ uptake have been well defined; however the identity of the proteins responsible for mitochondrial Ca2+ uptake and release were only recently identified. The molecular identification of the mitochondrial Ca2+ uniporter (MCU) (1, 2) and the mitochondrial Ca2+ efflux exchanger (Na+-Ca2+-Li+ exchanger (NCLX)) (3) has led to an explosion of new information about the regulation of mitochondrial Ca2+ and how this impacts cell function. This review will briefly summarize the data on mitochondrial Ca2+ uptake and release mechanisms that were defined during the 5 decades from 1960 to 2010 as well as the physiological and pathological consequences of changes in mitochondrial [Ca2+]. The bulk of the review will focus on new information obtained in the last decade concerning the identification and regulation of MCU and NCLX with an emphasis on genetic mouse models that provided insight into the physiological and pathophysiological regulation of mitochondrial Ca2+.
2.1. Ca2+ uptake and efflux mechanisms: the view from 1960 to 2010
It is well established that mitochondria can sequester large amounts of extramitochondrial Ca+2. Studies performed in the 1960s using isolated mitochondria demonstrated mitochondrial Ca2+ uptake that was dependent on the mitochondrial membrane potential (4, 5). This mitochondrial Ca2+ uptake, termed the mitochondrial Ca2+ uniporter (MCU), has been well characterized as reviewed previously (6, 7). Data showed that ruthenium red (RR) and the more specific ruthenium 360 (Ru360) can inhibit Ca2+ uptake. Na+ dependent and Na+-independent mitochondrial Ca2+ efflux pathways were also defined (8, 9). The relationship between mitochondrial Ca2+ uptake and extramitochondrial Ca2+ differs among different tissues, and mitochondria from many tissues were shown to have a sigmoidal relationship between extramitochondrial Ca2+ and Ca2+ uptake (10, 11).
Kirichok et al (12) patch-clamped the inner mitochondrial membrane and identified a Ca2+ selective channel that is sensitive to Ru360, a known inhibitor of MCU, demonstrating that MCU was indeed a channel. Fieni et al (13) performed patch clamp studies on mitochondrial membranes from different mouse tissues including heart, skeletal muscle, liver and kidney and found that the current density is lowest in heart, which was 30 times smaller than in skeletal muscle. They also found that IMCU was larger in neonatal cardiac tissue than adult.
2.2. Physiological and pathophysiological consequences of mitochondrial Ca2+
Soon after it was shown that mitochondria take up very large amounts of Ca2+ in the presence of an anion such as phosphate or acetate, Haworth et al (14) showed that there was a limit to mitochondrial Ca2+ uptake; when this limit (known as Ca2+ retention capacity) was exceeded, a non-specific pore (known as the mitochondrial permeability transition pore or PTP) opened, which resulted in release of the accumulated Ca2+ along with mitochondrial proteins and cofactors that were less than 1.5 kD. Opening of the PTP has been suggested to be a proximate cause of cell death in ischemia and reperfusion (I/R) and other diseases associated with mitochondrial Ca2+ overload (see (15–18)). The mechanism by which Ca2+ activates the PTP is still somewhat unclear, although data have suggested a role for Ca2+ modulation of complex V (19) as well as de-acylation of cyclophilin D (20), an activator of PTP. Thus mitochondria are key regulators of cell death and mitochondrial Ca2+ overload can precipitate mitochondria-mediated cell death.
In addition to activating the PTP and cell death pathways, physiological concentrations of mitochondrial Ca2+ regulate ATP production (21–24). When total mitochondrial Ca2+ levels are in the range of 10nmol/mg mitochondrial protein (25) Ca2+ uptake and efflux pathways in isolated mitochondria are matched and maintain extramitochondrial Ca2+ at a set point. This set point was initially proposed (26) to play a role in regulating cytosolic Ca2+ levels. However, in the steady state, the maintenance of cytosolic Ca2+ must dependent on the properties of the cell surface membrane. Furthermore, additional studies suggested that mitochondrial Ca2+ uptake and release pathways function primarily to regulate matrix Ca2+ levels, which regulate the activity of mitochondrial dehydrogenases (22) (see Figure 1). In isolated mitochondria, at total mitochondrial Ca2+ levels below 10nmol/mg mitochondrial protein, there is a linear relationship between free and total mitochondrial Ca2+ (27), and Ca2+ uptake in this range leads to an increase in free matrix Ca2+ and activation of the mitochondrial dehydrogenases. Denton and McCormack demonstrated that an increase in cytosolic Ca2+, as occurs with an increase in energy demand, leads to an increase in mitochondrial Ca2+ which activates 3 mitochondrial dehydrogenases: pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH) and α-ketoglutarate dehydrogenase (αKGDH) (22). Ca2+ activation of PDH occurs primarily via Ca2+ activation of PDH phosphatase which results in dephosphorylation and activation of PDH. Ca2+ appears to directly activate αKGDH and ICDH by reducing the Km for the substrate. Ca2+ activation of these dehydrogenases leads to an increase in NADH production, which provides substrate to the electron transport chain at complex I to enhance ATP production (Figure 1). ICDH also generates NADPH. Consistent with the concept that an increase in work leads to an increase in mitochondrial Ca2+ and a subsequent increase in NADH thereby matching energy demand and supply, Brandes and Bers (23) showed in cardiac trabeculae that with an increase in pacing, there was a close temporal relationship between the rise and fall in mitochondrial Ca2+ and NADH. Thus an increase in cytosolic Ca2+ and resultant work leads to Ca2+ entry into the mitochondria which activates ATP production, matching energy production to work. In further support of this hypothesis, Liu and O’Rourke (24) increased the pacing rate in adult guinea pig cardiomyocytes and with increased work found an increase in mitochondrial Ca2+, an increase in ATP hydrolysis but no change in [NADH]. Thus the increase in mitochondrial Ca2+ enhanced NADH production so there was no decline when electron transport was increased. When the experiment was repeated in the presence of Ru360, an inhibitor of MCU, the pacing induced increase in mitochondrial Ca2+ was reduced and net NADH oxidation was observed. They further showed a role of cytosolic Na+ in regulating mitochondrial Ca2+ and thereby regulating NADH. If the experiment was done in the presence of 15 mM Na+ (compared with 5 mM Na+), upon pacing there was less of an increase in mitochondrial Ca2+ and an oxidation in NADH, suggesting that Na+ dependent mitochondrial Ca2+ efflux plays a role in regulating mitochondria Ca2+ during increased work. Whether mitochondria have Ca2+ transients or whether they integrate the cytosolic Ca2+ has been debated and is discussed elsewhere (28).
Figure 1.
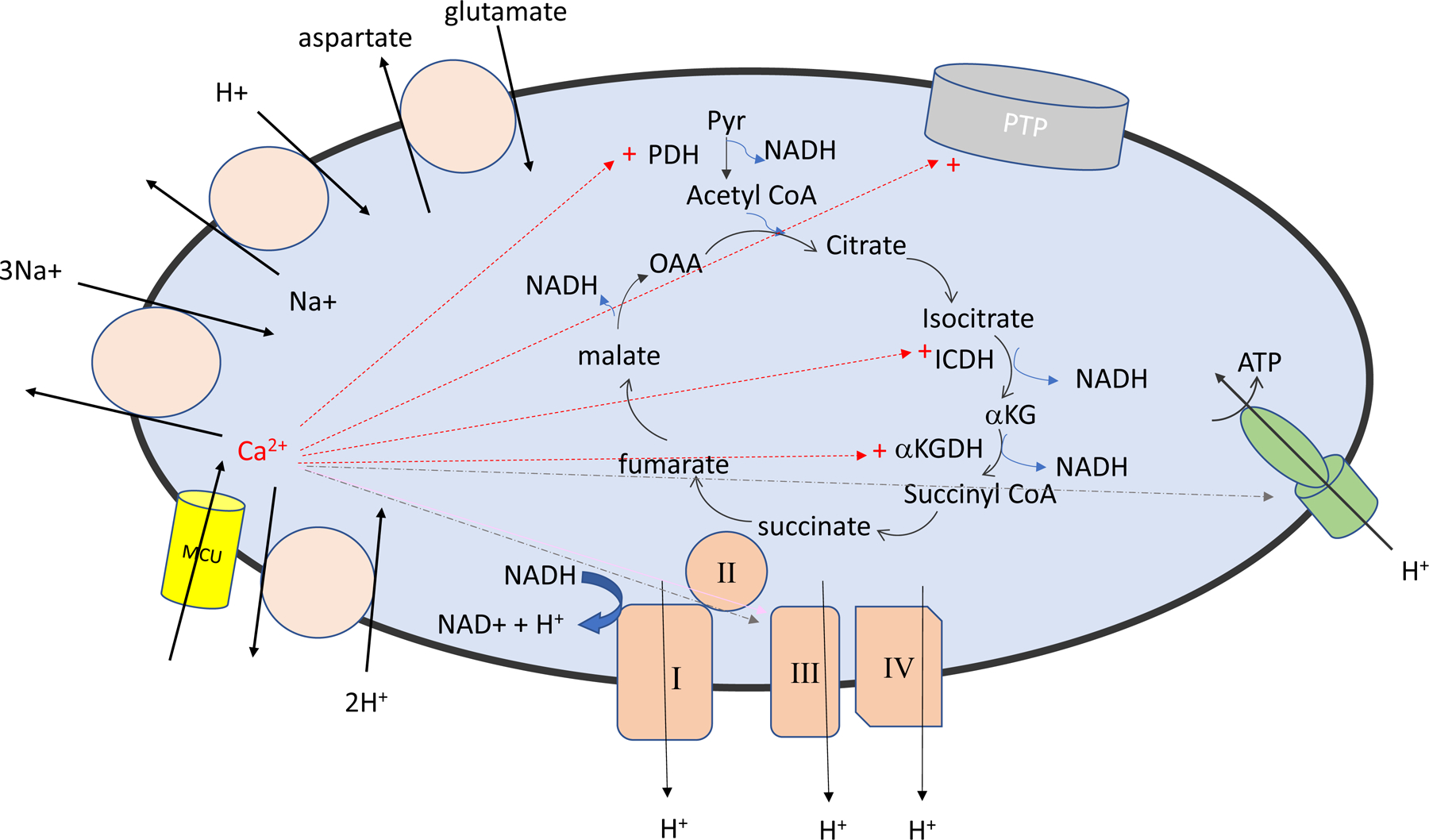
Illustrate mitochondrial Ca2+ uptake and efflux pathways and the mitochondrial Ca2+ activation of the mitochondrial sensitive dehydrogenases and the permeability transition pore (PTP). Red arrows indicate proteins/channels activated by Ca2+. The blue lines indicate enzyme reported to be activated by Ca+ which are disputed.
In addition to Ca2+ activation of the mitochondrial dehydrogenases, Ca2+ has also been suggested to activate mitochondrial electron transport complexes III and V (see (21) and Figure 1). However Wescott et al (29) recently reported that an increase in matrix Ca2+ only activates ATP production from NADH linked substrates via Ca2+ activated dehydrogenases, thereby questioning Ca2+ activation of complex III or V. Additional studies will be needed to resolve this discrepancy. It should be mentioned that cytosolic Ca2+ by activation of the aspartate-glutamate carriers (aralar and citrin), which function as part of the malate-aspartate shuttle to transport NADH from the cytosol to the mitochondria, can also increase mitochondrial NADH and stimulate electron transport (30).
3. Mitochondrial Ca2+ uptake:
Although mitochondrial Ca2+ uptake properties were established in the 1960 and 70s, it was not until 2011 that two groups independently identified the protein (CCD109A) responsible for mitochondrial Ca2+ uptake, which they renamed MCU (1, 2). It was quickly shown that MCU was part of a large complex consisting of multiple regulatory proteins. The MCU complex is comprised of MCU, EMRE (essential MCU regulator) and the EF hand proteins MICU (mitochondrial Ca uptake) 1, 2 and 3. The MCU paralog, MCUb, can also replace MCU in the complex and functions as a dominant negative protein. MCUR1 (MCU regulator 1) has also been proposed to be a scaffold connecting MCU and EMRE (31) and to regulate the Ca2+ sensitivity of the PTP (32); however others have suggested it is a cytochrome c oxidase assembly factor (33). Cryo-EM studies have suggested that the basic MCU complex consists of 4MCU, 4 EMRE, 1MICU1 and 1 MICU2 (34–37). Although MCU complexes are frequently shown with 2 MICU per MCU tetramer, a recent structure suggests 1 MICU1 (which can dimerize with MICU2) directly binds to the MCU tetramer (37). Figure 2A provides a cartoon of the MCU complex.
Figure 2.
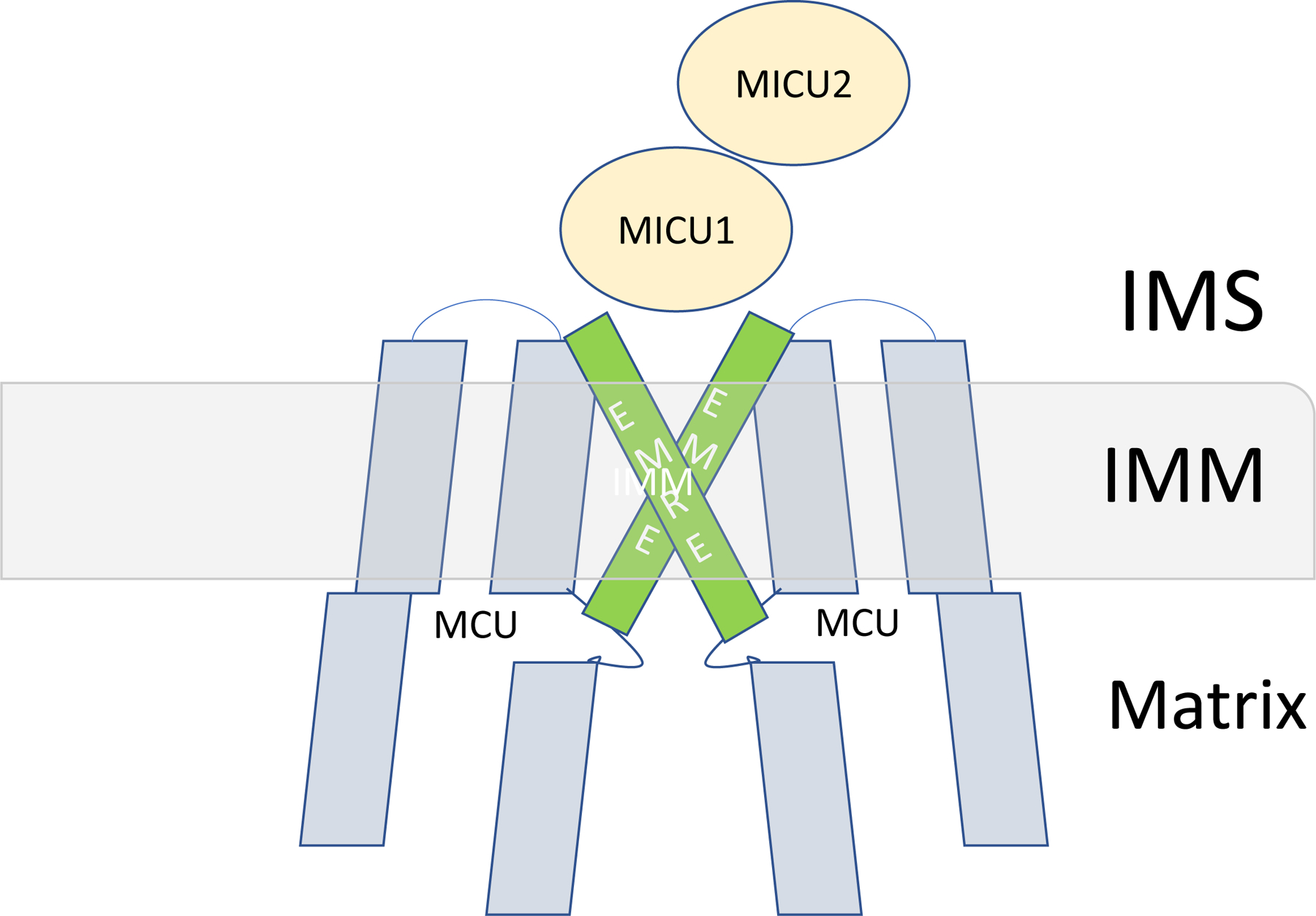

A) A schematic diagram of the MCU complex. B) Illustrates the differences in Ca2+ uptake occurring with differences in MCU regulators such as MICU1 and MCUb.
3.1. MCU:
As mentioned, studies by two groups independently identified MCU (previously known as CCD109a) as the pore forming unit of the mitochondrial Ca2+ uptake channel. De Stefani et al (2) screened the MitoCarta database and selected 89 proteins with 2 or more predicted transmembrane domains. S. cerevisae lacks MCU, thus providing another selection criteria. Twenty of the 89 transmembrane proteins were not present in S. cerevisae, which reduced the number of candidates to 20. After employing additional selection criteria, CCD109A was identified as a lead candidate. siRNA oligonucleotides were made and used to silence this putative MCU in Hela cells. Histamine was added to raise cytosolic Ca2+ and mitochondrial Ca2+ was measured with a mitochondrial encoded Ca2+ sensor in control and MCU silenced Hela cells; the MCU silenced Hela cells showed a much lower rise in mitochondrial Ca2+. Baughman et al used a very similar approach (1). A previous screen for mitochondrial transmembrane proteins that regulate mitochondrial Ca2+ uptake revealed MICU1 (previously known as CBARA1) as a regulator of mitochondrial Ca2+ uptake (38). They found that CCD109A was the 8th closest phylogenetic neighbor of MICU1. CCD109A was also found to be second highest in mRNA co-expression with MICU1 and CCD109A was found to be the top mitochondrial protein for co-expression with MICU1. They further showed in cultured cells that silencing MCU blocked mitochondrial Ca2+ uptake and that a point mutation in MCU (S259A) allowed mitochondrial Ca2+ uptake that was no longer blocked by the known MCU inhibitor, Ru360.
Taken together these two studies clearly established CCD109A as MCU. Soon after the identification of MCU, Pan et al (39) reported the first mouse model with germline deletion of MCU. Germline deletion of MCU was embryonic lethal in the mouse C57B6 strain background, but when crossed into an outbred CD mouse strain background, viable mice were obtained. However, the MCU-KO mice were born at less than Mendelian ratios (~10%). Mitochondria isolated from heart, brain and liver of MCU-KO mice did not take up Ca2+. In WT mitochondria, addition of high levels (e.g. 250 μM) of Ca2+ led to mitochondrial swelling associated with opening of the PTP; this Ca2+ induced swelling was not observed in mitochondria from MCU-KO mice. As the MCU-KO mitochondria did not accumulate Ca2+ and did not undergo PTP, it was expected that they would be protected from I/R induced cell death, as this has been proposed to be initiated by Ca2+ induced PTP opening. Surprisingly hearts from germline MCU-KO hearts did not have smaller infarcts following I/R injury (39).
As discussed, in addition to activation of PTP at high Ca2+ levels, an increase in matrix Ca2+ at physiological levels is reported to coordinate an increase in work with an increase in ATP production (21). Thus it was expected that MCU-KO mice would have an altered response to isoproterenol. However, in the germline MCU-KO mice, there was only a very modest difference in the contractile response to isoproterenol (40). Although WT hearts showed a slightly larger increase in dP/dtmax (measured with Millar catheter) after isoproterenol addition than KO mice, there was no significant difference in dP/dtmax between WT versus KO heart following isoproterenol (see Table). To further test whether loss of MCU leads to bioenergetic impairment, mice were subjected to a grip strength test and the MCU-KO mice did not perform as well as WT mice (39). Thus the germline MCU-KO mice are not protected from I/R injury and show only modest impairment and only under high work load.
Table.
Characterization of phenotype of mice with alterations in mitochondrial Ca2+ uptake.
| Germline Pan et al | DN-MCU Wu et al | inducible MCU-KO Kwong et al | inducible MCU-KO Luongo et al | inducible MCU-KO Altamimi et al | inducible MCU overexpression 1wk Lambert et al | inducible MCU overexpression 1 mo. Lambert et al | csMCUb overexpression Hou et al | |
|---|---|---|---|---|---|---|---|---|
| Mitochondrial Ca uptake PTP in isolated mitochondria |
no acute uptake no PTP |
no acute uptake NT |
no acute uptake no PTP |
no acute uptake no PTP |
ND ND |
70% decrease PTP reduced |
70% decrease PTP reduced |
no acute uptake no PTP |
| I/R | no protection | no protection | protected | protected | ND | no protected | protected | protected |
| Response to ISO Values are maximum in WT vs KO (or Tg) | dp/dt(max) mmHg/s WT12000; KO11000 | HR WT 750bpm; DN 550 bpm | Dp/dt(max) mmHg/s WT20000; KO 15000 | Dp/dt(max) mmHg/s WT10000; KO 8000 | no decrease in LVDP | Dp/dt(max) mmHg/s WT11000; Tg 6000 | Dp/dt(max) mmHg/s WT12; Tg 12 | ND |
| TAC | no difference vs WT | ND | no difference vs WT | ND | ND | ND | ND | ND |
PTP (permeability transition pore opening) measured by swelling. No PTP indicates the swelling was similar to that measured with CsA. TAC = transaortic constriction. ND = not determined. In Lambert et al (inducible MCUb at 1wk) the mice die minutes after induction of ischemia, so infarct size could not be measured.
Rasmussen et al developed a cardiac specific dominant negative MCU (DN-MCU) mouse with a mutation in the pore forming component of MCU (41). The dominant negative MCU gene was overexpressed in cardiac cells using the alpha myosin heavy chain promoter. Similar to the MCU-KO mice, mitochondria from these cardiac specific DN-MCU mice did not take up Ca2+ and did not undergo Ca2+-induced PTP. With increased pacing the DN-MCU mice showed a decrease in left ventricular developed pressure (LVDP) and other parameters of contractility, suggesting an impairment in matching energy supply and demand (42), and consistent with this, the DN-MCU mitochondria also exhibited increased phosphorylation of PDH at baseline. Similar to the germline MCU-KO heart, the DN-MCU mouse did not have smaller infarcts following I/R (see Table).
The Molkentin (43) and Elrod (44) labs generated and then independently studied a cardiac specific, inducible MCU-KO (csMCU-KO) mouse in which MCU was deleted by tamoxifen addition in the adult mouse. Similar to the germline MCU-KO and the DN-MCU mouse, cardiac mitochondria from these mice do not take up Ca2+ and do not undergo Ca2+-induced PTP. In contrast to the findings in the germline MCU-KO and the DN-MCU mouse, the inducible csMCU-KO hearts, with acute deletion of MCU in the adult, show reduced cell death following I/R. Also with acute deletion of MCU, there is a significant decrease in contractile response to isoproterenol in vivo. Both groups found following adrenergic stimulation that conditional MCU-KO hearts exhibited significantly less increase in dP/dtmax than WT hearts (see Table). Interestingly Kwong et al (43) found that with prolonged (60 min) addition of dobutamine, the contractile response was not different between the WT and csMCU-KO mice. With 60 minutes of dobutamine stimulation there was no difference in mitochondrial Ca2+ between WT and csMCU-KO, suggesting that with prolonged adrenergic stimulation, Ca2+ might enter the mitochondria by an alternative Ca2+ uptake mechanism. It is of note that this alternative Ca2+ uptake mechanism does not appear to be activated during ischemia in the csMCU-KO, as these hearts are protected from I/R injury, presumably due to reduced mitochondrial Ca2+ overload. This might suggest that the alternative Ca2+ uptake pathway requires a prolonged (60 min) period of both elevated cytosolic Ca2+ and a high mitochondrial membrane potential or it might be stimulated by an adrenergic-mediated signaling pathway (e.g. phosphorylation).
Altamini et al examined metabolism in an ex vivo perfused heart model using the inducible csMCU-KO mouse developed by Molkentin. In contrast to the in vivo studies, inducible loss of MCU did not compromise the contractile or energetic response to isoproterenol in an ex vivo model (45). In the ex vivo heart model perfused with physiological substrates (glucose and fatty acids) an increase in left ventricular developed pressure (LVDP) was observed at baseline before isoproterenol addition as well as after isoproterenol treatment. They showed that loss of MCU led to stimulation of fatty acid oxidation that improved energy reserves during isoproterenol stimulation. Consistent with these data, Kwong et al showed in skeletal muscle that under conditions of fatigue, acute deletion of MCU enhanced endurance and performance (46).
Summarizing the data on the mice with loss of MCU activity (see Table), with loss of MCU at birth or before birth, there is no protection following I/R, whereas mice with inducible deletion of MCU in adults exhibit reduced I/R injury. These data are consistent with an adaption occurring in the germline mice which appears to alter PTP such that cell death (47) is not reduced following I/R in spite of a presumed inhibition of mitochondrial Ca2+ uptake during ischemia. However, it should be noted that mitochondrial Ca2+ concentration during ischemia has not been measured in any of these models of MCU inhibition. The effect of adrenergic stimulation on contractile function also differs among the models. The germline MCU-KO hearts have little or no difference in the in vivo contractile response to isoproterenol. The mice in which the DN-MCU was expressed at birth show a decrease in heart rate (HR) response to adrenergic stimulation compared to WT. In the mice in which MCU is deleted in the adult, in vivo studies showed a very significant decrease in contractility in the MCU-KO hearts following acute adrenergic stimulation. However the decrease in contractility was not observed if dobutamine was administered in vivo for 1 hour or if isoproterenol was given in an ex vivo perfused heart. One might expect that if mitochondria cannot take up Ca2+ in response to adrenergic stimulation, that the initial contractile response would be normal (as the increase in cytosolic Ca2+ appears to be normal) but the mitochondria might be unable to enhance ATP production and there would be a decline in function with time. The variable response to adrenergic stimulation in the MCU-KO mice is complicated and additional studies are needed to clarify the reasons for the discrepancies.
3.2. MCUb:
MCUb is an inactive homologue of MCU with 50% sequence conservation (48). When MCUb is part of the tetramer with MCU, it acts as a dominant negative to reduce Ca2+ uptake (48). MCUb levels vary in different cell types and with disease, and it is thought to be a tissue specific regulator of MCU mediated Ca2+ uptake (48). Lambert et al (49) show that when MCUb displaces an MCU subunit from the tetramer, it decreases binding of MICU1, as MICU1 binds MCU but not MCUb. Huo et al (50) also showed that MCUb overexpression reduced the level of EMRE in the MCU complex.
Lambert et al (49) and Huo et al (50) both showed independently that MCUb levels in the MCU complex are increased following I/R. The two groups independently generated mice that overexpress MCUb and subjected them to I/R. Lambert et al generated a cardiac specific tamoxifen inducible mouse in which MCUb is overexpressed in the adult. When in vivo I/R was performed at one week after tamoxifen treatment to overexpress MCUb, 12 of the 13 mice subjected to LAD occlusion died within minutes. When I/R was done one month after tamoxifen treatment none of the mice died and infarct size compared to area at risk was lower in the MCUb overexpressing mice. The authors suggest that the deaths with I/R at one week after acute overexpression of MCUb are due to impaired energetics (due to reduced Ca2+ activation of ATP production in the non-ischemic area). It is suggested that after one month of MCUb overexpression, the energetic issues resolve, presumably due to some adaptation, and at this time, overexpression of MCUb reduces Ca2+ overload associated with I/R providing cardioprotection. These data suggest that if MCUb is overexpressed for 1 week, then the non-ischemic area of the heart cannot respond to an increase in work/energy demand, however if MCUb is overexpressed for 1 month non-ischemic myocardium is now able to increase contractility in response to stress. It will be interesting to understand the adaptations that occur between 1 week and 1 month of MCUb overexpression.
If MCU activity is inhibited for ~1 week, the heart seems to have a very impaired response to isoproterenol addition (see Table). In contrast, if MCU activity is inhibited for 1 month the isoproterenol response is more or less normal (no significant differences). Interestingly with germline loss of MCU, the contractile response to isoproterenol is similar to that observed with MCUb overexpression at 1 1 month, whereas with deletion of MCU in the adult the response to adrenergic stimulation is more similar to MCUb overexpression for 1 week. The mechanisms responsible for the various adaptations are unknown. It will be interesting to understand the details of these adaptations that correct the energetic imbalance.
If acute deletion or inhibition of MCU compromises energetics such that the mice cannot survive, this might suggest that acute inhibition of MCU would not be a useful therapeutic approach, unless the inhibition could be targeted exclusively to the ischemic tissue. One caveat is that these studies were done one week after tamoxifen injection and it is possible that the detrimental effects could be influenced by the tamoxifen treatment.
Huo et al (50) used a tetracycline transactivator (tTA) TG mouse line with cardiac-specific expression (α-myosin heavy chain, α-MHC) crossed with α-MHC tetracycline operator responsive TG mouse containing a MCUb cDNA (MCUb-TG) to generate an MCUb overexpressing mouse(MCUb-DTG) Overexpression of MCUb blocked mitochondrial Ca2+ uptake similar to what is observed in mitochondria isolated from MCU-KO or DN-MCU mice. When 6 week old mice were subjected to in vivo ischemia for 30 min followed by 24 hours of reperfusion, the MCUb overexpressing mice had significantly smaller infarcts than control (tTA- TG) mice. One might expect these MCUb overexpressing mice to be similar to the DN-MCU mice generated by Rasmussen et al (41); both mice overexpress a dominant negative MCU and in both mice the expression is regulated by α-MHC, so it would be turned on at birth. However, the DN-MCU mouse is not protected from I/R whereas the MCUb overexpressing mouse exhibits protection. The lack of protection from I/R in the DN-MCU mouse has been attributed to adaptations that occur as it is expressed from birth. One might expect that the overexpression of MCUb from birth would also lead to adaptations such that the mice overexpressing MCUb from birth might similarly lack protection; however that was not the case.
Huo et al (50) also generated a cardiac specific MCUb deletion mouse. Hearts from adult WT mice show no detectible MCUb protein, but consistent with the finding of Lambert et al (49) they find an increase in MCUb following I/R. They subjected the MCUb deletion mice to in vivo I/R and after 24 hours of reperfusion there was no difference in infarct size, presumably because there is little or no baseline expression of MCUb. They followed up these studies using a permanent occlusion model and found that the mice with cardiac specific MCUb deletion had exacerbated pathological remodeling, consistent with the concept that upregulation of MCUb following I/R is beneficial.
3.3. Mitochondrial Ca2+ uptake (MICU) proteins:
There are 3 EF hand proteins, MICU (mitochondrial Ca2+ uptake) 1, 2 and 3, that sense Ca2+ and regulate the activity of MCU (51, 52). MICU1 binds to the pore forming D ring of MCU and its interaction regulates Ca2+ entry as well as RR/Ru360 binding (53). Interestingly loss of MICU1 sensitizes MCU to Ru360 inhibition. These data combined with data showing tissue differences in MICU1 levels (54) provide an explanation for tissue differences in RR and Ru360 inhibition. Upon the discovery of MCU and the MICU1/2/3 EF hand proteins, it was suggested that these EF hand proteins might regulate Ca2+ uptake by MCU. MICU1 has been reported to bind directly to EMRE and MCU (but not to MCUb). MICU2 and MICU3 bind to MICU1 but cannot directly bind to MCU. Thus in the absence of MICU1, MICU2/3 cannot regulate MCU activity. MICU1 can form homodimers or heterodimers with MICU2 or MICU3. MICU1 homodimers have a Kd for Ca2+ binding of 300 nM whereas the heterodimers of MICU1 and MICU2 have a Kd for Ca2+ of 620 nM. The Kd for Ca2+ binding to MICU1 homodimers versus MICU1/2 heterodimers matches the different thresholds for Ca2+ uptake observed, suggesting that the threshold for Ca2+ uptake into the mitochondria is regulated by the proportion of MICU1 homodimers versus MICU1/2 heterodimers(55). MICU3 is highly expressed in neuronal tissue. Patron et al have overexpressed MICU1, 2 and 3 and show that MICU3 functions to enhance mitochondrial Ca2+ uptake (56).
MICU1:
MICU1 has been shown to reduce Ca2+ uptake at low extramitochondrial (0.5μM) Ca2+ levels but to enhance uptake at higher (15 μM) Ca2+ levels (57–59). Loss of MICU1 in cells and tissues leads to mitochondrial Ca2+ overload, presumably because Ca2+ enters at low extramitochondrial Ca2+ levels (57–60). Mallilankaraman et al (60) showed that downregulation of MICU1 in Hela cells led to an increase in basal mitochondrial Ca2+. Csordas et al (59) studied the effect of deletion of MICU1 on Ca2+ uptake in permeabilized mouse liver cells or Hela cells in the presence of thapsigargin to inhibit endoplasmic reticulum Ca2+ uptake and found that loss of MICU1 led to enhanced Ca2+ uptake at low Ca2+ concentrations (<1 μM) and reduced Ca2+ uptake at concentrations above ~3μM. MICU1 activation of Ca2+ uptake at Ca2+ levels above 3 μM is consistent with previous data showing that Ca2+ uptake into mitochondria exhibits cooperativity (7, 10). Consistent with a role for MICU1 in cooperativity, Csordas et al (59) showed that cooperativity was lost in cells expressing MICU1 with mutation in the EF hand.
Structural studies suggest that at low extramitochondrial Ca2+ concentrations (below ~300 nM) the EF hands of MICU1 and 2 are not bound with Ca2+ and are positioned to block Ca2+ entry via MCU. When Ca2+ rises above the Kd for Ca2+ binding to the EF hands of MICU1/2, the Ca2+ binding causes a conformational change that moves MICU1 from the channel allowing Ca2+ entry. A recent cryo-EM structure supports the concept that at low Ca2+ MICU1 occludes the MCU pore. At low Ca2+ the K/R ring of MICU1 seals the MCU pore (37). This study also shows that at high Ca2+ concentrations MICU1 no longer interacts with MCU and instead remains connected to the complex via its contacts with EMRE. These data suggesting a role for MICU1 in regulating Ca2+ threshold and cooperativity might seem at odds with the study of Wescott et al (29) which reported a no threshold for mitochondrial Ca2+ uptake. Some of the discrepancy appears to be related to the definition of “threshold”. As mentioned, mitochondrial Ca2+ uptake is often sigmoidal with low Ca2+ uptake at low extramitochondrial Ca2+, but with an marked increase in Ca2+ uptake at higher extramitochondrial Ca2+. The term threshold is often poorly defined and seems to be used to define the extramitochondrial Ca2+ values at which there is low Ca2+ uptake, but others, including Wescott, appears to define threshold at which there is no Ca2+ uptake.
Paillard et al (54) demonstrated that the ratio of MICU1/MCU varies between tissues and that the MICU1/MCU ratio regulates mitochondrial Ca2+ uptake. MICU1/MCU ratios are lower in heart than liver, and heart mitochondria take up Ca2+ faster at low Ca2+ concentration (3 μM) but slower at high (50 μM) Ca2+. They further show that overexpressing MICU1 in the heart transforms the mitochondrial Ca2+ uptake pattern in heart to that observed in liver and leads to cardiac contractile dysfunction. In addition to less MICU1, heart also has less MCUb compared to tissues such as liver. As illustrated in Figure 2B this would result in more Ca2+ uptake at low Ca2+ levels in heart compared to other tissues such as liver. In addition to different ratios of MICU1/MCU there are splice variants of MICU1 that alter the Ca2+binding to MICU. Skeletal muscle expresses a splice variant of MICU1, MICU1.1. This splice variant binds Ca2+ at a lower concentration and thereby activates MCU at a lower Ca2+ which is important for activation of mitochondrial dehydrogenases (61).
Antony et al (58) developed a mouse in the C57B6 background with whole body deletion of MICU1 and observed perinatal lethality in the mice; the mice were born at Mendelian ratios, but died within hours of birth. To avoid the perinatal lethality, they studied a model of liver specific MICU1 deletion and found that loss of MICU1 in the liver impairs liver regeneration following partial hepatectomy. Liu et al (57) used CRISPR/Cas9 to develop a mouse with deletion of MICU1 in an outbred CD strain mouse. Similar to the whole body mouse developed by Antony this mouse was born at Mendelian ratios and exhibited perinatal lethality; however in the outbred CD background some mice (~8%) survived. The surviving MICU1-KO mice were smaller than WT littermates. Mitochondria isolated from MICU1-KO mice at about ~1–2 weeks of age showed more Ca2+ uptake at a low extramitochondrial Ca2+ level of 0.5 μM than WT liver mitochondria; however at higher (16 μM) extramitochondrial Ca2+ levels the MICU1-KO mitochondria took up Ca2+ more slowly than WT mitochondria. These data are consistent with the concept that in liver mitochondria, MICU1 is important for regulating threshold (i.e. low Ca2+ uptake at low extramitochondrial Ca2+) and cooperativity. The MICU1-KO mice that survived gained weight in adulthood. Ca2+ uptake in mitochondria from older mice was evaluated and it was found that although there was still increased uptake at low Ca2+ in the MICU1-KO liver mitochondria, it was much less than that observed in mice at 1–2 week of age. An examination of other MCU regulatory proteins showed that at 1–2 week, the surviving MICU1-KO mice had lower levels of EMRE than WT mice and that the EMRE levels in 7 month old mice declined even further. These data suggest that a decline in EMRE levels occurs with MICU1 deletion and perhaps this decline allows the mice to survive by reducing mitochondrial Ca2+ overload.
Interestingly mutations and deletions in MICU1 have been reported in patients (62, 63). Human mutations in MICU1 have been associated with muscle dysfunction, ataxia and movement disorders. Lewis-Smith et al (63) reported a MICU1 mutation in patients that led to fatigue and muscle weakness. Mitochondria from these patients have decreased phosphorylation of pyruvate dehydrogenase (PDH) consistent with an increase in mitochondrial [Ca2+]. Debattisti et al (64) developed a skeletal muscle specific MICU1-KO mouse model and showed that these mitochondria had higher basal [Ca2+] but less Ca2+ uptake upon stimulation. They further show that these alterations in mitochondrial Ca2+ regulation result in muscle dysfunction, at least in part, due to impaired sarcolemmal membrane repair, suggesting that mitochondrial Ca2+ is important in membrane repair.
MICU1 also has been reported to have functions in addition to regulating MCU activity. Tufi et al (65) showed in fruit flies that loss of MICU1 is lethal. If MICU1 mediated death is caused by altering mitochondrial Ca2+ uptake one would expect that MICU1 lethality would be rescued by deleting MCU, as loss of MCU per se was not lethal in the fly. However, deletion of MCU did not rescue the lethality of MICU1 deletion. An alternative function for MICU1 has not been established, but MICU1 can localize with the mitochondrial cristae organizing system (MICOS) proteins in the mitochondria (66).
MICU2 and MICU3:
MICU2 is a paralogue of MICU1, but as MICU2 does not bind to MCU or EMRE directly, it cannot complex with MCU in the absence of MICU1 (55). Patron et al (56) reported that MICU1 and MICU2 have opposite effects on MCU mediated Ca2+ uptake; they suggest that MICU2 primarily functions to reduce MCU activity. Bick et al(67) showed that the transcript levels of MICU2 was increased in explanted human hearts with cardiomyopathies. To assess the role of MICU2 in the heart they developed MICU2-KO mice and found that the mice exhibited diastolic dysfunction. The authors suggest that the dysfunction is due to altered threshold due to loss of MICU2, but they also show that loss of MICU2 destabilizes the MCU complex and this further alters Ca2+ homeostasis.
MICU3 is highly expressed in neuronal tissue and enhances mitochondrial Ca2+ uptake. MICU3 associates with MICU1 but not with MICU2 (56). Overexpression of MICU3 in HeLa cells enhanced mitochondrial Ca2+ uptake following treatment with histamine (56). Ashrafi et al (68) showed that MICU3 is necessary in presynaptic mitochondria for the switch to oxidative metabolism.
3.4. EMRE:
Metazoan MCU does not conduct Ca2+ unless EMRE (Essential MCU Regulator) is present (69). A recent structural study shows that EMRE and MCU function in a 1:1 ratio and that metazoan MCU has a juxtamembrane loop that obstructs MCU, and EMRE functions to move this loop and allow Ca2+ transport (70). Kovacs-Bogdan et al (71) showed that MCU from metazoans does not reconstitute Ca2+ transport in S. cerevisiae unless EMRE is co-expressed.
MICU1 binds to EMRE and loss of EMRE leads to disruption of the MCU complex as measured by blue native gels (69, 72). To better understand the role of EMRE in vivo, Liu et al (72) used CRISPR-Cas9 to generate a mouse with loss of EMRE. Similar to what was observed in cell lines, mitochondria from EMRE-KO tissues did not take up Ca2+ and did not undergo PTP. The phenotype of these mice was remarkably similar to that of the germline MCU-KO mice; EMRE-KO mice were not protected from I/R injury and showed little or no impairment in energetic response to isoproterenol.
3.5. Regulation of MCU:
The genetic manipulation studies have made considerable progress in defining the function of MCU and its regulatory subunits. Comparison between tissues shows that the stoichiometry of the MCU complex can vary from organ to organ (54), but there is also evidence that the ratio between MCU and its regulatory subunits within a tissue is not fixed and can be altered under pathophysiologic conditions (49) and can be causally involved in pathological processes such as diabetic cardiomyopathy (73). Studies in the MICU1-KO mouse show that loss of MICU1 leads to changes in EMRE which appear to compensate for the loss of MICU1 (57). The mechanisms by which alterations in MCU and its regulatory proteins occur is an important area for future studies. One mechanism that can affect the MCU to MICU1 ratio is ROS generation by mitochondria, through the redox sensitive transcript factor SP1. Early evidence for this mitochondrial-to-nucleus signaling pathway in the heart was discovered by examination of the effects of a microRNA on mitochondrial function. miR-181c is a product of the nuclear genome, but in heart, its primary target is mt-COX1 in the mitochondrial matrix (74). Overexpression of miR-181c results in a reduction in mt-COX1 protein, an increase in ROS production, a decrease in MICU1 expression, and an increase in mitochondrial matrix Ca2+ concentration (75). Under these conditions, there is oxidation of SP1 at one or more cysteine residues, and SP1 is a transcription factor that regulates MICU1 expression. This process allows for an increase in mitochondrial dehydrogenase activity under conditions where electron transport is impeded by loss of a key electron transport component, and for nuclear regulation of mitochondrial Ca2+ homeostasis under conditions of impaired mitochondrial function. Further work will be needed to clarify additional mechanisms that can regulate the MCU to MICU1 ratio.
MCU can also undergo post translational modifications that can alter its activity. With increased ROS, cysteine-97 of MCU can undergo S-glutathionylation which leads to persistent activation of MCU (76). MCU has also been shown to undergo phosphorylation by CaMKII (77), although the physiological consequences of this are debated (78). It is likely that MCU and other members of the MCU complex are regulated by post-translational modifications and this is an important area for future studies.
3.6. Is MCU the only Ca influx pathway?
Deletion of MCU from mitochondria clearly ablates rapid mitochondrial Ca2+ uptake. However, MCU independent Ca2+ channels have been reported (79). Furthermore, the inducible MCU-KO hearts were able to increase mitochondrial Ca2+ and respond to dobutamine addition if they were treated for 1 hour (43), raising the possibility that an alternative Ca2+ uptake mechanism was activated. In addition, although most mitochondria lacking MCU have little or no measurable Ca2+ uptake, there are reports that loss of MCU does not completely block mitochondrial Ca2+ uptake and that a slow but measurable Ca2+ uptake remains (80). The identity of an alternative slow mitochondrial Ca2+ influx pathway is unclear. Ryanodine receptor 1 (RyR1) (81) have been suggested to be involved in mitochondrial Ca2+ transport, but this pathway has not been established. Uncoupler proteins (UCPs) (82) have been proposed as alternative mitochondrial Ca2+ uptake mechanisms, but this has been questioned (83). This is an area of active research and future studies should shed light on this question.
4. Mitochondrial Ca2+ efflux
In the steady-state the Ca2+ that enters the mitochondria via MCU must exit through one of the mitochondrial Ca2+ efflux mechanisms (see Figure 1). Two mitochondrial Ca2+ efflux mechanisms have been described: a Na+ dependent and a Na+-independent pathway (8, 9). In heart, the rate of efflux of the Na+ dependent pathway was measured as 3 nmol Ca2+/min/mg and the Na+ independent pathways as 2 nmol Ca/min/mg protein (8). The molecular identify of the Na+ dependent transporter was established in 2010. The identity of the Na+ independent mechanism is still debated.
4.1. NCLX
Calcium is extruded from the mitochondria via the mitochondrial Na+-Ca2+-Li+ transporter (NCLX) which couples the efflux of Ca2+ to the influx of Na+ (or Li+) (84). CGP37157 is a well-studied pharmacological inhibitor of NCLX. Matrix Na+ is reported to be lower than cytosolic Na+ (85) and thus the Na+ gradient would facilitate Ca2+ extrusion. The Na+ gradient is maintained by mitochondrial Na+-H+ exchange which extrudes Na+ using the inwardly directed H+ gradient as a driving force (85). NCLX transport is thought to be electrogenic, with 3 Na+ exchanging for 1 Ca2+ (86, 87); thus the large negative mitochondrial membrane potential coupled with the inwardly directed Na+ gradient provides a large driving force for extrusion of matrix Ca2+ (88). NCLX does not appear to operate at electrochemical equilibrium (88). With an electrogenic NCLX, in the presence of a normal mitochondrial membrane potential, it would only operate to extrude Ca2+. It has been suggested that under conditions of I/R, when the membrane potential is dissipated and cytosolic Na+ rises, that NCLX can operate to transport Ca2+ into the mitochondria (89).
Although it was long known that changes in extramitochondrial Na+ can modulate matrix Ca2+, the protein responsible for Ca2+ efflux was only identified in 2010 (3). As discussed in detail in the next section, alterations in cytosolic Na+, as occur with cardiac hypertrophy and heart failure, have been suggested to lead to a decrease in mitochondrial Ca2+ which could reduce activation of mitochondrial dehydrogenases and thus impair bioenergetics (90).
Mice with tamoxifen inducible-cardiac specific deletion of NCLX have been developed and they died within days after acute deletion of NCLX (91) because of acute mitochondrial Ca2+ overload. Mice with cardiac specific overexpression of NCLX have also been generated and hearts from these mice are protected from I/R injury, but interestingly these mice do not have impaired activation of mitochondrial Ca2+ activated dehydrogenases (91).
4.2. Ca2+-H+ exchange
Ca2+ can also be extruded from the mitochondria by a Na+-independent mechanism that has been suggested to occur via a Ca2+-H+ exchanger and uses the energy of the inwardly directed proton gradient. The alkaline pH of the matrix is maintained by electron transport. This exchange is thought to be electroneutral transporting 2H+ for 1 Ca2+ (7). The thermodynamic properties of the Ca2+-H+ exchanger are discussed elsewhere (92). Letm has been proposed as the Ca2+-H+ antiporter (93); however Letm has also been suggested to be a K+/H+ exchanger (94). Additional work will be needed to establish the identity of the Na+ independent Ca2+ efflux transporter. It is interesting that acute deletion of NCLX results in death of the mice, suggesting that NCLX is the major Ca2+ efflux pathway in heart and that with acute deletion of NCLX, the Ca2+-H+ exchanger cannot compensate.
5. Mitochondrial Ca transport as a therapeutic target:
Ca2+ overload is a trigger for PTP opening which has been suggested to be the primary mediator of cell death in the setting of myocardial ischemia (MI) (16). Several small molecule, cell permeable inhibitors of mitochondrial Ca2+ uptake (targeting MCU and MICU1) have been developed and evaluated as possible therapeutic agents to reduce cell death in the setting of Ca2+ overload (95–99). Deletion of MCU in the adult mouse reduces infarct size (43, 44), suggesting that acute inhibition of MCU might be cardioprotective. However acute inhibition of MCU by 1 week overexpression of MCUb was detrimental in an in vivo regional model of I/R, likely because the acute inhibition of MCU blocked the ability of the non-ischemic area to increase work (49). These data raise questions about the therapeutic use of small molecule inhibitors of MCU in MI. Cardiac specific overexpression of NCLX (I/R perfored 6 weeks after tamoxifen) reduced infarct size in an in vivo model of regional I/R (91) suggesting this might be a useful strategy. Overexpression of NCLX for 6 weeks did not impair the response to increased work (91).
Heart failure and hypertrophy are associated with ionic and metabolic changes and energetic deficits and as mitochondrial Ca2+ is important for regulating metabolism and matching ATP production with demand, it has been proposed that alterations in mitochondrial Ca2+ might play a role in the energetic deficits in heart failure. Neither the germline nor the inducible MCU-KO mice showed any difference in development of hypertrophy or heart failure (40, 43), suggesting that mitochondrial Ca2+ uptake is not required for hypertrophy or heart failure; although it is possible that with chronic Ca2+ overload, alternative pathways for Ca2+ entry develop, as 1 hour of adrenergic stimulation leads to an increase in mitochondrial Ca2+ (43).
In spite of the findings that MCU deletion had no effect on the development of transaortic constriction (TAC) induced heart failure in the mouse, there are a number of studies suggesting that alterations in mitochondrial Ca2+ have an important role in heart failure particularly in larger animal models such as the guinea pig. Studies have shown an increase in cytosolic Na+ during hypertrophy and heart failure which alters the equilibrium of NCLX leading to a decrease in matrix Ca2+ and this reduced mitochondrial Ca2+ impairs NADH production and energetics and, by reducing NADPH production, also impairs antioxidant capacity (100). Liu and O’Rourke (24) showed in control guinea pig cardiomyocytes than an increase in pacing leads to an increase in mitochondrial Ca2+ and no change in NADH; however in cardiomyocytes from failing hearts a similar increase in pacing resulted in a decrease in NADH and this decrease in NADH was blocked by addition of the NCLX inhibitor, CGP-37157. In further support of this hypothesis, Liu et al showed that contractile dysfunction and arrhythmias in the guinea pig model of heart failure were attenuated by chronic treatment with the CGP-37157 (101).
Ca2+ leak from sarcoplasmic reticulum (SR) ryanodine receptors has been suggested to play a role in contractile dysfunction associated with failing hearts. Leaky ryanodine receptors (RyR) result in Ca2+ release during diastole and reduced SR Ca2+ load. Interestingly the leaky ryanodine receptors have been reported to contribute to both an increase and a decrease in mitochondrial Ca2+ and both have been suggested to lead to energetic defects. Kohlhaas and Maack (102) show that plasma membrane Na+-Ca2+ exchange (NCX) mediated Ca2+ influx results in less mitochondrial Ca2+ uptake than occurs when an equivalent amount of Ca2+ is released from the SR. This is attributed to the localization of SR release channels in close proximity to mitochondria (103). They suggest that due to the leaky ryanodine receptors, the SR Ca2+ content is reduced resulting in less Ca2+ release from the SR on a beat-to-beat basis and therefore less mitochondrial Ca2+ uptake and less activation of ATP production. In contrast, Santulli et al (104) report in an MI model of heart failure that the SR diastolic Ca2+ leak leads to an increase in mitochondrial Ca2+ which leads to Ca2+ overload and mitochondrial dysfunction. They further show that expression of a RyR2 mutation that reduced the SR Ca2+ leak, blocked the increase in mitochondrial Ca2+ and improved ejection fraction in a post-MI model of heart failure.
In further support for a causal role for increased rather than decreased mitochondrial Ca2+ in mitochondrial dysfunction in heart failure, Luongo et al (91) report that hearts with overexpression of NCLX show improved contractile function at 4 weeks post-MI in a permanent occlusion model. They suggests that overexpression of NCLX is beneficial by reducing mitochondrial Ca2+ overload. Luongo et al (91) find that NCLX overexpression does change the basal matrix Ca2+. It is interesting to consider how overexpression might be beneficial. Does overexpression of NCLX lead to increased Ca2+ extrusion (which would require an increase in Na+ entry into the mitochondria) or does it just increase the rate of Ca2+ removal? One possibility is that overexpression of NCLX protects by more quickly removing Ca2+ from the matrix perhaps competing with some Ca2+ activated process (e.g. PTP opening) that is detrimental.
Thus, there are two opposing sets of data regarding mitochondrial Ca2+ and heart failure: one suggesting that low mitochondrial Ca2+ impairs energetics and another suggesting that elevated mitochondrial Ca2+ impairs energetics. Both too little and too much matrix Ca2+ can be detrimental. Possible explanations for the discrepancy include different models of heart failure or different species. It is also worth noting that Ca2+ handling in the mouse could be different than in species with a slower heart rate. Possible reasons for the discrepancy are discussed in a recent review (105).
Summary and Future Directions:
Since MCU was identified in 2011 there has been a flurry of new information about the complex that regulates Ca2+ uptake. Tissue specific uptake is regulated by altering the ratio of MCU to its regulatory units. Alterations in these regulatory subunits also play a role in physiology and in disease. The mechanisms responsible for regulating the ratio of these regulatory units to MCU are largely unknown and this is an important area for future research. What are the signals responsible for the differences in MICU1/MCU in heart versus liver? What are the signals that regulate the composition of MCU complex? Although it is clear that the MCU complex is the main mechanism responsible for rapid Ca2+ uptake into the mitochondria, there are suggestions that additional uptake mechanisms are present (43). What is the identity of these additional Ca2+ uptake mechanisms and how are they regulated? Details regarding the Ca2+ efflux pathways are also an area for future study. The identity of the Na+ independent Ca2+ efflux pathway and its role in regulating matrix Ca2+ is. Although great progress has been made in the past decade in elucidating mechanisms of mitochondrial Ca2+ transport, there is still much to learn.
References:
- 1.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, et al. 2011. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. 2011. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, et al. 2010. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A 107: 436–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deluca HF, Engstrom GW. 1961. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A 47: 1744–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vasington FD, Murphy JV. 1962. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem 237: 2670–7 [PubMed] [Google Scholar]
- 6.Lehninger AL. 1970. Mitochondria and calcium ion transport. Biochem J 119: 129–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gunter TE, Pfeiffer DR. 1990. Mechanisms by which mitochondria transport calcium. Am J Physiol 258: C755–86 [DOI] [PubMed] [Google Scholar]
- 8.Rizzuto R, Bernardi P, Favaron M, Azzone GF. 1987. Pathways for Ca2+ efflux in heart and liver mitochondria. Biochem J 246: 271–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholls DG, Crompton M. 1980. Mitochondrial calcium transport. FEBS Lett 111: 261–8 [DOI] [PubMed] [Google Scholar]
- 10.Scarpa A, Graziotti P. 1973. Mechanisms for intracellular calcium regulation in heart. I. Stopped-flow measurements of Ca++ uptake by cardiac mitochondria. J Gen Physiol 62: 756–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akerman KE, Wikstrom MK, Saris NE. 1977. Effect of inhibitors on the sigmoidicity of the calcium ion transport kinetics in rat liver mitochondria. Biochim Biophys Acta 464: 287–94 [DOI] [PubMed] [Google Scholar]
- 12.Kirichok Y, Krapivinsky G, Clapham DE. 2004. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427: 360–4 [DOI] [PubMed] [Google Scholar]
- 13.Fieni F, Lee SB, Jan YN, Kirichok Y. 2012. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun 3: 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haworth RA, Hunter DR. 1979. The Ca2+-induced membrane transition in mitochondria: II. Nature of the Ca2+ trigger site. Archives of Biochemistry and Biophysics 195: 460–67 [DOI] [PubMed] [Google Scholar]
- 15.Murphy E, Steenbergen C. 2011. What makes the mitochondria a killer? Can we condition them to be less destructive? Biochim Biophys Acta 1813: 1302–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bauer TM, Murphy E. 2020. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ Res 126: 280–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabo I, Zoratti M. 1992. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem 267: 2934–9 [PubMed] [Google Scholar]
- 18.Halestrap AP. 2009. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46: 821–31 [DOI] [PubMed] [Google Scholar]
- 19.Giorgio V, Burchell V, Schiavone M, Bassot C, Minervini G, et al. 2017. Ca2+ binding to F‐ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Reports 18: 1065–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amanakis G, Sun J, Fergusson M, S M, Liu C, et al. 2020. Cysteine 202 of Cyclophilin D is a site of multiple post-translational modifications and plays a role in cardioprotection. Cardiovasc Res In minor revision [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glancy B, Willis WT, Chess DJ, Balaban RS. 2013. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52: 2793–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denton RM, McCormack JG. 1980. The role of calcium in the regulation of mitochondrial metabolism. Biochem Soc Trans 8: 266–8 [DOI] [PubMed] [Google Scholar]
- 23.Brandes R, Bers DM. 2002. Simultaneous measurements of mitochondrial NADH and Ca(2+) during increased work in intact rat heart trabeculae. Biophys J 83: 587–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu T, O’Rourke B. 2008. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res 103: 279–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chalmers S, Nicholls DG. 2003. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem 278: 19062–70 [DOI] [PubMed] [Google Scholar]
- 26.Nicholls DG. 1978. The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem J 176: 463–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansford RG, Castro F. 1982. Intramitochondrial and extramitochondrial free calcium ion concentrations of suspensions of heart mitochondria with very low, plausibly physiological, contents of total calcium. J Bioenerg Biomembr 14: 361–76 [DOI] [PubMed] [Google Scholar]
- 28.O’Rourke B, Blatter LA. 2009. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol 46: 767–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wescott AP, Kao JPY, Lederer WJ, Boyman L. 2019. Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat Metab 1: 975–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amoedo ND, Punzi G, Obre E, Lacombe D, De Grassi A, et al. 2016. AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim Biophys Acta 1863: 2394–412 [DOI] [PubMed] [Google Scholar]
- 31.Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, et al. 2016. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep 15: 1673–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE. 2016. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A 113: E1872–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. 2015. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab 21: 109–16 [DOI] [PubMed] [Google Scholar]
- 34.Nguyen NX, Armache JP, Lee C, Yang Y, Zeng W, et al. 2018. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature 559: 570–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baradaran R, Wang C, Siliciano AF, Long SB. 2018. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature 559: 580–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoo J, Wu M, Yin Y, Herzik MA, Jr., Lander GC, Lee SY. 2018. Cryo-EM structure of a mitochondrial calcium uniporter. Science 361: 506–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan M, Zhang J, Tsai CW, Orlando BJ, Rodriguez M, et al. 2020. Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature 582: 129–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, et al. 2010. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467: 291–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan X, Liu J, Nguyen T, Liu C, Sun J, et al. 2013. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15: 1464–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmstrom KM, Pan X, Liu JC, Menazza S, Liu J, et al. 2015. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol 85: 178–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, et al. 2015. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A 112: 9129–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, et al. 2015. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun 6: 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, et al. 2015. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep 12: 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, et al. 2015. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep 12: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altamimi TR, Karwi QG, Uddin GM, Fukushima A, Kwong JQ, et al. 2019. Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve. J Mol Cell Cardiol 127: 223–31 [DOI] [PubMed] [Google Scholar]
- 46.Kwong JQ, Huo J, Bround MJ, Boyer JG, Schwanekamp JA, et al. 2018. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parks RJ, Menazza S, Holmstrom KM, Amanakis G, Fergusson M, et al. 2019. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc Res 115: 385–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, et al. 2013. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32: 2362–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, et al. 2019. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huo J, Lu S, Kwong JQ, Bround MJ, Grimes KM, et al. 2020. MCUb Induction Protects the Heart from Post-Ischemic Remodeling. Circ Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, et al. 2013. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8: e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, et al. 2014. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53: 726–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paillard M, Csordas G, Huang KT, Varnai P, Joseph SK, Hajnoczky G. 2018. MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca(2+) Flux and Sensitivity to Ru360. Mol Cell 72: 778–85 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paillard M, Csordas G, Szanda G, Golenar T, Debattisti V, et al. 2017. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca(2+) Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep 18: 2291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamer KJ, Grabarek Z, Mootha VK. 2017. High-affinity cooperative Ca(2+) binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep 18: 1397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D. 2019. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ 26: 179–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, et al. 2016. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep 16: 1561–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, et al. 2016. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat Commun 7: 10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, et al. 2013. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab 17: 976–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, et al. 2012. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151: 630–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, et al. 2016. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca(2+) Uptake Machinery of Skeletal Muscle. Mol Cell 64: 760–73 [DOI] [PubMed] [Google Scholar]
- 62.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, et al. 2014. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet 46: 188–93 [DOI] [PubMed] [Google Scholar]
- 63.Lewis-Smith D, Kamer KJ, Griffin H, Childs AM, Pysden K, et al. 2016. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol Genet 2: e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Debattisti V, Horn A, Singh R, Seifert EL, Hogarth MW, et al. 2019. Dysregulation of Mitochondrial Ca(2+) Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep 29: 1274–86 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tufi R, Gleeson TP, von Stockum S, Hewitt VL, Lee JJ, et al. 2019. Comprehensive Genetic Characterization of Mitochondrial Ca(2+) Uniporter Components Reveals Their Different Physiological Requirements In Vivo. Cell Rep 27: 1541–50 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gottschalk B, Klec C, Leitinger G, Bernhart E, Rost R, et al. 2019. MICU1 controls cristae junction and spatially anchors mitochondrial Ca(2+) uniporter complex. Nat Commun 10: 3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bick AG, Wakimoto H, Kamer KJ, Sancak Y, Goldberger O, et al. 2017. Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc Natl Acad Sci U S A 114: E9096–E104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ashrafi G, de Juan-Sanz J, Farrell RJ, Ryan TA. 2020. Molecular Tuning of the Axonal Mitochondrial Ca(2+) Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron 105: 678–87 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, et al. 2013. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342: 1379–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Y, Nguyen NX, She J, Zeng W, Yang Y, et al. 2019. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 177: 1252–61 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kovacs-Bogdan E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, et al. 2014. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci U S A 111: 8985–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu J, Syder N, Ghorashi N, Willingham T, Parks RJ, et al. 2020. EMRE is essential for mitochondrial calcium uniporter activity in a mouse model. JCI Insight in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ji L, Liu F, Jing Z, Huang Q, Zhao Y, et al. 2017. MICU1 Alleviates Diabetic Cardiomyopathy Through Mitochondrial Ca(2+)-Dependent Antioxidant Response. Diabetes 66: 1586–600 [DOI] [PubMed] [Google Scholar]
- 74.Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, et al. 2012. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res 110: 1596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Banavath HN, Roman B, Mackowski N, Biswas D, Afzal J, et al. 2019. miR-181c Activates Mitochondrial Calcium Uptake by Regulating MICU1 in the Heart. J Am Heart Assoc 8: e012919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, et al. 2017. Mitochondrial Ca(2+) Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol Cell 65: 1014–28 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Joiner ML, Koval OM, Li J, He BJ, Allamargot C , et al. 2012. CaMKII determines mitochondrial stress responses in heart. Nature 491: 269–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nickel AG, Kohlhaas M, Bertero E, Wilhelm D, Wagner M, et al. 2020. CaMKII does not control mitochondrial Ca(2+) uptake in cardiac myocytes. J Physiol 598: 1361–76 [DOI] [PubMed] [Google Scholar]
- 79.Bondarenko AI, Jean-Quartier C, Parichatikanond W, Alam MR, Waldeck-Weiermair M, et al. 2014. Mitochondrial Ca(2+) uniporter (MCU)-dependent and MCU-independent Ca(2+) channels coexist in the inner mitochondrial membrane. Pflugers Arch 466: 1411–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hamilton J, Brustovetsky T, Rysted JE, Lin Z, Usachev YM, Brustovetsky N. 2018. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J Biol Chem 293: 15652–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ryu SY, Beutner G, Kinnally KW, Dirksen RT, Sheu SS. 2011. Single channel characterization of the mitochondrial ryanodine receptor in heart mitoplasts. J Biol Chem 286: 21324–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. 2007. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol 9: 445–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brookes PS, Parker N, Buckingham JA, Vidal-Puig A, Halestrap AP, et al. 2008. UCPs--unlikely calcium porters. Nat Cell Biol 10: 1235–7; author reply 37–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kostic M, Sekler I. 2019. Functional properties and mode of regulation of the mitochondrial Na(+)/Ca(2+) exchanger, NCLX. Semin Cell Dev Biol 94: 59–65 [DOI] [PubMed] [Google Scholar]
- 85.Jung DW, Apel LM, Brierley GP. 1992. Transmembrane gradients of free Na+ in isolated heart mitochondria estimated using a fluorescent probe. Am J Physiol 262: C1047–55 [DOI] [PubMed] [Google Scholar]
- 86.Jung DW, Baysal K, Brierley GP. 1995. The sodium-calcium antiport of heart mitochondria is not electroneutral. J Biol Chem 270: 672–8 [DOI] [PubMed] [Google Scholar]
- 87.Dash RK, Beard DA. 2008. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J Physiol 586: 3267–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murphy E, Eisner DA. 2009. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res 104: 292–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Griffiths EJ, Ocampo CJ, Savage JS, Rutter GA, Hansford RG, et al. 1998. Mitochondrial calcium transporting pathways during hypoxia and reoxygenation in single rat cardiomyocytes. Cardiovasc Res 39: 423–33 [DOI] [PubMed] [Google Scholar]
- 90.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. 2006. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res 99: 172–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, et al. 2017. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 545: 93–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.De Stefani D, Rizzuto R, Pozzan T. 2016. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu Rev Biochem 85: 161–92 [DOI] [PubMed] [Google Scholar]
- 93.Tsai MF, Jiang D, Zhao L, Clapham D, Miller C. 2014. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol 143: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Austin S, Nowikovsky K. 2019. LETM1: Essential for Mitochondrial Biology and Cation Homeostasis? Trends Biochem Sci 44: 648–58 [DOI] [PubMed] [Google Scholar]
- 95.Di Marco G, Vallese F, Jourde B, Bergsdorf C, Sturlese M, et al. 2020. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell Rep 30: 2321–31 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arduino DM, Wettmarshausen J, Vais H, Navas-Navarro P, Cheng Y, et al. 2017. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol Cell 67: 711–23 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kon N, Murakoshi M, Isobe A, Kagechika K, Miyoshi N, Nagayama T. 2017. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov 3: 17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Woods JJ, Nemani N, Shanmughapriya S, Kumar A, Zhang M, et al. 2019. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent Sci 5: 153–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Woods JJ, Wilson JJ. 2019. Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr Opin Chem Biol 55: 9–18 [DOI] [PubMed] [Google Scholar]
- 100.Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, et al. 2010. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121: 1606–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, et al. 2014. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res 115: 44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kohlhaas M, Maack C. 2010. Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation 122: 2273–80 [DOI] [PubMed] [Google Scholar]
- 103.De La Fuente S, Fernandez-Sanz C, Vail C, Agra EJ, Holmstrom K, et al. 2016. Strategic Positioning and Biased Activity of the Mitochondrial Calcium Uniporter in Cardiac Muscle. J Biol Chem 291: 23343–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Santulli G, Xie W, Reiken SR, Marks AR. 2015. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A 112: 11389–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bertero E, Maack C. 2018. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ Res 122: 1460–78 [DOI] [PubMed] [Google Scholar]